
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Native mass spectrometric studies of IscSU reveal a concerted, sulfur-initiated mechanism of iron–sulfur cluster assembly†
Sophie P.
Bennett‡
a,
Jason C.
Crack‡
 *a,
Rita
Puglisi
b,
Annalisa
Pastore
b and
Nick E.
Le Brun
*a
*a,
Rita
Puglisi
b,
Annalisa
Pastore
b and
Nick E.
Le Brun
*a
aCentre for Molecular and Structural Biochemistry, School of Chemistry, University of East Anglia, Norwich Research Park, Norwich, NR4 7TJ, UK. E-mail: j.crack@uea.ac.uk; n.le-brun@uea.ac.uk
bThe Wohl Institute, King's College London, Denmark Hill Campus, London SE5 8AF, UK
First published on 15th November 2022
Abstract
Iron–sulfur (Fe–S) clusters are cofactors essential for life. Though the proteins that function in the assembly of Fe–S clusters are well known, details of the molecular mechanism are less well established. The Isc (iron–sulfur cluster) biogenesis apparatus is widespread in bacteria and is the closest homologue to the human system. Mutations in certain components of the human system lead to disease, and so further studies of this system could be important for developing strategies for medical treatments. We have studied two core components of the Isc biogenesis system: IscS, a cysteine desulfurase; and IscU, a scaffold protein on which clusters are built before subsequent transfer onto recipient apo-proteins. Fe2+-binding, sulfur transfer, and formation of a [2Fe–2S] was followed by a range of techniques, including time-resolved mass spectrometry, and intermediate and product species were unambiguously identified through isotopic substitution experiments using 57Fe and 34S. Under cluster synthesis conditions, sulfur adducts and the [2Fe–2S] cluster product readily accumulated on IscU, but iron adducts (other than the cluster itself) were not observed at physiologically relevant Fe2+ concentrations. Our data indicate that either Fe2+ or sulfur transfer can occur first, but that the transfer of sulfane sulfur (S0) to IscU must occur first if Zn2+ is bound to IscU, suggesting that it is the key step that initiates cluster assembly. Following this, [2Fe–2S] cluster formation is a largely concerted reaction once Fe2+ is introduced.
Introduction
A diverse range of cellular processes, including respiration, central metabolism and DNA repair, rely on proteins containing an iron–sulfur (Fe–S) cluster.1 This versatile prosthetic group can perform multiple functions including in electron transport, transcriptional regulation, redox catalysis, and sulfur donation.1,2 As well as being essential for virtually all bacteria, including pathogens, an increasing number of human diseases are now recognised as being associated with impaired maturation of Fe–S proteins.3,4 Amongst these is the neurodegenerative disorder Friedrich's ataxia, in which a component of the Fe–S cluster biogenesis machinery (frataxin) is deficient or carries mutations that alter its function5 or stability,6 resulting in impaired maturation of respiratory complexes I–III.7 An understanding of this machinery in both prokaryotes and eukaryotes is therefore essential.In bacteria, three well-known systems are responsible for Fe–S cluster assembly and post-translational insertion into apo-proteins: the isc and suf operons encode housekeeping assembly machineries that generate Fe–S clusters under normal and stress conditions, respectively.8,9 The nif operon encodes the system responsible for Fe–S cluster maturation specific to nitrogenase. The Isc assembly machinery, as exemplified by that encoded by the iscRSUA-hscBA-fdx operon in E. coli, is the most widespread system, and has direct orthologues in the eukaryotic mitochondrion.10
IscS and IscU are two essential components of the Isc Fe–S cluster biogenesis apparatus in E. coli. IscS is a cysteine desulfurase with a covalently attached pyridoxal phosphate (PLP) cofactor, which converts L-cysteine to alanine, generating sulfane sulfur (S0).11,12 Homologues of IscS are found in both Suf and Nif systems. The IscS desulfurase in E. coli has a more general role, producing S0 for other processes in addition to Fe–S cluster biogenesis. For example, TusA accepts S0 from IscS and supplies it to a range of biosynthetic pathways, such as molybdenum cofactor biosynthesis and thiomodifications of tRNA.13–15
The cysteine-derived S0 is stored on a conserved cysteine residue in IscS (Cys328 in the E. coli protein), generating a cysteine persulfide, and, in partnership with the scaffold protein IscU, a [2Fe–2S] cluster is assembled on IscU.16–19 A high resolution structure of IscS and IscU in complex revealed a heterotetramer consisting of a dimeric IscS core with two IscU proteins associated at the polar ends,17 see Fig. 1. The in vitro reconstitution of a [2Fe–2S] cluster on IscU from a reaction mixture containing IscS, cysteine, iron and reducing equivalents has been demonstrated.16,20,21 The in vivo source of iron for the cluster is unclear.22–25 IscX and CyaY (the bacterial orthologue of the eukaryotic protein frataxin) bind iron and associate with the same surface on IscS. The affinities of these proteins for IscS depend on the iron concentration, leading to the conclusion that accessible iron within the cell regulates Fe–S cluster biogenesis.22,26 A ferredoxin, Fdx, is also encoded by the isc operon and likely contributes the reducing equivalents needed to release sulfur from IscS to the cluster as sulfide (S2−).27,28
 | ||
| Fig. 1 Structure of (IscS)2(IscU)2 complex. Ribbon diagram representing the structure of Archaeoglobus fulgidus (IscS)2(IscU)2, with PLP and [2Fe–2S] cluster cofactors indicated in space filling mode (PDB 4EB5). | ||
The IscS-mediated assembly of a cluster on IscU is the starting point of a process by which Fe–S cluster proteins are matured. The subsequent steps involve transfer of the assembled [2Fe–2S] cluster from the scaffold to recipient apo-proteins. Additional proteins, such as the transfer proteins IscA and ErpA, and the chaperones HscA and HscB,9,29–32 are involved in this. Alternatively, the [2Fe–2S] cluster bound form of IscU can undergo a reductive coupling transformation, to generate a [4Fe–4S] cluster bound to a IscU dimer.33
IscU is isolated as a variable mixture of apo- and Zn2+-bound forms.34–36 Since Zn2+ and iron–sulfur clusters share similar coordinating ligands (e.g. Cys, His, Asp) in proteins, it has been proposed that zinc directly disrupts preassembled Fe–S clusters of the dehydratase class of enzymes (e.g. aconitase, fumarase).37 Like iron, the amount of ‘free’ intracellular zinc in bacteria is tightly regulated, and for Zn2+ rarely exceeds picomolar levels due to its toxic effects. Although the physiological role of Zn2+–IscU in E. coli remains unclear, binding appears to be tight (estimates of the Kd for the Zn2+–IscU complex range from micromolar38 to sub-picomolar34), and so it is possible that Zn2+–IscU may be relevant in vivo under certain conditions. Indeed, Li et al. recently demonstrated that zinc specifically inhibits IscU/IscA-mediated Fe–S assembly and transfer, rather than disrupting already assembled clusters.39
The precise nature of the events leading to the formation of a cluster on IscU, including Zn2+ removal, Fe2+ and sulfur delivery/insertion, and intermediates formed along the way to the final product have yet to be clearly established. Native (non-denaturing) mass spectrometry, in which non-covalent interactions are preserved, can provide high resolution mass data for protein–protein and protein–cofactor interactions40–44 and has been established recently as a powerful tool for investigating the Fe–S cluster machinery and elucidating chemistry involving protein-bound Fe–S clusters.45–51 Recently, native mass spectrometry was applied to Fe–S biogenesis through studies of the IscSU complex, leading to the identification of intermediate species and the proposal of an overall ‘iron-first’ mechanism.52
Here, through the application of native mass spectrometry together with other biophysical techniques, we provide further insight into the cluster assembly process involving IscS and IscU, by addressing questions relating to: the conformational flexibility of IscU, and its interaction with IscS; Fe2+- and S0-binding to IscU, and the importance of the presence of Zn2+ on IscU; the possibility of detecting intermediate Fe/S species during cluster assembly on IscU. Our data support the dynamic interconversion of IscU conformational states in the absence of IscS, and the effect of bound Zn2+ ions on this equilibrium. We also determine affinities of IscU for Fe2+ and for IscS, and demonstrate that Zn2+ binding prevents Fe2+-binding to IscU, thus inhibiting FeS assembly. Furthermore, the removal of Zn2+ requires IscS-mediated S0 transfer, and the ensuing mechanism of IscS-mediated [2Fe–2S] cluster assembly on IscU is largely concerted, with no significant accumulation of intermediates.
Materials and methods
Protein production
All proteins used in this study are from E. coli. Their corresponding DNA sequences were subcloned into a pET-24d vector, encoding fusion proteins with His-tagged glutathione-S-transferase (GST), and used to transform E. coli BL21 DE3. Cultures for the overproduction of IscU were grown in Luria Broth enriched medium containing 8.3 mg per L ZnSO4 (ref. 20) to help stabilize the folded form. IscS was expressed in the presence of 100 μM PLP (Sigma). The proteins were purified by affinity chromatography using a His-Trap column (GE Healthcare) and cleaved from the His-GST tag by tobacco virus protease (TEV) protease. The cleaved protein solution was reloaded onto a His-Trap column to separate the His-tagged GST from IscU.20Protein purity was confirmed by SDS-PAGE and liquid chromatography mass spectrometry (LC-MS). Protein concentrations were determined by absorbance at 280 nm using ε280 nm of 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 370 or 11460 M−1 cm−1 for IscS and IscU, respectively. IscU was treated with either a 25-fold excess of diethylene triamine pentaacetate (DTPA) to chelate Zn2+,53 or a 2-fold excess of ZnSO4 to produce a fully zinc-loaded form of IscU,34 before exchanging the protein into 50 mM Tris, 150 mM NaCl, pH 8.0 using a desalting column (PD10, Cytiva). Metal ion contents of IscU samples were determined by inductively coupled plasma-mass spectrometry (ICP-MS) using an iCAP-TQ (Thermofisher Scientific) instrument. Aerobically purified IscU contained Zn2+ (∼0.5 per protein), as previously observed,34,36 while DTPA-treated IscU contained ∼0.01 Zn2+ per protein.
370 or 11460 M−1 cm−1 for IscS and IscU, respectively. IscU was treated with either a 25-fold excess of diethylene triamine pentaacetate (DTPA) to chelate Zn2+,53 or a 2-fold excess of ZnSO4 to produce a fully zinc-loaded form of IscU,34 before exchanging the protein into 50 mM Tris, 150 mM NaCl, pH 8.0 using a desalting column (PD10, Cytiva). Metal ion contents of IscU samples were determined by inductively coupled plasma-mass spectrometry (ICP-MS) using an iCAP-TQ (Thermofisher Scientific) instrument. Aerobically purified IscU contained Zn2+ (∼0.5 per protein), as previously observed,34,36 while DTPA-treated IscU contained ∼0.01 Zn2+ per protein.
For LC-MS, a 20 μM aliquot of protein was diluted with an aqueous mixture of 2% (v/v) acetonitrile, 0.1% (v/v) formic acid, and loaded onto a Proswift RP-1S column (4.6 × 50 mm, Thermo Scientific) attached to an Ultimate 3000 uHPLC system (Dionex, Leeds, UK) with eluant infused into the electrospray ionisation (ESI) source of a Bruker micrOTOF-QIII mass spectrometer (Bruker Daltonics) operating in the positive ion mode. Processing and analysis of MS experimental data were carried out using Compass DataAnalysis v4.1 (Bruker Daltonik). Neutral mass spectra were generated using the ESI Compass v1.3 Maximum Entropy deconvolution algorithm. LC-MS of purified isolated IscU gave a mass of 13974 Da, which corresponds to the predicted mass of the apo-protein with a −2 Da mass shift, indicating that Zn2+ was lost under the denaturing conditions of the LC-MS experiment, and an intra-molecular disulfide bond had formed (see Table 1).
| Protein species | Predicted mass (Da) | Average observed massa (Da) | ΔMassb (Da) | 57Fe and 34S isotope shift (Da) |
|---|---|---|---|---|
| a The average observed mass is derived from at least three independent experiments, with standard deviation of ±1 Da. b The difference between the average observed and predicted masses. | ||||
| LC-MS | ||||
| IscU | 13976 |
13974 |
−2 | |
| IscS | 45288 |
45288 |
0 | |
|
||||
| Native MS | ||||
| IscU (aerobic) | 13976 |
13974 |
−2 | |
| IscU (anaerobic) | 13976 |
13976 |
0 | |
| (IscU)2 | 27952 |
27946 |
−6 | |
| Zn2+–IscU | 14039 |
14040 |
+1 | |
| Fe2+–IscU | 14030 |
14030 |
0 | |
| Zn2+–, Fe2+–IscU | 14092 |
14093 |
+1 | |
| (Fe2+)2–IscU | 14084 |
14084 |
0 | |
| [2Fe–2S]–IscU | 14150 |
14151 |
+1 | S: +4 |
| Fe: +2 | ||||
| S–IscU | 14008 |
14007 |
−1 | S: +2 |
| (S)2–IscU | 14040 |
14040 |
0 | S: +2 |
| (S)3–IscU | 14072 |
14072 |
0 | S: +4 |
| (S)4–IscU | 14104 |
14104 |
0 | S: +6 |
| Fe3+–, S2−–IscU | 14061 |
14061 |
0 | |
| Fe2+–, S2−–, S0–IscU | 14094 |
14094 |
0 | |
| Zn2+–, S–IscU | 14070 |
14071 |
+1 | |
| Zn2+–, (S)2–IscU | 14102 |
14103 |
+1 | |
| SO42−–IscU | 14074 |
14075 |
+1 | — |
| Zn2+–, SO42−–IscU | 14137 |
14138 |
+1 | — |
| (IscS)2 (with PLP bound) | 91038 |
91036 |
−2 | — |
Cluster reconstitution on IscU
All enzymatic IscU Fe–S cluster reconstitution experiments were performed in an anaerobic chamber (Belle technology) under nitrogen atmosphere (O2 < 2 ppm). Catalytic amounts of IscS were initially used because at higher concentrations, sulfur adducts of +32 Da accumulated on IscU to an extent that made cluster assembly much more difficult to follow. IscU concentrations were kept below 15 μM because higher concentrations were found to block the nebulizer needle when conducting native mass spectrometry. Hence, optimised reaction mixtures contained 0.2 μM IscS, 10 μM IscU, 20 μM ferrous ammonium sulfate, 50 μM L-cysteine and 2 mM DTT, unless otherwise stated. Isotopic substitution experiments were conducted using 34S-cysteine and 57Fe, as previously reported.46,54 The use of ammonium acetate as volatile buffer was found to inhibit cluster reconstitution, possibly due to competition for Fe2+-binding, or for the cysteine binding site on IscS, and so ammonium formate was used instead. Cluster assembly was also followed by measuring A456 nm as a function of time using a Jasco V550 spectrophotometer,20,21 or by circular dichroism measurements made using a Jasco J-810 CD spectropolarimeter.55The effect on cluster assembly of pre-loading of IscU with S0 or Fe2+, or Zn2+/Fe2+, was investigated. Apo-IscU (∼200 μM) containing S0 was prepared as previously described.56 Briefly, IscU was treated with 10 μM IscS and 3 mM L-cysteine under aerobic conditions and incubated at 37 °C for 1 h, prior to desalting (PD10, Cytiva). S0 loading was confirmed by routine LC-MS. Fe2+-loading was achieved by treating apo-IscU with a 30-fold excess of ferrous ammonium sulfate under anaerobic conditions. The solution was incubated at an ambient temperature for 15 min, prior to desalting (PD10, Cytiva). For sequential Zn2+/Fe2+ treatment, IscU was first treated with a 30-fold excess of zinc sulfate in the presence of 3 mM DTT under anaerobic conditions. The resulting solution was incubated at an ambient temperature for 15 min, prior to desalting (PD10, Cytiva). The sample was then treated with a 30-fold excess of ferrous ammonium sulfate, as described above, then desalted.
IscU treated with S0, Fe2+ or sequential Zn2+/Fe2+ was then used as the sole source of sulfur or iron for in vitro Fe–S assembly. Where IscU (∼50 μM) served as the sulfur source, Fe–S assembly was initiated following the addition of 0.17 mM ferrous ammonium sulfate in the presence of absence of 2 mM DTT, GSH, or L-cysteine. Where IscU (∼50 μM) served as the iron source, Fe–S assembly was initiated following the addition of 5 μM IscS and 2 mM L-cysteine. The buffer was 100 mM Tris, pH 7.5. Fe–S cluster assembly was followed by UV-visible absorbance. IscU-bound Fe2+ was determined via Ferrene™ (Merck)57 and bound Zn2+ was determined via 4-(2-pyridylazo)resorcinol (Merck).58
For non-denaturing (native) mass spectrometry measurements, Isc proteins were buffer exchanged at room temperature in an anaerobic chamber into ammonium formate or ammonium acetate, volatile buffers that assist with desolvation of proteins as they transition into the gas phase. Samples were then loaded into a 500 μL gas-tight syringe and infused directly in a Bruker micrOTOF-QIII mass spectrometer (Bruker Daltonics) operating in the positive ion mode. The ESI-TOF was calibrated online using ESI-L Low Concentration Tuning Mix (Agilent Technologies) and, where necessary, subsequently re-calibrated offline in the 4000–8000 m/z range using a 4.6 mM caesium perfluoroheptanoate mix. Data were acquired over the m/z range of 600–3000 for IscU with acquisition controlled by Bruker qTOFControl software, with parameters as follows: dry gas flow 4 L min−1, nebulise gas pressure 0.8 Bar, dry gas 180 °C, capillary voltage 3000 V, offset 500 V, quadrupole voltage 5 V, collision RF 650 Vpp. The m/z acquired data were averaged over the given time period and the neutral mass spectrum calculated using the ESI Compass v1.3 Maximum Entropy (MaxEnt) deconvolution algorithm, taking into account all charge states detected in the 600–3000 m/z region. Average resolution for this range was 20000 FWHM. For IscS and IscS complexes, a m/z range of 4000–8000 was used to acquire data, with parameters as follows: dry gas flow 4 L min−1, nebulise gas pressure 0.8 Bar, dry gas 180 °C, capillary voltage 3000 V, offset 500 V, quadrupole voltage 5 V, collision RF 2650 Vpp, collision cell voltage 10 eV, isCID energy 120 eV. Deconvolution was performed as described above but over the mass range 4000–8000 m/z. Average resolution for this range was 10500 FWHM. Isotopic exchange experiments with 57Fe and 34S were performed as previously described.45,5434S was supplied as 34S-cysteine, synthesised using a thermostable cysteine synthase as previously described.54
Determination of Fe2+-binding affinity via competition assay
The dissociation constant (Kd) for Fe2+-binding to apo-IscU was determined using an Fe2+-binding competition assay employing the divalent metal ligand mag-fura-2, as previously described.48,59 Mag-fura-2 forms a 1:1 complex with Fe2+, resulting in a shift of absorbance maximum from 366 nm (metal free) to 325 nm (Fe2+ bound).59 Briefly, apo-IscU (at ∼5 μM or 12 μM) was mixed with 5 μM mag-fura-2 in 1 mL of buffer (20 mM HEPES, 150 mM NaCl, pH 7.5, or 200 mM ammonium formate, pH 8.0) under anaerobic conditions. Incremental amounts of Fe2+ (as ferrous ammonium sulfate) were added until binding saturation was achieved. Binding data were simultaneously fitted with Dynafit using eqn (1) and (2):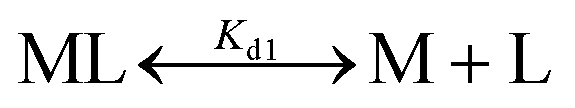 | (1) |
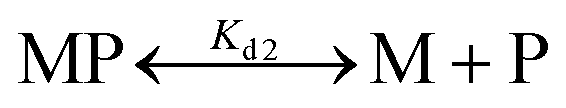 | (2) |
The effect of ESI on the Fe2+-binding affinity of IscU
Solutions of apo-IscU (15.5 μM), in 200 mM ammonium formate, 0.94 mM DTT, pH 8, were mixed with increasing concentrations of iron(II) formate (0–100 IscU/Fe2+ molar ratios). Samples were loaded into a 500 μL gas-tight syringe (Hamilton) and infused directly into the ionisation source of the mass spectrometer. Data were acquired (750–4000 m/z region) for 5 min, averaged, and the neutral mass spectrum calculated (9000–38000 Da). For the determination of apparent dissociation constants, ion counts for Fe2+–IscU were compared with the combined ion counts for apo-IscU, and IscU +32 Da (S or 2O), +64 Da (Zn), and +54 Da (Fe). At least two datasets were acquired, expressed as fractional saturation, then averaged. The response of Fe2+–IscU in the average data set was fitted using eqn (2), from which an apparent Kd for Fe2+ binding under conditions of ESI-MS was estimated. A stock solution of iron(II) formate (67 mM) was prepared under anaerobic conditions by reacting ferrous ammonium sulfate with excess sodium bicarbonate, resulting in Fe(II)CO3 precipitate. This was isolated, reacted with formic acid, and diluted in 200 mM ammonium formate pH 8.0. The solution of formate was further diluted to 1.7 mM just prior to use.
IscS-free reconstitution of Fe–S–IscU
The lack of cluster intermediate species during IscS-catalysed reconstitutions, might have been due to a component of the reaction mixture affecting ionisation; thus, we sought to simplify the reconstitution by pre-generating a source of S0. 23.4 mM elemental sulfur (S0) was solubilised overnight in 2 mL of 200 mM ammonium acetate pH 7.0 containing 36.6 mM DTT and 200 mM NH4OH (final ratio of S0 to DTT, 1:1.6).60–62 The resulting solution was used as a low molecular weight IscS mimic (DTT-S), see Scheme S1.† To initiate reconstitution, IscU (10.5 μM) in 200 mM ammonium formate pH 8.0 was pre-treated with a 54-fold excess of DTT-S (0.56 mM, final conc.) for ∼50 s and then, immediately before infusion, the sample was treated with an ∼18-fold excess (0.18 mM, final conc.) of (NH4)2Fe(II)(SO4)2 in DTT (1.8 mM, final conc.) and mixed. Data were acquired from ∼20 s post iron addition for up to 5 min, averaged (every 0.3 min), and the neutral mass spectrum calculated (9000–38000 Da). Zn2+–IscU served as an internal standard and the ion counts combined with those for apo-IscU, and IscU +32 Da (S), +54 Da (Fe), +86 Da (Fe–S), +108 Da (Fe2), +118 Da (Fe–S2), +153 Da (DTT), +174 Da (Fe2S2). At least two datasets were acquired, expressed as fractional saturation, and then averaged.
IscS–IscU interaction measurements
To evaluate the Kd of the IscSU complex by native mass spectrometry, an aliquot of purified protein (as isolated IscU or IscS) was first exchanged into 250 mM ammonium acetate pH 8.0 using desalting columns (PD MiniTrap G25, Cytiva). The protein concentration was determined by absorbance and working solutions (200 μL) were prepared by combining aliquots of IscS and IscU to achieve the desired concentration or IscS/IscU ratio, then infused directly into the source of a Waters Synapt XS (Waters Inc.) mass spectrometer operating in the positive ion mode. Data were acquired and processed with Mass Lynx v4.2 (Waters Inc.) over the m/z range of 3000 to 8000 m/z with parameters as follows: dry gas flow 4 L min−1, nebuliser gas pressure 6 Bar, dry gas 150 °C, source temperature 80 °C, capillary voltage 3100 V, offset 30 V, cone voltage (isCID) 150 V, trap collisional energy 10 eV, and transfer collision energy 2 eV. Spectra were averaged, and the neutral mass spectrum calculated (90–125 kDa) using the MaxEnt1 deconvolution algorithm of MassLynx v4.2 (Waters Inc.). At least two datasets were acquired, expressed as fractional saturation, and then averaged prior to fitting with Dynafit (Biokin), as previously described.22 The mass spectrometer was calibrated with sodium iodide. Average resolution for the measured range was 20000 FWHM.
Results
IscU exists as an equilibrium between folded and disordered states in the absence of IscS
Native mass spectrometry was used to study IscU. Mass spectra for apo-IscU and Zn2+–IscU under aerobic conditions are shown in Fig. 2. Zn2+–IscU exhibited two dominant charge states of 7+ and 8+, which were also present in the spectrum of apo-IscU, along with a second envelope of charge states at lower m/z values (charge states 9+ to 17+, Fig. 2A). This indicated the presence of two conformational states of this form of the protein. Deconvolution across all charge states gave a spectrum with apo-IscU as the major species (at a mass of 13974 Da, corresponding to oxidised IscU containing one disulfide bond). The two apo-IscU charge state envelopes were also deconvoluted separately (Fig. S1†), revealing that the more highly charged ions arise from apo-IscU, while the 7+ and 8+ charge states arise from apo-IscU and the small component of Zn2+-bound IscU present in the sample.
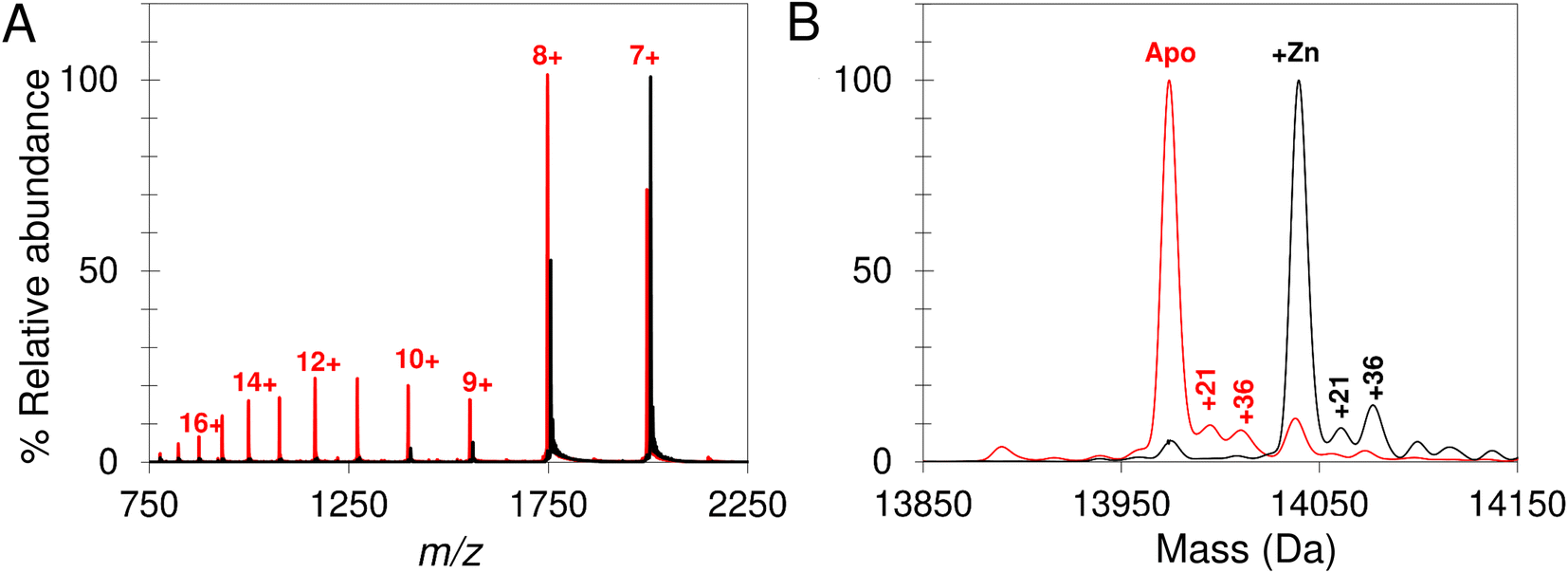 | ||
| Fig. 2 Apo- and Zn2+–IscU have different charge distributions. The acquired m/z spectrum (A) and deconvoluted neutral mass spectrum (B) for apo-IscU (<1% Zn2+, in red) and Zn–IscU (100% Zn2+, black) by non-denaturing mass spectrometry, in 200 mM ammonium formate, pH 8.0. Charge states for IscU are noted in red (A). Adducts masses, noted in Daltons, correspond to Na (+21) and Cl or Na/O (+36) (B). | ||
The similarity between the higher charge state envelop and that observed using LC-MS (where the protein was unfolded), indicates that this form of the protein is less well folded. This is also consistent with the tendency of folded proteins to exhibit lower charge states compared to unfolded forms because fewer amino acid residues are available for protonation. In addition to monomeric oxidised IscU, a dimeric form of apo-IscU was also observed by native MS, containing an inter-molecular disulfide (Fig. S2A†). Addition of excess DTT and measurement after 1 min revealed the almost complete loss of the dimeric form, and an increase in reduced monomeric IscU in the D state (Fig. S2B†). Re-measurement after 5 min indicated the re-equilibration of the sample with an increase in S state relative to D state monomeric IscU (Fig. S2C and D†).
Apo-IscU was then prepared under anaerobic conditions (in preparation for cluster reconstitution) and native MS recorded. Surprisingly, a peak at +64 Da was found to dominate the deconvoluted mass spectrum, which could arise from either Zn2+ or two sulfurs bound to IscU. Reducing agents such as DTT or TCEP typically remove sulfur adducts but, in this case, there was no effect on the +64 Da adduct. Furthermore, LC-MS did not reveal any significant covalent adducts of IscU (sulfur adducts, multiples of +32 Da, are covalently attached as persulfides and survive LC-MS conditions).50,51,63 Thus, we conclude that the IscU +64 Da peak is not due to sulfur, and must be due to Zn2+, even though analysis had demonstrated that the protein contained ≤0.01 Zn2+ per protein (Fig. 3).
 | ||
| Fig. 3 Anaerobic/aerobic cycling of IscU modulates Zn2+-binding. Deconvoluted neutral mass spectrum of apo-IscU prepared under anaerobic conditions (red), exposed to atmospheric O2 for 4 hours (grey) and made anaerobic again by incubation in an anaerobic chamber for 2 h (black). Inset is a plot of the apo-IscU mass region, illustrating that a −2 Da shift occurs open exposure to O2, which is reversed upon return to anaerobic conditions. | ||
The unexpected prevalence of the Zn2+-bound form of IscU in the spectrum of apo-IscU under anaerobic conditions, compared to what was observed under aerobic conditions (Fig. 2), suggested that the redox state of the protein could be important. Thus, an anaerobically treated IscU sample was exposed to air for 4 h and the native mass spectrum re-measured (Fig. 3). Following introduction of air, the peak due to apo-IscU was observed to be the only major species. Exposure of Zn2+-loaded IscU to air had no effect on the mass spectrum, suggesting that the effect is confined to samples principally containing apo-IscU.
The O2-exposed IscU sample was then returned to the anaerobic glove box for 2 h (Fig. 3); the resulting deconvoluted native mass spectrum was similar to that recorded initially for the anaerobic sample, with the Zn2+-form once again the more prevalent. The mass of the apo-IscU peak following O2 exposure (13974 Da) indicated oxidation of the protein via the introduction of one disulfide bond, resulting in a −2 Da mass shift. Removal of O2 essentially restored the peak to the position of fully reduced IscU (see inset Fig. 3). These data illustrate that these different forms of IscU can be cycled.
Further evidence that the adduct is due to Zn2+ was derived from the m/z spectrum of the aerobic, oxidised sample (corresponding to a mix of S and D states), for which charge states between 500 and 2500 m/z were observed, while under anaerobic conditions the most intense peaks were the 7+ and 8+ charge states (corresponding to the S state).
To rationalise the above observations, it is important to acknowledge that mass spectrometry under native conditions is not a quantitative technique in terms of comparing signal intensities between two species. This is because of the variability in ionisation efficiency, where one species may ionise much more readily than another. The IscU data could arise from the much more efficient ionisation of the Zn2+-bound form relative to the apo-form, or more specifically, reduced apo-IscU. For closely related species, very different ionisation efficiencies would be unusual. In this case, it is known that Zn2+-binding to IscU stabilizes the structured state, and so the changes in IscU due to Zn2+-binding are more significant than only the binding of a metal ion. The formation of an intra-molecular disulfide bond upon exposure to O2 could stabilise IscU.
Another possibility is that reduced IscU, which binds Zn2+ with high affinity,34 is able to scavenge Zn2+ from the anaerobic glove box environment, or from buffer components/DTT (e.g. ref. 64). In such a situation, the apparent loss of Zn2+–IscU upon exposure to O2 could result from displacement of Zn2+ upon oxidation of coordinating Cys thiols to a disulfide.65 However, similar anaerobic/aerobic experiments with IscU containing 0.5 or 1 Zn2+ per protein did not result in significant changes in the spectrum, indicating that displacement of Zn2+ upon exposure of IscU to O2 does not alone account for the observed mass changes above. Thus, the behaviour of IscU-derived peaks in native MS spectra upon redox cycling most likely arises from the suppression of reduced IscU ionisation by Zn2+–IscU in combination with some degree of scavenging of Zn2+.
Overall, we conclude that in mixtures of reduced, apo-IscU and Zn2+–IscU, native MS signals due to the latter are more intense than predicted from its solution abundance. Both structured (S) and disordered (D) conformational states of IscU are present under the conditions of the native mass spectrometry experiment. Zn2+–IscU is structured, while apo-IscU exists as an equilibrium mixture of S and D forms, consistent with previous studies that showed that Zn2+ stabilises the folded conformation of IscU.34,36,66
Fe2+-binding to IscU
To determine the stability of Fe2+–IscU, we used the well characterised Fe2+–chelator mag-fura-2 in a competition assay (Fig. 4A), in which binding was monitored by optical titration, as previously described.48,59 (Fig. 4B). The resulting dissociation constants for Fe2+–mag-fura-2 and Fe2+–IscU were Kd = 2.0 ± 0.2 μM and Kd = 3.5 ± 0.2 μM (Fig. 4B), respectively, in close agreement with previously published binding constants for mag-fura-2 and IscU from prokaryotic or eukaryotic sources.59,67–69 | ||
| Fig. 4 Determination of the binding affinity of Fe2+–IscU. (A) Spectra showing Fe2+ binding to mag-fura-2 (5 μM) in competition with apo-IscU (5 μM) in 20 mM HEPES, 150 mM NaCl pH 7.5, leading to a transition from apo- (λmax 366 nm) to Fe2+–mag-fura-2 (λmax 325 nm). Arrows indicate direction of absorbance changes for apo- and Fe2+–mag-fura-2, [L] and [ML], respectively. An equivalent titration of mag-fura-2 alone with Fe2+ was performed as a control experiment from which the affinity of Fe2+ for mag-fura-2 was confirmed as Kd = 2.0 (±0.2) μM. (B) Absorbance changes at measured at 325 nm (red points) and 366 nm (black points) following the addition of increasing concentrations of Fe2+ to mag-fura-2 in competition with 5.0 μM (closed circles, solid lines) and 12.4 μM (open triangles, dashed lines). The data were simultaneously fitted in Dynafit using a simple binding equation, see Results, giving an apparent Kd of 3.5 (±0.2) μM for IscU. (C) Deconvoluted mass spectrum of apo-IscU (9 μM) titrated with Fe2+–formate and ionised from 200 mM ammonium formate pH 8. We note that a proportion of apo-IscU remains at the end of the titration in an oxidised form that appears unable to bind Fe2+. The species at 14044 Da is unknown. (D) Plot of fractional intensity due to Fe2+–IscU as a function of Fe2+–formate added. The dissociation constant for Fe2+–IscU under these conditions was Kd ∼ 73 μM. | ||
Next, we sought to determine the affinity of apo-IscU for Fe2+ under native MS conditions, to evaluate whether the presence of high concentrations of formate interferes with Fe2+-binding (Fig. S3†). Thus, similar experiments as those above were performed, except that 200 mM ammonium formate pH 8.0 was used. No effect of changing the buffer was observed when Fe2+ was titrated in to ammonium formate solutions containing mag-fura-2 alone (Fig. S3A and B†), or containing apo-IscU (Fig. S3C–F†), confirming that formate does not compete for Fe2+ under these conditions.
We then attempted to measure Fe2+-binding directly using native MS. Here, apo-IscU in ammonium formate was titrated with increasing amounts of Fe2+, and the formation of Fe2+–IscU followed by native MS (Fig. 4C). The apparent affinity constant for Fe2+–IscU under these conditions was determined as Kd = 73 ± 13 μM (Fig. 4D), ∼20-fold lower affinity than that measured by in solution competition assay (see above). Thus, while there are many examples in which behaviour of proteins in the gas phase closely mimics that in solution, in this case there is a clear effect on the affinity of IscU for Fe2+ in the gas phase. The consequence of this is that it is more difficult than expected to observe Fe2+-bound forms of IscU by native MS.
Addition of a 10-fold excess of Fe2+ over apo-IscU resulted in a small peak that could be due to Zn2+/Fe2+-bound IscU, along with sulfate adducts (Fig. 5). At a 50-fold excess of iron, the peak due to a possible Zn2+/Fe2+ adduct was much more clearly resolved, while at a 100-fold excess of iron, IscU species with one and two irons bound (at 14030 and 14083 Da, respectively) were observed along with a loss of the Zn2+/Fe2+ peak. The m/z spectrum of IscU with a 100-fold excess of Fe2+ contained the lower charge states (7+, 8+) only, indicating stabilization of IscU in the S state by Fe2+-binding (Fig. S4†).
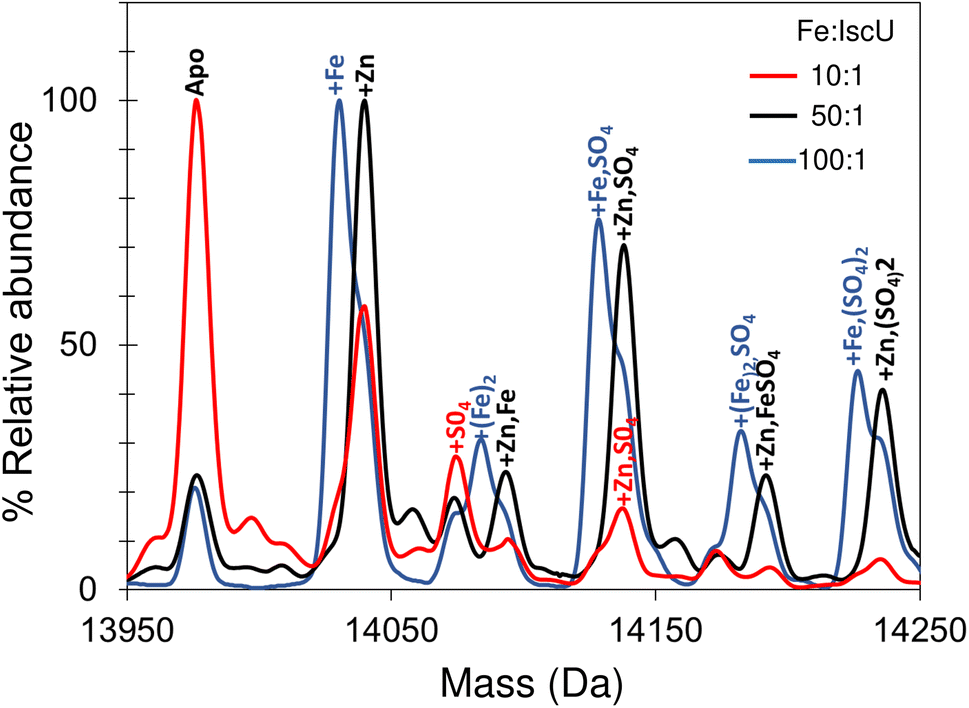 | ||
| Fig. 5 Iron binding to IscU. Incubation of IscU in 200 mM ammonium formate, with 2 mM DTT and a 5, 50 and 100-fold excess of ferrous ammonium sulfate. Fe2+-bound forms of IscU, along with sulfate adducts, are detected. | ||
Interactions between IscS and IscU
We then looked into the interactions between IscU and IscS. The Kd of the IscU–IscS complex in solution has been determined to be 1–2 μM by isothermal calorimetry,19,70 consistent with the dynamic role of these two proteins play in Fe–S cluster assembly (IscU–IscS) and Fe–S delivery (IscU). Under native MS conditions, the m/z spectrum for as isolated IscS (Fig. S5†) contained four principal charge states (17+ to 20+), which upon deconvolution gave a mass of 91036 Da, in close agreement with that expected for a dimer of IscS with PLP bound to each protomer (Fig. S5B,†Table 1). Addition to IscS of a 2-fold excess of IscU resulted in significant changes in the m/z spectrum. The deconvoluted spectrum (spanning 90–125 kDa) revealed the presence of a new species, corresponding to dimeric IscS in complex with up to two IscU molecules in their structured form, Fig. 6A.
 | ||
| Fig. 6 ESI-MS investigation of complex formation between IscS and IscU. (A) Deconvoluted mass spectra of IscS over the mass range 90–125 kDa, showing the presence of the IscS dimer (black spectrum). Addition of IscU at a 2:1 excess (as indicated) gave rise to a series of IscU–IscS complexes in which the IscS dimer is bound by 1–2 IscU molecules. Asterisks indicate unknown species that may be due to complexes of IscS with degraded forms of IscU. (B) Plots of fractional intensity of the two IscU–IscS complexes, as indicated, as a function of IscU concentration. Solid lines show fits of the data to a sequential binding model for 1–2 IscU per IscS dimer. The binding of one or two IscU molecules to dimeric IscS occurred with a similar affinity, Kd of 3.26 ± 0.46 μM. (C)–(E) Representative deconvoluted mass spectra at increasing ratios of IscU to IscS showing the formation/decay of the IscU–IscS complexes, as indicated. An adduct species (+154 or +308 Da) is present in each of the spectra, including that of the IscS dimer, indicating that it originates from IscS. The precise nature of the adduct is unknown, but it is likely to arise from β-mercaptoethanol or DTT hetero-disulfides with IscS, as previously observed.22 IscS (8 μM) and IscU were in 250 mM ammonium acetate pH 8. | ||
Titration of IscS with increasing concentrations of as isolated IscU resulted in the gradual formation of dimeric IscS complex containing 1 or 2 IscU molecules ((IscS)2(IscU) or (IscS)2(IscU)2, Fig. 6B–E). (IscS)2(IscU) formed readily at low levels of IscU (IscU/IscS ≈ 0.5) and maximised at IscU/IscS ≈ 1.0, with the concomitant loss of un-complexed dimeric IscS. The (IscS)2(IscU)2 complex was evident at IscU/IscS ≈ 1.0 and became the dominant species at an IscU/IscS ≈ 8.0 Fig. 6C–E. The data were analysed according to a sequential binding model. The resulting data fit revealed that binding of one or two IscU molecules to dimeric IscS occurred with a similar affinity, Kd = ∼3 μM, consistent with solution studies (Fig. 6B).70 As the binding behaviour is well described by a single average dissociation constant, there is no evidence for cooperativity, suggesting that the IscS dimer contains two equivalent but independent binding sites for IscU.
To investigate if IscS had a preference for apo-IscU or Zn2+–IscU, they were separately added to IscS and native mass spectrometry measurements performed at fixed IscS–IscU ratios. Deconvoluted spectra revealed that both monomeric apo- and Zn2+-forms of IscU interact with the IscS dimer to form either (IscS)2IscU and/or (IscS)2(IscU)2 complexes (Fig. S6†). We also investigated whether the presence of IscS influences the binding of Fe2+ to IscU. A 100-fold excess of iron (as described above) was added to apo-IscU in the presence of IscS, both at catalytic concentration and at a 2:1 ratio of IscU:IscS (but in the absence of substrate cysteine). Up to two irons per IscU monomer were observed (Fig. S7†), as in the absence of IscS, indicating that the presence of IscS does not significantly affect the affinity of IscU for iron. Thus, the stability of the IscS–IscU complex(es) and binding of metal ions on IscU do not significantly affect one another. Similarly, binding of IscU to one IscS of the dimer does not affect binding of IscU at the other.
Reconstitution of [2Fe–2S] cluster on IscU monitored by native mass spectrometry
Initially, volatile buffer compositions and protein concentration that supported Fe–S cluster reconstitution were established and then optimized. Briefly, catalytic quantities of IscS were used initially (IscS:IscU ratio of 1:50), with apo-IscU in ammonium formate. Under these conditions cluster formation was partially inhibited compared to that observed in a typical Tris-buffered reconstitution, as monitored by absorbance/CD spectroscopy (Fig. 7A and 8A).70 Volatile buffers containing acetate were found to inhibit Fe–S cluster assembly, consistent with acetate inhibition of other PLP dependent enzymes;71 we note Lin et al. reported a 4-fold decrease in the desulfurase activity of IscS exchanged into ammonium acetate.52
 | ||
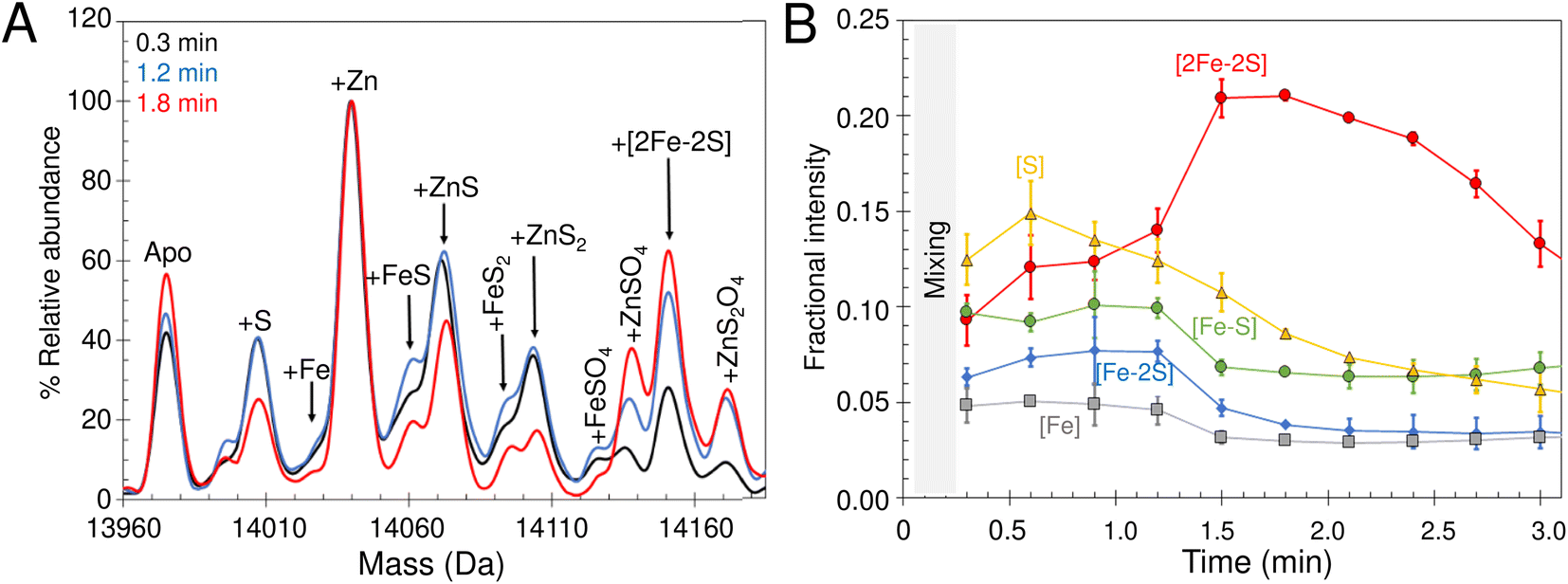
| Fig. 7 Synthesis of a [2Fe–2S] cluster on IscU. (A) Reconstitution of apo-IscU (10 or 5 μM, as indicated) in 50 mM Tris, 150 mM NaCl, pH 8.0, (darker green) or 200 mM ammonium formate pH 8.0 (lighter green), followed at 456 nm and plotted as ε456 nm to account for protein concentration difference. (B) Time-resolved deconvoluted mass spectra of the reconstitution reaction in the IscU region measured over 25 min. Starting and final spectra are in black and red, respectively, with intervening spectra in grey. (C) m/z spectrum measured after 25 min of reconstitution of IscU (containing 0.01 Zn2+ per protein) in 200 mM ammonium formate buffer, pH 8.0 with 10 μM IscU, 20 μM Fe, 50 μM cysteine, 2 mM DTT and 0.2 μM IscS. Charge states due to [2Fe–2S] IscU indicated. (D) The [2Fe–2S] peak for IscU reconstituted using natural abundance cysteine and iron (black spectrum), 57Fe (blue) and 34S Cys (red). Mass shifts due to the incorporation of 57Fe and 34S are indicated. | ||
 | ||
| Fig. 8 Non IscS-mediated formation of [2Fe–2S] cluster on IscU reveals assembly intermediates. (A) Deconvoluted mass spectrum of apo-IscU under conditions of spontaneous self-assembly. IscU was pre-treated for ∼50 s with a 54-fold excess of DTT-S generated by dissolving S0 in 200 mM ammonium formate buffer containing DTT (final ratio of S0 to DTT, 1:1.6) and then, immediately before infusion, the IscU sample was treated with a ∼18-fold excess (180 μM) of Fe2+ and mixed. Data were acquired from ∼20 s post iron addition. Major species are indicated. (B) Temporal behaviour of Fe–S cluster intermediates suggests [S] precedes the appearance of [Fe–S] and [Fe–S2], which in turn precedes the appearance of the [2Fe–2S] cluster. The gradual loss of [2Fe–2S] IscU beyond 2 min, as noted above, is consistent with the increased lability of the cluster in ammonium formate. | ||
Because only catalytic amounts of IscS were used in reconstitution reactions, and the added difficulties in resolving small increases in mass on large protein complexes (such as IscSU), native MS measurement conditions were optimised to favour the ionisation of free IscU, over the IscSU complex. At the start of the reconstitution, peaks corresponding to apo-IscU (13976 Da) and Zn2+–IscU (14039 Da) were observed (Fig. 7B, Table 1), along with various adducts. Over the course of the reconstitution (25 min), peaks due to apo- and Zn2+–IscU were significantly reduced and a mass gain of +175 Da (relative to apo-IscU) was observed, which corresponds to a [2Fe–2S] cluster (Fig. 7B and C). The charge state distribution observed for the product (Fig. 7C) was very similar to that observed for Zn2+–IscU, indicating that the cluster stabilises the folded form of the protein. Measurements over longer time periods revealed the gradual loss of the cluster-bound form of IscU, suggesting significant lability of the cluster.72
Reconstitution experiments were repeated as above but with 57Fe in place of natural abundance iron (56Fe), or 34S-cysteine as a source of sulfur in place of natural abundance (32S) cysteine.51,54 Cluster assembly in both cases was slower (Fig. S8†), possibly due to kinetic isotope effects,73,74 but generated the product [2Fe–2S] cluster with mass shifts, relative to the natural abundance spectrum, of +2 Da or +4 Da, respectively (Fig. 7D). The data demonstrate the incorporation of two irons and two sulfides into the nascent cluster. The time-resolved data did not reveal any clear intermediates of cluster formation, though the prevalence of sodium adducts of IscU tended to obscure regions of the spectrum where Fe/S complexes might be observed. Furthermore, under the conditions employed here, dimeric [4Fe–4S] IscU33 was not observed, even after prolonged incubation.
Thus, native MS provided unambiguous evidence of the incorporation of a [2Fe–2S] cluster into IscU, apparently without accumulating intermediates, resulting in a shift of the protein's conformational equilibrium towards the structured form. The cluster is labile and no evidence of reductive coupling of two [2Fe–2S] clusters to form a [4Fe–4S] cluster was observed.
Intermediates of [2Fe–2S] cluster assembly on IscU with an IscS mimic
The lack of cluster intermediate species in the above reconstitution experiments may be a consequence of the apparent poor ionisation efficiency of apo-IscU and adducts from a complex reconstitution solution. Thus, we sought to simplify the reconstitution. Iron-sulfur clusters spontaneously assemble in vitro under anaerobic conditions if inorganic S2− and Fe2+ ions are combined with an apo-form of an Fe–S protein in the presence of a thiol based reductant. Elemental sulfur (S0) was dissolved in ammonium formate buffer containing DTT to generate a low molecular weight IscS mimic (DTT-S) compatible with native mass spectrometry. IscU was pre-treated with excess DTT-S and then, immediately before infusion, with excess Fe2+. Peaks consistent with the presence of S, [Fe–S], [Fe–S2] and [2Fe–2S] adducts appeared during the reaction time course (Fig. 8A). The temporal behaviour indicated that S precedes the appearance of [Fe–S] and [Fe–S2], which in turn precedes the appearance of the [2Fe–2S] cluster (Fig. 8B). The gradual loss of [2Fe–2S] IscU beyond 2 min, as noted above, is consistent with the increased lability of the cluster in ammonium formate. These results give us a clear temporal sequence of the species that appear first during the reaction. The reciprocal reaction, in which Fe2+ was added first, gave a constant low level of the same IscU intermediates, with only [2Fe–2S]–IscU increasing during the reaction time course (not shown).Intermediates of [2Fe–2S] cluster assembly on IscU probed by discontinuous assay
In an alternative approach, IscU (<0.1 Zn2+ per IscU) reconstitution reactions in Tris buffer were allowed to proceed for increasing time periods, prior to buffer exchange into ammonium acetate. This had the effect of quenching the reconstitution reaction and removing low molecular weight products and reactants (that could potentially interfere with IscU ionisation). Samples were then immediately infused into the ESI-MS to capture newly formed intermediates and products associated with IscU. Fig. 9A shows the deconvoluted spectrum of IscU at specific time points during the first 15 min of the cluster reconstitution, which, based on absorbance data (Fig. 7), is close to completion.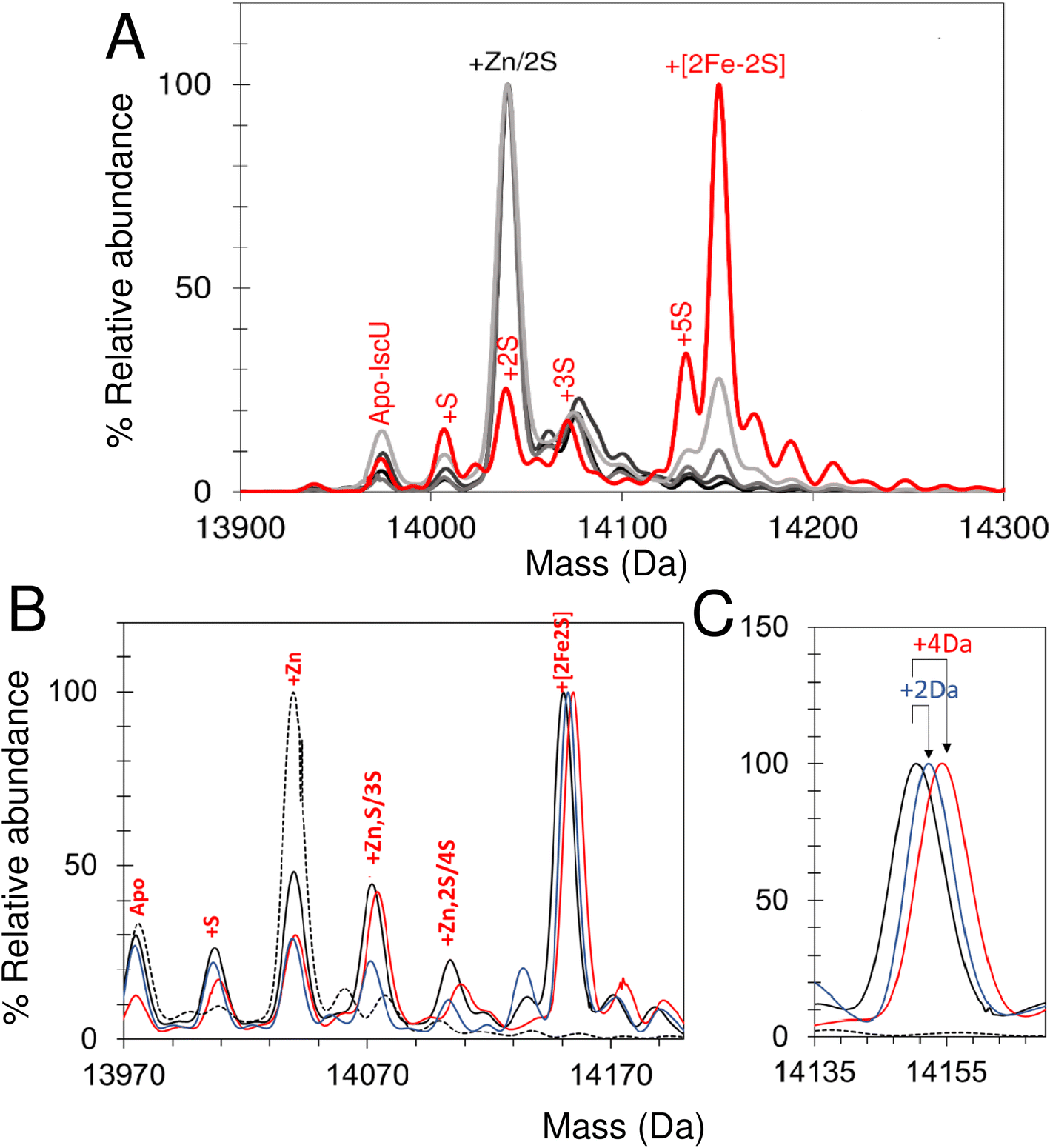 | ||
| Fig. 9 Assembly of [2Fe–2S] IscU monitored by discontinuous assay. (A) Reconstitution of apo-IscU in 50 mM Tris, 150 mM NaCl, pH 8 for increasing time periods and exchange into 250 mM ammonium acetate, pH 8 prior to native mass spectrometric measurement. Starting and end (15 min) spectra are in black and red, respectively. Intervening spectra are in increasingly light grey (1–5 min). (B) Deconvoluted mass spectra of IscU reconstituted in Tris buffer for 15 min before buffer exchange into 200 mM ammonium formate, pH 8.0. DTT-reduced apo-IscU (black dotted line), 56Fe and 32S-cysteine reconstitution (black), 57Fe reconstitution (blue), and 34S reconstitution (red). (C) Expanded view of the [2Fe–2S] cluster region. | ||
After 2.5 min, the peak associated with a [2Fe–2S] cluster bound form of IscU was clearly observed. Adducts of apo-IscU containing Zn2+ or S (multiples of +32 Da, as previously reported75) were also observed. After 15 min, [2Fe–2S]–IscU at 14150 Da was the most abundant species. As observed for continuous assay experiments above, the peak due to Zn2+–IscU (in this sample, <10% of total IscU) was significantly reduced during the reaction. Because these samples were buffer-exchanged before injection into the instrument, it was possible to determine whether the decrease in the abundance of the Zn2+–IscU peak was due to dissociation of Zn2+ from IscU. ICP-MS analysis of identical samples showed that, after 15 min reconstitution, very little Zn2+ (<0.01 Zn2+ per IscU) was protein-bound, thus demonstrating that Zn2+ is indeed displaced during the cluster assembly reaction.52
Isotopic substitution using 34S-cysteine as sulfur source assisted in the assignment of intermediate species (Fig. 9B and C and S9†). All IscU adducts were shifted to a heavier mass in the 34S reaction. The single S adduct was shifted by the expected +2 Da to +34 Da. The Zn2+–IscU peak, which was present at the start and significantly decreased as the reaction proceeded, would not be expected to shift, unless it overlapped with a peak due to an adduct of 2S. A shift was observed, but only of +2 Da, indicating that the peak arises from overlapping peaks due to Zn2+/2S adducts of IscU. The shifts observed for high mass adduct peaks at +96 Da and + 128 Da indicated that they, too, result from overlapping Zn2+/S species (Fig. S9†). We note that persulfide exchange could lead to the accumulation of Sn, where n ≥ 2.
An equivalent isotope substitution experiment with 57Fe revealed no shifts in any of the apo-IscU adducts over the course of the reconstitution (Fig. 9B). Thus, in this discontinuous assay, the only stable iron species detected in complex with IscU was that incorporated into the product [2Fe–2S] cluster. We note that labile Fe2+ ions may be lost from IscU during the buffer exchange process, and that apparent Fe2+-binding affinity is lower in the ESI experiment.
Persulfide forms of IscU are competent for cluster assembly
The above experiments revealed persulfide forms of IscU are readily formed. To investigate whether such species can accumulate in the absence of iron, assays were conducted (in ammonium formate) with varying concentrations of cysteine and IscS. Cysteine at 5-fold (as in reconstitution reactions), and at 20-fold excess over IscU, was used, with catalytic amounts of IscS (IscU:IscS 50:1) (Fig. S10A†). In additional experiments a higher concentration of IscS (IscU:IscS 2:1) was also employed to increase the availability of sulfur. In all cases, S adducts similar to those observed above (and confirmed using 34S-cysteine, Fig. S10B†) were observed, demonstrating that S transfer from IscS to IscU can also occur in the absence of iron. At higher cysteine concentration, adducts of IscU with one and two cysteines were also detected, Fig. S10A.† Also noteworthy was that, in the presence of IscS/cysteine, apo-IscU was the highest abundance peak (rather than Zn2+–IscU), indicating the displacement of Zn2+ from IscU in the presence of IscS/cysteine, and/or improved ionisation efficiency of apo-IscU under these conditions. A reconstitution reaction with the higher concentration of IscS was also carried out (Fig. S11†), confirming that [2Fe–2S] assembly on IscU occurs under these conditions.
The above experiments indicate that transfer of S0 from IscS to IscU can readily occur in the absence of iron. Previous reports suggested that such species are off pathway products that are unable to facilitate [2Fe–2S] assembly.56 To investigate this further, the cluster assembly process, followed by UV-visible absorbance, was broken down into two steps: IscS-mediated addition of S0, and subsequently addition of iron and reductant (Fig. S12†). Cysteine (5-fold excess) and catalytic amounts of IscS (IscU:IscS 50:1) were added to IscU under aerobic conditions, and unbound small molecules removed by gel filtration, resulting in persulfide-containing IscU (Fig. S12A†). Under anaerobic conditions, Fe2+ alone (Fig S12B†) and in combination with reductant (DTT) was added to persulfide-IscU. The inclusion of reductant resulted in the rapid (within 5 min) formation of [2Fe–2S] (Fig. S12C†). Alternative reductants GSH and cysteine were also employed, in both cases resulting in cluster formation but over a longer period (Fig. S12D and E†). In the case of cysteine, further reaction was observed, presumably because of turnover by IscS.
Overall, the data above demonstrate that S adducts readily accumulate on IscU both in the presence and absence of Fe, and are available and fully competent for [2Fe–2S] cluster assembly (Fig. S12F†).
Effect of Zn2+ on [2Fe–2S] cluster assembly on IscU
In order to assess the effect of bound Zn2+ on cluster assembly, reconstitution experiments as described above (i.e. with IscU in the presence of catalytic quantities of IscS) were carried out with fully Zn2+-loaded IscU. These resulted in a significantly reduced rate of assembly, but still yielded [2Fe–2S] IscU (Fig. 10A and B). This is consistent with some previous reports,76,77 but not all.34 Native mass spectra revealed formation of a [2Fe–2S] cluster bound to IscU without Zn2+ (Fig. 10C), indicating that Zn2+ was displaced during cluster assembly, as was also reported in a recent native MS study.52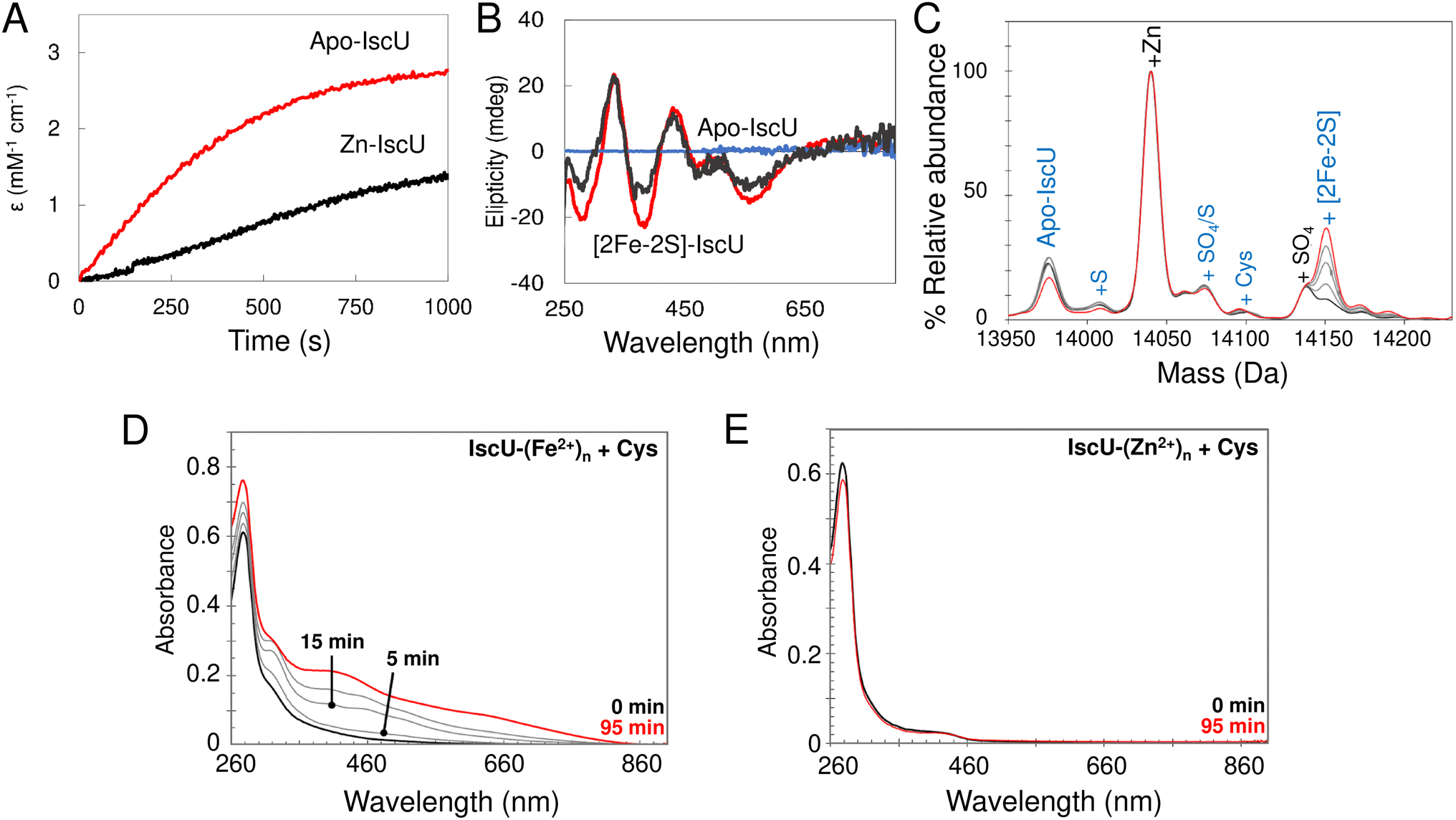 | ||
| Fig. 10 The effect of Zn2+ on the reconstitution of [2Fe–2S] IscU. (A) and (B) Reconstitution of apo-IscU (red) and Zn2+–IscU (grey) in 50 mM Tris, 150 mM NaCl, pH 8.0, monitored by absorbance (A) or CD (B) spectroscopies. (C) Zn2+–IscU (10 μM) in 200 mM ammonium formate buffer, pH 8.0 was reconstituted using 20 μM Fe2+, 50 μM cysteine, 2 mM DTT and 0.2 μM IscS. The reaction was directly infused into the MS once the IscS was added. The starting spectrum is in black and the end spectrum (at 25 min) is in red (intervening spectra in grey). Adducts of apo-IscU and Zn–IscU are described in blue and black, respectively. (D) IscU pre-loaded with excess Fe2+ ions retained 0.68 (±0.06) Fe2+/IscU post-desalting, and supported Fe–S cluster assembly in the presence of 5 μM IscS and 2 mM L-cysteine. (E) IscU pre-loaded with excess Zn2+ ions, then Fe2+ ions, retained 1.65 (±0.35) Zn2+/IscU, but contained no detectable Fe2+ ions post desalting. The addition of 5 μM IscS and 2 mM L-cysteine failed to support Fe–S cluster assembly, consistent with the lack of Fe2+ ions. | ||
The displacement of Zn2+ may be of physiological importance. To probe this further, cluster reconstitution experiments were performed, separately, with apo-IscU and fully loaded Zn2+–IscU in regular reconstitution Tris buffer. First, a 30-fold excess of Fe2+ was added anaerobically to each. Unbound excess Fe2+ was subsequently removed by gel filtration. Catalytic IscS and cysteine (3 mM) was then added and cluster assembly followed by UV-visible absorbance. For the sample originally containing apo-IscU, the protein was found to contain ∼0.7 Fe2+ per protein following gel filtration, and clear evidence of cluster assembly was apparent at 20 min (Fig. 10D). For the sample originally containing Zn2+–IscU, no bound Fe2+ was detected following gel filtration and cluster assembly was thus not observed (Fig. 10E).
These data demonstrate that, while Zn2+ inhibits cluster assembly on IscU, it does not abolish it; this is because the cation can be displaced from IscU. However, a 30-fold excess of Fe2+ had no effect on Zn2+-binding, indicating that it is the transfer of S0 from IscS that promotes displacement. Furthermore, the data indicate that Fe2+ binds at the same (or similar) site as that occupied by Zn2+.
Discussion
Iron–sulfur clusters spontaneously assembly in vitro under anaerobic conditions if S2− and Fe2+/3+ ions are combined with apo-protein in the presence of a thiol-based reductant. In contrast, living organisms coordinate the availability of potentially toxic iron and sulfide ions via a multiprotein machinery that de novo synthesizes Fe–S clusters on a dedicated scaffold protein. Crystal structures of the sulfide-generating cysteine desulfurase, IscS, alone and in complex with apo- and [2Fe–2S]-forms of the scaffold protein IscU, are available.11,14,17 These provide key insight into the mechanism of inter-molecular sulfur transfer and protein–protein interactions that facilitate the successful formation of [2Fe–2S]–IscU. However, despite the apparent simplicity, the precise mechanistic sequence of events of Fe–S cluster assembly remains elusive.The application of native MS has provided extraordinary mechanistic insight into Fe–S cluster proteins,45,46,48 but major challenges in using this technique to investigate intermediates in the Fe–S assembly process exist, including efficient ionisation of large IscS–IscU complexes (∼105 to 120 kDa), and difficulties in achieving sufficient resolution to distinguish the addition of single atoms/ions (e.g. S32, S34, Fe56, Fe57) to large complexes. For this reason, we optimised the experiments to enable detection of IscU and Fe/S adduct species.
While the work described here was underway, Lin et al.52 reported their native MS study of IscSU-mediated Fe–S assembly, in which they identified potential Fe–S cluster intermediates and products associated with IscU. Here we have implemented a complementary set of native MS experiments. Our data are broadly consistent with the previous study, but offer significant further insights into the enigmatic process of Fe–S cluster assembly through the use of 34S and 57Fe isotopes to investigate S- and Fe-binding/transfer, and the behaviour of Zn2+–IscU.
Conformational flexibility of IscU and Zn2+- and Fe2+-binding
Various biophysical approaches, including NMR spectroscopy, have shown IscU to be a dynamic protein that readily interconverts between two different conformations on a second time scale in vitro:34,66,77,78 one conformation (S-form) is largely structured, the other (D-form) is dynamically disordered. We note that Sato et al. have recently demonstrated that the interconversion between S- and D-forms of IscU is a fundamental requirement for the assembly and transfer of Fe–S clusters in vivo.79 The presence of Zn2+ ions, or Fe2+ ions (and indeed a [2Fe–2S] cluster), shifted the equilibrium between D- and S-forms in favour of the S-form (Fig. S13†), consistent with previous solution studies.20,38,53,78,80 The presence of Zn2+ ions also reduces the tendency of apo-IscU to dimerise and stabilises monomeric IscU in the S-form. Li et al. recently demonstrated that zinc inhibits IscU/IscA-mediated Fe–S assembly and transfer, rather than disrupting already assembled clusters,39 and data presented here are consistent with this (Fig. 10). We also note that IscU is a substrate for degradation by ClpXP and FtsH proteases, and it is possible that Zn2+–IscU may be more resistant to the effects of these proteases than apo-IscU.81E. coli IscU is isolated containing significant amounts of Zn2+. Here we showed that Zn2+ is displaced from IscU during cluster assembly, and that this is dependent on the presence of IscS and Cys, where S0/S2− is generated, consistent with previous reports.52 This occurred even in the absence of Fe2+ (where Fe–S assembly could not occur). It is possible that transfer of the persulfide formed on IscS–Cys321 during the process of cluster assembly triggers the displacement of Zn2+, perhaps by altering the Zn2+ binding environment of IscU to generate a less optimal, lower affinity site.
In contrast to zinc, the ‘free’ iron pool is 10–30 μM under aerobic conditions, rising several fold under anaerobic conditions.82,83 We have determined the Kd of Fe2+ for E. coli IscU to be ∼3.5 μM, consistent with literature values for IscU homologues.59,67–69 Taken together, these values suggest that Fe2+–IscU should exist as a major species in vivo under aerobic and anaerobic conditions, although we are unaware of any successful retrieval of Fe2+–IscU from an in vivo source.
Interestingly, we found that the Kd of Fe2+-binding under the conditions of the native MS experiment was significantly higher (∼73 μM), accounting for why high concentrations of Fe2+ were necessary to observe Fe2+-bound forms by MS.52 Thus, it appears that something occurs during ionisation/transfer to the gas phase that affects the affinity of IscU for Fe2+. This is not typical of protein–metallocofactor interactions measured by native MS, and does not appear to be general for divalent metal ion binding, because Zn2+-binding was not affected. One possibility is that the Fe2+-binding site features a water as a coordinating ligand, which may be lost during ionisation/transfer to the gas phase, where the solvent is removed. A metal binding site lacking a coordinating ligand would be expected to exhibit lower affinity, as observed.
IscS–IscU interactions
Native MS experiments revealed that both apo- and Zn2+–IscU can from a largely structured complex with IscS, with no significant difference in stability.52 As the concentration of IscU was increased, we observed the sequential formation of (IscS)2IscU and (IscS)2(IscU)2. The former maximised at IscU/IscS ≈ 1.0, with the concomitant loss of un-complexed dimeric IscS, whereas the (IscS)2(IscU)2 complex was evident at IscU/IscS ≥ 1.0. The data indicate that dimeric IscS has two equivalent, independent (non-cooperative) binding sites for IscU (Kd ∼ 3 μM). We note that, in vivo, IscS and IscU concentrations are approximately equal (∼15 μM each84), suggesting a mix of (IscU)(IscS)2 and (IscU)2(IscS)2, with almost no free (IscS)2.Mechanism of [2Fe–2S] cluster assembly
The biological assembly of Fe–S clusters requires a multiprotein machinery that de novo synthesizes a Fe–S cluster on a dedicated scaffold protein (e.g. IscU) prior to transfer to client proteins. Escherichia coli possesses two distinct systems, the ISC (iscRSUA-hscBA-fdx-iscX) and SUF (sufABCDSE) systems, which are utilised during standard growth or conditions of oxidative stress/iron limitation, respectively. In a Δsuf background, Tanaka et al. found that iscSU represents the minimal requirement for biosynthesis under anaerobic conditions.85 The human pathogen, H. pylori, a micro-aerobe, naturally contains a minimalist Fe–S system (nifSU) that is essential for viability and is functionally exchangeable for E. coli Fe–S assembly systems.86 Minimalist Fe–S assembly systems likely evolved before their more complex counterparts, e.g. ISC,87 suggesting that Fe acquisition is likely an inherent property of the IscSU complex or its constituent parts, at least under anaerobic conditions.Our reconstitutions, conducted with catalytic quantities of IscS, were monitored by time dependent native MS, using both continuous and discontinuous assays. As noted above, the intensity of the Zn2+–IscU peak decreased with a concomitant increase in the amount of [2Fe–2S]2+ IscU. No significant Fe–S intermediates were observed on IscU. A range of IscU–S adducts, but no Fe2+-only IscU species, were detected: in all cases, apo-IscU was observed to transition directly to [2Fe–2S]–IscU during the reaction time course. To gain access to Fe–S cluster assembly intermediates, it was therefore necessary to adopt an approach in which S0 transfer was not optimised, achieved here using a low molecular weight IscS mimic. Peaks consistent with the presence of IscU-bound [S], [Fe–S], [Fe–S2] and [2Fe–2S] were observed during the reaction time course. The temporal behaviour indicates that [S] precedes the appearance of [Fe–S] and [Fe–S2], which in turn precedes the appearance of the [2Fe–2S] cluster.
Overall, the data indicate that the IscS-catalysed assembly reaction on IscU is largely concerted, such that intermediate species do not readily accumulate to detectable levels. Intermediates were observed in the absence of IscS, where an IscS-mimic was employed, probably because the reaction without IscS was sub-optimal, allowing for some accumulation of intermediates.
Based on our observations and previous work, a working model for Fe–S assembly (Fig. 11) starts with apo-IscU (or Zn2+–IscU) docking to a vacant site on the IscS dimer. Although, anchored by intra-molecular salt bridges and hydrogen bonds, IscU remains dynamic, ensuring access to the Fe–S assembly site, as well as the inter-molecular catalytic centre. Hence, all subsequent steps in the assembly processes most likely take place on the IscU–IscS complex. Upon binding, and in the presence of substrate cysteine, IscS donates two successive sulfane (S0) atoms to IscU17via a persulfide generated by IscS on Cys321 and involving non-redox transfer of the S0 (trans-persulfidation).19
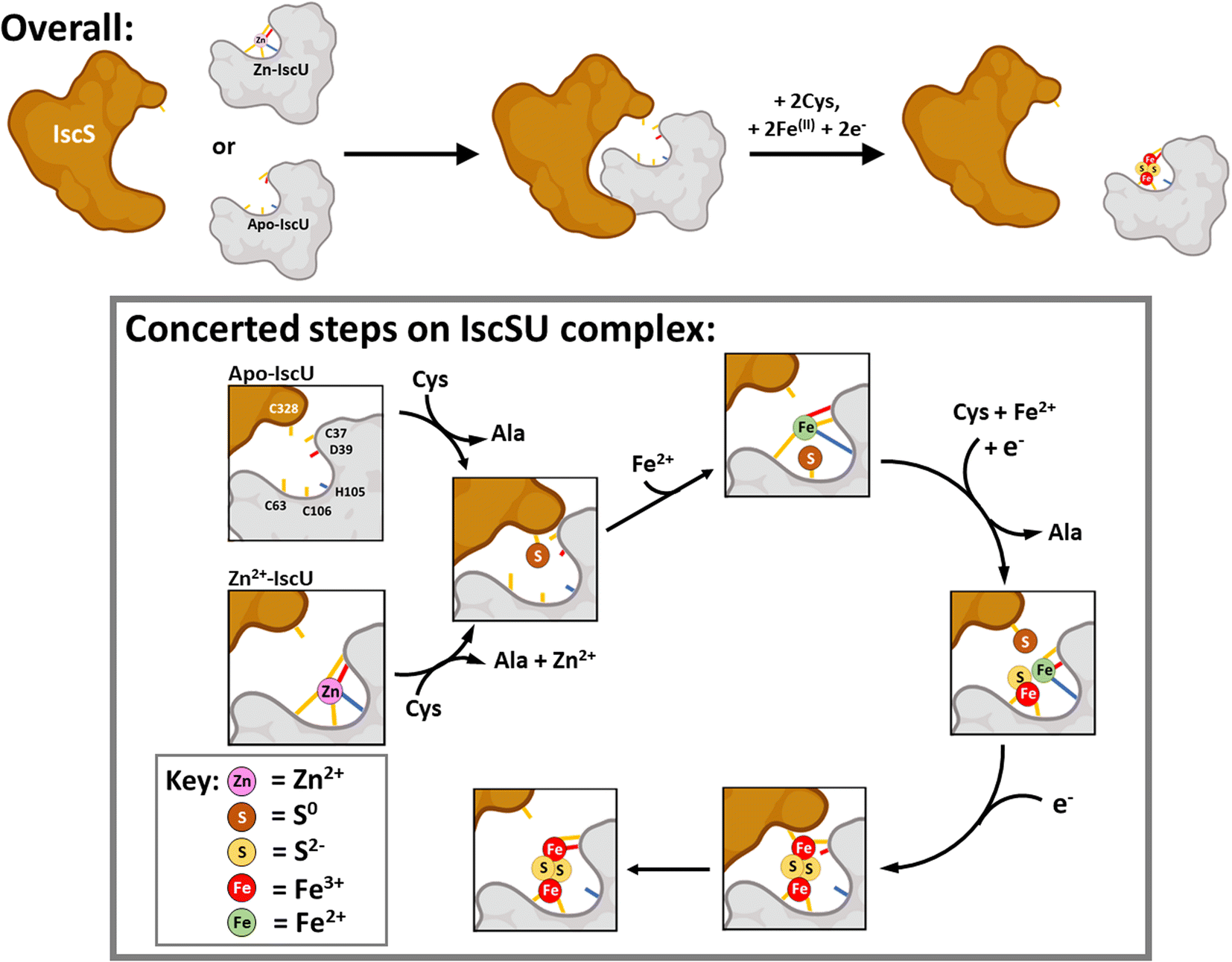 | ||
| Fig. 11 Scheme describing the proposed mechanism of assembly of [2Fe–2S] on IscU in complex with IscS. The difficulty in detecting intermediates of cluster assembly points to a conserved mechanism,17 in which assembly of a [2Fe–2S] goes to completion in preference to the initiation of cluster formation on another IscU molecule, with the effect that assembly intermediates don't accumulate. Details of S- and Fe-binding/transfer are speculative, based on data presented here and previously. The Cys residue of IscU to which S0 is transferred from IscS is not known. Evidence for the involvement of the residues equivalent to Cys37 and Cys106 in the human homologue IscU is available.53,88,89 Inspection of the structure of IscS/IscU suggests that Cys37 is the most likely residue for initial transfer, prior to arrival at Cys106. | ||
Our results suggest that IscS-mediated addition of S0 to IscU creates a site that has lower affinity for Zn2+, permitting displacement by Fe2+ ions at the assembly site, probably involving persulfide-ligated Fe2+. A crystal structure of the Archaeoglobus fulgidus IscS–IscUD35A complex containing a partly oxidised [2Fe–2S] intermediate with a unique μ2–η3 sulfido ligation indicates that persulfide coordination of iron at the cluster site is possible.17
Overall, four electrons are required to reduce two S0 to S2− ions for the synthesis of one [2Fe–2S]2+ cluster. The oxidation of the incoming Fe2+ ions to Fe3+ ions may provide two of the four required electrons, but we note that Fe2+ in the absence of IscS was not efficient in reducing S0 species on IscU. Alternatively, or additionally, electrons are supplied by the ferredoxin encoded by fdx (note that Fdx is not essential in vivo under anaerobic conditions,85 and that electrons can be alternatively supplied, e.g. by Cys and/or GSH). The exact sequence of events, including the order in which Fe2+-binding/sulfur transfer to IscU occurs, and how Fdx binding on IscS facilitates electron transfer to S0 is not clear. The time-resolved data reported here suggest that S0 is first transferred and one Fe2+ then binds, followed by a second S0/Fe2+ to generate the product cluster.
Previous structural studies of A. fulgidus IscS–IscUD35A revealed a cluster coordination consisting of the three Cys residues of IscU along with Cys321 of IscS.17 Molecular dynamic simulations suggested the dissociation of the complex involves the recruitment of IscU Asp35 as a cluster ligand, displacing Cys33, which in turn displaces IscS–Cys321, leading to the dissociation of [2Fe–2S]2+–IscU from the IscS–IscU complex.
Overall, our observations are broadly consistent with those of Lin et al.52 in that Fe–S assembly can be initiated in vitro from Fe2+ or S0 bound forms of IscU. However, our data point to IscSU mechanism of Fe–S cluster assembly that is concerted, in which either iron or sulfur can bind first. In support of this are the following observations: (1) cluster Fe/S intermediate species are not readily observed on ‘free’ IscU during IscS-mediated [2Fe–2S] assembly, indicating that once assembly begins on the IscSU complex, it goes to completion before cluster assembly is initiated on another IscSU complex; (2) sulfur adducts are readily observed on ‘free’ IscU both under cluster assembly conditions, and when Fe2+ is absent (Fig. S11†), as reported by others,19,75 consistent with facile transfer from IscS to IscU. Transferred sulfur can subsequently support cluster assembly. Thus, Fe2+-binding is not a pre-requisite for S0 transfer; (3) IscU pre-loaded with Fe2+ can also support cluster assembly when IscS/cysteine are introduced.
While the order of Fe2+-binding and sulfur transfer does not appear to matter in vitro, it seems likely that the process of cluster assembly on IscU is controlled by sulfur availability, such that sulfur transfer is the key step. The presence of Zn2+ on IscU entirely inhibits Fe2+-binding and prevents the formation of dimeric IscU. However, IscS/cysteine promotes the removal of Zn2+ from Zn2+–IscU,52 facilitating Fe2+-binding and cluster assembly. This must involve/require S0 transfer and points to sulfur transfer as the key initiating step of assembly. This is consistent with the regulation of Isc Fe–S cluster assembly by CyaY at the level of S0 generation/transfer.81,90 The supply of IscS-generated S0 in the cell is carefully regulated, by proteins such as CyaY and IscX,22,26 which tune the activity of IscS through competition binding. Tight regulation of S0 generation/transfer is consistent with this being the key committed step, rather than reversible Fe2+-binding (where Fe2+ is supplied by an iron donor, or more likely from the chelatable iron pool), in cluster assembly.
Concluding remarks
We have applied native mass spectrometry alongside more traditional methods to gain insight into key aspects of the properties and behaviour of the Fe–S cluster scaffold protein IscU, and in particular the chemistry taking place during IscU-mediated [2Fe–2S] cluster assembly. Two conformations of IscU were readily detected, corresponding to the previously characterised S- and D-states. IscU was isolated in a Zn2+-bound form that existed exclusively in the S-state, but Zn2+ could be removed using a high affinity chelator, or through the assembly of a [2Fe–2S] cluster on IscU. Cluster assembly was followed using time-dependent mass spectrometry, with a lack of observable Fe–S intermediates on ‘free’ IscU suggesting a concerted reaction. Although Fe–S cluster assembly can be initiated in vitro from Fe2+- or S0-bound forms of IscU, when Zn2+ is bound to IscU, sulfur transfer is required to initiate cluster assembly through the displacement of Zn2+, which enables Fe2+ to bind at the assembly site. The proposed sulfur-initiated assembly process is consistent with the allosteric regulation of IscS sulfur production/transfer by CyaY and IscX.Data availability
Data supporting the conclusions of this study are available in the main paper with additional experimental data given in the ESI.†Author contributions
JCC, AP and NLB conceived the study and contributed to experimental design. SPB, JCC and RP contributed to protein production and purification. SPB and JCC carried out the Fe–S cluster assembly experimental work. SPB, JCC and NLB analysed the data. JCC and NLB wrote the paper with assistance from RP and AP. All authors have approved the final version of the manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the Biotechnology and Biological Sciences Research Council through grants BB/S001018/1, BB/T017708/1, BB/R013578/1 (to NLB), and BB/S001832/1 (to AP). Further support was provided by a British Mass Spectrometry Society Research Support Grant (to JCC), and by UEA through the purchase/upgrade of the ESI-MS instrument.References
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS PubMed.
- H. Beinert, R. H. Holm and E. Munck, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- T. A. Rouault, BioMetals, 2019, 32, 343–353 CrossRef CAS PubMed.
- H. Ye and T. A. Rouault, Biochemistry, 2010, 49, 4945–4956 CrossRef CAS PubMed.
- M. Pandolfo and A. Pastore, J. Neurol., 2009, 256(1), 9–17 CrossRef CAS.
- R. Puglisi, Biomedicines, 2022, 10, 425 CrossRef CAS.
- K. Bürk, Cerebellum Ataxias, 2017, 4, 4 CrossRef PubMed.
- J. Frazzon and D. R. Dean, Curr. Opin. Chem. Biol., 2003, 7, 166–173 CrossRef CAS PubMed.
- B. Roche, L. Aussel, B. Ezraty, P. Mandin, B. Py and F. Barras, Biochim. Biophys. Acta, 2013, 1827, 455–469 CrossRef CAS PubMed.
- R. Lill and S. A. Freibert, Annu. Rev. Biochem., 2020, 89, 471–499 CrossRef CAS PubMed.
- J. R. Cupp-Vickery, H. Urbina and L. E. Vickery, J. Mol. Biol., 2003, 330, 1049–1059 CrossRef CAS.
- M. Iciek, A. Bilska-Wilkosz and M. Gorny, Acta Biochim. Pol., 2019, 66, 533–544 CAS.
- W. Zhang, A. Urban, H. Mihara, S. Leimkuhler, T. Kurihara and N. Esaki, J. Biol. Chem., 2010, 285, 2302–2308 CrossRef CAS PubMed.
- R. Shi, A. Proteau, M. Villarroya, I. Moukadiri, L. Zhang, J. F. Trempe, A. Matte, M. E. Armengod and M. Cygler, PLoS Biol., 2010, 8, e1000354 CrossRef PubMed.
- T. S. Tanabe, S. Leimkühler and C. Dahl, in Advances in Microbial Physiology, ed. R. K. Poole, Academic Press, 2019, vol. 75, pp. 233–277 Search PubMed.
- J. N. Agar, L. Zheng, V. L. Cash, D. R. Dean and M. K. Johnson, J. Am. Chem. Soc., 2000, 122, 7856–7862 CrossRef.
- E. N. Marinoni, J. S. de Oliveira, Y. Nicolet, E. C. Raulfs, P. Amara, D. R. Dean and J. C. Fontecilla-Camps, Angew. Chem., Int. Ed., 2012, 51, 5439–5442 CrossRef CAS PubMed.
- A. D. Smith, J. Frazzon, D. R. Dean and M. K. Johnson, FEBS Lett., 2005, 579, 5236–5240 CrossRef CAS PubMed.
- H. D. Urbina, J. J. Silberg, K. G. Hoff and L. E. Vickery, J. Biol. Chem., 2001, 276, 44521–44526 CrossRef CAS PubMed.
- F. Prischi, C. Pastore, M. Carroni, C. Iannuzzi, S. Adinolfi, P. Temussi and A. Pastore, Protein Expression Purif., 2010, 73, 161–166 CrossRef CAS.
- M. Marengo, R. Puglisi, S. Oliaro-Bosso, A. Pastore and S. Adinolfi, Methods Mol. Biol., 2021, 2353, 79–95 CrossRef PubMed.
- S. Adinolfi, R. Puglisi, J. C. Crack, C. Iannuzzi, F. Dal Piaz, P. V. Konarev, D. I. Svergun, S. Martin, N. E. Le Brun and A. Pastore, Front. Mol. Biosci., 2017, 4, 97 CrossRef.
- J. H. Kim, J. R. Bothe, R. O. Frederick, J. C. Holder and J. L. Markley, J. Am. Chem. Soc., 2014, 136, 7933–7942 CrossRef CAS.
- G. Layer, S. Ollagnier-de Choudens, Y. Sanakis and M. Fontecave, J. Biol. Chem., 2006, 281, 16256–16263 CrossRef CAS PubMed.
- C. Iannuzzi, S. Adinolfi, B. D. Howes, R. Garcia-Serres, M. Clemancey, J. M. Latour, G. Smulevich and A. Pastore, PLoS One, 2011, 6, e21992 CrossRef CAS PubMed.
- S. Adinolfi, C. Iannuzzi, F. Prischi, C. Pastore, S. Iametti, S. R. Martin, F. Bonomi and A. Pastore, Nat. Struct. Mol. Biol., 2009, 16, 390–396 CrossRef CAS PubMed.
- J. H. Kim, R. O. Frederick, N. M. Reinen, A. T. Troupis and J. L. Markley, J. Am. Chem. Soc., 2013, 135, 8117–8120 CrossRef CAS PubMed.
- R. Yan, S. Adinolfi and A. Pastore, Biochim. Biophys. Acta, 2015, 1854, 1113–1117 CrossRef CAS PubMed.
- L. Loiseau, C. Gerez, M. Bekker, S. Ollagnier-de Choudens, B. Py, Y. Sanakis, J. Teixeira de Mattos, M. Fontecave and F. Barras, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13626–13631 CrossRef CAS.
- B. Blanc, C. Gerez and S. Ollagnier de Choudens, Biochim. Biophys. Acta, 2015, 1853, 1436–1447 CrossRef CAS.
- R. Puglisi, R. Yan, S. Adinolfi and A. Pastore, Front. Mol. Biosci., 2016, 3, 48 Search PubMed.
- R. Puglisi and A. Pastore, FEBS Lett., 2018, 592, 4011–4019 CrossRef CAS.
- J. N. Agar, C. Krebs, J. Frazzon, B. H. Huynh, D. R. Dean and M. K. Johnson, Biochemistry, 2000, 39, 7856–7862 CrossRef CAS PubMed.
- C. Iannuzzi, M. Adrover, R. Puglisi, R. Yan, P. A. Temussi and A. Pastore, Protein Sci., 2014, 23, 1208–1219 CrossRef CAS PubMed.
- J. Liu, N. Oganesyan, D. H. Shin, J. Jancarik, H. Yokota, R. Kim and S. H. Kim, Proteins, 2005, 59, 875–881 CrossRef CAS PubMed.
- T. A. Ramelot, J. R. Cort, S. Goldsmith-Fischman, G. J. Kornhaber, R. Xiao, R. Shastry, T. B. Acton, B. Honig, G. T. Montelione and M. A. Kennedy, J. Mol. Biol., 2004, 344, 567–583 CrossRef CAS PubMed.
- F. F. Xu and J. A. Imlay, Appl. Environ. Microbiol., 2012, 78, 3614–3621 CrossRef CAS.
- P. Roy, M. A. Bauman, H. H. Almutairi, W. G. Jayawardhana, N. M. Johnson and A. T. Torelli, ACS Chem. Biol., 2018, 13, 591–599 CrossRef CAS PubMed.
- J. Li, X. Ren, B. Fan, Z. Huang, W. Wang, H. Zhou, Z. Lou, H. Ding, J. Lyu and G. Tan, Appl. Environ. Microbiol., 2019, 85, e01967–18 CAS.
- N. Morgner and C. V. Robinson, Curr. Opin. Struct. Biol., 2012, 22, 44–51 CrossRef CAS.
- C. Schmidt and C. V. Robinson, FEBS J., 2014, 281, 1950–1964 CrossRef CAS.
- S. Mehmood, T. M. Allison and C. V. Robinson, Annu. Rev. Phys. Chem., 2015, 66, 453–474 CrossRef CAS PubMed.
- F. D. Kondrat, G. R. Kowald, C. A. Scarff, J. H. Scrivens and C. A. Blindauer, Chem. Commun., 2013, 49, 813–815 RSC.
- A. C. Leney and A. J. Heck, J. Am. Soc. Mass Spectrom., 2017, 28, 5–13 CrossRef CAS PubMed.
- J. C. Crack and N. E. Le Brun, Chem.–Eur. J., 2019, 25, 3675–3684 CrossRef CAS PubMed.
- J. C. Crack, A. J. Thomson and N. E. Le Brun, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3215–E3223 CrossRef CAS PubMed.
- M. Jia, S. Sen, C. Wachnowsky, I. Fidai, J. A. Cowan and V. H. Wysocki, Angew. Chem., Int. Ed., 2020, 59, 6724–6728 CrossRef CAS PubMed.
- M. T. Pellicer Martinez, J. C. Crack, M. Y. Stewart, J. M. Bradley, D. A. Svistunenko, A. W. Johnston, M. R. Cheesman, J. D. Todd and N. E. Le Brun, eLife, 2019, 8, e47804 CrossRef PubMed.
- R. Puglisi, E. Boeri Erba and A. Pastore, FEBS J., 2020, 287, 2428–2439 CrossRef CAS PubMed.
- J. C. Crack, E. Gray and N. E. Le Brun, Dalton Trans., 2021, 50, 7887–7897 RSC.
- J. C. Crack and N. E. Le Brun, Methods Mol. Biol., 2021, 2353, 231–258 CrossRef.
- C. W. Lin, J. W. McCabe, D. H. Russell and D. P. Barondeau, J. Am. Chem. Soc., 2020, 142, 6018–6029 CrossRef CAS PubMed.
- S. Gervason, D. Larkem, A. B. Mansour, T. Botzanowski, C. S. Muller, L. Pecqueur, G. Le Pavec, A. Delaunay-Moisan, O. Brun, J. Agramunt, A. Grandas, M. Fontecave, V. Schunemann, S. Cianferani, C. Sizun, M. B. Toledano and B. D'Autreaux, Nat. Commun., 2019, 10, 3566 CrossRef PubMed.
- J. C. Crack, M. Y. Y. Stewart and N. E. Le Brun, Biol. Methods Protoc., 2019, 4, bpy015 CrossRef PubMed.
- F. Bonomi, S. Iametti, D. Ta and L. E. Vickery, J. Biol. Chem., 2005, 280, 29513–29518 CrossRef CAS PubMed.
- M. Nuth, T. Yoon and J. A. Cowan, J. Am. Chem. Soc., 2002, 124, 8774–8775 CrossRef CAS.
- J. C. Crack, A. A. Gaskell, J. Green, M. R. Cheesman, N. E. Le Brun and A. J. Thomson, J. Am. Chem. Soc., 2008, 130, 1749–1758 CrossRef CAS.
- C. E. Sabel, J. L. Shepherd and S. Siemann, Anal. Biochem., 2009, 391, 74–76 CrossRef PubMed.
- A. V. Rodrigues, A. Kandegedara, J. A. Rotondo, A. Dancis and T. L. Stemmler, BioMetals, 2015, 28, 567–576 CrossRef CAS PubMed.
- M. R. Kurek, W. P. Gilhooly, G. K. Druschel, M. D. O'Beirne and J. P. Werne, Chem. Geol., 2018, 481, 18–26 CrossRef CAS.
- A. S. Nashef, D. T. Osuga and R. E. Feeney, Anal. Biochem., 1977, 79, 394–405 CrossRef CAS.
- G. S. Rao and G. Gorin, J. Org. Chem., 1959, 24, 749–753 CrossRef CAS.
- J. C. Crack, C. J. Hamilton and N. E. Le Brun, Chem. Commun., 2018, 54, 5992–5995 RSC.
- H. Hernandez, K. S. Hewitson, P. Roach, N. M. Shaw, J. E. Baldwin and C. V. Robinson, Anal. Chem., 2001, 73, 4154–4161 CrossRef CAS.
- X. Wu, N. H. Bishopric, D. J. Discher, B. J. Murphy and K. A. Webster, Mol. Cell. Biol., 1996, 16, 1035–1046 CrossRef CAS PubMed.
- J. H. Kim, M. Tonelli, T. Kim and J. L. Markley, Biochemistry, 2012, 51, 5557–5563 CrossRef.
- I. Bertini, J. A. Cowan, C. Del Bianco, C. Luchinat and S. S. Mansy, J. Mol. Biol., 2003, 331, 907–924 CrossRef CAS PubMed.
- J. D. Cook, K. C. Kondapalli, S. Rawat, W. C. Childs, Y. Murugesan, A. Dancis and T. L. Stemmler, Biochemistry, 2010, 49, 8756–8765 CrossRef CAS.
- M. W. Foster, S. S. Mansy, J. Hwang, J. E. Penner-Hahn, K. K. Surerus and J. A. Cowan, J. Am. Chem. Soc., 2000, 122, 6805–6806 CrossRef CAS.
- F. Prischi, P. V. Konarev, C. Iannuzzi, C. Pastore, S. Adinolfi, S. R. Martin, D. I. Svergun and A. Pastore, Nat. Commun., 2010, 1, 95 CrossRef PubMed.
- C. H. Tai, S. R. Nalabolu, T. M. Jacobson, D. E. Minter and P. F. Cook, Biochemistry, 1993, 32, 6433–6442 CrossRef CAS.
- M. Adrover, B. D. Howes, C. Iannuzzi, G. Smulevich and A. Pastore, Biochim. Biophys. Acta, 2015, 1853, 1448–1456 CrossRef CAS PubMed.
- H. Gu and S. Zhang, Molecules, 2013, 18, 9278–9292 CrossRef CAS PubMed.
- N. Nakai and M. L. Jensen, Geochim. Cosmochim. Acta, 1964, 28, 1893–1912 CrossRef CAS.
- A. D. Smith, J. N. Agar, K. A. Johnson, J. Frazzon, I. J. Amster, D. R. Dean and M. K. Johnson, J. Am. Chem. Soc., 2001, 123, 11103–11104 CrossRef CAS PubMed.
- Z. Dai, M. Tonelli and J. L. Markley, Biochemistry, 2012, 51, 9595–9602 CrossRef CAS PubMed.
- J. L. Markley, J. H. Kim, Z. Dai, J. R. Bothe, K. Cai, R. O. Frederick and M. Tonelli, FEBS Lett., 2013, 587, 1172–1179 CrossRef CAS.
- J. H. Kim, M. Tonelli and J. L. Markley, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 454–459 CrossRef CAS PubMed.
- S. Sato, Y. Matsushima, M. Kanazawa, N. Tanaka, T. Fujishiro, K. Kunichika, R. Nakamura, H. Tomioka, K. Wada and Y. Takahashi, Mol. Microbiol., 2021, 115, 807–818 CrossRef CAS PubMed.
- S. Adinolfi, F. Rizzo, L. Masino, M. Nair, S. R. Martin, A. Pastore and P. A. Temussi, Eur. J. Biochem., 2004, 271, 2093–2100 CrossRef CAS.
- E. L. Mettert and P. J. Kiley, Annu. Rev. Microbiol., 2015, 69, 505–526 CrossRef CAS PubMed.
- K. Keyer and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13635–13640 CrossRef CAS.
- N. A. Beauchene, E. L. Mettert, L. J. Moore, S. Keles, E. R. Willey and P. J. Kiley, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12261–12266 CrossRef CAS PubMed.
- G. W. Li, D. Burkhardt, C. Gross and J. S. Weissman, Cell, 2014, 157, 624–635 CrossRef CAS PubMed.
- N. Tanaka, M. Kanazawa, K. Tonosaki, N. Yokoyama, T. Kuzuyama and Y. Takahashi, Mol. Microbiol., 2016, 99, 835–848 CrossRef CAS.
- U. Tokumoto, S. Kitamura, K. Fukuyama and Y. Takahashi, J. Biochem., 2004, 136, 199–209 CrossRef CAS.
- P. S. Garcia, F. D'Angelo, S. Ollagnier de Choudens, M. Dussouchaud, E. Bouveret, S. Gribaldo and F. Barras, Nat. Ecol. Evol., 2022, 6, 1564–1572 CrossRef.
- A. Parent, X. Elduque, D. Cornu, L. Belot, J. P. Le Caer, A. Grandas, M. B. Toledano and B. D'Autreaux, Nat. Commun., 2015, 6, 5686 CrossRef CAS.
- B. Srour, S. Gervason, M. H. Hoock, B. Monfort, K. Want, D. Larkem, N. Trabelsi, G. Landrot, A. Zitolo, E. Fonda, E. Etienne, G. Gerbaud, C. S. Muller, J. Oltmanns, J. B. Gordon, V. Yadav, M. Kleczewska, M. Jelen, M. B. Toledano, R. Dutkiewicz, D. P. Goldberg, V. Schunemann, B. Guigliarelli, B. Burlat, C. Sizun and B. D'Autreaux, J. Am. Chem. Soc., 2022, 144, 17496–17515 CrossRef CAS PubMed.
- D. di Maio, B. Chandramouli, R. Yan, G. Brancato and A. Pastore, Biochim. Biophys. Acta, 2017, 1861, 3154–3163 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04169c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |