
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A self-optimised approach to synthesising DEHiBA for advanced nuclear reprocessing, exploiting the power of machine-learning†
Thomas
Shaw
 ,
Adam D.
Clayton
,
Ricardo
Labes
,
Thomas M.
Dixon
,
Sarah
Boyall
,
Oliver J.
Kershaw
,
Richard A.
Bourne
and
Bruce C.
Hanson
*
,
Adam D.
Clayton
,
Ricardo
Labes
,
Thomas M.
Dixon
,
Sarah
Boyall
,
Oliver J.
Kershaw
,
Richard A.
Bourne
and
Bruce C.
Hanson
*
Institute of Process Research and Development, School of Chemistry & School of Chemical and Process Engineering, University of Leeds, LS2 9JT, UK. E-mail: B.C.Hanson@Leeds.ac.uk
First published on 3rd November 2023
Abstract
In an effort to advance the development of hydrometallurgical reprocessing of used nuclear fuel across the globe, this work sets out to explore and identify an optimised, cost effective pathway to synthesise the ligand DEHiBA (N,N-di-(2-ethylhexyl)isobutyramide). Currently, very few chemical suppliers stock and distribute this specialist ligand, designed for selective uranium chelation and extraction from nuclear fuel. The current high cost of DEHiBA therefore restricts access to essential large-scale testing of this promising ligand designed to advance nuclear reprocessing. This work utilises an automated flow reactor platform for the efficient optimisation of four synthetic routes to DEHiBA. These optimisations focus on optimising cost, reagent efficiency, yield, and productivity target functions by exploiting the power of machine-learning algorithms for rapid process development. Ultimately, we have identified an efficient and cost-effective solvent-free route to DEHiBA from isobutyric anhydride and di-2-ethylhexylamine for <£100 (current prices) per litre of DEHiBA in reagent costs enabling affordable access to litres of this material for subsequent testing. The exothermic nature of this reaction required a tubular flow reactor to control the reaction and mitigate this safety risk. This enabled the continuous production of crude DEHiBA with the capability to achieve yields >99%, at a purity of 76%, and a process mass intensity of 1.29 g g−1, whilst alternative conditions demonstrated productivities >75 kg L−1 h−1, all whilst maintaining a high level of process control with outlet temperatures not exceeding 35 °C.
Introduction
The demand for nuclear energy has surged in recent years due to a global drive toward sustainability. In line with this, the UK government stated in its 2020 energy white paper that nuclear energy would play a key role in decarbonising Britain over the next 30 years,1 which has resulted in multi-billion pound investments in large nuclear reactors at Hinkley and Sizewell. The recent rise in interest towards nuclear fission as a means to not only decarbonise power, but other, “more difficult to decarbonise industries”,2–4 brings the question of how sustainable is nuclear fission as a whole? The clean energy benefits and low carbon emissions associated with energy production from nuclear power is indisputable.5,6 Yet, an open fuel cycle remains popular for most, despite its unsustainable nature.7–9 To improve the sustainability of nuclear fission the appropriate management of used nuclear fuel is vital for nuclear fission to be sustainable for future generations over a long time-frame. Currently, two overarching routes exist for the management of used nuclear fuel: direct disposal in geological facilities, and nuclear reprocessing/recycle.10 For nuclear fission to be truly sustainable, the employment of a closed fuel cycle that avoids wasting this valuable resource, rich in elements from across the periodic table, is critical.10–13 Yet, at present, few reprocessing facilities are in operation and these only employ the limited, but mature PUREX (Plutonium Uranium Reduction EXtraction) process for the selective extraction of uranium and plutonium.14–17 This selective extraction is achieved through a series of liquid–liquid extractions and is governed by the ligand tri-n-butyl phosphate (TBP).18–20The selectivity of hydrometallurgical reprocessing is defined by organic ligands like TBP, due to their affinity for chelation of specific metal ions.21,22 To enhance the capability of nuclear reprocessing, researchers have developed and tested a host of different ligands over a series of decades.23–26 Dialkylamide and diamide ligands are two successful groups of ligands for the removal of long-lived radionuclides, with the dialkylamide, DEHiBA (N,N-di-(2-ethylhexyl)isobutyramide) proving to be an ideal replacement for TBP.11,27–29 DEHiBA offers improved selectively for uranium devoid of extracting plutonium,30–32 thus enhancing proliferation resistance for the GANEX (Grouped Actinide Extraction) process.33,34 Notably, the GANEX second cycle flowsheets employ more complex ligands like the diamides to facilitate transuranic extraction downstream.35–37 Recent publications from Hall et al. highlight the continued interest and relevance of DEHiBA for the selective extraction of uranium, their research focussing on process intensification of uranium extraction, ensuring performance under various conditions.38,39
The specialist nature of DEHiBA makes it a high cost research material with limited suppliers. This is not only detrimental to the economics of advanced reprocessing, but also restricts access to research in this field, in particular large-scale performance testing that is necessary to further the technology readiness level. A less costly approach is to synthesise ligands like DEHiBA in-house using procedures outlined in the available literature,40 with Thiollet and Musikas being frequently referenced.41 This approach employs isobutyryl chloride (iBCl), triethylamine, and di(2-ethylhexyl)amine (DiEHA) to yield DEHiBA, via an effective method in a lab environment. Unfortunately, iBCl is currently more costly than alternative materials such as isobutyric acid (iBA), whilst also being highly toxic, halogenated, and violently reacting with water, thus being unfavourable for scale-up. These issues are avoidable through alternative synthetic pathways (Scheme 1). An ideal manufacture process for DEHiBA would minimise the use of toxic/hazardous reagents, avoid halogenated reagents and solvents whilst minimising process mass intensity (PMI) in order to reduce safety concerns and reduce environmental impact. The most desirable condition would minimise reagent cost, produce little trade-off between productivity and reaction mass efficiency (RME), whilst being high yielding, minimising by-products to reduce downstream purification costs.
 | ||
| Scheme 1 Four synthetic pathways to DEHiBA, (a–f) are the different routes that have been investigated in this work, where routes (c–e) represent one synthetic pathway but with different solvent systems. | ||
To identify the optimum route and conditions to DEHiBA, each process will be optimised and compared in this work, traditionally this can take months if not years to find an optimised solution depending on the chemistry and objectives chosen. However, recent advances at the interface between chemistry, chemical engineering and computer science has led to the development of platforms capable of rapid process development and optimisation.42–48 The emergence and success of these technologies has been largely driven and adopted by academia with the chemical industry to save valuable time and money needed for chemical development.49–51 Self-optimising flow reactors that utilise machine-learning algorithms in conjunction with online/inline analysis and a feedback loop are one of these technologies that has gained considerable interest of late.47,52 These advanced chemical reactors, automatically adjust operating conditions depending on the reaction outcome in order to optimise reaction performance.52–58 As traditional process optimisation campaigns are renowned for being research and labour intensive, the utilisation of these more efficient, machine learning driven technologies (Fig. 1), enables the reduction of chemical consumption, labour, risk, and optimisation time-lines.53,59,60 Furthermore, a continuous flow process facilitates continuous manufacture with enhanced process control, offering high product throughput with the beneficial ease of real-time process monitoring.47
 | ||
| Fig. 1 An illustration of a self-optimising flow reactor platform for the automated process optimisation of chemical reactions. | ||
In this work we have employed self-optimising flow reactor platforms to optimise key process metrics of the four synthetic pathways to DEHiBA. This data has enabled identification of the best performing route for the large-scale manufacture of DEHiBA in continuous flow. Tubular flow was utilised for its ease of access to more expansive operating windows such as pressures over 200 bar and temperatures well in excess of typical boiling points when compared to batch chemistry.61 This facilitated process intensification of each route and enhanced reaction kinetic understanding, all whilst enhancing process control and safety by minimising reaction volumes for controlling exothermic reactions. Batch chemistry was used as a screening tool to ensure reaction feasibility and suitability in continuous flow and was not optimised in this work. This work focussed on identifying the most cost effective, reagent efficient conditions that are suitable for large-scale continuous manufacture. The productivity of each process is of importance and has been optimised in this work, here Pareto fronts have been identified to understand any trade-offs between process metrics for each route.52 This is particularly important if there is a trade-off between cost or reagent efficiency and productivity, therefore Pareto fronts aid the identification of conditions that balance these metrics and enhance the understanding of each route, supporting the decision making process for scale-up. A combination of single- and multi-objective optimisation algorithms have been exploited to optimise product yield, reaction mass efficiency (RME), space–time yield (STY), and reagent cost per moles of DEHiBA produced. A range of high performing reaction conditions across the four synthetic pathways have been identified in this work to inform key decisions for an optimised synthesis of DEHiBA prior to future scale-up plans. This work further validates the power of self-optimising flow platforms by identifying a scalable, high-performance route to DEHiBA, a specialist ligand for advanced nuclear reprocessing, via the rapid optimisation of four synthetic pathways.
Results and discussion
Overview of optimisations and comparisons
This work compares routes (a–f) using the process metrics yield, RME, STY, and reagent cost throughout. It is improbable that one route and condition will provide the lowest cost whilst maximising yield, RME, and STY, therefore conditions that minimise trade-offs between key process metrics are optimum for scale-up. This section provides an overview of how each route has been optimised, whilst the parameter bounds for each optimisation are detailed in the ESI.† The solvated routes (a–d) were optimised at 0.01 mol dm−3 of DEHiBA to minimise chemical consumption and facilitate fair comparison between routes due to the effect of concentration on STY, a concentration limitation with route (b) defined the reaction concentration for (a–d). In Fig. 3, the minimum residence time was limited to 0.5 minutes for ease of STY comparison between routes (a–d). Maximum/minimum theoretical limits for each route are defined in the 2D metric comparison plots.As the ultimate driving force behind this work was to identify a cost-effective process to synthesise litres to tonnes of DEHiBA on demand, RME was utilised as the optimisation objective to achieve this. Simply minimising the amount of reagent used whilst maximising the amount of product meant that the algorithm located low reagent costs per mole of DEHiBA formed, omitting changes in reagent cost over time. The low cost of iBA added complexity to this for route (f), therefore reagent cost was instead minimised during this optimisation. Maximising STY was another key objective for this work due to its large role to play in the economics of a manufacture process.
Each optimisation utilised Latin hypercube sampling to initiate the optimisation,62 following this Bayesian optimisation was employed to maximise yield for (a–e) to ensure setup and parameter space feasibility.48,53,63 The core optimisation for each route was facilitated by the Thompson sampling efficient multi-objective optimization (TSEMO) algorithm to maximise STY and RME or reagent cost.55,64–68
The discussion for each route focusses on comparing the RME, STY Pareto fronts, reagent costs, and product yield to aid identification of optimum conditions. Comparisons of key process metrics for the different routes are illustrated by Fig. 3, whilst Fig. 4 compares the RME relative to the conditions explored for each route. STY, yield, and reagent cost, plots in the format of Fig. 4 can be found in the ESI† with additional process data such as Table S2.
Route (a). Acyl chloride route: synthesis from isobutyryl chloride
The most common synthetic route to DEHiBA found in the literature employs isobutyryl chloride (iBCl),40,41,69–71 as a highly reactive starting material capable of yielding the product in a single step (Scheme 1). The prevalence of this route meant that route (a) was the obvious starting point for this research. This chemistry was transitioned into continuous flow and optimised for comparison with the other routes in this work. Concerns with the environmental footprint, reactivity and toxicity of iBCl when considering scale-up are unfavourable, additionally the demanding safeguards required could heighten the cost of this route and burden plant design compared to alternative routes. Beneficially however, the utilisation of continuous flow here improves process safety via enhanced process control and improved heat transfer properties, enabling greater regulation of exothermic, runaway reactions.The reaction screening of route (a) was trialled in batch prior to a transition to continuous flow to verify the suitability of the reaction. Incompatibility issues were encountered with the majority of common solvents due to the precipitation of triethylamine hydrochloride, this forced the adoption of chloroform to solubilise this salt and ensure homogeneity. Again, this further reduced the desire to implement this route for large-scale manufacture due to the environmental drawbacks of chloroform.72,73
Multiple flow setups were investigated for the optimisation of route (a) (see ESI†), however the setup detailed in Fig. 2 proved most suitable, with many conditions reaching the maximum theoretical STY (374 g L−1 h−1), but falling slightly short of the maximum theoretical RME (69.4%). The fast reaction kinetics for this route granted almost no trade-off between RME and STY (Fig. 3), whereby the data formed a right angle along a STY of 374 g L−1 h−1 and an RME of 65% (Table 1). No trade-off was observed beyond 300 g L−1 h−1, however a gain in RME to 68% was observed around 280–300 g L−1 h−1, this equated to a reduction in reagent cost from £37 per mol to £35 per mol but at the loss of 70–90 g L−1 h−1. This breakthrough in RME was achieved with sub stoichiometric equivalents of iBCl and triethylamine at 150 °C. The preference for short residence times was clear, with product yield typically diminishing as residence time increased, likely due to the increased exposure of iBCl to elevated temperature. Ultimately this benefitted reaction performance due to minimal trade-off between these key process metrics.
 | ||
| Fig. 2 (Left) This illustrates the flow reactor setups for the self-optimisation of the solvated routes (a–d), where a reaction concentration of 0.01 M (DiEHA in the reactor) was maintained. All reagents in black text are used in all setups, whilst the colour coded reagents refer to the setups for each specific route as defined in Scheme 1 and the dashed boxes atop the setups. (Right) The solvent-free routes (e) and (f) were set up as illustrated, utilising solvent dilution pumps to enable quantitative online analysis. | ||
 | ||
Fig. 3 Space–time yield and reagent cost vs. reaction mass efficiency data demonstrating Pareto fronts for the solvated routes (a)  , (b) , (b)  , & (c) , & (c)  , where the dashed lines indicate the maximum theoretical limits for the respective process metrics. , where the dashed lines indicate the maximum theoretical limits for the respective process metrics. | ||

|
It was observed that the minimum residence time of 0.5 minutes limited the performance of route (a), therefore shorter residence times as low as 12 seconds were explored. This again resulted in product yields up to 99+%, no loss in RME, and product throughputs up to 944 g L−1 h−1 (Fig. S9, ESI†). This further highlights the rapid reaction kinetics of route (a) even at 0.01 mol dm−3. These shorter residence times also provided minor performance improvements, with RMEs of 66% even at 900+ g L−1 h−1, potentially due to the reduced exposure of iBCl to elevated temperature.
Six promising reaction conditions have been identified as candidates for scale up in Table 1. The most cost-effective condition, £35 per mol equated to the optimum RME of 67.8%, and a STY of 285 g L−1 h−1. Whilst the best STY and RME at 374 g L−1 h−1 and 64.8% respectively required just 35 °C, and a slight excess of iBCl and triethylamine. This reaction appears to be kinetically limited at 35 °C due to the need for greater equivalents to achieve similar performance to reactions beyond 100 °C. Whereas the final two conditions at 12 second residence times demonstrate some loss in performance due to the rapid reaction time despite temperatures well in excess of 35 °C.
In summary, route (a) demonstrates rapid reaction kinetics at this concentration, favouring shorter residence times with optimal temperature depending on the available equivalents of iBCl and triethylamine (Fig. 4). High yields were achieved throughout the optimisation and the insignificant trade-off between RME and STY provided hard to beat process metrics at this concentration, thus route (a) is a convenient and effective route for the lab scale synthesis of DEHiBA. Nevertheless, the hazards and halogenated nature of iBCl and chloroform introduce avoidable complications and concerns. Therefore, the following sections explore and optimise alternative routes to identify an optimised route to DEHiBA to improve access to large-scale reprocessing testing that is currently economically restrictive if purchasing ligands from commercial suppliers.
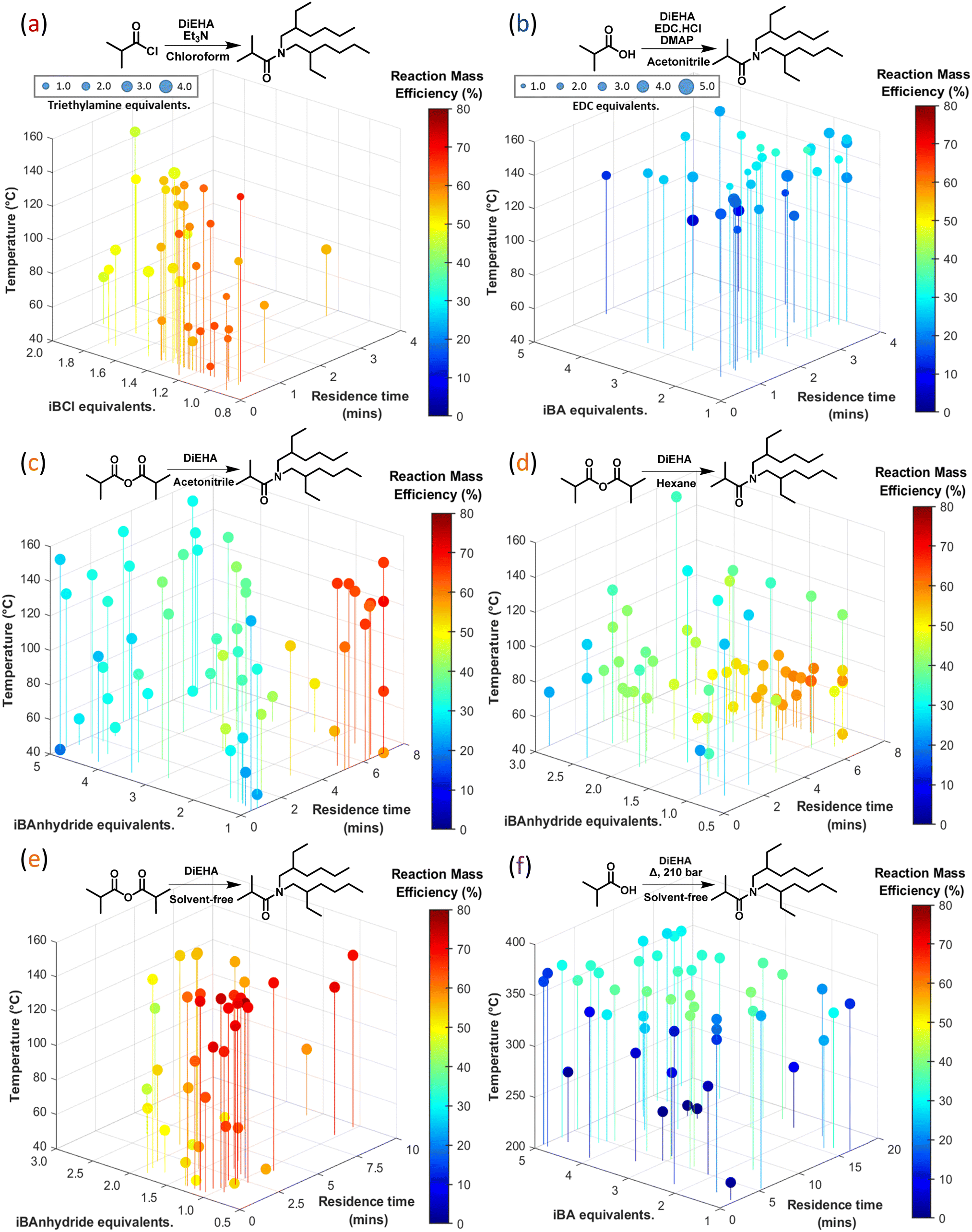 | ||
| Fig. 4 Six 4/5D plots demonstrating the reaction mass efficiency for routes (a–f), the synthetic route for each is defined above the plots for ease of comparison. A consistent colour bar is illustrated throughout, ranging between 0–80%, whilst the x, y, z and size ranges are subject to the parameter space for each optimisation, finally a reduced dataset has been presented for clarity. Whilst Fig. S2–S4 in the ESI† illustrate reagent cost, space–time yield and DEHiBA yield respectively for these reactions. | ||
Route (b). The coupling reagent approach: EDC.HCl mediated synthesis
Coupling reagents are popular in the pharmaceutical industry for amide/peptide and ester formations, with their highly effective and robust nature seemingly outweighing their inherent atom inefficiency.74 This popularity is prevalent even for the large-scale manufacture of pharmaceuticals as highlighted by Dunetz et al.75 where popular coupling reagents like thionyl chloride, 1,1′-carbonyldiimidazole (CDI) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC.HCl) are discussed and compared. The dominance of EDC.HCl for amide bond formations in the pharmaceutical industry is largely due to its ease of purification and applicability to a broad range of substrates. These characteristics are beneficial for the successful manufacture of troublesome tertiary amides and may prove effective for the sterically bulky DEHiBA target in this case. Further to these benefits, Pfizer's publication discussing amide bond formations in continuous flow,76 identifies EDC.HCl as one of the few coupling reagents suitable for continuous flow. Therefore, route (b) was attempted and optimised in continuous flow to assess this alternative route to DEHiBA that starts with isobutyric acid (iBA), a more benign and cost-effective raw material than iBCl.Batch screening led to the adoption of acetonitrile (MeCN) as the reaction solvent due to comparatively good product yields and the limited solubility of EDC.HCl in most organic solvents (see ESI†).77 Further solubility limitations with DiEHA solutions required the addition of iBA to exceed 0.1 mol dm−3. Additive screening in batch led to the addition of 4-dimethylaminopyridine (DMAP) to the flow setup, as 5 mol% saw product yields rise from 50 to 99+% at room temperature.
The optimisation of route (b) in continuous flow required the exploration of a wider equivalent range to route (a), as ≥2 equivalents of both EDC.HCl and iBA was necessary to achieve comparable product yields. This inefficient reagent excess was extreme when compared to a batch setup, whereby equimolar amounts of DiEHA, iBA and EDC.HCl, yielded almost complete conversion to DEHiBA, although a 6 hour reaction time was required. The setup also demonstrated a requirement for temperatures >75 °C, else little or no product formation for residence times as long as 10 minutes, whilst the best conditions showed a preference for >140 °C (Table 2). We hypothesise that the elevated temperature required by this setup encourages the formation of N-acylurea, an unwanted, unreactive by-product formed by the rearrangement of O-acylisourea (Scheme S3, ESI†).78 This hypothesis was confirmed via kinetic batch studies, where an increase in temperature led to the loss of product yield (Fig. S11, ESI†).

|
Although a number of competitive yields (>90%) were achieved here in continuous flow the typical requirement for large reagent excesses proved to be detrimental to the RME and hence results in an increased reagent cost for this route. The best RMEs (33–35%, 59% of the theoretical maximum) were achieved when using approximately 2 equivalents of EDC.HCl and iBA, this led to uncompetitive reagent costs as low as £77 per mol. These poor RME and reagent cost metrics result in an uneconomic manufacture route with this setup.
As it was unclear why an excess of iBA and EDC.HCl is needed in continuous flow but not in batch, further kinetic studies investigated the effect of concentration on the batch reaction. It was observed that over a concentration range of 0.15 to 0.01 mol dm−3, a drop in yield from 99+% to 42% was observed respectively (Fig. S11, ESI†). We hypothesise that this is due to the reduction in the rate of collision between DiEHA and the activated intermediate of iBA (O-acylisourea) as concentration reduces, whereas the rate of rearrangement from the O-acylisourea to N-acylurea is unaffected by this change and remains constant at the same temperature. Therefore, as concentration reduces, so too does the yield due to increased by-product formation. We expect that the RME would improve if this optimisation was conducted at a greater concentration. To improve the competitiveness of this reaction we focussed on increasing the reservoir concentration of EDC.HCl as this largely limited the reaction concentration. The addition of bases such as di-isopropyl ethylamine (DIPEA) resulted in a dramatic improvement in solubility of EDC.HCl, unfortunately this led to a 60–70% reduction in reaction yield in addition to further setup challenges. Consequently, attempts to increase the reaction concentration were abandoned and work progressed to routes (c–f) in hope these would provide competitiveness. Ultimately, the current flow setup limitations mean this reaction is better suited to batch chemistry if RME or reagent cost are important process metrics.
Six conditions that exhibit the most promise for this route are detailed in Table 2. The lack of competitiveness of this route led to the pursuit of other synthetic routes before further continued on the optimisation of route (b) to increase the reaction concentration. The next route utilises isobutyric anhydride directly, removing the need for EDC.HCl.
Routes (c) & (d). A direct route from isobutyric anhydride–acetonitrile and hexane solvated reactions
The direct combination of DiEHA and isobutyric anhydride (iBAnhydride) for the synthesis of DEHiBA is pursued here. Theoretically this synthetic pathway has the capability to outperform the reagent cost and RME of routes (a) and (b), with the added benefits that iBAnhydride is a readily available, cost-effective reagent, with reduced hazards relative to iBCl whilst also being ideal for continuous flow due to the enhanced heat transfer that facilitates the control of exothermic reactions. Though it should be noted that for each equivalent of iBAnhydride consumed an equivalent of iBA is produced, burdening the purification process.Preliminary batch studies were limited due to the nature of the exothermic, runaway reactions. No solubility issues were encountered unlike routes (a) and (b), opening the reaction to a range of solvents and facilitating a quick transition of this reaction to continuous flow. The diverse solvent compatibility of this reaction enabled the pursuit of two optimisation setups to evaluate the effect of solvent on this chemistry.
Acetonitrile and hexane, routes (c) and (d) respectively were chosen as the solvents for this comparison. The optimisation of route (c) showcased excellent conversion even at low equivalents (<2), with nearly half of the 116 conditions attaining yields ≥90%. The best conditions for RME and reagent cost exhibited a distinct preference for 100–130 °C, low iBAnhydride equivalents (1.0–1.8), and longer residence times (up to seven minutes). Shorter residence times were typically poorer yielding unless combined with greater equivalents, though this largely reduced the RME and added cost for the price of improving productivity. Consequently, this gave rise to a significant trade-off between RME and STY for this route, a stark contrast to route (a). In this case the cost for improving the STY dramatically reduces the RME, for example a maximum RME of 70.4% at a STY of 24 g L−1 h−1, reduced dramatically to 45.6% for a minor increase in STY to 51 g L−1 h−1 or an RME of 26.5% to achieve a STY of 326 g L−1 h−1. This trade-off was uncompetitive with route (a), as illustrated in Fig. 3, if both productivity and cost are valued as important.
Route (c) is however the most cost-effective manufacture route to DEHiBA with reagent costs as low as £26 per mol. Still, a reasonable STY is necessary for continuous large-scale manufacture to be viable. Therefore, this setup requires further optimisation before it can be scaled up.
Six of the best conditions for route (c) are defined in Table 3 covering a range of performance metrics for a more holistic understanding of this route. The trade-off between RME and STY is further emphasised by this data, whilst the high temperature and equivalent dependence for improved STY is best highlighted by this data.
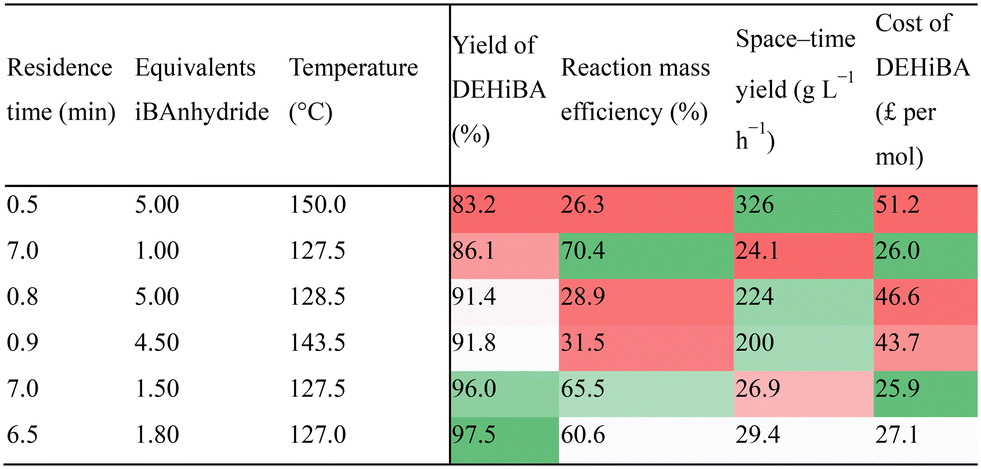
|
The second optimisation of this synthetic pathway utilised hexane as the solvent (route (d)) to improve process understanding in an attempt to improve the RME, STY Pareto front. Solvent often plays a highly influential role in promoting and controlling chemical reactions,79,80 such as influencing key process metrics like cost and purity. Therefore, the understanding of solvent effect on a given reaction is of great interest for process optimisation and cost minimisation. In this case the change in solvent results not only results in a shift in the conditions required to achieve the optimum RME but also a difference in the optimum RME. Fig. 4 illustrates this change, whereby the reaction in hexane (route (d)) favours a lower temperature of 70 °C to achieve the optimum RME, with losses in yield for similar conditions but greater temperature. The optimisation of route (d) was completed in less than 48 hours, emphasising the power of self-optimising flow reactor platforms by relocating process optima for a new solvent system.
Overall, routes (c) and (d) largely demonstrated similar trends for the process metrics of interest, with the exception of the difference in temperature preference between these routes. Route (d) did however underperform with respect to (c), potentially due to the reduced parameter space for the optimisation of route (d), although the STY, RME Pareto front trends are in agreement with one another (Fig. S15, ESI†). Table 4 defines six of the best performing conditions for route (d) for contrast with Table 3. Route (c) proves to be the most promising candidate to compete with route (a), therefore further optimisation of this route is needed to improve the productivity.

|
Comparison of solvated routes (a), (b), (c) and (d)
To summarise the performance of routes (a–d), this section will briefly compare these routes. Route (a) demonstrated near perfect performance with little to no trade-off between RME and STY resulting in unmatched performance when considering all process metrics equally. Route (a) even proved highly successful for 12 second residence times, producing excellent product throughput given the concentration in addition to near perfect RME. The downfalls of route (a) arose from iBCl as it is around 3 times the price of iBAnhydride, limiting the reagent cost to £35 per mol whilst route (c) was capable of £26 per mol. Further to this the hazards of iBCl outweighed those of iBAnhydride, these factors are of great importance when moving towards large-scale manufacture as they add cost and risk. Unfortunately however, the large trade-off between RME and STY for route (c) largely hinders its performance making it unfavourable to scale-up at this stage despite the low reagent cost involved with the synthesis. In an attempt to improve this the setup of route (c) was further optimised, the results of which led to route (e), a solvent-free synthesis that utilises the same synthetic pathway as routes (c) and (d) but with massive gains in concentration and hence reaction kinetics. Route (a) was not scaled up in a similar way due to the corrosive nature of the chemistry and safety concerns with these neat materials.Route (b) was inspired by the pharmaceutical industry, with the hope that EDC.HCl would afford an alternative, competitive manufacture route to DEHiBA. Unfortunately, the performance of this route was far from competitive due to limitations of this chemistry in continuous flow and the current setup for this optimisation. The batch synthesis of route (b) was however very reagent and temperature efficient requiring little to no reagent excess for near complete conversion at room temperature, although 6 hour reaction times were necessary.
Towards scale-up: a solvent-free synthesis of route (e)
In an effort to enhance the performance and scalability of route (c/d), process intensification led to a productive and efficient solvent-free synthesis with close to perfect PMI, albeit disregarding purification. The flow setup was tailored towards the optimisation via the inclusion of solvent pumps following the reactor, a continuous manufacture platform would not need these (Fig. 2). Undoubtedly, the elimination of solvent removes an element of process control in the form of a heat sink, however the excellent heat transfer of the tubular flow platform facilitates the pursuit of this chemistry. This undertaking carried too much risk in a batch reactor so was conducted in continuous flow to enable sufficient cooling after the reactor, this ensured a high level of control over the outlet temperature of the crude product and no runaway reactions were observed even without solvent dilution.Initially the optimisation of this route was confined to 1–10 minute residence times, however this route now exhibited a preference for short residence times to achieve optimum performance, therefore this limit was extended to 0.5 minutes part way through the optimisation. Both these optimisations yielded excellent process metrics, demonstrating an extensive improvement over the solvated syntheses. The shift in optimisation parameters to 0.5 minute residence times resulted in a STY improvement from 37.1 and 74.7 kg L−1 h−1, with RMEs of 72.2 and 72.6% respectively, plus reagent costs at £25 per mol (Table 5), demonstrating no loss in RME at these STYs. STY was the most notable improvement of route (e) over routes (c/d), this was achieved due to the greater reaction concentration and the preference towards shorter reaction times, a consequence of the concentration increase.
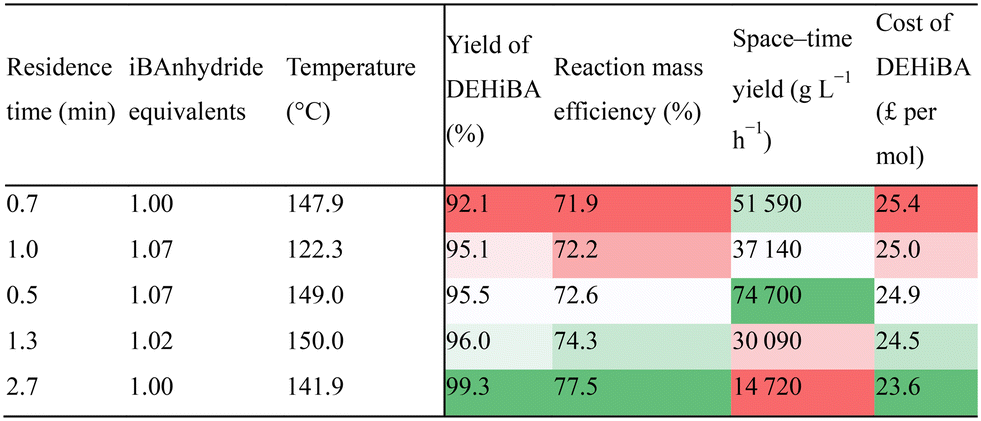
|
A further illustration of the enhanced performance is the optimum RME of 77.5%, this equated to the lowest reagent cost per mole of DEHiBA at £23.60, and practically complete product conversion, with both RME and reagent cost also close to their theoretical maxima. A 2.7 minute residence time was however required for this, resulting in a STY of 14.7 kg L−1 h−1, a large improvement over route (c), but a far from the optimum STY. Advantageously, the STY, RME trade-off for route (e) presented a considerably different 2D profile to routes (c/d) (Fig. 5), due to the shift in preference to shorter residence times for optimum overall performance. This minimised the RME loss with increasing STY, but notably the trade-off was more pronounced at lower STY as demonstrated by Fig. 4 with an RME loss of 5%. The minimisation of this trade-off is highly desirable, facilitating an efficient, low cost, and highly productive manufacture route to DEHiBA.
 | ||
Fig. 5 Space–time yield vs. reaction mass efficiency data with Pareto fronts for routes (e)  and (f) and (f)  , with their maximum theoretical RME limits as dashed lines. , with their maximum theoretical RME limits as dashed lines. | ||
 | ||
Fig. 6 Reagent cost and DEHiBA yield metrics vs. reaction mass efficiency comparison plots for routes (a)  , (b) , (b)  , (e) , (e)  and (f) and (f)  . . | ||
The optimum conditions for this route were all identified above 100 °C, with roughly 1 equivalent of iBAnhydride, and a variety of relatively short residence times. These conditions have led to process metrics that possess exceptional performance, with STY figures in the tens of kilograms per litre per hour in comparison with the solvated routes that were only capable of hundreds of grams per litre per hour. Finally, the RME here outperforms all other routes and provides a low reagent cost, with single reaction conditions capable of desirable RME and STY.
The optimum RME and STY conditions were utilised in two continuous runs on the flow platform but without dilution solvent to manufacture 5 litres of crude DEHiBA without the presence of biphenyl. Yields were determined via an external standard to be 99.7% and 97.2% for the optimum RME of 77.5% and STY of 74.7 kg L−1 h−1 respectively throughout the 32 and 5 hour continuous runs. The slight improvement in yield led to an improvement in other process metrics, especially STY for the second run up to 75.8 kg L−1 h−1 however the PMIs are most noteworthy for these runs at 1.29 and 1.36 g g−1 respectively due to the slight excess of iBAnhydride and elimination of solvent facilitating the lessened environmental impact of this process.
Route (f). Solvent-free direct thermal amidation
Route (f) is an alternative solvent-free route with the potential to achieve a RME of 94.4% and costs as low as £18.80 per mol due to the low cost of iBA, with water as the by-product of this coupling. Therefore, this direct thermal amidation was screened and optimised in the search for another competitive route to DEHiBA, especially as the solvent-free synthesis of route (a) presented unnecessary risks and precipitation issues that required the omission or replacement of triethylamine whilst not offering cost or RME improvements.Ideally the preparation of an amide bond would proceed through the direct coupling of carboxylic acid and amine, the only by-product being water. Unfortunately a large energy barrier must be overcome to achieve this, such as temperatures in excess of 150 °C. Often, such temperatures are too extreme and cause chemical degradation, thus limiting the applicability of this methodology, therefore this route is not common-practice for the creation of amide bonds.74 In this work the stainless steel reactor tubing was pressurised to 210 bar to access temperatures up to 370 °C, this ensured that the reagents would not undergo a phase change to guarantee accurate residence times and reproducibility.
Preliminary temperature screening in the continuous flow reactor identified minimal product formation below 250 °C, whilst temperatures above 370 °C required greater reactor pressure. The crude product from these reactions eluted at 25 °C despite reactor temperatures of up to 370 °C, whilst the emission of gas was noted after the final BPR (back pressure regulator) and was identified as gaseous carbon dioxide and propane, products of the degradation of iBA. Additional signals were observed during the analysis and were confirmed to be the thermal degradation products of DEHiBA: N-(2-ethylhexyl)isobutyramide and 3-methylheptane (Fig. S19, ESI†), the formation of which increased with relation to increasing temperature and residence time. This degradation adds complexity to the purification of DEHiBA as this amide may interfere with the extraction of uranium.
Two reactor volumes were employed for the optimisation of this route to explore a wide residence time range between 1 and 18 minutes. A wide equivalent range (1–5) proved necessary as the reaction demonstrated poor conversion at low iBA equivalents. Yields of up to 77.8% were achieved, with the highest yielding conditions proving to be the most cost effective at £27.20 per mol, despite the need for 5 equivalents of iBA. The low reagent cost of iBA results in a lesser relationship between RME and reagent cost, instead reagent cost reduced with increasing yield. This difference is also due to the poorer conversions achieved by this route.
Overall, route (f) performed poorly at the shortest residence times, with the best STY conditions requiring high equivalents of iBA and leading to poor yields, RMEs and reagent costs. Consequently, a large trade-off between STY and the other process metrics is apparent and largely affects the performance of this route. This chemistry demonstrates a preference for temperatures between 330 and 370 °C, with the best yields demanding 340–350 °C, roughly 5 equivalents of iBA, and a range of residence times between 5–13 minutes. Interestingly as the temperature neared 370 °C, lower residence times granted better yields, likely due to the reduced thermal degradation of DEHiBA.
Table 6 defines the best conditions for route (f), highlighting the best process metrics and conditions along the key Pareto-fronts. In conclusion, route (f) offers a reagent cost competitive route to DEHiBA, however the additional capital for equipment, heating and safeguards require significant consideration when comparing to other routes. Additionally, the poor STY of route (f) compared to route (e) results in an uncompetitive manufacture route despite the low reagent cost.

|
Conclusions
This research has utilised an advanced, fast-paced approach to successfully optimise four synthetic pathways to DEHiBA in continuous flow, whereby a favourable manufacture route to DEHiBA has been identified for large scale manufacture. Initial optimisation work, routes (a–d), employed solvent to control these reactions and their concentration for fair cross-comparison. Pareto fronts for key process metrics were analysed and provided insight into the performance of each route by highlighting optimum and scalable reaction conditions. Routes (a) and (c) were most promising in the search for a cost-effective, scalable route to DEHiBA, whereas route (b) underperformed in continuous flow, and the solvent choice for route (d) resulted in performance loss compared to (c).Due to limitations and issues with route (a), route (c) was developed into route (e) via the mitigation of solvent, facilitated by the excellent heat transfer properties of tubular flow this enhanced all key process metrics, yielding a very desirable manufacture route. The increased reaction concentration and lower reagent cost of iBAnhydride in comparison to iBCl resulted in a route capable of manufacturing DEHiBA for just £23.60 per mol (not bulk costs), with product throughputs up to 75.8 kg L−1 h−1. Most advantageously, the trade-off between RME and STY has been suppressed due to the improved reaction kinetics, where only a 5% loss in RME is observed along the Pareto front, to contrast, route (c) suffers from a 44% loss. Notably, further process optimisation of route (a) could improve competitiveness through base optimisation and reaction concentration, however this was not pursued here due to the increased risk and environmental footprint driving us away from this chemistry. The solvent-free process in route (e) has largely aided develop a cleaner more economical well-rounded and scalable manufacture route.
Route (f) was designed and optimised as another solvent-free route to DEHiBA, possessing the potential to be the most cost effective and atom efficient route by directly coupling iBA and DiEHA with temperatures up to 370 °C and a pressure of 210 bar. Thermal degradation of iBA resulted in RME losses, whilst the relatively slow reaction kinetics resulted in uncompetitive STYs with route (e). Fortunately, the low cost of iBA afforded reagent costs as low as £27.20 per mol, however the requirement for relatively extreme operating conditions burdens the capital and operational cost, whilst the degradation products from the thermal amidation add further complexity to purification, thus scalability is less favourable.
The methodology used to optimise these routes aids to improve access to specialist chemicals like DEHiBA to promote large-scale testing and advance the technology readiness level of advanced nuclear reprocessing technologies like GANEX. This methodology largely reduces process optimisation timelines and overall costs, whilst providing holistic understandings of each process in the lead up to large-scale manufacture. The application of this approach to other promising ligands for nuclear reprocessing will aid improve accessibility to these specialist molecules that require large scale testing in order to advance nuclear reprocessing so that a more diverse range of radionuclides can be recovered from used nuclear fuel in the future. In this work continuous flow has facilitated the safe exploration of these routes, with the large datasets gathered in this research available in the ESI† for further use by researchers, whereby alternative conditions may be preferred for their own synthesis of DEHiBA.
Future work
As this work solely focusses on the formation of the amide bond by exploring more economically and environmentally friendly route to DEHiBA, to further improve the economics the manufacture of DiEHA (di-2-ethylhexylamine) should be investigated to improve its cost. Additionally, this work does not include purification work which will add cost and complexity to the manufacture, thus future work should investigate and add to this work to provide a complete manufacture for DEHiBA along with cost and PMI for this.Experimental
Chemicals
All of the following commercially available compounds were purchased and used without further purification. N,N-Di-(2-ethylhexyl)isobutyramide (DEHiBA; >99%) was purchased from Technocomm Ltd. Di(2-ethylhexyl)amine (DiEHA, 99%), isobutyric acid (iBA; 99+%) and triethylamine (99%) were purchased from Acros Organics. 4-Dimethylaminopyridine (DMAP; 99%), acetonitrile (MeCN; HPLC grade), N,N-dimethylformamide (DMF; extra pure), hexane (97%) and chloroform (99+%) were purchased from Fisher Scientific Ltd. Isobutyric anhydride (iBAnhydride, 99%), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC.HCl; 99%), and isobutyryl chloride (iBCl; 97%) were purchased from Fluorochem. Biphenyl (99+%) was purchased from Merck Life Science UK Ltd.Reagent costs were acquired as of March 2022 and can be found in the ESI.†
The self-optimising flow reactor platform
Reagents were pumped using JASCO PU-2080 dual piston HPLC pumps and flow paths were mixed using Swagelok SS-100-3 tee-pieces. Reactors of desired volumes were made from either PFA, PTFE or 316 stainless steel tubing (1/16′′ OD), these were fitted to a cylindrical aluminium block and heated via a Eurotherm 3200 temperature controller. Sampling was achieved using a VICI Valco EUDA-CI4W sample loop (4-port) with 0.5–0.06 μL injection volume. Reactions were maintained under fixed back pressures using an Upchurch Scientific back pressure regulators (100/250 psi), whilst route (e) employed the Tescom™ 26-1762-22 control pressure regulator to achieve a pressure of 210 bar. Quantitative analysis was performed on an Agilent 1260 Infinity II series HPLC instrument fitted with an Agilent Poroshell 120 EC-C18 reverse phase column (5 cm length, 4.6 mm ID and 2.7 μm particle size), HPLC method can be found in the ESI.† The automated platform was controlled using a custom written MATLAB program, the optimisation algorithms were also written and implemented in MATLAB. Calibration curves were obtained to quantify the analysis using biphenyl as the internal standard.MATLAB was used to control pump flow rates, reactor temperature and sampling. For each iteration the reactor was allowed to stabilize at the desired operating temperature; the pumps were set to the required flow rates and left for three reactor volumes to reach steady state; then finally, the sampling valve was triggered alongside HPLC analysis. To minimize the duration and material consumption per iteration: (i) pump flow rates were reduced to a minimum during the heating/cooling of the reactor; (ii) initial LHC experiments were sorted in order of increasing temperature; (iii) sequential LHC experiments were started whilst analysis of the previous experiment was running. Responses for each objective were calculated from HPLC chromatograms and used to inform the optimisation algorithm of the reactions' outcome and generate the next set of reaction conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TS would like to thank the EPSRC and GREEN CDT for funding this research, EP/S022295/1, with special thanks to ADC, RAB and BCH for their invaluable guidance. RAB was supported by the Royal Academy of Engineering under the Research Chairs and Senior Research Fellowships scheme. This work was funded, in part, by the EPSRC project “Cognitive Chemical Manufacturing” EP/R032807/1.Notes and references
- UK Government, UK Energy White Paper: Powering Our Net Zero Future, UK Government, London, 2020 Search PubMed.
- IAEA, Nuclear Technology Review, https://IAEA.org, 2020 Search PubMed.
- A. Peakman and B. Merk, Energies, 2019, 12, 3664 CrossRef CAS.
- P. Ekins, Energy Policy, 2004, 32, 1891–1904 CrossRef.
- J. B. Greenblatt, N. R. Brown, R. Slaybaugh, T. Wilks, E. Stewart and S. T. McCoy, Annu. Rev. Environ. Resour., 2017, 42, 290–308 CrossRef.
- A. Cherp, V. Vinichenko, J. Jewell, M. Suzuki and M. Antal, Energy Policy, 2017, 101, 612–628 CrossRef.
- M. Gong, Y. Gao, L. Koh, C. Sutcliffe and J. Cullen, Int. J. Prod. Econ., 2019, 217, 88–96 CrossRef.
- Z. Liu, Z. Deng, S. J. Davis, C. Giron and P. Ciais, Nat. Rev. Earth Environ., 2022, 3, 217–219 CrossRef.
- C. Poinssot, S. Bourg, N. Ouvrier, N. Combernoux, C. Rostaing, M. Vargas-Gonzalez and J. Bruno, Energy, 2014, 69, 199–211 CrossRef.
- P. Wattal, Prog. Nucl. Energy, 2017, 101, 133–145 CrossRef CAS.
- M. Carrott, C. Maher, C. Mason, M. Sarsfield, D. Whittaker and R. Taylor, Solvent Extr. Ion Exch., 2023, 41, 88–117 CrossRef CAS.
- J. M. Pearce, Sustainability, 2012, 4, 1173–1187 CrossRef.
- C. W. Forsberg, Prog. Nucl. Energy, 2009, 51, 192–200 CrossRef.
- I. Denniss and A. Jeapes, in The nuclear fuel cycle from ore to wastes, 1996 Search PubMed.
- K. L. Nash and G. J. Lumetta, Advanced separation techniques for nuclear fuel reprocessing and radioactive waste treatment, Elsevier, 2011 Search PubMed.
- R. Herbst, P. Baron and M. Nilsson, in Advanced separation techniques for nuclear fuel reprocessing and radioactive waste treatment, Elsevier, 2011, pp. 141–175 Search PubMed.
- M. F. Simpson and J. D. Law, in Nuclear Energy, Springer, 2018, pp. 187–204 Search PubMed.
- E. Aneheim, C. Ekberg, A. Fermvik, M. R. S. J. Foreman, B. Grűner, Z. Hájková and M. Kvičalová, Solvent Extr. Ion Exch., 2011, 29, 157–175 CrossRef CAS.
- G. Modolo, A. Wilden, A. Geist, D. Magnusson and R. Malmbeck, Radiochim. Acta, 2012, 100, 715–725 CrossRef CAS.
- R. S. Herbst, P. Baron and M. Nilsson, in Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, 2011, pp. 141–175, DOI:10.1533/9780857092274.2.141.
- J. M. Mckibben, Radiochim. Acta, 1984, 36, 3–16 CrossRef CAS.
- W. B. Lanham and T. C. Runion, Purex Process for Plutonium and Uranium Recovery (Technical Report), https://OSTI.GOV, 1949 Search PubMed.
- A. Geist, J.-M. Adnet, S. Bourg, C. Ekberg, H. Galán, P. Guilbaud, M. Miguirditchian, G. Modolo, C. Rhodes and R. Taylor, Sep. Sci. Technol., 2021, 56, 1866–1881 CrossRef CAS.
- T. Lyseid Authen, J.-M. Adnet, S. Bourg, M. Carrott, C. Ekberg, H. Galán, A. Geist, P. Guilbaud, M. Miguirditchian and G. Modolo, Sep. Sci. Technol., 2022, 57, 1724–1744 CrossRef CAS.
- R. Garbil, S. Bourg, A. Geist, J.-M. Adnet, C. Rhodes, B. C. Hanson, C. Davies and D. Diaconu, EPJ Nucl. Sci. Technol., 2020, 6(35), 1–8 Search PubMed.
- A. Geist, R. Taylor, C. Ekberg, P. Guilbaud, G. Modolo and S. Bourg, Procedia Chem., 2016, 21, 218–222 CrossRef.
- J. Martinez-Val, P&T Roadmap proposal for Advanced Fuel Cycles leading to a Sustainable Nuclear Energy - Syntheses Report, Euratom, 2008 Search PubMed.
- R. Taylor, W. Bodel, L. Stamford and G. Butler, Energies, 2022, 15, 1433 CrossRef CAS.
- A. Rout, K. Venkatesan, M. Antony and P. V. Rao, Sep. Sci. Technol., 2016, 51, 474–484 CrossRef CAS.
- M. Carrott, A. Geist, X. Heres, S. Lange, R. Malmbeck, M. Miguirditchian, G. Modolo, A. Wilden and R. Taylor, Hydrometallurgy, 2015, 152, 139–148 CrossRef CAS.
- M. Miguirditchian, C. Sorel, B. Camès, I. Bisel, P. Baron, D. Espinoux, J. Calor, C. Viallesoubranne, B. Lorrain and M. Masson, proceedings from GLOBAL 2009, 2009, pp. 1032–1035 Search PubMed.
- R. Taylor, M. Carrott, H. Galan, A. Geist, X. Hères, C. Maher, C. Mason, R. Malmbeck, M. Miguirditchian, G. Modolo, C. Rhodes, M. Sarsfield and A. Wilden, Procedia Chem., 2016, 21, 524–529 CrossRef.
- J. Drader, G. Saint-Louis, J. M. Muller, M. C. Charbonnel, P. Guilbaud, L. Berthon, K. M. Roscioli-Johnson, C. A. Zarzana, C. Rae, G. S. Groenewold, B. J. Mincher, S. P. Mezyk, K. McCann, S. G. Boyes and J. Braley, Solvent Extr. Ion Exch., 2017, 35, 480–495 CrossRef CAS.
- A. Leoncini, J. Huskens and W. Verboom, Chem. Soc. Rev., 2017, 46, 7229–7273 RSC.
- M. Carrott, K. Bell, J. Brown, A. Geist, C. Gregson, X. Hères, C. Maher, R. Malmbeck, C. Mason and G. Modolo, Solvent Extr. Ion Exch., 2014, 32, 447–467 CrossRef CAS.
- K. Bell, C. Carpentier, M. Carrott, A. Geist, C. Gregson, X. Hérès, D. Magnusson, R. Malmbeck, F. McLachlan and G. Modolo, Procedia Chem., 2012, 7, 392–397 CrossRef CAS.
- R. Taylor, M. Carrott, H. Galan, A. Geist, X. Hères, C. Maher, C. Mason, R. Malmbeck, M. Miguirditchian and G. Modolo, Procedia Chem., 2016, 21, 524–529 CrossRef.
- G. B. Hall, N. P. Bessen, P. R. Zalupski, E. L. Campbell, T. S. Grimes, D. R. Peterman and G. J. Lumetta, Solvent Extr. Ion Exch., 2023, 1–19 Search PubMed.
- G. B. Hall, E. L. Campbell, N. P. Bessen, T. R. Graham, H. Cho, M. RisenHuber, F. D. Heller and G. J. Lumetta, Inorg. Chem., 2023, 62, 6711–6721 CrossRef CAS.
- G. M. Gasparini and G. Grossi, Solvent Extr. Ion Exch., 1986, 4, 1233–1271 CrossRef CAS.
- G. Thiollet and C. Musikas, Solvent Extr. Ion Exch., 1989, 7, 813–827 CrossRef CAS.
- N. Cherkasov, Y. Bai, A. J. Expósito and E. V. Rebrov, React. Chem. Eng., 2018, 3, 769–780 RSC.
- B. Lin, J. L. Hedrick, N. H. Park and R. M. Waymouth, J. Am. Chem. Soc., 2019, 141, 8921–8927 CrossRef CAS PubMed.
- C. J. Taylor, A. Baker, M. R. Chapman, W. R. Reynolds, K. E. Jolley, G. Clemens, G. E. Smith, A. J. Blacker, T. W. Chamberlain, S. D. R. Christie, B. A. Taylor and R. A. Bourne, J. Flow Chem., 2021, 11, 75–86 CrossRef CAS.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. Zondag and T. Noël, Chem. Rev., 2021, 122, 2752–2906 CrossRef PubMed.
- S. Langner, F. Häse, J. D. Perea, T. Stubhan, J. Hauch, L. M. Roch, T. Heumueller, A. Aspuru-Guzik and C. J. Brabec, Adv. Mater., 2020, 32, 1907801 CrossRef CAS.
- M. Christensen, L. P. Yunker, F. Adedeji, F. Häse, L. M. Roch, T. Gensch, G. dos Passos Gomes, T. Zepel, M. S. Sigman and A. Aspuru-Guzik, Commun. Chem., 2021, 4, 112 CrossRef.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS.
- N. Holmes, G. R. Akien, A. J. Blacker, R. L. Woodward, R. E. Meadows and R. A. Bourne, React. Chem. Eng., 2016, 1, 366–371 RSC.
- A. D. Clayton, L. A. Power, W. R. Reynolds, C. Ainsworth, D. R. Hose, M. F. Jones, T. W. Chamberlain, A. J. Blacker and R. A. Bourne, J. Flow Chem., 2020, 10, 199–206 CrossRef.
- H. L. Carter, A. W. Connor, R. Hart, J. McCabe, A. C. McIntyre, A. E. McMillan, N. R. Monks, A. K. Mullen, T. O. Ronson and A. Steven, React. Chem. Eng., 2019, 4, 1658–1673 RSC.
- A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Chem. Eng. J., 2018, 352, 277–282 CrossRef CAS.
- A. D. Clayton, J. A. Manson, C. J. Taylor, T. W. Chamberlain, B. A. Taylor, G. Clemens and R. A. Bourne, React. Chem. Eng., 2019, 4, 1545–1554 RSC.
- M. I. Jeraal, N. Holmes, G. R. Akien and R. A. Bourne, Tetrahedron, 2018, 74, 3158–3164 CrossRef CAS.
- P. Müller, A. D. Clayton, J. Manson, S. Riley, O. S. May, N. Govan, S. Notman, S. V. Ley, T. W. Chamberlain and R. A. Bourne, React. Chem. Eng., 2022, 7, 987–993 RSC.
- A. D. Clayton, E. O. Pyzer-Knapp, M. Purdie, M. F. Jones, A. Barthelme, J. Pavey, N. Kapur, T. W. Chamberlain, A. J. Blacker and R. A. Bourne, Angew. Chem, Int. Ed., 2023, 62, e202214511 CrossRef CAS PubMed.
- O. J. Kershaw, A. D. Clayton, J. A. Manson, A. Barthelme, J. Pavey, P. Peach, J. Mustakis, R. M. Howard, T. W. Chamberlain and N. J. Warren, Chem. Eng. J., 2023, 451, 138443 CrossRef CAS.
- D. Fabry, E. Sugiono and M. Rueping, React. Chem. Eng., 2016, 1, 129–133 RSC.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Science, 2018, 361, 1220–1225 CrossRef PubMed.
- S. V. Ley, Catal. Sci. Technol., 2016, 6, 4676–4677 RSC.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem, Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- V. R. Joseph and Y. Hung, Stat. Sin., 2008, 171–186 Search PubMed.
- P. Jorayev, D. Russo, J. D. Tibbetts, A. M. Schweidtmann, P. Deutsch, S. D. Bull and A. A. Lapkin, Chem. Eng. Sci., 2022, 247, 116938 CrossRef CAS.
- E. Bradford, A. M. Schweidtmann and A. Lapkin, J. Glob. Optim., 2018, 71, 407–438 CrossRef.
- S. T. Knox, S. J. Parkinson, C. Y. Wilding, R. A. Bourne and N. J. Warren, Polym. Chem., 2022, 13, 1576–1585 RSC.
- A. D. Clayton, A. M. Schweidtmann, G. Clemens, J. A. Manson, C. J. Taylor, C. G. Niño, T. W. Chamberlain, N. Kapur, A. J. Blacker and A. A. Lapkin, Chem. Eng. J., 2020, 384, 123340 CrossRef CAS.
- P. Sagmeister, F. F. Ort, C. E. Jusner, D. Hebrault, T. Tampone, F. G. Buono, J. D. Williams and C. O. Kappe, Adv. Sci., 2022, 9, 2105547 CrossRef.
- F. Wagner, P. Sagmeister, C. E. Jusner, T. G. Tampone, V. Manee, F. G. Buono, J. D. Williams and C. O. Kappe, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-gb117.
- C. Musikas, Inorg. Chim. Acta, 1987, 140, 197–206 CrossRef CAS.
- D. R. Prabhu, G. R. Mahajan and G. M. Nair, J. Radioanal. Nucl. Chem., 1997, 224, 113–117 CrossRef CAS.
- N. Condamines and C. Musikas, Solvent Extr. Ion Exch., 1988, 6, 1007–1034 CrossRef CAS.
- D. R. Joshi and N. Adhikari, J. Pharm. Res. Int., 2019, 28, 1–18 Search PubMed.
- T. Welton, Proc. R. Soc. A, 2015, 471, 20150502 CrossRef.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10–17 CrossRef CAS.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- B. Li, G. A. Weisenburger and J. C. McWilliams, Org. Process Res. Dev., 2020, 24, 2311–2318 CrossRef CAS.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- C. Wang, Q. Yan, H.-B. Liu, X.-H. Zhou and S.-J. Xiao, Langmuir, 2011, 27, 12058–12068 CrossRef CAS PubMed.
- M. H. Abraham, P. L. Grellier, J.-L. M. Abboud, R. M. Doherty and R. W. Taft, Can. J. Chem., 1988, 66, 2673–2686 CrossRef CAS.
- T. Welton and C. Reichardt, Solvents and solvent effects in organic chemistry, John Wiley & Sons, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00357d |
| This journal is © The Royal Society of Chemistry 2024 |