
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tracking sodium cobaltate formation pathways and its CO2 capture dynamics in real time with synchrotron X-ray diffraction†
Federico Hector
Cova
 *a and
Maria Valeria
Blanco
*b
*a and
Maria Valeria
Blanco
*b
aALBA CELLS, Carrer de la Llum 2-26, Barcelona, 08290 Cerdanyola del Vallés, Spain. E-mail: fcova@cells.es
bAragon Institute of Nanosciences and Materials, University of Zaragoza, C/ Pedro Cerbuna, 12 50009, Zaragoza, Spain. E-mail: mariavaleria.blanco@unizar.es
First published on 2nd November 2023
Abstract
Na-based high temperature CO2 solid sorbents hold great potential for capturing the CO2 emitted by large stationary sources. However, to benefit from these materials in schemes of CO2 capture, simple synthesis procedures together with a comprehensive understanding of their behaviour under operative conditions is essential. In this work, we use time-resolved in situ synchrotron X-ray diffraction coupled with Rietveld analysis to investigate the synthesis and high temperature CO2 capture dynamics of NaCoO2 solid sorbent. NaCoO2 was synthesized via two different routes, from CaCO3·H2O and Na2CO3 and from Co3O4 and Na2CO3 reactants, and a comparative analysis of the temperature-dependent phase transformations occurring during each synthesis reaction and their effect on the final product allowed to identify the most efficient synthesis route. The reaction mechanism between NaCoO2 and CO2 in the temperature range between 50 °C and 750 °C at 1 bar of CO2 is also provided. The results on the fundamental aspects underpinning NaCoO2 synthesis and its CO2 capture dynamics under realistic operative conditions are key for the design and development of affordable CO2 solid sorbents.
Greenhouse gas emissions are pinpointed as the key driver of global warming and therefore responsible for the changes observed in the climate, which are unprecedented in thousands of years and which have set in motion far-reaching massive effects on ecosystems, bringing immeasurable ecological, economical and social negative impacts.1–5 Among greenhouse gases of varying lifetimes contributing to climate change, the great persistence displayed by CO2 renders its warming practically irreversible for more than 1000 years.6 Because of this, strong and sustained reductions in emissions of CO2 are an imperious need.
Society is confronted with the important objective of achieving carbon neutrality on a global scale. While there is widespread agreement that electromobility is leading the charge in decarbonizing transportation, finding viable solutions for other industrial sectors proves to be a significant challenge. In cases where electrification is not feasible, it becomes essential to explore technological advancements that can effectively prevent CO2 emissions from being released into the atmosphere. Considering that power plants contribute to 40% of human-caused CO2 emissions,7 the development and implementation of engineering solutions capable of capturing CO2 from high-temperature flue gases in large stationary sources, such as power stations and industrial plants, emerge as a promising avenue for mitigating CO2 gas emissions.
Many ceramic compounds are able to react with CO2 at high temperatures, and this occurs via a two-step temperature dependent gas–solid reaction. At low temperatures, CO2 reacts at the surface of the material producing a carbonated shell, and at high temperatures the reaction proceeds through the bulk of the particles. The captured carbon can be later utilized as a valuable feedstock for industrial chemical production. Within ceramic compounds, Lithium containing materials have shown high CO2 sorption capacity, together with good reversibility over a wide temperature operation range.5
Li4SiO4 is one of the most studied systems for high temperature CO2 capture applications,8–10 exhibiting a high CO2 sorption capacity of 0.367 gCO2 gsorbent−1, low regeneration temperatures (<750 °C), and remarkable sorption/desorption cycling stability, which ensures a long operational lifetime under realistic conditions.11 However, despite Li4SiO4 excellent CO2 capture properties, the growing demand of Li to fulfil the needs of other technological applications, such as Lithium-ion batteries, along with the fact that it is not highly abundant has given rise to a serious concern about Li long-term availability12 and creates the need to explore more accessible, cheap and sustainable materials for CO2 sorbents.
Partial replacements of lithium by sodium in Li4SiO4 ceramic in the form of solid solutions of Li4−xNaxSiO4 have shown an enhancement of CO2 absorption kinetics compared to pure Li4SiO4, together with increased CO2 absorption.13 This indicates that Na atoms act as active sites for CO2 capture reactions. Furthermore, complete replacements of Li by Na, leading to the Na4SiO4 structure, have shown improved CO2 capture performance in terms of high temperature sorption and desorption compared with Li4SiO4.14
Na2CO3, Na2ZrO3 and NaCoO2 are promising options for obtaining lithium-free CO2 solid sorbents.15–19 A recent study showed the great ability of NaCoO2 to catalyze the conversion of CO to CO2 and to subsequently chemisorb the latter.20 NaCoO2 compounds are comprised of hexagonal CoO2 blocks and a Na layer, forming a layered oxide material. Notably, the Na layer exhibits a high degree of vacancy, leading to variations in the crystal structure depending on the Na content. Such unique structure consisting of layered hexagonal blocks with alkaline elements is expected to allow for easy diffusion and improved chemisorption of CO2, and the introduction of structural defects can be used as an strategy to modify the diffusion processes. Therefore, the crystalline structure of the material plays a crucial role in enhancing the diffusion of sodium during the CO2 chemisorption process. Furthermore, the presence of cobalt in this ceramic may induce catalytic activity in the CO oxidation reaction. Based on these findings, efforts have been dedicated to understand the chemical reactions that this material undergo at high temperatures.18–20
Results from ex situ X-ray diffraction studies suggest that upon carbonation NaCoO2 would transform to Na2CO3 and cobalt oxides, Co3O4 and CoO. While these findings provide support for the reactivity of NaCoO2 towards CO2, the presence of such cobalt oxides suggests a change of cobalt valence, indicating a complex CO2 capture process involving not only surface and diffusion reactions but also electronic exchanges. This ex situ evidence provokes the need of a comprehensive understanding on how NaCoO2 behaves in the presence of CO2 in the temperature range that is of interest for CO2 capture applications, which is critical to evaluate its performance under real working conditions and to determine its regeneration potential. This requires the use of in situ advanced characterization techniques.
X-ray diffraction analysis (XRD) has been widely used for identifying crystallographic phases after high-temperature CO2 experiments, and in situ XRD experiments have proven to be particularly beneficial for monitoring the chemical reactions and morphological changes that sorbents undergo upon carbonation.21–25 Moreover, the advanced capabilities of synchrotron facilities, including their high brilliance and detection capabilities, have enabled the performance of time-resolved synchrotron XRD studies. This cutting-edge approach allows for the dynamic behavior of materials at elevated temperatures to be tracked with a resolution of seconds, allowing to reveal the appearance of intermediate phases and the occurrence of re-conversion processes which would not be possible to detect using standard laboratory sources.26–29 Hence, time-resolved high temperature synchrotron analysis on the carbonation reactions of NaCoO2 sorbent would bring unique insights on its reaction mechanism with CO2 under operative conditions and would provide solid grounds to analyze its potential use as CO2 solid sorbent for real applications.
Owing to its promising properties as a CO2 sorbent and its excellent performance as cathode for rechargeable sodium batteries,30–32 there is also a general interest in improving NaCoO2 synthesis conditions. Among the many reported synthesis methods, the solid-state route is a well-established and simple procedure to synthesize NaCoO2 structures. Reddy and co-workers have shown to obtain hexagonal P2-NaCoO2 through the calcination of mixed stoichiometric amounts of CH3COONa, Co(CH3COO)2·4H2O and glycine at 800 °C. Also, in a recent work,33 P2-NaCoO2 was synthesized by solid state reaction from Na2CO3 and Co3O4 heat treated at 850 °C for 24 h under air flow. Other works used reactive Na2O2 and Co3O4 as starting reactants and performed different heat treatments at temperatures ranging from 450 °C to 750 °C.34 Hence, due to the multiple methods that have been reported for the synthesis of NaCoO2 and the need to use cheap and benign precursors, more thorough investigations are required.
To provide a comprehensive assessment of the potential use of NaCoO2 as CO2 sorbent material, this study focuses on two main aspects. Firstly, we perform a comparative investigation of two thermally driven synthesis routes for obtaining this material at the lower possible temperature from simple reactants using time-resolved synchrotron powder X-ray diffraction coupled with Rietveld analysis. Later, we analyze in real time the structural changes of NaCoO2 under CO2 flow in the temperature range from 30 °C to 750 °C. In situ X-ray diffraction coupled with Rietveld analysis allowed to identify reaction products and to propose a simple reaction mechanism, which is different from the one proposed on the basis of previous ex situ XRD results. The results from this work are crucial for the efficient synthesis of NaCoO2 and provide solid grounds of its reaction mechanism as CO2 sorbent and its regeneration capability.
1. Experimental
1.1. In situ synthesis of NaCoO2
NaCoO2 was synthesized via two solid-state routes. In the first synthesis route, powders of Na2CO3 (99.5%, Sigma Aldrich) and CoCO3·xH2O (99.9%, Sigma Aldrich) were used as reactants, whereas in the second synthesis NaCoO2 was produced from Na2CO3 and Co3O4 (99.9%, Sigma Aldrich). Based on preliminary results, 20 wt% excess of the sodium source was added to the mixture to compensate Na losses due to sublimation, which is in line with previous publications.20 Reactant powders were mechanically mixed in a Retsch shaker for 5 minutes, then loaded into a 1 mm quartz capillaries and heated up under synthetic air flow from room temperature to 800 °C at a ramp rate of 5 °C min−1. Temperature inside the capillary was measured constantly during the experiment using a thermocouple in direct contact with the sample. Another thermocouple was located on the outside of the capillary of control the temperature of the hot nitrogen stream provided by the gas blower to heat the system. In the first synthesis 30 min isothermal conditions were kept after each 50 °C increment, whereas in the second synthesis the temperature was constantly increased without any isothermal step.Time-resolved in situ synchrotron X-ray powder diffraction measurements were performed during both synthesis reactions. A sketch of the experimental setup can be found elsewhere.27 Experiments were performed at the High-energy X-ray Diffraction Beamline ID31 of the European Synchrotron Radiation Facility (ESRF). Data collection time was 2 seconds, and the time resolution was of 30 seconds. Data was acquired using a Pilatus Dectris 2 M CdTe detector, at a wavelength was of 0.1771 Å and 0.1589 Å, and with a beamsize of 0.6 mm × 0.6 mm (V × H). Data was processed using pyFAI package35 and Rietveld analysis was performed using Fullprof suite software.36
1.2. In situ NaCoO2 CO2 capture
NaCoO2 powder was loaded into a 1 mm quartz capillary. The inlet of the capillary was connected to a gas rig system that allowed the feed of a 10 mL min−1 flow of CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2 (50:50) gas mixture to the sample and a continuous monitoring of the gas pressure. The capillary was horizontally placed on top of a gas blower that enabled the controlled heating and cooling of the sample in the temperature range from 30 °C to 750 °C at a ramp rate of 5 °C min−1.
:N2 (50:50) gas mixture to the sample and a continuous monitoring of the gas pressure. The capillary was horizontally placed on top of a gas blower that enabled the controlled heating and cooling of the sample in the temperature range from 30 °C to 750 °C at a ramp rate of 5 °C min−1.
2. Results
2.1. NaCoO2 from Na2CO3 and CoCO3·xH2O
In Fig. 1a is displayed a contour plot with the X-ray diffraction patterns collected during the high-temperature synthesis reaction of NaCoO2 from monoclinic Na2CO3 (m-Na2CO3) and CoCO3·xH2O. The miller indices of the main phases present at different stages of the synthesis process are indicated within the graph. The evolution of temperature during the synthesis is depicted in Fig. 1b. The vertical axis of Fig. 1b is scaled in order to match the evolution of the diffractograms presented in Fig. 1a.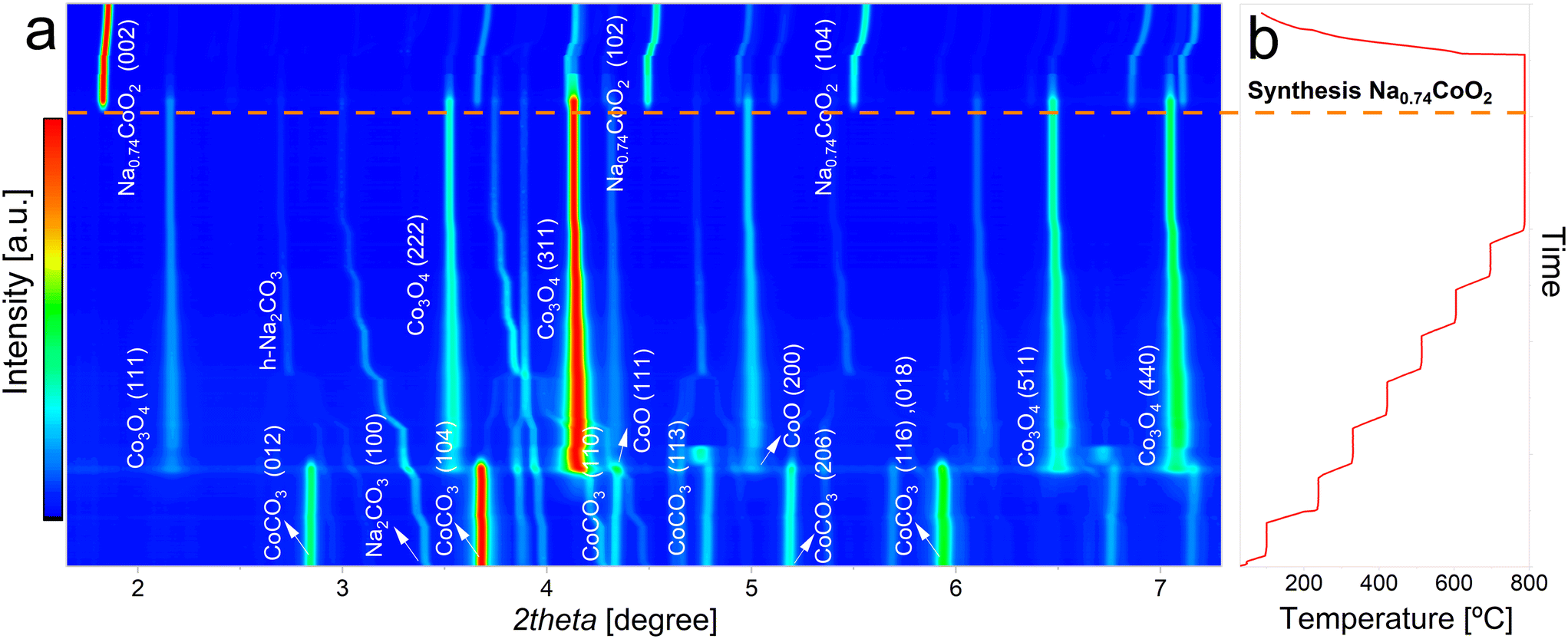 | ||
| Fig. 1 a) Contour plot of the XRD patterns collected during the synthesis of NaCoO2 from CoCO3 and m-Na2CO3 precursors, b) evolution of the temperature and time during the synthesis process. | ||
As temperature increases during the initial heating process, the reflections associated with the starting phases undergo a noticeable shift towards lower angles. This shift is a result of the thermal expansion of the respective lattices due to the temperature rise.
A first phase transition is observed at 240 °C. At this temperature, CoCO3 decomposes into Co3O4 and CoO. This reaction must be accompanied by a release of CO2. Interestingly, CoO rapidly reacts with oxygen coming from the synthetic air stream and is converted to Co3O4. According to previous reports,37 at 240 °C this intermediate reaction that results in the formation of CoO is not thermodynamically favoured against the one that results on its complete transformation into Co3O4. However, the presence of CoO is clear in the diffraction patterns. This is an indication that the conversion is being affected by kinetic and/or diffusion limitations, which introduce this intermediate reaction. In this regard, it is worth noting that previous thermogravimetric analysis conducted on the heating of CoCO3 (ref. 38) in air atmosphere based on the assumption of the chemical reaction 6CoCO3 + O2 → 2Co3O4 + 6CO2 showed weight losses higher than the theoretical ones. These results could be explained considering the intermediate transition of CoCO3 to Co3O4 + CoO + CO2.
At 240 °C a coexistence of four phases, m-Na2CO3, CoCO3·xH2O, CoCO3 and CoO, is evidenced. At slightly higher temperatures, the reflections belonging to the CoCO3 phase completely vanish. Further heating until 480 °C provokes a phase transition of Na2CO3 from monoclinic to an hexagonal crystal structure (h-Na2CO3). This high temperature phase transition was previously reported to occur above 400 °C (ref. 39) and has shown reversibility upon cooling.40 At temperatures above 480 °C, the only phases present are h-Na2CO3 and Co3O4.
At 770 °C, both phases are expected to react to form NaCoO2 through the following reaction:
| 2Na2CO3 + 4CoCO3 + O2 → 4NaCoO2 + 6CO2 |
However, the in situ XRD results showed that the phase Na0.74CoO2 is formed, as it can be observed both from the refined crystal structure (Fig. 2f) and from comparison with previous results,41 indicating the following reaction:
| 0.74Na2CO3 + 2CoCO3 + 0.63O2 → 2Na0.74CoO2 + 2.74CO2 |
 | ||
| Fig. 2 Experimental X-ray diffraction patterns and results of the fittings performed using the Rietveld method for data collected at: a) 50 °C, b) 300 °C, c) 500 °C, d) 700 °C, e) 770 °C and f) 780 °C during NaCoO2 synthesis process. Values of the refined parameters are presented at Tables S1A–F in ESI.† | ||
This synthesis occurs immediately, the formation of a phase with vacancies in the Na site has been widely reported in previous experiments.20 At 800 °C no further phase transformations are observed, and no residues from h-Na2CO3 and Co3O4 are evidenced, meaning that the reaction is completed. Once the synthesis is completed, the sample was cooled down at a rate of 10 °C min−1. The shifts to the right of the peaks corresponding to the Na0.74CoO2 phase are due to cooling down of the sample.
In Fig. 2 are shown selected XRD patterns corresponding to six different stages of the synthesis process, together with their respective Rietveld refinement fitting results. Starting Na2CO3 and CoCO3·xH2O exhibit a monoclinic C12/m1 (JCPDS 01-077-2082) and rhombohedral R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c (JCPDS 01-078-0209) structures, respectively. At 300 °C these two structures are still preserved, but the cobalt carbonate suffers a disproportion to cubic Co3O4Fdm (JCPDS 01-076-1802) and cubic CoO Fmm (JCPDS 00-009-0402). At this stage the phase percentages obtained by Rietveld analysis yielded the following results: 28.8 wt% Na2CO3, 31.6 wt% CoCO3, 16.9 wt% Co3O4 and 22.7 wt% CoO. Further heating leads to the complete transformation of cobalt-based phases to Co3O4, with a phase composition of 48.4 wt% Na2CO3 and 51.6 wt% Co3O4, and Na2CO3 transforms from monoclinic C12/m1 to hexagonal P63/mmc structure (JCPDS 01-009-8623).
c (JCPDS 01-078-0209) structures, respectively. At 300 °C these two structures are still preserved, but the cobalt carbonate suffers a disproportion to cubic Co3O4Fdm (JCPDS 01-076-1802) and cubic CoO Fmm (JCPDS 00-009-0402). At this stage the phase percentages obtained by Rietveld analysis yielded the following results: 28.8 wt% Na2CO3, 31.6 wt% CoCO3, 16.9 wt% Co3O4 and 22.7 wt% CoO. Further heating leads to the complete transformation of cobalt-based phases to Co3O4, with a phase composition of 48.4 wt% Na2CO3 and 51.6 wt% Co3O4, and Na2CO3 transforms from monoclinic C12/m1 to hexagonal P63/mmc structure (JCPDS 01-009-8623).
Further heating up to 600 °C results in a significant growth h-Na2CO3 grain domains, as it can be observed from the more defined and pronounced diffraction peaks of Fig. 2d. In Fig. 2e are depicted the XRD pattern corresponding to a phase coexistence between Co3O4, h-Na2CO3 and NaCoO2, with a phase percentage of 64.0 wt%, 15.6 wt% and 20.4 wt%, respectively. Fig. 2f shows the state of the system at 780 °C, where the reaction is not fully completed but most of the system has transformed to Na0.74CoO2. The reported experimental results demonstrate that NaCoO2 (Na0.74CoO2) can be synthesized at 780 °C, which is a temperature 70 °C lower than the reported in the literature using the same reactants.20
2.2. In situ NaCoO2 synthesis from Na2CO3 and Co3O4
NaCoO2 was also synthesized from Na2CO3 and Co3O4 powders. This synthesis route was previously used by Krasutskaya and co-workers.42 In the proposed method, powder precursors were first mixed and grinded using an agate mortar and the resulting powder mixture was pressed at 40 MPa to produce pellets, which were then fired at 860 °C in air for 12 h. In their work, the authors used a Na:Co ratio of 1.2× :1. This excess of Na2CO3 in the starting mixture compensates for the Na2O loss during the high temperature heat treatment. In another work,43 NaCoO2 was synthesized from the same reactants, but using a procedure in which the powder mixture was grinded manually in an agate mortar for 2 h, then loaded into an alumina crucible and subsequently sintered three times at 860 °C in air. In such synthesis procedure, intermediate grinding was performed in a moisture-free atmosphere inside a glove box to obtain an homogeneous composition. Therefore, the total sintering time was of 36 h.
In this work, the synthesis was performed by heating the mixture of oxides with a ramp of 5 °C min−1 without any isothermal step. In Fig. 3 it can be observed that m-Na2CO3 and Co3O4 are the main phases present from room temperature until 200 °C, temperature at which Co3O4 evolves towards the formation of Co2O3. The initial composition is presented in Fig. 4a and the coexistence of the three mentioned phases is clearly observed in Fig. 4b. The presence of Co2O3 phase was not expected, since this phase is thermodynamically unstable, however, Fig. 4c indicates a significant amount of this phase at 450 °C. As can be seen from Fig. 3, the temperature range at which this particular phase exists is narrow, and Co2O3 swiftly reverts to Co3O4 upon further heating. At 550 °C, Fig. 4d this reversion is fully completed, leading to Co3O4 becoming the dominant phase in the system. From this temperature onwards, Na2CO3 and Co3O4 are the main components, although now m-Na2CO3 has transitioned to h-Na2CO3, as it was reported in the previous synthesis process. Finally, reflections corresponding to Na0.74CoO2 phase emerge at 690 °C as observed in Fig. 4e. At 700° only 12 wt% of Na0.74CoO2 is present on the sample, and after 20 min of isothermal conditions at this temperature, Fig. 4f, the synthesis reaction reaches completion. After 1 h of isothermal conditions the system remains stable, with 3.6 wt% of h-Na2CO3 detectable as impurity.
 | ||
| Fig. 3 a) Contour plot of the XRD patterns collected during the synthesis of NaCoO2, starting from m-Na2CO3 and Co3O4 powders, b) temperature evolution during the solid-state synthesis of NaCoO2. | ||
 | ||
| Fig. 4 Experimental X-ray diffraction patterns and results of the fittings performed using the Rietveld method for data collected at: a) 50 °C, b) 350 °C, c) 450 °C, d) 550 °C, e) 700 °C and f) 700 °C after 1 h of isothermal conditions during NaCoO2 synthesis process. Values of the refined parameters are presented at Tables S2A–F in ESI.† | ||
According to the results, Na0.74CoO2 is formed according to the following chemical reaction:
| 0.74Na2CO3 + 0.667Co3O4 + 0.297O2 → 2Na0.74CoO2 + 0.74CO2 |
Which reveals again the presence of Na vacancies on the reaction product.
The synthesis of Na0.74CoO2 from m-Na2CO3 and Co3O4 reactants shows a Na0.74CoO2 formation temperature of about 100 °C lower compared with the solid state route involving the use of Na2CO3 and CoCO3·H2O precursors. Comparing the in situ XRD data of both synthesis processes, it becomes clear that the difference observed in the synthesis temperature of Na0.74CoO2 comes from the need of an intermediate stage in the first synthesis method, in which the CoCO3 transitions towards Co3O4, a step that requires higher temperature and energy input.
Overall, the lower synthesis temperature observed in the reaction between Na2CO3 and Co3O4 suggests a more efficient and potentially cost-effective route for the production of sodium cobaltate compared to the solid-state method involving m-Na2CO3 and CoCO3·H2O as precursors. It must also be emphasized that the reported synthesis temperature for a sodium cobalate from m-Na2CO3 and Co3O4 is 150 °C lower than temperatures reported in the literature using the same reactants.
2.3. CO2 capture
Previous research on sodium and lithium ceramics suggests that a reaction between NaCoO2 and CO2 would occur according to the following route:19,20| 2NaCoO2 + CO2 → Na2CO3 + Co2O3 |
However, Co2O3 reaction product is a highly unstable compound according to thermodynamics and has been rarely observed outside very particular conditions,44,45 so its formation during the absorption process is highly unlikely, although it may be possible for it to appear as an intermediate product for shorts amounts of time before decomposing into O2 and Co3O4.
In a study conducted by Vera et al.,20 the dynamic thermogram of NaCoO2 under CO2 flow was measured and displayed a continuous weight gain between 150 °C and 740 °C. This weight gain was divided into two temperature ranges: (i) 180–415 °C, corresponding to the superficial CO2 chemisorption process, and (ii) 415–740 °C, corresponding to the bulk CO2 chemisorption process. The authors analyzed ex situ XRD samples of CO2NaCoO2 isothermal reaction products and observed significant structural modifications of NaCoO2 at 400 °C, which were attributed to partial sodium release and subsequent formation of Co3O4. As the temperature increased further, between 450 °C and 700 °C, reflections belonging to NaCoO2 phase gradually disappeared, diffraction peaks corresponding to Na2CO3 phase emerged and reflections corresponding to Co3O4 phase became stronger. Interestingly, at temperatures exceeding 700 °C, the authors evidenced the presence of CoO phase. These findings contradicted previous literature proposing the presence of a different cobalt oxide, Co2O3, based on the assumption that cobalt retains its +3 valence state. Hence, Vera and coworkers claim that the valence of cobalt changes from +3 to +2, as indicated by the presence of Co3O4 and CoO detected by XRD analysis and that therefore, cobalt would reduce with increasing temperature, which must be accompanied by an oxygen release.
The carbonation reaction mechanism of NaCoO2 provided by Vera et al. is presented in the following:
| 3Co2O3 → 2Co3O4 + 1/2O2 400 °C < T < 700 °C |
| Co3O4 → 3CoO + 1/2O2T > 700 °C |
In this work, dynamical chemical reactions occurring during standard thermograms in CO2 sorption analysis were analyzed. For this, synthesized NaCoO2 was subjected to continuous heating at a ramp of 5 °C min−1 under a CO2:N2 gas flow. Fig. 5a displays the evolution of the X-ray diffraction patterns upon heating, and Fig. 5b shows the corresponding temperature evolution.
 | ||
| Fig. 5 a) Contour plot displaying XRD data corresponding to NaCoO2 dynamic heating under CO2 gas flow until 750 °C, b) temperature evolution during NaCoO2 carbonation process. | ||
The contour plot of Fig. 5a sheds light on the high-temperature reactions taking place during CO2 absorption, and reveals the occurrence of a single step process in which NaCoO2 reacts to form Co3O4 and Na2CO3. Importantly, no Co2O3 is evidenced. Therefore, it can be stated that Co2O3 would not form as an intermediate compound previous to the formation of Co3O4, as it was claimed by previous authors. These findings are consistent with theoretical results,44 which indicate that Co2O3 compound is thermodynamically highly unstable. Also, it must be remarked that the appearance of CoO phase is not observed from our experimental data, which corresponds to temperatures up to 750 °C. It is worth noting that the presented results do not contradict the previous reports by Vera et al.20 since they have reported the presence of CoO only in samples that had been subjected to temperatures higher than 750 °C, which is the maximum temperature reached in this experiment.
Rietveld refinement results allowed to track the composition of the system in real time. The analysis of the XRD patterns during the CO2 capture process was performed and is presented in Fig. 6. As it is possible to observe, Na0.74CoO2 remains stable until approximately 100 °C, temperature at which the absorption reaction starts. At this temperature, marked with a vertical dashed line in the figure, the formation of Co3O4 and Na2CO3 is observed. From Fig. 6 it is possible to see a previous jump in the reaction at t = 37 min. This is an artifact caused by the need to keep the temperature on hold for a long time due to a beam loss during the experiment (in Fig. 5 and 6 the time loss caused by the beam drop is compressed to make the figures more readable). The reaction keeps advancing very slowly until the system reaches 400 °C, temperature at which the reaction rate starts to grow. To make it easier to follow with the reaction, weight fractions were transformed into molar fractions using the following equation:
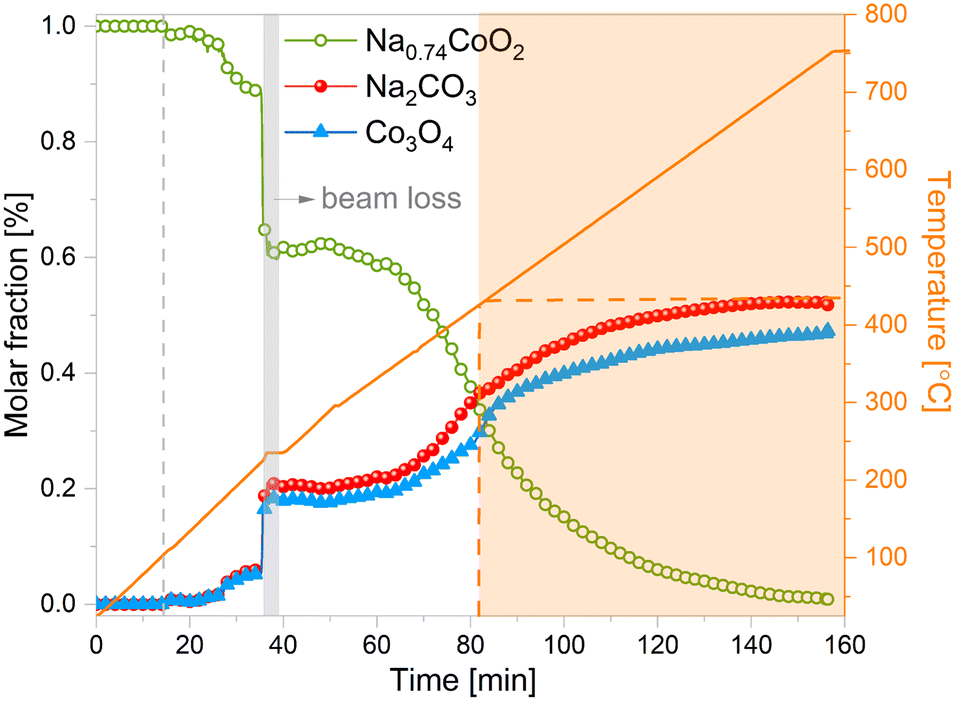 | ||
| Fig. 6 Evolution of temperature and sample weight percent during NaCoO2 carbonation reaction according to results from Rietveld analysis. | ||
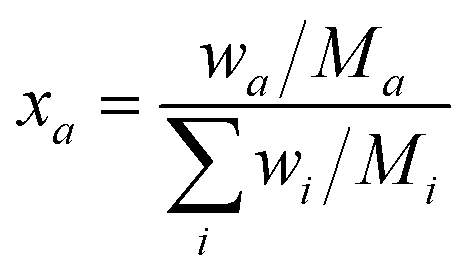
At 400 °C, the CO2 capture rate increases until reaching 550 °C. At this temperature, the system CO2 capture dynamics slows again showing an asymptotic behaviour. Previous works have reported that at temperatures under 400 °C there is process of surface adsorption, however our results indicate that at these temperatures there is already some formation of Co3O4 and Na2CO3. This is an indication that the actual process occurring is actually a chemisorption and that the reason why the reaction does continue is because of kinetic limitations.
Based on the time-resolved in situ results provided in this work, the proposed temperature-dependent reaction mechanism for Na0.74CoO2 carbonation process in the temperature range of 50 °C < T < 750 °C is the following:
| 4Na0.74CoO2 + 1.48CO2 → 1.48Na2CO3 + 1.33Co3O4 + 0.59O2 |
The main difference between the proposed reaction mechanism and the one that has been proposed by previous authors is the absence of Co2O3 phase, which was not observed at any point during the absorption process.
3. Conclusions
In this work, we analyzed the solid-state synthesis of NaCoO2via two different routes. In the first route, the starting reactants were Na2CO3 and CoCo3·nH2O, while in the second case powders of Co3O4 and Na2CO3 were used as precursors. In both cases, the starting powder mixtures were heated up from room temperature to 700–800 °C and the evolution of the chemical reactions taking place at high temperatures was tracked time-resolved synchrotron powder X-ray diffraction.Results revealed that the synthesis of NaCoO2 from Na2CO3 and CoCo3·nH2O involves a two step process, in which CoCo3·nH2O first decomposes to Co3O4 and CoO, and the latter readily reacts with oxygen, resulting in its rapid conversion to Co3O4. Then, Co3O4 reacts with Na2CO3 to form NaCoO2. In the second route, NaCoO2 is formed following a single step process from Na2CO3 and Co3O4. However, a reversible reaction of Co3O4 to form Co2O3 was evidenced. Co2O3 reaction product is unstable thermodynamically and therefore was rapidly converted to Co3O4. Overall, the second synthesis route allowed to obtain NaCoO2 at the lowest temperature of 700 °C.
The CO2 capture dynamics of the resulting NaCoO2 was also analyzed in the temperature range from 30 °C to 750 °C by heating the sample under controlled CO2 flow. Time-resolved in situ X-ray diffraction data allowed to propose a reaction mechanism for the gas–solid reaction in which NaCoO2 reacts with CO2 to form Na2CO3 and Co3O4 as solid reaction products. These results contradict the previously proposed reaction mechanism for NaCoO2 carbonation reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the European Synchrotron Radiation Facility for beamtime allocation, Florian Rusello and Tiago Couthinio are acknowledged for their help with the preparation of the experimental setup.Notes and references
- J. P. McCarty, Conserv. Biol., 2001, 15, 320–331 CrossRef.
- K. L. O'Brien and R. M. Leichenko, Glob. Environ. Change, 2000, 10, 221–232 CrossRef.
- H. G. Bohle, T. E. Downing and M. J. Watts, Glob. Environ. Change, 1994, 4, 37–48 CrossRef.
- W. N. Adger and P. M. Kelly, Mitig. Adapt. Strateg. Glob. Chang., 1999, 4, 253–266 CrossRef.
- Y. Zhang, Y. Gao, H. Pfeiffer, B. Louis, L. Sun, D. O'Hare and Q. Wang, J. Mater. Chem. A, 2019, 7, 7962–8005 RSC.
- S. Solomon, J. S. Daniel, T. J. Sanford, D. M. Murphy, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci., 2010, 107, 18354–18359 CrossRef CAS.
- A. Alonso, J. Moral-Vico, A. Abo Markeb, M. Busquets-Fité, D. Komilis, V. Puntes, A. Sánchez and X. Font, Sci. Total Environ., 2017, 595, 51–62 CrossRef CAS.
- M. T. Izquierdo, A. Saleh, E. Sánchez-Fernández, M. M. Maroto-Valer and S. García, Ind. Eng. Chem. Res., 2018, 57, 13802–13810 CrossRef CAS.
- K. Wang, W. Li, Z. Yin, Z. Zhou and P. Zhao, Energy Fuels, 2017, 31, 6257–6265 CrossRef CAS.
- A. Nambo, J. He, T. Q. Nguyen, V. Atla, T. Druffel and M. Sunkara, Nano Lett., 2017, 17, 3327–3333 CrossRef CAS.
- Y. Hu, W. Liu, Y. Yang, M. Qu and H. Li, Chem. Eng. J., 2019, 359, 604–625 CrossRef CAS.
- P. Greim, A. A. Solomon and C. Breyer, Nat. Commun., 2020, 11, 4570 CrossRef CAS.
- V. L. Mejía-Trejo, E. Fregoso-Israel and H. Pfeiffer, Chem. Mater., 2008, 20, 7171–7176 CrossRef.
- A. Sanna and M. M. Maroto-Valer, Ind. Eng. Chem. Res., 2016, 55, 4080–4088 CrossRef CAS.
- T. Cai, X. Chen, J. Zhong, Y. Wu, J. Ma, D. Liu and C. Liang, Chem. Eng. J., 2020, 395, 124139 CrossRef CAS.
- T. Zhao, E. Ochoa-Fernández, M. Rønning and D. Chen, Chem. Mater., 2007, 19, 3294–3301 CrossRef CAS.
- M. Z. Memon, G. Ji, J. Li and M. Zhao, Ind. Eng. Chem. Res., 2017, 56, 3223–3230 CrossRef CAS.
- E. Vera, B. Alcantar-Vazquez, Y. Duan and H. Pfeiffer, RSC Adv., 2016, 6, 2162–2170 RSC.
- E. Vera, Y. Duan and H. Pfeiffer, Energy Technol., 2019, 7, 1980301 CrossRef.
- E. Vera, B. Alcántar-Vázquez and H. Pfeiffer, Chem. Eng. J., 2015, 271, 106–113 CrossRef CAS.
- R. Molinder, T. P. Comyn, N. Hondow, J. E. Parker and V. Dupont, Energy Environ. Sci., 2012, 5, 8958–8969 RSC.
- M. T. Dunstan, S. A. Maugeri, W. Liu, M. G. Tucker, O. O. Taiwo, B. Gonzalez, P. K. Allan, M. W. Gaultois, P. R. Shearing, D. A. Keen, A. E. Phillips, M. T. Dove, S. A. Scott, J. S. Dennis and C. P. Grey, Faraday Discuss., 2016, 192, 217–240 RSC.
- A. Biasin, C. U. Segre and M. Strumendo, Cryst. Growth Des., 2015, 15, 5188–5201 CrossRef CAS.
- J. Manuel Valverde, A. Perejon, S. Medina and L. A. Perez-Maqueda, Phys. Chem. Chem. Phys., 2015, 17, 30162–30176 RSC.
- J. M. Valverde and S. Medina, Phys. Chem. Chem. Phys., 2017, 19, 7587–7596 RSC.
- M. V. Blanco, K. Kohopää, I. Snigireva and F. Cova, Chem. Eng. J., 2018, 354, 370–377 CrossRef CAS.
- F. Cova, G. Amica, K. Kohopää and M. V. Blanco, Inorg. Chem., 2019, 58, 1040–1047 CrossRef CAS.
- M. V. Blanco, P. M. Abdala, F. Gennari and F. Cova, React. Chem. Eng., 2021, 6, 1974–1982 RSC.
- M. L. Grasso, M. V. Blanco, F. Cova, J. A. González, P. A. Larochette and F. C. Gennari, Phys. Chem. Chem. Phys., 2018, 20, 26570–26579 RSC.
- B. V. R. Reddy, R. Ravikumar, C. Nithya and S. Gopukumar, J. Mater. Chem. A, 2015, 3, 18059–18063 RSC.
- P. Kehne, C. Guhl, Q. Ma, F. Tietz, L. Alff, R. Hausbrand and P. Komissinskiy, J. Power Sources, 2019, 409, 86–93 CrossRef CAS.
- N. Bucher, S. Hartung, J. B. Franklin, A. M. Wise, L. Y. Lim, H.-Y. Chen, J. N. Weker, M. F. Toney and M. Srinivasan, Chem. Mater., 2016, 28, 2041–2051 CrossRef CAS.
- S. Hwang, Y. Lee, E. Jo, K. Y. Chung, W. Choi, S. M. Kim and W. Chang, ACS Appl. Mater. Interfaces, 2017, 9, 18883–18888 CrossRef CAS.
- Y. Lei, X. Li, L. Liu and G. Ceder, Chem. Mater., 2014, 26, 5288–5296 CrossRef CAS.
- J. Kieffer and D. Karkoulis, J. Phys.: Conf. Ser., 2013, 425, 202012 CrossRef.
- J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef CAS.
- C.-B. Wang, H.-K. Lin and C.-W. Tang, Catal. Lett., 2004, 94, 69–74 CrossRef CAS.
- H. Du, L. Jiao, Q. Wang, Q. Huan, L. Guo, Y. Si, Y. Wang and H. Yuan, CrystEngComm, 2013, 15, 6101–6109 RSC.
- A. Arakcheeva and G. Chapuis, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 601–607 CrossRef.
- Y. J. Min, S.-M. Hong, S. H. Kim, K. B. Lee and S. G. Jeon, Korean J. Chem. Eng., 2014, 31, 1668–1673 CrossRef CAS.
- C. T. Lin, D. Chen, A. Maljuk and P. Lemmens, J. Cryst. Growth, 2006, 292(2), 422–428 CrossRef CAS.
- N. S. Krasutskaya, A. I. Klyndyuk, L. E. Evseeva and S. A. Tanaeva, Inorg. Mater., 2016, 52, 393–399 CrossRef CAS.
- I. F. Gilmutdinov, I. R. Mukhamedshin, F. Rullier-Albenque and H. Alloul, J. Phys. Chem. Solids, 2018, 121, 145–150 CrossRef CAS.
- P. N. Shanbhag, R. K. Biswas, S. K. Pati, A. Sundaresan and C. N. R. Rao, ACS Omega, 2020, 5, 29009–29016 CrossRef CAS PubMed.
- F.-C. Kong, Y.-F. Li, C. Shang and Z.-P. Liu, J. Phys. Chem. C, 2019, 123, 17539–17547 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00459g |
| This journal is © The Royal Society of Chemistry 2024 |