
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Demonstration and experimental model validation of the DME synthesis by reactive distillation in a pilot-scale pressure column†
Malte
Gierse
 ac,
Innokentij
Bogatykh
bd,
Benedikt
Steinbach
a,
Jörg
Sauer
c,
Jens-Uwe
Repke
d and
Ouda
Salem
*a
ac,
Innokentij
Bogatykh
bd,
Benedikt
Steinbach
a,
Jörg
Sauer
c,
Jens-Uwe
Repke
d and
Ouda
Salem
*a
aFraunhofer Institute for Solar Energy Systems ISE, Heidenhofstr. 2, 79110 Freiburg, Germany. E-mail: ouda.salem@ise.fraunhofer.de
bASG Analytik-Service AG, Trentiner Ring 30, Neusäss, 86356, Germany
cKarlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
dProcess Dynamics and Operations Group, Technische Universität Berlin, Sekr. KWT 9, Straße des 17. Juni 135, Berlin 10623, Germany
First published on 30th May 2023
Abstract
The dehydration of methanol to produce the hydrogen carrier and alternative fuel dimethyl ether (DME) is an equilibrium limited reaction, resulting in a relatively complex and expensive production process. A promising method for process intensification is reactive distillation (RD), as this allows the synthesis and purification of DME in a single unit operation. However, existing kinetic models for liquid phase DME synthesis have never been validated in an industrially relevant reactive distillation environment, preventing a detailed model-based design of industrial-scale applications. In this work, a pilot-scale pressure distillation column was used to successfully demonstrate the feasibility of the process involving pure and crude MeOH feed using the catalyst Amberlyst 36. Based on the measured composition and temperature profiles, a kinetic model could successfully be validated for the RD system. A process simulation model was developed in Aspen Plus to analyze an industrial-scale process and validated on the pilot scale. Hereby the influences of column size, methanol feed purity and catalyst selection were examined in detail.
Introduction
With a production capacity of approximately 5 Mt per annum1 DME is an important chemical produced globally in large industrial plants. It is mainly used as a propellant or liquified petroleum gas (LPG) blend. In the power-to-X (PtX) context, DME is gaining scientific interest due to its attractive physicochemical properties as an additive and a potential green alternative for fossil LPG. DME is also an environmentally benign hydrogen carrier with an excellent technical hydrogen capacity of 26.1 wt%. Due to analogies in the physicochemical properties of DME and CO2, a closed DME/CO2 cycle for global point-to-point H2 trade can be established.2 These emerging applications will lead to a vast increase in the worldwide DME production capacity. For the defossilization of the LPG sector alone, projections estimate the production capacity for renewable DME to exceed 40 MT per annum by 2050,3 implying a fourfold increase.4 Locations with high abundance of cheap renewable electricity are often accompanied by poor infrastructural conditions and high maintenance cost.5 Consequently, besides the ability of using a CO2-rich syngas as a feed, simple and highly integrated processes with high conversion efficiencies are desired. Usually, DME is produced in a two-step process by first synthesizing methanol from fossil syngas and then performing the gas phase methanol dehydration to DME and subsequent DME purification. Alternatively, the direct route of DME production faces the challenge of a complex downstream process and excessive water production when using CO2-rich feedstocks.6 Reactive distillation presents an energy efficient approach, which reduces the number of unit operations into a single unit. In this process, methanol is dehydrated to DME and water while removing the products in situ in a reactive distillation unit. Apart from simplifying the process, the heat demand can be significantly reduced in contrast to the conventional process for the following reasons:5• The reaction occurs in the liquid phase. Thus, no energy intensive evaporation of the MeOH feed is required.
• The distillation column reboiler duty is reduced by integrating the exothermic MeOH dehydration heat into the column.
• Since water is produced and separated in the RD column, water containing crude MeOH may directly be used as feedstock, thus neglecting the crude MeOH distillation step necessary in the conventional indirect production route.
In scientific literature, several authors published simulation-based investigations of the reactive distillation concept for DME production. The RADFRAC model of Aspen Plus was used by several authors for the simulation of RD DME production.7–10 Besides the examination of a single RD column achieving a full MeOH conversion in a single unit operation, process concepts extending the RD column by additional unit operations such as a gas-phase fixed bed reactor or an additional distillation column were evaluated.9,11,12 A simulation of a reactive dividing wall column was performed by Kiss et al.12 and Gor et al.7 Along with stationary simulations, dynamic process intensification has also been investigated.10,13 Within all of these investigations, different kinetic models were applied for process simulation. While some authors use their own developed kinetic models, others adopted literature kinetic models to their required working range.
Besides process simulations, the concept of DME RD has also been demonstrated experimentally. Di Stanislao et al.14 conducted experiments at an operating pressure of 8 bar in a DN 50 pressure distillation column using KATAPAK catalytic packing equipped with Amberlyst 35. However, only a MeOH conversion of 71% and a DME purity of 90 mol% could be achieved. Su et al.15 also used Amberlyst 35 in a pressure distillation column operated at 6.4 bar. However, this work was not able to achieve a full conversion of the MeOH feed and the DME product purity did not exceed 58 wt%. The full conversion of MeOH to produce pure DME in a single unit operation – the main objective of applying RD – has never been demonstrated experimentally. Furthermore, none of the kinetic models available in literature was validated in an RD column.
The main objective of this work is to demonstrate the production of norm compliant DME in a single unit operation using the commercial IER catalyst Amberlyst 36 (A36). Besides using a pure MeOH feedstock, the effect of using water containing crude MeOH (raw MeOH) as a feedstock will be evaluated. Furthermore, the applicability of the kinetic model proposed by Gierse et al.16 based on kinetic measurements in a fixed-bed reactor, will be critically assessed under the actual process conditions of a reactive distillation process. A process simulation of an industrial-sized RD column will be conducted to investigate the influence of major design parameters on the RD process performance. Based thereon, the influence of the water content of the feed and the catalyst selection on process design and process performance will be discussed.
Physicochemical characteristics of the RD system
The chemical system studied in this publication is the DME synthesis by dehydration of MeOH according to the following equation:6| 2CH3OH ⇌ CH3OCH3 + H2O ΔH0R = −23.5 kJ mol−1 | (1) |
In the ternary system DME–MeOH–H2O, DME is the light boiler and H2O is the heavy boiler. According to the boiling temperatures of the components, DME is removed from the top of the column as distillate and H2O is the bottoms product, i.e. the liquid removed from the reboiler. The reaction educt MeOH is the middle boiler and thus held in the reactive section in the middle of the column. Accordingly, in the top of the column, DME needs to be separated from MeOH and in the bottom section, H2O is separated from MeOH. Considering the relative volatilities, the thermal fractionation of the H2O–MeOH system presents the more challenging separation task compared to the DME–MeOH system. Since MeOH enriches the reactive section in the middle of the column, the temperature in the reactive section is in proximity to the MeOH boiling point at the respective column pressure. This system-immanent phase equilibrium implies a coupling of the reaction temperature with the column pressure. Consequently, the high temperatures of the conventional synthesis could only be achieved at excessive pressures. In turn, at moderate column pressures, significantly lower reaction temperatures are demanded compared to the conventional gas phase reaction as illustrated in Fig. 1.
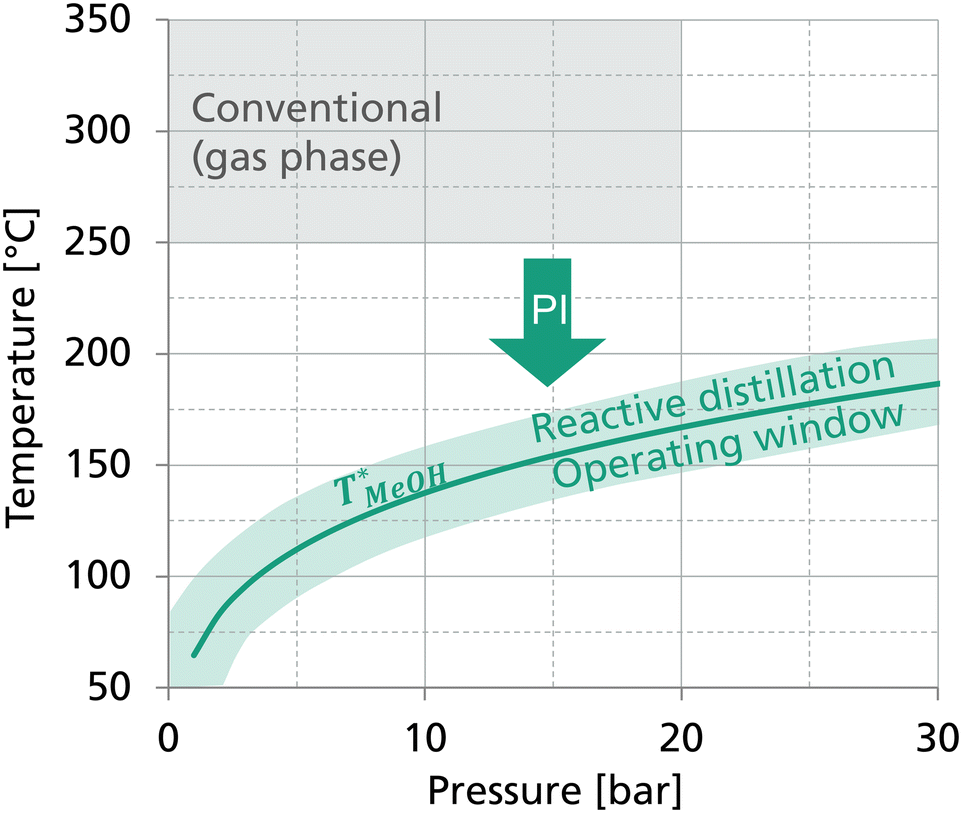 | ||
| Fig. 1 Boiling point curve of MeOH and resulting operating window of a process-intensified (PI) reactive distillation process for DME synthesis compared to the operating conditions of the conventional gas phase reaction. Illustration from Gierse et al.16 | ||
The ternary system exhibits a miscibility gap between DME and H2O at low MeOH fractions. However, since MeOH is the middle boiler, mixtures with high H2O and DME content do not occur in the reactive distillation column and the process is not affected by the miscibility gap.
Material and methods
Materials
Synthesis grade MeOH (99.9%, Bestchem GmbH) was obtained in industrial barrels. Besides using pure MeOH, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar mixture of MeOH and deionized H2O obtained in the in-house laboratory was used as a feedstock. A36 was purchased from Sigma Aldrich in its wet form. To avoid the loss of catalyst through the mesh of the KATAMAX® catalytic packing, the catalyst was sieved to remove the fine fraction. The coarse fraction was filled into the pockets of the packing in a partially dried, pourable state. The catalytic packing was only filled partially, leaving room for the volume expansion due to catalyst swelling. Subsequently, the filled catalytic packing was exposed to H2O in a horizontal position, to reduce mechanical stress on the catalyst during the swelling process. The overall catalyst mass in the RD column was 0.213 kg (referring to its dry state, reference drying conditions: 50 mbar, 100 °C for 24 hours).
:1 molar mixture of MeOH and deionized H2O obtained in the in-house laboratory was used as a feedstock. A36 was purchased from Sigma Aldrich in its wet form. To avoid the loss of catalyst through the mesh of the KATAMAX® catalytic packing, the catalyst was sieved to remove the fine fraction. The coarse fraction was filled into the pockets of the packing in a partially dried, pourable state. The catalytic packing was only filled partially, leaving room for the volume expansion due to catalyst swelling. Subsequently, the filled catalytic packing was exposed to H2O in a horizontal position, to reduce mechanical stress on the catalyst during the swelling process. The overall catalyst mass in the RD column was 0.213 kg (referring to its dry state, reference drying conditions: 50 mbar, 100 °C for 24 hours).
Pilot-scale pressure distillation column
The reactive distillation process was realized in a pilot-scale pressure distillation column from ILUDEST Destillationsanlagen GmbH shown in Fig. 2. The stainless steel DN50 column has a total height of 4.4 m, a packing height of 2.45 m and can be operated up to a pressure of 16 bara. | ||
| Fig. 2 Simplified process flow diagram of the pilot reactive distillation setup including positions of sample positions, thermocouples and packed height of the three sections. | ||
The top of the column is equipped with a coiled tube condenser tempered by a cryostat (Huber AG). The condenser was operated at 0 °C, implying a subcooling of the distillate. The subcooling was necessary to achieve high DME purity in the distillate. Downstream the condenser, the distillate was split by a time-controlled, pneumatic three-way valve. The reflux ratio was set by the respective reflux and withdrawal time and was controlled by a PID controller to achieve a desired target temperature in the top stage of the column. The distillate was continuously transferred to the distillate storage vessel. The natural circulation evaporator of the column was heated electrically to allow a direct measurement of the heat demand. The heat input was controlled by a PID controller to achieve a desired temperature at the bottom of the reactive zone. The bottoms product was continuously transferred to the bottoms product storage vessel to achieve a constant reboiler filling level. Both the distillate and the bottoms product storage vessel were equipped with a hydrostatic level indicator, allowing the derivation of the respective incoming volume flows. To allow an adiabatic operation, the column was jacketed by isolated mantle heaters, operated at the current column temperature. The temperature profile along the column was measured by 12 axially distributed Pt-100 elements with direct contact to the medium. Along the column, sampling ports for the withdrawal of gaseous samples were constructed on three different positions.
The column can be classified by three zones with individual packing configurations to account for the local hydraulic conditions as determined beforehand based on process simulations:
1. Upper rectifying section for the separation of DME and MeOH, equipped with 9 Montz A3-1200 structured packings (grey) with a height of 50 mm per element.
2. The reactive section for the incorporation of the catalyst, equipped with 8 Montz KATAMAX® catalytic packing with a height of 150 mm per element (orange). One layer of Montz A3-1500 (blue) was added between two corresponding elements of catalytic packing to account for the low separating efficiency of the catalytic packings on this scale and to increase the number of theoretical stages in the reactive zone.
3. The lower rectifying section for the separation of H2O and MeOH was equipped with 9 Montz A3-1500 (blue) structured packings with a height of 50 mm per element.
The feedstock was stored under pressure in a vessel and dosed by a gear pump controlled by a Coriolis mass flow meter (Krohne). The feed stream was preheated to a temperature of 50 °C in a heat exchanger and subsequently introduced to the column above the reactive zone. Small amounts of N2 (grade 5.0) were used for the level indicators of the column reboiler, feed and product vessels as well as for the pressure control of the distillation column.
The operating temperature of A36 is limited to 150 °C for thermal stability reasons of the sulphonated groups and to avoid leaching according to the manufacturer's datasheet.18 Due to the vapor–liquid equilibrium (VLE) inside the column, the column pressure was adapted to prevent a temperature rise above this threshold in the reactive zone of the column. In the measurement campaign, column pressure, feed composition and feed mass flow were varied during the experiments. The experimental conditions of all measured operating points are presented in the results section.
For RD column start-up, the column reboiler was filled with H2O and heated with total reflux mode. As soon as the temperature rise approached the reactive section, MeOH was fed to the column to keep the temperature throughout the reactive section below 150 °C. After a significant amount of DME was produced and the temperature in the upper rectifying section approached the dew point of DME, the desired reflux ratio was set. Since the start-up procedure was very time-consuming (>3 h), the column was operated continuously without shut down for several days and nights. The time required until a new steady state was reached after changing the operating point was at least 1 h (changing the RR) or more than 3 h (changing the feed flow). Every operating point was held at constant conditions for at least 30 minutes to ensure steady state was achieved.
Analytical methods
The three sampling ports were connected to a multi-position valve via heated capillaries to withdraw samples from the gas phase at the selected stages. The gas stream of the selected sampling port was diluted with N2 and transferred to an MKS Multigas™ 2030 on-line FT-IR spectrometer with an optical path length of 5.11 m, while the other two sampling capillaries were dead ended. After switching the sample port on the multiposition valve, typically 30 s of retention time were required until the new concentration was achieved in the FT-IR. Consequently, the position was varied in an interval of 1 minute. Within the time frame of steady state operation, every sampling point was analysed multiple times to ensure a reproducible measurement of the gas phase composition. Details of this column analysis system developed by ASG Analytik Service AG can be found in Bogatykh et al.19 In addition to the FT-IR measurements, an online gas chromatograph (Agilent 8860, thermal conductivity detector) was used to analyse the composition of the distillate product and to validate the FT-IR measurements.Simulation
A profound and validated simulation model of a reactive distillation process is required to allow the design of the process, optimize process parameters, and evaluate the key performance indicators of the reactive distillation process and thus to lay the foundation for a techno-economic analysis of the process concept. The RD process implemented in Aspen Plus is shown in Fig. 3. Since the RD already integrates synthesis and product purification, the process flowsheet is relatively simple. Compared to the experiments, only a product-to-feed heat exchanger to recover the heat of the water by-product stream was added to the flowsheet for heat integration. | ||
| Fig. 3 Process flowsheet used for the process simulation. | ||
The column pressure was adjusted by a design specification so that a certain maximum temperature is reached on the lowest stage of the reactive section. Since this stage is the one with the highest stage temperature, this maximum temperature is not exceeded in any other catalyst containing stage. To achieve the desired DME purity, the reflux ratio was adjusted by another design specification so that a distillate purity of >99.9 mol% DME was reached. The distillate to feed ratio of the column is set to 0.5 mol mol−1 in the simulation program, corresponding to a full conversion of MeOH. The water purity does not need to be specified, since the satisfaction of the constraints for DME purity and distillate to feed ratio only allow operating points with pure water in the bottoms product. The influence of the design parameters (number of stages of each section, feed stage, total catalyst mass) on the process are evaluated in the results section. In any configuration, the total catalyst mass in the RD column was distributed equally over all stages of the reactive section. The pressure drop over the column was neglected, since the comparatively high operating pressure leads to low gas velocities and thus low pressure losses.
Simulation approach
Modelling distillation columns, two approaches can be distinguished. The equilibrium stage model assumes the vapor and liquid streams leaving the stage to be in phase equilibrium with each other. In contrast, the non-equilibrium model or so-called rate-based model aims to consider the actual transport rates based on mass and heat transfer between vapor and liquid phase. Hereby, a diffusion model such as the Maxwell–Stefan approach is required.20 Opposed to conventional distillation columns, the reaction needs to be modelled on each reactive stage in RD columns. Hereby an equilibrium-based reaction model can be applied, assuming an instantaneously reached chemical equilibrium. Alternatively, a kinetic approach can be applied, considering the reaction rate based on a kinetic model.DME reactive distillation is characterized by a slow reaction, according to the definition of Schoenmakers and Bessling,21 since the reaction needs significantly more time than the typical residence time on each stage to reach the chemical equilibrium. The high relative volatility of the components leads to a rather low number of required separation stages. Consequently, an RD process in DME production is limited by the reaction rather than the separation.
Thus, the equilibrium stage model was used in combination with a kinetic approach for modelling the reaction in the scope of this work. The RD was simulated with the RADFRAC model of the simulation environment Aspen Plus® incorporating the reaction kinetics on each reactive stage. By default, RADFRAC only allows for the implementation of power-law type kinetic rate equations. To allow the modelling of other extended mathematical expressions, the respective rate equation was implemented by a Fortran subroutine. In the simulation, the reaction was assumed to proceed in the liquid phase only.22 Consequently, on each reactive stage, the reaction rate was calculated based on the respective stage temperature, liquid molar fraction of the components and catalyst mass per stage.
Reaction kinetics and equilibrium
The experimental part of this work is conducted using the ion exchange resin (IER) A36. IER are known to exhibit a distinct swelling behaviour, leading to a significant increase in volume and mass when exposed to a liquid solvent such as methanol or H2O. Due to the permanent liquid flow and the surface tension of the medium the IER particles are expected to be permanently contacted to the liquid phase. Consequently, in an RD process the IER will always be in the swollen state defined by the liquid composition of the respective stage. Thus, to correctly model the reaction in the RD column, the liquid composition of the respective stage needs to be coupled with a kinetic model measured under liquid conditions.In an earlier publication of our group,16 A36 and CAT400 were identified as the two most promising catalysts for RD by screening various catalysts. Furthermore, a novel kinetic model accounting for the RD operating conditions was proposed based on experimental kinetic measurements in a fixed-bed profile reactor. During these kinetic experiments, the reaction pressure was significantly higher than in the VLE of the RD process to guarantee pure liquid operating conditions. However, it was experimentally proven that the pressure has no influence on the reaction rate, since the reaction takes place in the incompressible liquid phase.16
Due to the higher operating temperature of Treverlyst CAT400 and the corresponding higher pressure rating requirements of the distillation column, A36 was chosen for the experimental part of this work. The intrinsic kinetic rate equation introduced by Gierse et al.16 is based on a Langmuir–Hinshelwood approach extended by a water inhibition term given by eqn (2) and the corresponding parameters given in Table 1. The catalyst mass mcat is referring to the water-free dry state of the catalyst.
 | (2) |
 | (3) |
 | (4) |
 | (5) |
| Parameter | Unit | A36 | CAT400 |
|---|---|---|---|
| k 0 | mol kgKat−1 s−1 | 8.089 × 109 | 5.973 × 1010 |
| E A | kJ mol−1 | 91.56 | 101.98 |
| K W1 | — | −4.2255 | 0.4118 |
| K W2 | K | −2360.9 | −345.2587 |
Internal mass transfer limitations were identified to be negligible in the kinetic study, even at the highest reaction rate, i.e. conditions of maximum educt concentration and temperature. Under the reactive distillation conditions applied in this work, the reaction rate was always lower than the maximum rate within the kinetic study. Since the diffusion coefficient in liquid phase is independent of the pressure, the internal diffusion in the catalyst particle is not affected by the reactive distillation conditions. Thus, in reactive distillation no internal mass transport limitation is to be expected either. Consequently, the kinetic equation was found fully applicable to the reactive distillation conditions applied in this work without the consideration of internal or external mass transport.
In the scope of this work, the weight hourly space velocity (WHSV) is defined as the ratio of feed mass flow of MeOH and the catalyst mass in the RD column. Thus, WHSV indicates the amount of MeOH that needs to be converted per catalyst mass.
 | (6) |
Thermodynamics and physical properties
The used thermodynamic model was regressed based on binary and ternary experimental VLE data of the DME–MeOH–H2O system and is described in detail by Ye et al.26 It incorporates the Peng–Robinson equation of state with Wong–Sandler mixing rules and the UNIFAC-PSRK model for the calculation of the activity coefficients. The small amounts of N2 used for the level indicators of the column were not considered in the simulation model. A detailed list of all models used for the calculation of thermodynamic and physical properties in Aspen Plus® is shown in the ESI† SI4.Data analysis and generation of continuous column profiles
The experimental column profile data of each steady-state operating point consists of the gas phase analysis on three positions and the temperature measurement on 14 positions. However, a continuous temperature and liquid composition profile is required to allow the evaluation of literature kinetic models and calculate the DME production rate according to the kinetic model. A continuous temperature profile was achieved by spline interpolation of the discrete experimental temperature data. Due to the low axial resolution of the gas phase analysis, a simple interpolation was considered inappropriate. Instead, the gas and liquid phase composition exhibiting a bubble or dew point equivalent to the measured temperature were calculated by modelling of the component system's VLE.Considering the degrees of freedom, this procedure is only possible for binary systems, or ternary systems where the fraction of one component is known. As experimentally confirmed by the gas phase measurement at the sampling port below the reactive zone, no DME was present in the lower rectifying section. This can be explained by to the absence of catalyst in this section of the column and the low boiling temperature of DME. Consequently, the composition in this section could directly be calculated based on the measured temperature and the thermodynamic modelling of the binary MeOH–H2O system. In the reactive zone, however, all three components are present. Consequently, one component must be estimated to determine the composition. Hereby, the water fraction was interpolated linearly between the sampling ports #2 and #3. This assumption is going to be validated below in the results section. The residual fraction of DME and MeOH was then calculated based on the measured temperature and the thermodynamic modelling of the ternary system. Following this methodology, continuous temperature and liquid composition profiles were calculated based on the discrete experimental data. Vice versa, the dew temperature of the measured gas phase was calculated on each sampling position to allow a comparison with the measured temperature and consequently evaluate the consistency of the measured data. Hereby it was found that the temperature measurement and dew temperature of the analysed gas phase are consistent, providing confidence in the measured data. More details about this methodology are available in the ESI† SI1.
Results and discussion
Experimental results
In total, 19 experiments were carried out over a total time on stream (TOS) of 150 h. Table 2 summarizes the key operating parameters of all the performed experiments in the pilot-scale RD column. A detailed table with all experimental data, including the temperature profile and all gas phase compositions, is included in the ESI† SI2. The temperatures, column pressure, gas compositions, feed mass flow and reboiler duty were explicitly measured.| # Exp. | p column bara | ṁ Feed kg h−1 | x MeOH,Feed mol mol−1 | RRcalc. [−] | x DME,distillate [mol mol−1] | x H2O,bottoms [mol mol−1] | X MeOH [−] |
![[Q with combining dot above]](https://www.rsc.org/images/entities/i_char_0051_0307.gif) reboiler [W]
reboiler [W] |
condenser [W] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 12.0 | 0.30 | 1 | 12.8 | 98.0% | 98.0% | 98.1% | 433.6 | 408.9 |
| 2 | 12.0 | 0.40 | 1 | 14.0 | 98.7% | 96.8% | 97.8% | 613.4 | 581.2 |
| 3 | 12.0 | 0.20 | 1 | 9.1 | 92.9% | 98.5% | 95.6% | 231.2 | 215.3 |
| 4 | 12.0 | 0.20 | 1 | 11.0 | 96.9% | 99.2% | 98.1% | 257.4 | 241.2 |
| 5 | 12.0 | 0.20 | 1 | 10.7 | 96.7% | 99.3% | 98.0% | 251.7 | 235.8 |
| 6 | 12.0 | 0.30 | 1 | 10.4 | 96.9% | 99.0% | 98.0% | 370.1 | 345.5 |
| 7 | 12.0 | 0.30 | 1 | 5.8 | 86.5% | 98.9% | 92.3% | 259.6 | 236.9 |
| 8 | 12.0 | 0.30 | 1 | 5.2 | 80.2% | 98.9% | 88.6% | 257.0 | 235.0 |
| 9 | 12.0 | 0.30 | 1 | 4.3 | 77.4% | 99.0% | 86.9% | 230.4 | 208.9 |
| 10 | 12.5 | 0.30 | 1 | 10.3 | 96.4% | 98.0% | 97.3% | 367.3 | 343.5 |
| 11 | 12.5 | 0.40 | 1 | 12.6 | 96.7% | 98.4% | 97.6% | 584.0 | 552.2 |
| 12 | 12.5 | 0.40 | 1 | 8.9 | 88.8% | 98.3% | 93.3% | 477.7 | 448.0 |
| 13 | 12.5 | 0.40 | 1 | 8.6 | 89.5% | 98.4% | 93.8% | 462.5 | 432.6 |
| 14 | 12.5 | 0.50 | 1 | 7.2 | 80.9% | 98.1% | 88.8% | 554.0 | 517.8 |
| 15 | 12.5 | 0.50 | 1 | 11.0 | 92.6% | 97.9% | 95.2% | 683.9 | 644.1 |
| 16 | 12.5 | 0.40 | 1 | 13.9 | 98.3% | 98.7% | 98.5% | 619.8 | 587.3 |
| 17 | 12.5 | 0.40 | 1 | 20.7 | 99.9% | 95.8% | 97.8% | 856.2 | 824.1 |
| 18 | 12.5 | 0.50 | 0.5 | 17.5 | 97.2% | 98.4% | 96.3% | 642.9 | 584.7 |
The reflux ratio is defined by the reflux mass flow divided by the withdrawal mass flow and can be estimated by dividing the reflux time interval by the withdrawal time interval of the three-way valve:
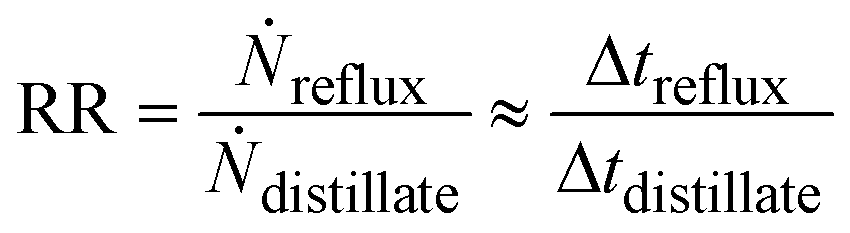 | (7) |
The distillate was analysed with respect to potential side products. For all experiments, no side product formation was detected, confirming the results of the kinetic measurements previously executed in a fixed-bed reactor.16 In the distillate and the bottoms product, the only identified impurity was unseparated MeOH. Due to the limited column height, the number of packings in the upper rectifying section had to be reduced to a minimum to leave room for the reactive section. Consequently, the DME distillate purity in most experiments is below 99 mol%. Yet, to demonstrate the production of norm compliant DME, experiment E17 was conducted with a particularly high RR.
For clarity, a few general remarks applying to all measured operating points will be made based on the experimental data of the exemplary experiment E1 as shown in Fig. 4. Furthermore, the continuous profile calculated by the methodology described in the simulation chapter is presented.
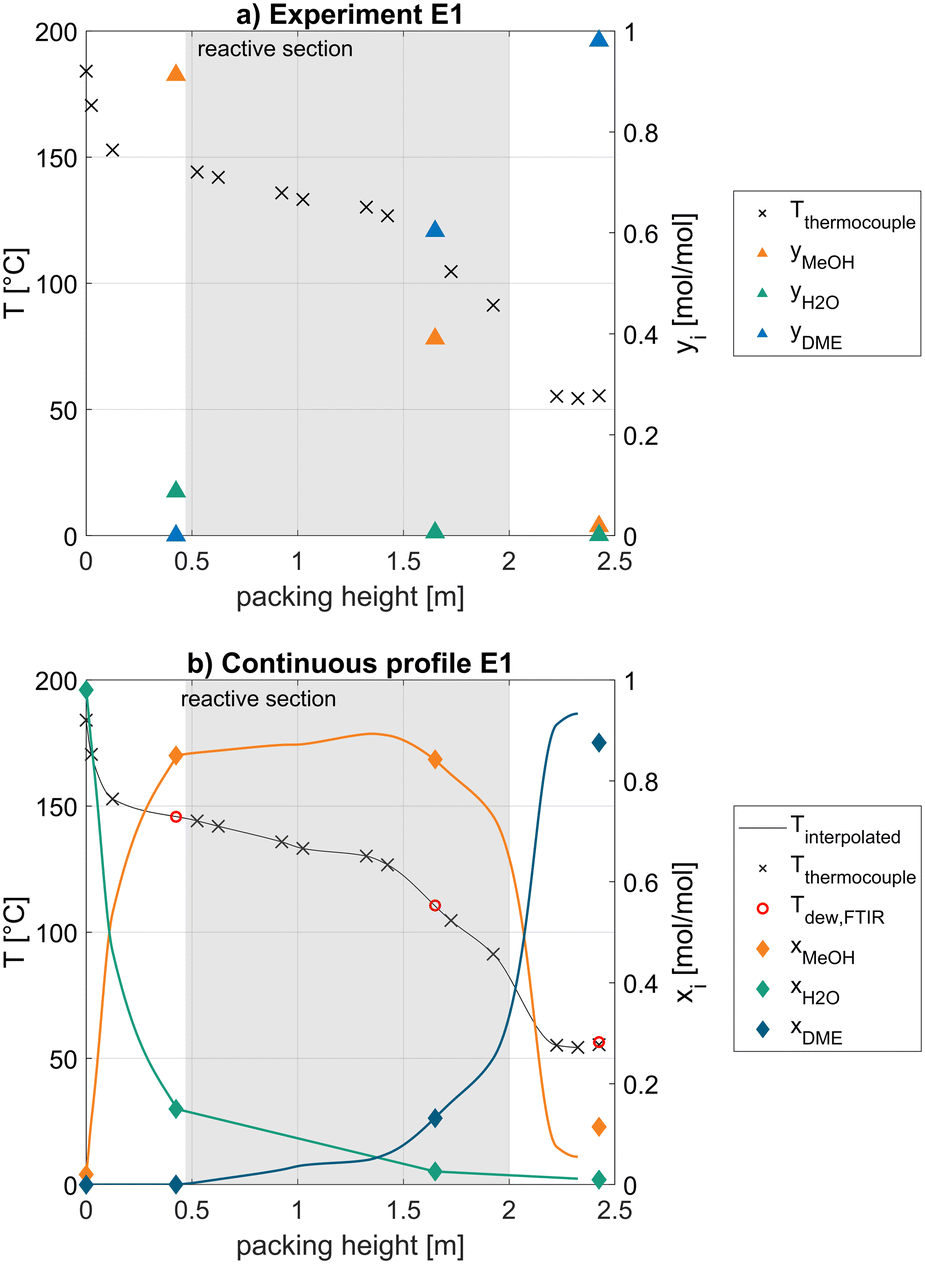 | ||
| Fig. 4 a) Discrete measured temperature and gas phase composition along the packing height of the column; b) calculated continuous temperature obtained by interpolation and liquid phase composition profile obtained from measured gas phase composition according to the methodology described in the simulation chapter; exemplary representation for experiment #1; operating pressure 12.0 bar, feed mass flow 0.3 kg h−1 pure MeOH. | ||
At this exemplary operating point, the reboiler of the column operates at a temperature of 184 °C, corresponding to a water purity of 98 mol% at the operating column pressure of 12 bar. Above the reboiler a sharp temperature drop over a short column height is visible, representing an effective separation of water and MeOH, as can also be seen at the gas phase analysis (a) at 0.425 m, where only 9 mol% water are present. In the interpolated continuous profile (b), this corresponds to an equilibrium liquid concentration of 15 mol% water. The dew temperature of the measured gas phase composition at this position (red circle) agrees very well with the measured temperature (blue line), indicating a strong consistency of the measurements.
At the lower end of the reactive section at 0.47 m, the composition is rich in MeOH and low in water which is beneficial from a kinetic perspective, since water has a strong inhibition effect on the reaction rate.16 The lower and middle part of the reactive section (0.5–1.5 m) are characterized by a linear temperature decrease with increasing column height. In the upper third of the reactive section, above 1.5 m, the slope of the temperature profile is decreasing significantly which can be explained by an increasing concentration of the low-boiler DME. This observation is reinforced considering the high DME fraction of 60 mol% in the gas phase (a) at 1.65 m. In the calculated continuous column profile (b), this corresponds to an equilibrium liquid concentration of only 13 mol%. Between the measured composition at 0.425 m and 1.65 m, the linear interpolation of the water fraction is visible. In contrast, the DME and MeOH fraction are calculated based on the measured temperature. At the sampling position at 1.625 m, a high consistency between the measured temperature and the dew point of the gas phase analysis is clear. Overall, the whole reactive section is characterized by a high liquid MeOH mole fraction of >73 mol%. While this is beneficial from a kinetic perspective, the steep temperature decrease in the upper third of the reactive section significantly lowers the reaction rate in this region of the column. In an industrial process realization, this behaviour should be avoided by improving the separation in the upper rectifying section, e.g., increasing the number of theoretical separation stages. As a result, less DME would be present in the reactive section, thus decreasing the temperature gradient within the reactive packing and thereby increasing the reaction rate. Due to the limited column height, this was not possible as it would not leave enough space for the reactive section.
In the upper rectifying section, the temperature is decreasing further to 54 °C due to the purification of DME by rectification. The gas phase analysis shows a high distillate purity of 98 mol% DME. A slightly higher temperature of the top thermocouple compared to the second uppermost thermocouple can be observed. This is most likely due to an unreached thermodynamic equilibrium in the upper rectifying section, resulting from a short liquid–gas contact time. For this reason, the match between calculated composition profile and measured composition is not ideal in this section of the column.
Fig. 5 shows the temperature profiles of the experiments E1, E2 and E4. These experiments are characterized by different feed mass flows, resulting in different WHSV. At the same time, a comparable distillate and bottoms purity, thus MeOH conversion is maintained by varying RR.
 | ||
| Fig. 5 Measured temperature profiles of the experiments E1, E2 and E4 along the packing height of the column. The feed mass flow is varied and the reflux ratio is adapted in order to achieve a comparable distillate purity. | ||
The three temperature profiles have comparable bottoms and distillate temperatures, however the temperatures in the reactive section differ significantly. While the profiles show a linear temperature decrease between 0.5–1.5 m and an identical temperature at 0.5 m, the slope of the temperature profiles decreases with increasing feed mass flow. This leads to a flatter temperature profile and consequently a higher average temperature in the reactive section enabling higher reaction rates required for the conversion of the higher feed mass flow. Furthermore, an increasing RR with increasing WHSV is evident. This shows that the conversion of more feedstock by a higher average reaction temperature is only enabled by an increase of the RR. Vice versa, by reducing the WHSV through a lower feed mass flow, the RR can be reduced. From an industrial perspective, this implies a conflict of objectives when designing the RD: increasing the column size results in higher investment cost, however, at a constant feed mass flow the RR decreases, thus leading to a lower energy demand in the column reboiler and consequently reduced operating costs. To solve this conflict of objectives, rigorous process optimization is required to identify an appropriate column design. The same trend can be observed when comparing the reboiler duties of E10 with E11 or E13 with E15.
Fig. 6 compares the experiments E10 and E18 to examine the influence of water in the feed. The crude MeOH feed mass flow in E18 is 0.5 kg h−1, corresponding to 0.32 kg h−1 of MeOH considering the MeOH concentration of 50 mol%. Thus, the amount of MeOH is comparable to experiment E10 with 0.3 kg h−1 of pure MeOH feed.
 | ||
| Fig. 6 Comparison of the experiments E10 (pure MeOH feed) and E18 (crude MeOH feed). Measured temperature profile (a) and calculated liquid composition profile (b) of both experiments along the packing height of the column. | ||
The crude MeOH experiment E18 shows a significantly higher temperature in the reactive section than E10, due to a flatter temperature profile. As discussed before, this is the consequence of the higher RR of E18 compared to E10 (17.5 vs. 10.3). The higher average temperature in the reactive section is required to maintain a high reaction rate despite the higher water content due to the crude MeOH feed. As shown in Fig. 6b) the liquid profile of E18 generally shows a higher water concentration throughout the reactive section. Particularly in the upper part of the reactive section, the water concentration is considerably higher since the water in the crude MeOH is fed directly on top of the reactive section. To reduce the water concentration in the reactive section, a lower feed position could be beneficial. At the lower end of the reactive section, E18 shows a higher water mole fraction than E10, while the bottoms product purity of E18 and E10 is nearly identical. Consequently, the MeOH–H2O separation in the lower rectifying zone is more effective in E18, which can also be explained by the higher RR. The reboiler duty of E18 is significantly higher than E10 (642.9 W vs. 367.3 W), due to the higher required RR.
This measurement proves the feasibility of a one-step process to produce purified DME from crude MeOH under industrially relevant conditions. This presents a major process simplification, allowing to omit the crude MeOH distillation step and reduce the investment cost of the process within the system boundary of syngas to DME. From an industrial perspective, it is important to note the energy demand of the RD increases when using crude MeOH. However, this implementation can lead to a total process efficiency increase compared to a DME synthesis based on pure MeOH feed, as no energy demand is required for the omitted crude MeOH distillation column. Process simulations including economic aspects are therefore required.
Model validation
A key challenge in modelling RD is the precise description of the reaction kinetics. Particularly the transfer of the kinetic model from laboratory kinetic reactors to the actual RD column often yields unsatisfying results.24,27 In the following, the calculated continuous column profiles were used to evaluate various literature kinetic models in terms of their suitability to describe the actual kinetic behaviour in the RD column. Hereby, the temperature and liquid composition on each column height increment are used to calculate the reaction rate profile and evaluate the overall DME production rate by integrating over the whole reactive section. By comparing the simulated DME production rate to the actual withdrawn mole flow of DME in the distillate product, the suitability of the examined literature kinetic models for their application in RD can be compared.Fig. 7 compares the measured DME distillate flow with the simulated overall DME production rate predicted by the respective literature kinetic model in a parity plot. The model of Lei et al.11 shows an extreme underestimation of the amount of produced DME, predicting less than half of the actually measured DME distillate for all conducted experiments. In contrast, the kinetic models proposed by An et al.8 and Hosseininejad et al.,28 respectively overestimate the reaction rate significantly. Hereby it needs to be considered, that An and Hosseininejad use Amberlyst 35 instead of A36 as catalyst, which might lead to slight deviations in the kinetic behaviour. However, both catalysts show a very similar acid capacity and were reported to show nearly identical MeOH conversion in a catalyst screening.28 More likely, the small water concentration range considered in the kinetic studies of An et al. and Hosseininejad et al. (0–13 mol% and 0–20 mol%, respectively) leads to a deviation in reaction rate due to the required extrapolation of their kinetic model. Summarizing all experiments, a temperature range of 78–151 °C and a water concentration range of 0.18–28.5 mol% was examined in the reactive section.
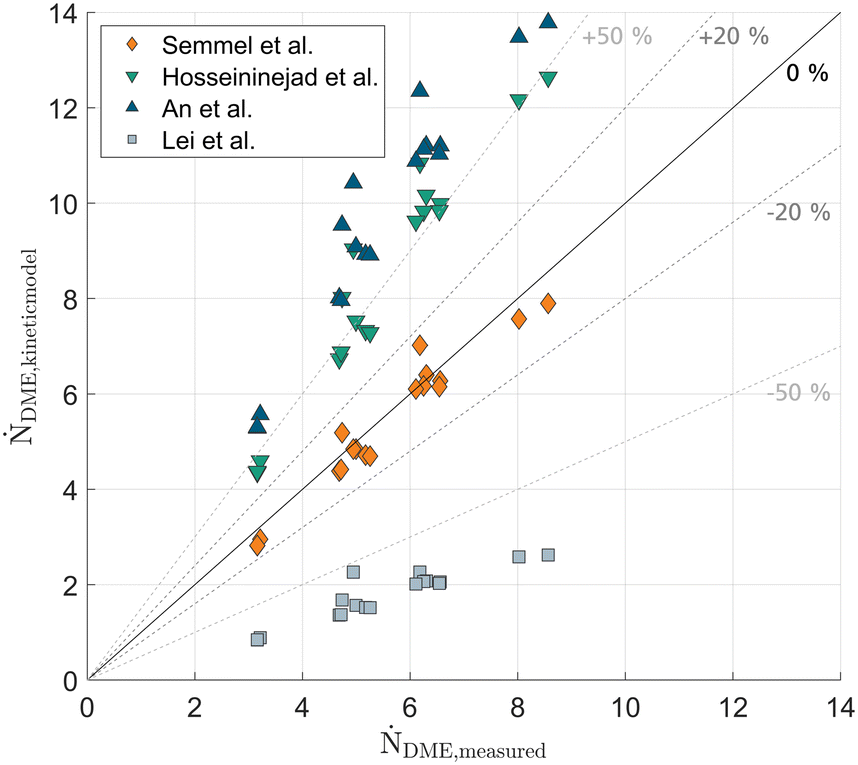 | ||
| Fig. 7 Measured distillate flow rate ṄDME,measured over simulated overall reaction rate ṄDME,kinetic model evaluated based on calculated continuous column profiles and different kinetic models.8,11,16,28 Parity plot for all measured experimental datapoints E1–E18. | ||
Compared to the other literature models, the model proposed by Gierse et al.16 predicts the measured distillate stream very well and thus presents a suitable model for describing the kinetics of DME reaction over the large temperature and concentration range appearing in RD columns. Experiment E18 with crude MeOH feed and a significantly higher water concentration in the reactive section is also described well by the kinetic model. While a slight scattering of the measurements can be observed, TOS of the catalysts at the respective experiment has no influence on whether the distillate flow is higher or lower than predicted by the kinetic model. In the case of a catalyst deactivation, experiments at a later point in the measurement campaign would tend to show lower distillate flows than predicted by the model. As this was not observed, no significant catalyst deactivation was observed over the total TOS of 150 h. This verdict should be confirmed by performing longer time-on-stream (TOS) operation.
Importantly, the great agreement of the kinetic model – despite the significantly different operating conditions in RD and kinetic reactor – cannot be taken for granted. The kinetic model for A36 was derived in a fixed-bed profile reactor operated under a pressure of 40 bar to guarantee a reaction solely in the liquid phase at unambiguous conditions. In contrast, the significantly lower pressure in the RD leads to the reaction occurring under conditions where the gas and liquid phase coexist. The light-boiler DME, formed at the active site inside the catalyst pores, could potentially lead to a partial evaporation inside the catalyst pores which in turn would hamper the internal mass transport inside the catalyst, as reported by Datsevich et al.29 Furthermore, fluid dynamic non-idealities in the catalytic packing of the RD, such as liquid bypassing of the catalyst or insufficient catalyst usage by non-wetted or stagnant zones of the catalyst could lead to significantly reduced reaction rates under RD conditions.24 The observations based on the experiments presented in this work are free of such influences, and the kinetic model by Gierse et al.16 could be successfully validated for the application in RD. At industrial scale, fluid dynamic non-idealities are less likely to occur compared to the comparatively small column diameter used in this work. Therefore, the application of the kinetic model for an industrial design of RD columns was found feasible.
To further validate the whole process simulation model applying the kinetic model of Gierse et al. in the reactive section, the continuous column profiles of experiment E1 with the column profiles acquired by the Aspen Plus process simulation are compared in Fig. 8. Hereby, the parameters applied in the experiment (catalyst mass, feed stage, feed flow, feed composition and temperature, and RR) were applied in the process model. The number of stages of each zone in the simulation was set to Nrect,upper = 4, Nrect,lower = 3, and Nreactive = 7 to match the experimental data. The number of theoretical stages in each experiment could only be estimated, as the separation efficiency of the packing could not be stated by the manufacturer at the conditions applied in this study. Furthermore, the separation efficiency depends on the hydraulic conditions at the specific location of the packing and at each experiment.
A very good fit for each component liquid composition profile as well as the temperature profile can be observed along the whole packing height of the column. Particularly, the water composition profile in the reactive section is indeed almost ideally linear, as assumed in the methodology for calculating the continuous profiles.
 | ||
| Fig. 8 Comparison between experiment and process simulation; experimentally determined continuous temperature and liquid composition profile of experiment E1 (solid lines) and simulated temperature and liquid composition profile (dashed lines) at the conditions applied in the experiment; for the simulation, the stage numbers of the RD column were set to Nrect,upper = 4, Nrect,lower = 3, Nreactive = 7 process model. The number of stages of each zone in the simulation was set to Nrect,upper = 4, Nrect,lower = 3, and Nreactive = 7 to match the experimental data. The number of theoretical stages in each experiment could only be estimated, as the separation efficiency of the packing could not be stated by the manufacturer at the conditions applied in this study. Furthermore, the separation efficiency depends on the hydraulic conditions at the specific location of the packing and at each experiment. | ||
Yet, it needs to be emphasized, that the operating conditions in the pilot-scale reactive distillation column differ from the conditions in an industrial scale RD plant: due to the limited size of the pilot-scale distillation column used in this work and the height required for the two rectifying sections, only a relatively low catalyst mass could be introduced in the reactive section. At the same time, the feed mass flow could not be reduced indefinitely to ensure appropriate F-factors and avoid a dewetting of the packings.30 Consequently, the operating points examined in this work are characterized by a rather high WHSV, requiring a high RR. As mentioned above, this operating point is likely to be non-optimal for the plant economics due to a high energy demand of the process. For this reason, the following chapter examines the RD process based on the validated process simulation at a wider range of operating conditions, including the economically more favourable operating range with lower RR.
Process analysis
In the following, a large-scale RD process to produce DME with a production capacity of 100000 ton per year is simulated using the design parameters given below. The RADFRAC column was simulated according to the flowsheet in Fig. 3. A sensitivity study of the process was conducted to define the main influencing parameters on the reboiler duty of the RD column, as shown in Fig. 9. These can be distinguished by parameters related to the reaction kinetics and parameters related to the distillation. The base case configuration of the sensitivity study is shown in brackets:
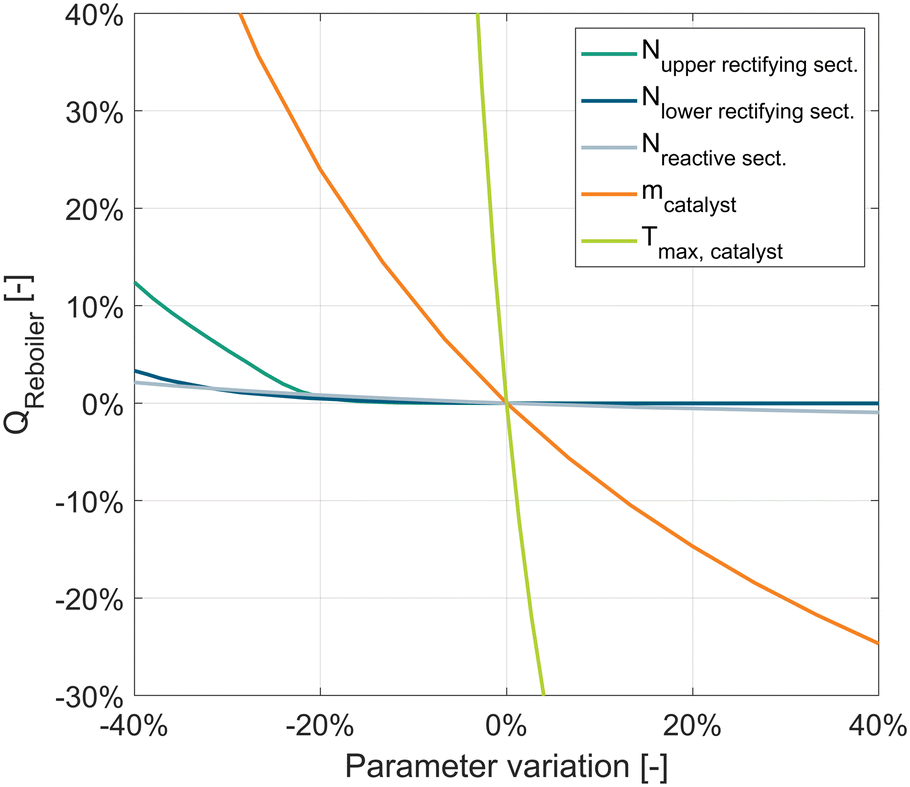 | ||
| Fig. 9 Sensitivity study of kinetic parameters and distillation parameters on the reboiler duty of the RD column. | ||
1. Kinetic parameters.
a. Catalyst mass (19.2 t).
b. Max. operating temperature of the catalyst (150 °C).
2. Distillation parameters.
a. Number of stages in upper rectifying section (10).
b. Number of stages in lower rectifying section (14).
c. Number of stages in reactive section (25).
The modification of the operating temperature of the catalyst was achieved by variation of the column pressure.
The sensitivity study shows that the parameters influencing the reaction kinetics, i.e., temperature and catalyst mass, are the main limiting factors for the design of the reactive distillation process. The number of stages in the two rectifying sections must not fall below the required stage number, otherwise the reflux ratio needs to be increased to achieve the desired purity and consequently the reboiler duty is increased. In this case, the RR is dominated by the distillation. A further increase in the number of rectifying stages beyond the minimum does not reduce the reboiler duty. In this case, the RR is dominated by the reaction. The number of stages in the reactive section (while keeping the same catalyst mass in the RD column) shows a very small sensitivity over the whole parameter range examined.
In contrast, the kinetic parameters show a significantly higher sensitivity than the distillation parameters. An increase of the catalyst mass leads to a reduced reboiler duty of the column. This can be explained in accordance with the experiments: an increase of the catalyst mass leads to a reduced WHSV and consequently a lower reaction temperature and a less flat temperature profile is required which can be achieved by a lower RR. The curve of the variation of the catalyst mass per stage represents a proportional manipulation of the reaction rate and is synonymous for a kinetic model with respectively higher or lower pre-exponential factor. The maximum operating temperature of the catalyst shows a drastically higher sensitivity since the reaction temperature affects the reaction rate exponentially. However, this parameter represents a theoretical reflection as no catalyst with higher operating temperature and identical kinetic behaviour exists. Nonetheless, it shows that the use of catalysts with higher operating temperature presents a strong lever for improving the process efficiency.
Overall, it can be derived, that reaction kinetics is the key to optimizing the energy efficiency of the DME RD process. If the number of stages in both rectifying sections is large enough, which is already achieved at comparably low stage numbers, the required RR is dominated by the reaction and the reboiler duty can only be reduced by increasing the reaction rate.
Fig. 10 shows the specific reboiler duty per ton of produced DME in dependence of the feed stage NFeed for different water mole fractions xH2O,Feed of the feed. The absolute methanol flow of the feed was kept constant at 17.5 t h−1, while an additional water mass flow was added to achieve the respective molar fraction. The catalyst mass in the RD column was 64 t, resulting in a WHSV of 0.27 h−1. The used catalyst for the simulation was A36 and the maximum operating temperature in the reactive section was set to 130 °C by varying the column pressure as described in the simulation chapter. The resulting operating pressure of all operating points considered in Fig. 10 was between 6.4–7.2 bar depending on the feed stage and the feed composition.
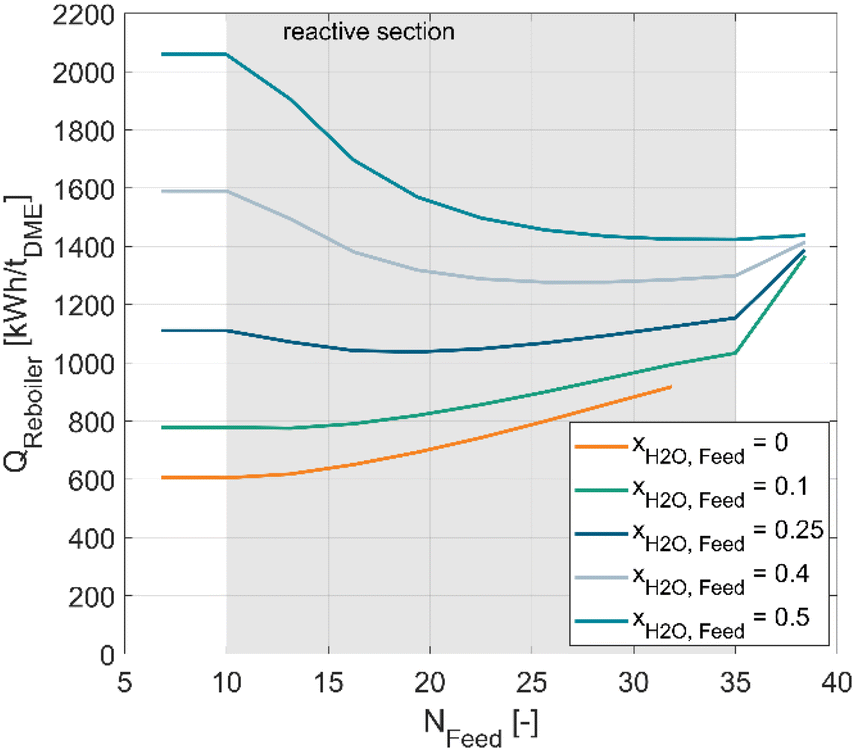 | ||
| Fig. 10 Specific reboiler duty per ton of produced DME in dependence of the feed stage NFeed for various molar water fractions in the feed. WHSV = 0.27. Nrect,upper = 10, Nrect,lower = 14, Nreactive = 25. Use of A36 in the reactive section with Tmax = 130 °C. | ||
The simulations show a clear increase of the specific reboiler duty with increasing water mole fraction of the feed. This trend agrees well with the experimental measurements, where the specific reboiler duty increases roughly by a factor of two when comparing a crude MeOH feed (50 mol% H2O) with a pure MeOH feed. Furthermore, it can be seen, that the optimal feed stage varies with the feed composition, resulting in a lower optimal feed position with increasing water fraction of the feed. For a pure MeOH feed, the top reactive stage (10) is the optimal feed stage. In contrast, a feed stage of 35 is optimal for a feed with 50% H2O. With this feed composition, feeding at stage 10 would lead to a 45% higher energy demand. This behaviour underlines the importance of a proper simulation-based design of the RD column.
As mentioned in the simulation chapter the kinetic parameters for the two catalysts A36 and CAT400 are available. While CAT400 exhibits a lower acid capacity and consequently is less active than A36 at identical conditions, it offers a higher thermal stability leaving potential for an increase in reaction rate. In the following, the influence of the catalyst choice on the RD process performance is examined. The maximum operating temperature was set to 130 °C for A36 and to 160 °C for CAT400, representing a lower temperature than the respective manufacturers specification to allow a long-term stability of the catalysts. Fig. 11 compares A36 and CAT 400 with respect to the specific energy demand per ton of DME as a function of the WHSV of the RD column. For both pure MeOH (a) and crude MeOH feed (b), the specific reboiler duty increases with increasing WHSV, as already discussed in the sensitivity study and confirmed by the experiments. Consequently, the energy demand of the RD process strongly dependents on the dimensioning of the RD column. Moreover, all operating points with complete MeOH conversion can be found along the operating lines in the graphs. This behaviour leads to a conflict of objectives between operating costs and investment costs that can only be solved by rigorous techno-economic evaluation. The optimal feed stage for CAT400 for pure and crude MeOH was determined analogously to Fig. 10, details are shown in the ESI† SI5.
 | ||
| Fig. 11 Specific reboiler duty per ton of produced DME as a function of the WHSV of the RD column. Comparison between A36 (orange) and Treverlyst CAT 400 (green). Results for pure MeOH feed (top) and crude MeOH feed (bottom) with 50 mol% H2O and 50 mol% MeOH. Nrect,upper = 10, Nrect,lower = 14, Nreactive = 25. NFeed = 10 in case of pure MeOH feed for both A36 and CAT400. In case of crude MeOH feed NFeed = 35 (A36) or NFeed = 25 (CAT400). | ||
Comparing the two catalysts for pure MeOH feed, a clearly lower reboiler duty using CAT 400 can be identified throughout the WHSV range. At WHSV = 0.4 and WHSV = 1.4 the reboiler duty is reduced by 63% and 81%, respectively when using CAT 400 instead of A36. This strong reduction is possible through the significantly higher reaction rate achievable due to the 30 °C higher operating temperature of CAT 400 and the strong sensitivity of the reboiler duty to the operating temperature as discussed in the sensitivity study. Similarly, for a specific reboiler duty of 750 kW h per tonDME, 74% less catalyst mass and consequently a 74% smaller reactive section is required for the full conversion of the same amount of MeOH. The column pressure is in the range of 13.4–15 bar for CAT400 and 6.6–8.0 for A36, depending on the WHSV. This presents a disadvantage of CAT400 and implies another conflict of objectives that can only be answered in a techno-economic evaluation: while the increased temperature stability of CAT400 allows a higher reaction rate and thus a smaller column, the wall thickness of the column would need to be increased to account for the higher operating pressure.
Regarding a crude MeOH feed, analogous conclusions can be drawn with CAT 400 allowing a reduction of the reboiler duty by 68% and 66% at a WHSV of 0.8 and 0.4, respectively. Compared to pure MeOH feed, the reboiler duty is roughly twice as high when using a crude MeOH feed for both catalysts. While this presents a significant increase in energy demand, it needs to be evaluated in view of the energy demand saved by omitting the crude MeOH distillation. For the purification of a CO2-based crude MeOH, a specific energy demand of QMeOH = 391.4 kW h per tonMeOH has been reported by Nyári et al.31 corresponding to 544.6 kW h per tonDME considering the stoichiometry of the DME synthesis. The dashed line in Fig. 11b) shows the resulting energy demand when adding the energy demand for the crude MeOH distillation column to the reboiler duty of the RD column operated with pure MeOH feed. The curves of the energy demand show that for CAT 400 below a WHSV of 0.7 h−1 feeding the crude MeOH directly to the RD column requires less energy than purifying the crude MeOH in a dedicated distillation column and then feeding the pure MeOH to the RD column. Above WHSV = 0.7 h−1, using crude MeOH for the RD feed is less energy efficient. For A36, using a dedicated crude MeOH distillation column presents the more efficient process configuration throughout the entire examined WHSV range. However, it needs to be emphasized that regardless of the energy demand, feeding crude MeOH to the RD also has the benefit of reducing the investment cost of the plant, since a whole distillation column can be omitted. In order to entirely evaluate these two process options, further investigations by rigorous process simulation including techno-economic considerations are required.
Conclusions and outlook
DME is witnessing an increasing interest as a PtX product due to its outstanding characteristics as an LPG alternative and as an environmentally benign global hydrogen carrier. For DME production, reactive distillation presents a promising process intensification technique, which reduces both the energy demand and the investment cost of the process. In the present work, a pilot-scale pressure distillation column was equipped with Amberlyst 36 catalyst to demonstrate the technical feasibility of the reactive distillation process under industrially relevant conditions. A total of 18 experiments were conducted in which the pressure, reflux ratio and feed mass flow were varied. It was observed that various feed flows can be converted with the identical distillate purity by adjusting the reflux ratio. However, this leads to an increased specific energy demand of the column reboiler, since the reflux ratio needs to be increased to flatten the temperature profile in the reactive section. Furthermore, the effect of using crude MeOH feed instead of pure MeOH was examined experimentally. Hereby, a significant increase in the reboiler duty was observed for crude methanol operation. This was due to the fact that the reflux ratio needs to be increased considerably to maintain a high reaction rate, despite the high concentration of the inhibiting water in the reactive section. Based on continuous column profiles obtained by coupling the experimental data with VLE modelling, the different kinetic models could be applied at the reactive distillation process conditions. Hereby it was validated that the model proposed by Gierse et al.16 was the only model, among those presently discussed in the literature for this system, that is capable of predicting the measured amount of DME in the distillate stream. A simulation of the complete DME RD process was done by implementing this kinetic model in combination with an experimentally validated thermodynamic phase equilibrium model into Aspen Plus.Using this validated process model, the design parameters were evaluated with respect to their influence on the reboiler duty. It was found that the kinetic parameters are the key influencing factors for reducing the energy demand of the process. This can be achieved either by increasing the amount of catalyst, using more active catalyst, or by raising the operating temperature. Furthermore, it was concluded that the reboiler duty is not sensitive to the number of stages. The influence of feeding crude MeOH to the RD was rigorously examined by process simulation complementary to the experimental validation. It was shown that with increasing feed water fraction, the reboiler duty increases and the optimal feed stage shifts towards lower reactive stages. The experimental trend of a reduced energy demand with decreasing WHSV could be verified by the simulations for the examined catalysts Amberlyst 36 and CAT 400 as well as with pure and crude MeOH. CAT 400, which was evaluated by simulation only, shows a significantly higher performance, in general, which leads to a potential reduction in energy demand by up to 81% compared to using the same mass of Amberlyst 36 catalyst. This effect is primarily attributed to the higher thermal stability of CAT 400, which allows for higher temperature operation and consequently higher reaction rates in the reactive section. This should be further investigated in experiments and validated. When using crude MeOH (50 mol% H2O) instead of pure MeOH, the DME purity can be maintained, but the energy demand of the RD process roughly doubles. This could be relevant in integrated PtX plants where MeOH and DME production from renewable feedstock are coupled. Also, the direct use of crude MeOH in the RD column, instead of using a dedicated MeOH column, saves energy. Thus, the overall energy demand of DME production, starting from crude MeOH, can be lowered. The amount of energy savings depends on the process design and is a matter of current evaluation using a complete process flow sheet-based, techno-economic analysis which considers an expanded system boundary. Only then can a quantitative comparison between Amberlyst 36 and CAT 400 be drawn, and a profound statement on whether the omission of the dedicated crude MeOH distillation column is beneficial can be made. Moreover, the process concept of RD can be developed further, e.g., by interconnection with a separate fixed-bed reactor in order to reduce the column size and thus the overall plant cost. The optimization, evaluation and comparison of these process concepts using state-of-the-art techno-economic evaluations are a matter of current research in our group. The findings there will be addressed in future publications. With the experimentally validated process model developed in this work, a scientifically profound foundation has been developed for the industrial design and engineering of RD processes for large scale DME production.
Author contributions
Conceptualization: Malte Gierse, Innokentij Bogatykh, Ouda Salem. Methodology: Malte Gierse, Innokentij Bogatykh, Benedikt Steinbach, Ouda Salem. Project administration: Ouda Salem. Supervision: Ouda Salem, Jörg Sauer, Jens-Uwe Repke. Writing – original draft: Malte Gierse.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Deutsche Bundesstiftung Umwelt (DBU) is gratefully acknowledged for funding of the work of Malte Gierse (20020/662). Special thanks are dedicated to the group Power to Liquids at Fraunhofer Institute for Solar Energy Systems and the team of ASG Analytik-Service AG.References
- Methanol Institute, DME: An Emerging Global Fuel, https://www.methanol.org/wp-content/uploads/2016/06/DME-An-Emerging-Global-Guel-FS.pdf, (accessed 2 June 2020) Search PubMed.
- P. Schühle, R. Stöber, M. Gierse, A. Schaadt, R. Szolak, S. Thill, M. Alders, C. Hebling, P. Wasserscheid and O. Salem, Dimethyl ether/CO2 - a hitherto underestimated H2 storage cycle, Energy Environ. Sci., 2023 10.1039/D3EE00228D.
- S. H. Nikos Xydas and LPG-DME Blends Workshop, WLPGA rLPG/rDME Activities, Zurich, 2022 Search PubMed.
- Fact Sheet, rDME Overview, 2022, https://www.aboutdme.org/aboutdme/files/cclibraryfiles/filename/000000004180/rDME_Fact_Sheet_General.pdf, (accessed 13 February 2023) Search PubMed.
- T. Cholewa, M. Gierse, F. Mantei, R. Güttel and O. Salem, Process Intensification Strategies for Power-to-X Technologies, ChemEngineering, 2022, 6, 13 CrossRef CAS.
- M. Gierse, R. E. Ali, M. Ouda, A. Schaadt, J. Sauer and C. Hebling, in Power to Fuel, ed. G. Spazzafumo, Elsevier, 2021, pp. 123–151 Search PubMed.
- N. K. Gor, N. A. Mali and S. S. Joshi, Intensified reactive distillation configurations for production of dimethyl ether, Chem. Eng. Process., 2020, 149, 107824 CrossRef CAS.
- W. An, K. T. Chuang and A. R. Sanger, Dehydration of Methanol to Dimethyl Ether by Catalytic Distillation, Department of Chemical and Materials Engineering, University of Alberta, Edmonton, 2004 Search PubMed.
- C. S. Bîldea, R. Győrgy, C. C. Brunchi and A. A. Kiss, Optimal design of intensified processes for DME synthesis, Comput. Chem. Eng., 2017, 105, 142–151 CrossRef.
- J. Liu, L. Gao, J. Ren, W. Liu, X. Liu and L. Sun, Dynamic Process Intensification of Dimethyl Ether Reactive Distillation Based on Output Multiplicity, Ind. Eng. Chem. Res., 2020, 59(45), 20155–20167 CrossRef CAS.
- Z. Lei, Z. Zou, C. Dai, Q. Li and B. Chen, Synthesis of dimethyl ether (DME) by catalytic distillation, Chem. Eng. Sci., 2011, 66, 3195–3203 CrossRef CAS.
- A. A. Kiss and D. J.-P. Suszwalak, Innovative dimethyl ether synthesis in a reactive dividing-wall column, Comput. Chem. Eng., 2012, 38, 74–81 CrossRef CAS.
- I. Bogatykh, C. Hoffmann, V. Kozachynskyi, M. Illner, T. Osterland, T. Wilharm and J.-U. Repke, Insights into Dynamic Process Intensification for Reactive Distillation Columns, Chem. Eng. Process., 2022, 177, 108978 CrossRef CAS.
- M. Di Stanislao, A. Malandrino, R. Patrini, C. Pirovano, A. Viva and E. Brunazzi, DME synthesis via catalytic distillation: Experiments and simulation, Comput.-Aided Chem. Eng., 2007, 1077–1082 CAS.
- W.-B. Su, J.-H. Hwang, H.-Y. Huang and T.-K. Chang, Dehydration of methanol to dimethyl ether in a dual-catalyst system catalytic distillation tower, J. Taiwan Inst. Chem. Eng., 2016, 59, 86–90 CrossRef CAS.
- M. Gierse, L. Steiner, M. Bontrup, J. Sauer and O. Salem, Catalyst screening and reaction kinetics of liquid phase DME synthesis under reactive distillation conditions, Chem. Eng. J., 2022, 140525 Search PubMed.
- M. Müller and U. Hübsch, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012, p. 39 Search PubMed.
- Lenntech, Product Data Sheet Amberlyst 36, https://www.lenntech.com, (accessed 1 September 2021) Search PubMed.
- I. Bogatykh, M. Illner, F.-J. Nagler, H. Stein, T. Osterland and J.-U. Repke, Low-interference real-time at-line spectroscopic composition analysis for chemical plants, Meas. Sci. Technol., 2023, 34, 55902 CrossRef.
- R. Taylor and R. Krishna, Modelling reactive distillation, Chem. Eng. Sci., 2000, 55, 5183–5229 CrossRef CAS.
- H. G. Schoenmakers and B. Bessling, Reactive and catalytic distillation from an industrial perspective, Chem. Eng. Process., 2003, 42, 145–155 CrossRef CAS.
- M. Sakuth, D. Reusch and R. Janowsky, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- M. Schmitt, H. Hasse, K. Althaus, H. Schoenmakers, L. Götze and P. Moritz, Synthesis of n-hexyl acetate by reactive distillation, Chem. Eng. Process., 2004, 43, 397–409 CrossRef CAS.
- M. Schmitt, S. Blagov and H. Hasse, Mastering the Reaction Is the Key to Successful Design of Heterogeneously Catalyzed Reactive Distillation: A Comprehensive Case Study of Hexyl Acetate Synthesis, Ind. Eng. Chem. Res., 2008, 47, 6014–6024 CrossRef CAS.
- P. Moritz and H. Hasse, Fluid dynamics in reactive distillation packing Katapak®-S, Chem. Eng. Sci., 1999, 54, 1367–1374 CrossRef CAS.
- K. Ye, H. Freund and K. Sundmacher, Modelling (vapour+liquid) and (vapour+liquid+liquid) equilibria of {water (H2O)+methanol (MeOH)+dimethyl ether (DME)+carbon dioxide (CO2)} quaternary system using the Peng–Robinson EoS with Wong–Sandler mixing rule, J. Chem. Thermodyn., 2011, 43, 2002–2014 CrossRef CAS.
- E. von Harbou, A. Yazdani, M. Schmitt, C. Großmann and H. Hasse, Reaction Kinetics for Reactive Distillation Using Different Laboratory Reactors, Ind. Eng. Chem. Res., 2013, 52, 624–637 CrossRef CAS.
- S. Hosseininejad, A. Afacan and R. E. Hayes, Catalytic and kinetic study of methanol dehydration to dimethyl ether, Chem. Eng. Res. Des., 2012, 90, 825–833 CrossRef CAS.
- L. B. Datsevich, Alternating motion of liquid in catalyst pores in a liquid/liquid–gas reaction with heat or gas production, Catal. Today, 2003, 79-80, 341–348 CrossRef CAS.
- H. Z. Kister, Distillation Design, 1st edn, 1992, ISBN: 9780070349094 Search PubMed.
- J. Nyári, M. Magdeldin, M. Larmi, M. Järvinen and A. Santasalo-Aarnio, Techno-economic barriers of an industrial-scale methanol CCU-plant, J. CO2 Util., 2020, 39, 101166 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3re00200d |
| This journal is © The Royal Society of Chemistry 2023 |