
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the catalytic vapor-phase dehydration of lactic acid to acrylic acid: a systematic review
Lin
Huang
 ,
Ming Hui
Wai
and
Sibudjing
Kawi
*
,
Ming Hui
Wai
and
Sibudjing
Kawi
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, Engineering Drive 4, 117585, Singapore. E-mail: chekawis@nus.edu.sg
First published on 12th January 2023
Abstract
Acrylic acid is one of the fastest growing monomers. Acrylic acid and its derivatives are very versatile monomers for many industrial and commodity chemicals such as absorbents, detergents, dispersants, and flocculants. The catalytic dehydration of lactic acid is a promising approach for the renewable, efficient, economic, and green production of acrylic acid. The understanding and control of lactic acid reaction pathways and catalytic selectivity are of critical importance for the rational design of new generations of catalyst systems. This review summarizes the developments of heterogeneous catalyst systems applied in the vapor-phase dehydration of lactic acid to acrylic acid to date with our constructive comments and appropriate discussion. We systematically and comparatively present the catalytic performance of various types of heterogeneous catalyst systems including sulfate salts, phosphate salts, nitrate salts, hydroxyapatites, and modified zeolites. We also deal with crucial factors controlling the catalytic selectivity, possible catalytic active species involved, and reaction mechanisms proposed by different groups.
 Lin Huang | Dr. Lin Huang graduated with his BS in chemistry from Nanjing University, China. He obtained his DEA and doctorate in physical chemistry from Université Lyon 1, France. He successively joined Dalian Institute of Chemical Physics, China; Institute of Chemical and Engineering Sciences, Singapore; and National University of Singapore. His research interests include surface organometallic chemistry, catalysis towards Fischer–Tropsch, CO2 hydrogenation to methanol, hydroformylation, carbon–carbon coupling, ethanol steam reforming, and dehydration of lactic acid. |
 Ming Hui Wai | Ming Hui Wai graduated with B. Tech (Chemical Engineering) in 2014 and M. Eng (Chemical Engineering) from the National University of Singapore in 2018. He worked in the design of hydroxyapatite catalyst for CO2 hydrogenation to methane and dry reforming of methane. His current research is on the design of hydroxyapatite catalyst for CO2 hydrogenation to methanol and its coupling with zeolite membrane for enhanced methanol yield. |
 Sibudjing Kawi | Prof. Sibudjing Kawi obtained his Bachelor, Master, PhD degrees, and Postdoc from the Univ. Texas @ Austin, Univ. Illinois @ Urbana-Champaign, Univ. Delaware, and Univ. California @ Davis, respectively, before he joined National University of Singapore. His research focuses on the integration of catalyst with membranes for tackling energy and environmental challenges (especially CO2 and H2 challenges) via the valorization of biomass waste, CO2, and natural gas. He has published >400 papers (citations >22 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000,
000, Introduction
Acrylic acid (AA) and its derivatives are very versatile monomers for many industrial and commodity chemicals such as absorbents, detergents, dispersants, and flocculants.1–4 The market of AA is one of the fastest growing markets of monomers.1–4 The global market of AA reached a value of ca. 6.60 million tons in 2021.5 The industry is projected to grow at a compound annual growth rate of ca. 4.7% in the forecast period of 2022–2027 to reach a value of USD 20.19 billion by 2027,5 which is comparable to the other fast growing markets of monomers such as glycolic acid, levulinic acid, maleic anhydride, and adipic acid.6–9 Currently, AA is still derived from petroleum resources. To satisfy the demand for alternatives based on renewable resources and/or biotechnological processes, lactic acid (LA) has been identified as a preferable source of AA via the chemical approach.1,3 The catalytic conversion of LA to produce AA is viewed to be a renewable alternative to the widely used production of AA from propene.1–4 LA is mainly produced by fermentation from biomass such as starches, glucose, and sucrose,3,4,10 keeping in view the less costly biochemical approach than chemical ones.11As early as 1935, Burns et al. published the production of alkyl acrylate from alkyl lactate in two steps in an academic paper, which proceeded via an alkyl α-acetoxypropionate as the primary product.12 The dehydration of α-hydroxy-acid derivatives is difficult. However, dehydration may be eased by acetylating the hydroxy group and eliminating acetic acid via pyrolysis. In such a two-step process, alkyl lactate first reacted with acetic anhydride to give alkyl α-acetoxypropionate in 78–92% yield in the presence of H2SO4 as the catalyst in the liquid phase. The resultant alkyl α-acetoxypropionate then pyrolyzed to alkyl acrylate in 61–88% yield at 400–600 °C in the vapor phase. This work lays the foundation for research on the two-step process of production of AA from LA.13–19 In 1946, Filachione and Fisher disclosed for the first time the production of α-acetoxypropionic acid as the first step product from LA in a patent.13 In a batch reactor, LA reacted with acetic anhydride or acetyl chloride or glacial acetic acid to give α-acetoxypropionic acid in 42–75% yield in the presence of H2SO4, HCl, or acetic acid in the liquid phase. Later on, the research on the first step was followed up by Lilga et al., Miller et al., and Beerthuis et al.17–19 To avoid the use of acetic anhydride or acetyl chloride, which results in the formation of a corrosive mixture of acids in water, Lilga et al. esterified LA with glacial acetic acid in the presence of H2SO4 to produce α-acetoxypropionic acid in >90% yield in a batch reactor.17 The unreacted acetic acid was recycled for the reaction. To achieve a high reaction efficiency with secure product separation from the catalyst, Miller et al. adopted a continuous reactive distillation system containing an Amberlyst ion exchange resin as an acid catalyst for the production of α-acetoxypropionic acid from LA and acetic acid.18 The yield of α-acetoxypropionic acid could reach 96%. Beerthuis et al. comparatively tested various solid acid catalysts such as TeO2, Nb2O5, Mo–V–Te–Nb–O mixed oxide, microporous NaY, microporous HY, mesoporous HY, p-toluenesulfonic acid-functionalized glucose, sulfonic acid-functionalized activated carbon, sulfonated silica, sulfonated activated carbon, sulfonated graphene, sulfated zirconia, Amberlyst 70, and Nafion NR50 for the production of α-acetoxypropionic acid from LA and acetic acid in a batch reactor.19 After 3 h of reaction at 100 °C and 1 atm, sulfonic acid-functionalized activated carbon, sulfonated graphene, Amberlyst 70, and Nafion NR50 produced higher LA conversion at 41–44% with selectivity toward α-acetoxypropionic acid at 83–90%. Amberlyst 70 was not deactivated at all after five cycles of 3 h of reaction, whereas sulfonic acid-functionalized activated carbon, sulfonated graphene, and Nafion NR50 more or less deactivated from the second reaction cycle. Catalyst deactivation was shown to result from the leaching of –SO3H groups from these three catalysts. From the kinetic trends of the reactions over all the solid acid catalysts studied, 3 h of catalytic reactions was far from being enough to reach the chemical equilibrium. As long as the reaction is prolonged, LA conversion is supposed to increase to an optimal value over the four more active solid acid catalysts. In light of the research advances of the first step,13,17–19 the catalytic esterification of LA with acetic acid in the presence of either H2SO4 or Amberlyst ion exchange resin is selective for the production of α-acetoxypropionic acid, producing a high yield of AA.17,18 Until 1972, Arpe and Grosspietsch published for the first time the thermolysis of α-acetoxypropionic acid to AA as the second step product from LA in a patent.14 In a continuous reactor, α-acetoxypropionic acid thermolyzed to AA in 82–90% yield in the presence of Ba3(PO4)2, Ca3(PO4)2, or Li3PO4/Ca3(PO4)2 as a heterogeneous precatalyst at 300–360 °C and 6–10 torr in the vapor phase. Until now, little has been reported on the research of the second step.15,16 In the absence of the catalyst, the thermolysis of α-acetoxypropionic acid is by no means selective for the production of AA, resulting in a negligible yield of AA.15,16 The catalytic thermolysis of α-acetoxypropionic acid in the presence of alkali or/and alkaline-earth phosphates is deemed to be selective for the production of AA, giving a high yield of AA.14
The invention of vapor-phase dehydration of LA to AA was published by Holmen in a patent in 1958.20 As addressed above, the research on the two-step process of conversion of LA to AA was not simple in the early period. The first step could only deliver α-acetoxypropionic acid in 42–75% yield using a homogeneous acid catalyst before 1960.13 The second step was not successful in the selective production of AA using alkali or/and alkaline-earth phosphates as a precatalyst until the early 1970s.14 While the direct process of dehydration of LA to AA remained challenging, the two-step process of conversion of LA to AA appeared disadvantageous. Alternatively, the development of processes for the production of AA from other starting materials was restricted by either the high cost and expense of the processes or the trouble of catalyst handling.20 For example, the oxidation of acrolein could produce AA using Ag2O. But Ag2O was expensive and reduced to the metal during the reaction, requiring reconversion to Ag2O prior to reuse. The hydrolysis of ethylene cyanohydrin or acrylonitrile could lead to the production of AA in a few steps. But the cost and operational expense were high. The pyrolysis of polymeric β-lactone produced from ketene and formaldehyde also involved several separate reactions. In the meantime, the acetylene-based process was known in the presence of CO and water or alcohol. But the recovery of expensive catalysts presented a major problem. Although the dehydration of hydraacrylic acid (i.e., β-hydroxypropionic acid) was a simple and economic process, the starting material was neither low in cost nor readily available in quantity. In contrast, LA (i.e., α-hydroxypropionic acid) was much readily and potentially available. However, its dehydration had never been found to be efficient. In addition, it was known that LA readily converts to lactides or poly(LA) (PLA) at moderate temperatures, and to AD, CO, and water or alcohol at higher temperatures. The same decomposition to AD and CO was known when LA was heated in the presence of an acid such as H2SO4 or H3PO4. In such a context, Holmen was determined to explore new and efficient processes for the dehydration of LA to AA with heterogeneous catalyst systems. He successfully devised a catalytic process for the vapor-phase dehydration of LA to AA using alkali and alkaline-earth sulfates and phosphates such as Na2SO4/CaSO4, Ba3(PO4)2, Ca3(PO4)2/Na4P2O7, CaSO4/Na4P2O7, CaSO4/CsH2PO4, CaSO4/LaPO4, and CaSO4/CsH2PO4/LaPO4 as heterogeneous precatalysts.20 Through the vapor-phase dehydration process, the AA yields of 42–68% were achieved from aqueous 10–50% solutions of LA over these salts in a fixed-bed reactor at 400–425 °C and 1 atm. According to his catalyst screening, many other materials such as pumice, silicophosphoric acid, H3PO4, WO3, TiO2, Na2WO4, Na2MoO4, NaVO3, MoO3, SiO2, Al2O3, NiMoO2, and ZnMoO3, which are traditionally active for other dehydration reactions, brought about little or no AA in vapor-phase dehydration of LA.20 The successful catalytic vapor-phase dehydration of LA to AA probably consists of the generation of lactate salt as the reaction intermediate and the proper acidity (or acid strength) of a reaction system.21–23 The generation of lactate salt is presumably the prerequisite.21,22 Under this prerequisite, the corresponding acidity (or acid strength) of the reaction system is presumably required to be moderate or low such that the lactate salt could stabilize.23 Under a more acidic condition, the lactate salt readily dehydrates to acrylate salt that subsequently polymerizes.23 In Holmen's catalyst screening for the vapor-phase dehydration of LA, the alkali and alkaline-earth sulfate and phosphate systems perform well, probably in which lactate salts are efficiently generated from LA and the alkali and alkaline-earth sulfates and phosphates under the reaction conditions and the acid strengths of these salts are moderate or low.24,25 Those perform poorly, probably because they either contain no alkali or alkaline-earth cation or possess a high acid strength.
The liquid-phase dehydration of LA to AA was reported first by Odell et al. in 1985.26 At 180–250 °C and at 1 atm or below in the presence of [PtH(PEt3)3]OH as a precatalyst in aqueous solution in a batch rector, the hydrothermal reactions of LA produced AA, propionic acid (PA), β-hydroxypropanoic acid, pyruvic acid, acetic acid, acetone, and ethanol. Under the optimal reaction conditions (230 °C, aqueous 10% LA and reaction time = 2 h), an LA conversion at 85%, and yields of AA, PA, β-hydroxypropanoic acid, and acetic acid at 3.4, 43, 24, and 3.0%, respectively, were obtained. The results show that such a homogeneous LA dehydration reaction is unselective for the production of AA.
Later on, the dehydration of LA to AA in supercritical water was investigated by Mok et al. and further by Lira and McCrackin.27,28 The reaction was reportedly operated at 320–400 °C and 310–345 bar in a continuous reactor.27,28 The investigation by Mok et al. used the typical reaction conditions of 385 °C, 345 bar, initial aqueous 0.1 M LA, and a residence time of 28 s.27 The essential and important reaction results are illustrated in Fig. 1 and 2. In the absence of the catalyst, the reaction gave an LA conversion at 41% with selectivities toward (AA + PA), acetaldehyde (AD), CO, CO2, and H2 at 27, 34, 37, 18, 27, and 13%, respectively. The reaction data suggested that the non-catalytic LA dehydration (to AA and PA) occurs with the concurrent LA decarbonylation (to AD and CO) and LA decarboxylation (to AD, CO2, and H2). As the concentration of added H2SO4 as a precatalyst rose up to 10 mM, the LA conversion increased together with increased selectivities toward AD and CO, decreased selectivities toward (AA + PA), CO2, and H2, and decreased AA and PA yields. In the presence of 10 mM H2SO4, 100% LA conversion was attained with the formation of AD and CO as the predominant products and of AA, CO2, and H2 as the minor products. This clearly indicated that the addition of strong acid promotes LA decarbonylation, which is detrimental to LA dehydration and decarboxylation. In the presence of NaOH instead of H2SO4 as a precatalyst, the LA conversion decreased together with decreased selectivities toward (AA + PA) and CO and increased selectivities toward AD, CO2, and H2, as the concentration of added NaOH rose. The reaction data with NaOH hinted that the addition of base tends to inhibit LA dehydration and decarbonylation in favor of LA decarboxylation. The simultaneous increases in the selectivities toward AD, CO2, and H2 with increasing NaOH concentration suggested that the catalytic activity for LA decarboxylation increases with increasing NaOH concentration. The fall in the selectivity toward (AA + PA) may be due to the higher catalytic activity for LA decarboxylation.
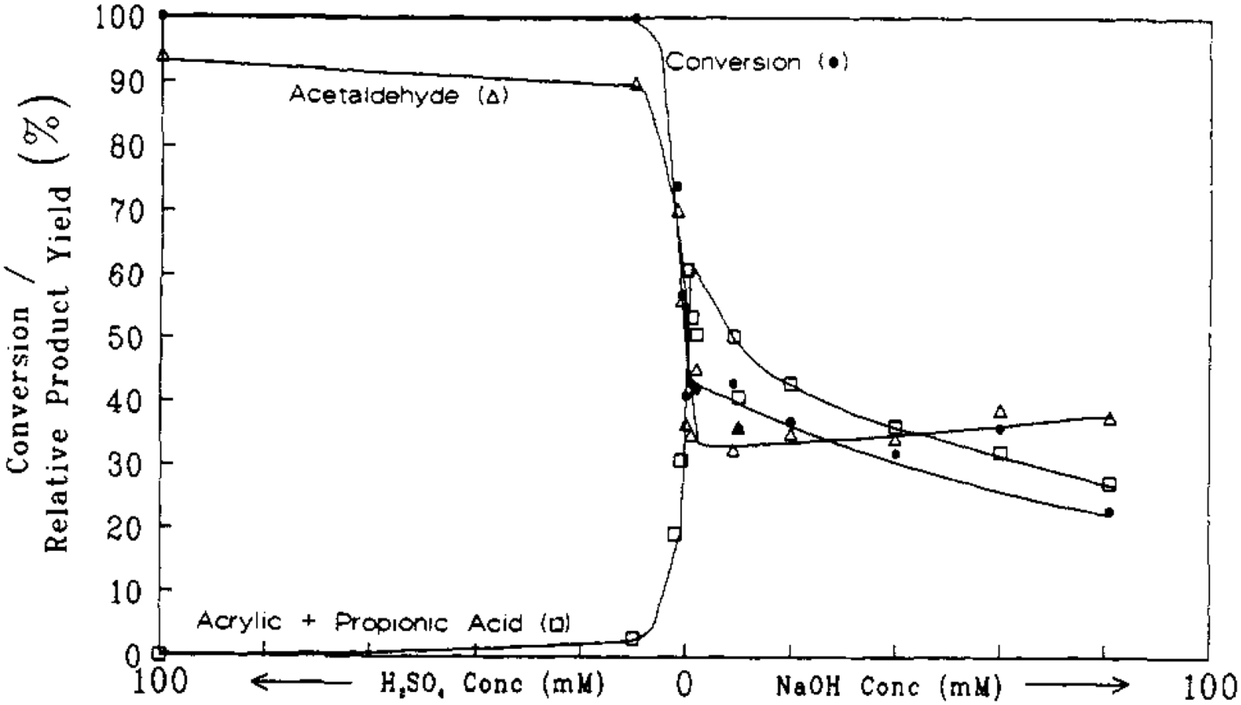 | ||
| Fig. 1 Effect of acid and base concentration on the conversion and selectivities toward liquid products from reactions of LA in supercritical water at 385 °C, 345 bar, a residence time of 28 s with an initial aqueous 0.1 M LA. Reprinted with permission from ref. 27, copyright 1989 American Chemical Society. | ||
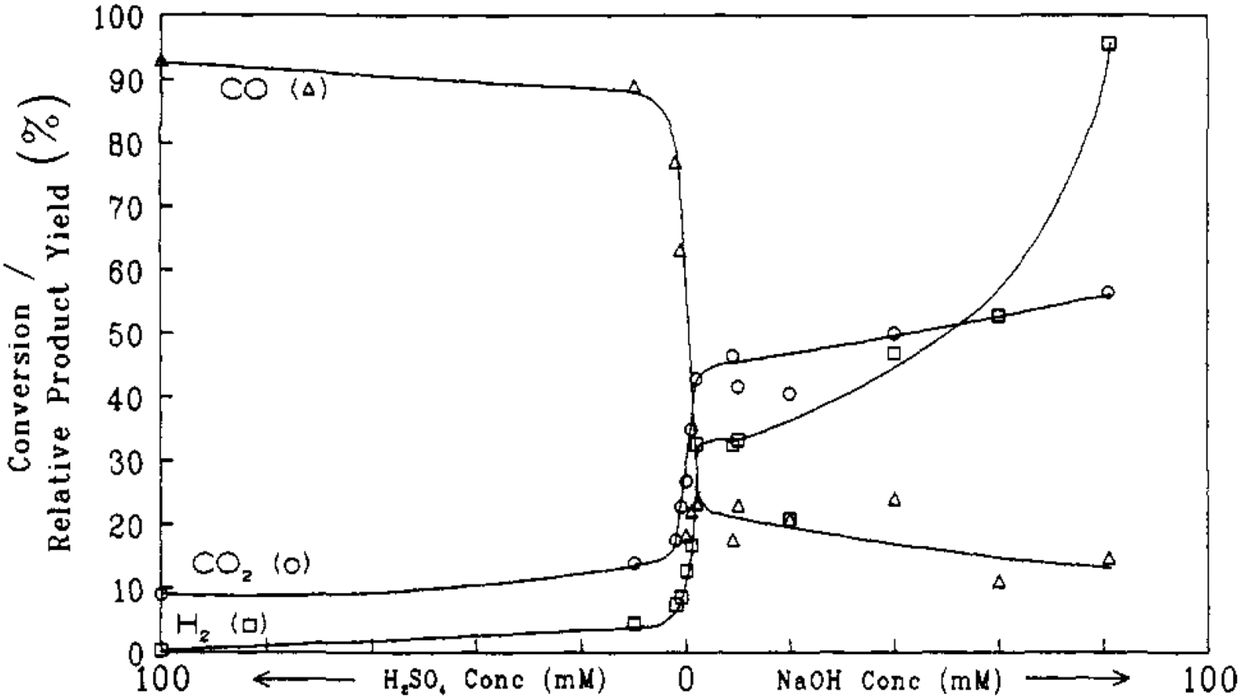 | ||
| Fig. 2 Effect of acid and base concentration on the selectivities toward gaseous products from reactions of LA in supercritical water at 385 °C, 345 bar, a residence time of 28 s with an initial aqueous 0.1 M LA. Reprinted with permission from ref. 27, copyright 1989 American Chemical Society. | ||
In the investigation by Lira and McCrackin, H3PO4, NaOH, and Na2HPO4 were tested in detail as precatalysts with initial aqueous 0.4 M LA.28 With the concentration of added H3PO4, the LA conversion increased together with increased selectivity toward AD, decreased selectivity toward AA, and unchanged AA yield. In the presence of 0.16 M H3PO4, an LA conversion of 59% was produced with an AA yield of 7.8% and selectivities toward AA and AD of 13 and 49%, respectively. This implied that that the addition of medium acid somewhat increases the AA yield while promoting LA decarbonylation. Under the typical reaction conditions in the presence of NaOH (360 °C, 322 bar, pH = 2.74 and residence time = 107 s), an LA conversion of 24% was obtained with selectivities toward AA and AD of 45 and 8.8%, respectively. It is noted that an optimal AA yield in the presence of NaOH is inferior to an AA yield in the absence of the catalyst under equivalent reaction conditions, in agreement with that reported by Mok et al.27 Under the optimal reaction conditions in the presence of Na2HPO4 (360 °C, 316 bar, 0.02 M Na2HPO4, and residence time = 102 s), an LA conversion of 21% was achieved with selectivities toward AA and AD of 53 and 6.8%, respectively. Compared to the case with no catalyst, the addition of Na2HPO4 obviously increases the selectivity toward AA at the expense of selectivity toward AD while somewhat decreasing the LA conversion and somewhat increasing the AA yield.
In 2009, non-catalytic dehydration of LA to AA in supercritical water at high temperatures and pressures was reported by Aida et al.29 The reaction was investigated at 450 °C and 400–1000 bar in a continuous reactor. Increasing the reaction pressure led to a decrease in the LA conversion together with increased AA yield and decreased AD yield. At 450 °C, 1000 bar, and a residence time of 1.0 s, the reaction produced an LA conversion at 35% with selectivities toward AA, PA, and AD at 37, 1.2, and 31%, respectively, from aqueous 0.05 M LA. The reaction efficiency for the production of AA is tremendously increased compared to the cases with lower temperature and pressure of supercritical water.27,28 In the meantime, increasing the reaction pressure resulted in an increase in the kinetic rate constants of both the dehydration reaction (k1) and the decarbonylation and decarboxylation reaction (k2). But k1 exhibited a greater increase than k2. The reaction data and kinetic analysis consistently showed that the selectivity of the dehydration reaction increases with increasing reaction pressure. This work significantly indicated that the production of AA can be enormously enhanced by increasing the temperature and pressure of supercritical water. The enhanced dehydration reactivity was hypothesized to result from an increase in the water density owing to the increased pressure, which may strengthen the activation of LA by water.29
From the above research on the dehydration of LA in supercritical water, the liquid-phase dehydration of LA to AA in supercritical water can proceed either in a non-catalytic or catalytic way. The dehydration reaction is catalyzed by neither acid nor base.27,28 The addition of either acid or base inhibits the dehydration reaction.27,28 Under non-catalytic conditions, LA dehydrates via self-H+ donation to its hydroxyl group intermolecularly. LA acts both as the reactant and Brønsted acid in the dehydration process. No catalytic role of either acid or base in the dehydration reaction, combined with the observed first-order rate law, made Mok et al. propose an intramolecular elimination mechanism.27 Since the AA yield and k1 increase with increasing water density, Aida et al. estimated that water should take part in the dehydration process, and thus proposed a reaction mechanism with a water-containing six-membered ring transition state.29 However, this mechanism is inconsistent with the obtained first-order rate law in the kinetic studies.27–29 Herein, it is suggested to undergo a modified reaction mechanism with the intermolecular elimination as shown in Scheme 1. This is because the increase in the water density is naturally accompanied by an increase in the LA density, which may increase the reaction rate.
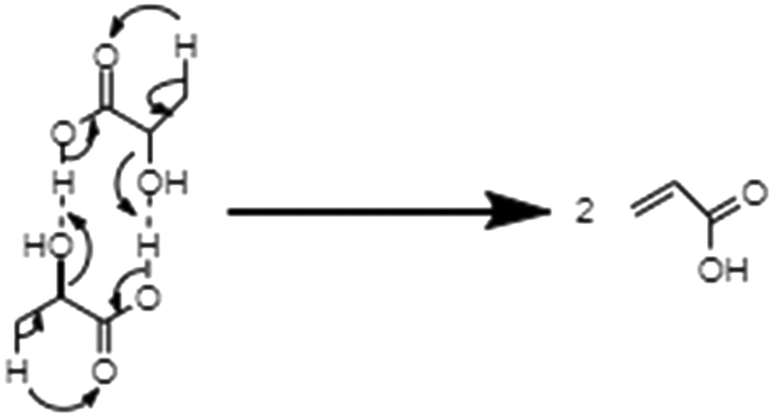 | ||
| Scheme 1 Modified elimination mechanism for the non-catalytic dehydration of LA in supercritical water. | ||
As long as an alkali or alkaline-earth cation is present in an LA dehydration system, the dehydration reaction is exposed to catalytic conditions. This is because lactate salt is generated in situ from LA and alkali or alkaline-earth cation under reaction conditions and suggestively acts as the catalytic active species.21,22 Under catalytic conditions, LA acts both as the reactant and Brønsted acid that assists in the dehydration process with lactate salt as the catalyst.23 The catalytic dehydration reaction turns out predominant over the non-catalytic dehydration reaction.23 However, the observed AA yields in the presence of the sodium salt systems, including NaOH and Na2HPO4, are similar to that in the absence of the catalyst.28 This is probably associated with the chemical instability of unsupported sodium lactate as the catalyst under an acidic condition of LA dehydration. It is demonstrated that unsupported sodium lactate tends to dehydrate to sodium acrylate which subsequently polymerizes under LA dehydration conditions.23i.e., the unsupported catalyst easily deactivates during LA dehydration. Therefore, the NaOH and Na2HPO4 systems probably fail to perform in the liquid-phase LA dehydration to AA.28
Based on all these reaction studies, the liquid-phase dehydration of LA to AA is inefficient and uneconomic to meet the requirements of industrial applications.
The catalytic process of the vapor-phase dehydration of LA to AA has been attracting considerable interest of researchers for the sake of not only high efficiency but also green chemical manufacture.1–4,23 It is a simple continuous vapor-phase reaction process using heterogeneous catalyst systems. Almost full LA conversion and ca. 80% selectivity toward AA can be reached with effective catalyst systems.3,4 After the reaction, all products are conveniently separated and collected, and the solid catalyst system stays in the fixed-bed reactor without contamination with the products. Moreover, because the main by-products are the low boiling point AD and gaseous CO/CO2, the cost and expense of the process are low in operation and separation. Evidently, the process of vapor-phase dehydration of LA to AA possesses incomparable advantages over the process of liquid-phase dehydration of LA to AA in LA conversion, selectivity toward AA, economy, and environmental protection. As compared with the two-step process of conversion of LA to AA, the process of vapor-phase dehydration of LA to AA has marked advantages in economy and environmental protection. Even if the two-step process of conversion of LA to AA uses heterogeneous catalyst systems in both the steps, the separation and recovery of acetic acid from the products render the process complicated and less attractive. In conversion and selectivity, the two-step process of conversion of LA to AA can realize excellent conversions of LA and α-acetoxypropionic acid and an excellent selectivity toward AA,14,18 which may be slightly advantageous over the process of vapor-phase dehydration of LA to AA. The excellent selectivity toward AA is attributed to the prevention of formation of lactides or PLA from LA thanks to the acetylation of LA. But integrally, the process of vapor-phase dehydration of LA to AA is advantageous over the two-step process of conversion of LA to AA, taking into account all the evaluation aspects. The characteristics of the three different routes for conversion of LA to AA are compared in Table 1.
| Route | Reaction conditions | Catalyst systems | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| Two-step process | 1st step: α-acetoxypropionic acid preparation from LA and acetic acid either in a batch or continuous reactor at 20–100 °C and ≤1 atm | 1st step: H2SO4, Amberlyst resin, Nafion NR50, –SO3H-containing activated carbon, –SO3H-containing graphene | Excellent conversion and selectivity toward AA | Separation and recovery of acetic acid, –SO3H leaching, acid contamination | 14, 17–19 |
| 2nd step: AA production from α-acetoxypropionic acid thermolysis at 300–360 °C and <80 torr | 2nd step: Ba3(PO4)2, Ca3(PO4)2, Li3PO4/Ca3(PO4)2 | ||||
| Liquid-phase dehydration | (1) In aqueous solution a 180–250 °C and ≤1 atm in a batch reactor | (1) [PtH(PEt3)3]OH | No catalyst required in supercritical water | Low conversion and selectivity toward AA, high cost, fast catalyst deactivation, catalyst contamination | 26–29 |
| (2) In supercritical water at 320–450 °C and 310–1000 bar in a continuous reactor | (2) Nil | ||||
| Vapor-phase dehydration | Thermolysis at 350–450 °C and ca. 1 atm in a continuous reactor | Na2SO4/CaSO4, Ba3(PO4)2, HAP, K0.97Na0.03ZSM-5, neutral KNO3/silica, KNO3/SiO2–Al2O3, etc. | Excellent conversion, fair to good selectivity to AA, low cost, clean process | Formation of lactides or PLA from LA | 1–4, 20, 22, 23 |
Vapor-phase dehydration has doubtless been the preferable choice for the conversion of LA to AA. Major pathways of LA conversion were earlier summarized by Miller and coworkers, as shown in Scheme 2.30 Under dehydration conditions, LA can undergo a series of complex reactions to corresponding products such as dehydration to AA, AA reduction to PA, decarbonylation to AD and CO, decarboxylation to AD and CO2, condensation to 2,3-pentanedione (2,3-PD), and polymerization to lactides or PLA. An acidic condition promotes LA decarbonylation to AD and is unfavorable for AA production.27,28,31–33 A basic condition facilitates LA decarboxylation to AD and impedes LA dehydration to AA.27,28 As long as the generation of H2 occurs under reaction conditions, it certainly results in AA reduction to PA.23,28 LA readily polymerizes to lactides or PLA at moderate temperatures.20 At lower temperatures (280–300 °C), a weakly acidic environment benefits LA condensation to 2,3-PD.34 Only at higher temperatures (350–450 °C), a weakly acidic environment favors LA dehydration to AA.21–23,27–29,34 It is hence evident that the control of the catalytic selectivity of LA dehydration to AA is a tough challenge. While advances have been progressively made in this area, the understanding and control of crucial factors affecting the reaction kinetics (e.g., acid–base property of catalyst, structure of catalyst, and reaction intermediate) are yet to be satisfied. Systematically reviewing the development of catalytic vapor-phase dehydration of LA will promote the thorough study of crucial factors controlling the catalytic selectivity in the community of catalytic research to fulfill the breakthroughs in this area.
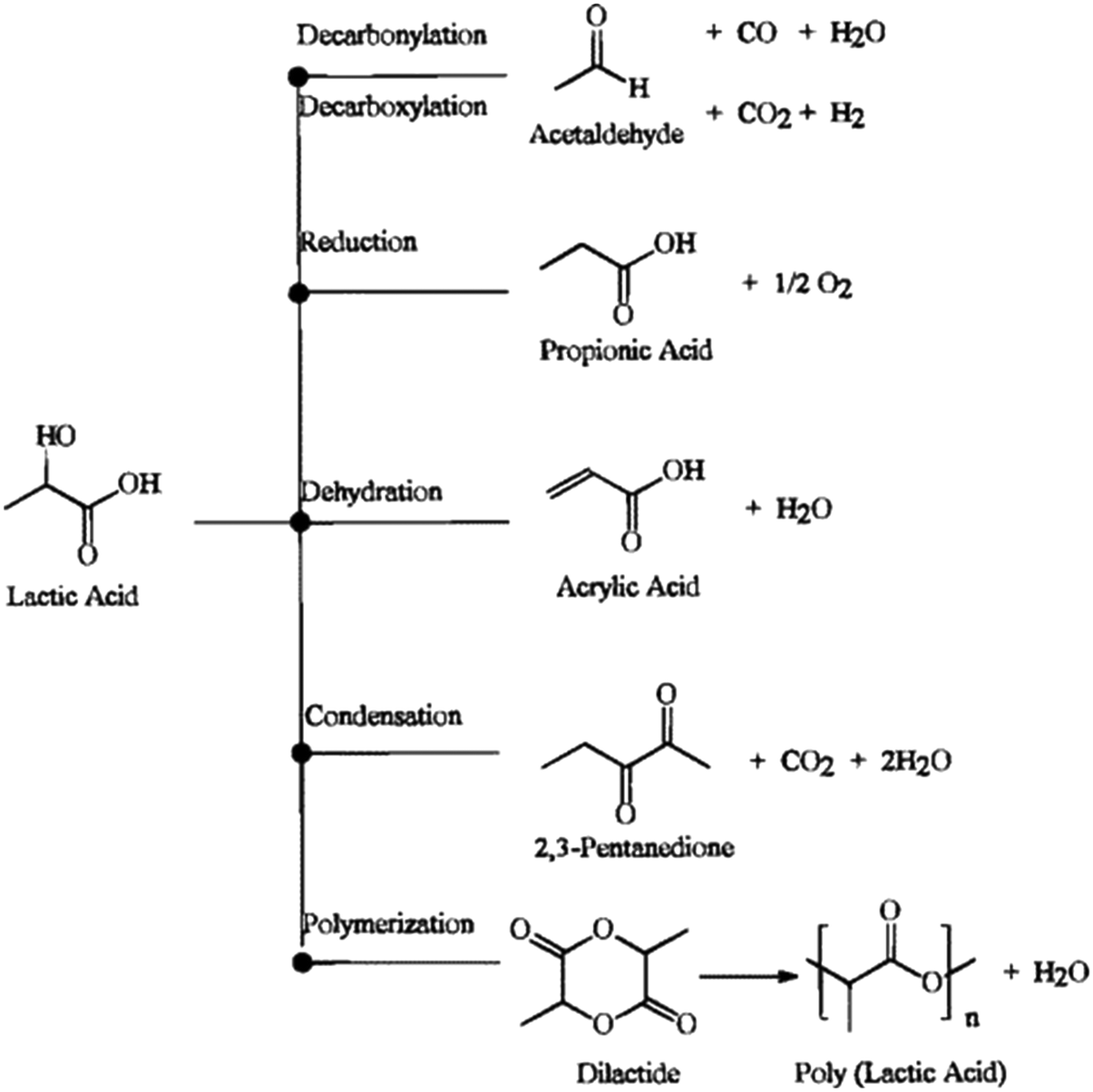 | ||
| Scheme 2 Major pathways of LA conversion. Reprinted with permission from ref. 30, copyright 1998 American Chemical Society. | ||
This review systematically summarizes the developments of catalytic vapor-phase dehydration of LA to AA. We present the research advances of various types of heterogeneous catalyst systems to date in chronological order with our critiques. Meanwhile, we deal with crucial factors controlling the catalytic selectivity, possible catalytic active species involved, and reaction mechanisms in play proposed by different groups. We expect to bring an explicit overview of the catalytic vapor-phase dehydration of LA to AA with the rational design of effective heterogeneous catalyst systems to readers.
Heterogeneous catalyst systems
Holmen addressed for the first time the catalytic vapor-phase dehydration of LA and alkyl lactates to AA and acrylates in his patent.20 He reported an AA yield of 68% with a mixture precatalyst consisting of Na2SO4 and CaSO4 at 400 °C and 1 atm. His pioneering work stressed sulfate and phosphate salts as effective catalyst systems for the vapor-phase dehydration of LA and alkyl lactates to AA and acrylates. Since then, many groups have been working to investigate potential heterogeneous catalyst systems for the vapor-phase dehydration of LA to AA and to shed light on the reaction mechanism in play.1–4,23 We classify the heterogeneous catalyst systems documented in the literature into five types, i.e., sulfate salts, phosphate salts, nitrate salts, hydroxyapatites (HAPs), and modified zeolites.Sulfate salts
As early as 1958, Holmen reported the first work on sulfate salt catalyst systems for the vapor-phase dehydration of LA to AA in his patent.20 An Na2SO4/CaSO4 (1/25 molar ratio) material was prepared by mixing a concentrated aqueous solution of Na2SO4 with finely divided CaSO4 to form a stuff paste, which was dried in a layer. This system resulted in a maximum AA yield of 68% from an aqueous 10% LA solution in a fixed-bed reactor at 400 °C, 1 atm, and a weight hourly space velocity of LA (WHSVLA) of 0.026 h−1. In the meantime, he reported four mixed sulfate/phosphate salt systems, i.e., CaSO4/Na4P2O7 (25/1 molar ratio), CaSO4/CsH2PO4 (25/1 molar ratio), CaSO4/LaPO4 (25/1 molar ratio), and CaSO4/CsH2PO4/LaPO4 (25/0.04/1 molar ratio), which could produce AA yields of 51, 54, 42, and 58%, respectively, from aqueous 50% LA at 425 °C, 1 atm, and a WHSVLA of 0.20 h−1. The satisfactory catalytic results for the conversion of LA to AA may be attributed to the generation of lactate salt, which Tam et al. thought of as the reaction intermediate21 and Huang et al. suggested as the catalytic active species22 under the reaction conditions. The content and chemical stability of lactate salt may determine the catalytic performance. At the reaction temperature of 400–425 °C, it may be assumed that either sulfate or phosphate salt displaces smoothly with LA to generate lactate salt and its conjugate acid as follows.21,22| nC2H4(OH)COOH + Mn+ → n[C2H4(OH)COO]M + nH+ |
A second report on sulfate salt systems for the vapor-phase dehydration of LA to AA appeared nearly forty years later. Tam et al. presented the catalytic and IR studies of sodium salt systems for LA conversion to 2,3-PD and AA.21 The precatalysts were prepared by the impregnation of low surface area (LSA) SiO2 (BET area of 7.2 m2 g−1) with various sodium salts, including Na2SO4, NaOH, NaCl, NaNO3, Na2SiO3, Na3PO4·12H2O, and sodium lactate at a salt loading of 1.0 mmol on 1.0 g of SiO2(LSA). Using Na2SO4/SiO2(LSA) as a precatalyst, a vapor-phase LA dehydration reaction gave rise to an LA conversion of 47% with yields of AA, PA, 2,3-PD, and AD at 3.9, 3.1, 2.4, and 20%, respectively, from aqueous 34% LA at 350 °C, 0.5 MPa, and a WHSVLA of 1.1 h−1. The very low production of 2,3-PD and AA was ascribed to no generation of sodium lactate, which they thought of as the reaction intermediate. The IR analysis of Na2SO4/SiO2(LSA) under LA vapor showed that LA and the supported Na2SO4 did not give sodium lactate at 150–350 °C.21 Nevertheless, a careful examination of their surface IR spectra reported shows that a tiny new band appears at about 1600 cm−1 from 300 °C upward and grows at 350 °C. The 1600 cm−1 band corresponds to the main feature of sodium lactate. This additional information is a hint that sodium lactate is able to form from LA and the supported Na2SO4 to a tiny extend at 300–350 °C and to a greater extent at higher temperatures. It was the first time that lactate salt was put forward as the reaction intermediate of LA dehydration to 2,3-PD and AA. At ≤350 °C, Na2SO4/SiO2(LSA) leads to the very low production of 2,3-PD and AA, presumably in that sodium lactate hardly forms from LA and the supported Na2SO4, which coincides with the comparative acidities of LA (pKa = 3.86 at 25 °C) and H2SO4 (pKa1 = −3, pKa2 = 1.99 at 25 °C).35 However, their reaction and IR results at higher temperatures (≥400 °C) were not reported to check if the AA yield and the sodium lactate amount could increase and thus to verify their hypothesis with the case of Na2SO4/SiO2 (LSA).
In 2008, Zhang et al. reported a mixed sulfate/phosphate salt system, which was utilized to catalyze the vapor-phase dehydration of LA to AA.36 This sulfate salt-based system made use of phosphate salts as promoters, in which CaSO4/CuSO4/Na2HPO4/KH2PO4 with a mass ratio of 150/13.8/2.5/1.2 was prepared by mixing these salts in a concentrated aqueous solution, following drying and calcination. Using CO2 as the carrier gas, an optimal AA yield of 64% was achieved from aqueous 26% LA at 330 °C and a WHSVLA of 0.42 h−1 over this system. Under such optimal reaction conditions, the yields of PA and AD were found to be 12 and 17%, respectively. In this case, the AA yield of 64% is satisfactory. As far as the generation of lactate salt as the reaction intermediate is concerned in explaining this reaction result, CaSO4 or CuSO4 or KH2PO4 may hardly accept an H+ from LA to form the lactate salt at 330 °C, based on the comparative acidities of LA (pKa = 3.86 at 25 °C),35 H2SO4 (pKa1 = −3, pKa2 = 1.99 at 25 °C),35 and H4P2O7 (pKa1 = 1.5, pKa2 = 2.4, pKa3 = 6.6, pKa4 = 9.3 at 25 °C).37 Only Na2HPO4 may provide for the formation of sodium lactate.37 Na2HPO4 readily condenses to Na4P2O7 at this reaction temperature.38,39 The resultant Na4P2O7 readily accepts an H+ from LA to give the lactate salt and Na3HP2O7, which is in accordance with the comparative acidities of LA and H4P2O7.
In 2014, Peng et al. reported the vapor-phase dehydration of LA to AA over various metal sulfates such as Na2SO4, Al2(SO4)3, NiSO4, ZnSO4, MgSO4, BaSO4, and CaSO4.24 They examined the relationship between the catalytic activity and the acid strength or Hammett acidity function (H0) over these systems. Under the equivalent reaction conditions, the AA yield over these systems increased in the order CaSO4 > BaSO4 > MgSO4 > NiSO4 > ZnSO4, > Al2(SO4)3 > Na2SO4. The acid strength of these systems increased in the order Al2(SO4)3 > ZnSO4, > NiSO4 > MgSO4 > BaSO4 > CaSO4 > Na2SO4. Except for Na2SO4, the increase in the AA yield coincides with the decrease in the acid strength. This indicates that the AA yield increases with decreasing acid strength in the definite range. Through the study of this correlation, they deduced that only moderate acid strength facilitates LA dehydration to AA and that the acid strength or H0 of sulfate salt in favor of conversion of LA to AA lies between 3.3 and 4.8. BaSO4 and CaSO4 were found to result in better catalytic activity for the production of AA owing to their moderate acid strength. Under the optimal reaction conditions (400 °C, aqueous 20% LA, and WHSVLA = 0.38 h−1), LA conversion of 100% and selectivity toward AA of 66% were achieved over BaSO4. Meanwhile, the selectivities toward PA, 2,3-PD, and AD were 9.4, 1.7, and 20, respectively. Nevertheless, the catalytic stability of these systems is unsatisfactory. After 80 h of reaction on stream over BaSO4, the selectivity toward AA declined by 34% while the LA conversion decreased by 10%. Under the equivalent reaction conditions, the LA conversion dramatically dropped from 100 to 75% and the selectivity toward AA maintained at ca. 70% after 5 h of reaction over CaSO4. Amid all the sulfate salt systems studied, the BaSO4 and CaSO4 systems outperform the other sulfate salt systems in terms of AA yield, possibly owing to the better chemical stability of the in situ generated barium and calcium lactates against their dehydration to acrylates followed by acrylate polymerization under the acidic reaction conditions. Comparatively, the barium lactate is more chemically stable than the calcium lactate based on the better catalytic stability of the BaSO4 system. From this work, the nature of cation in lactate salt presumably has a critical effect on the catalytic activity and stability of lactate salt.
In 2017, Li et al. presented their work on the vapor-phase dehydration of LA to AA over NH3-promoted BaSO4.40 BaSO4 was prepared from H2SO4 and Ba(OH)2 by precipitation. When aqueous NH3 was used as an alkaline agent at pH = 5 in the preparation of BaSO4, the resultant BaSO4 system displayed good catalytic performance, producing LA conversion of 100% and selectivity toward AA of 81% from aqueous 20% LA at 400 °C and a WHSVLA of 0.53 h−1. They attributed this unexpected catalytic performance to a balance between the acid and base sites present on the catalyst surface. However, both the LA conversion and the selectivity to AA obviously decreased with the reaction time within 24 h. The deactivated catalyst system could be easily regenerated after 10 h of calcination in air. In the same year, Lyu and Wang published a paper on the structure–activity relationship in the vapor-phase dehydration of LA to AA over BaSO4.41 BaSO4 was prepared from (NH4)2SO4 and BaCl2·2H2O by precipitation or by precipitation, followed by ultrasonic treatment. Combined with BET, SEM, XRD, and photoluminescence studies, their reaction results suggested that BaSO4 with smaller crystals and more defects has higher catalytic activity and selectivity toward the production of AA. BaSO4 prepared by precipitation gave rise to an LA conversion of 70% with selectivities toward AA and AD of 66 and 23%, respectively, from aqueous 20% LA at 350 °C and a WHSVLA of 0.48 h−1. Using ultrasonic treatment and ethanol as the solvent in the preparation of BaSO4, imperfect small crystals with more defects were formed, which led to an increase in the LA conversion to 78% and selectivity toward AA to 79% and to decrease in the selectivity toward AD to 13%. The difference in the catalytic activity and selectivity could be attributed to the difference in the acid amount. With increasing crystal defect and surface area, much more acid sites were observed with BaSO4 prepared by ultrasonic treatment with ethanol. Moreover, the acid sites of all the catalyst systems in this work were of weak to medium strength, which were suitable for dehydration of LA to AA. Accordingly, they believed that the crystal defects provide the active acid sites for the selective dehydration of LA to AA. Under the same reaction conditions over BaSO4 prepared by ultrasonic treatment with ethanol, the LA conversion fell from 83 to 65% and the selectivity toward AA declined from 82 to 60% at steady state during 48 h of reaction on stream.
From the papers of Peng et al., Li et al., and Lyu and Wang, the sulfate salt systems studied are all unsupported. The fact that the fair or good AA yields are obtained at the initial stage of the reaction suggests that the sulfate salts convert to lactate salts efficiently. During an LA dehydration reaction, the catalytic stability may be strongly related to the chemical stability of lactate salt as the reaction intermediate or catalytic active species.22,23 An aqueous 20% LA solution is rather acidic. Its pH or H0 is estimated to be 1.7 based on pKa = 3.86 at 25 °C. Under such an acidic condition, unsupported lactate salt is demonstrated to tend to dehydrate to acrylate salt, which in turn readily polymerizes as follows.23
n[C2H4(OH)COO]M → n[CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOO]M + nH2O CHCOO]M + nH2O |
| m{n[CH2CHCOO]M} → {n[CH2CHCOO]M}m |
Table 2 summarizes the catalytic performance of the above-described sulfate salt-based systems at the initial stage in the vapor-phase dehydration of LA from the literature. Where catalytic stability is concerned, we evaluate the “stability” based on both the variation of conversion, AA yield, and selectivity toward AA with reaction time, and WHSVLA. Herein, we set the standards for the stability of a catalyst system in terms of loss of AA yield during 50 h of reaction at steady state and a WHSVLA of 0.5 h−1 as follows. If the loss of AA yield is maintained within 10%, then the stability is regarded to be good. If the loss of AA yield is found to be beyond 25%, then the stability is considered to be poor. Given a grade for the stability of a catalyst system, the higher WHSVLA, the more loss of AA yield is allowable.
| Precatalyst | T (°C) | WHSVLA (h−1) | Conversion (%) | AA yield (%) | Selectivity (%) | Stability | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| AA | PA | 2,3-PD | AD | |||||||
| Na2SO4/CaSO4 (1/25 molar ratio) | 400 | 0.026 | — | 68 | — | — | — | — | — | 20 |
| CaSO4/Na4P2O7 (25/1 molar ratio) | 425 | 0.20 | — | 51 | — | — | — | — | — | 20 |
| CaSO4/CsH2PO4 (25/1 molar ratio) | 425 | 0.20 | — | 54 | — | — | — | — | — | 20 |
| CaSO4/LaPO4 (25/1 molar ratio) | 425 | 0.20 | — | 42 | — | — | — | — | — | 20 |
| CaSO4/CsH2PO4/LaPO4 (25/0.04/1 molar ratio) | 425 | 0.20 | — | 58 | — | — | — | — | — | 20 |
| Na2SO4/SiO2(/LSA) | 350 | 1.1 | 47 | 3.9 | 8.3 | 6.6 | 5.1 | 43 | — | 21 |
| CaSO4/CuSO4/Na2HPO4/KH2PO4 (150/13.8/2.5/1.2 mass ratio) | 330 | 0.42 | — | 64 | — | — | — | — | — | 36 |
| Na2SO4 | 400 | 0.38 | 30 | 8.1 | 27 | 24 | 1.3 | 23 | — | 24 |
| Al2(SO4)3 | 400 | 0.38 | 100 | 9.4 | 9.4 | 5.5 | 0.7 | 79 | — | 24 |
| MgSO4 | 400 | 0.38 | 85 | 53 | 62 | 6.2 | 1.2 | 29 | — | 24 |
| NiSO4 | 400 | 0.38 | 100 | 19 | 19 | 9.3 | 1.3 | 54 | — | 24 |
| ZnSO4 | 400 | 0.38 | 45 | 12 | 26 | 13 | 1.5 | 55 | — | 24 |
| CaSO4 | 400 | 0.38 | 100 | 69 | 69 | 7.6 | 0.9 | 21 | Poor | 24 |
| BaSO4 | 400 | 0.38 | 100 | 66 | 66 | 9.4 | 1.7 | 20 | Poor | 24 |
| BaSO4 | 350 | 0.53 | 70 | 57 | 82 | 4.5 | 0.2 | 9.2 | — | 40 |
| BaSO4 | 400 | 0.53 | 100 | 81 | 81 | 4.5 | 0.3 | 12 | Poor | 40 |
| BaSO4 | 350 | 0.48 | 78 | 62 | 79 | — | — | 13 | Poor | 41 |
| BaSO4 | 350 | 1.0 | 66 | 51 | 77 | 2.0 | — | 16 | — | 41 |
From the limited available information regarding conversion, AA yield, selectivity, and stability (Table 2), it can be seen that the unsupported sulfate salt systems are active and selective for the production of AA at 400 °C at the initial stage.20,24,36,40,41 However, their catalytic stability is unsatisfactory.24,40,41 This may be mainly due to the chemical instability of the unsupported lactate salt as the reaction intermediates or catalytic active species under an acidic condition of LA dehydration. It is necessary to note that during LA dehydration, unsupported lactate salt is exposed to the vapor of acidic LA. An aqueous LA solution is rather acidic. Aqueous 10% LA and 100% LA solutions are estimated to have pH or H0 values of 1.9 and 1.4, respectively, based on the pKa (= 3.86 at 25 °C) of LA. It is demonstrated that unsupported lactate salt, once produced from unsupported sulfate salt and LA, readily dehydrates to acrylate salt, which then readily polymerizes under such an acidic condition.23 The chemical instability of unsupported lactate salt can cause the fast deactivation of an unsupported sulfate salt system. To improve the chemical stability of lactate salt, it is suggested to support sulfate salts on inorganic materials such as silica and SiO2–Al2O3, which can prevent the occurrence of lactate salt dehydration to acrylate salt via the interaction between the OH group of lactate salt and silica or SiO2–Al2O3.22,23
As far as the stabilization of lactate salt is concerned, the nature of cation in lactate salt may affect the occurrence of lactate salt dehydration to acrylate salt followed by acrylate salt polymerization under an acidic condition of LA dehydration. An unsupported BaSO4 system is more effective than unsupported other sulfate salt systems for the production of AA,24 possibly in that the in situ generated barium lactate is more stable under acidic reaction conditions. The copresence of Ca2+ may promote the stabilization of an unsupported lactate salt catalyst like sodium or caesium or lanthanum lactate under acidic reaction conditions.20
To date, the nature of catalytic active species and reaction mechanism with sulfate salt systems have not been dealt with yet in the literature. According to the reported cases, sulfate salt is supposed not to be activated until ca. 400 °C before entering the catalytic state. The reaction intermediate or true catalytic species for the vapor-phase dehydration of LA to AA is presumably lactate salt.21,22,42 Certainly, the lactate salt does not easily form from LA and sulfate salt at lower temperatures, as evidenced by the IR study of Tam et al.21 This is consistent with the comparative acidities of LA (pKa = 3.86 at 25 °C) and H2SO4 (pKa1 = −3, pKa2 = 1.99 at 25 °C),35 which accounts for LA and sulfate salt hardly producing lactate salt and HSO4− at lower temperatures. Nevertheless, it is envisioned that a series of chemical equilibria could be established during the reaction of LA over sulfate salt at higher temperatures (≥400 °C) as follows.
| C2H4(OH)COOH + SO42− ⇌ C2H4(OH)COO− + HSO4− |
| 2HSO4− ⇌ S2O72− + H2O |
| C2H4(OH)COOH + S2O72− ⇌ C2H4(OH)COO− + HS2O7− |
| C2H4(OH)COOH + HS2O7− ⇌ C2H4(OH)COO− + H2S2O7 |
| H2S2O7 ⇌ H2SO4 + SO3 |
Upon the evaporation of H2SO4 as a conjugate acid derivative, the solid acidity of a sulfate salt system depletes. Eventually, the H0 of the sulfate salt system may be dependent on the lactate salt only.
Phosphate salts
Phosphate salt system-catalyzed vapor-phase dehydration of LA to AA likewise started with the work disclosed by Holmen in his patent in 1958.20 Over Ba3(PO4)2 prepared from BaCl2 and (NH4)2HPO4 by precipitation, an AA yield of 48% was achieved from aqueous 50% LA at 425 °C and a WHSVLA of 0.34 h−1. Over Ca3(PO4)2/Na4P2O7 (25/1 molar ratio) prepared by physical mixing, the reaction gave an AA yield of 50% under similar reaction conditions (425 °C, aqueous 50% LA and WHSVLA = 0.36 h−1). In 1986, another patent was published by Paparizos et al. regarding AlPO4 system-catalyzed vapor-phase dehydration of LA and/or ammonium lactate to AA.45 At 340 °C and a WHSVLA of 0.16 h−1, the LA conversion and the AA yield attained were 100 and 43%, respectively, from aqueous 22% LA over AlPO4, which was pretreated with an aqueous 14% NH3 solution. At the same time, the yields of PA and AD were found to be 3.2 and 35%, respectively. Three years later, Sawicki was granted a patent on the vapor-phase dehydration of LA to AA over base-modified SiO2-supported phosphate salts.31 With NaHCO3-modified SiO2-supported NaH2PO4 systems prepared by impregnation, the AA yield was found to be dependent on the pH of NaHCO3 prior to impregnation on the SiO2-supported NaH2PO4. At an optimal pH (= 5.9), 350 °C, and a WHSVLA of 0.11 h−1, the system gave rise to optimal AA yield of 42% and lower AD yield of 24% from aqueous 20% LA. Sawicki estimated that in converting LA to AA, the number and strength of acid sites on the surfaces of heterogeneous catalyst systems influence the catalytic selectivity toward AA, and the either too high or too low surface acidity favors the formation of AD rather than AA.31Through the exploratory work, these early researchers were aware that only weakly acidic catalyst systems are favorable for the production of AA, whereas more acidic or more basic catalyst systems are beneficial for the formation of AD via LA decarbonylation or decarboxylation. Actually, the catalyst systems studied are by far less acidic than the LA source used in their work in terms of pH or H0. The acidity of the LA aqueous solutions may determine the Brønsted acidity of the reaction systems so that the weaker solid acid sites of the catalyst systems could not act significantly. In the presence of these weaker solid acids, phosphate salt displacement with LA to result in lactate salts as the catalytic active species can proceed smoothly. Although the low rates of feed of LA have the drawbacks in efficiency and economy, these early explorations lay an important foundation for later research on phosphate salts and HAPs toward the vapor-phase dehydration of LA to AA.
Later on, Tam et al. and Gunter et al. studied the conversion of LA to 2,3-PD and AA over supported phosphate salts.21,34,38,46 In the catalyst preparation, phosphate salts such as Na3PO4·12H2O, NaH2PO4·H2O, Na2HPO4, and Na4P2O7 were mixed with supports such as activated carbon, SiO2 (BET area of 7.2 m2 g−1), and amorphous SiO2–Al2O3 (93/7 mass ratio, BET area of 5.1 m2 g−1) by impregnation. In the catalytic investigation, it was discovered that low temperatures (<320 °C) and elevated pressures favor 2.3-PD formation, whereas high temperatures and low pressures benefit AA and AD production.34 At 350 °C and 0.5 MPa, the LA conversion was 33%, and the yields of AA, PA, 2.3-PD, and AD were 9.8, 1.5, 7.0, and 5.2%, respectively, from aqueous 34% LA over Na3PO4/SiO2–Al2O3.38 Similar reaction results were obtained over Na2HPO4/SiO2–Al2O3, whereas much lower LA conversion and product yields were produced over NaH2PO4/SiO2–Al2O3.38 It was also noted that Na3PO4/SiO2 and Na4P2O7/SiO2 resulted in comparable catalytic results in the reaction at 300–350 °C, 0.5 MPa, and a WHSVLA of 1.1 h−1.21 For the first time, the catalytically active species present on supported inorganic salt systems for the vapor-phase dehydration of LA was discussed.38 The reactivity of LA with a variety of sodium phosphates supported on SiO2 as precatalysts at 25–350 °C was monitored by IR spectroscopy. Except NaH2PO4, Na2HPO4 or Na3PO4 turned out to be able to accept an H+ from LA with the formation of its conjugate acid and sodium lactate on SiO2 to a certain extent, in conformity with the acid–base chemistry of these compounds.35 At the same time that this surface reaction proceeds, LA polymerization occurs to give lactides or PLA. By combining the IR and 31P NMR studies, pyrophosphate (P2O74−) was identified as an important surface species. In the case with Na3PO4/SiO2, the resultant Na2HPO4 condensed to Na4P2O7 at reaction temperatures, consistent with the reactivity of Na2HPO4.47 In the case with Na2HPO4/SiO2, Na2HPO4 condensed to Na4P2O7, which then accepted an H+ from LA at reaction temperatures. The results could explain similar catalytic properties over the supported Na2HPO4, Na3PO4, and Na4P2O7 systems.21,38 It was noticed that LA conversion was dependent on the amount of sodium lactate generated as the reaction intermediate. However, the selectivity toward 2,3-PD and AA remained rather low despite the activity for LA conversion not being bad over all these systems. The main reason is probably why LA polymerization consumes much LA, which shifts the LA-sodium lactate chemical equilibria toward LA so that the selectivity of production of 2,3-PD and AA decreases. The resultant Na3HP2O7 stays in the solid phase because of the high melting point of pyrophosphate salt (≥600 °C)47,48 and thereby rarely continues reacting with LA to give Na2HP2O7 and sodium lactate. This impedes the chemical equilibrium shift toward sodium lactate.
It is necessary to note that the supported phosphate salt systems use the very low surface area SiO2 and SiO2–Al2O3 as the supports in the work of Tam et al. and Gunter et al. The in situ-generated supported sodium lactate inevitably faces a difficulty in chemical stability during LA dehydration. Due to the lack of efficient interaction between the sodium lactate and the support, sodium lactate is believed to easily dehydrate to sodium acrylate that subsequently polymerizes, which causes catalyst deactivation. To prevent the occurrence of sodium lactate dehydration, it is suggested to use high surface area supports to make supported phosphate salts as precatalysts. Only chemically stable lactate salt can produce good catalytic stability in LA dehydration.
More than a decade later, the research on phosphate salt-based systems toward vapor-phase dehydration of LA to AA was developed by Zhang et al.39 In the catalyst preparation, they incorporated sodium phosphates into zeolites by impregnation. At 340 °C and a WHSVLA of 1.3 h−1, an AA yield of 57% was achieved from aqueous 34% LA over Na2HPO4/NaY (14% Na2HPO4 loading, Si/Al = 2.5 molar ratio). Under the same reaction conditions, the AA yield dropped by 61% after 28 h of reaction on stream. The 31P NMR study of the fresh and spent catalyst samples suggested that Na2HPO4 condenses to Na4P2O7 after the thermal treatment of Na2HPO4/NaY at 120 °C and the resultant Na4P2O7 transforms to Na3HP2O7 after reaction. The results offered indirect evidence that sodium lactate forms from LA and Na4P2O7 on NaY during the reaction. The IR spectra of the spent catalyst samples exhibited a band at 1581–1560 cm−1. The band position increased with increasing Na2HPO4 loading on NaY from 2 to 18%. The lowest band at about 1560 cm−1 may be assigned to the ν(CO) stretching vibration of poly(sodium acrylate) rather than sodium lactate. The highest band at 1581 cm−1 should be attributed to a mixture of sodium lactate and poly(sodium acrylate). In view of the 31P NMR and IR results, it is inferred that the in situ-generated sodium lactate presumably acts as the catalytic active species is unstable on NaY and quickly dehydrates to sodium acrylate that subsequently polymerizes during reaction so that a catalyst systemsdeactivates fast.23 The work was followed up using nanocrystallite NaY (NaY-n) as the support.49 The catalytic activity and selectivity for the production of AA were obviously enhanced. Under the same reaction conditions as above, an AA yield as high as 74% was obtained from aqueous 34% LA over Na2HPO4/NaY-n (12% Na2HPO4 loading, Si/Al = 1.6 molar ratio). They attributed the enhanced catalytic performance mainly to the short pore channel and high external surface area of NaY-n, which facilitates quick product departure from the catalyst surface. However, the catalytic stability was little improved compared to that over Na2HPO4/NaY. Under the same reaction conditions, the AA yield dropped by 53% after 28 h of reaction on stream. The reasons are probably that the Na2HPO4/NaY precatalysts prepared by the incorporation of Na2HPO4 into NaY lack the interaction between Na2HPO4 and NaY in structure to stabilize the in situ-generated sodium lactate under the LA dehydration conditions on the one hand, and the high acid strength of NaY makes the resultant sodium lactate readily dehydrate to sodium acrylate, followed by sodium acrylate polymerization on the other hand.23 Eventually, the catalyst system is deactivated.
In 2013, Lingoes and Collias disclosed a series of phosphate salt-based systems for the vapor-phase dehydration of LA to AA in a patent.50 The phosphate salt-based systems were prepared by mixing two desired salts in a concentrated aqueous solution, followed by drying and calcination. At 350 °C and a WHSVLA of 0.94 h−1 with aqueous 20% LA, Ba(NO3)2/K2HPO4 with K/Ba = 40/60 molar ratio resulted in an LA conversion of 77 % and AA and PA yields of 72 and 3.9 %, respectively, and Ca2P2O7/KH2PO4 with K/Ca = 25/75 molar ratio led to LA conversion of 65% and AA yield of 53%. In contrast, K2HPO4 gave rise to an LA conversion of 98 % and AA and PA yields of 11 and 15 %, respectively, and Ba3(PO4)2 produced an LA conversion of 52 % and AA and PA yields of 24 and 2.6 %, respectively. It was noted that after 21.6 h of reaction on stream over Ba(NO3)2/K2HPO4, the LA conversion fell by 14% while the selectivity toward AA remained stable. The catalytic stability was thus assessed to be fair. From this work, the combination of Ba2+ (or Ca2+) and K+ may help improve the chemical stability of potassium lactate in situ generated from K2HPO4 under an acidic condition of LA dehydration. This appears to enable overcoming the shortcoming of monometallic cation in an unsupported salt system. A concerted effect of Ba2+ (or Ca2+) and K+ on the stabilization of potassium lactate against potassium lactate dehydration to potassium acrylate followed by potassium acrylate polymerization can be assumed.
In 2016, a study of ZSM-5-supported Na2HPO4 for the vapor-phase dehydration of LA to AA was reported by Zhang and coworkers.51 In the catalyst preparation, the parent H-ZSM-5 (Si/Al = 25 molar ratio, surface area of 278 m2 g−1) was subjected to treatments with NaOH and subsequently with Na2HPO4. The resultant Na2HPO4/ZSM-5 exhibited obviously decreased acid strength based on the NH3-TPD analysis. Under the optimized reaction conditions of 350 °C and a high WHSVLA of 2.6 h−1, LA conversion of 97% and high selectivity toward AA of 78% were produced from aqueous 30% LA over the best catalyst system 0.5P/ZSM-5-0.5AT. The selectivities toward PA, 2,3-PD, and AD were 0.8, 2.1, and 18, respectively. Under the same reaction conditions, the LA conversion decreased by 13% and the selectivity toward AA declined by 21% after 53 h of reaction on stream, as shown in Fig. 3. The catalytic stability was thus assessed to be good. The good catalytic stability may benefit from the proper interaction between Na2HPO4 and H-ZSM-5, which may result in the formation of the stable supported sodium lactate. This case is of interest for the application of ZSM-5 in supported phosphate salts for LA dehydration to AA because the adverse effect of high acid strength of ZSM-5 on the stabilization of the in situ generated sodium lactate catalyst is challenged.
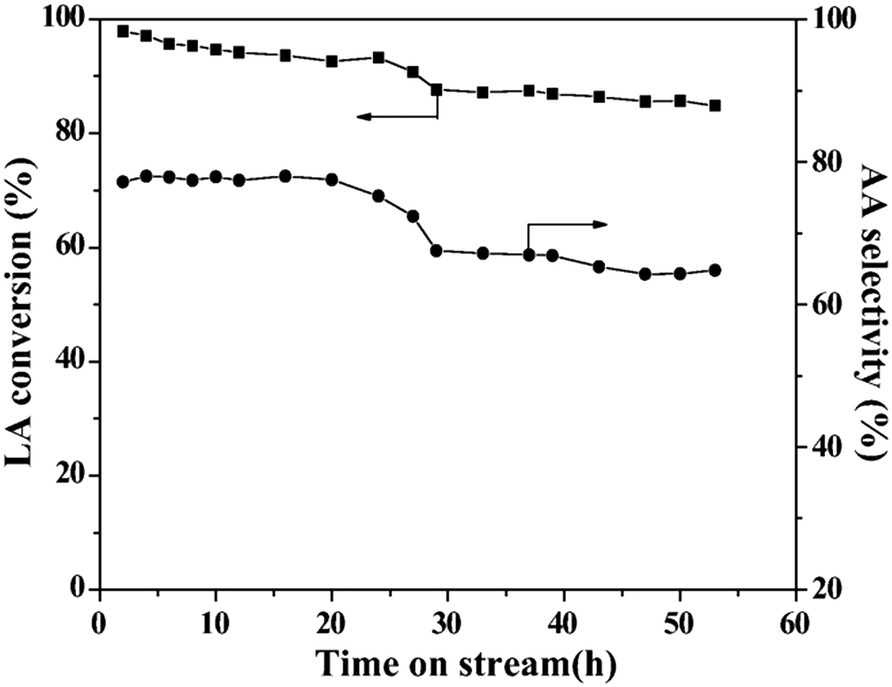 | ||
| Fig. 3 Stability test in vapor-phase dehydration of LA at 350 °C and a WHSVLA of 2.6 h−1 over 0.5P/ZSM-50.5AT. Reprinted with permission from ref. 51, copyright 2016 Elsevier Inc. | ||
During this period, the area of catalysis by phosphate salt-based systems for the vapor-phase dehydration of LA to AA was developed by several other groups as well.52–59 Tang et al. communicated the investigation of barium phosphate systems including Ba2P2O7, Ba3(PO4)2, and Ba2P2O7/Ba3(PO4)2 (1/1 molar ratio).52 The three barium phosphate precatalysts were prepared from BaCl2·2H2O and Na4P2O7 or Na3PO4 by precipitation. The Ba2P2O7 system displayed better catalytic performance with LA conversion of 100% and selectivity toward AA of 76% from aqueous 20% LA at 400 °C and a WHSVLA of 0.34 h−1. However, this system exhibited poor catalytic stability, the LA conversion falling by 6.3% and the selectivity toward AA dropping by 20% after 25 h of reaction on stream. They also reported on H3PO4-modified Sr2P2O7 systems.53 Under the optimized reaction conditions (400 °C, aqueous 20% LA, N2 flow rate = 3 mL min−1 and WHSVLA = 0.33 h−1), LA conversion of 100% and selectivity toward AA of 72% were produced over Sr2P2O7-0.10% H3PO4 within 4 h of reaction. After 4 h of reaction, the selectivity toward AA dramatically decreased while the LA conversion slightly fell. The catalytic instability of the unsupported phosphate systems likewise may reflect the easy transformation of unsupported phosphate salt to poly(acrylate salt) under the acidic condition of LA dehydration.23
Blanco et al. investigated the relationship between selectivity toward AA and acid–base properties over a series of alkaline-earth phosphates such as Ca2P2O7, Sr2P2O7, Sr3(PO4)2, Ba2P2O7, and Ba3(PO4)2.54 Sr3(PO4)2 (or Ba3(PO4)2) was prepared from (NH4)2HPO4 or Na2HPO4 and Sr(NO3)2 (or Ba(NO3)2) by precipitation at pH = 9. Ca2P2O7 (or Sr2P2O7 or Ba2P2O7) was prepared from Na4P2O7 or (NH4)2HPO4 and Ca(NO3)2·4H2O (or Sr(NO3)2 or Ba(NO3)2) by precipitation (pH adjusted to 10 when using (NH4)2HPO4). The acid–base property measurements revealed that alkaline-earth phosphates possess a high proportion of acid and base sites with the same strength. For the first time, they established the correlation between selectivity toward AA and solid acid–base balance that the optimal selectivity is 48% at an acidity/basicity ratio of 1.3, above which the selectivity decreases as the acidity/basicity ratio increases. However, the acid–base properties of the phosphate salt systems are determined before the catalytic reaction. The actual catalyst acidity certainly depletes when lactate salt forms with the evolution of H3PO4 (boiling point at 212 °C) as a conjugate acid during catalytic reaction at 350 °C. It is difficult to accurately establish the correlation between the selectivity toward AA and the catalyst acid–base properties. In addition, the IR spectra of the spent catalyst samples testified the presence of sodium and calcium lactates with a 1605 cm−1 band, which they interpreted as the formation of the reaction intermediates.
Ghantani et al. presented their work on the highly efficient and selective nonstoichiometric calcium pyrophosphate systems for the vapor-phase dehydration of LA to AA.55 The calcium pyrophosphates they prepared from Ca(NO3)2 and different sodium phosphates such as NaH2PO4·2H2O, Na2HPO4·2H2O, and Na3PO4·12H2O by precipitation had a Ca/P molar ratio from 1.02 to 0.76, corresponding to different pH values of the synthesis solutions. Calcium pyrophosphate with Ca/P = 0.76 molar ratio was the most effective system, giving LA conversion of 100% and selectivity toward AA of 74% from aqueous 50% LA at 375 °C and a WHSVLA of 1.5 h−1. They associated the higher selectivity toward AA to the higher acidity and the lower basicity of this system. In the IR study of reactivity of LA with calcium pyrophosphate, the adsorption of LA on the calcium pyrophosphate at 150 °C produced two bands at 1748(s) and 1600(s) cm−1. The former may be ascribed to lactides or PLA and the latter to calcium lactate. The results revealed that H+ transfer readily occurs from LA to calcium pyrophosphate to give the adsorbed calcium lactate and that LA polymerization strongly competes with the H+ transfer of LA in the LA–calcium lactate chemical equilibria over calcium pyrophosphate. Therefore, the higher catalyst acidity apparently has the drawback of triggering LA polymerization.
In 2016, Nagaraju et al. reported on cerium phosphate systems for use in vapor-phase dehydration of LA to AA.56 A series of cerium phosphates were synthesized from Ce(NO3)3·6H2O and H3PO4 by precipitation on varying the Ce/P molar ratio in the range from 0.5 to 3.0. Under the optimized reaction conditions (380 °C, aqueous 20% LA, and WHSVLA = 0.35 h−1), cerium phosphate with Ce/P = 2.5 molar ratio gave rise to good initial catalytic performance with LA conversion of 100% and AA yield of 64%. The catalytic stability test showed that LA conversion dropped by 13% and the selectivity toward AA slightly decreases after 20 h of reaction on stream under the same reaction conditions. The thermogravimetric (TG) and elemental analyses of the spent catalyst sample indicated the presence of carbonaceous species over the catalyst surface, which Nagaraju et al. thought may cause the deactivation of the catalyst system.
In 2020, Nekkala et al. disclosed magnesium hydrogen phosphate systems (MgHP) applied in the vapor-phase dehydration of LA to AA.57 A series of MgHP precatalysts were prepared from Mg(NO3)2·6H2O and NH4OH with different Mg/P molar ratios at varying calcination temperatures. Under the typical reaction conditions (360 °C, aqueous 20% LA, and WHSVLA = 0.62 h−1), an MgHP system with Mg/P = 1.1 molar ratio precalcined at 500 °C produced LA conversion of 100% and selectivity toward AA of 89%. After 23 h of reaction on stream at 360 °C, the AA yield fell by 21%, which is indicative of fast catalyst deactivation. The NH3-TPD and pyridine adsorption IR studies suggested that the effectiveness of the MgHP system is not linked to the strength of the acid sites but is associated with the presence of Lewis acid sites. However, the association of the catalytic performance with the presence of Lewis acid sites is ambiguous. Only a weak IR band of adsorbed pyridine at 1445 cm−1 was observed on almost all the precatalyst samples with Ce/P = 0.5–3.0 molar ratio. Moreover, the Lewis acid sites are identified by ex situ rather than in situ pyridine adsorption IR analysis. According to Murphy et al., NaY is able to convert to HY in water at 200 °C,60 which implies that the Lewis acid sites could convert to Brønsted acid sites under LA dehydration conditions. The possible key factor affecting the production of AA is the extent of formation of cerium lactate as the reaction intermediate.
Guo et al. investigated the application of lanthanum phosphate nanorods (LaP) in vapor-phase dehydration of LA to AA.58 A series of LaP precatalysts were synthesized from LaNO3·6H2O and NH4H2PO4 by precipitation using n-butylamine as the shape-directing agent. The catalytic properties were adjusted by varying the n-butylamine/La molar ratio. Under the optimized reaction conditions (350 °C, aqueous 20% LA, and WHSVLA = 1.2 h−1), LaP-3 (n-butylamine/La = 3 molar ratio, La/P = 0.52 molar ratio) brought about LA conversion of 67% and selectivity toward AA of 50%. During 30 h of reaction on stream at 350 °C and a WHSVLA of 0.46 h−1 with aqueous 20% LA, the AA yield declined by 7.5% from 40 to 37% at steady state (from the 6th h), which indicates fair catalytic stability. Through the NH3-TPD and pyridine adsorption IR studies, they found that the use of n-butylamine not only affects the porosity of LaP but also modifies the acid–base properties. A better catalytic stability over LaP was associated with a higher concentration of Lewis acid sites. Both the LA conversion and the selectivity toward AA were observed to increase with increasing density of Lewis acid sites. The ex situ pyridine adsorption IR study indicated that Lewis acid sites are present as the sole solid acid sites on LaP. The basic sites were believed to have an adverse effect on the catalytic stability over LaP. Nevertheless, the favorable effect of Lewis acid sites on the catalytic properties is not convincing. The Lewis acid sites are analyzed by ex situ rather than in situ pyridine adsorption IR means. Under the LA dehydration conditions, the Lewis acid sites may convert to Brønsted acid sites.60 The total acidity of LaP measured by NH3-TPD can only represent the acid property of LaP before the catalytic reaction. The actual Lewis acidity cannot be accurately quantified under the reaction conditions.
Lastly, further research on lanthanum phosphates toward the vapor-phase dehydration of LA to AA was published by Nekkala et al.59 A series of lanthanum phosphate precatalysts was prepared from LaNO3·6H2O and H3PO4 by precipitation at pH ≈ 10. The catalytic properties were tuned by varying the La/P molar ratio and calcination temperature. Under the optimized reaction conditions (360 °C, aqueous 30% LA, and WHSVLA = 0.96 h−1), the catalytic performance with LA conversion of 100% and selectivity toward AA of 65% was achieved over a lanthanum phosphate with LA/P = 0.35 molar ratio, which was precalcined at 500 °C. After 24 h of reaction on stream, the AA yield fell by 19% from 80 to 65% at steady state (from the 8th h). The catalyst system was thus assessed to be poor in the catalytic stability.
Comparing the cases reported by Guo et al., and Nagaraju et al., it is found that the different LA/P molar ratios lead to different catalytic stabilities. The better catalytic stability with LA/P = 0.52 molar ratio may be related to a better chemical stability of lanthanum lactate generated in situ in this environment against lanthanum lactate dehydration to lanthanum acrylate followed by lanthanum acrylate polymerization.
Table 3 summarizes the catalytic performance of the above-described phosphate salt-based systems at the initial stage in the vapor-phase dehydration of LA from the literature. From the comparative data regarding LA conversion, selectivity, and stability, barium and calcium phosphate systems behave prominently among the phosphate salt systems studied,20,51,52,55 and pyrophosphate salt systems outperform the other phosphate salt systems studied.20,52,55,56 For barium and calcium phosphate systems, the in situ generated barium and calcium lactates as the catalytic active species are assumed to more active and selective for the production of AA on the one hand and to be more resistant to lactate dehydration to acrylate (which leads to acrylate polymerization, i.e., catalyst deactivation) on the other hand. For pyrophosphate salt systems, it may be beneficial to in situ generate lactate salt as the catalytic active species. The use of unsupported phosphate salts or phosphate salts incorporated on the low surface area support faces easy catalyst deactivation. Based on the reported cases, neither unsupported nor supported phosphates systems are satisfactory in terms of catalytic stability. Under an acidic condition of LA dehydration, lactate salt generated in situ from unsupported metal salt readily dehydrates to acrylate salt, which then polymerizes, as demonstrated by Huang et al.23 Supporting phosphate salt on high surface area materials may offer an efficient way to stabilize in situ generated lactate salt via the interaction between lactate salt and support. To date, high surface area material-supported phosphate salt systems have been seldom reported for use in the vapor-phase dehydration of LA to AA.39,49
| Precatalyst | T (°C) | WHSVLA (h−1) | Conversion (%) | AA yield (%) | Selectivity (%) | Stability | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|
| AA | PA | 2,3-PD | AD | |||||||
| Ba3(PO4)2 | 425 | 0.34 | — | 48 | — | — | — | — | — | 20 |
| Ca3(PO4)2/Na4P2O7 (25/1 molar ratio) | 425 | 0.36 | — | 50 | — | — | — | — | — | 20 |
| AlPO4 | 340 | 0.16 | 100 | 43 | 43 | 3.2 | — | 35 | — | 45 |
| NaHCO3–NH2PO4/SiO2 | 350 | 0.11 | 85 | 42 | 49 | — | — | 29 | — | 31 |
| Na3PO4/SiO2(93%)–Al2O3(7%) | 350 | — | 33 | 9.8 | 30 | 4.5 | 21 | 13 | — | 38 |
| Na2HPO4/SiO2(93%)–Al2O3(7%) | 350 | — | 33 | 9.7 | 29 | 8.2 | 25 | 19 | — | 38 |
| NaH2PO4/SiO2(93%)–Al2O3(7%) | 350 | — | 8.7 | 1.5 | 17 | 20 | 15 | 29 | — | 38 |
| Na3PO4/SiO2(LSA) | 350 | 1.1 | 95 | 20 | 21 | 7.1 | 14 | 16 | — | 21 |
| Na4P2O7/SiO2(LSA) | 350 | 1.1 | 80 | 17 | 21 | 3.8 | 21 | 23 | — | 21 |
| Na2HPO4/NaY (Si/Al = 2.5 molar ratio) | 340 | 1.3 | 78 | 57 | 72 | 1.3 | 5.9 | 5.4 | Poor | 39 |
| Na2HPO4/NaY-n (Si/Al = 1.6 molar ratio) | 340 | 1.3 | 94 | 74 | 80 | 1.1 | 5.3 | 5.4 | Poor | 49 |
| Ba(NO3)2/K2HPO4 (K/Ba = 40/60 molar ratio) | 350 | 0.94 | 77 | 72 | 92 | 0 | — | — | Fair | 50 |
| Ca2P2O7/KH2PO4 (K/Ca = 25/75 molar ratio) | 350 | 0.94 | 65 | 53 | 76 | 0 | — | — | — | 50 |
| Ba3(PO4)2/K2HPO4 (K/Ba = 25/75 molar ratio) | 350 | 0.94 | 97 | 41 | 42 | 5 | — | — | — | 50 |
| K2HPO4 | 350 | 0.94 | 98 | 11 | 11 | 15 | — | — | — | 50 |
| Ba3(PO4)2 | 350 | 0.94 | 52 | 24 | 48 | 5 | — | — | — | 50 |
| 0.5P/ZSM-5-0.5AT (Si/Al = 25 molar ratio) | 350 | 2.6 | 97 | 76 | 78 | 0.8 | 2.1 | 18 | Good | 51 |
| Ba2P2O7 | 400 | 0.34 | 100 | 76 | 76 | 2.8 | 2.1 | 14 | Poor | 52 |
| Ba2P2O7/Ba3(PO4)2 (1/1 molar ratio) | 400 | 0.34 | 81 | 44 | 54 | 3.9 | 1.1 | 11 | Poor | 52 |
| Sr2P2O7–0.10%H3PO4 | 400 | 0.33 | 100 | 72 | 72 | — | — | 21 | Poor | 53 |
| Cax(P2O7)y (x/y = 0.76 ) | 375 | 1.5 | 100 | 74 | 74 | — | — | 10 | — | 55 |
| Cex(PO4)y (x/y = 2.5) | 380 | 0.35 | 100 | 64 | 64 | 7.4 | — | — | Poor | 56 |
| MgHP (Mg/P = 1.1 molar ratio) | 360 | 0.62 | 100 | 89 | 89 | — | — | — | Poor | 57 |
| LaP-3 | 350 | 1.2 | 67 | 34 | 50 | 3.1 | 0.2 | 21 | Fair | 58 |
| LaP (La/P = 0.35 molar ratio) | 360 | 0.96 | 100 | 65 | 65 | — | — | — | Poor | 59 |
When using dihydrogen phosphate (H2PO4−) salt as a precatalyst, H2PO4− tends to condense to a mixture of polyphosphate (PO3−)n and hexametaphosphate (P6O186−) salts at reaction temperatures, which are little reactive with LA to give lactate salt.38 As such, the use of dihydrogen phosphate salt leads to low activity for the production of AA probably due to the lack of lactate salt. When using hydrogen phosphate (HPO42−), orthophosphate (PO43−), and pyrophosphate salts as precatalysts, several chemical equilibria exist at reaction temperatures as follows.
| C2H4(OH)COOH + PO43− ⇌ C2H4(OH)COO− + HPO42− |
| 2HPO42− ⇌ P2O74− + H2O |
| C2H4(OH)COOH + P2O74− ⇌ C2H4(OH)COO− + HP2O73− |
| C2H4(OH)COOH + HP2O73− ⇌ C2H4(OH)COO− + H2P2O72− |
| C2H4(OH)COOH + H2P2O72− ⇌ C2H4(OH)COO− + H3P2O7− |
| C2H4(OH)COOH + H3P2O7− ⇌ C2H4(OH)COO− + H4P2O7 |
| 3H4P2O7 ⇌ 2H5P3O10 + H2O |
| H4P2O7 + H2O ⇌ 2H3PO4 |
Nitrate salts
Gunter et al. were the first to publish catalytic work on nitrate salt-based systems for the vapor-phase dehydration of LA to 2,3-PD and AA (in 1995).46 In the catalyst preparation, NaNO3 was supported on SiO2 (LSA, BET area of 7.2 m2 g−1) or physically mixed SiO2–Al2O3 (93/7 mass ratio, BET area of 5.1 m2 g−1) by impregnation with an NaNO3 loading of 1.0 mmol on 1.0 g support. At 300 °C, 0.5 MPa and a WHSVLA of 1.1 h−1 with aqueous 34 % LA, the LA conversion was 34% and the yields of AA, PA, 2.3-PD, and AD were 5.6, 0.8, 12, and 4.0%, respectively, over NaNO3/SiO2(LSA). Over NaNO3/SiO2–Al2O3, the LA conversion reached 61% and the yields of AA, PA, 2.3-PD, and AD obtained were 11, 3.7, 17, and 7.6%, respectively. The results indicated that a lower temperature favors 2,3-PD production rather than AA production on the one hand and a low acidity of support promotes the catalysis of the NaNO3 system for LA conversion and AA production on the other hand. The catalytic work of nitrate salt-based systems was followed up by this group.21,42 At 350 °C, 0.5 MPa, and a WHSVLA of 1.1 h−1, NaNO3/SiO2(LSA) led to an LA conversion at 99% with yields of AA, PA, 2.3-PD, and AD at 16, 7.9, 12, and 22%, respectively, from aqueous 34% LA.21 Nevertheless, the catalytic stability of this system is poor.21 In fact, this group demonstrated through the IR study that on model SiO2 wafers, the supported sodium salts including NaOH, NaNO3, Na2SiO3, and Na3PO4, when exposed to LA vapor at 150–300 °C, authentically transform to sodium lactate with the concurrent formation of their conjugate acids as follows.21,32| C2H4(OH)COOH + Na+ → C2H4(OH)COONa + H+ |
In 2014, Zhang et al. reported on SBA-15 silica (SBA-15)- and fumed silica (FS)-supported NaNO3 systems for the production of AA and 23-PD from LA.65 The precatalysts were prepared by impregnation without subsequent calcination. Prior to catalytic reaction, the precatalysts were subjected to 3 h of N2 treatment at the reaction temperature. Using either high surface area SBA-15 or medium surface area FS as support, a similar AA yield was produced at 340–370 °C. The uncalcined NaNO3/SBA-15 gave rise to a higher 23-PD yield than the uncalcined NaNO3/FS. Under the optimized reaction conditions (360 °C, aqueous 34% LA, and WHSVLA = 5.2 h−1), yields of AA, 23-PD, and AD at 45, 25, and 13%, respectively, were obtained over the uncalcined NaNO3/SBA-15 with a high NaNO3 loading of 23%. In the IR study, the surface IR spectrum on the uncalcined NaNO3/SBA-15 exhibited a broad band centered at 1565(s) cm−1 together with bands at 1470(sh), 1417(m), and 1375(sh) cm−1 after 15 min of reaction at 360 °C. Zhang et al. assigned this spectrum to the supported sodium lactate, which they thought of as the real catalytic species for LA conversion to AA and 2,3-PD. They thus deduced that NaNO3 on the uncalcined system transforms to sodium lactate smoothly under the reaction conditions, which could perhaps explain the much better initial catalytic results for the production of AA and 2,3-PD over the uncalcined NaNO3/SBA-15 than over the uncalcined Na4SiO4/SBA-15. Under the equivalent reaction conditions, the LA conversion was 74% and the yields of AA and 2,3-PD were 20 and 10%, respectively, over the uncalcined Na4SiO4/SBA-15. Meanwhile, they believed that Na4SiO4 transforms to sodium lactate with difficulty. However, by examining the surface spectrum on the uncalcined NaNO3/SBA-15, it is perceived that the broad 1565(s) cm−1 band should be interpreted as the νCO stretching vibration of poly(sodium acrylate) as the major surface species23,63,64 rather than sodium lactate.21,42,66 This broad band may comprise the signature feature of surface sodium lactate as the minor surface species at about 1595 cm−1. The IR observations implicate that under the reaction conditions, NaNO3 on the uncalcined system readily transforms to sodium lactate and subsequently the resultant sodium lactate is prone to dehydrating to sodium acrylate, which converts to poly(sodium acrylate) as the major surface species. Both with high NaNO3 loading and without calcination, the lack of proper interaction between NaNO3 and the SBA-15 surface may render the resultant sodium lactate unstable during the reaction. This will probably cause the fast dehydration of the sodium lactate to sodium acrylate, followed by sodium acrylate polymerization, i.e., fast catalyst deactivation,23 although the catalytic stability of this system has not been reported.
In 2019, Huang et al. reported on (SiO2–Al2O3)-supported alkali nitrate systems including KNO3/SiO2–Al2O3 and NaNO3/SiO2–Al2O3 for the vapor-phase dehydration of LA to AA.22 The precatalysts were prepared by the impregnation of sodium-form amorphous SiO2–Al2O3 with Si/Al = 36 molar ratio (SiO2–Al2O3(36)) with alkali nitrates at a KNO3 or NaNO3 loading of 8.8%.28 The precatalysts are slightly acidic according to the surface acidity data acquired by NH3-TPD. But whether the surface acidity is retained during the vapor-phase LA dehydration at 350 °C is uncertain. HNO3 evaporation following KNO3 or NaNO3 displacement with LA (to give potassium or sodium lactate and HNO3) under the reaction conditions likely diminishes the surface acidity of the KNO3/SiO2–Al2O3(36) or NaNO3/SiO2–Al2O3(36) system. At 350 °C and a WHSVLA of 0.46 h−1 with aqueous 20% LA, KNO3/SiO2–Al2O3(36) led to better catalytic performance than NaNO3/SiO2–Al2O3(36) in terms of LA conversion (97% versus 89%) and selectivity toward AA (43% versus 37%), which shows the advantage of K+ over Na+ in catalysis promotion. Under the reaction conditions, both the systems exhibited excellent catalytic stability in more than 50 h of reaction on stream. Furthermore, KNO3/SiO2–Al2O3(36) resulted in an LA conversion at 67% with yields of AA, PA, and 2.3-PD at 37, 3.3, and 7.4%, respectively, from aqueous 20% LA at 350 °C and a WHSVLA of 1.8 h−1. After 90 h of reaction on stream, the catalytic performance remained almost unchanged, indicative of the good catalytic stability, as shown in Fig. 4. It was noticed that the selectivity toward AA slightly increased from 44 to 55% with increasing WHSVLA from 0.92 to 1.8 h−1 over 0.50 g of the catalyst sample, which suggests that the production of AA is favored in kinetics. In the IR monitoring study with LA and reaction intermediates on SiO2–Al2O3(36), it was observed that a potassium or sodium salt readily displaces with LA and the resultant potassium lactate with a characteristic feature at 1607–1593 cm−1 is quite stable at 350 °C. Meanwhile, the IR spectrum of the spent KNO3/SiO2–Al2O3(36) or NaNO3/SiO2–Al2O3(36) catalyst sample in the long-term vapor-phase dehydration of LA to AA displayed a marked band of potassium lactate at 1595 cm−1. From the combined catalytic and IR results, Huang et al. suggested for the first time that the in situ generated supported lactate salt acts as the true catalytic species for the dehydration of LA to AA.22
 | ||
| Fig. 4 LA conversion and selectivities toward chief products in the vapor-phase dehydration of LA at 350 °C and a WHSVLA of 1.8 h−1 over KNO3/SiO2–Al2O3(36). | ||
To shed light on the influences of surface acidity and interaction between alkali salt or base and support on the catalyst stabilization and deactivation, Huang et al. worked on a series of different KNO3 systems including unsupported KNO3, acidic KNO3/silica, and neutral KNO3/silica. Lastly, they disclosed neutral catalysts, i.e., neutral, high surface area silica-supported potassium lactate derived in situ from KNO3 for the vapor-phase dehydration of LA to AA.23 The catalytic and acid property studies illustrated that the surface acidity has a significant effect on the catalytic performance of the KNO3/silica system. The acidic KNO3/S15 system incurs the formation of more coke, which is detrimental to the catalytic selectivity and stability for the production of AA. The neutral KNO3/AS300, KNO3/S15cal, and KNO3/SBA-15 systems enable the formation of coke under control, which is beneficial for the catalytic selectivity and stability for the production of AA. The catalytic and TG studies showed that the interaction between KNO3 and silica brings forth a striking promotion on the catalytic performance of a KNO3 system. The unsupported KNO3 system performs poorly in catalytic stability for the production of AA, with the AA yield dropping quickly. The neutral KNO3/silica systems perform well in catalytic stability for the production of AA. The LA conversion of 91–97% decreases by upto 12%, and the selectivity toward AA of 28–37% increases by up to 32% at steady state during 72–99 h of reaction at 350 °C and a WHSVLA of 0.46 h−1. The typical case with KNO3/SBA-15 is shown in Fig. 5. The IR monitoring and catalytic studies suggested that the effects of surface acidity and interaction between potassium salt or base and silica on the catalytic performance are associated with the content and stability of the in situ generated potassium lactate as the catalytically active species. With the lack of surface acid and interaction between potassium salt or base and silica, the unsupported KNO3 less efficiently reacts with LA to produce potassium lactate at 200 °C or below. The resultant potassium lactate readily dehydrates to potassium acrylate at above 200 °C, which easily polymerizes at 300–350 °C, thus causing fast catalyst deactivation. With the absence of surface acid but with the interaction between potassium salt or base and silica, the reactions relating to the production of potassium lactate proceed efficiently and are dominant at 350 °C on neutral KNO3/silica. The content and stability of the potassium lactate could be secured while the formation of poly(potassium acrylate) could be prevented. The chemistry of the neutral KNO3/silica under LA dehydration conditions is illustrated in Scheme 3. Both with the presence of surface acid and with the interaction between potassium salt or base and silica, the reactions not favoring the production of potassium lactate are dominant at 22–350 °C on the acidic KNO3/silica, such that the irreversible potassium acrylate polymerization occurs and incurs quick catalyst deactivation. In accordance with the good catalytic stability of a neutral KNO3/silica system, the three critical steps (KNO3 displacement with LA to potassium lactate; potassium lactate dehydration to potassium acrylate; and AA formation and potassium lactate regeneration from LA and potassium acrylate) in the catalytic process was demonstrated to proceed smoothly on neutral KNO3/silica. It was deduced that LA acts both as the reactant and Brønsted acid in the catalytic process of LA dehydration with potassium lactate. The catalysis of potassium lactate is assisted by LA as the Brønsted acid. This work included for the first time that the dehydration of LA to AA proceeds smoothly with a neutral heterogeneous catalyst.23 This is Clear evidence of non-extra acid catalysis for the dehydration of LA to AA. This work meanwhile suggested that potassium lactate acts as the catalytic active species for LA decarbonylation apart from LA dehydration over neutral KNO3/silica.23
 | ||
| Fig. 5 LA conversion, AA yield and selectivities toward chief products in the vapor-phase dehydration of LA at 350 °C and a WHSVLA of 0.46 h−1 over KNO3/SBA-15. | ||
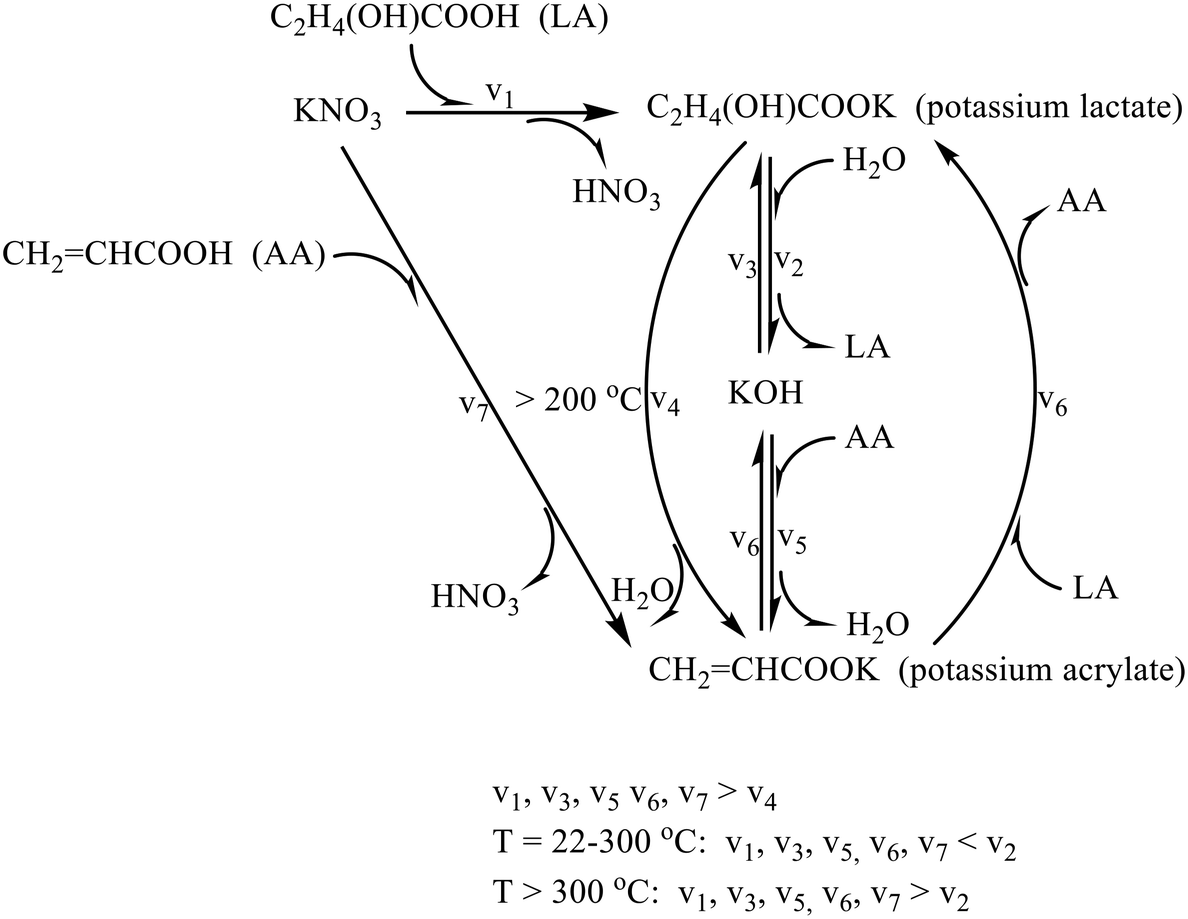 | ||
| Scheme 3 Suggested chemistry of the neutral KNO3AS300 or KNO3/S15cal under LA dehydration conditions. | ||
From the above cases with KNO3/SiO2–Al2O3(36), NaNO3/SiO2–Al2O3(36), and KNO3/silica studied by Huang et al., it is envisioned that as long as a supported lactate salt is efficiently produced and stabilized during catalytic LA dehydration, it can be expected to display stable catalytic performance for the production of AA. The IR spectrum of a spent catalyst sample is evidence of the catalyst stabilization. If the bands of lactate salt are obviously present in the spectrum, then the catalyst is assumed to be stable during reaction. But instead, if the bands of poly(acrylate salt) are observed in the spectrum, then the catalyst is considered to have already been deactivated during the reaction. When lactate salt dehydration and hydrolysis get controlled in rate and lactate salt dehydration becomes the rate-limiting step in the catalytic process of LA dehydration to AA, it is assumed that resultant acrylate salt timely displaces with excess LA to produce AA and regenerate lactate salt without acrylate salt polymerization, which can secure the content and stability of lactate salt during reaction.23 It is the case with either the neutral KNO3/silica or slightly acidic KNO3/SiO2–Al2O3(36) or NaNO3/SiO2–Al2O3(36).22,23 However, the stabilization of the supported lactate salt is affected by the surface acidity. The polymerization of acrylate salt can be induced by a higher surface acidity.23 Note that under LA dehydration conditions where LA itself serves as a Brønsted acid, the neutral silica-supported potassium lactate is able to tolerate the acidity of aqueous 20% LA (H0 = 1.7).23
The fact that the SiO2–Al2O3(36)-supported lactate salt behaves as well as the neutral silica-supported potassium lactate in the stabilization during the reaction may be attributed to the lower surface Brønsted acidity of SiO2–Al2O3(36) (at least not higher than the Brønsted acidity of aqueous 20% LA). Provided that a support with a higher acidity than that of aqueous 20% LA is used, the stabilization of the supported lactate salt will encounter a challenge. Apart from the effect of the surface acidity of catalyst systems, the effect of LA acidity on the stabilization of the supported lactate salt ought to be equally considered. The use of concentrated aqueous LA with a lower H0 (H0 = 1.4 for aqueous 100% LA) may render the supported lactate salt unstable under reaction conditions. The reaction may preferably choose dilute aqueous LA to assist in the catalysis of in situ generated lactate salt.
Table 4 summarizes the catalytic performance of the above-described nitrate salt-based systems at the initial stage in the vapor-phase dehydration of LA from the literature. The use of nitrate salts as the catalyst precursors provides for the easy formation of lactate salts acting as the catalytic active species because of the low boiling point of HNO3, which favors the shift of the chemical equilibria of nitrate salt displacement with LA toward lactate salts.21–23 From the comparative reaction data, it is admitted that to achieve a higher AA yield, an optimal reaction temperature should be chosen at 350 °C. It is deduced that the important factors affecting the catalytic performance, include the support acidity, support surface area, and nitrate salt loading. When a more acidic material is used as a support of nitrate salt, the obtained catalyst system fails to generate the stable supported lactate salt under reaction conditions and thus deactivates quickly, just like in the case of KNO3/S15.23 The use of either a neutral or less acidic support can remedy this issue, which is clearly embodied in the cases of KNO3/S15cal, KNO3/AS300, KNO3/SBA-15, KNO3/SiO2–Al2O3(36), and NaNO3/SiO2–Al2O3(36).22,23 These systems display stable catalytic performance in the long-term vapor-phase dehydration of LA to AA at 350 °C, the supported lactate salt being detected as the catalytically active species.22,23 Under the prerequisite of use of a neutral or less acidic support, the proper interaction between nitrate salt and the support becomes critically important in preventing the occurrence of lactate salt dehydration to acrylate salt since the unsupported lactate salt is prone to dehydrating to acrylate salt under the acidic condition of LA dehydration.23 The proper nitrate salt–support interaction is dependent on the choices of support surface area and nitrate salt loading. As long as high support surface area or low nitrate salt loading is adopted, the proper nitrate salt–support interaction can be met for achieving stable catalytic performance for the production of AA, as shown in the cases of KNO3/S15cal, KNO3/AS300, KNO3/SBA-15, KNO3/SiO2–Al2O3(36), and NaNO3/SiO2–Al2O3(36).22,23 The choice of either low support surface area or high nitrate salt loading may cause failure in the interaction. The comparison of the catalytic stabilities at 350 °C over NaNO3/SiO2(LSA) and over KNO3/S15cal or KNO3/AS300 shows that the inferiority in the catalytic stability over NaNO3/SiO2(LSA) lies in the low surface area of SiO2(LSA), which disables the proper NaNO3–SiO2(LSA) interaction to to make the in situ generated supported sodium lactate stable. Probably because of the high surface area of either S15cal or AS300, which enables the proper interaction between KNO3 and S15cal or AS300, either KNO3/S15cal or KNO3/AS300 leads to the good catalytic stability in the long-term vapor-phase dehydration of LA to AA at 350 °C.23
| Precatalyst | T (°C) | WHSVLA (h−1) | Conversion (%) | AA yield (%) | Selectivity (%) | Stability | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| AA | PA | 2,3-PD | AD | |||||||
| NaNO3/SiO2(LSA) | 300 | 1.1 | 59 | 12 | 20 | 3.7 | 41 | 16 | — | 21 |
| NaNO3/SiO2(LSA) | 350 | 1.1 | 99 | 16 | 16 | 8.0 | 12 | 22 | Poor | 21 |
| NaNO3/SiO2(LSA) | 300 | 1.1 | 34 | 5.6 | 16 | 2.4 | 35 | 12 | — | 46 |
| NaNO3/SiO2(93%)–Al2O3(7%) | 300 | 1.1 | 61 | 11 | 18 | 6.1 | 28 | 12 | — | 46 |
| NaNO3/SBA-15 | 360 | 5.2 | 91 | 38 | 42 | 1.6 | 27 | 12 | — | 65 |
| NaNO3/FS | 360 | 5.2 | 90 | 37 | 41 | 3.1 | 17 | 10 | — | 65 |
| KNO3 | 350 | 0.46 | 65 | 24 | 37 | 11 | 1.1 | 24 | Poor | 23 |
| KNO3/SiO2–Al2O3(36) | 350 | 1.8 | 67 | 37 | 55 | 4.9 | 11 | — | Good | 22 |
| NaNO3/SiO2–Al2O3(36) | 350 | 0.46 | 89 | 33 | 37 | 5.7 | 2.7 | — | Good | 22 |
| KNO3/S15 | 350 | 0.46 | 81 | 25 | 30 | 16 | 1.5 | — | Poor | 23 |
| KNO3/S15cal | 350 | 0.46 | 95 | 26 | 28 | 8.9 | 2.0 | 18 | Good | 23 |
| KNO3/AS300 | 350 | 0.46 | 91 | 30 | 33 | 5.0 | 4.3 | — | Good | 23 |
| KNO3/SBA-15 | 350 | 0.46 | 95 | 35 | 37 | 4.0 | 4.5 | 20 | Good | 23 |
HAPs
HAPs, i.e., calcium phosphates with apatite structure are well known as an important class of acid–base catalysts.1–3,67–72 HAPs have also widely been studied in the field of medicine for applications in bone integration, dental implants, and vectorization of medicines.73–75 HAPs have a stoichiometric formula of Ca10(PO4)6(OH)2 (Ca/P = 1.67 molar ratio). By varying the Ca/P molar ratio in the range of 1.50–1.67, nonstoichiometric HAPs would gain unusual surface acid–base properties. This salient feature is well embodied in acid–base catalysis for alcohol conversion.67–72 Furthermore, catalysis with HAPs has rapidly become a subject of interest for dehydration, oxidation, dehydrogenation, and heavy alcohol synthesis.1–3,72,76–80The earliest research on HAPs in the area of catalytic LA dehydration was reported as a preliminary test by Gunter et al. in 1994.34 Their HAP was obtained by the calcination of bovine teeth in air at 800 °C to remove organic matter. The resultant material had a surface area of 6 m2 g−1. Its MAS 31P NMR spectrum essentially gave a single peak, illustrating the homogeneity of the material At 320 °C, 0.5 MPa, and a WHSVLA of 0.80 h−1; the LA dehydration reaction gave an LA conversion of 13% with yields of AA, PA, 2,3-PD, and AD of 7.0, 0.89, 2.7, and 2.1%, respectively, from aqueous 34% LA over this natural HAP. Under the same reaction conditions, relatively stable product yields were achieved during 100 h of reaction on stream. The low yields of AA and 2,3-PD are probably due to the lower reaction temperature used. The long-term stable product yields with the natural HAP could foresee potential catalytic stability of synthetic HAPs toward LA dehydration. This work using the natural material as the precatalyst lays the foundation for the later application of synthetic HAPs in the vapor-phase dehydration of LA to AA.
In 2012, two patent publications reported the application of synthetic HAPs in the vapor-phase dehydration of LA to AA.81,82 Dongare et al. disclosed the synthesis of a series of HAPs and NaHAPs from Ca(NO3)2 and (NH4)2HPO4 by precipitation with varying pH and Ca/P molar ratio, and the process for conversion of LA to AA using these HAPs and NaHAPs.81 At 375 °C and a WHSVLA of 1.5 h−1 over HAPs or NaHAPs with Ca/P = 1.5–1.9 molar ratio, an LA conversion of 100% could be gained with a selectivity toward AA of 50–70% from aqueous 50% LA. They asserted some advantages of HAP systems: (1) the process provides for 100% conversion of LA, (2) the process is capable of converting high concentrations of LA, (3) the process leads to the minimal production of by-products such as AD, and (4) the catalyst is promoter-free. Onda et al. disclosed their results of AA production from LA dehydration using HAPs with varying Ca/P molar ratio.82 Their HAPs were synthesized from P2O5 and Ca(NO3)2 by hydrothermal treatment. Under the optimized reaction conditions (350 °C, aqueous 38% LA, and WHSVLA = 0.44 h−1), the LA conversion and the AA yield could reach 91 and 72%, respectively, over HAP with Ca/P = 1.67 molar ratio. After 60 h of reaction on stream under the same conditions, the AA yield decreased to 50%, indicating poor catalytic stability. From this case, the synthetic HAPs may have an issue of chemical stability in structure. HAPs must face the stabilization of in situ generated calcium lactate against calcium lactate dehydration to calcium acrylate followed by calcium acrylate polymerization, assuming that calcium lactate acts as the catalytic active species. To meet the needs of catalytic applications, the research might want to consider more factors preventing catalyst deactivation, e.g., acid and coke tolerance during LA dehydration. In such a sense, the rational design of new classes of HAPs is of critical importance.
In 2013, a paper of Ghantani et al. appeared, reporting a series of very stable HAP systems for the vapor-phase dehydration of LA to AA.83 Their HAPs were synthesized from Ca(NO3)2·4H2O and (NH4)2HPO4 by precipitation with a Ca/P molar ratio of 1.3–1.89 by varying the pH values of Ca(NO3)2·4H2O and (NH4)2HPO4 aqueous solutions. The HAP structure of the synthesized samples with different Ca/P molar ratios was confirmed by the XRD and IR analyses. The Ca/P molar ratio of 1.3 was found to be optimal to the HAP series utilized for the LA dehydration reaction. At 375 °C and a WHSVLA of 1.5 h−1, LA conversion of 100% and AA yield of 60% could be produced with an AD yield of 17% from aqueous 50% LA over HAP with Ca/P = 1.3 molar ratio. Under the reaction conditions, it was claimed that LA conversion of 100% remained and the selectivity toward AA declined by 5% only after 300 h of reaction on stream. The results exhibited unprecedented catalytic stability of heterogeneous catalyst systems toward LA dehydration to AA. In their IR experiment of reactivity of LA with HAP, two bands at 1755(s) and 1604(s) cm−1 appeared upon LA adsorption on an HAP wafer at room temperature and both of them rose in intensity at the expense of intensity of the LA band as the temperature was increased to 200 °C. The former can be due to lactides or PLA and the latter to calcium lactate generated from calcium phosphate displacement with LA. The IR observations implicated that H+ transfer is prone to taking place from LA to calcium phosphates with the generation of calcium lactate and that LA polymerization is rather competitive with calcium phosphate displacement with LA over HAP. The very stable surface calcium lactate is likely responsible for the excellent catalytic stability of the HAP system, similar to the cases with potassium or sodium lactate generated in situ on KNO3/S15cal, KNO3/AS300, KNO3/SBA-15, KNO3/SiO2–Al2O3(36), and NaNO3/SiO2–Al2O3(36).22,23 Ghantani et al. thus regarded the in situ generated surface calcium lactate as the reaction intermediate in the catalytic cycle. It seems that the use of the HAP disables the control of LA polymerization while enabling tolerating coke deposition.
The successful case of HAPs in the catalytic stability reported by Ghantani et al. provides a significant foundation for the stabilization of catalytic active components in HAPs systems toward LA dehydration to AA. It is envisioned that as catalyst precursors, HAPs could play two independent roles under LA dehydration conditions, i.e., (1) precatalysts of catalytic active components and (2) supports of catalytic active components. While the reaction proceeds, calcium phosphate displacement with LA to produce calcium lactate occurs only on the HAP surface, i.e., at the LA–HAP interface. The resultant calcium lactate as the catalytic active species may be comfortably situated in the original apatite crystal structure. The unreacted calcium phosphate in the HAP bulk phase may play the role of a support of calcium lactate. The supported calcium lactate thus generated in situ can be expected to possess unusual chemical stability against its leaching and its dehydration to calcium acrylate that causes catalyst deactivation under an acidic condition of LA dehydration.
A year later, Xu and coworkers published their research on the catalytic behavior of HAPs in the vapor-phase dehydration of LA to AA.84 Their HAPs were prepared from Ca(NO3)2 and (NH4)2HPO4 by precipitation with varying Ca/P molar ratio and calcination temperature. Consistent catalytic performance with that reported previously81–83 was shown in terms of LA conversion and selectivity toward AA. Under the optimized reaction conditions (360 °C, aqueous 36% LA, and WHSVLA = 2.1 h−1), an LA conversion of 70% was obtained with selectivities toward AA, PA, 2,3-PD, and AD of 71, 4, 1, and 16%, respectively, over HAP with Ca/P = 1.62 molar ratio. However, the catalytic stability of their HAP systems was noted to be unsatisfactory—the LA conversion dropped by 8% and the selectivity toward AA remained unchanged in the first 8 h of reaction over HAP with Ca/P = 1.62 molar ratio. In this work, the correlation between the catalytic performance and surface acidity and basicity was put forwarded in HAP catalysis for the dehydration of LA to AA. The AA formation rate showed a volcano-type dependence on the acidity/basicity ratio (Fig. 6), which suggested cooperative acid–base catalysis operating during the dehydration of LA to AA. A follow-up of the work was reported by this group in 2020 on a comparative study of dehydration of alkyl lactates and LA for AA production over HAPs.85 At 360 °C and a WHSVLA of 1.0 h−1, an LA conversion of 95% was produced with selectivities toward AA, PA, and AD of 53, 23, and 22%, respectively, from aqueous 36% LA over HAP with Ca/P = 1.60 molar ratio. In the first 8 h of reaction, the LA conversion decreased by 3.6% and the selectivity toward AA remained unchanged at steady state. The catalytic stability is thus assessed poor.
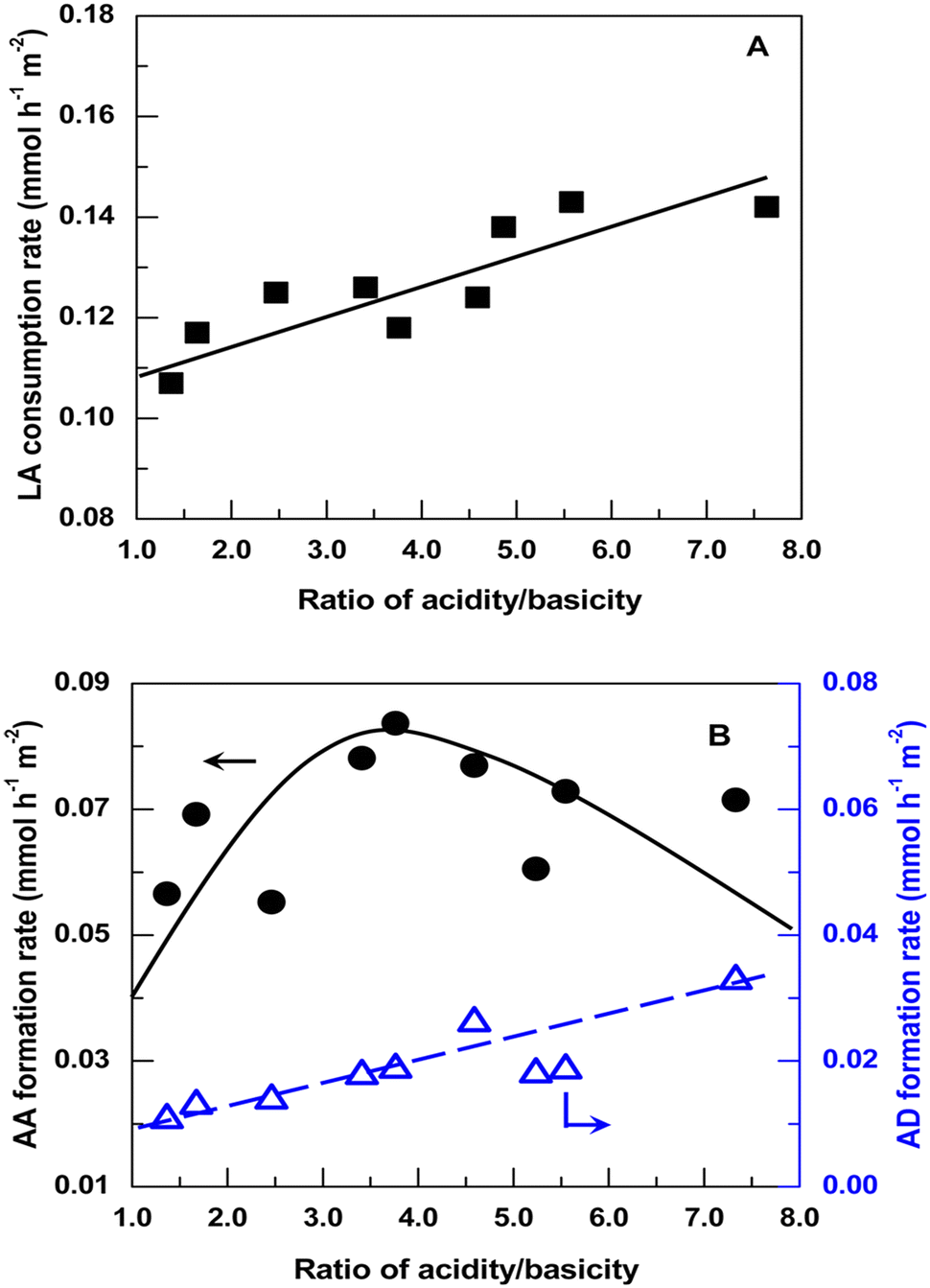 | ||
| Fig. 6 Dependences of specific catalytic rates of (A) LA consumption, and (B) AA and AD formation on the surface acidity/basicity ratio of HAPs. Reprinted with permission from ref. 84, copyright 2014 American Chemical Society. | ||
Matsuura et al. reported on various HAPs applied in AA production from vapor-phase LA dehydration.86,87 Their HAPs were synthesized from Ca(NO3)2·4H2O and H3PO4 by hydrothermal treatment.86,87 Their optimal nonstoichiometric HAP led to higher AA yield of 78% and lower AD yield of 6.0% from aqueous 38% LA at 350 °C and a WHSVLA of 0.44 h−1.86 Under the same reaction conditions, the initial AA yield of 82% declined by 12% after 50 h of reaction on stream, which is indicative of its fair catalytic stability. Furthermore, it was observed that amid the HAP systems studied, a higher acidity accelerates the formation of AD, and a higher basicity facilitates the production of unidentified byproducts, whereas moderate acidity and basicity favor the production of AA.87 This somewhat supports the suggestion by Xu and coworkers of the cooperative acid–base catalysis for the dehydration of LA to AA.84
From the work of Xu and coworkers and Matsuura et al., the moderate acidity and basicity is another important concept for understanding and controlling the catalytic behavior of HAP systems in the dehydration of LA to AA. Where the acidity is concerned, the H0 of a good performance HAP system is supposed to exceed that of LA such that the in situ generated calcium lactate could maintain a good chemical stability against its dehydration to calcium acrylate. Under a more acidic condition in which the H0 of an HAP itself is inferior to that of LA, the calcium lactate may be at risk to dehydrate to calcium acrylate followed by calcium acrylate polymerization, which leads to catalyst deactivation.23
Recently, Li et al. studied in detail the deactivation and regeneration of HAPs for use in the vapor-phase dehydration of LA to AA.88 Their HAPs were prepared from Ca(NO3)2·4H2O and H3PO4 by hydrothermal treatment with varying Ca/P molar ratio. An HAP system with Ca/P = 1.57 molar ratio displayed good catalytic stability during 140 h of reaction on stream at 350 °C and a WHSVLA of 0.46 h−1 with aqueous 40% LA—the LA conversion of 95% decreased by 5.3%, the selectivity toward AA of 74% declined by 12%, and the selectivity toward AD of 15% remained almost unchanged at the end of 140 h. From 140 to 250 h, the selectivity toward AD and lactides obviously increased with the concurrent marked decreases in the LA conversion and the selectivity toward AA. By N2 adsorption–desorption, XRD, 13C NMR, TGA, NH3-TPD, and CO2-TPD, they proposed a two-step coke-induced catalyst deactivation mechanism: (1) the formation of aliphatic coke by the deposition of low carbon products and (2) the formation of aromatic coke by the condensation of lactides. Although the formation of much coke during catalytic LA dehydration to AA is recognized by many groups, it does not seem that the formation of much coke significantly affects the catalytic activity and stability. This point is also reflected in this case where the HAP system performs well in catalytic stability for the production of AA and the control of AD during 140 h of reaction.88 The deposition of coke can be viewed as an unimportant factor causing catalyst deactivation in LA dehydration to AA.
Lastly, Wojcieszak et al. presented F−-substituted HAPs that enabled improving the production of AA while enabling reducing the formation of AD from LA dehydration.89 Their HAPs were prepared from Ca(NO3)2 and (NH4)2HPO4 by precipitation with varying Ca/P molar ratio. The original HAPs were modified by the substitution of surface OH− with F−, which was assumed to enable preventing the formation of HPO42− from PO43− in the presence of water in view of the IR study of propyne adsorption. They estimated that the restriction of formation of HPO42− could keep the formation of AD under control because HPO42− is presumably responsible for the LA decarbonylation pathway.89 Under the typical reaction conditions (375 °C, aqueous 58% LA and WHSVLA = 0.98 h−1), an F−-substituted HAP system with a substitution ratio of 2 (i.e., Ca-XAP-F4) produced increased AA yield by 24% and decreased AD yield by 26%, as compared with the reaction data over an F−-substituted HAP system with a substitution ratio of 0.5 (i.e., Ca-XAP-F-OH). The latter gave rise to increased AA yield by 41% and decreased AD yield by 15% as compared with the reaction data over a standard HAP system (i.e., Ca-HAP-S). The results significantly illustrate the effect of F− on the catalytic performance of HAP systems. From this study, it is preferably considered that the substitution of surface OH− with F− would lead to the easy formation of calcium lactate as the catalytic active species for LA dehydration to AA as well as the restriction of formation of HPO42− from PO43− on HAPs. During the catalytic reaction, CaF2 may readily displace with LA to calcium lactate with concomitant H+ transfer from LA to surface F−. The formation of HF, followed by its easy evaporation, definitely promotes the shift of the displacement chemical equilibrium toward calcium lactate, thus producing more calcium lactate on HAP. After the evolution of HF, how well the Ca-XAP-F4 system performs in a long-term LA dehydration reaction is unknown. Even if the formation of HPO42− can apparently be restricted by the presence of F−, the authentic restriction is questionable under reaction conditions. Under an acidic condition of LA dehydration, surface PO43− readily accepts an H+ from LA to give lactate and HPO42− based on the comparative acidities of LA (pKa = 3.86 at 25 °C) and H3PO3 (pKa1 = 2.15, pKa2 = 7.21, pKa3 = 12.3 at 25 °C),35 as discussed in the part for phosphate salts.
Table 5 summarizes the catalytic performance of the above-described HAP systems at the initial stage in the vapor-phase dehydration of LA from the literature. The use of HAPs is capable of controlling the selectivity toward AD and thus increasing the selectivity and stability for the production of AA. The advantage of HAPs is their nonstoichiometric chemical compositions with varying Ca/P molar ratio, which allows tuning the surface acid–base properties for the dehydration of LA to AA. The difference in the catalytic stability of the HAP systems reported by the different groups probably lies in the different HAP structural properties, which are affected by the sample preparation processes. The structural stability of HAPs may determine the catalytic stability of calcium lactates in situ generated as the catalytic active species. Not only the lactate salt is suggested by several groups as the reaction intermediate with phosphate salts as precatalysts but the calcium lactate is also verified by Ghantani et al. as the reaction intermediate with HAPs as the precatalysts. From the cases reported by Ghantani et al. and Li et al.,83,88 it is believed that high-quality HAPs possess a particular structural stability. The in situ generated calcium lactate may be well located in the parent apatite crystal structure. The unreacted calcium phosphate in an HAP system may play the role of support of calcium lactate. Such a supported calcium lactate catalyst is assumed to be very stable against its leaching and its dehydration to calcium acrylate, which causes catalyst deactivation under an acidic condition of LA dehydration. On the other hand, the moderate acidity and basicity of HAPs may provide a proper environment for the stabilization of the calcium lactate under LA dehydration conditions. Owing to the lower acidity of HAPs than that of LA, as indirectly revealed by the studies of Xu and coworkers and Matsuura et al.,84,87 calcium lactate dehydration may be prevented such that the supported calcium lactate could maintain its long-term catalytic stability. In addition, HAPs appear to have strong resistance to catalyst deactivation caused by coke formation.88
| Precatalyst | T (°C) | WHSVLA (h−1) | Conversion (%) | AA yield (%) | Selectivity (%) | Stability | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| AA | PA | 2,3-PD | AD | |||||||
| HAP | 320 | 0.80 | 13 | 7.0 | 54 | 6.8 | 21 | 16 | Good | 34 |
| HAP (Ca/P = 1.5 molar ratio) | 375 | 1.5 | 100 | 60 | 60 | — | — | 33 | — | 81 |
| NaHAP (Ca/P = 1.5 molar ratio) | 375 | 1.5 | 100 | 70 | 70 | — | — | 25 | — | 81 |
| HAP (Ca/P = 1.67 molar ratio) | 350 | 0.44 | 91 | 72 | 66 | — | — | — | Poor | 82 |
| HAP (Ca/P = 1.3 molar ratio) | 375 | 1.5 | 100 | 60 | 60 | — | — | 17 | Excellent | 83 |
| HAP (Ca/P = 1.62 molar ratio) | 360 | 2.1 | 70 | 50 | 71 | 4.0 | 1.0 | 16 | Poor | 84 |
| HAP (Ca/P = 1.60 molar ratio) | 360 | 1.0 | 95 | 50 | 53 | 23 | — | 22 | Poor | 85 |
| HAP (Ca/P = 1.55 molar ratio) | 350 | 0.44 | 90 | 78 | 87 | 0.67 | 0.44 | 6.7 | Fair | 86 |
| HAP (Ca/P = 1.57 molar ratio) | 350 | 0.46 | 95 | 70 | 74 | 2.0 | 1.0 | 17 | Good | 88 |
| Ca-HAP-S (Ca/P = 1.62 molar ratio) | 375 | 0.99 | 98 | 27 | 28 | 0.5 | — | 28 | — | 89 |
| Ca-XAP-F-OH (Ca/P = 1.66 molar ratio) | 375 | 0.99 | 96 | 38 | 40 | 4.3 | — | 24 | — | 89 |
| Ca-XAP-F4 (Ca/P = 1.73 molar ratio) | 375 | 0.99 | 93 | 47 | 50 | 2.4 | — | 18 | — | 89 |
Modified zeolites
Zeolites have long been applied in many catalytic reactions because of the salient solid acid–base properties and shape-selectivity function.90,91 Toward the dehydration of LA, modifying zeolites with alkali, alkaline-earth, and rare-earth elements is generally aimed at lowering the acidity (or acid strength) and enhancing the basicity (or base strength) of zeolites to meet the catalytic needs of high AA production.The first example of modified zeolites for use in the vapor-phase dehydration of LA to AA was communicated by Huang and coworkers in 2008, who acquired enhanced catalytic performance on NaY zeolites modified with rare-earth metals, including La, Ce, Sm, and Eu.92 Their modified NaY systems were prepared via the impregnation of NaY (Si/Al = 4.5 molar ratio) with aqueous nitrate salt solution. La(NO3)3/NaY led to better catalytic performance. At 350 °C and a WHSVLA of 1.2 h−1, LA conversion of 100% and selectivity toward AA of 56% were obtained from aqueous 38% LA over La(NO3)3/NaY with 2% La loading. They ascribed the higher selectivity toward AA to decreased acid density, increased surface area, and pore size on the modified NaY. Afterward, the work of modification of NaY was extended to alkali and alkaline-earth metals by this group.32,93,94 A series of K-modified NaY systems with Si/Al = 2.5 molar ratio such as KNO3/NaY, K2SO4/NaY, KHPO4/NaY, KOH/NaY, KCl/NaY, KBr/NaY, and KI/NaY was prepared by impregnation and tested in the vapor-phase dehydration of LA to AA at 325 °C.91 These systems displayed obviously increased AA yield compared to that over NaY. Typically, KI/NaY gave rise to LA conversion of 98% and selectivity toward AA of 68% from aqueous 29% LA at 325 °C and a WHSVLA of 0.93 h−1. The increased catalytic performance was explained by an electronic effect of K.90,91 The influence of the catalyst precursor on the catalytic performance is reflected in the effect of the surface base strength. The AA yield over these systems increased in the order KI/NaY > KBr/NaY > KCl/NaY > KF/NaY > NaY, which corresponds to the order of increase in the surface base strength measured by CO2-TPD except for NaY.32 Meanwhile, the four supported sodium halide systems had about the same acid strength based on the NH3-TPD study.32 This seems to account for that properly increasing the base strength by use of different catalyst precursors favoring the production of AA. However, the catalytic stability of these alkali metal-modified NaY systems is unsatisfactory.32 Under the same reaction conditions over KI/NaY, the LA conversion of 98% slightly decreased, whereas the selectivity toward AA of 68% dropped to 42% in the first 6 h. Among the alkaline-earth metals, Mg, Ca, Sr, and Ba, Ba was found to be most favorable for the production of AA.95 Using Ba(NO3)2/NaY (2% Ba loading, Si/Al = 4.5 molar ratio) prepared by impregnation, the LA conversion of 100% and selectivity toward AA of 45% were produced from aqueous 38% LA at 325 °C and a WHSVLA of 1.2 h−1. By the studies of XRD, NH3-TPD, and CO2-TPD, they speculated that medium base sites are mainly responsible for the formation of AA.94
From the above cases studied by Huang and coworkers, the acid strength of NaY may affect the catalytic stability of the modified NaY systems. The use of NaY with low Si/Al molar ratio brings about a high acid strength.95,96 Although the modification of NaY with alkali and/or alkaline-earth metals reduces the acid strength, as shown by the NH3-TPD data,90–92 the actual acid strength of the modified NaY can increase as the reaction proceeds with the formation of lactate salt as the catalytic active species or the neutralization of base by LA. If the H0 of a catalyst system is lower than that of LA, the in situ generated lactate salt may dehydrate to acrylate salt, which subsequently polymerizes under a higher acid strength, which may cause catalyst deactivation.23 As such, the observed poor catalytic stability is probably due to the high acid strength of these modified NaY systems. To improve the catalytic stability, it is advisable to tune the H0 of the catalyst systems to above the H0 of LA (H0 ≥ 1.4 for an aqueous LA solution). In doing so, it is preferable to reduce the acid strength of the parent NaY by increasing the Si/Al molar ratio.
For the purpose of suppressing decarbonylation of LA to AD in favor of selective LA dehydration to AA, the alkalization of NaY was carried out to reduce the acidity and increase the basicity by Lari et al.97 The parent NaY was partially dealuminated, followed by treatment with NaOH. The resultant alkalized NaY showed reduced acidity by 27Al and 23Na MAS NMRs and increased basicity by CO2-TPR. As a result of the alkalization of NaY, the modified NaY systems displayed significantly improved catalytic activity, selectivity, and stability for the production of AA compared to the unmodified (NaY) or dealuminated NaY (NaY-DA) system. At 350 °C and a high WHSVLA of 5.8 h−1, the typical modified NaY system NaY-DA-0.15 gave rise to increased LA conversion from 84 to 95%, increased selectivity toward AA from 46 to 72%, and decreased selectivity toward AD from 38 to 20%, respectively, from aqueous 10% LA. The comparative results significantly indicate that the increase in the basicity of NaY plays a critical role in the improvement of the initial catalytic performance. After 6 h of reaction, however, the LA conversion dropped by 32% and the selectivity toward AA remained unchanged, implying that the catalytic stability is far from being satisfactory. Although the Si/Al molar ratio of the parent NaY is increased from 3.6 to 5.7 after the dealumination, the acid strength of NaY-DA-0.15 is believed to be still rather high, which is not supposed to be good enough to prevent sodium lactate dehydration to sodium acrylate from taking place during LA dehydration to AA. In addition, as the surface sodium salt or base displaces with LA to produce sodium lactate and conjugate acid, the basicity of NaY-DA-0.15 is deemed to gradually fall till vanishing. As such, the alkalized NaY does not appear to act significantly at steady state.
Later on, another case of alkalization of NaY was reported by Zhang et al. who studied K- and Ca-comodified NaY for the vapor-phase dehydration of LA to AA.98 The parent NaY with Si/Al = 5.1 molar ratio was impregnated or etched with KOH, followed by further impregnation with Ca(NO3)2. The resultant alkalized NaY systems KOH–Ca–NaY and KOH–Ca–ENaY had both reduced acid and reduced base strengths with respect to the unmodified NaY based on the NH3-TPD and CO2-TPD studies. The K- and Ca-comodified NaY systems exhibited remarkably improved catalytic selectivity toward AA with the concomitant greatly decreased selectivity toward AD. At 350 °C and a WHSVLA of 0.46 h−1, KOH–Ca–NaY resulted in LA conversion of 100% and selectivities toward AA, PA, 2,3-PD, and AD of 84, 2.7, 0.4, and 1.5%, respectively, from aqueous 20% LA. Ca2+ was suggested to play an important role in shifting the dynamic chemical equilibria in favor of the dehydration reaction and thus ensuring high selectivity toward AA at the initial stage. After 55 h of reaction on stream under the same conditions, LA conversion remained unchanged while the selectivity toward AA dropped by 35%, evidently indicating fast catalyst deactivation. The explanation for the catalytic instability in this case can be made in the same manner as that for the cases reported by Huang and coworkers and Lari et al.32,92–94,97 Although the alkalization diminishes the acid strength of the parent NaY, the actual acid strength of the alkalized NaY system can be restored to that of the parent NaY after the salt and base displacements with LA to lactate salts. With such a high acid strength, the in situ generated lactate salts as the catalytic active species probably readily dehydrate to acrylate salts that subsequently polymerize.
Other than the above research on the modification of Y zeolites, the work on alkali metal-modified ZSM-5 in this area was published first by Yuan et al.99 They studied the effects of alkali metal in modified ZSM-5 and surface acid–base properties on the catalytic performance in the vapor-phase dehydration of LA to AA.99 In the catalyst preparation, commercial NaZSM-5 (Si/Al = 75 molar ratio, surface area of 287 m2 g−1) was precalcined at 550 °C and ion exchanged with aqueous nitrate salt solution. Amid the alkali metal-containing ZSM-5 systems studied, the KZSM-5 system was more effective for the production of AA. The catalytic performance of the alkali metal-modified ZSM-5 systems in terms of AA yield increased in the order as KZSM-5 > CsZSM-5 > RbZSM-5 > NaZSM-5 > LiZSM-5. At 365 °C and a WHSVLA of 0.39 h−1, KZSM-5 resulted in LA conversion of 96% and selectivities toward AA, PA, 2,3-PD, and AD of 76, 0.91, 0.85, and 14%, respectively, from aqueous 40% LA. Under the same reaction conditions, the LA conversion decreased by 8.2% and the selectivity toward AA remained stable after 60 h of reaction on stream, as shown in Fig. 7, which is assessed as good catalytic stability. By NH3-TPD and CO2-TPD, KZSM-5 and CsZSM-5 were found to have lower acid and lower base strengths than NaZSM-5. It was therefore suggested that the synergistic effect of weaker acid and weaker base sites of ZSM-5 leads to the improvement of catalytic performance in LA dehydration to AA. In this work, a lower catalyst acid strength may be secured because of the choice of the higher Si/Al molar ratio for the parent ZSM-5. Under this lower acid strength, the in situ generated lactate salts as the true catalyst may better withstand dehydration to acrylate salts and thus may maintain the good catalytic stability for the AA production.
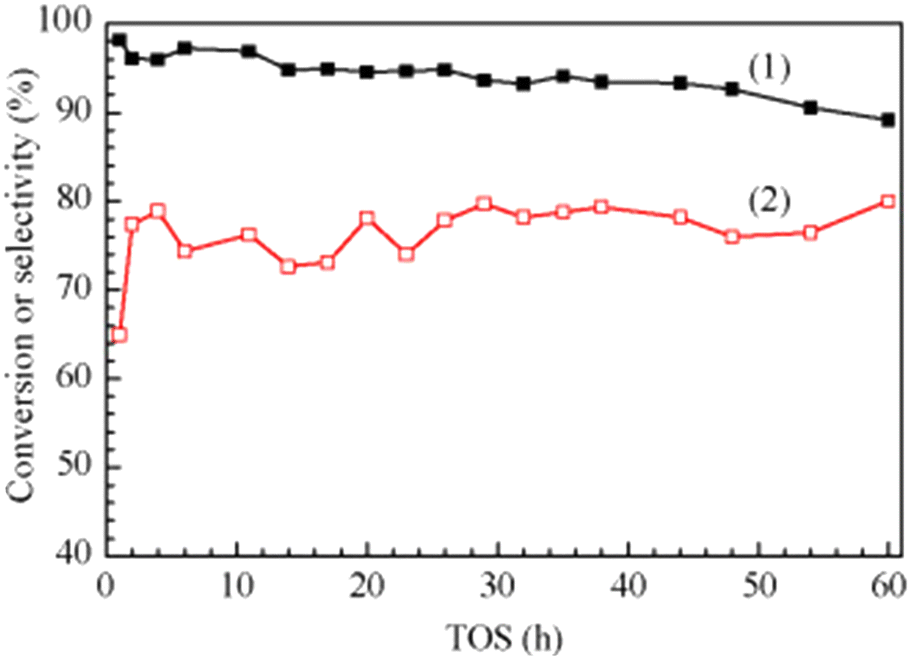 | ||
| Fig. 7 Stability test in the vapor-phase dehydration of LA at 365 °C and a WHSVLA of 0.34 h−1 over KZSM-5 (Si/Al = 75 molar ratio). (1) LA conversion and (2) selectivity toward AA. Reprinted with permission from ref. 99, copyright 2015 Elsevier Inc. | ||
More comprehensive work on alkali metal-modified zeolites for use in the vapor-phase dehydration of LA to AA was published by Xu and coworkers.100–103 The zeolite types studied included β, ZSM-5, ZSM-11, ZSM-5/ZSM-11, ZSM-22, ZSM-35, and MCM-22. These zeolites were modified through alkali cation exchange with K+, Rb+, and Cs+.100–102 In the process of alkali cation exchange, a commercial or as-synthesized zeolite was precalcined at 420 °C to remove any organic residues. Then, the precalcined zeolite was converted to its sodium form (NaZ) by ion exchange with aqueous NaNO3 solution. Finally, the NaZ was ion exchanged with aqueous alkali nitrate solution. The alkali cation exchange degree was varied by adjusting the concentration of aqueous alkali nitrate. The reaction kinetics and the relationship of catalytic performance with zeolite type, Si/Al molar ratio, alkali cation exchange degree, and surface acid–base properties were investigated. Amid the various K+-exchanged zeolites (KxNa1−xZ_Si/Al, x = 0.90–0.98) with similar K/Al molar ratios, the KxNa1−xβ and KxNa1−xZSM-5 systems (x = 0.90–0.98) were more efficient for the production of AA.102 Given a zeolite type, the catalytic performance was shown to change with varying Si/Al molar ratio and alkali cation exchange degree.100–102 Lowering the Si/Al molar ratio increased the catalytic selectivity toward AA and the AA yield (at Si/Al ≥ 27 molar ratio for KNaβ, at Si/Al ≥ 22 molar ratio for KNaZSM-5).102 Increasing the alkali cation exchange degree increased the catalytic selectivity toward AA and AA yield (at K/Na ≤ 16 molar ratio for KNaβ_20, at Rb/Na ≤ 19 molar ratio for RbNaβ_20, at Cs/Na ≤ 4.3 molar ratio for CsNaβ_20, and at K/Na ≤ 8.1 molar ratio for KNaZSM-5_27).100–102 Meanwhile, the lowering of the Si/Al molar ratio was accompanied by the enhancement in the surface acid strength and the fall in the surface base strength,102 and the increase in the alkali cation exchange degree led to a fall in the surface acid strength and an enhancement in the surface base strength.100–102 A balance between the surface acidity and basicity was postulated to elucidate the correlation between the catalytic performance in the vapor-phase dehydration of LA to AA and the catalyst acid–base properties (Fig. 8).100–102 By varying the Si/Al molar ratio, the AA yield was found to increase with increasing acidity/basicity ratio (up to 47 for KNaβ, up to 75 for KNaZSM-5). By varying the alkali cation exchange degree, the AA yield was observed to increase with decreasing acidity/basicity ratio (up to 13 for KNaβ_20, up to 0.29 for RbNaβ_20, up to 0.13 for CsNaβ_20, and up to 156 for KNaZSM-5_27).100–102 In support of the cooperative acid–base bifunctional catalysis for the vapor-phase dehydration of LA to AA, the additive effects of acidic (CO2, acetic acid, and trifluoroacetic acid) and basic (NH3) molecules on the catalytic performance of the K0.89Na0.11ZSM-5_27 system were studied.103 While the addition of either CO2 or acetic acid showed no effect on the catalytic performance, the addition of either NH3 or trifluoroacetic acid caused a significant decline in both the catalytic activity and selectivity for AA production due to the poisoning of the surface acid sites by NH3 or base sites by trifluoroacetic acid. The results further suggested that both acidic and basic sites at the catalyst surface are involved in catalyzing the LA dehydration reaction. The K0.97Na0.03ZSM-5_14 system showed better catalytic performance.102 At 360 °C and a WHSVLA of 2.1 h−1, the reaction produced an LA conversion of 96% with selectivities toward AA, PA, 2,3-PD, and AD of 81, 0, 0, and 17%, respectively, from aqueous 36% LA. Under the same reaction conditions, the LA conversion and the selectivity toward AA fell by 20 and 24%, respectively, whereas the selectivity toward AD remained unchanged after 80 h of reaction on stream, as shown in Fig. 9. This case definitely shows good catalytic stability.
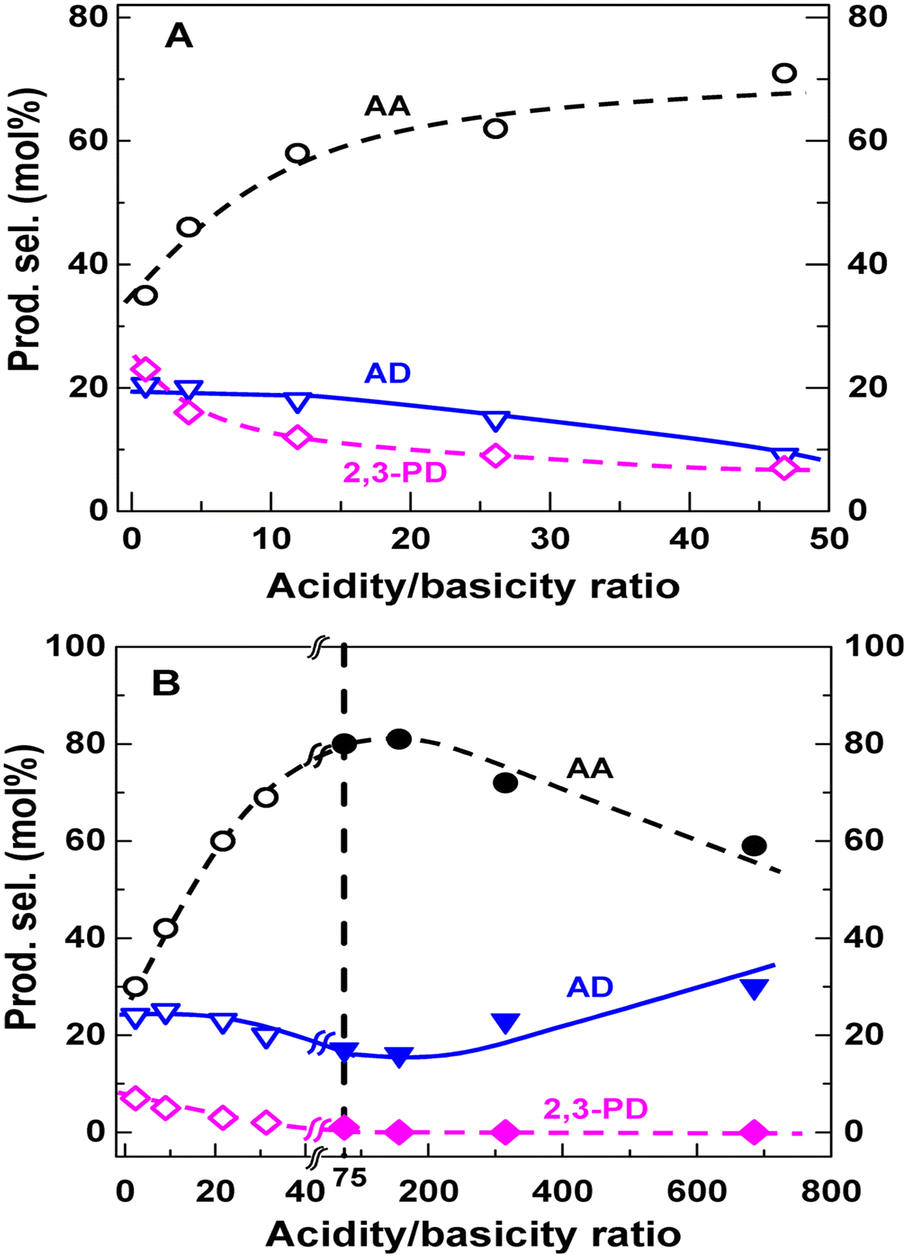 | ||
| Fig. 8 Correlations between the selectivity toward (AA (○,●), AD (▽,▼), and 2,3-PD (◇,◆)) at TOS = 9–10 h and the surface acidity/basicity ratio of (A) K0.90+mNa0.10−mβ_y of y = 22–111, and (B) K0.90+mNa0.10−mZSM-5_y of y = 36–75 (open symbols) and KxNa1−xZSM-5_27 of x = 0–0.97 (solid symbols). Reprinted with permission from ref. 102, copyright 2017 American Chemical Society. | ||
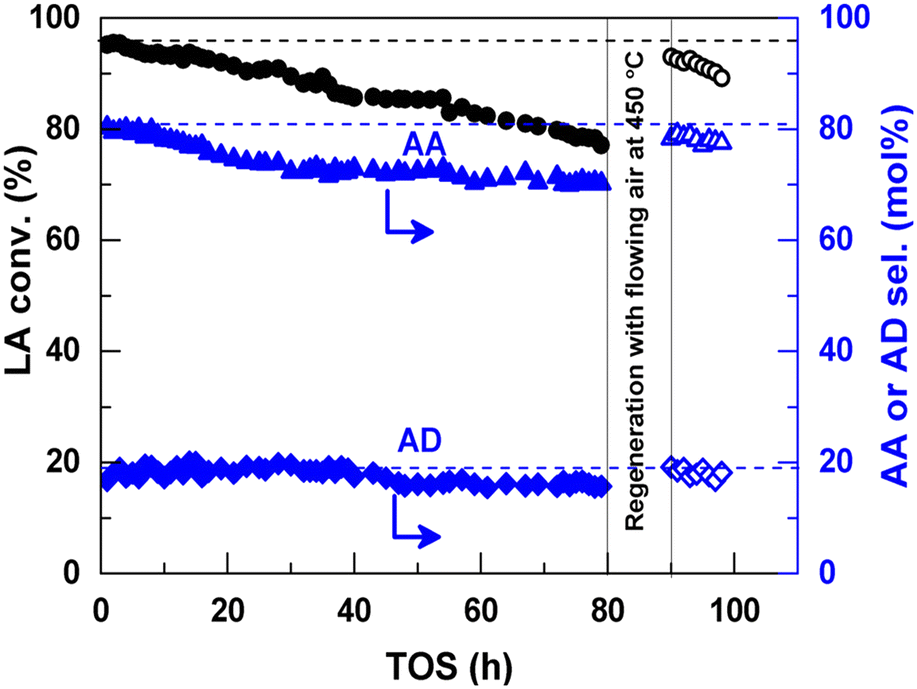 | ||
| Fig. 9 Stability and regeneration performance by the time course toward LA conversion (●,○), selectivity toward AA (▲,△), and selectivity toward AD (◇,◆) at 360 °C over K0.97Na0.03ZSM-5_27. The precatalyst worked for 80 h and then switched to an air flow, and the temperature was increased to 450 °C for 10 h for catalyst regeneration, followed by switching back to the reaction feed at 360 °C for further reaction. Reprinted with permission from ref. 102, copyright 2017 American Chemical Society. | ||
Interestingly and surprisingly, the K0.97Na0.03ZSM-5_14 system, prepared from the parent ZSM-5_14 with Si/Al = 14 molar ratio corresponding to a high acid strength (H0 < 3.0),104 displays the high catalytic activity and good catalytic stability for the production of AA.102 The catalytic stability is inconsistent with that with the KZSM-5 reported by Yuan et al.99 This implies that the in situ generated lactate salts as the true catalysts in the K0.97Na0.03ZSM-5_14 system could stabilize against lactate salt dehydration to acrylate salts followed by acrylate salt polymerization under high acid strength. During the long-term LA dehydration, the actual acid strength of the catalyst system is likewise deemed to be restored to the that of the parent ZSM-5_14, although the modification with K reduces the acid strength, as shown by the NH3-TPD data.100 The difference where the K0.97Na0.03ZSM-5_14 is made from the KZSM-5 and the K-modified NaY in the catalyst stabilization deserves in-depth study.
More recently, Czekaj and coworkers reported on the modification of synthetic β and natural clinoptilolite (CLI) zeolites with transition metals, including Fe, Cu, Co, and Sn toward the vapor-phase dehydration of LA to AA.105,106 At first, the influence of the preparative method of modified zeolites on the catalytic performance was examined.105 Feβ, Cuβ, and Coβ were prepared from Naβ with Si/Al = 13–38 molar ratio using both the ion exchange and sonication methods. Under the typical reaction conditions (370 °C, aqueous 40% LA, and WHSVLA = 3.5 h−1), Naβ led to LA conversion of 99% and selectivities toward AA, PA, 2,3-PD, and AD of 0, 0, 14, and 40%, respectively, which signifies that the parent Naβ system is active almost only for LA decarbonylation to AD. When using ion exchange, Feβ resulted in an AD yield of 100%, Cuβ gave rise to LA conversion of 100%, and selectivities toward AA, PA, 2,3-PD, and AD of 0, 0, 15, and 79%, respectively, and Coβ led to LA conversion of 100% and selectivities toward AA, PA, 2,3-PD, and AD of 13, 0, 38, and 0%, respectively. When using sonication, Feβ(s) led to the complete conversion of LA to PA, Cuβ(s) gave rise to LA conversion of 100%, and selectivities toward AA, PA, 2,3-PD, and AD of 62, 8.9, 0, and 0%, respectively, and Coβ(s) resulted in LA conversion of 99% and selectivities toward AA, PA, 2,3-PD, and AD of 35, 28, 20, and 0%, respectively. For the first time, this contribution communicated an effective transition metal-based catalyst system for the dehydration of LA to AA.105 The two preparative methods led to a distinct difference in the reaction outcomes. Ion exchange favors LA decarbonylation to AD or LA condensation to 2,3-PD, whereas sonication facilitates LA dehydration to AA or PA. The reason why the different reaction routes arise by the different preparative methods is unclear. The influence of the different preparative methods on the structure and composition of β was not reported.105
Next, the effect of zeolite hierarchization on the catalytic performance was studied by the ion exchange method.106 The comparative reaction results indicated that LA decarbonylation and LA condensation dominate using Naβ (Si/Al = 38 molar ratio), whereas LA dehydration dominates using Naβ(h) (after hierarchization). At 370 °C and a WHSVLA of 3.5 h−1, Naβ(h) led to LA conversion of 99% and selectivities toward AA, PA, 2,3-PD, and AD of 17, 30, 19, and 0%, respectively, from aqueous 40% LA.
The marked discrepancy in the reaction results after hierarchization was presumably attributed to partial degradation of the zeolite structure incurred by hierarchization, which facilitates the access of the reactant to previously inaccessible zeolite channels in which Na+ is located as the active site for LA dehydration, as depicted in Scheme 4.106 Regardless of hierarchization, the catalytic activity over β or CLI-modified with the transition metals for LA dehydration is inferior. The Snβ(h) system is most active for LA dehydration among all these transition metal-modified β and CLI systems. At 370 °C and a WHSVLA of 3.5 h−1 Snβ(h) led to LA conversion of 99% and selectivities toward AA, PA, 2,3-PD, and AD of 15, 19, 31, and 30%, respectively, from aqueous 40% LA. Without hierarchization, all the β systems modified with the transition metals mainly led to increased catalytic activity for LA decarbonylation; SnCLI or FeCLI produced increased catalytic activity for LA dehydration to PA, while CuCLI or CoCLI resulted in increased catalytic activity for LA dehydration to AA. After hierarchization, all the β(h) systems modified with the transition metals primarily resulted in improved catalytic activity for LA decarbonylation except that Feβ(h) produced increased catalytic activity for LA dehydration to PA; SnCLI(h) or FeCLI(h) gave rise to improved catalytic activity for LA dehydration to PA and for LA condensation. However, these reaction data were not interpreted due to the lack of coherent analysis on the structures and compositions of the β and CLI systems studied.106
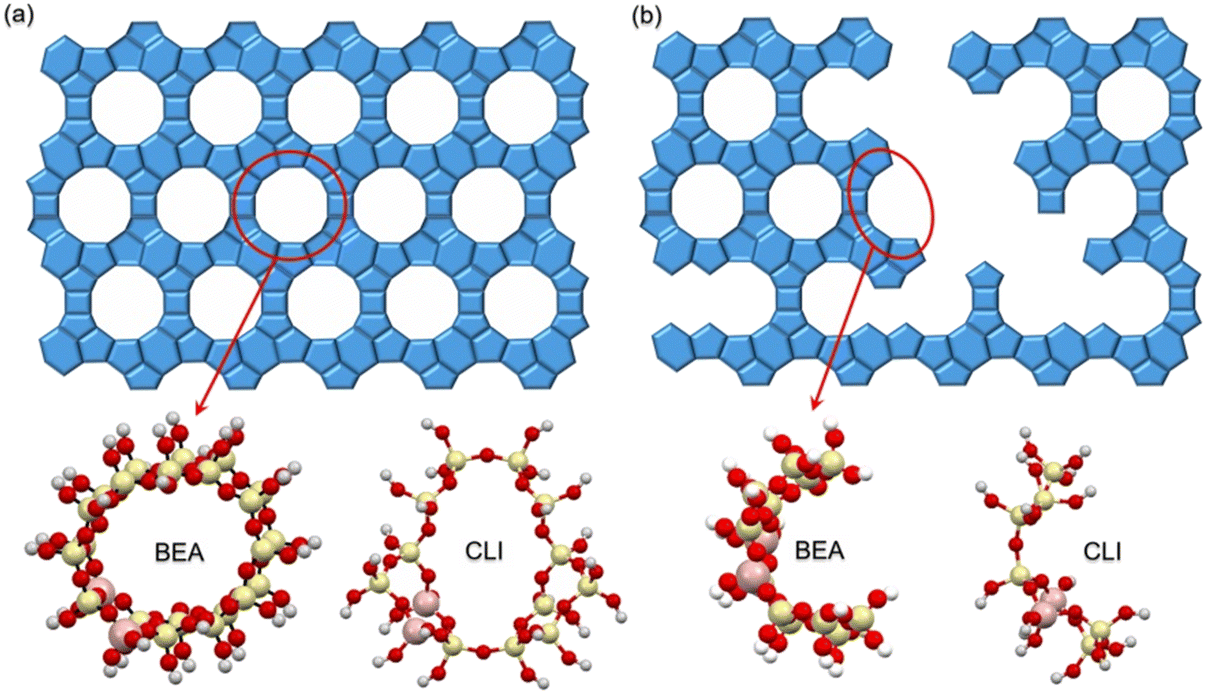 | ||
| Scheme 4 Clusters used for BEA and CLI: (a) ideal structure and (b) hierarchical structure. Reprinted with permission from ref. 106, copyright 2022 Elsevier Inc. | ||
Table 6 summarizes the catalytic performance of the above-described modified zeolite systems at the initial stage in the vapor-phase dehydration of LA from the literature. According to the catalytic screening of commercial zeolite systems in the sodium form toward this reaction at 350 °C by Lari et al. (Fig. 10),97 the magnitude of catalytic performance in terms of AA yield at the initial stage follows the order FAU-3 (Si/Al = 3 molar ratio) > MOR-10 > LTL-2.9 > MFI-5-15 > β-12.5, while the magnitude of AD yield has the order MFI-5-15 > MOR-10 > β-12.5 > FAU-3 > LTL-2.9. Typically, NaY-3 gives rise to AA and AD yields of 54 and 26%, respectively, and ZSM-5-15 leads to AA and AD yields of 4.0 and 96%, respectively. With NaY, the selectivity toward AA decreases with increasing Si/Al molar ratio, whereas the selectivity toward AD follows a reverse trend. With ZSM-5, the selectivity toward AA is very low with respect to the selectivity toward AD irrespective of the Si/Al molar ratio. Probably in view of the better catalytic activity for the production of AA, NaY zeolites, especially NaY-3, has been selected and widely investigated for the dehydration of LA to AA.
| Precatalyst | T (°C) | WHSVLA (h−1) | Conversion (%) | AA yield (%) | Selectivity (%) | Stability | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| AA | PA | 2,3-PD | AD | |||||||
| NaY (Si/Al = 4.5 molar ratio) | 350 | 1.2 | 100 | 35 | 35 | 1.6 | — | 16 | — | 92 |
| La(NO3)3/NaY (2% La, Si/Al = 4.5 molar ratio) | 350 | 1.2 | 100 | 56 | 56 | 1.6 | — | 12 | — | 92 |
| NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 96 | 14 | 15 | — | — | 11 | Poor | 93 |
| 3.5K/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 99 | 41 | 41 | 3.5 | 11 | 1.4 | Poor | 93 |
| NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 96 | 35 | 36 | — | 8.0 | 22 | — | 32 |
| KNO3/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 98 | 57 | 58 | 2.6 | 10 | 6.8 | Poor | 32 |
| K2SO4/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 95 | 49 | 52 | — | — | 20 | — | 32 |
| K2HPO4/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 95 | 48 | 50 | — | 6.5 | 6.0 | — | 32 |
| K2C2O4/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 94 | 46 | 49 | — | 6.5 | 15 | — | 32 |
| K2CO3/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 95 | 45 | 47 | — | 4.9 | 15 | — | 32 |
| KOH/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 96 | 42 | 44 | — | — | 20 | — | 32 |
| KI/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 98 | 67 | 68 | — | 7.9 | — | Poor | 32 |
| KBr/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 97 | 58 | 60 | — | 8.1 | 9.2 | Poor | 32 |
| KCl/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 97 | 52 | 54 | 3.9 | 10 | 10 | Poor | 32 |
| KF/NaY (Si/Al = 2.5 molar ratio) | 325 | 0.93 | 91 | 36 | 40 | — | — | 17 | Poor | 32 |
| NaY (Si/Al = 4.5 molar ratio) | 325 | 1.2 | 99 | 22 | 22 | 1.4 | 0 | 23 | — | 94 |
| Mg(NO3)2/NaY (Si/Al = 4.5 molar ratio) | 325 | 1.2 | 99 | 12 | 12 | 0 | 0 | 12 | — | 94 |
| Ca(NO3)2/NaY (Si/Al = 4.5 molar ratio) | 325 | 1.2 | 100 | 31 | 31 | 0 | 0 | 19 | — | 94 |
| Sr(NO3)2/NaY (Si/Al = 4.5 molar ratio) | 325 | 1.2 | 100 | 44 | 44 | 0.8 | 0 | 14 | — | 94 |
| Ba(NO3)2/NaY (Si/Al = 4.5 molar ratio) | 325 | 1.2 | 100 | 45 | 45 | 1.0 | 4.2 | 26 | — | 94 |
| NaY (Si/Al = 3 molar ratio) | 350 | 6.1 | 96 | 44 | 46 | — | — | 36 | Poor | 97 |
| NaY-DA (Si/Al = 3 molar ratio) | 350 | 6.1 | 99 | 71 | 72 | — | — | 20 | Poor | 97 |
| NaY-DA-0.15 (Si/Al = 3 molar ratio) | 350 | 6.1 | 95 | 68 | 72 | — | — | 20 | Poor | 97 |
| NaY (Si/Al = 2.6 molar ratio) | 350 | 0.46 | 89 | 56 | 63 | 4.3 | 0.10 | 15 | Poor | 98 |
| KOH–NaY (Si/Al = 2.6 molar ratio) | 350 | 0.46 | 78 | 34 | 43 | 1.9 | 0.60 | 5.6 | Poor | 98 |
| (Si/Al = 2.6 molar ratio) | 350 | 0.46 | 68 | 48 | 70 | 2.6 | 0.20 | 6.7 | Poor | 98 |
| KOH–Ca–NaY (Si/Al = 2.6 molar ratio) | 350 | 0.46 | 100 | 84 | 84 | 2.7 | 0.40 | 1.5 | Poor | 98 |
| KOH–Ca–ENaY (Si/Al = 2.6 molar ratio) | 350 | 0.46 | 100 | 83 | 83 | 2.3 | 1.0 | 2.8 | Poor | 98 |
| NaZSM-5 (Si/Al = 75 molar ratio) | 350 | 0.39 | 94 | 49 | 52 | 0.82 | 0.45 | 17 | — | 99 |
| KZSM-5 (Si/Al = 75 molar ratio) | 350 | 0.39 | 91 | 64 | 70 | 0.64 | 0.66 | 15 | Good | 99 |
| LiZSM-5 (Si/Al = 75 molar ratio) | 350 | 0.39 | 98 | 37 | 38 | 0.56 | 0.74 | 32 | — | 99 |
| RbZSM-5 (Si/Al = 75 molar ratio) | 350 | 0.39 | 92 | 52 | 56 | 1.9 | 3.4 | 15 | — | 99 |
| CsZSM-5 (Si/Al = 75 molar ratio) | 350 | 0.39 | 85 | 53 | 62 | 0.82 | 0.45 | 15 | — | 99 |
| Naβ (Si/Al = 20 molar ratio) | 360 | 2.1 | 100 | 26 | 26 | 1.0 | 3.0 | 32 | — | 100 |
| K0.94Na0.06β (Si/Al = 20 molar ratio) | 360 | 2.1 | 98 | 55 | 56 | 2.0 | 5.0 | 23 | Poor | 100 |
| Kβ (Si/Al = 20 molar ratio) | 360 | 2.1 | 100 | 41 | 41 | 2.0 | 3.0 | 21 | — | 100 |
| Rb0.95Na0.05β (Si/Al = 20 molar ratio) | 360 | 2.1 | 96 | 66 | 69 | 2.0 | 12 | 14 | — | 101 |
| Rbβ (Si/Al = 21 molar ratio) | 360 | 2.1 | 95 | 53 | 56 | 4.0 | 22 | 8.0 | — | 101 |
| Cs0.90Na0.10β (Si/Al = 20 molar ratio) | 360 | 2.1 | 98 | 67 | 69 | 3.0 | 16 | 10 | — | 101 |
| Csβ (Si/Al = 22 molar ratio) | 360 | 2.1 | 86 | 44 | 52 | 6.0 | 25 | 7.0 | — | 101 |
| NaZSM-5 (Si/Al = 14 molar ratio) | 360 | 2.1 | 99 | 58 | 59 | — | — | — | — | 102 |
| K0.97Na0.03ZSM-5 (Si/Al = 14 molar ratio) | 360 | 2.1 | 96 | 78 | 81 | 0 | 0 | 17 | Good | 102 |
| Naβ (Si/Al = 13–38 molar ratio) | 370 | 3.5 | 99 | 0 | 0 | 0 | 14 | 40 | — | 105 |
| Feβ(s) (Si/Al = 13–38 molar ratio) | 370 | 3.5 | 100 | 0 | 0 | 100 | 0 | 0 | — | 105 |
| Cuβ(s) (Si/Al = 13–38 molar ratio) | 370 | 3.5 | 100 | 62 | 62 | 8.9 | 0 | 0 | — | 105 |
| Coβ(s) (Si/Al = 13–38 molar ratio) | 370 | 3.5 | 99 | 35 | 28 | 8.9 | 20 | 0 | — | 105 |
| Naβ(h) (Si/Al = 38 molar ratio) | 370 | 3.5 | 99 | 17 | 17 | 30 | 19 | 0 | — | 106 |
| Snβ(h) (Si/Al = 38 molar ratio) | 370 | 3.5 | 99 | 15 | 15 | 19 | 31 | 30 | — | 106 |
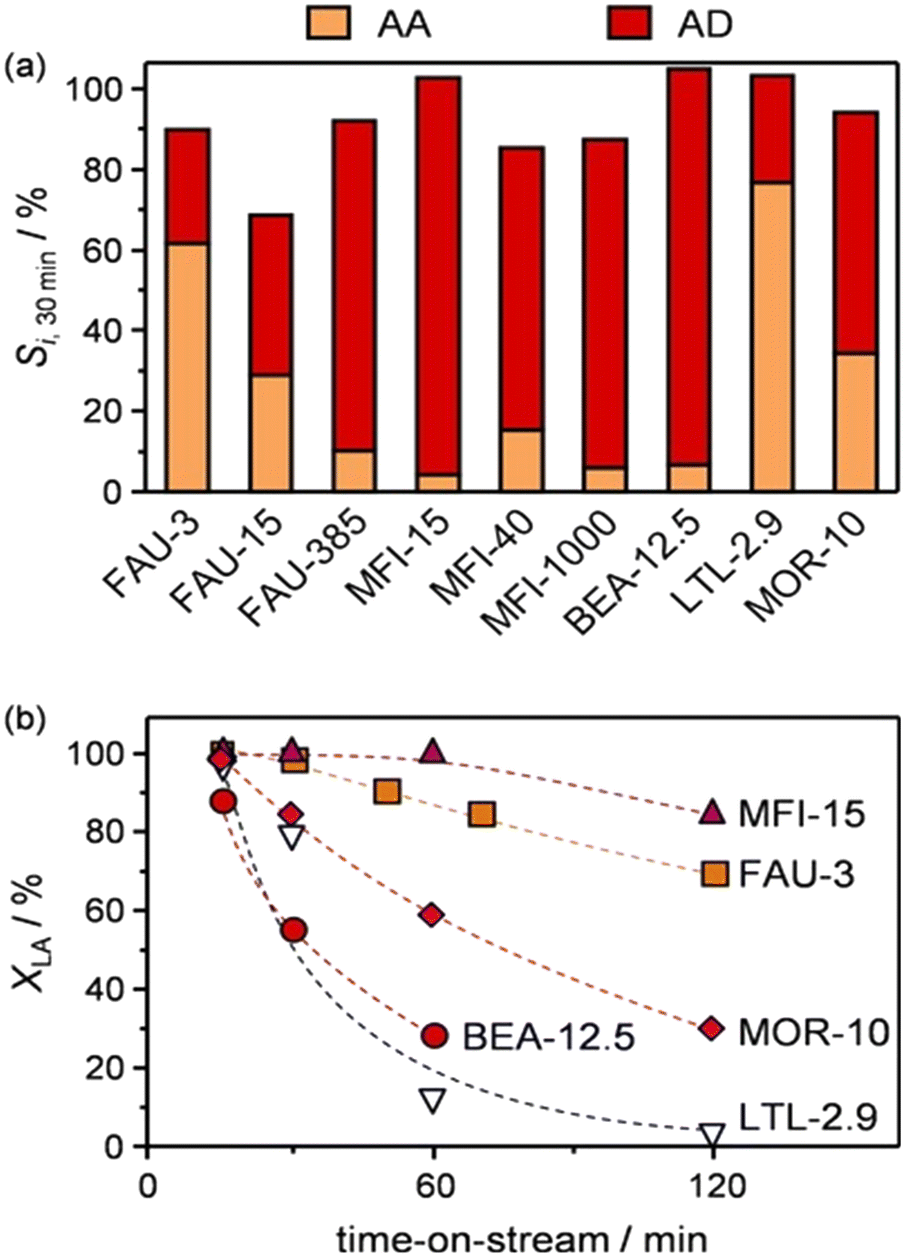 | ||
| Fig. 10 (a) Selectivities toward products at 30 min of reaction, and (b) evolution of LA conversion during 2 h of reaction over sodium-form zeolites with different framework topologies and Si/Al ratios. Reprinted with permission from ref. 97, copyright 2016 John Wiley and Sons. | ||
From the comparative catalytic data between the sodium form and modified zeolite systems in Table 6, most of the modified zeolite systems are able to bring about an enhancement in the AA yield at the initial stage to a different extent by at least 15% with the alkali metal-modified systems,32,97–102 by at most 10% with the alkaline-earth metal-modified systems,94 by at most 21% with the rare-earth metal-modified systems,92 and by at least 106% with the transition metal (Cu and Co)-modified systems.105 Although the catalytic stability is more or less improved with the modified zeolite systems compared to the sodium form zeolite systems, the obtained results are unsatisfactory. It is consistently admitted from all the available reports that the modified zeolites possess a decreased acid strength (or acidity). Apparently, the increased catalytic activity for the production of AA may be attributed to the decreased surface acid strength (or acidity).
On the other hand, the catalytic role of lactate salt in the vapor-phase dehydration of LA over a supported alkali salt is demonstrated.21,22 Once lactate salt forms from LA and initial inorganic base or salt on the zeolite surface under the reaction conditions, the initial surface acid–base properties are definitely changed and are no longer meaningful to the dehydration reaction. As the reaction proceeds, the content of the initial inorganic base or salt gradually falls in the favor of formation of lactate salt. Eventually, the initial inorganic base or salt completely converts to lactate salt. The nature of the catalyst only depends on the properties of zeolite, lactate salt, and zeolite–lactate salt interaction. In the real sense, the acid–base properties of initial modified zeolites cannot represent the acid–base characters of actual catalysts under LA dehydration conditions.
From the comparative catalytic stabilities of the various zeolite systems in Table 6, the modified zeolite systems hardly maintain good catalytic stability during the vapor-phase dehydration of LA due to the high acid strength of zeolites. The H0 of zeolites is generally much lower that of an aqueous LA solution.95,96,104 Under an LA acid strength (H0 ≥ 1.4), a neutral or slightly acidic supported lactate salt catalyst can remain stable.22,23 Under a higher acid strength of support (H0 < 1.4), supported lactate salt may dehydrate to acrylate salt, followed by acrylate salt polymerization. Even if the acid strength of a modified zeolite is reduced via alkalization, it can be increased to that of the parent zeolite after the generation of lactate salt with the neutralization of the base by LA during LA dehydration.
Inspired by the work of Sobuś and Czekaj,106 it is necessary to stress on the effect of diffusion of reactant molecules inside zeolite channels on the dynamics of LA dehydration to AA. The parent Naβ systems are not active for LA dehydration but predominantly for LA decarbonylation,106 presumably implying that LA fails to contact Na+ in the extensive zeolite channels to form sodium lactate as the catalytic active species for LA dehydration due to the limitation of internal mass transfer. Zeolite hierarchization allows the partial degradation of a zeolite crystal structure, providing for faster diffusion of LA into the zeolite extensive channels to react with Na+.106 As a result, the Naβ(h) system is no longer active for LA decarbonylation but substantially for LA dehydration,106 which supports the assumption from another angle that lactate salt acts as the catalytic active species for the dehydration of LA to AA.21,22
Catalytic performance evaluation
From the available literature, there has been continued interest in the exploration of new catalyst systems for the vapor-phase dehydration of LA to AA since it is challenging to understand and control the catalytic selectivity. By simply comparing the catalytic performance of the five types of heterogeneous catalyst systems, it is hard to draw general conclusions. Further classifying sulfate, phosphate, and nitrate salt systems into unsupported and supported forms is helpful for clearer evaluation, as shown in Table 7.| Type of catalyst system | Reaction temperature (°C) | Conversion (%) | Selectivity toward AA (%) | Stability | Ref. |
|---|---|---|---|---|---|
| Sulfate salts | 350–400 | 30–100 | 9.4–82 | Poor | 24, 40, 41 |
| Phosphate salts | 350–400 | 67–100 | 50–89 | Poor to fair | 52, 53, 56–59 |
| Supported phosphate salts | 340–350 | 78–97 | 72–80 | Poor to good | 39, 49, 50 |
| Nitrate salts | 350 | 65 | 37 | Poor | 23 |
| Supported nitrate salts | 350 | 67–99 | 16–55 | Poor to good | 21–23 |
| HAPs | 350–375 | 70–100 | 60–87 | Poor to excellent | 82–86, 88 |
| Modified zeolites | 325–360 | 68–100 | 41–84 | Poor to good | 32, 93, 97–100, 102 |
Sulfate and phosphate salt-based systems require higher temperatures to generate lactate salts as the catalytic active species probably due to the higher boiling points of H2SO4 and H3PO4. Nitrate salt-based systems easily give rise to a higher quantity of lactate salts because of the easy evaporation of HNO3. Amid the unsupported salt systems, the unsupported nitrate salt systems are inferior in terms of both LA conversion and selectivity toward AA. All the three types of unsupported salt systems perform poorly in terms of catalytic stability due to the easy lactate salt dehydration to acrylate salt, followed by acrylate salt polymerization. Amid the supported salt systems, the catalytic activity and selectivity of the supported nitrate salt systems are improved compared to those of the unsupported nitrate salt systems. It seems that the three types of supported salt systems can achieve good catalytic stability depending on the lactate salt–support interaction. If an acidic support is used, it is difficult for the supported salt system to stabilize because the acidic support can induce lactate salt dehydration to acrylate salt followed by acrylate salt polymerization and cause catalyst deactivation.23
Amid all the five types of catalyst systems, the HAP systems exhibit the best catalytic stability apart from the good catalytic activity and selectivity. An HAP system may play the role of both catalytic active component and support. During an LA dehydration reaction over HAP, part of the calcium phosphate in the HAP displaces with LA to give calcium lactate as the catalytic active species, which may be well situated in the parent apatite crystal structure. The unreacted calcium phosphate in the HAP may act as the support. Such a supported calcium lactate catalyst can be viewed to be very stable against its leaching and its dehydration to calcium acrylate, which causes catalyst deactivation under an acidic condition of LA dehydration. The moderate acidity and basicity of HAP provides a proper environment for the stabilization of calcium lactate. By contrast, although many modified zeolite systems show good initial catalytic activity and selectivity, they hardly maintain good catalytic stability due to the high acid strength of zeolites,95,96,104 which can cause the dehydration of in situ generated lactate salts to acrylate salts followed by acrylate salt polymerization. Even if the acid strength of a modified zeolite is reduced via alkalization, it can be increased to that of the parent zeolite after the generation of lactate salt with the neutralization of the base by LA during LA dehydration.
On the other hand, when working on modified zeolites, the factors that influence the internal diffusion of the reactant molecules affecting the dynamics of LA dehydration to AA inside microporous zeolites should be considered. To overcome the limitation of mass transfer, subjecting microporous zeolites to hierarchization enables the crystal structures to partially degrade and thus allows the reactant to easily diffuse into the extensive channels to react with metal cations to form lactate salts, which increases the catalytic selectivity of LA dehydration to AA.
Reaction mechanism
Although a number of research papers by a few groups have reported the mechanisms of catalytic vapor-phase LA dehydration,21–23,32,57,58,60,83,84,100 the mechanistic study toward the catalytic reaction is still superficial. There are two types of mechanisms reported for the catalytic reaction. One is a reaction pathway with lactate salt as the catalytic active species21–23 and the other type describes a reaction pathway with solid acid as the catalytic active site.32,57,58,60,83,84,100 Herein, we present five typical mechanistic studies selected from the available literature.Miller and coworkers earlier studied the vapor-phase LA condensation mechanism with model SiO2-supported sodium salts by means of IR, NMR, and GC/MS analyses of surface species.21,38,42 They sought to characterize the catalytic active species starting with the various supported sodium salts such as NaOH, NaNO3, Na2SiO3, Na3PO4, Na2HPO4, sodium lactate, NaCl, and Na2SO4 in the vapor-phase condensation of LA at 280–350 °C. The resultant sodium lactate intermediate was suggested to be the actual catalyst for 2,3-PD formation because the catalytic activity for 2,3-PD formation was associated with the presence of the supported sodium lactate under the reaction conditions.21,42 They proposed the following two-stage reaction pathway for the catalytic condensation of LA with sodium lactate.21
Stage (1): initial sodium lactate generation
| C2H4(OH)COOH + NaX → C2H4(OH)COONa + HX↑ |
Stage (2): sodium lactate-catalyzed 2,3-PD production from LA
| 2C2H4(OH)COOH + C2H4(OH)COONa → C2H5COCOCH3 + 2H2O + CO2 + C2H4(OH)COONa |
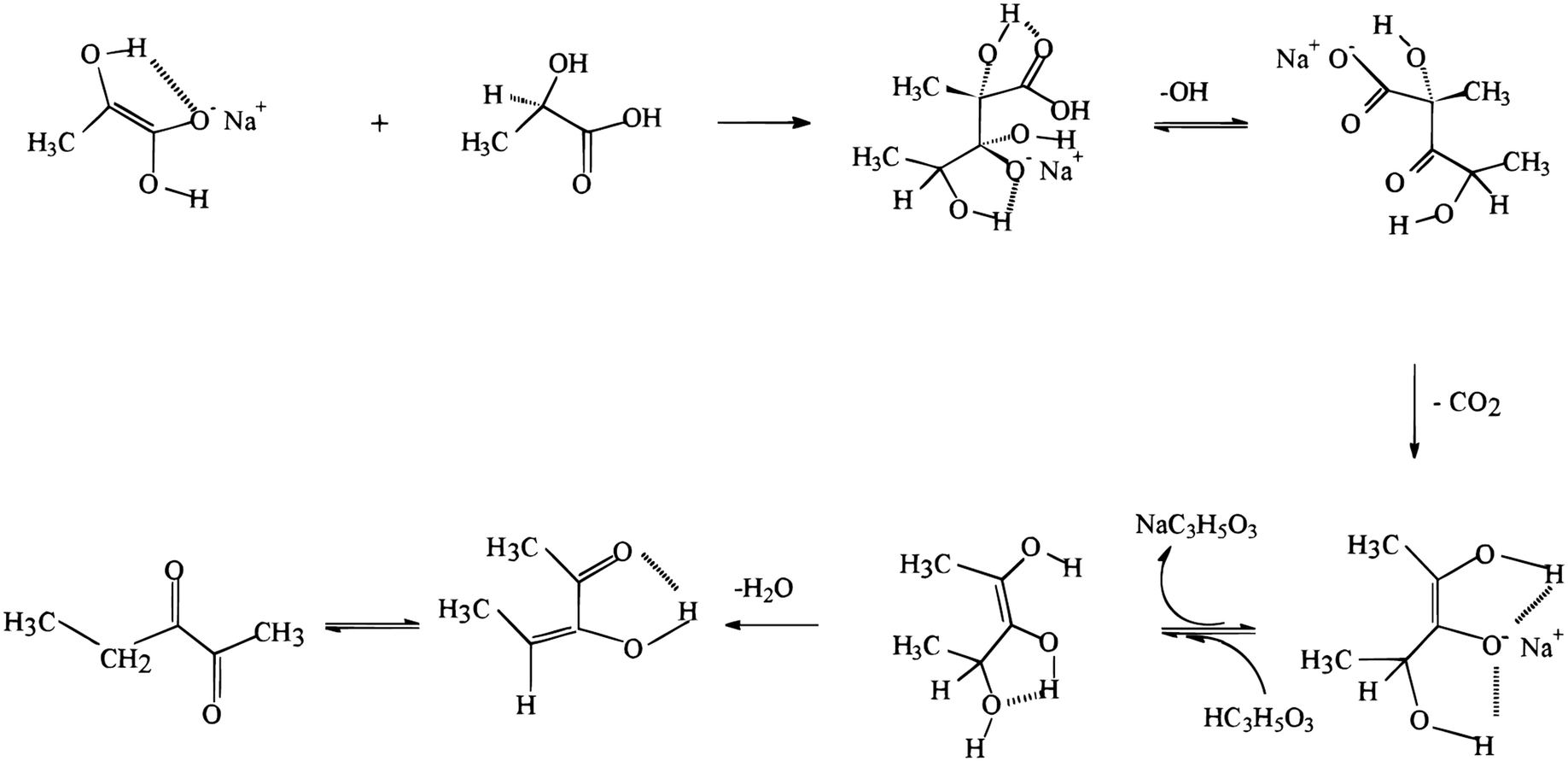 | ||
| Scheme 5 Proposed mechanism for the vapor-phase LA condensation to 2,3-PD with sodium lactate. Reprinted with permission from ref. 21, copyright 1997 American Chemical Society. | ||
In 2014, Xu and coworkers reported the speculated vapor-phase LA dehydration mechanism with Kβ based on their study of alkali cation-exchanged β zeolites relating to the catalytic performance acid–base property dependence.100 Kβ was regarded as the catalytic active site in Scheme 6. In the catalytic cycle, Kβ first displaces with LA to give potassium lactate and Hβ, as evidenced by others.21,22,38 Then, the resultant potassium lactate undergoes the attack of the hydroxyl group by the H+ of Hβ before the dehydration to potassium acrylate with the regeneration of Hβ. Finally, the resultant potassium acrylate exchanges with the H+ of the regenerated Hβ to produce AA with the regeneration of Kβ. Apparently, the speculated mechanism lacks the basis of reactivity of potassium lactate and acrylate with Hβ. Hβ is supposed to possess a higher acid strength (H0 < 1.4)104 than LA (H0 ≥ 1.4). It is hard to imagine that under a more acidic condition of LA dehydration, the potassium lactate dehydration and potassium acrylate hydrolysis could proceed. In light of the latest study by Huang et al., potassium lactate is prone to hydrolyzing to LA, which then readily polymerizes, and potassium acrylate readily polymerizes in the presence of acidic KNO3/silica.23
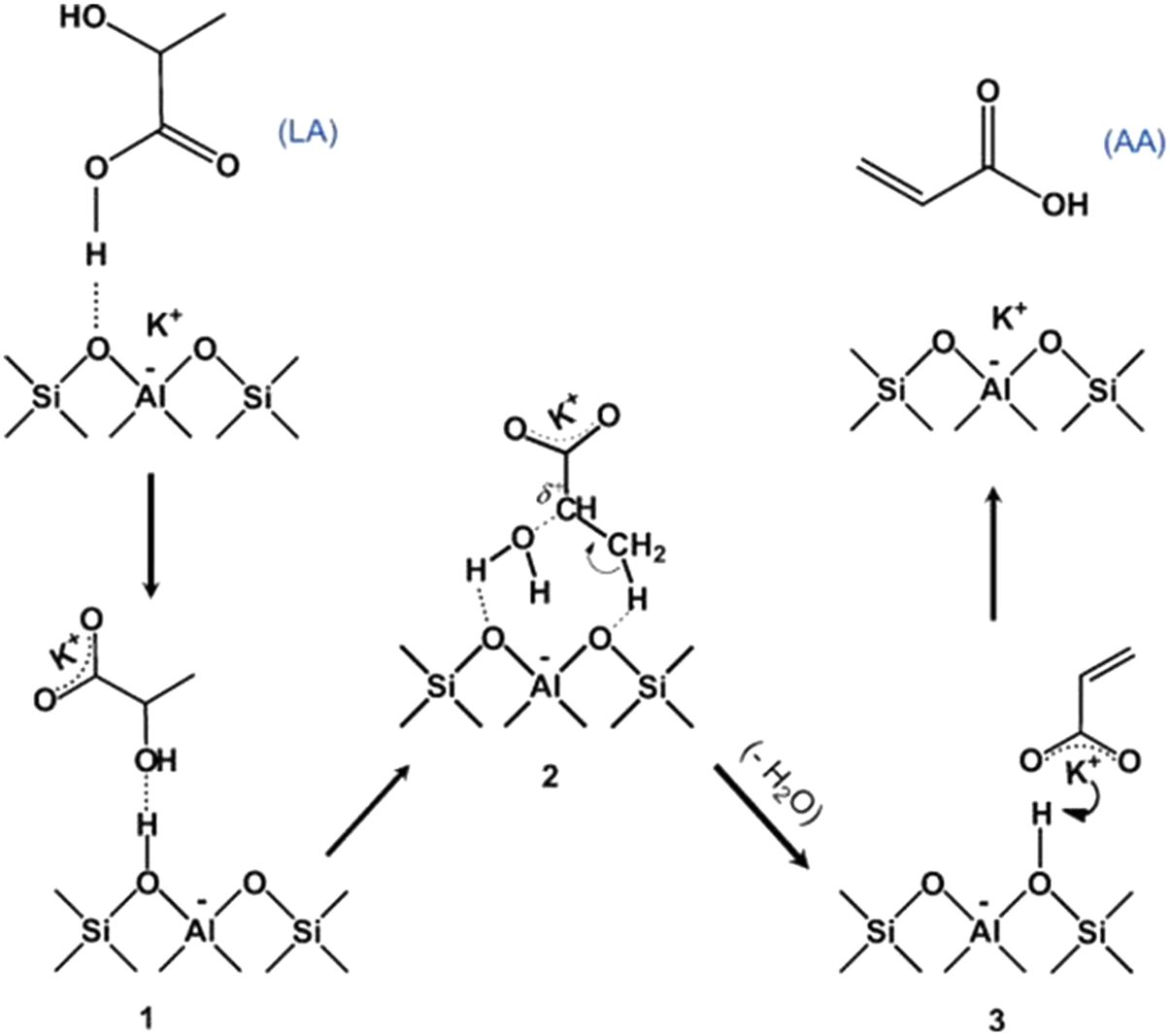 | ||
| Scheme 6 Possible mechanism for AA production from LA with Kβ. Reprinted with permission from ref. 100, copyright 2014 John Wiley and Sons. | ||
At the same time, Xu and coworkers proposed another mechanism with HAP to account for the cooperative acid–base catalysis for selective LA dehydration to AA over the HAPs.84 Calcium phosphate was suggested as the catalytically active site, as shown in Scheme 7. This is inconsistent with the IR study of Ghantani et al. wherein calcium lactate was observed as the stable reaction intermediate upon the adsorption of LA on HAP.83 The stable reaction intermediate is reasonably viewed as the catalytically active species. In the catalytic cycle, LA first donates the H+ of its carboxyl group to the surface oxygen of calcium phosphate to generate the surface acidic OH and calcium lactate. Then, the resultant calcium lactate is activated by the surface acidic OH for dehydration. Next, the calcium lactate dehydrates to calcium acrylate with the concomitant regeneration of the surface acidic OH. Finally, the resultant calcium acrylate exchanges with the H+ of the surface acidic OH to produce AA with the regeneration of calcium phosphate. Similar to the catalytic cycle with Kβ, the Brønsted acid site of calcium phosphate was suggested to assist the dehydration of the in situ generated calcium lactate and the hydrolysis of the resultant calcium acrylate. Either in the catalytic cycle with Kβ or HAP, the Lewis acid site accepts the H+ of LA to convert to the Brønsted acid site, which acts as the catalyst. However, the cooperative acid–base catalysis is not displayed in the catalytic cycle with HAP. Calcium phosphate or HAP is supposed to possess a lower acid strength25 than LA. It is known that the H+ of LA plays an efficient role in the dehydration of lactate salt and the hydrolysis of acrylate salt over a neutral catalyst system.23 Whether the Brønsted acid site of calcium phosphate or the H+ of LA plays this role over HAP needs to be testified.
In 2016, Murphy et al. published a mechanistic study on the dehydration of LA and methyl lactate on NaY via a combination of reactivity and IR spectroscopic measurements.60 The acid strength of NaY was found to have a substantial effect on the catalytic activity for LA decarbonylation because the variation was observed over orders of magnitude due to the use of more acidic materials. This may prove to be true for LA dehydration. Their IR study of pyridine adsorption suggested that NaY converts to HY in water at 200 °C. They therefore claimed that the Brønsted acid sites generated in situ with the assistance of water act as the primary active sites for the LA dehydration pathway. They proposed the vapor-phase LA dehydration mechanism with HY as the true catalyst, as shown in Scheme 8. In the first step, NaY displaces with LA to generate sodium lactate and HY via ion-exchange. Next, the hydroxyl group of the resultant sodium lactate is activated by the H+ of the HY as the Brønsted acid site. Subsequently, the activated sodium lactate dehydrates to sodium acrylate with the regeneration of HY. In the final step, AA is released via reverse ion exchange between the sodium acrylate and the HY. However, the role of HY as the active site in this catalytic cycle is questionable. HY is supposed to possess a higher acid strength95,96 than LA. Under a more acidic condition of LA dehydration, sodium lactate is prone to hydrolyzing to LA, which then readily polymerizes, and sodium acrylate readily polymerizes.23 Throughout the mechanistic study, no experimental evidence was provided in the presence of HY either that sodium lactate dehydrates to sodium acrylate or that sodium acrylate hydrolyzes to AA, i.e., steps (2) and (3) did not prove to be true in the presence of HY.
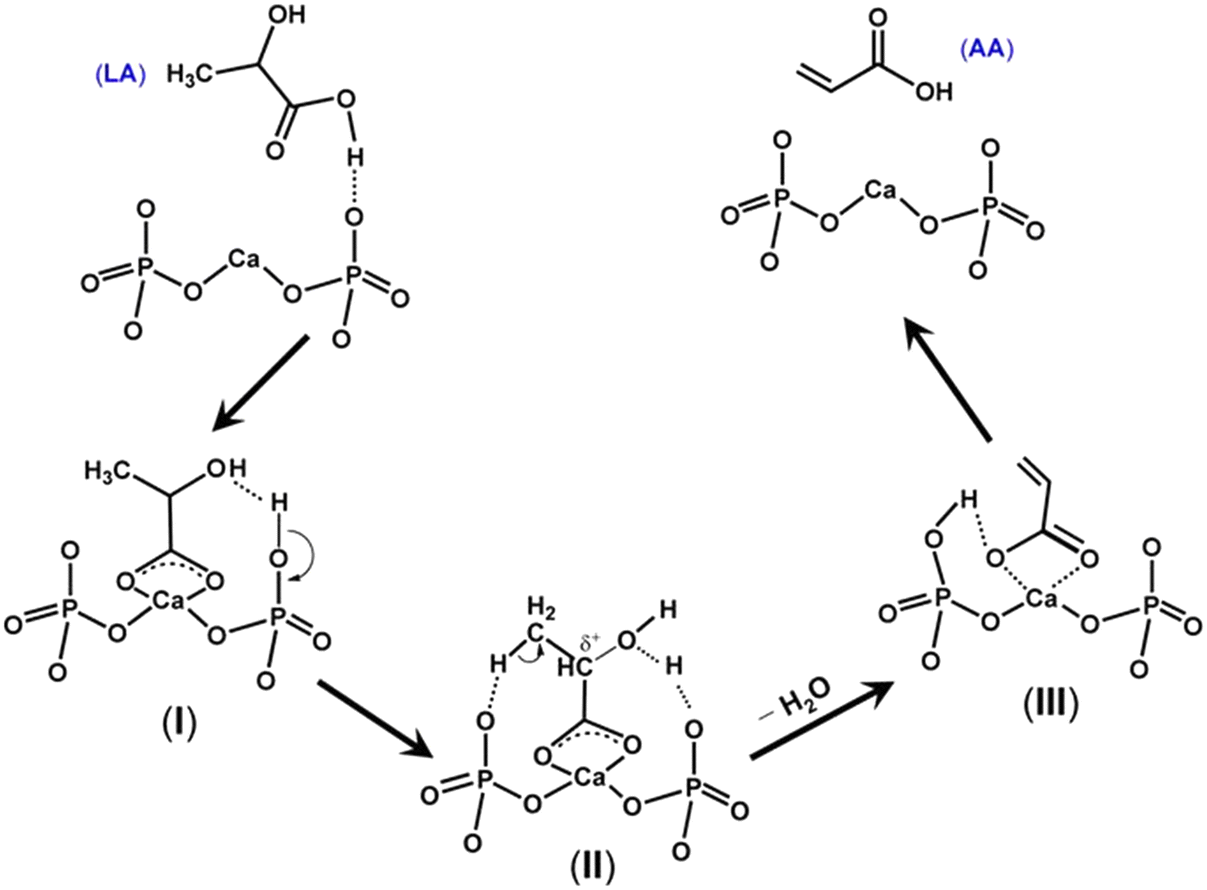 | ||
| Scheme 7 Possible mechanism for AA production from LA with HAP. Reprinted with permission from ref. 84, copyright 2014 American Chemical Society. | ||
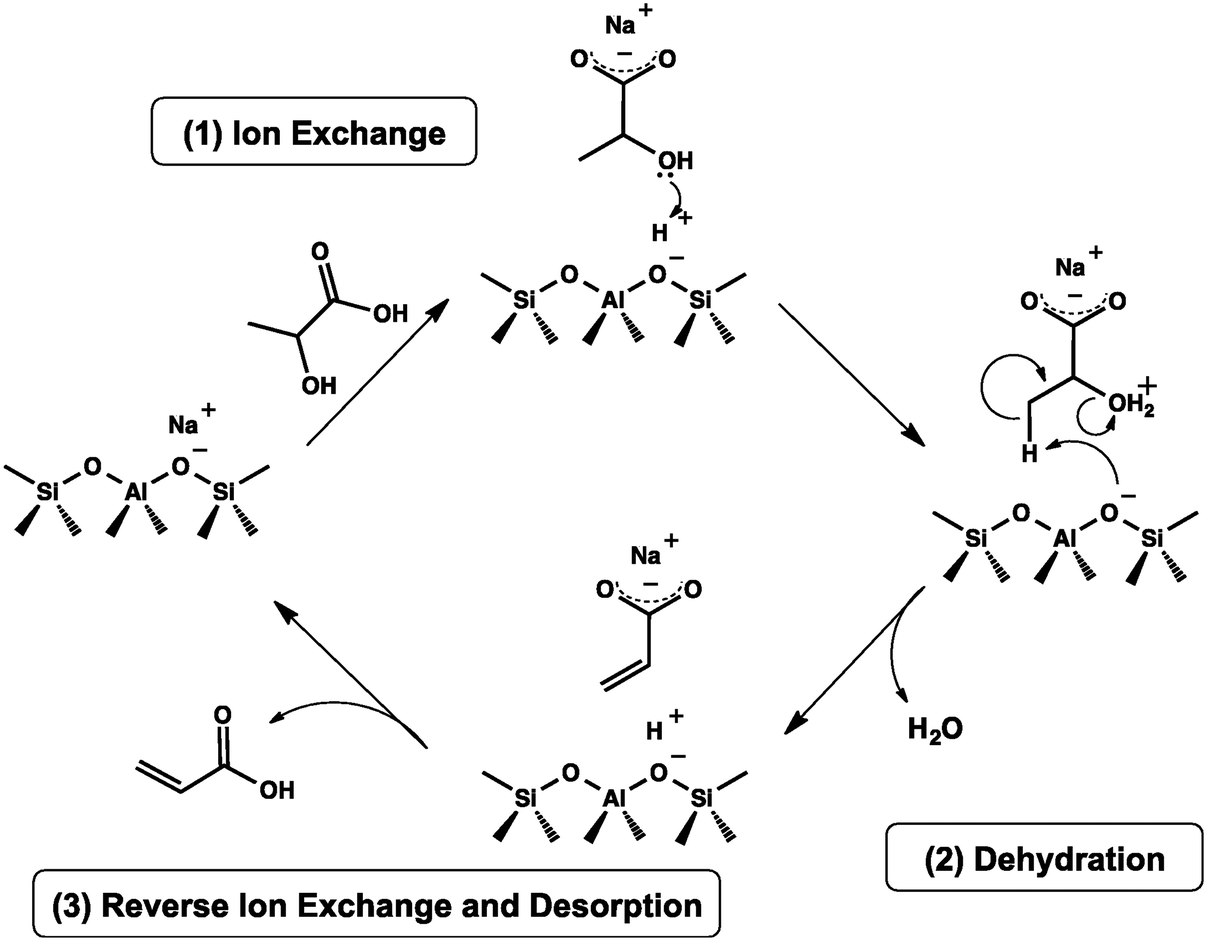 | ||
| Scheme 8 Proposed mechanism for the dehydration of LA on NaY. (1) Ion exchange between the adsorbed LA and sodium balancing cation, creating a surface Brønsted acid site and adsorbed sodium lactate, (2) Brønsted acid site-catalyzed dehydration, and (3) reverse ion exchange between adsorbed sodium acrylate and surface H+ leading to product desorption. Reprinted with permission from ref. 60, copyright 2016 Elsevier Inc. | ||
Huang et al. lastly reported a mechanism for vapor-phase LA dehydration with neutral KNO3/silica, which was studied via a combination of catalytic and IR spectroscopic experiments.23 To unveil the reaction pathway and the stabilization of the catalytic active species, they monitored the reactions of LA on KNO3 and KNO3/silica and behavior of the resultant unsupported or supported potassium lactate as the catalytic active species under dehydration conditions by IR spectroscopy. To better understand the step of AA production and potassium lactate regeneration in the catalytic cycle, they followed the reactions between LA and potassium acrylate on KBr and silicas under dehydration conditions by IR spectroscopy. Combining the catalytic and IR results, they assumed that LA acts as the Brønsted acid itself to promote the step of potassium lactate dehydration to potassium acrylate on the one hand and displaces with potassium acrylate to fulfil the step of AA production and potassium lactate regeneration on the other hand. They deduced that the catalytic process of LA dehydration to AA involves the following three critical steps starting with a KNO3 precatalyst: (1) KNO3 displaces with LA to potassium lactate; (2) potassium lactate dehydrates to potassium acrylate; and (3) AA forms and potassium lactate regenerates from LA and potassium acrylate. All the three steps indispensably determine the catalytic selectivity and stability of potassium lactate derived from the KNO3 precatalyst during LA dehydration to AA as these steps are related to the reaction intermediate chemistry. Scheme 9 illustrates the reaction mechanism over neutral KNO3/silica. The three critical steps in the catalytic LA dehydration proceed smoothly over neutral systems such as KNO3/AS300, KNO3/S15cal, and KNO3/SBA-15 whereas they fail to be fulfilled over acidic systems such as KNO3/S15. Correspondingly, the neutral systems perform well in catalytic activity and stability for the vapor-phase dehydration of LA to AA, whereas the acidic system does poorly. In this mechanism, KNO3 efficiently displaces with LA to generate potassium lactate as the catalyst in the first step. Then, LA as the Brønsted acid lends its H+ to the potassium lactate for assisting its dehydration to potassium acrylate. As soon as the potassium lactate dehydration step finishes, the resultant potassium acrylate quickly displaces with LA to release AA and regenerate potassium lactate as the final step. Presumably due to the adverse effect of the interaction between the OH group of potassium lactate and silica on the potassium lactate dehydration,23 the potassium lactate dehydration step is the rate-limiting step in the catalytic cycle. Since the reactions relating to the production of potassium lactate are dominant over potassium lactate hydrolysis and dehydration over neutral KNO3/silica, the supported potassium lactate catalyst is stable for long-term vapor-phase LA dehydration. In light of the similar LA reactivity, intermediate chemistry and catalytic performance over the slightly acidic KNO3/SiO2–Al2O3(36) and NaNO3/SiO2–Al2O3(36) to those over the neutral KNO3/silica,22,23 the proposed reaction mechanism is deemed to apply to slightly acidic (SiO2–Al2O3)-supported alkali salt systems.
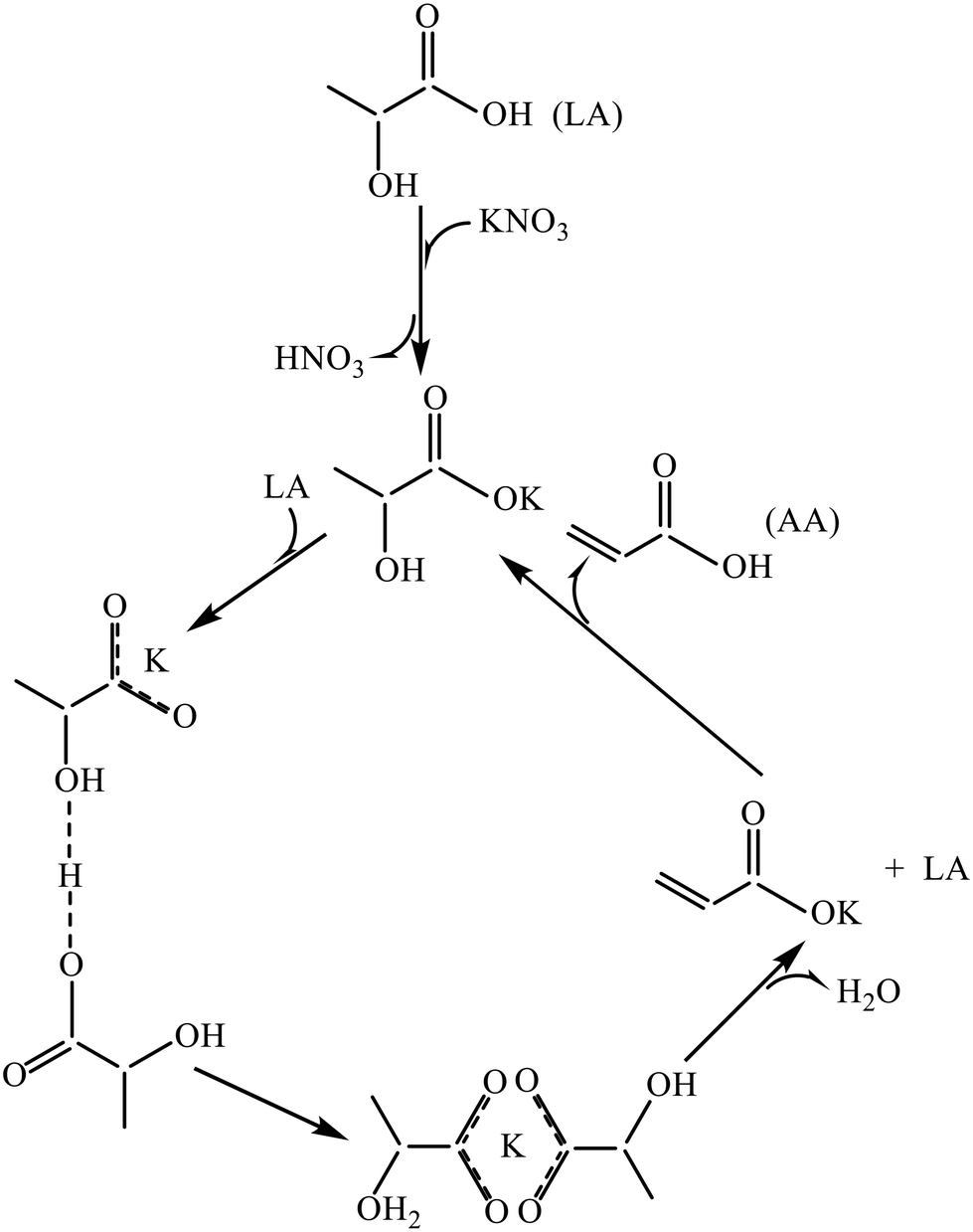 | ||
| Scheme 9 Proposed mechanism for the vapor-phase dehydration of LA with potassium lactate generated in situ over neutral KNO3/silica. | ||
From the reported mechanisms for catalytic vapor-phase LA dehydration, it is commonly recognized that lactate salt is present as an intermediate in the catalytic cycles.21–23,32,57,58,60,83,84,100 This signifies that the in situ generated lactate salt plays an essential role in the reaction pathways. The discrepancy in the proposed mechanisms is whether the lactate salt dehydration and the acrylate hydrolysis are promoted by the H+ of LA21–23 or by the Brønsted acid site of a catalyst system.32,57,58,60,83,84,100 Further study needs to be conducted for elucidation.
Conclusions and outlook
In the investigation of a catalytic reaction, experimental conditions, including reactor design, reaction operation, product collection, reaction data acquisition, and catalyst preparation and treatments, have a significant influence on the reaction results. Since the pioneering design of the process of catalytic vapor-phase dehydration of LA by Holmen,20 the investigations of catalytic vapor-phase LA dehydration have been done using down-flow fixed-bed reaction apparatus. From both the patent and paper literature, the operation conditions in the reaction investigations are basically comparable and consistent in the different laboratories. From the available reaction data, the use of the unsupported catalyst systems is undesirable in terms of catalytic stability due to the chemical instability of the in situ generated lactate salt as the true catalyst under LA dehydration conditions. The use of the more acidic supported catalyst systems including the modified zeolites scarcely generates the stable supported lactate salts under LA dehydration conditions, which easily causes catalyst deactivation. The use of more acidic materials like zeolites as supports is challenging. Under the prerequisite of use of neutral or less acidic supported catalyst systems, the proper interaction between lactate salt and the support is critically important in preventing the occurrence of lactate salt dehydration to acrylate salt followed by acrylate salt polymerization and thus ensuring the catalytic stability of the supported lactate salts. Among all the five types of heterogeneous catalyst systems, the HAP type outperforms the other types in terms of catalytic selectivity and stability, probably because of the moderate acidity and basicity of HAPs on the other hand and the particular stabilization of the in situ generated calcium lactate on the unreacted calcium phosphate in the HAP crystal structure on the other hand. Where the same type of heterogeneous catalyst systems are concerned, the use of the different precursors leads to the distinct difference in the catalytic performance. The difference probably lies in the effects of metal cation on the catalytic performance and the stabilization of the in situ generated lactate salt, as well as in the effect of solid acidity on the stabilization of the in situ generated lactate salt. The unsupported barium and calcium monometallic salt systems are more active than the unsupported other monometallic salt systems for the production of AA, possibly in that the in situ generated barium and calcium lactates are more stable against their dehydration to acrylates followed by acrylate polymerization under acidic reaction conditions. The copresence of Ba2+ or Ca2+ may bring about a concerted promotion on the catalytic activity and stability of the Na+ or K+ or Cs+ or La3+-containing unsupported lactate salts under acidic reaction conditions.When comparatively looking back at dehydration reactions of LA and other compounds,107 researchers should be aware of the particularity of LA dehydration. A dehydration reaction usually proceeds in the presence of acid except for LA.27–29,107 Due to the higher acid strength (H0 = 1.4) that LA itself possesses, LA dehydration, LA decarbonylation, and LA decarboxylation occur either in the liquid or vapor phase in a non-catalytic way.27,34 The addition of stronger acid can only strengthen LA decarbonylation, which does not favor AA production.27,31–33 The addition of base can trigger LA decarboxylation, which weakens LA dehydration and decarbonylation.27,28 In this sense, the necessity of solid acid in catalysis for the vapor-phase dehydration of LA is questionable. Miller and coworkers have put forward a reaction pathway for the SiO2(LSA)-supported sodium lactate catalyzed 2,3-PD production from LA vapor regardless of solid acid.21 Huang et al. have suggested that the lactate salt generated in situ from KNO3 or NaNO3 acts as the true catalytic species for the dehydration of LA to AA.22 They have recently studied the neutral, high surface area silica-supported potassium lactate derived in situ from KNO3.23 Their neutral KNO3/silica systems possess the long-term stable catalytic performance. They have proposed the mechanisms for the vapor-phase dehydration of LA to AA and decarbonylation of LA to AD over neutral KNO3/silica regardless of solid acid. It is deduced that LA acts both as the reactant and Brønsted acid that assists in the catalysis of potassium lactate and that the dehydration of LA to AA proceeds smoothly with a neutral heterogeneous catalyst. This is clear evidence of non-extra acid catalysis for the dehydration of LA to AA. From the work of these two groups, extra acid is unnecessary toward LA dehydration as long as a suitable catalyst system is present.
For the vapor-phase dehydration of LA to AA, using a stronger solid acid can reinforce LA decarbonylation to AD. To seek the inhibition of LA decarbonylation in favor of AA production, researchers have been properly increasing basicity (or base strength) on a catalyst system. In this aspect, introducing basic sites in the catalyst system should be considered. However, it is questioned how the basic sites take part in the reaction process and play a synergistic role with existing acid sites in catalysis for the dehydration reaction. When lactate salt forms from LA and initial inorganic base or salt on the catalyst surface under the reaction conditions, the surface acid–base properties of the catalyst system are certainly modified. The surface basicity (or base strength) decreases as the reaction proceeds so that the initial measurements of the acid–base properties of the catalyst system are trivial for the explanation of the reaction results.
An alternative way of modifying the catalyst systems to favor AA production is related to the use of organic bases. Murphy et al. have tackled the neutralization of Brønsted acid sites generated during the dehydration of methyl lactate over NaY with pyridine for the suppression of decarbonylation pathway.108,109 Brønsted acid sites are identified as the catalytic active sites for the decarbonylation reaction.60 Through combined kinetic and transmission IR spectroscopic investigations, they demonstrated that introducing pyridine to the reaction feed increases the selectivity toward acrylates while inhibiting the formation of byproducts, including AD and coke, and that the reduced decarbonylation speed is directly related to pyridine-quenching Brønsted acid sites.108 The increase in the selectivity toward acrylates arises from the inhibition of side reaction pathways including decarbonylation and coke formation, both of which are catalyzed by the in situ generated surface Brønsted acid sites, rather than the acceleration of the dehydration pathway.109 In the absence and presence of pyridine in the feed, catalyst deactivation proceeds through a drastically different mechanism. The formation of coke is the primary cause of catalyst deactivation in the pyridine-free feed, whereas when pyridine is used, the accumulation of intact bulky and high boiling point acid/base complexes, e.g., pyridinium acrylate and pyridinium lactate in the zeolite pores, limits the access of the reactant to the catalytic sites. Regeneration at 330–450 °C in inert atmosphere does not have any effect on the catalyst system that has deactivated in the pyridine-free feed, but partially restores the activity of the spent catalyst system in the pyridine-spiked feed. It is expected that porous catalyst systems with more open structures that facilitate desorption of the bulky acid/base complexes are more resistant to deactivation.
Meanwhile, a better understanding of different reaction pathways and possible reaction intermediates in the LA dehydration process is helpful for the rational design of new generations of catalyst systems for the vapor-phase dehydration of LA to AA. Further in-depth investigations should be conducted to gain insights into the chemistry of reaction intermediates and the reaction mechanism. As far as lactate salt as the true catalytic species is concerned, the in-depth research on acido basic, electronic, and geometric factors controlling catalytic selectivity and stability should be focused. As the catalytic selectivity and stability of HAP systems are attractive, the compositional variation of HAPs can be expected to bring about a significant impact on the catalytic properties in the vapor-phase dehydration of LA to AA. The composition of HAPs is extremely variable because not only they are rarely stoichiometric but also each constitutive element can be substituted to a certain extent without losing the crystal structure: Ca2+ can be replaced with other metal cations such as Na+, Sr2+, Ba2+, Pb2+, Mg2+ and Zn2+; PO43− may be substituted with other oxyacid anions such as CO32−, AsO43−, SiO44−, VO43−, SO42− and HPO42−; and OH− can be replaced with halogenic anions including F−, Cl−, Br− and I−.89
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National University of Singapore's Green Energy Program (WBS: A-0005323-05-00) and FRC MOE T1 (WBS: A-0009184-00-00) and from the A*STAR LCERFI Project (Award ID: U2102d2011) is acknowledged.References
- M. Dusselier, P. van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- T. Bonnotte, S. Paul, M. Araque, R. Wojcieszak, F. Dumeignil and B. Katryniok, ChemBioEng Rev., 2018, 5, 34–56 CrossRef CAS.
- E. V. Makshina, J. Canadell, J. Krieken, E. Peeters, M. Dusselier and B. F. Sels, ChemCatChem, 2019, 11, 180–201 CrossRef CAS.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. Martin Alonso, R. Mariscal and M. López Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- The Acrylic Acid Market Online, https://www.procurementresource.com/reports/acrylic-acid-industry-report, (accessed December 2022).
- The Glycolic Acid Market Online, https://www.marketsandmarkets.com/Market-Reports/glycolic-polyglycolic-acid-market-1090.html, (accessed December 2022).
- The Levulinic Acid Market Online, https://www.marketdataforecast.com/market-reports/levulinic-acid-market, (accessed December 2022).
- The Maleic Anhydride Market Online, https://www.grandviewresearch.com/industry-analysis/maleic-anhydride-market, (accessed December 2022).
- The Adipic Acid Market Online, https://www.grandviewresearch.com/industry-analysis/adipic-acid-market, (accessed December 2022).
- P. Maki-Arvela, I. Simakova, T. Salmi and D. Murzin, Chem. Rev., 2014, 114, 419–427 CrossRef PubMed.
- G. Juodeikiene, D. Vidmantiene, L. Basinskiene, D. Cernauskas, E. Bartktiene and D. Cizeikiene, Catal. Today, 2015, 239, 11–16 CrossRef CAS.
- R. Burns, D. T. Jones and P. D. Ritchie, J. Chem. Soc., 1935, 400–406 RSC.
- E. M. Filachione and C. H. Fisher, US Pat., 2399595, 1946 Search PubMed.
- H. J. Arpe and H. Grosspietsch, DE2046411, 1972.
- J. Galoci, M. Procházka and E. Klinotová, Collect. Czech. Chem. Commun., 1983, 48, 1729–1733 CrossRef CAS.
- G. Chuchani, R. M. Dominguez, A. Herize and R. Romero, J. Phys. Org. Chem., 2000, 13, 757–764 CrossRef CAS.
- M. A. Lilga, T. A. Werpy and J. E. Holladay, US Pat., 0110974, 2004 Search PubMed.
- D. J. Miller, C. T. Lira, L. Peereboom and A. K. Kolah, US Pat., 0150619, 2013 Search PubMed.
- R. Beerthuis, M. Granollers, D. R. Brown, H. J. Salavagione, G. Rothenbergand and N. R. Shiju, RSC Adv., 2015, 5, 4103–4108 RSC.
- R. E. Holmen, US Pat., 2859240, 1958 Search PubMed.
- M. S. Tam, G. C. Gunter, R. Craciun, D. J. Miller and J. E. Jackson, Ind. Eng. Chem. Res., 1997, 36, 3505–3512 CrossRef CAS.
- L. Huang, D. S. Theng, L. Zhang, L. Chen, C. Wang and A. Borgna, ACS Omega, 2019, 4, 8146–8166 CrossRef CAS PubMed.
- L. Huang, D. S. Theng, L. Zhang, L. Chen, C. Wang, F. Gao and A. Borgna, Dalton Trans., 2022, 51, 15912–15932 RSC.
- J. Peng, X. Li, C. Tang and W. Bai, Green Chem., 2014, 16, 108–111 RSC.
- D. I. Collias, J. E. Velasquez, J. E. Godlewski, J. C. Hayes and W. D. Laidig, WO148929, 2016.
- B. Odell, G. Earlam and D. J. Cole-Hamiton, J. Organomet. Chem., 1985, 290, 241–248 CrossRef CAS.
- W. S.-L. Mok, M. J. Antal Jr. and M. Jones Jr., J. Org. Chem., 1989, 54, 4596–4602 CrossRef CAS.
- C. T. Lira and P. J. McCrackin, Ind. Eng. Chem. Res., 1993, 32, 2608–2613 CrossRef CAS.
- T. M. Aida, A. Ikarashia, Y. Saito, M. Watanabe, R. L. Smith Jr. and K. Arai, J. Supercrit. Fluids, 2009, 50, 257–264 CrossRef CAS.
- M. S. Tam, R. Craciun, D. J. Miller and J. E. Jackson, Ind. Eng. Chem. Res., 1998, 37, 2360–2366 CrossRef CAS.
- R. A. Sawicki, US Pat., 4729978, 1988 Search PubMed.
- P. Sun, D. Yu, Z. Tang, H. Li and H. Huang, Ind. Eng. Chem. Res., 2010, 49, 9082–9087 CrossRef CAS.
- B. Katryniok, S. Paul and F. Dumeignil, Green Chem., 2010, 12, 1910–1913 RSC.
- G. C. Gunter, D. J. Miller and J. E. Jackson, J. Catal., 1994, 148, 252–260 CrossRef CAS.
- The Aqion Online, https://www.aqion.de/site/125, (accessed December 2022).
- J. Zhang, J. Lin and P. Cen, Can. J. Chem. Eng., 2008, 86, 1047–1053 CrossRef CAS.
- S. Greenfield and M. Glift, Analytical Chemistry of the Condensed Phosphates, Pergamon, New York, 1975 Search PubMed.
- G. C. Gunter, R. Craciun, M. S. Tam, J. E. Jackson and D. J. Miller, J. Catal., 1996, 164, 207–219 CrossRef CAS.
- J. Zhang, Y. Zhao, M. Pan, X. Feng, W. Ji and C.-T. Au, ACS Catal., 2011, 1, 32–41 CrossRef CAS.
- X. Li, Z. Chen, P. Cao, W. Pu, W. Zou, C. Tang and L. Dong, RSC Adv., 2017, 7, 54696–54705 RSC.
- S. Lyu and T. Wang, RSC Adv., 2017, 7, 10278–10286 RSC.
- D. C. Wadley, M. S. Tam, P. B. Kokitkar, J. E. Jackson and D. J. Miller, J. Catal., 1997, 165, 162–171 CrossRef CAS.
- The Wikipedia Online, https://en.wikipedia.org/wiki/Sodium_bisulfate, (accessed December 2022).
- The Softschools Online, https://www.softschools.com/formulas/chemistry/pyrosulfuric_acid_uses_properties_structure_formula/241, (accessed December 2022).
- C. Paparizos, W. D. Shaw and S. R. Dolhyj, EU Pat., 0181718, 1986 Search PubMed.
- G. C. Gunter, R. H. Langford, J. E. Jackson and D. J. Miller, Ind. Eng. Chem. Res., 1995, 34, 974–980 CrossRef CAS.
- The Wikipedia Online, https://en.wikipedia.org/wiki/Tetrasodium_pyrophosphate, (accessed December 2022).
- The Wikipedia Online, https://en.wikipedia.org/wiki/Disodium_pyrophosphate, (accessed December 2022).
- J. Zhang, Y. Zhao, X. Feng, M. Pan, J. Zhao, W. Ji and C.-T. Au, Catal. Sci. Technol., 2014, 4, 1376–1385 RSC.
- J. V. Lingoes and D. L. Collias, WO155245, 2013.
- X. Zhang, L. Lin, T. Zhang, H. Liu and X. Zhang, Chem. Eng. J., 2016, 284, 934–941 CrossRef CAS.
- C. Tang, J. Peng, G. Fan, X. Li, X. Pu and W. Bei, Catal. Commun., 2014, 43, 231–234 CrossRef CAS.
- C. Tang, J. Peng, X. Li, Z. Zhai, N. Jiang, W. Bai, H. Gao and Y. Liao, RSC Adv., 2014, 4, 28875–28882 RSC.
- E. Blanco, P. Delichere, J. M. M. Millet and S. Loridant, Catal. Today, 2014, 226, 185–191 CrossRef CAS.
- V. C. Ghantani, M. K. Dongare and S. B. Umbarkar, RSC Adv., 2014, 4, 33319–33326 RSC.
- N. Nagaraju, V. P. Kumar, A. Srikanth, N. P. Rajan and K. V. R. Chary, Appl. Petrochem. Res., 2016, 6, 367–377 CrossRef CAS.
- N. Nekkala, P. Balla, S. Ginjupalli, H. Mitta, S. K. Hussain, B. Ponnala and K. V. R. Chary, J. Nanosci. Nanotechnol., 2021, 21, 1537–1548 CrossRef CAS PubMed.
- Z. Guo, D. S. Theng, K. Y. Tang, L. Zhang, L. Huang, A. Borgnaa and C. Wang, Phys. Chem. Chem. Phys., 2016, 18, 23746–23754 RSC.
- N. Nekkala, P. Balla, S. R. Ginjupalli, P. K. Seelam, S. K. Hussain, B. Ponnala and V. R. C. Komandur, Biomass Convers. Biorefin., 2022, 12, 3535–3546 CrossRef CAS.
- B. M. Murphy, M. P. Letterio and B. Xu, J. Catal., 2016, 339, 21–30 CrossRef CAS.
- The Wikipedia Online, https://en.wikipedia.org/wiki/Pyrophosphoric_acid, (accessed December 2022).
- W. Sun, X.-L. Li and C.-M. Tang, Wuli Huaxue Xuebao, 2016, 32, 2327–2336 Search PubMed.
- B. Grabowska and M. Holtzer, Arch. Metall. Mater., 2009, 54, 427–437 CAS.
- S. Khanlari and M. A. Dubé, Ind. Eng. Chem. Res., 2015, 54, 5598–5603 CrossRef CAS.
- J. Zhang, X. Feng, Y. Zhao, W. Jia and C.-T. Au, J. Ind. Eng. Chem., 2014, 20, 1353–1358 CrossRef CAS.
- G. Cammas, M. Morssli, E. Fabregue and L. Bardet, J. Raman Spectrosc., 1991, 22, 409–413 CrossRef.
- J. A. S. Bett, L. G. Christner and W. K. Hall, J. Am. Chem. Soc., 1967, 89, 5535–5541 CrossRef CAS.
- J. A. S. Bett and W. K. Hall, J. Catal., 1968, 10, 105–113 CrossRef CAS.
- S. J. Joris and C. H. Amberg, J. Phys. Chem., 1971, 75, 3167–3171 CrossRef CAS.
- C. L. Kibby and W. K. Hall, J. Catal., 1973, 29, 144–159 CrossRef CAS.
- C. L. Kibby and W. K. Hall, J. Catal., 1973, 31, 65–73 CrossRef CAS.
- T. Tsuchida, J. Kubo, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, J. Catal., 2008, 259, 183–189 CrossRef CAS.
- K. Kurashina, G. L. de Lange, C. de Puttert and K. de Groot, Biomaterials, 1984, 5, 215–220 CrossRef CAS PubMed.
- H. Zhou and J. Lee, Acta Biomater., 2011, 7, 2769–2781 CrossRef CAS PubMed.
- N. Zhang, T. Gao, Y. Wang, Z. Wang, P. Zhang and J. Liu, Mater. Sci. Eng., C, 2015, 46, 158–165 CrossRef CAS PubMed.
- S. D. Sebti, R. Tahir, R. Nazih, A. Saber and S. Boulaajaj, Appl. Catal., A, 2002, 228, 155–159 CrossRef CAS.
- T. Hara, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2003, 44, 6207–6210 CrossRef CAS.
- M. Gruselle, T. Kanger, R. Thouvenot, A. Flambard, K. Kriis, V. Mikli, R. Traksmaa, B. Maaten and K. Tonsuaadu, ACS Catal., 2011, 1, 1729–1733 CrossRef CAS.
- K. Zhao, B. Qiao, Y. Zhang and J. Wang, Chin. J. Catal., 2013, 34, 1386–1394 CrossRef CAS.
- J. Lan and Z. Zhang, J. Ind. Eng. Chem., 2015, 23, 200–205 CrossRef CAS.
- M. K. Dongare, S. B. Umbarkar and S. T. Lomate, WO156921, 2012.
- A. M. Onda, Y. Matsuura and K. Yanagisawa, US Pat., 8772539, 2012 Search PubMed.
- V. C. Ghantani, S. T. Lomate, M. K. Dongare and S. B. Umbarkar, Green Chem., 2013, 15, 1211–1217 RSC.
- B. Yan, L.-Z. Tao, Y. Liang and B.-Q. Xu, ACS Catal., 2014, 4, 1931–1943 CrossRef CAS.
- Z.-H. Liu, B. Yan, Y. Liang and B.-Q. Xu, Mol. Catal., 2020, 494, 17417–17428 Search PubMed.
- Y. Matsuura, A. Onda and K. Yanagisawa, Catal. Commun., 2014, 48, 5–10 CrossRef CAS.
- Y. Matsuura, A. Onda, S. Ogo and K. Yanagisawa, Catal. Today, 2014, 226, 192–197 CrossRef CAS.
- C. Li, Q. Zhu, Z. Cui, B. Wang and T. Tan, Ind. Eng. Chem. Res., 2019, 58, 53–59 CrossRef CAS.
- R. Wojcieszak, T. Bonnotte, S. Paul, B. Katryniok and F. Dumeignil, Front. Chem., 2020, 8, 421 CrossRef CAS PubMed.
- B. Yilmaz and U. Müller, Top. Catal., 2009, 52, 888–892 CrossRef CAS.
- V. Verdoliva, M. Saviano and S. De Luca, Catalysts, 2019, 9(3), 248 CrossRef.
- H. Wang, D. Yu, P. Sun, J. Yan, Y. Wang and H. Huang, Catal. Commun., 2008, 9, 1799–1803 CrossRef CAS.
- P. Sun, D. Yu, K. Fu, M. Gu, Y. Wang, H. Huang and H. Ying, Catal. Commun., 2009, 10, 1345–1349 CrossRef CAS.
- J. Yan, D. Yu, P. Sun and H. Huang, Chin. J. Catal., 2011, 32, 405–411 CrossRef CAS.
- H. Otouma, Y. Arai and H. Ukihashi, Bull. Chem. Soc. Jpn., 1969, 42, 2449–2453 CrossRef CAS.
- M. Ikemoto, K. Tsutsumi and H. Takahashi, Bull. Chem. Soc. Jpn., 1972, 45, 1330–1334 CrossRef CAS.
- G. M. Lari, B. Puértolas, M. S. Frei, C. Mondelli and J. Pérez-Ramirez, ChemCatChem, 2016, 8, 1507–1514 CrossRef CAS.
- L. Zhang, D. S. Theng, Y. Du, S. Xi, L. Huang, F. Gao, C. Wang, L. Chen and A. Borgna, Catal. Sci. Technol., 2017, 7, 6101–6111 RSC.
- C. Yuan, H. Liu, Z. Zhang, H. Lu, Q. Zhu and Y. Chen, Chin. J. Catal., 2015, 36, 1861–1866 CrossRef CAS.
- B. Yan, L.-Z. Tao, Y. Liang and B.-Q. Xu, ChemSusChem, 2014, 7, 1568–1578 CrossRef CAS PubMed.
- B. Yan, A. Mahmood, Y. Liang and B.-Q. Xu, Catal. Today, 2016, 269, 65–73 CrossRef CAS.
- B. Yan, L.-Z. Tao, A. Mahmood, Y. Liang and B.-Q. Xu, ACS Catal., 2017, 7, 538–550 CrossRef CAS.
- B. Yan, Z.-H. Liu, Y. Liang and B.-Q. Xu, Ind. Eng. Chem. Res., 2020, 59, 17417–17428 CrossRef CAS.
- S.-H. Chai, H.-P. Wang, Y. Liang and B.-Q. Xu, Green Chem., 2007, 9, 1130–1136 RSC.
- N. Sobuś, B. Michorczyk, M. Piotrowski, Ł. Kuterasiński, D. K. Chlebda, J. Łojewska, R. J. Jędrzejczyk, P. Jodłowski, P. Kuśtrowski and I. Czekaj, Catal. Lett., 2019, 149, 3349–3360 CrossRef.
- N. Sobuś and I. Czekaj, Catal. Today, 2022, 387, 172–185 CrossRef.
- The Wikipedia Online, https://en.wikipedia.org/wiki/Dehydration_reaction, (accessed December 2022).
- B. M. Murphy, M. P. Letterio and B. Xu, ACS Catal., 2016, 6, 5117–5131 CrossRef CAS.
- B. M. Murphy, M. P. Letterio and B. Xu, ACS Catal., 2017, 7, 1912–1930 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |