
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the chemical space of phenyl sulfide oxidation by automated optimization†
Pia
Mueller
 *a,
Aikaterini
Vriza
a,
Adam D.
Clayton
a,
Oliver S.
May
bc,
Norman
Govan
b,
Stuart
Notman
b,
Steven V.
Ley
c,
Thomas W.
Chamberlain
a and
Richard A.
Bourne
*a
*a,
Aikaterini
Vriza
a,
Adam D.
Clayton
a,
Oliver S.
May
bc,
Norman
Govan
b,
Stuart
Notman
b,
Steven V.
Ley
c,
Thomas W.
Chamberlain
a and
Richard A.
Bourne
*a
aInstitute of Process Research and Development, School of Chemistry & School of Chemical and Process Engineering, University of Leeds, LS2 9JT, UK. E-mail: p.mueller@leeds.ac.uk; r.a.bourne@leeds.ac.uk
bDefence Science and Technology Lab, Porton Down, Salisbury, SP4 0JQ, UK
cYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 10th January 2023
Abstract
Automated platforms allow for fast and efficient optimisation of single and multi-objective chemical systems. Herein, we report the application of automated optimisation platforms for the chemical screening of sulfide oxidations in flow. The identification of different optima for each substrate highlights the requirement of rapid, individual experimental optimisation for successful compound screening.
The rise in digitalisation and automation of chemical processes has led to novel, fast evolving experimental platforms capable of high throughput experimentation (HTE) and self-optimisation using a variety of machine learning algorithms.1,2 HTE is often performed in small vials or microfluidic systems, screening a variety of catalysts and substrates at often fixed conditions (temperature, reaction time).3,4 This is contrasted by self-optimising systems5 or design of experiments,6 which aim to map the response surfaces of chemical space to develop process understanding and locate global optima. A combination of both approaches or the integration of chemically informed models, such as density functional theory (DFT) simulations, is also possible.7 This data driven revolution is contrasted by traditional chemical screening, in which a set of favourable experimental conditions is found for a model substrate, and then applied to several further, often more complex molecules.8 Higher complexity of molecules is linked to additional functionalities that influence electronic or steric effects. A good catalytic system needs to show a tolerance towards additional functionalities as this can drastically effect reactivity.9 In one-point screenings often those are excluded due to negative results or will be retested under new synthetic routes.10 Understanding the mechanistic differences by an automated screening can potentially lead to a time reduction and novel compound discovery without additional expert knowledge.11
Only a few instances are published that combine chemical or solvent screening with algorithm supported optimisation.12,13 More recently, Kershaw et al. extended multi-objective optimisations to mixed variables screening, including ligands and solvents, allowing continuous and discrete variables to be optimised simultaneously, for one substrate.14 Simon et al. screened several nitro aromatic substrates in heterogeneous hydrogenation to evaluate the tolerance and stability of the catalysts by varying conditions in set steps, finding enough data to give an informed benchmarking. While these studies exceed a simple, one-point screening, they are not focussed on finding individual optima for different substrates.12
For this study the oxidation of organic sulfides to sulfoxides is chosen due to its appeal in pharmaceutical chemistry (Fig. 1).15 Furthermore, oxidation of organic sulfides containing β-chloro or β-hydroxyl substituents is widely used in sulfur-mustard decontamination studies.16
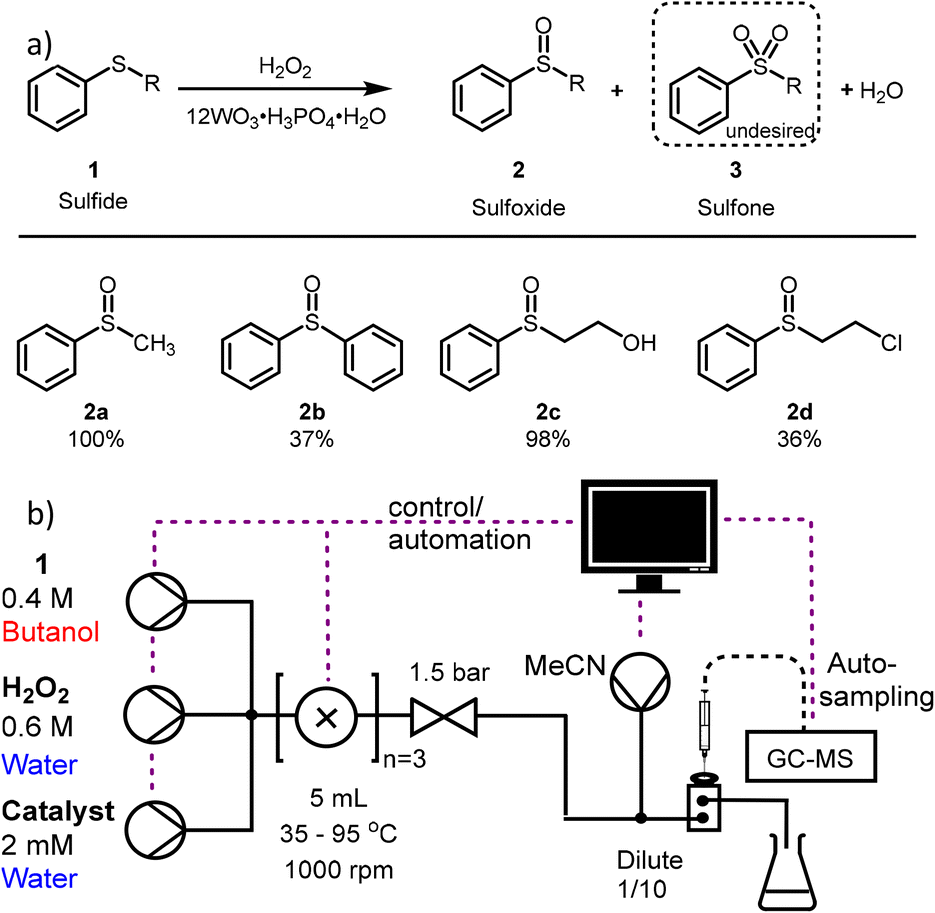 | ||
| Fig. 1 Reaction and experimental scheme for sulfide oxidation a) schematic sulfide oxidation with hydrogen peroxide and phosphotungstic acid as catalyst including optimised yields for the formed sulfoxides. b) The dilution is added allowing direct sampling to the GCMS. Samples are taken by an automated liquid sampler (Shimadzu AOC-6000). | ||
Whilst standard experimental studies have previously been able to find optima for one objective by changing one variable at a time,17 self-optimized studies are able to correlate variables in a coherent functionality to multiple objectives.18,19 A wide range of heterogeneous and homogenous catalytic studies20,21 and electrochemical reaction routes22,23 have been performed on the selective oxidation of organic sulfides achieving fast, selective oxidation in minutes or even seconds at close to room temperatures.
In this study we selected a homogenous phosphotungstic acid catalyst, which has been shown to exhibit catalytic activity for selective oxidations, owing to the presence of Brønsted acid sites and proton channelling.24 Whilst not being the catalyst with highest reactivity, this comparatively low-cost catalyst demonstrates good stability, enables a green solvent system to be used (water with butanol as organic sulfide carrier),17,25 and can potentially be recycled via phase separation.26 In this study a more complex experimental set-up is chosen to highlight the versatility of the optimisation of automated continuous flow platforms. Usually, these optimisations are performed in tubular reactors, assuming plug flow conditions, using one liquid phase or 2 liquid phases as slugs.27,28 Herein, the biphasic reaction with varying phase ratios requires active mixing to increase the extent of contact between the phases. For this purpose, mini-continuously stirred tank reactor (CSTR) cells were found effective.29 Using these cells for automated optimisations in this way constitutes a new field which has the potential to expand the breadth of flow chemistry conditions currently accessible.30,31
The experimental study was performed on a previously described flow platform using an on-line GCMS fitted with an automated liquid sampling system for determining conversion and selectivity (Fig. 1b). Flow rates, reaction temperature and sampling were monitored by the MATLAB interface, which used the calibrated mass spectrum peak areas for analysis.19 (more information can be found in the ESI,† S1.2–S1.4).
Data collection was automatically performed during a time span of ∼24 hours. The stability of the system was tested using diphenylsulfide (DPS). Following 10 replications a standard deviation of 1.7% of the yield was observed (ESI,† S2.3). Each compound optimisation was initialised using a Latin hypercube (LHC, size = 9, for 2n + 1, n = 4 parameter) for maximal experimental distribution.32 Subsequent experiments were then designed using a Bayesian Optimisation with an Adaptive Expected Improvement algorithm (BOAEI).
The oxidation of methyl phenyl sulfide (TA) is a common model reaction in literature due to its commercial availability and low toxicity. As the first model substrate, the catalytic system was able to selectively oxidise TA to the corresponding sulfoxide (2a).
The optimisation converged on long residence times (9–10 min) and a temperature above 75 °C (Fig. 2). An excess of hydrogen peroxide of 1.5 to 2.5 equivalents to TA was seen as advantageous. This is on average slightly below what can be found in literature (typically used equiv. of 2.5 or an excess), indicating that the presence of a catalyst and water as a solvent increases the reactivity.33 Compared to the reaction rates the decomposition of hydrogen peroxide was negligible in our system. Similar 3D scatter plots as in Fig. 2 for the other 3 substrates can be found in the ESI,† S2.1. The optimisations for all 4 compounds were successful in finding a global optimum defined by a maximal yield which could not be improved by 5 subsequent runs (see ESI,† S2.1). However, in the given boundaries the system was unable to achieve full conversion for DPS (2b) and CEPS (2d). For DPS, we propose this is due to the 2nd phenyl ring sterically hindering the reaction and decreasing water solubility (see ESI,† S3), leading to lower reactivity in this bi-phasic system. For CEPS the low yield was in contrast caused by the formation of side products. The mass spectrometry identified the elimination product, allyl phenylsulfide, and its oxidised sulfoxides and sulfone. This suggests a parallel oxidation of the eliminated sulfide. Additional signals were seen for some experimental runs but due to the low analytical response could not be quantified or qualified (ESI,† S1.4).
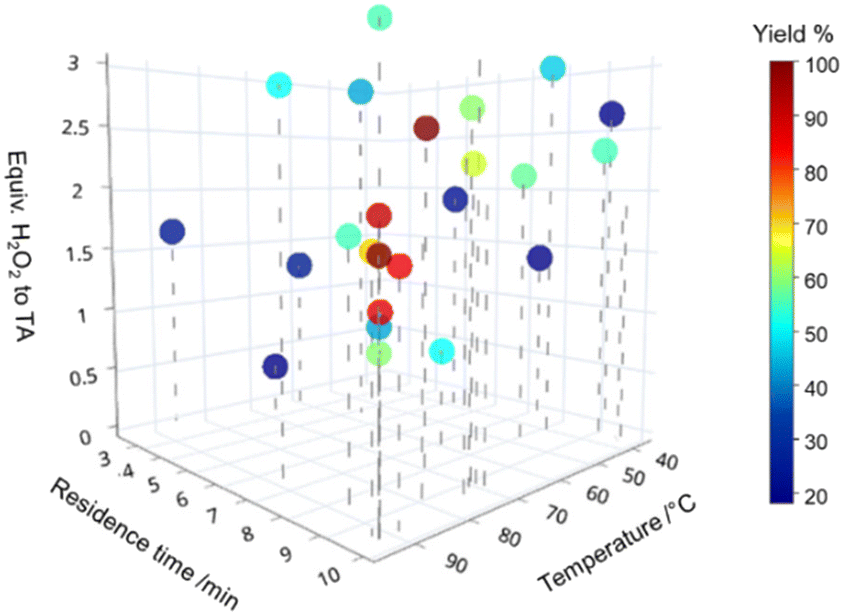 | ||
| Fig. 2 Optimisation results for the selective oxidation to sulfoxide. An interactive graph can be found here: https://chart-studio.plotly.com/~pmueller2209/18/. A total of 30 experiments were performed succeeding in finding 98% yield at 100% conversion for TA. | ||
With the more complex formation of side products, the BOAEI algorithm correlated with the need to perform an increased number of experimental iterations to find global optima than in the previous optimisations of HEPS and TA.
The accuracy of the trained surrogate models by the BOAEI algorithm for each sulfide was tested by predicting the response (yield) for each experiment and comparing the result to the experiments in a Pareto plot (S2.4†). A close agreement between BAOEI models and experiments were found. Due to this synergy, a simulation of the bounds was performed to show the surface response of each parameter on the yield. Fig. 3 shows the response for the optimisation parameters paired in comparison for each sulfide. The 4-dimensionality was broken down by only showing the response at the fixed optima of the other 2 parameters (for further explanations see ESI,† S2.5).
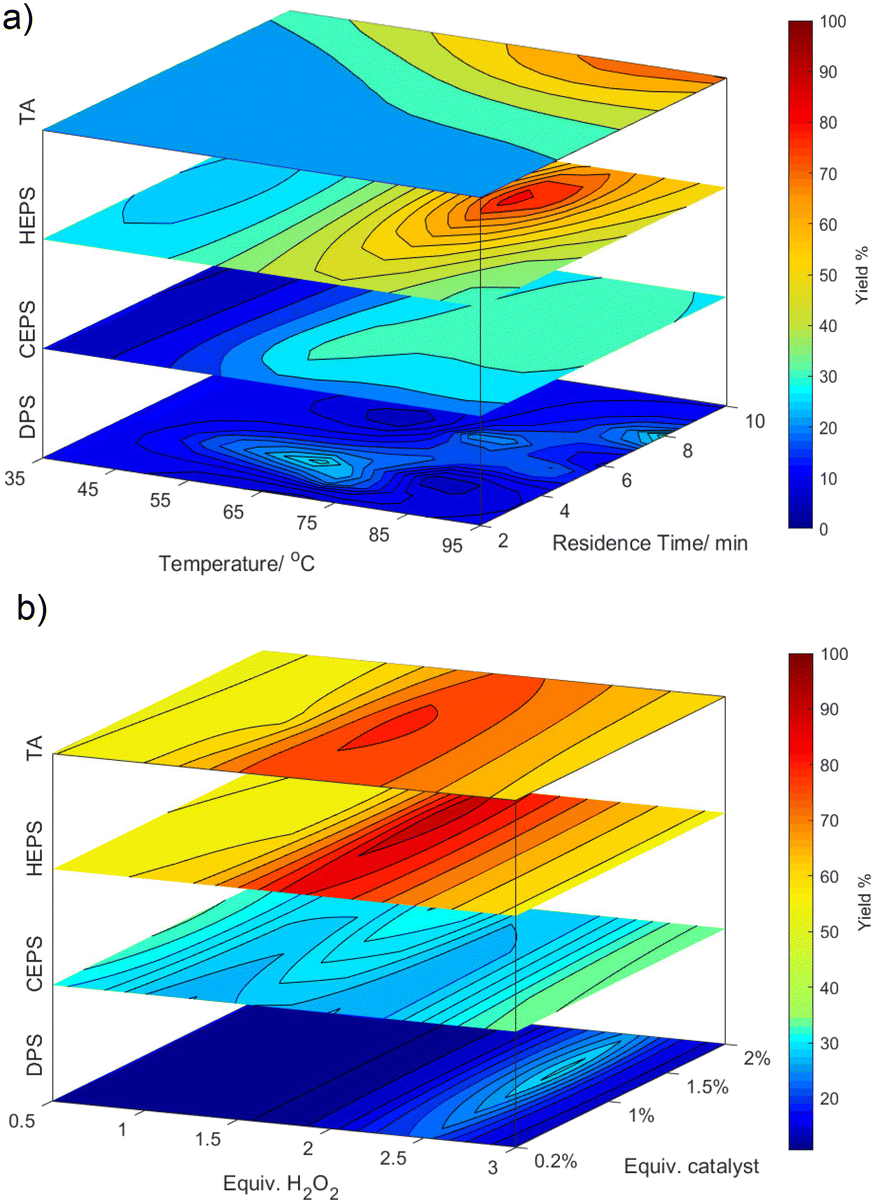 | ||
| Fig. 3 Stacked surface response of yield towards the experimental conditions simulated by the BOAEI algorithm. a) Yield response in between bounds of temperature and residence time at fixed conditions for equiv. of H2O2 and catalyst at each experimental sulfide optima. b) Yield response in between bounds of equiv. of H2O2 and catalyst at fixed conditions for temperature and residence time at each experimental sulfides optima. | ||
The surface response for HEPS and at lower yields CEPS, show a localised optimum, while for TA the optimum is clearly at the upper bounds for the temperature and residence time. Whereas with DPS there is a band in the optimisation which balances between the desired oxidation and over-oxidation. In accordance with rate laws, temperature and residence time impact the yield significantly (up to 60% deviation in yield in the given bounds). In comparison, the influence of equivalents of hydrogen peroxide and catalyst on the sulfide is lower (maximal 30%). Interestingly, the zone in which the hydrogen peroxide equivalent leads to optimal yields is quite narrow but moves from 2 for HEPS and TA, over 2.6 for DPS and to above 2.9 for CEPS.
This is likely correlated with the lower selectivity of the reactions. The catalyst ratio shows a slight increase in optimal yields between 1 and 1.5%. The ratio of the catalyst stream to the sulfide did not show a clear trend as catalytic activity was counteracted by the dilution of the system through the 3rd stream.
Focusing on the optima the 4 varied process conditions lead to a multi-dimensional problem often visualised using a parallel coordinate plot. Fig. 4 shows such a graph for the four screened sulfides. The conditions necessary for each optimum (dark lines) and their standard deviation (light lines) are shown. Small deviations relate to high impact of variation as seen for residence time and temperature for TA, HEPS and CEPS and are in alignment with the algorithm simulated surface responses (Fig. 4). The difference in reactivity of DPS and CEPS can be easily seen by comparing the associated total conversion with the yield of the formed sulfoxide. While DPS has a low conversion, CEPS is able to convert in the given bounds to up to 85%, but due to the less selective nature leading only to low optimal yields as well. The phosphotungstic acid catalyst is known for selective oxidation by providing additional acid sites. For the non-chlorinated sulfides, selectivities of 94% were seen at the optimum, for CEPS a maximum selectivity of 80% was found at the highest yields. A likely explanation is that the formation of hydrochloric acid leads to a further decrease in the pH making the hydrogen peroxide more likely to decompose or over oxidise to the corresponding sulfone. An indication of this is the significant formation of ethyl-phenyl-sulfone seen in the GCMS trace. Comparing our novel full chemical space screening method towards common best point screenings, the increase in chemical reaction understanding is clear. When only looking at the optima, the traditional approach of finding and optimising for a model substrate, in our case TA, and testing the best conditions for DPS and HEPS resulted in final yields of 32% and 70% respectively. Being 5–25% below the optimised screening defined global optima, making it a compelling case as a viable alternative to traditional screenings.
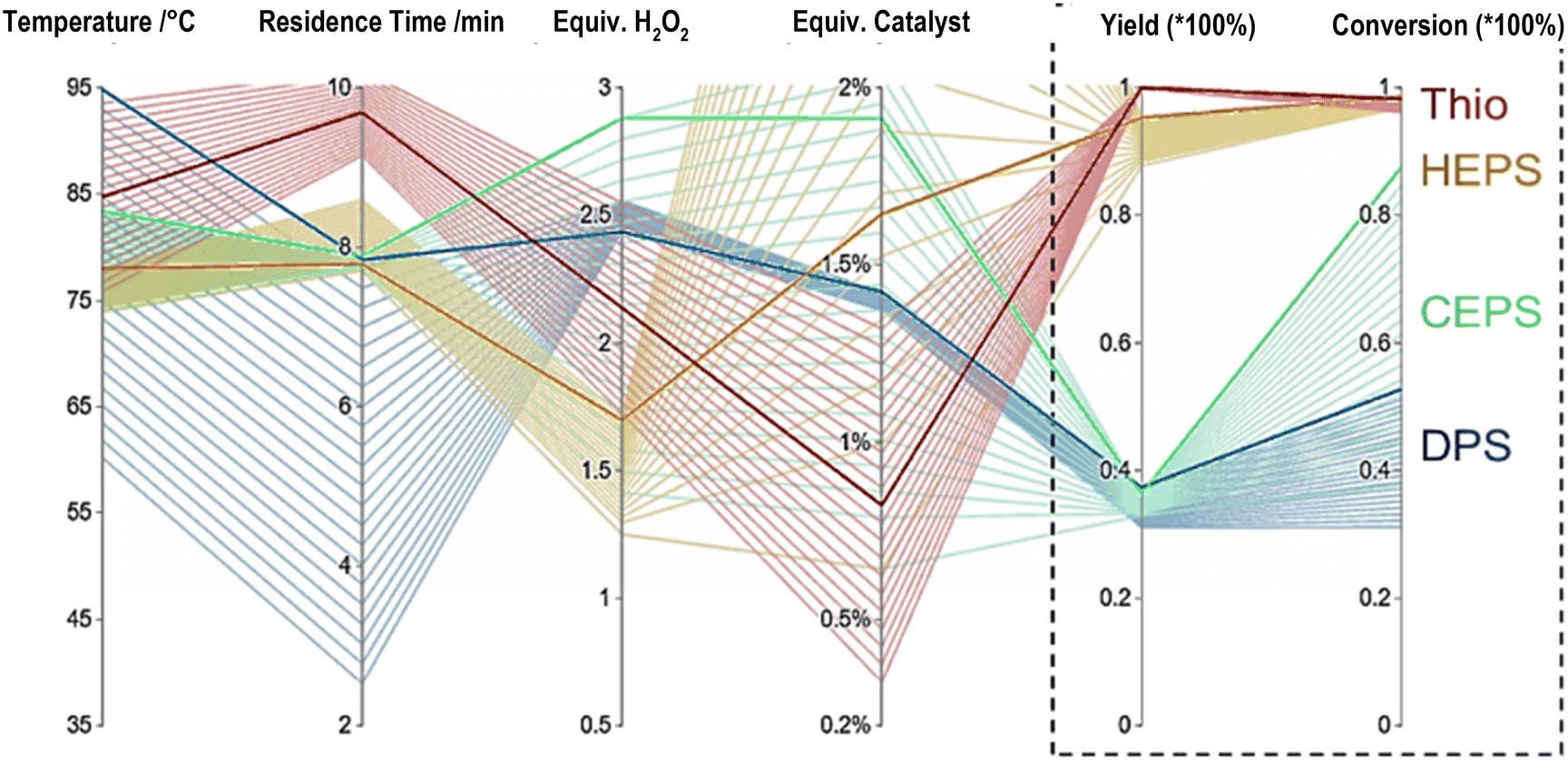 | ||
| Fig. 4 Parallel coordinate plot of screened sulfoxide optima. Main optima for each of the for sulfide are shown in darker colours while the standard deviation of 5% or the top 3 results from the optimal yield were taken to show the influence of each input condition and the corresponding conversion. Small variation of the lighter coloured lined means higher significands of the parameter, while a broad range signifies a less defining influence. | ||
The real advantage of the proposed system is the use of a fully automated system equipped with a smart algorithm finding optima in less than 20 experiments for each substrate, which were continuously performed without human interference. This autonomous approach reduced experimental time and allowed for data collection overnight.
Advances in automated optimisation allow routine tasks to be handled by the equipment and gain a deeper process understanding without prior knowledge. This approach was utilised in the work to successfully screen the catalysed oxidation of four sulfides ranging from relatively simple model compound thioanisole to mustard simulant chloroethylphenylsulfide. The experimental focus lays on the complexity of the process, using organic and aqueous streams mixed in mini CSTR cascades varying in total 4 parameters in the optimisation. The study found different optima for the oxidation of each substrate in less than 25 experiments and a total of less than 24 hours of experimental time for each of them, showcasing the power of modern Bayesian algorithm in finding global optima. While temperature and residence time proved to have a clear and significant influence on all compounds, optimal equivalents of hydrogen peroxide were found for each species and did increase in correlation with the amount of formed by-products per sulfide. In the absence of β-chloro substituents, the catalyst achieved selective oxidisation to sulfoxide. However, no clear trend emerged since varying the catalyst concentration resulted in a dilution effect which impacted the total reaction yield. Overall, the automated screening delivered reliable global optima with up to 25% more yield than using the one-point screening based on the model compound optimisation. An increased process understanding could be gained by simulating the surface response and parameter sensitivity of optimal points. Herein, an approach is suggested that is superior to the traditional screening approach, providing a faster and more reliable way to detect the optimal reaction conditions for four structurally diverse sulfides.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PM thanks Defence Science and Technology Laboratory and University of Leeds for funding. ADC thanks UCB, AstraZeneca and University of Leeds for funding. SVL thanks the ACS Arthur Cope Award for funding. RAB was supported by the Royal Academy of Engineering under the Research Chairs and Senior Research Fellowships scheme.Notes and references
- C. Avila, C. Cassani, T. Kogej, J. Mazuela, S. Sarda, A. D. Clayton, M. Kossenjans, C. P. Green and R. A. Bourne, Chem. Sci., 2022, 13, 12087–12099 RSC.
- A. D. Clayton, L. A. Power, W. R. Reynolds, C. Ainsworth, D. R. J. Hose, M. F. Jones, T. W. Chamberlain, A. J. Blacker and R. A. Bourne, J. Flow Chem., 2020, 10, 199–206 CrossRef.
- A. Buitrago Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 49–53 CrossRef CAS PubMed.
- S. Langner, F. Häse, J. D. Perea, T. Stubhan, J. Hauch, L. M. Roch, T. Heumueller, A. Aspuru-Guzik and C. J. Brabec, Adv. Mater., 2020, 32, 1907801 CrossRef CAS PubMed.
- R. Liang, X. Duan, J. Zhang and Z. Yuan, React. Chem. Eng., 2022, 7(3), 590–598 RSC.
- S. N. Politis, P. Colombo, G. Colombo and D. M. Rekkas, Drug Dev. Ind. Pharm., 2017, 43, 889–901 CrossRef CAS PubMed.
- D. Frey, J. H. Shin, C. Musco and M. A. Modestino, React. Chem. Eng., 2022, 7, 855–865 RSC.
- A. Talla, B. Driessen, N. J. W. Straathof, L.-G. Milroy, L. Brunsveld, V. Hessel and T. Noël, Adv. Synth. Catal., 2015, 357, 2180–2186 CrossRef CAS.
- R. Labes, C. Battilocchio, C. Mateos, G. R. Cumming, O. de Frutos, J. A. Rincón, K. Binder and S. V. Ley, Org. Process Res. Dev., 2017, 21, 1419–1422 CrossRef CAS.
- Y. Chen, M. Leonardi, P. Dingwall, R. Labes, P. Pasau, D. C. Blakemore and S. V. Ley, J. Org. Chem., 2018, 83, 15558–15568 CrossRef CAS PubMed.
- E. Bradford, A. M. Schweidtmann and A. Lapkin, J. Glob. Optim., 2018, 71, 407–438 CrossRef.
- K. Simon, P. Sagmeister, R. Munday, K. Leslie, C. A. Hone and C. O. Kappe, Catal. Sci. Technol., 2022, 12(6), 1799–1811 RSC.
- B. J. Reizman and K. F. Jensen, Chem. Commun., 2015, 51, 13290–13293 RSC.
- O. J. Kershaw, A. D. Clayton, J. A. Manson, A. Barthelme, J. Pavey, P. Peach, J. Mustakis, R. M. Howard, T. W. Chamberlain, N. J. Warren and R. A. Bourne, Chem. Eng. J., 2022, 138443 Search PubMed.
- A. S. Surur, L. Schulig and A. Link, Arch. Pharm., 2019, 352, 1800248 Search PubMed.
- S. L. Bartelt-Hunt, D. R. U. Knappe and M. A. Barlaz, Crit. Rev. Environ. Sci. Technol., 2008, 38, 112–136 CrossRef CAS.
- M. Jereb, Green Chem., 2012, 14, 3047–3052 RSC.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
- P. Müller, A. D. Clayton, J. Manson, S. Riley, O. S. May, N. Govan, S. Notman, S. V. Ley, T. W. Chamberlain and R. A. Bourne, React. Chem. Eng., 2022, 7, 987–993 RSC.
- G. Vernet, M.-S. Salehi, P. Lopatka, S. K. Wilkinson, S. K. Bermingham, R. Munday, A. O'Kearney-McMullan, K. Leslie, C. A. Hone and C. Oliver Kappe, Chem. Eng. J., 2021, 416, 129045 CrossRef CAS.
- S. Doherty, J. G. Knight, M. A. Carroll, J. R. Ellison, S. J. Hobson, S. Stevens, C. Hardacre and P. Goodrich, Green Chem., 2015, 17, 1559–1571 RSC.
- N. Amri and T. Wirth, J. Org. Chem., 2021, 86, 15961–15972 CrossRef CAS PubMed.
- G. Laudadio, N. J. W. Straathof, M. D. Lanting, B. Knoops, V. Hessel and T. Noël, Green Chem., 2017, 19, 4061–4066 RSC.
- F. Can, X. Courtois and D. Duprez, Catalysts, 2021, 11, 703 CrossRef CAS.
- G. Anilkumar and S. Saranya, Green Organic Reactions, Springer Nature, 2021 Search PubMed.
- B. Zeiler, A. Bartl and W.-D. Schubert, Int. J. Refract. Hard Met., 2021, 98, 105546 CrossRef CAS.
- C. A. Hone and C. O. Kappe, Chem.: Methods, 2021, 1, 454–467 CAS.
- C. P. Breen, A. M. K. Nambiar, T. F. Jamison and K. F. Jensen, Trends Chem., 2021, 3, 373–386 CrossRef.
- M. R. Chapman, M. H. T. Kwan, G. King, K. E. Jolley, M. Hussain, S. Hussain, I. E. Salama, C. González Niño, L. A. Thompson, M. E. Bayana, A. D. Clayton, B. N. Nguyen, N. J. Turner, N. Kapur and A. J. Blacker, Org. Process Res. Dev., 2017, 21, 1294–1301 CrossRef CAS.
- L. A. Power, A. D. Clayton, W. R. Reynolds, D. R. J. Hose, C. Ainsworth, T. W. Chamberlain, B. N. Nguyen, R. A. Bourne, N. Kapur and A. J. Blacker, React. Chem. Eng., 2021, 6, 1806–1810 RSC.
- F. Guan, N. Kapur, L. Sim, C. J. Taylor, J. Wen, X. Zhang and A. J. Blacker, React. Chem. Eng., 2020, 5, 1903–1908 RSC.
- V. R. Joseph and Y. Hung, Stat. Sin., 2008, 18, 171–186 Search PubMed.
- K. Sato, M. Hyodo, M. Aoki, X.-Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469–2476 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00552b |
| This journal is © The Royal Society of Chemistry 2023 |