
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of support effects during ethanol steam reforming over a Ni/sepiolite catalyst†
Marinela D.
Zhurka
a,
James A.
Anderson
a,
Alan J.
McCue
 b,
Angeliki A.
Lemonidou
c and
Panagiotis N.
Kechagiopoulos
*a
b,
Angeliki A.
Lemonidou
c and
Panagiotis N.
Kechagiopoulos
*a
aChemical Processes & Materials Group, School of Engineering, University of Aberdeen, Aberdeen, AB24 3UE, UK. E-mail: p.kechagiopoulos@abdn.ac.uk
bDepartment of Chemistry, University of Aberdeen, Aberdeen, AB24 3UE, UK
cLaboratory of Petrochemical Technology, Department of Chemical Engineering, Aristotle University of Thessaloniki, GR-54124 Thessaloniki, Greece
First published on 12th July 2023
Abstract
Sustainable hydrogen production can be achieved efficiently by steam reforming of bio-ethanol. The use of low-cost and abundant minerals as catalyst supports can further improve the sustainability of the process. In this work, a kinetic study of ethanol steam reforming is presented using a Ni catalyst supported on natural sepiolite. Focus is placed on probing the effect of the support on the reaction mechanism, which is found to depend on the catalyst calcination temperature and degree of hydration of the sepiolite. Results suggest the presence of more than one adsorption sites where both oxygenates and water can competitively adsorb, when the catalyst has not been exposed to temperatures higher than 500 °C. This bifunctional mechanism is further found to be affected by the feed steam/carbon ratio. Thermally pre-treating the catalyst at 550 °C leads to an irreversible removal of support silanol groups that hinders the adsorption of reactants on sepiolite. Hydrating the non-thermally treated catalyst prior to experiments through steam exposure enhances the density of support adsorption sites leading to kinetic performances in line with those over inert supports such as SiO2. Steam reforming of acetaldehyde, a major product of ethanol steam reforming, is also carried out leading to similar observations, building a consistent kinetic picture of the reaction over Ni/sepiolite.
1. Introduction
The basis of energy production systems of the world is fossil fuels, resulting in well recognised negative environmental impact. A promising alternative to fossil fuels is hydrogen, which is a clean energy carrier and can be derived sustainably from biomass.1 The primary source of hydrogen production is natural gas or coal, leading to high CO2 emissions. Recent studies have shown that biomass derived oxygenated compounds are economically attractive and environmentally friendly energy sources.2 Bio-ethanol is an ideal candidate to produce H2 through catalytic reforming (ethanol steam reforming – ESR), as it can be formed from renewable feedstocks (energy crops, forestry or agro-industry waste) via fermentation and hydrolysis procedures.3 Further advantages of bio-ethanol relate to its low toxicity and safe handling (storage and transportation). Ethanol can further be regarded as a model compound of alcohols in the aqueous fraction of bio-oil, the liquid product of biomass pyrolysis.4The main products of ESR are H2 and CO2 according to the overall reaction (1) seen in Table 1, with hydrogen production further influenced by the water gas shift (WGS) reaction (2). By-products can also be formed, such as methane, carbon monoxide, ethylene, acetaldehyde, acetic acid, and acetone, according to reactions (3) to (10).5 The reaction pathways of ESR presented in the literature show significant differences, depending on catalyst5 and operating conditions,6 further affecting hydrogen production and product selectivities. Several research groups presented kinetic studies of ESR,7,8 suggesting, initially, dehydrogenation (3) and dehydration reactions (8) to occur, favoured by the basic and acidic features of the catalyst, respectively, and the overall balance and strength of such sites.9 Acetaldehyde can form methane through decarbonylation (5), which can further undergo steam reforming (6) and (7). Some catalysts reported in the literature can also promote the reaction of acetaldehyde with water to produce acetic acid (9),10,11 which can decompose to methane and carbon dioxide (10). Transition metals also tend to form encapsulating coke through the polymerization of ethylene (13), which blocks the active sites, and filamentous coke via the Boudouard reaction (11), which does not block active sites but can lead to metal segregation from the support.12
| (1) CH3CH2OH + 3H2O ⇄ 6H2 + 2CO2 | (2) CO + H2O ⇄ CO2 + H2 |
| (3) CH3CH2OH ⇄ CH3CHO + H2 | (4) CH3CH2OH ⇄ CH4 + CO + H2 |
| (5) CH3CHO ⇄ CH4 + CO | (6) CH4 + H2O ⇄ CO + 3H2 |
| (7) CH4 + 2H2O ⇄ CO2 + 4H2 | (8) CH3CH2OH ⇄ C2H4 + H2O |
| (9) CH3CHO + H2O ⇄ CH3COOH + H2 | (10) CH3COOH ⇄ CH4 + CO2 |
| (11) 2CO ⇄ CO2 + C | (12) CH4 → C + 2H2 |
(13)  |
(14) CH3CHO + 3H2O ⇄ 5H2 + 2CO2 |
| (15) 2CH3CHO → CH3CH(OH)CH2CHO | (16) CH3CH(OH)CH2CHO + H2O → CH3COCH3 + CO2 + 2H2 |
Many studies have suggested Ni as a suitable metal for ethanol steam reforming due to its high activity, and low cost in comparison to noble metals.13 Carbon deposition remains a major challenge for Ni, although the nature of the support and operating conditions can prove critical in overcoming this limitation. Thus, basic supports, high operating temperature and high steam to carbon ratio are preferred as these can reduce coking without negatively impacting activity.
Currently, the interest in catalysts supported on minerals such as olivine, dolomite, attapulgite and sepiolite has increased due to their abundance, low cost, and high surface area. Sepiolite, a phyllosilicate mineral, is an interesting catalyst support characterised by its porous inner structure and fibrous morphology. Sepiolite has been widely studied as a catalyst support for a variety of hydrotreating reactions,14–17 where doping with potassium or lanthanum,18 incorporating metal oxides19 and preparing the catalyst via precipitation20,21 have further been employed to enhance its performance. Sepiolite has also been proven recently to be a favorable support for steam reforming reactions with its structural features contributing to high adsorption capacity with high stability.22,23 The steam reforming of bio-oil and its model compounds has specifically been reported on Ni/sepiolite, however the focus of these works was on performance analysis and not on the elucidation of the kinetics of the reaction.24,25 Ni supported on natural sepiolite was further shown to exhibit better activity during furfural steam reforming when compared to traditional supports (SiO2, MgO and Al2O3).26
The present study reports on an extensive kinetic analysis of ESR over Ni/sepiolite under a wide range of experimental conditions in a fixed bed reactor. Particular focus is placed on probing the effect of the support on the reaction mechanism, which is found to depend on the catalyst calcination temperature and degree of hydration of the sepiolite. Acetaldehyde kinetic experiments are also carried out to corroborate the findings of the work, building a consistent kinetic picture of the ESR reaction over Ni/sepiolite.
2. Experimental
2.1. Sample preparation
The Ni/sepiolite catalyst with nominal 10 wt% Ni to the final weight of the solid was prepared by a precipitation method using Ni(NO3)2·6H2O (Merck, reagent grade) as precursor and untreated/fresh sepiolite (Tolsa S.A., surface area 135 m2 g−1) as support. Chemical analysis of the material indicated a composition of SiO2 (66.19%), H2O (15.95%), MgO (13.16%), Na2O (1.71%), Al2O3 (1.66%), CaO (0.56%), K2O (0.41%) and Fe2O3 (0.36%). The solution of nickel nitrate was firstly added to a suspension of the support material, adjusting the pH to 3.5 by the addition of nitric acid. This solution was heated to 90 °C for 30 min before addition of an urea solution, which resulted in the controlled precipitation of nickel hydroxide over the support. The suspension was filtered after 48 h and washed repeatedly with distilled water. The sample was dried at 110 °C in air for 16 h, heat treated in a N2 flow (100 cm3 min−1) at 300 °C for 3 h and then reduced in a 3.5% H2 in Ar flow (100 cm3 min−1) at 500 °C for 5.5 h,27 before cooling to ambient temperature in a flow of N2 and storing. Further details are provided in previous studies.28,292.2. Reactor setup
The kinetic experiments were carried out in a fully automated reaction system by PID Eng & Tech (Micro Activity-effy unit). The reactants (a water/ethanol or water/acetaldehyde mixture) were fed by an HPLC pump (Gilson 307). This feed was channelled through an evaporator, operating at 150 °C, and further mixed with N2 before entering the reactor. An internally passivated stainless steel (SS316) fixed bed reactor (9.1 mm i.d., total length 304.8 mm) was used, heated by a single-zone furnace providing an isothermal region of 5 cm. The catalyst bed was diluted with α-Al2O3 granules (1.5 mm average size) at 1/9 catalyst/alumina weight ratio and placed within two layers of quartz wool on top of a fitted porous plate. The α-Al2O3 diluent was independently verified to be inert at the test conditions. A Liquid/Gas separator was used to collect the liquid reactions products, while gas products were analysed on-line in a HP5890 GC, equipped with a TCD detector and MS-5A and HS-T columns. Liquids were analysed offline in a Thermo Scientific TRACE 1300 GC with an FID detector and a WAXMS A column.2.3. Experimental conditions and parameters
Ahead of the experiments, the catalyst was treated in situ under a flow of N2 at 500 °C for 1 h, and then reduced at 500 °C using a 5% H2 in N2 flow of 100 cm3 min−1 for 1 h. Reaction temperature was varied from 300 to 600 °C with a steam/carbon (S/C) ratio equal to 3 molH2O molC−1 (section 3.2) at total pressure of 1.7 bar and partial pressures of ethanol and water of 0.15 and 0.90 bar, respectively, balanced by N2. All pressure values reported herein refer to absolute pressure, with total values measured upstream of the catalyst bed. Volumetric flowrates were all measured at normal conditions. The space time effect was examined by varying the feed flow from 97 to 330 cm3 min−1, with a S/C ratio of 3, over a fixed catalyst mass (W/Ft0,Eth/Acet from 58 to 349 gcat s gEth/Acet−1) at 400 °C and steady pressure of 1.8 bar and partial pressures of ethanol and water of 0.21 and 1.29 bar, respectively, balanced by N2 (section 3.3). The partial pressure of water was varied from 0.26 to 1.56 bar at a total pressure of 1.9 bar and partial pressure of ethanol of 0.13 bar at 400 °C (section 3.4.1). Correspondingly, the partial pressure of ethanol was varied from 0.06 to 0.37 bar at a total pressure of 1.9 bar and partial pressure of water of 0.74 bar at 400 °C (section 3.4.2). Additional partial pressure variation experiments were carried out at the same temperature to investigate the effect of catalyst thermal pre-treatment carrying out the latter at 550 °C instead of 500 °C (see section 3.4.3 for details), and degree of hydration of the support subjecting the catalyst to a flow of steam prior testing (see section 3.4.4 for details). The behaviour of acetaldehyde versus ethanol as a reactant was also investigated, given the recognised role of the former as an important intermediate in ESR. A mass of 80 mg Ni/sepiolite catalyst (sieved to a 300–500 μm fraction) was used in all experiments, resulting in a catalyst bed of apparent packing density of 0.4 g cm−3.Standard criteria by Mears30 and Weisz–Prater31 were applied to ensure measurements were conducted under explicit kinetic control. Atomic C, H and O mass balance closure in all tests was in the order of 100 ± 5%. The results presented in the following sections are expressed in terms of the following parameters, and are further compared with thermodynamic equilibrium, calculated via Gibb's free energy minimisation within the Aspen Plus software using the Peng–Robinson equation of state.
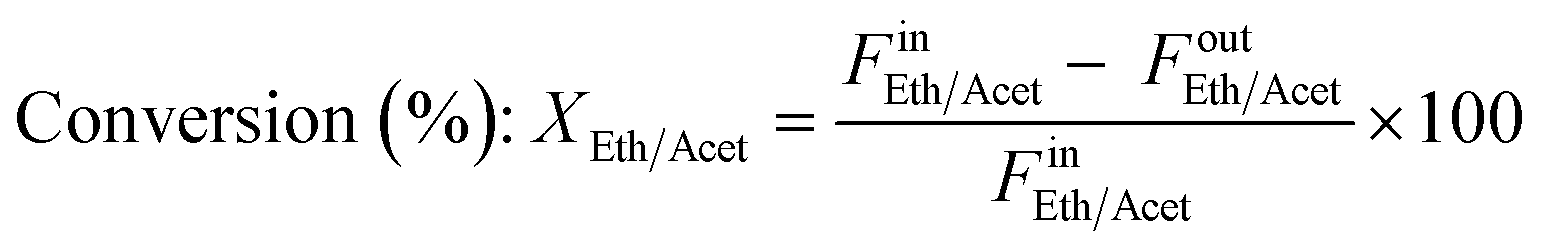
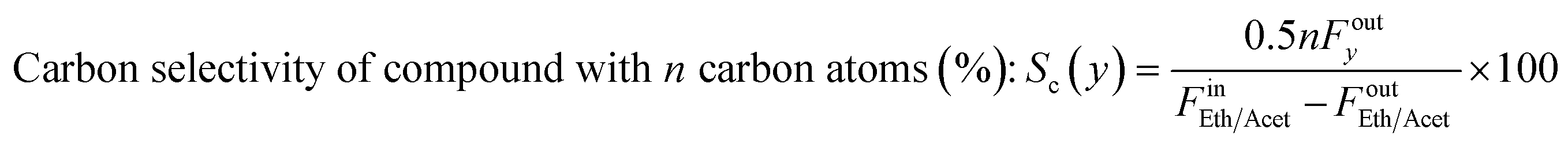
where Fin/out is the inlet/outlet molar flow of the respective compound (mol s−1) and m the number of hydrogen moles produced by the oxygenate's stoichiometric steam reforming, equal to 6 and 5 for ethanol and acetaldehyde, respectively. Turnover Frequency (TOF) was defined as the moles of ethanol or acetaldehyde converted per mole of Ni active sites per second and was calculated according to the below equation:
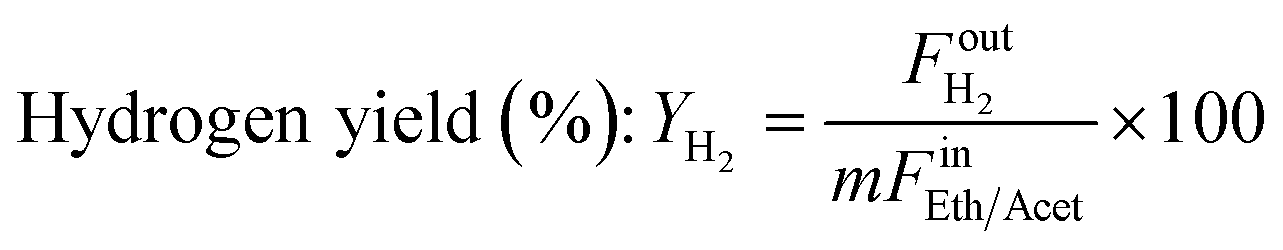
where wcat is the catalyst weight, wNi the nominal weight composition of nickel (10%), D the metal dispersion of nickel calculated via H2 TPD measurements (see below), and AWNi the atomic weight of nickel.
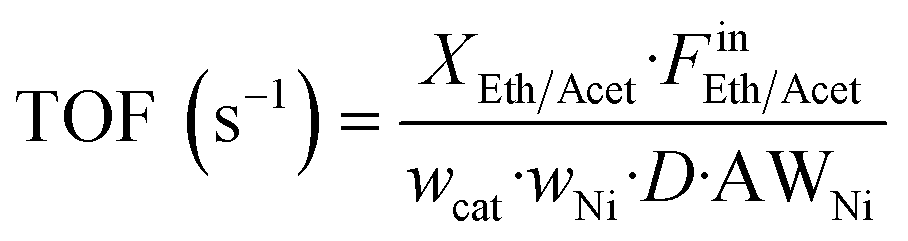
2.4. Characterisation of fresh catalyst samples
TPD and TPR experiments have been conducted to characterize the metal surface of the catalyst and investigate the optimal reduction conditions, respectively. A TPDRO 1100 instrument was used with a TCD detector, with a trap bed placed before the detector to remove the moisture. Prior each experiment, samples were fully oxidised under a flow of 5% O2 in He with a heating rate of 10 °C min−1 up to 600 °C. For the H2-TPD analysis the samples were re-reduced in situ by a flow of H2 at 500 °C for 1 h, then the system was purged with He for 20 min at 30 °C. The chemisorption step took place under a flow of 10% H2/Ar for 1 h at 100 °C, before measuring the desorbed H2 while heating until 600 °C with a 10 °C min−1 rate. Quantified H2 desorbed was used to obtain the active metal dispersion D defined as the ratio of the number of exposed Ni surface atoms to the total number of Ni atoms present in the sample. For the TPR the samples were firstly dried at 200 °C for 30 min under N2 flow, then the system was cooled down and the temperature was increased again until 800 °C with a 10 °C min−1 temperature ramp, using a 5% H2/N2 flow. For Thermogravimetric Analysis (TGA) experiments, a small amount of the sample (10 to 15 mg) was placed in an Al-based sample cup of a TG 209 F3 Tarsus apparatus (NETZSGH, Germany) and heated up to 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C at a rate of 10°C min−1 under a flow of air (50cm3 min−1). XRD patterns were obtained at room temperature on a Panalytical powder diffractometer to identify crystalline phases apparent. The diffraction patterns were recorded over an angular range of 10° < 2θ < 90° with a step-size of 0.013°. A BET surface area analyser Micromeritics Tristar 3000 was used to determine the total surface area of solid samples by using N2 as adsorbate at liquid nitrogen temperature. Prior all characterisation analyses, fresh reduced samples were thermally treated under a flow of N2 at 500 °C (or 550 °C) to simulate the procedures followed during catalytic testing.
°C at a rate of 10°C min−1 under a flow of air (50cm3 min−1). XRD patterns were obtained at room temperature on a Panalytical powder diffractometer to identify crystalline phases apparent. The diffraction patterns were recorded over an angular range of 10° < 2θ < 90° with a step-size of 0.013°. A BET surface area analyser Micromeritics Tristar 3000 was used to determine the total surface area of solid samples by using N2 as adsorbate at liquid nitrogen temperature. Prior all characterisation analyses, fresh reduced samples were thermally treated under a flow of N2 at 500 °C (or 550 °C) to simulate the procedures followed during catalytic testing.
3. Results and discussion
3.1. Characterization of Ni/sepiolite catalyst and sepiolite
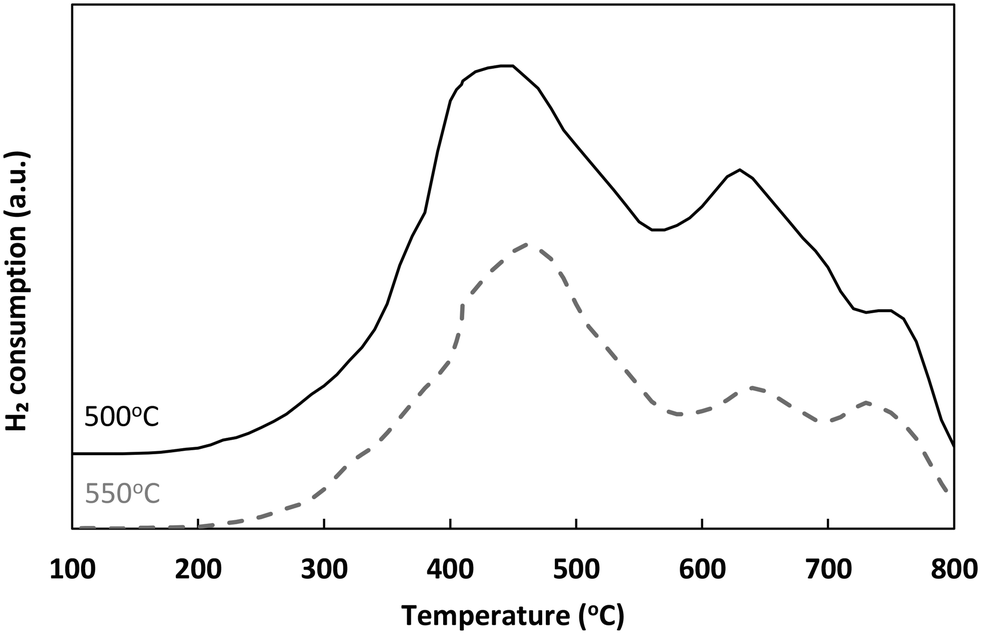 | ||
| Fig. 1 H2-TPR profiles of Ni/sepiolite thermally treated at 500 °C and 550 °C. | ||
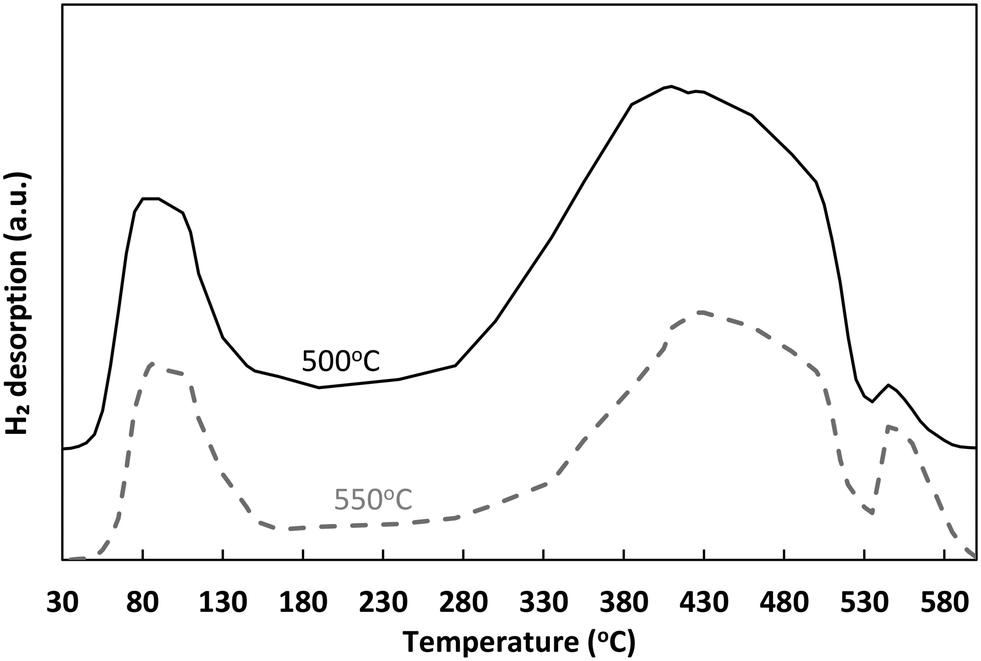 | ||
| Fig. 2 H2-TPD profiles of Ni/sepiolite thermally treated at 500 °C and 550 °C. | ||
3.2. Effect of temperature
The effect of temperature for ESR over Ni/sepiolite is presented in Fig. 3. For all results presented in this and following sections, unless otherwise stated, the catalyst sample used has been reduced and heat treated at 500 °C. Ethanol conversion increases from 13 to 58% as temperature rises, with the hydrogen yield following closely the trend, reaching a maximum value of 46% at the highest temperature (600 °C) studied (Fig. 3a). Thermodynamics predict conversion to be 100% at these temperatures and much higher hydrogen yield values than those experimentally observed, indicative that the results were not limited by equilibrium. Based on the high selectivities of CH3CHO, CH4 and CO (Fig. 3c and e), it is evident that ethanol dehydrogenation and decomposition reactions dominate below 400 °C. The low H2 yield and CO2 selectivity further indicate that ethanol reforming and water gas shift reactions do not participate significantly at these low temperatures. The increasing CH4 selectivity up to 400 °C, combined with the concurrent decreasing CO selectivity and overall low H2 yield, indicate the presence of the methanation reaction in line with prior studies on ethanol steam reforming over Ni catalysts.13 The increase in CO2 selectivity and hydrogen yield, while CH4, CO and CH3CHO selectivities decrease as the temperature rises from 400 to 600 °C, suggest the progressive promotion of WGS and reforming reactions. Trends are similar to those presented in our previous work on ESR over Ni/SiO2,40 indicative of the overall dominant role of the Ni metal in the reaction mechanism. To further examine this fact, the temperature effect on the steam reforming of ethanol over pure sepiolite was studied (Fig. S6 in ESI†). Very low conversions, below 8% were observed with selectivity to C2H4 being above 90% across the temperature range. Clearly, sepiolite alone cannot activate the cleavage of the C–C bond in ethanol, suggesting that operation at 450 °C or higher at the presence of Ni is needed to ensure the efficient activation of reactants.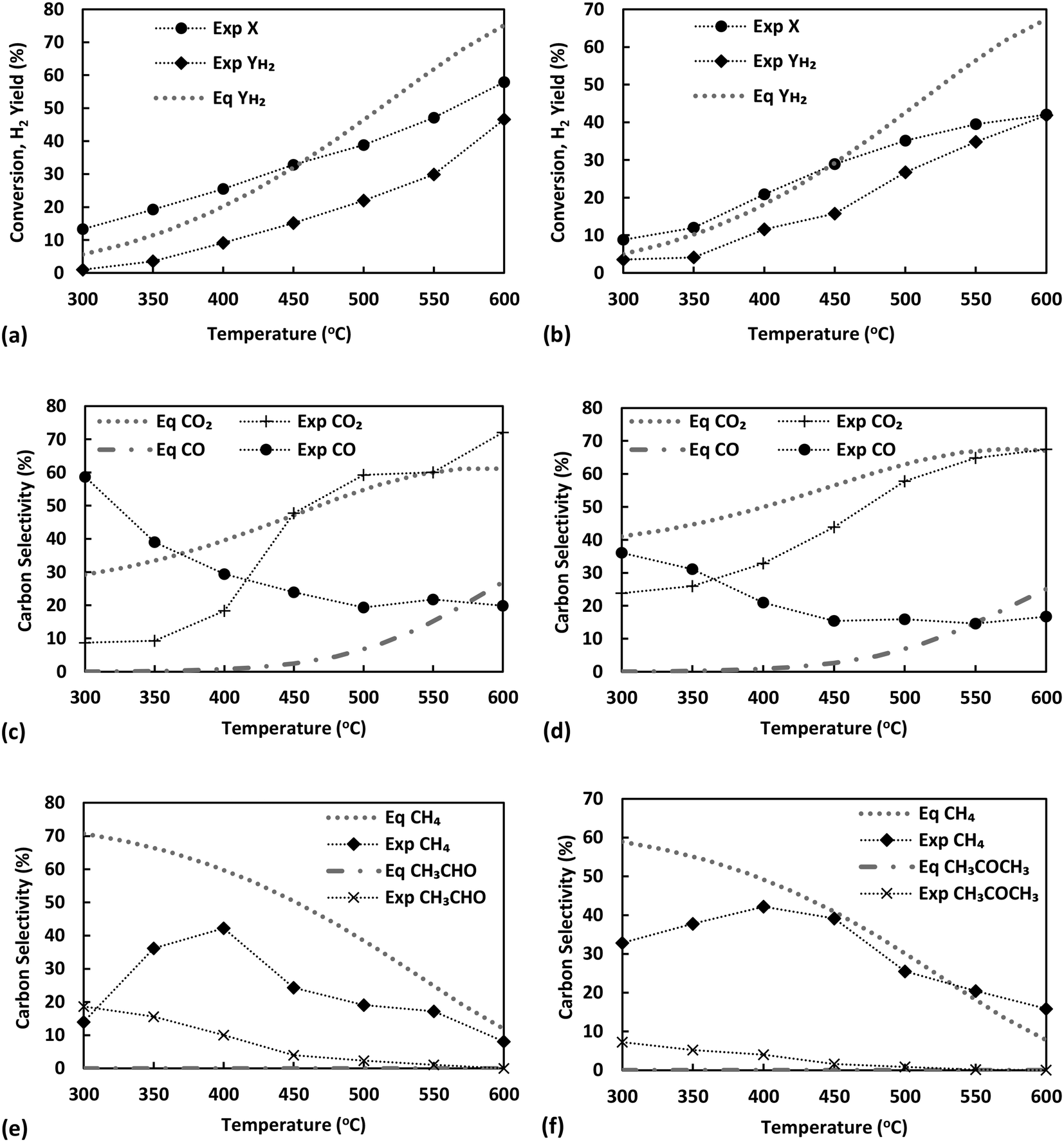 | ||
| Fig. 3 Temperature effect on (top) conversion and H2 yield for ESR (a) and ASR (b), (middle) carbon selectivities of CO2, CO for ESR (c) and ASR (d), and (bottom) carbon selectivities of CH4 and CH3CHO for ESR (e) and CH4 and CH3COCH3 for ASR (f) compared with the equilibrium (W/FEth/Acet = 91.8 gcat s gEth/Acet−1, P = 1.7 bar, S/C = 3) (lines connecting experimental points are visual aids). | ||
Considering that acetaldehyde is an important intermediate in the reaction pathway of ESR, acetaldehyde steam reforming (reaction (14) in Table 1) was also studied under equivalent conditions, ensuring partial pressures were the same as with the ethanol runs. As with ethanol, conversion and H2 yield are observed to increase with temperature but are again far from thermodynamic equilibrium (Fig. 3b). At temperatures higher than 450 °C, acetaldehyde conversion appears to follow a different trend to that observed at lower temperatures. This could indicate that thermal decomposition of acetaldehyde is taking place, possibly upstream of the catalyst bed, resulting in the direct production of CH4 and CO2 in the gas phase. The changing trend in the selectivities of these two compounds at this temperature range further support this (see below.) It is worthy to note, though, the lower conversion of acetaldehyde compared to ethanol at temperature below 450 °C, which indicates that the latter's conversion does not necessarily proceed via acetaldehyde, particularly considering that it is the initial C–H bond scissions towards ethoxy and acetaldehyde that are typically considered as rate-limiting.8 Indeed, in our previous work over Ni/SiO240 it was demonstrated that at temperatures over 450 °C the dominant conversion pathway of ethanol most likely involves a different surface reaction intermediate, which, in an independent microkinetic modelling study from our group, was suggested to be 1-hydroxyethyl.41 The H2 yield achieved is approximately equal to the case of ethanol, however, given the metric's definition in acetaldehyde steam reforming reaction, this actually indicates a lower hydrogen production, as ethanol has a higher hydrogen content of 3 moles of H2 per carbon atom compared to 2.5 for acetaldehyde.
The products of acetaldehyde steam reforming are CO, CO2, CH4, CH3COCH3 and H2 (Fig. 3d and f). The carbon selectivities of CO, CO2 and CH4 indicate that the reaction network shares similarities to that of ESR. Decomposition and methanation reactions are clearly favoured at the lower temperatures with WGS having low activity, as indicated by the high and opposing trends of CH4 and CO selectivities as the temperature rises to 450 °C. Reforming reactions towards hydrogen production dominate above that point, further enhanced by the WGS, clearly evident by the rising CO2 selectivity and increasing H2 yield. As commented above, under these conditions, where the S/C ratio is kept constant, results suggest a prominent role of the active metal in the reaction mechanism, with Ni being a well-known methanation and steam reforming catalyst.
An important difference between ASR and ESR is the detection of acetone at measurable quantities in the liquid products of ASR. Acetone may be a product of aldol condensation of acetaldehyde to 3-hydroxybutanal (reaction (15) in Table 1),42–44 the latter undergoing further dehydrogenation and decarboxylation to CH3COCH3, CO2 and H2 (reaction (16) in Table 1). The aldol condensation of acetaldehyde over MgO has been particularly well-examined,45,46 while the use of sepiolite as a catalyst for condensation reactions due to its structure, consisting of octahedral sheets mainly of MgO, has also been a matter of investigation.47 In the current work, larger amounts of acetone were observed at lower temperatures, potentially explaining also the relatively higher CO2 selectivity in this temperature range in comparison to ESR. Nonetheless, this enhanced CO2 productivity at low temperatures, where WGS has been demonstrated to be low in activity, could also originate from the presence of surface acetate species generated from adsorbed acetaldehyde.48
3.3. Effect of space time
The effect of contact time on the catalytic performance of ethanol and acetaldehyde steam reforming over Ni/sepiolite at 400 °C and S/C = 3 is presented in Fig. 4. Conversion and H2 yield, plotted as a function of W/F (gcat s gEth−1 and gcat s gAcet−1) in panel a, are seen in both cases to rise with contact time. For ethanol, the conversion follows a linear trend spanning from 16 to 67% across the W/F range studied. Acetaldehyde conversion spans from 18 to 55% across the same range, however appears to follow different slopes depending on W/F value. Considering the selectivity trends discussed below, namely the very high acetone production, the varying conversion trend of acetaldehyde can be attributed to a pronounced presence of condensation reactions at the higher W/F values.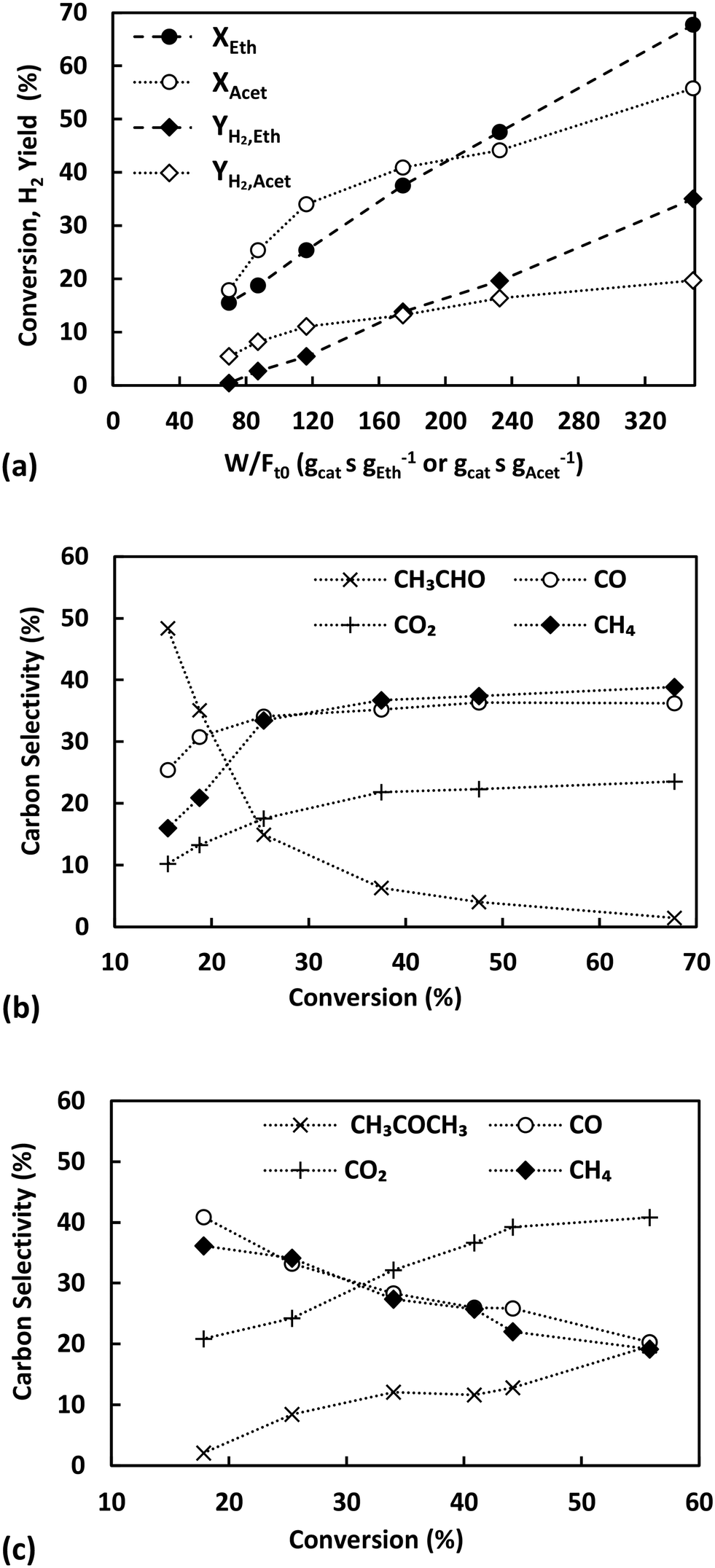 | ||
| Fig. 4 W/Ft0 effect on ethanol and acetaldehyde conversion and H2 yield at 400 °C (a) and carbon selectivities of CO, CO2, CH4, and CH3CHO or CH3COCH3versus ethanol (b) and acetaldehyde (c) conversion (P = 1.8 bar, S/C = 3) (lines are visual aids). | ||
Fig. 4b shows the evolution of product selectivities with varying ethanol conversion. The selectivity of CH3CHO is seen to increase as conversion (and W/F) decreases and would clearly reach a finite value if conversion approached zero, evidence that acetaldehyde is a primary product of ESR. Opposite trends are observed for CO, CO2 and CH4, which decrease with conversion's decrease, suggesting these are secondary products. The dehydrogenation of ethanol to acetaldehyde is responsible for its primary formation, while decomposition of the later or ethanol itself to CO and CH4 and their subsequent reforming and shift reactions to COx give rise to their secondary production. The effect of space time over Ni/sepiolite is analogous to that observed over Ni/SiO240 indicative of the active metal driving the overall reaction pathways under these conditions.
To examine the impact of the support, similar W/F variation experiments at 400 °C were carried out over natural sepiolite under steam reforming (Fig. S7 in ESI†) and decomposition (Fig. S8 in ESI†) conditions. During decomposition, very low conversions were observed (0.5–4% depending on W/F tested), well below those achieved over Ni/sepiolite, and indicative of limited support activity in activating ethanol. The only product identified was C2H4 suggesting that sepiolite enables the dehydration of ethanol. When steam reforming was tested over natural sepiolite slightly higher conversions were observed (1–6% for same W/F range), but still much lower than with Ni/sepiolite. Selectivity to C2H4 again was very high, above 90% across all W/F values tested. Nonetheless, traces of CO2 were also detected in the gas products, most likely originating from the gasification of carbon deposits by steam, as C2H4 is a well-known coke precursor. In all cases, the very high selectivity to ethylene is indicative of sepiolite not being able to activate the cleavage of the C–C bond in ethanol. In the following sections these findings and the role of the sepiolite support in explaining observed catalytic trends will be further elaborated.
Finally, Fig. 4c presents the effect of space time on product selectivities for ASR, where significant differences can be observed in relation to ESR. At lower conversion (and W/F) the selectivities of CO and CH4 are high and approximately equal at 40%. The CO/CH4 ratio in the product stream remains almost one for all space times tested, indicative of the dominant role of the acetaldehyde decomposition reaction (reaction (5) in Table 1). As contact time and conversion decrease, the selectivities of CO and CH4 increase tending to non-zero values, linked to their primary formation from acetaldehyde decomposition. In contrast, CH3COCH3 is clearly a secondary reaction product of ASR as its selectivity is reduced with a decrease in conversion, reaching an almost zero value at the lowest conversion studied. As discussed, its formation most likely originates from an acetaldehyde aldol condensation reaction network proceeding in parallel to ASR. Finally, the selectivity of CO2 is influenced by the multiple pathways that involve the species, with its creation affected by acetate decomposition, acetone formation, WGS reaction and methane steam reforming reactions. Nonetheless, the decreasing trend of its selectivity as conversion decreases and the nature of the above reactions suggests CO2 to be a secondary reaction product of ASR.
3.4. Effect of partial pressure of reactants
51 a slightly negative order with respect to water was observed in agreement with steam-derived intermediates not participating in the kinetically determining step. However, current results on the Ni/sepiolite catalyst suggest a critical role of the support in the kinetic mechanism, which appears to be consistent for both ethanol and acetaldehyde reactions.
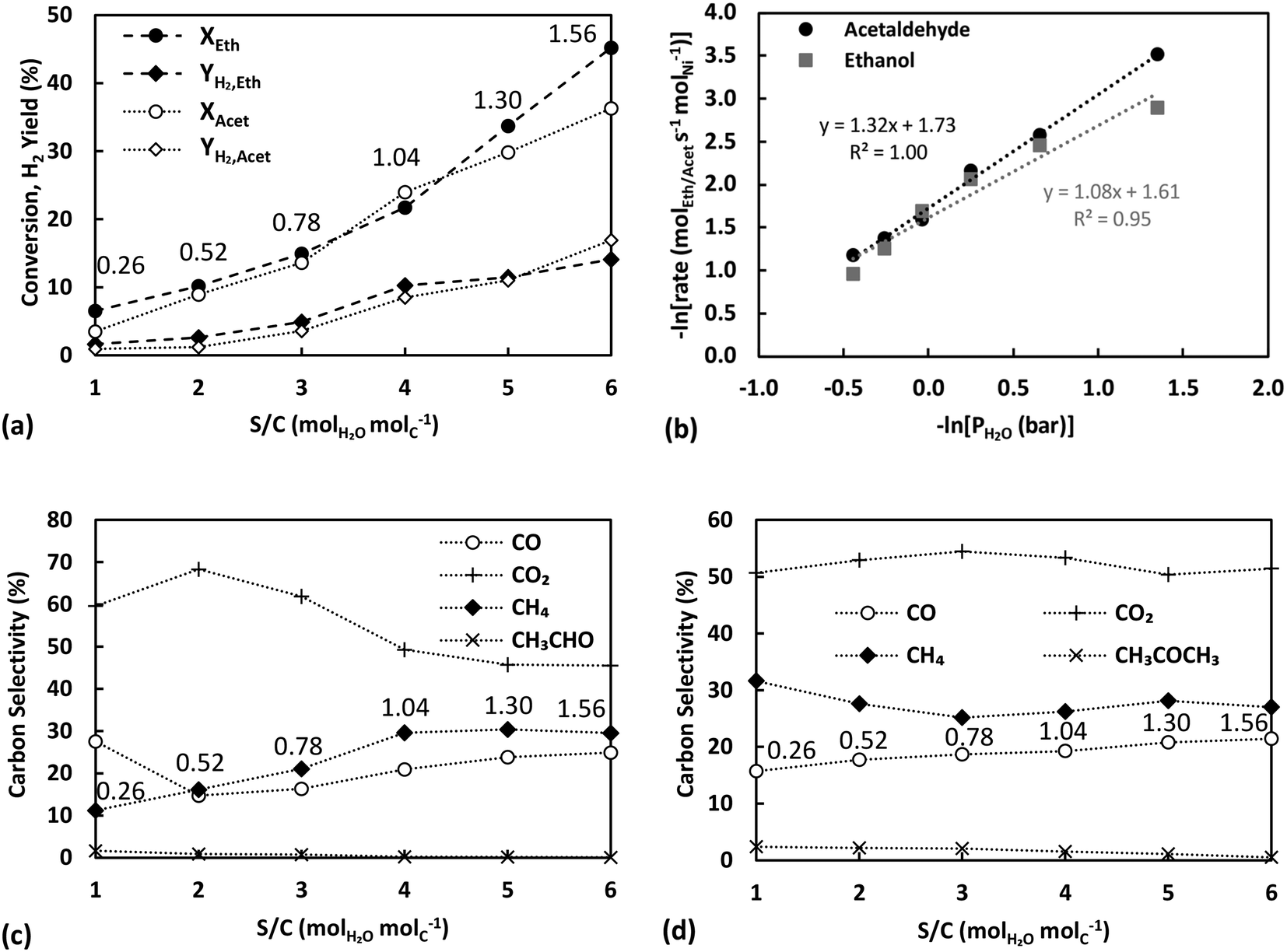 | ||
| Fig. 5 Partial pressure effect of water on (top) ethanol and acetaldehyde conversion and H2 yield (a) and logarithm of reaction rate of ESR and ASR with respect to logarithm of water partial pressure (b), and (bottom) carbon selectivities of ESR products (c) and ASR products (d) at 400 °C (P = 1.9 bar, Vtot = 213 cm3 min−1). For panels (a), (c) and (d) the partial pressure effect is presented as a S/C variation with numbers on plots annotating the equivalent partial pressures of water in bar (panels a, c, and d: lines are visual aids). | ||
Further evidence of support participation in the kinetic mechanism is provided by the carbon selectivities of ESR plotted against S/C variation in Fig. 5c. Apart from S/C = 1, where the supply of water is sub-stoichiometric for ESR, increasing the partial pressure of water and the equivalent S/C ratio is seen to lead to a decrease in the selectivity of CO2 and conversely a rise in those of CO and CH4. The much lower selectivity towards acetaldehyde in Fig. 5c in comparison to that in Fig. 3e or 4b can be attributed to the comparatively much lower partial pressure of ethanol used in these experiments. In all cases, the observed selectivity profiles contrast with the conventionally expected behaviour from an increase in the S/C ratio and the associated promotion of reforming and WGS reaction pathways.40 The carbon selectivities observed during ASR, presented in Fig. 5d, display milder trends, although still in line with the findings on ESR, with CH4 and CO selectivities showing a slight increase at the higher S/C ratios. A complex interplay between the metal and the support is evident, where the availability of water appears to have a rate controlling influence on ethanol or acetaldehyde conversion but at the same time does not lead to a promotion of secondary reaction pathways.
These findings are tentatively attributed to the structure of sepiolite (Fig. 6), consisting of six SiO4 tetrahedra linked together to form a polymeric silica layer.52 Between the silica layers there are MgO6 octahedra arranged to give threefold strips leaving channels parallel to the fibre axis containing water molecules. The external surface contains OH groups originating from the exposed sides of the tetrahedral and the octahedral sheets, in the form of Si–OH and Mg–OH. Additionally, Si–OH groups are produced when the Si–O–Si bridges linking the silicate layers are broken, leaving terminal OH groups. It is known that acid media, which was used for the catalyst preparation, may attack the sepiolite structure, and is expected to increase the number of Si–OH groups.29 These OH groups are crucial as precipitation–deposition of the nickel precursor appears to consume such hydroxyls. This preferential reaction indicates that most, if not all, of the precursor nucleation occurs at these locations and, hence, that Ni is placed on the top part of the tetrahedral sheets and inside the open channels.28 Indeed, the nature of the exposed metal faces was determined by infrared (IR) CO adsorption in previous work revealing the formation of layered Ni silicate precursors following precipitation and indicating the production of mostly 2D Ni (100) crystal planes on the catalyst upon reduction.29
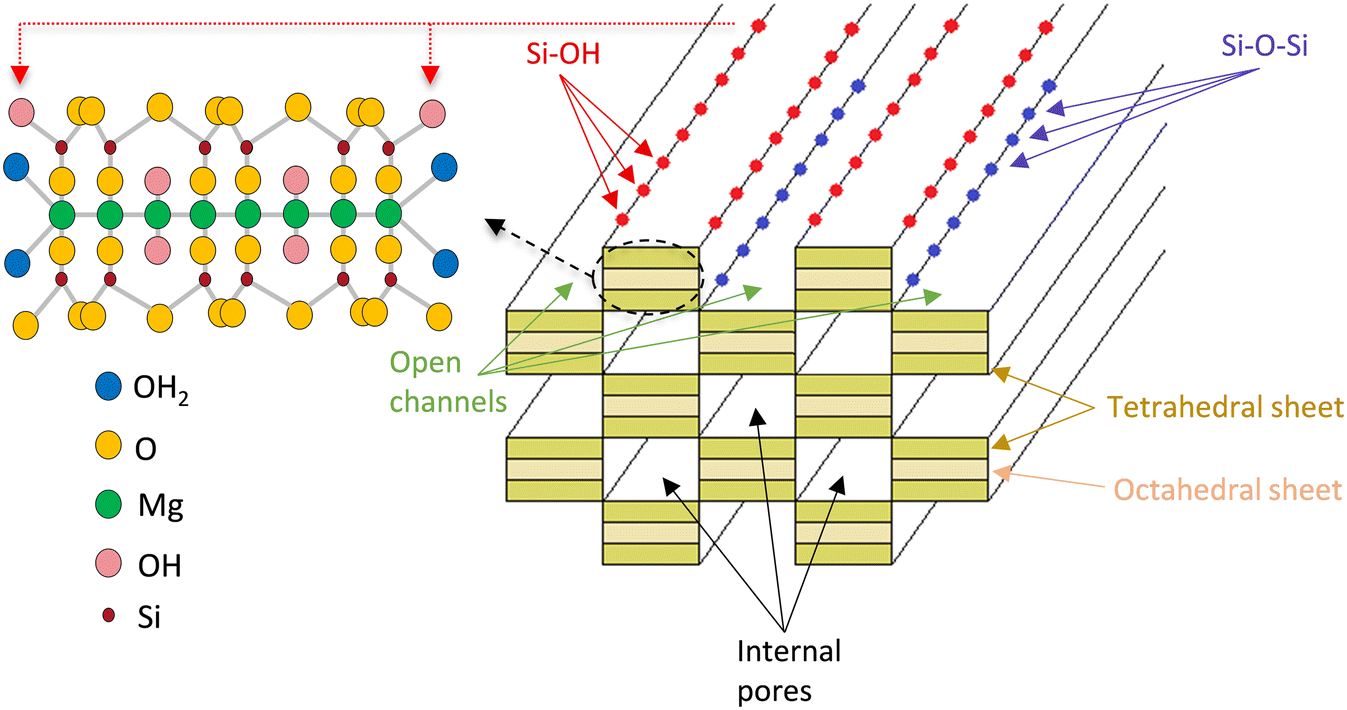 | ||
| Fig. 6 Schematic representation of sepiolite structure showing open channels, internal pores, molecular and crystalline structure. | ||
Furthermore, sepiolite's hydrophilic surface is recognized as responsible for increased adsorbent behavior towards polar molecules such as water, ethanol and acetaldehyde.53 The hydrophilicity of sepiolite depends on the high density of the silanol groups (Si–OH) that can form hydrogen bonds with the hydroxyl groups of ethanol and water.54,55 It has indeed been suggested during phenol steam reforming on a Ni catalyst supported over mixed Ti and Zn oxides of perovskite structure that the high concentration of surface OH of the support was accountable for dual site adsorption.56 Phenol's functional group (OH) was found to interact not only with the metal active sites but also with the surface hydroxyl groups leading to the adsorption of phenol on the support. Additionally, experimental studies on ethanol57 and acetic acid58 reforming on Ni/biochar indicated that O-containing functional groups of the support could affect the catalytic performance. The ethanol decomposition and steam reforming experiments over natural sepiolite discussed in the previous section and in Fig. S6 to S8 in the ESI† further demonstrate the adsorption and even dehydration of ethanol on the support.
Considering the above and combined TOF and selectivity trends observed in the present work, a bifunctional mechanism is proposed that is summarized below, over two types of sites; the sepiolite surface sites (silanol groups), designated with S, and the Ni metal sites, designated with *. In the simplified network below most reactions should be considered multi-step pathways.
| CH3CH2OH/CH3CHO + S → CH3CH2OH⋯S/CH3CHO⋯S | (1) |
| H2O + S → H2O⋯S | (2) |
| CH3CH2OH⋯S + 2* → CH3CHO⋯S + H2 + 2* | (3) |
| CH3CH2OH⋯S + w* → CH4 + CO + H2 + w* + S | (4) |
| CH3CHO⋯S + x* → CH4 + CO + x* + S | (5) |
| CH3CH2OH⋯S + H2O⋯S + y* → 2CO + 4H2 + y* + 2S | (6) |
| CH3CHO⋯S + H2O⋯S + z* → 2CO + 3H2 + z* + 2S | (7) |
| CO* + H2O⋯S + * → CO2 + H2 + 2* + S | (8) |
As the partial pressure of water increases, more water adsorbs on sepiolite's adsorption sites, eventually activating and forming steam intermediates (such as OH). The addition of water could also hydrate sepiolite's surface possibly forming new silanol groups, through the conversion of siloxane groups (Si–O–Si).60 Therefore, this could enhance the surface diffusion of ethanol/acetaldehyde towards the metal–support interface, indicating that the migration process of the oxygenates proceeds by binding to neighbouring silanols. As a result, a higher coverage of the oxygenates occurs on the metal–support interface, leading to higher conversion but lacking the secondary reactions like the WGS. This competitive process on the support and the interface could be responsible for the enhancement of the TOF with increasing partial pressure of water. In the case of acetaldehyde, the parallel aldol condensation pathway leading to the independent formation of CO2 potentially further affects the observed trends.
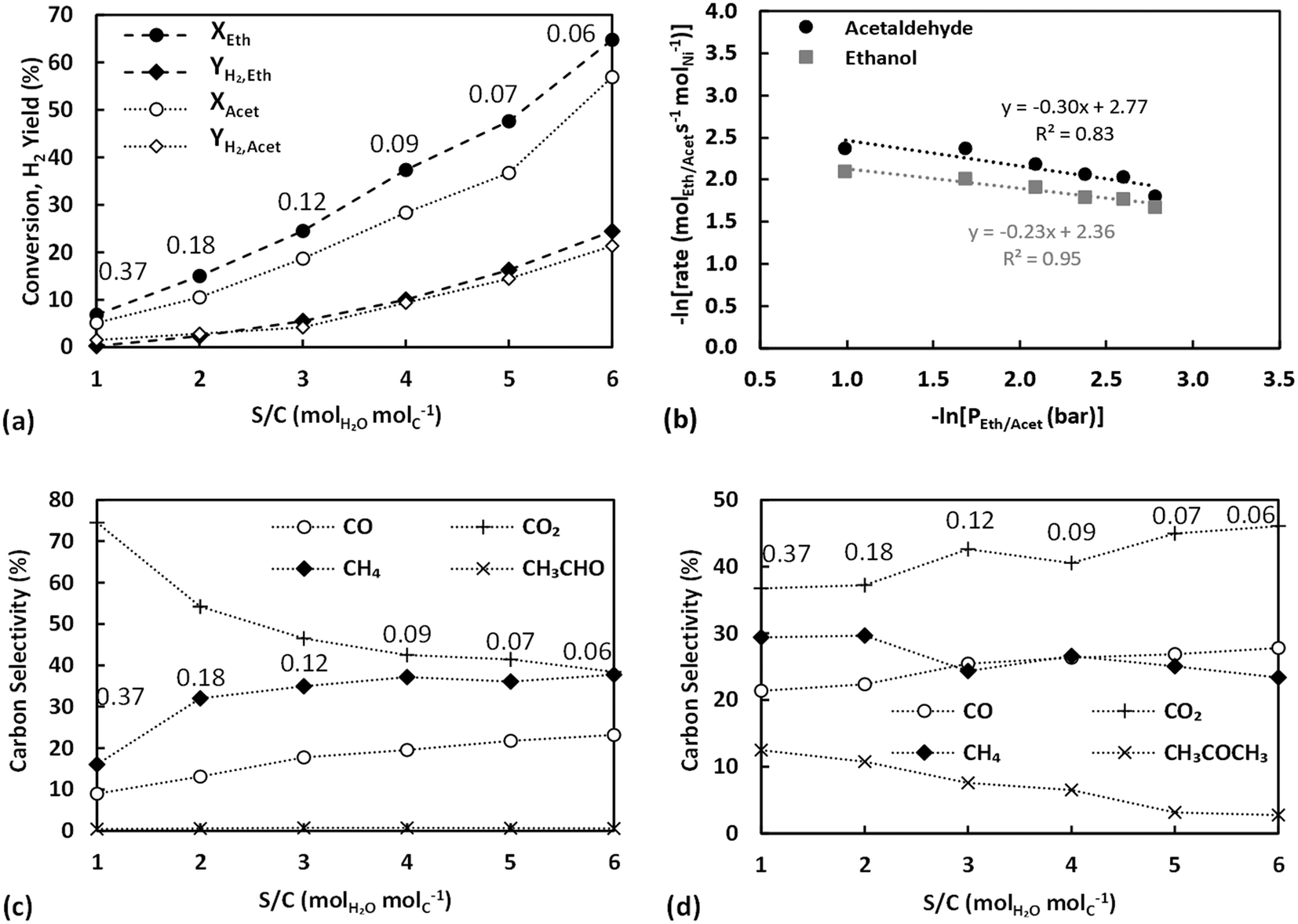 | ||
| Fig. 7 Partial pressure effect of ethanol and acetaldehyde on (top) the respective conversion and H2 yield (a) and logarithm of reaction rate of ESR and ASR with respect to logarithm of ethanol and acetaldehyde partial pressure (b), and (bottom) carbon selectivities of ESR products (c) and ASR products (d) at 400 °C (P = 1.9 bar, Vtot = 160 cm3 min−1). For panels (a), (c) and (d) the partial pressure effect is presented as a S/C variation with numbers on plots annotating the equivalent partial pressures of ethanol or acetaldehyde in bar (panels a, c, and d: lines are visual aids). | ||
The observed trend is consistent with the observations made in the previous section, further supporting the presence of a bifunctional mechanism. In these experiments the partial pressure of water was kept constant, but the increasing partial pressure of each oxygenate could promote water's substitution by the ethanol/acetaldehyde on sepiolite's surface (through hydrogen bonds formed between the oxygenates and the silanol groups). At high oxygenate to water ratio, a low surface diffusion rate of the oxygenates towards the metal–support interface would be observed, due to the discontinuous support active sites (silanols). Subsequently, a decreased coverage of the oxygenates on the metal–support interface would be expected, explaining the drop in the TOF. The latter can possibly also be further negatively affected by the rising availability of water to adsorb on and saturate the metal active surface, as shown previously.40 However, as the oxygenate to water ratio decreases, more silanol groups are expected eventually leading to an increased coverage of the metal–support interface by ethanol or acetaldehyde.
Fig. 7c presents the carbon selectivities of ESR in terms of S/C ratio for these experiments. Similar behaviour, as that discussed in section 3.4.1, is observed, with the selectivity to CO2 decreasing and the selectivities of CH4 and CO increasing with higher S/C. Along the lines of the previous discussion, the rise of the oxygenate to water ratio leads to a lower ethanol/acetaldehyde concentration, accompanied also with a high water concentration, at the metal–support interface explaining the attained selectivities. In these experiments, even the sub-stoichiometric S/C = 1 point follows the discussed trend, which can possibly be related to the much higher partial pressure of water compared to the previous experiments (0.74 bar versus 0.26 bar). For ASR carbon selectivities (Fig. 7d), the trends with S/C are less significant, possibly due to lower adsorption of acetaldehyde on the silanol groups. However, they can still be considered to be in line with the discussed support effects, as the carbonyl-containing group of acetaldehyde could also interact with the silanol groups.64 Specifically, the proposed higher active metal coverage with water as the partial pressure of the oxygenates increases agrees with the rising selectivity to acetone and decreasing selectivity to CO, both linked with the rise in water-derived surface intermediates.
Fig. 8a presents the ethanol conversion and the H2 yield achieved by varying the ethanol and water partial pressure plotted versus the S/C ratio. Decreasing the partial pressure of ethanol (increasing S/C), the ethanol conversion and H2 yield are seen to increase steadily, whereas increasing the partial pressure of water (and S/C) resulted in a mild decrease of conversion with H2 yield still displaying a slight rise. The results are very much in line with those using Ni/SiO2,40 which, given the inert support used, can be considered characteristic of metal-dominated pathways. Ethanol conversion in experiments where partial pressures were varied is overall lower than that achieved with the untreated catalyst (Fig. 7a), however this can be justified by the reduced dispersion of the thermally pre-treated catalyst. The conversion achieved during experiments where the water partial pressure was varied is actually higher than that with the untreated catalyst (Fig. 5a), despite the reduced active metal area, and is largely constant, evidencing the kinetic independence of the reaction to steam and its intermediates40 once support effects have been eliminated. This is further demonstrated by the TOF values achieved for these runs, (Fig. 8b), where a positive reaction order for ethanol and a mildly negative order for water are seen, evidence of ethanol dissociation/dehydrogenation being rate limiting and catalyst surface saturation by water having a negative effect on the rate.
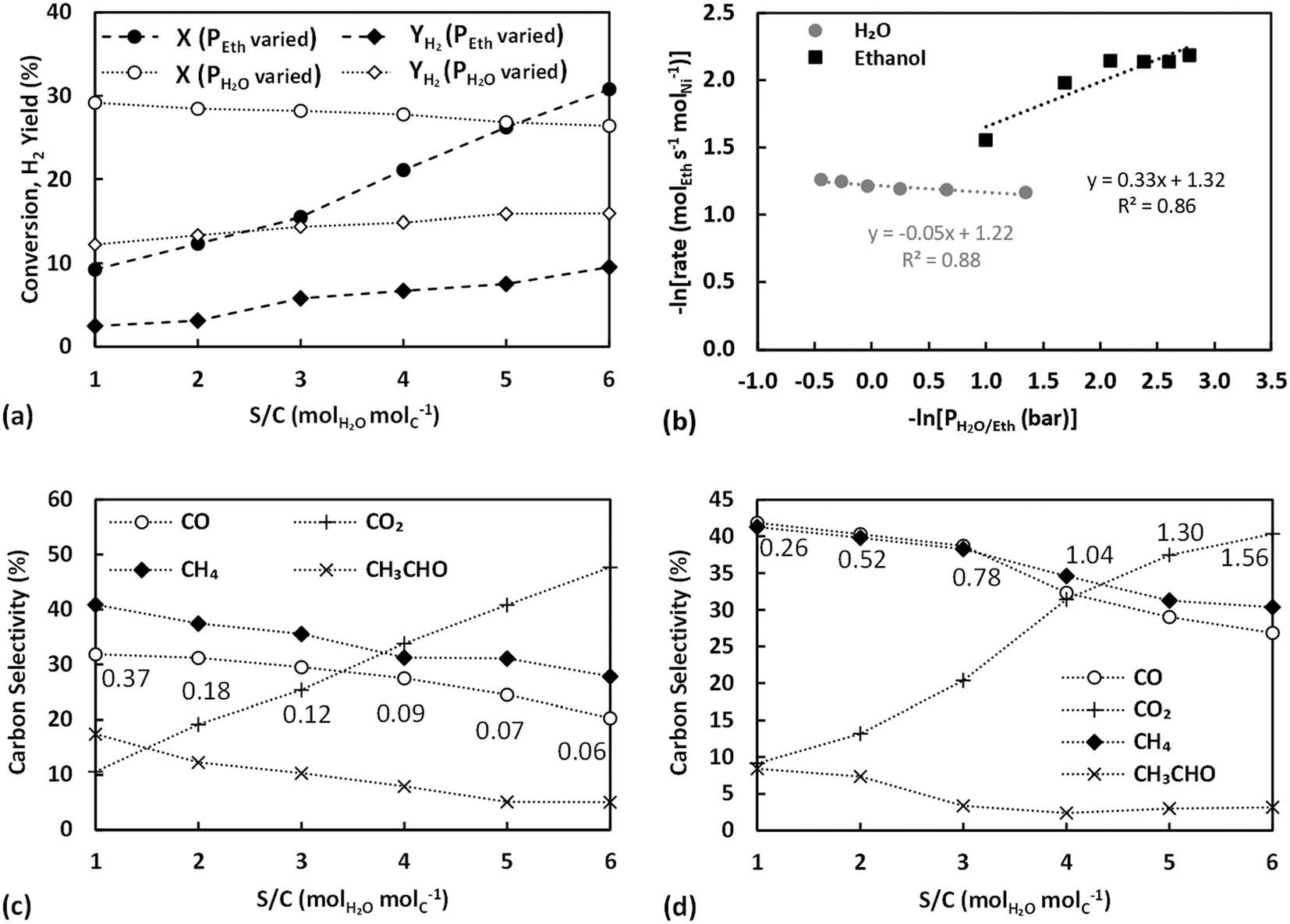 | ||
| Fig. 8 Partial pressure effect (top) of ethanol and water on the respective conversion and H2 yield (a) and logarithm of reaction rate of ESR with respect to logarithm of water and ethanol partial pressure (b), and (bottom) partial pressure effect of ethanol (c) and water (d) on carbon selectivities of ESR products at 400 °C, on thermally pre-treated Ni/sepiolite catalyst at 550 °C (P = 1.9 bar, VEthanoltot = 160 cm3 min−1, VWatertot = 213 cm3 min−1). For panels (a), (c) and (d) the partial pressure effect is presented as a S/C variation with numbers on plots annotating the equivalent partial pressures of ethanol or water in bar (panels a, c, and d: lines are visual aids). | ||
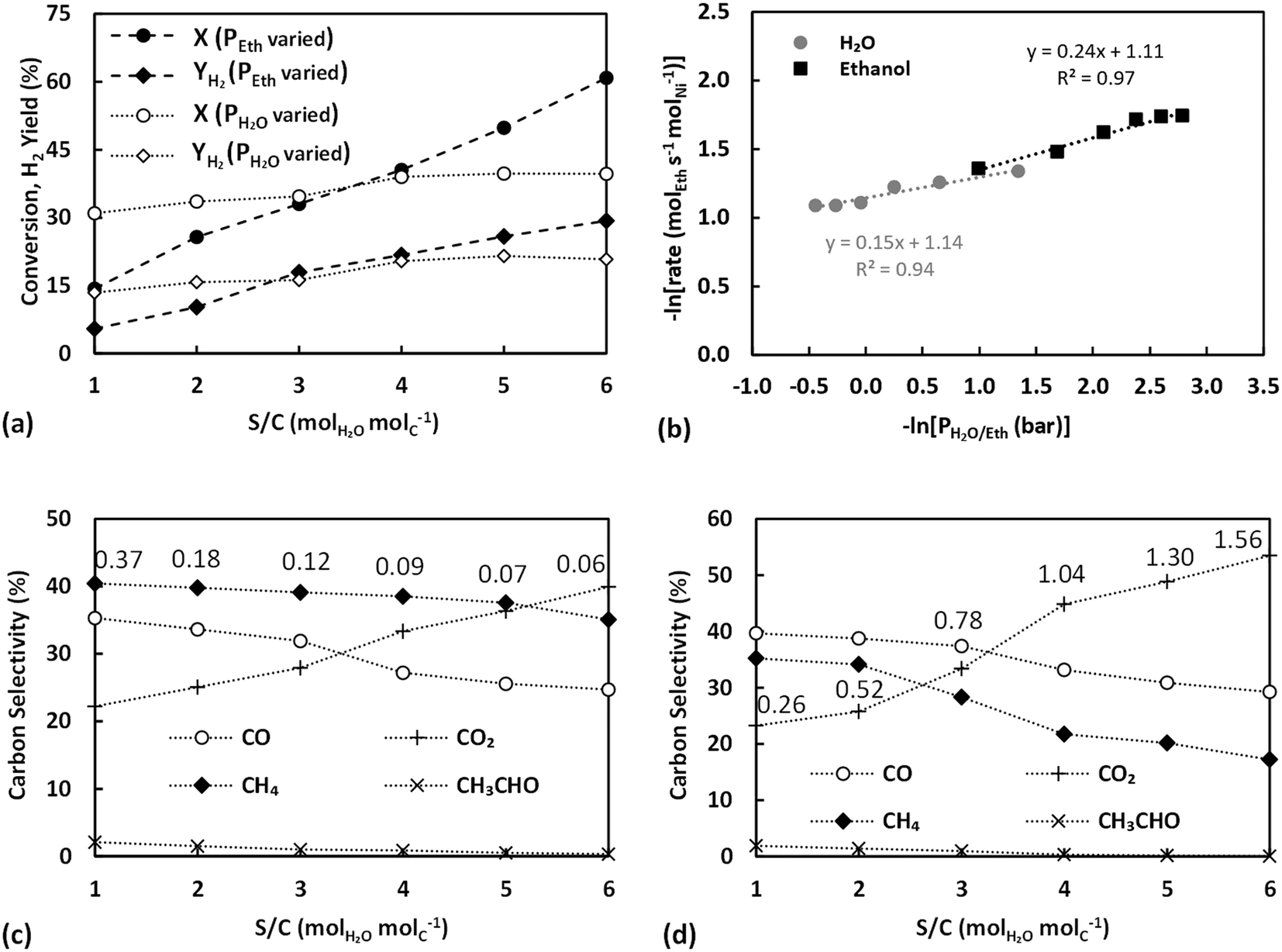
The TOF values obtained for all partial pressure variation experiments are further compared between each other and against those over Ni/SiO2 (taken from Zhurka et al.40) in Table 2 and in graphical form in Fig. S9 in the ESI.† As has been shown, the thermal treatment above 540 °C results in the destruction of the structure of sepiolite and the irreversible removal of surface silanol groups,65 which should lead to reduced adsorption of reactants on sepiolite. Given the 2D growth of Ni during precipitation for the untreated catalyst, the thermal pretreatment possibly also revealed more sites of lower coordination, restructuring Ni towards 3D particles. It is important to note that the TOF obtained in these experiments, where water partial pressure was varied, is always higher than that with the untreated catalyst and only at the highest S/C is the untreated catalyst able to achieve TOF values close to those of the thermally treated sample. This is strong evidence that Ni is indeed primarily deposited on the external surface of the sepiolite, in line with the discussion in section 3.4.1 and the observations of Anderson et al.27 Hence, even though the thermally treated catalyst has less Ni sites due to the sintering of the metal (based on the slightly decreased Ni dispersion value), these sites are potentially of higher activity. The very similar trends and values achieved for the thermally treated catalyst in comparison to Ni/SiO2 further support this claim.65 Other studies where sepiolite was used as catalyst support,22,26 have involved much higher calcination temperatures (≥600 °C) resulting in permanent removal of surface OH.
| P H2O | S/C | Base case | Treated at 550 °C | Hydrated at 400 °C | Ni/SiO240 |
P Eth | S/C | Base case | Treated at 550 °C | Hydrated at 400 °C | Ni/SiO240 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (bar) | (molEth s−1 molNi−1) | (bar) | (molEth s−1 molNi−1) | ||||||||
| 0.26 | 1 | 0.056 | 0.314 | 0.263 | 0.321 | 0.06 | 6 | 0.188 | 0.113 | 0.176 | 0.172 |
| 0.52 | 2 | 0.087 | 0.306 | 0.285 | 0.297 | 0.07 | 5 | 0.170 | 0.118 | 0.177 | 0.172 |
| 0.78 | 3 | 0.127 | 0.303 | 0.295 | 0.280 | 0.09 | 4 | 0.167 | 0.119 | 0.180 | 0.191 |
| 1.04 | 4 | 0.185 | 0.299 | 0.331 | 0.271 | 0.12 | 3 | 0.148 | 0.118 | 0.198 | 0.221 |
| 1.30 | 5 | 0.288 | 0.289 | 0.338 | 0.264 | 0.18 | 2 | 0.134 | 0.139 | 0.229 | 0.256 |
| 1.56 | 6 | 0.386 | 0.284 | 0.337 | 0.255 | 0.37 | 1 | 0.123 | 0.211 | 0.258 | 0.357 |
The carbon selectivities obtained in these experiments are finally presented in Fig. 8c and d, respectively. In both cases, CO2 selectivity increased with S/C ratio while the selectivities of CH4, CO and CH3CHO all decreased, in very good agreement with previous works all evidencing that an excess of water favours the secondary reactions. Results are indicative of the water gas shift reaction and the oxidation of CHx surface species being accelerated by the abundance of steam derived intermediates, namely O and OH surface species.
 | ||
| Fig. 9 Partial pressure effect (top) of ethanol and water on the respective conversion and H2 yield (a) and logarithm of reaction rate of ESR with respect to logarithm of water and ethanol partial pressure (b), and (bottom) partial pressure effect of ethanol (c) and water (d) on carbon selectivities of ESR products at 400 °C, on Ni/sepiolite hydrated in situ at 400 °C (P = 1.9 bar, VEthanoltot = 160 cm3 min−1, VWatertot = 213 cm3 min−1). For panels (a), (c) and (d) the partial pressure effect is presented as a S/C variation with numbers on plots annotating the equivalent partial pressures of ethanol or water in bar (panels a, c, and d: lines are visual aids). | ||
More importantly, a positive order for ethanol is again obtained (Fig. 9b) in contrast to the results over the untreated catalyst sample (section 3.4.2), further supporting the proposal that in the experiments over the untreated catalyst the decreased silanol group concentration on sepiolite's surface is determining the performance. The pre-treatment procedure led to a high degree of hydration of sepiolite's surface, increasing the concentration of silanol groups, enhancing surface diffusion of the oxygenates towards the metal–support interface, where C–C bond scission and reforming reactions occur. For the case of the water partial pressure variation experiments, a positive order is still obtained over the hydrated catalyst (Fig. 9b), but with a much milder slope in comparison to the untreated catalyst. Potentially, the hydration process led to increased adsorption and activation of steam, or the high partial pressure of ethanol used in these runs was still able to displace a small fraction of the water. Nonetheless, as also seen in Table 2 the TOF values obtained are in the range of the thermally treated catalyst and even closer to those over Ni/SiO2, hence again strongly indicate that ethanol activation is proceeding largely unhindered.
Selectivities obtained further support these observations, (Fig. 9c and d), since in both experiments where partial pressure is varied CO2 selectivity is found to increase, whereas CH4 and CO selectivities are seen to decrease, again in agreement with a mechanism where secondary reactions are promoted by steam derived intermediates. It can be concluded that the hydration procedure resulted in the enhancement of the surface diffusion of the oxygenates onto sepiolite's surface towards the metal–support interface, at the same time preventing catalyst particle sintering, achieving as such optimal catalytic performance both in terms of selectivity and TOF.
4. Conclusions
Kinetic measurements of the ethanol and acetaldehyde steam reforming reactions over a Ni/sepiolite catalyst were carried out at a wide variety of operating conditions. Results obtained over catalyst samples supported on natural sepiolite and exposed to temperatures up to 500 °C suggested a critical role of the support. A positive reaction order for water and a negative reaction order for both ethanol and acetaldehyde were obtained, in contrast to conventional observations on other hydrocarbons and oxygenates reforming. A bifunctional mechanism was proposed with both steam and oxygenates being able to adsorb on the support, namely on sepiolite silanol groups. The abundance of the latter was further suggested to influence surface diffusion of the oxygenates towards the active metal. Results over catalyst samples subjected to thermal, or hydration pre-treatment processes corroborated this conclusion. In both cases, a positive reaction order for the oxygenate and an almost zero reaction order for water were obtained in line with metal dominated pathways over inert supports. Thermal pre-treatment at 550 °C or above leads to the removal of surface silanol groups, preventing the adsorption of the reactants on the support. Through the full hydration of sepiolite before each experiment the increase of surface silanol groups was suggested to enhance the surface diffusion of the oxygenates towards the metal–support interface. TOF values obtained with pre-treated catalyst samples are higher than the untreated cases and similar to those obtained with inert SiO2 supported Ni, further concluding that the majority of Ni is deposited on the external surface of sepiolite and not in its channels. Finally, ethanol and acetaldehyde were found to follow similar reforming pathways, consisting of primary decomposition or dehydrogenation followed by secondary water gas shift and reforming, with a parallel aldol condensation pathway for acetaldehyde leading further to acetone formation.Author contributions
MDZ: investigation, writing – original draft. JAA: resources, supervision. AJM: resources, writing – review & editing. AAL: resources, writing – review & editing. PNK: conceptualization, methodology, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.References
- D. Tang, G. L. Tan, G. W. Li, J. G. Liang, S. M. Ahmad, A. Bahadur, M. Humayun, H. Ullah, A. Khan and M. Bououdina, State-of-the-art hydrogen generation techniques and storage methods: A critical review, J. Energy Storage, 2023, 64, 107196 CrossRef.
- C. K. R. Pocha, W. Y. Chia, Silvanir, T. A. Kurniawan, K. S. Khoo and K. W. Chew, Thermochemical conversion of different biomass feedstocks into hydrogen for power plant electricity generation, Fuel, 2023, 340, 127472 CrossRef CAS.
- S. H. Mohd Azhar, R. Abdulla, S. A. Jambo, H. Marbawi, J. A. Gansau, A. A. Mohd Faik and K. F. Rodrigues, Yeasts in sustainable bioethanol production: A review, Biochem. Biophys. Rep., 2017, 10, 52–61 Search PubMed.
- M. N. Uddin, K. Techato, J. Taweekun, M. Mofijur, M. G. Rasul, T. M. I. Mahlia and S. M. Ashrafur, An overview of recent developments in biomass pyrolysis technologies, Energies, 2018, 11, 3115 CrossRef CAS.
- P. Rosha, A. Jelle and H. Ibrahim, Recent advances in hydrogen production through catalytic steam reforming of ethanol: Advances in catalytic design, Can. J. Chem. Eng., 2023, 1–21 Search PubMed.
- W. H. Chen, P. P. Biswas, H. C. Ong, A. T. Hoang, T. B. Nguyen and C. Di Dong, A critical and systematic review of sustainable hydrogen production from ethanol/bioethanol: Steam reforming, partial oxidation, and autothermal reforming, Fuel, 2023, 333, 126526 CrossRef CAS.
- S. Crowley and M. J. Castaldi, Mechanistic insights into catalytic ethanol steam reforming using isotope-labeled reactants, Angew. Chem., Int. Ed., 2016, 55, 10650–10655 CrossRef CAS PubMed.
- D. Zanchet, J. B. O. Santos, S. Damyanova, J. M. R. Gallo and J. M. C. Bueno, Toward understanding metal-catalyzed ethanol reforming, ACS Catal., 2015, 5, 3841–3863 CrossRef CAS.
- Y. Guan, Y. Li, R. A. van Santen, E. J. M. Hensen and C. Li, Controlling reaction pathways for alcohol dehydration and dehydrogenation over FeSBA-15 catalysts, Catal. Lett., 2007, 117, 18–24 CrossRef CAS.
- G. Busca, T. Montanari, C. Resini, G. Ramis and U. Costantino, Hydrogen from alcohols: IR and flow reactor studies, Catal. Today, 2009, 143, 2–8 CrossRef CAS.
- L. Barattini, G. Ramis, C. Resini, G. Busca, M. Sisani and U. Costantino, Reaction path of ethanol and acetic acid steam reforming over Ni-Zn-Al catalysts, Flow reactor studies, Chem. Eng. J., 2009, 153, 43–49 CrossRef CAS.
- K. Shi, X. An, X. Wu and X. Xie, Modification strategies for enhancing anti-coking of Ni-, Co-based catalysts during ethanol steam reforming: A review, Int. J. Hydrogen Energy, 2022, 47, 39404–39428 CrossRef CAS.
- S. Bepari and D. Kuila, Steam reforming of methanol, ethanol and glycerol over nickel-based catalysts-A review, Int. J. Hydrogen Energy, 2020, 45, 18090–18113 CrossRef CAS.
- S. K. Maity, B. N. Srinivas, V. V. D. N. Prasad, A. Singh, G. M. Dhar and T. S. R. P. Rao, Studies on sepiolite supported hydrotreating catalysts, Stud. Surf. Sci. Catal., 1998, 113, 579–590 CrossRef CAS.
- S. Inoue, T. Takatsuka, Y. Wada, S. Nakata and T. Ono, A new concept for catalysts of asphaltene conversion, Catal. Today, 1998, 43, 225–232 CrossRef CAS.
- A. J. Aznar, E. Gutiérrez, P. Díaz, A. Alvarez and G. Poncelet, Silica from sepiolite: Preparation, textural properties, and use as support to catalysts, Microporous Mater., 1996, 6, 105–114 CrossRef CAS.
- X. L. Zhai, M. Y. Jia and Y. S. Shen, Structure and properties of Ni/Sepiolite catalyst, Chin. J. Chem., 2005, 23, 557–561 CrossRef CAS.
- L. Yang, H. Fang, S. Xiong, P. Liu and H. Luo, Catalytic properties of nickel/sepiolite promoted with potassium and lanthanum in adiponitrile hydrogenation under mild conditions, RSC Adv., 2016, 6, 60933–60939 RSC.
- A. Guerrero-Torres, C. P. Jiménez-Gómez, J. A. Cecilia, C. García-Sancho, J. J. Quirante-Sánchez, J. M. Mérida-Robles and P. Maireles-Torres, Influence of the incorporation of basic or amphoteric oxides on the performance of Cu-based catalysts supported on sepiolite in furfural hydrogenation, Catalysts, 2019, 9, 315 CrossRef.
- D. Jovanovic, R. Radovic and L. Mares, Nickel hydrogenation catalyst for tallow hydrogenation and for the selective hydrogenation of sunflower seed oil and soybean oil, Catal. Today, 1998, 43, 21–28 CrossRef CAS.
- C. P. Jiménez-Gómez, J. A. Cecilia, R. Moreno-Tost and P. Maireles-Torres, Selective furfural hydrogenation to furfuryl alcohol using Cu-based catalysts supported on clay minerals, Top. Catal., 2017, 60, 1040–1053 CrossRef.
- T. Liang, Y. Wang, M. Chen, Z. Yang, S. Liu, Z. Zhou and X. Li, Steam reforming of phenol-ethanol to produce hydrogen over bimetallic Ni-Cu catalysts supported on sepiolite, Int. J. Hydrogen Energy, 2017, 42, 28233–28246 CrossRef CAS.
- P. D. Holmes, A. R. Thornhill and D. K. Nicholas, US Pat. and Trademark Office, 3379505, 1968 Search PubMed.
- S. Liu, M. Chen, L. Chu, Z. Yang, C. Zhu, J. Wang and M. Chen, Catalytic steam reforming of bio-oil aqueous fraction for hydrogen production over Ni-Mo supported on modified sepiolite catalysts, Int. J. Hydrogen Energy, 2013, 38, 3948–3955 CrossRef CAS.
- M. Chen, X. Li, Y. Wang, C. Wang, T. Liang, H. Zhang, Z. Yang, Z. Zhou and J. Wang, Hydrogen generation by steam reforming of tar model compounds using lanthanum modified Ni/sepiolite catalysts, Energy Convers. Manage., 2019, 184, 315–326 CrossRef CAS.
- S. Sayas and A. Chica, Furfural steam reforming over Ni-based catalysts, Influence of Ni incorporation method, Int. J. Hydrogen Energy, 2014, 39, 5234–5241 CrossRef CAS.
- J. A. Anderson, M. T. Rodrigo, L. Daza and S. Mendioroz, Influence of the support in the selectivity of nickel/clay catalysts for vegetable oil hydrogenation, Langmuir, 1993, 9, 2485–2490 CrossRef CAS.
- J. A. Anderson and M. Galan-Fereres, Precursor-support interactions in the preparation of sepiolite-supported Ni and Pd catalysts, Clay Miner., 1999, 34, 57–66 CrossRef CAS.
- J. Anderson, S. Falconer and M. Galán-Fereres, Ni/sepiolite hydrogenation catalysts Part 1: Precursor–support interaction and nature of exposed metal surfaces, Spectrochim. Acta, Part A, 1997, 53, 2627–2639 CrossRef.
- M. A. Vannice, Kinetics of Catalytic Reactions, Springer, 2005 Search PubMed.
- P. B. Weisz and C. D. Prater, Interpretation of measurements in experimental catalysis, Adv. Catal., 1954, 6, 143–196 CAS.
- G. T. Wurzler, R. C. Rabelo-Neto, L. V. Mattos, M. A. Fraga and F. B. Noronha, Steam reforming of ethanol for hydrogen production over MgO - Supported Ni-based catalysts, Appl. Catal., A, 2016, 518, 115–128 CrossRef CAS.
- F. Wang, L. Xu and W. Shi, Syngas production from CO2 reforming with methane over core-shell Ni@SiO2 catalysts, J. CO2 Util., 2016, 16, 318–327 CrossRef CAS.
- S. Sayas, N. Vivó, J. F. Da Costa-Serra and A. Chica, Toluene steam reforming over nickel based catalysts, Int. J. Hydrogen Energy, 2021, 46, 17472–17480 CrossRef CAS.
- M. Menor, S. Sayas and A. Chica, Natural sepiolite promoted with Ni as new and efficient catalyst for the sustainable production of hydrogen by steam reforming of the biodiesel by-products glycerol, Fuel, 2017, 193, 351–358 CrossRef CAS.
- Y. Grillet, J. M. Cases, M. Francois, J. Rouquerol and J. E. Poirier, Modification of the porous structure and surface area of sepiolite under vacuum thermal treatment, Clays Clay Miner., 1988, 36, 233–242 CrossRef CAS.
- C. Serna, J. L. Ahlrichs and J. M. Serratosa, Folding in sepiolite crystals, Clays Clay Miner., 1975, 23, 452–457 CrossRef CAS.
- H. Nagata, S. Shimoda and T. Sudo, On dehydration of bound water of sepiolite, Clays Clay Miner., 1974, 22, 285–293 CrossRef CAS.
- Z. A. Alothman, A review: Fundamental aspects of silicate mesoporous materials, Materials, 2012, 5, 2874–2902 CrossRef CAS.
- M. D. Zhurka, A. A. Lemonidou, J. A. Anderson and P. N. Kechagiopoulos, Kinetic analysis of the steam reforming of ethanol over Ni/SiO2 for the elucidation of metal-dominated reaction pathways, React. Chem. Eng., 2018, 3, 883–897 RSC.
- F. Tijani Ahmed Afolabi, C. Li and P. N. Kechagiopoulos, Microkinetic modelling and reaction pathway analysis of the steam reforming of ethanol over Ni/SiO2, Int. J. Hydrogen Energy, 2019, 44, 22816–22830 CrossRef.
- T. Nishiguchi, T. Matsumoto, H. Kanai, K. Utani, Y. Matsumura, W. J. Shen and S. Imamura, Catalytic steam reforming of ethanol to produce hydrogen and acetone, Appl. Catal., A, 2005, 279, 273–277 CrossRef CAS.
- T. Umegaki, K. Kuratani, Y. Yamada, A. Ueda, N. Kuriyama, T. Kobayashi and Q. Xu, Hydrogen production via steam reforming of ethyl alcohol over nano-structured indium oxide catalysts, J. Power Sources, 2008, 179, 566–570 CrossRef CAS.
- R. Padilla, M. Benito, L. Rodríguez, A. Serrano, G. Muñoz and L. Daza, Nickel and cobalt as active phase on supported zirconia catalysts for bio-ethanol reforming: Influence of the reaction mechanism on catalysts performance, Int. J. Hydrogen Energy, 2010, 35, 8921–8928 CrossRef CAS.
- T. Yan, W. Dai, G. Wu, S. Lang, M. Hunger, N. Guan and L. Li, Mechanistic insights into one-step catalytic conversion of ethanol to butadiene over bifunctional Zn-Y/Beta zeolite, ACS Catal., 2018, 8, 2760–2773 CrossRef CAS.
- G. Pomalaza, M. Capron, V. Ordomsky and F. Dumeignil, Recent breakthroughs in the conversion of ethanol to butadiene, Catalysts, 2016, 6, 203 CrossRef.
- A. Corma and R. M. Martín-Aranda, Alkaline-substituted sepiolites as a new type of strong base catalyst, J. Catal., 1991, 130, 130–137 CrossRef CAS.
- M. C. Sanchez-sanchez, R. M. N. Yerga, D. I. Kondarides, X. E. Verykios and J. L. G. Fierro, Mechanistic aspects of the ethanol steam reforming reaction for hydrogen production on Pt, Ni and PtNi catalysts supported on γ-Al2O3, J. Phys. Chem., 2010, 114, 3873–3882 CrossRef PubMed.
- M. Patel, T. K. Jindal and K. K. Pant, Kinetic study of steam reforming of ethanol on Ni-based ceria-zirconia catalyst, Ind. Eng. Chem. Res., 2013, 52, 15763–15771 CrossRef CAS.
- N. R. Peela and D. Kunzru, Steam reforming of ethanol in a microchannel reactor : Kinetic study and reactor simulation, Ind. Eng. Chem. Res., 2011, 50, 12881–12894 CrossRef CAS.
- M. D. Zhurka, A. A. Lemonidou and P. N. Kechagiopoulos, Elucidation of metal and support effects during ethanol steam reforming over Ni and Rh based catalysts supported on (CeO2)-ZrO2-La2O3, Catal. Today, 2021, 368, 161–172 CrossRef CAS.
- M. F. Brigatti, E. Galan and B. K. G. Theng, in Developments in Clay Science, 2006, vol. 1, pp. 19–86 Search PubMed.
- H. Marsh and F. Rodríguez-Reinoso, Activated Carbon, 2006, vol. 2 Search PubMed.
- Y. Lv, F. Hao, P. Liu, S. Xiong and H. Luo, Improved catalytic performance of acid-activated sepiolite supported nickel and potassium bimetallic catalysts for liquid phase hydrogenation of 1,6-hexanedinitrile, J. Mol. Catal. A: Chem., 2017, 426, 15–23 CrossRef CAS.
- N. Volle, F. Giulieri, A. Burr, S. Pagnotta and A. M. Chaze, Controlled interactions between silanol groups at the surface of sepiolite and an acrylate matrix: Consequences on the thermal and mechanical properties, Mater. Chem. Phys., 2012, 134, 417–424 CrossRef CAS.
- K. S. Baamran and M. Tahir, Ni-embedded TiO2-ZnTiO3 reducible perovskite composite with synergistic effect of metal/support towards enhanced H2 production via phenol steam reforming, Energy Convers. Manage., 2019, 200, 112064 CrossRef CAS.
- A. T. F. Afolabi, P. N. Kechagiopoulos, Y. Liu and C. Z. Li, Kinetic features of ethanol steam reforming and decomposition using a biochar-supported Ni catalyst, Fuel Process. Technol., 2021, 212, 106622 CrossRef CAS.
- Y. Wang, Z. Zhang, S. Zhang, Y. Wang, S. Hu, J. Xiang and X. Hu, Steam reforming of acetic acid over Ni/biochar catalyst treated with HNO3: Impacts of the treatment on surface properties and catalytic behaviors, Fuel, 2020, 278, 118341 CrossRef CAS.
- W. Cai, F. Wang, C. Daniel, A. C. Van Veen, Y. Schuurman, C. Descorme, H. Provendier, W. Shen and C. Mirodatos, Oxidative steam reforming of ethanol over Ir/CeO2 catalysts: A structure sensitivity analysis, J. Catal., 2012, 286, 137–152 CrossRef CAS.
- S. L. Warring, D. A. Beattie and A. J. McQuillan, Surficial siloxane-to-silanol interconversion during room-temperature hydration/dehydration of amorphous silica films observed by ATR-IR and TIR-Raman spectroscopy, Langmuir, 2016, 32, 1568–1576 CrossRef CAS PubMed.
- O. A. Olafadehan, A. A. Ayoola, O. O. Akintunde and V. O. Adeniyi, Mechanistic kinetic models for steam reforming of concentrated crude ethanol on Ni/Al2O3 catalyst, J. Eng. Sci. Technol., 2015, 10, 633–653 Search PubMed.
- M. A. Christiansen and D. G. Vlachos, Microkinetic modeling of Pt-catalyzed ethylene glycol steam reforming, Appl. Catal., A, 2012, 431, 18–24 CrossRef.
- J. Wei and E. Iglesia, Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts, J. Catal., 2004, 224, 370–383 CrossRef CAS.
- M. A. Natal-Santiago, J. M. Hill and J. A. Dumesic, Studies of the adsorption of acetaldehyde, methyl acetate, ethyl acetate, and methyl trifluoroacetate on silica, J. Mol. Catal. A: Chem., 1999, 140, 199–214 CrossRef CAS.
- A. Torró-Palau, J. C. Fernández-García, A. C. Orgilés-Barceló, M. M. Pastor-Blas and J. M. Martín-Martínez, Structural modification of sepiolite (natural magnesium silicate) by thermal treatment: Effect on the properties of polyurethane adhesives, Int. J. Adhes. Adhes., 1997, 17, 111–119 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi/org/10.1039/d3re00181d |
| This journal is © The Royal Society of Chemistry 2023 |