
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-fouling flow reactors for nanomaterial synthesis
Maximilian O.
Besenhard
 ,
Sayan
Pal
,
Georgios
Gkogkos
and
Asterios
Gavriilidis
*
,
Sayan
Pal
,
Georgios
Gkogkos
and
Asterios
Gavriilidis
*
Department of Chemical Engineering, University College London, London, WC1E 7JE, UK. E-mail: a.gavriilidis@ucl.ac.uk
First published on 1st February 2023
Abstract
The potential of flow chemistry for nanomaterial synthesis has been amply demonstrated in the last decade. Robust and reproducible synthetic protocols, scalability, high-throughput screening and novel process conditions are the main drivers to “go flow”. Not always acknowledged, is how reactor fouling restricts the operation of flow reactors making it a bottleneck for nanomaterial flow synthesis. Even though remarkable achievements in process and reactor design have been made, providing solutions to minimise and prevent fouling, there is no single non-fouling flow reactor suitable for nanomaterial synthesis at all relevant synthetic protocols. There are, however, reactor designs and operations that can prevent fouling, depending on the nature of the synthesis, i.e., the particle formation mechanism and kinetics. Although the expression “fouling” is used generically, neither its causes, nor its circumvention can be generalised. Therefore, this review describes the diverse origins and consequences of flow reactor fouling for wet-chemical nanomaterial syntheses, and most importantly, showcases the variety of reactor designs and operations to mitigate or prevent fouling. The reactor characteristics are discussed with respect to the critical synthetic conditions aiming to guide the customisation of non-fouling flow reactors for a broad variety of continuous nanomaterial syntheses. For a successful flow synthesis, the flow reactor elements must be selected considering the possible fouling causes and consequences and how to mitigate (or at least monitor and detect) fouling, while providing the synthetic conditions required.
Introduction
Nanoparticles (NPs) remain at the forefront of new technologies, which explains the enduring interest in synthetic strategies to upscale production or optimise their properties. Their dimensions exceeding single atoms by only 1–2 orders of magnitude yield unique optical, electronic, and/or magnetic features, as well as a high surface to volume ratio. This has paved the way for advances in electronics, sensor technology, biomedicine, catalysis, separation processes and many more.1–6The nanomaterial properties differ from their bulk equivalent, evidencing that nanoparticles are not thermodynamically favoured and that their formation is governed by reaction and particle formation kinetics rather than thermodynamic equilibria. Therefore, the properties of nanoparticles synthesised via wet chemical routes (which is the focus of this review) are highly sensitive to the process conditions. These conditions, also referred to as synthetic conditions, include mixing times, mass and heat transfer rates, temperatures, pH values, reagent concentrations and the sequence and timing of their addition, etc., as well as their spatio-temporal profile in the reactor.
For one century, batch processes have been the prevailing method to produce nanoparticles. As batch reactors provide only limited control of process conditions, it is not surprising that nanoparticle syntheses in batch suffer from poor reproducibility and/or batch-to-batch variability, operative constraints (e.g., in terms of mixing and heating/cooling rates),7 and varying operator skills. This makes up-scaling of lab procedures challenging and hinders the transition to pilot production and ultimately full-scale production. This is crucial to bring research across the so called “Valley of Death” (the gap between research and commercialisation) in high-cost, and high-risk areas such as nanotechnology.8,9
Flow reactors can overcome these scalability limitations as they offer good control of process conditions and yield as well as increased production volumes via longer operation times – and not larger reactors. The progress of flow chemistry for nanoparticle syntheses during the last decade has been remarkable as the field transitioned from proof of principle studies10 to screening11,12 and fully automated synthesis platforms13–15 as well as large scale production.16–19 The growing interest in robust nanomaterial flow syntheses of different material classes (from lipids to metals) requiring different synthetic routes, as well as the various flow chemistry applications (from discovery to production), resulted in a great diversity of flow reactors. This “flow reactor jungle” comprises microchip silicon and glass reactors, capillary-based systems using standard tubing and connectors, stainless steel tube and plate reactors, and many more. Although these reactors differ in scale, material, and operation mode, they share one problem when used for nanoparticle synthesis, i.e., fouling.
The reason why nanoparticle (wet chemical) syntheses are prone to foul reactors is the phase transition occurring, i.e., solid matter is formed from solution. This transition forms nanoparticles but is also likely to form solid wall depositions, not necessarily in the form of particles. Hence, the established small (development & screening) and large (manufacturing) scale flow reactors are likely to foul when used to synthesise nanoparticles.
The flow reactors utilised to synthesise nanoparticles are well documented, with several recent reviews on flow synthesis in general20–24 and specialisation on drug nanonization,25,26 lipid-based nanoparticles,27,28 noble metal nanoparticles,29,30 metal oxide,31,32 and magnetic nanoparticles.33,34 Although reactor fouling is recognised as a challenge, there is limited awareness of its severity (see Fig. 1 for fouling examples). Fouling is the bottleneck for many nanoparticle flow syntheses hindering long term operation, which makes it challenging to use flow reactors to screen synthetic conditions and produce nanoparticles at large scales. Unfortunately, there is no universal “one for all” strategy for its prevention.
 | ||
| Fig. 1 Flow reactors that fouled during the synthesis of (a) silver,35 (b) lipid,36 (c) iron oxide,37 (d) palladium,18 (reproduced from ref. 18 and 35–37 with permission from the Royal Society of Chemistry, © 2017, 2015, 2021 & 2023) and (e and f) gold nanoparticles (reproduced from ref. 38 and 39 with permission from Elsevier, © 2022 & 2018). | ||
Therefore, this review discusses the origin and consequences of fouling in the context of nanoparticle synthesis, flow reactor designs minimising and preventing fouling, considering the respective synthetic procedures as well as their limitations, and highlights recent developments for single and multiphase systems.
In addition to showcasing the flow reactors used for fouling-mitigating nanoparticle synthesis, we discuss their design characteristics with respect to the particle formation kinetics. This aims to guide the design of non-fouling systems by providing options for suitable flow reactor elements to meet the critical process parameters considering the particle formation kinetics. Avoiding fouling requires a successful interplay between the synthetic procedure chemistry and reactor design, with the latter being the focus hereinafter.
Nature and consequences of fouling
Definition of fouling with regard to nanoparticle synthesis
Fouling is a term which reactor engineers commonly define as “the accumulation of unwanted material on solid surfaces” or “the unwanted deposition on surfaces”. The associations with “unwanted”, however, vary when designing crystallisers, heat exchangers, bioreactors, membrane separators, fluidised bed reactors, and other reactors.40–43Commonly distinguished are macro (caused by coarse matter) and micro fouling. The latter has several classifications, including the five classes defined by Epstein in the 1980s (considering heat exchangers),44 including: I crystallisation fouling (surface crystallisation of dissolved solutes); II particulate fouling (deposition of particulate phase(s) present); III chemical reaction fouling (deposits of reaction product, impurity, or intermediate formed directly on the wall or in solution prior to wall deposition, including precipitations); IV corrosion fouling (deposits formed by corroding wall material, most commonly at metallic walls); V biofouling (surface attachment of living organisms). Other classes described are: VI solidification fouling (accumulation by solidification/freezing of a component dissolved, similar to crystallisation fouling);45 VII gas bubble clogging (micro-specific fouling category describing the trapping of gas bubbles blocking channels).43 The categories listed here (other classes are used too) are neither 100% descriptive, nor exclusive, i.e., classifying fouling into a single class is not always possible. As this review focusses on wet-chemical nanoparticle syntheses only, fouling hereinafter refers to the classes I–III only. Although this review briefly discusses the fouling problems due to microparticles that form when non-stabilised nanoparticles agglomerate, the focus is on nanoparticle solutions only. We refer to other literature for problems due to larger particles not following the fluid streamlines, i.e., have a Stokes number >1.46–49
To better distinguish the fouling origin and consequences for flow reactors, we distinguish between local (small axial reactor fraction affected), or traversed (significant axial reactor fraction affected), and fouling affecting the surface only (depositions at the reactor wall not extending radially into the channel) or being constrictive (depositions extending from the reactor wall reducing the channel cross section), see Fig. 2.
 | ||
| Fig. 2 Fouling classification in tubular reactors considering (a) axial and (b) radial characteristics. | ||
Some obvious reasons to avoid fouling are the costs associated with the yield reduction (nanoparticles are valuable materials, not only if costly reagents are used) and to guarantee unimpeded reactor operation. For example, constrictive fouling requires higher inlet pressure to maintain the flow rate set. Once the pressure becomes sufficient to break up constrictions, the flow rate exceeds the set value, at least temporarily, hampering accurate flow rate control. If pressure constraints inherent to every flow reactor system (including the pumps), however, are significant, pressure cannot break up constrictions and their growth causes clogging.50
Even if fouling does not impede the operation, it can affect the synthetic conditions severely. Examples are surface and/or constrictive fouling inducing changes of the heat transfer through reactor walls, resulting in varying spatial temperature profiles (crucial for temperature sensitive particle formation mechanisms, e.g., for thermal decomposition synthesis),51,52 or unwanted reactions involving the deposits (crucial if depositions are catalytically active and promote precursor decomposition or particle growth, e.g., for noble and transition metal nanoparticle syntheses).53–57 The higher reactor wall area-to-volume of flow reactors, compared to their batch equivalent, makes nanoparticle synthesis in flow inherently vulnerable to fouling-induced changes of the synthetic conditions and impedes reactor operation.
Small and large scale flow reactors for nanoparticle synthesis are almost exclusively operated in steady state mode (i.e., the synthetic conditions do not change in time) for process and material development and production. Therefore, it is important to clarify that, strictly speaking, if fouling changes the synthetic conditions (i.e., a continuous alteration of the reactor wall) a steady state operation is practically not possible. Hence, advantages of flow reactors for high-throughput experimentation and large scale production via long operation times would be compromised if fouling is present.
Reactor fouling during nanoparticle synthesis
The variety of synthetic procedures cannot be described by a unified nanoparticle formation concept. What most wet-chemical syntheses have in common though is that solutions containing the soluble reagents (e.g., the precursor, reducing or oxidation agents, acids, bases, etc.) are mixed to react and form solid nanoparticles. A simplified, hence not always applicable, nanoparticle formation mechanism (see Fig. 3) comprises the following steps: i) precursor(s) decompose forming monomers/the particles' building blocks; ii) these building blocks cluster and form solid nuclei nanoparticles which iii) grow via incorporating more building blocks and/or coalescence; iv) the nanoparticles become stabilised either sterically or electrostatically to prevent particle agglomeration and aggregation. Changes in the particles' crystal structure and stoichiometric compositions during or post these steps are common. | ||
| Fig. 3 Typical steps during NP formation. | ||
The timescales for the synthesis and its steps can vary from ≪1 s (e.g., for rapid precipitation or when using strong reducing agents) to ≫1 h (e.g., for thermal decomposition synthesis) and depend on the synthesis.56,58 Knowledge of the particle formation and stabilisation kinetics is vital not only to design a flow reactor facilitating the synthetic conditions required, but also to avoid or minimise the likelihood and severity of fouling.
During precursor decomposition, chemical reaction fouling is commonly causing surface fouling. This happens either locally or transversally depending on decomposition kinetics. When precursor decomposition is initiated by a mixing process (e.g., after adding reducing or oxidation agents, or an acid or base changing the pH), surface fouling is observed post/downstream the addition.39 If precursor decomposition is temperature or radiation induced, fouling is likely to occur during or after the corresponding exposure. Once particles form from solution, the monomers/particles' building blocks energetically favour (at least partially) the solid state. Hence, particle formation is likely accompanied by crystallisation fouling, which can become constrictive quickly at high precursor concentrations. As long as particle growth is not completed and reactive by-products or intermediates (including the particles' building blocks) remain in solution, e.g., while growth proceeds through surface integration, surface fouling via chemical reaction fouling remains likely. If growth occurs via coalescence, particulate fouling is likely to cause constrictive fouling.59 Similarly, insufficient particle stabilisation can promote constrictive particulate fouling. In addition, it can cause excessive agglomeration yielding particles exceeding the nanometre range with Stokes numbers >1, which are likely to accumulate locally and constrict channels.
There is no obvious route to prevent fouling during flow syntheses, even when the nanoparticle formation kinetics are known. The different causes of fouling during each particle formation step have different consequences (see Table 1) and require different prevention measures with the reactor design playing a significant role.
| Formation step | Kinetics | Dominant fouling | Examples |
|---|---|---|---|
| Precursor decomposition | Fast | Local, surface | NaBH4 reduction methods,60 hot injection61 |
| Precursor decomposition | Slow | Traversed, surface | Slow thermal decomposition,62 low concentration or mild reducing agent53,63 |
| Particle formation | Fast | Local, surface or constrictive | Co-precipitation,64 flash nano precipitation65 |
| Particle formation | Slow | Traversed, surface | Polydopamine synthesis,66 Stoeber process for SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 67,68 67,68 |
| Surface integration | Fast | Local, surface | Methods using high precursor concentrations |
| Surface integration | Slow | Traversed, surface | Seed mediated or surface catalytic growth54 |
| Coalescence | Fast | Local, constrictive | Rapid noble metal reduction methods56,69 |
| Coalescence | Slow | Traversed, surface or constrictive | Coalescent growth of clustered nanoparticles or nanoflowers70,71 |
Designing flow reactors that provide the synthetic conditions required and prevent fouling, however, is not trivial and might require a compromise between the two. The following sections present the state-of-the-art of fouling-mitigating single and multiphase flow reactors for nanoparticle synthesis.
Single phase reactors
Single phase flow reactors which are most commonly used in flow chemistry, are inherently prone to fouling as the reactions involved in precursor decomposition, particle formation and growth occur in contact with the reactor wall. Several methods and reactors have been reported to prevent fouling and subsequent clogging, which are discussed in the following sections.Fouling prevention via tuning particle and wall surface chemistry
In this section we discuss fouling prevention induced by unfavourable reactor wall/particulate phase interactions. Controlling the interaction between the particles or intermediate products with the reactor walls can be hugely beneficial in prevention of reactor fouling in nanoparticle synthesis. Controlling the electrostatic interaction between the particles (and charged reagents and/or building blocks) and the reactor wall by means of ionic strength and pH variation in aqueous particle formation environments can generate the repulsion required.43,72–74 Varying the pH value changes not only the zeta potential of nanoparticles, but also the reactor wall material charge, hence the zeta potential difference and repulsion between the two. Plastics, glass and metal exhibit a positive zeta potential at low pH values which becomes negative at high pH values,75,76 with no electrostatic protection inherent to the wall in between, i.e., at the isoelectric point. Fouling is likely when nanoparticles, precursor, or other reagents are charged opposite to the reactor wall.39 Examples show that at low pH values, nanoparticles (and intermediates) were electrostatically attracted to the surface, whereas a pH increase resulted in a net negative charge of the particles and the reactor surface causing electrostatic repulsion between them,72,74,76 which has been beneficial in prevention of particle deposition,77 and fouling prevention.74,76 This highlights the importance and the potential of pH tuning (where the synthetic protocol allows) for fouling prevention in single phase systems.The stabilisation of nanoparticles can also help to avoid particle agglomeration and prevent particle accumulation at the reactor wall (see Fig. 4a). Suspensions of bare nanoparticles or poorly stabilised nanoparticles are more susceptible to agglomeration and aggregation, hence particle accumulation at the reactor wall surface. Stabilisation of gold nanoparticles with citric acid and polyvinyl pyrrolidone have been reported to prevent particle aggregation.78 Resistance to reactor fouling has been observed after particle stabilisation, for example for flow syntheses of iron oxide stabilised via tetraethylammonium hydroxide.79 However, nanoparticle formation and stabilisation are likely to occur at different timescales (with stabilisation being usually slower). Hence, fouling due to agglomeration or adhesion of not yet stabilised nanoparticles at the wall may still occur during the initial stages of the synthesis. It is also reported that colloidally stable suspensions create a particle monolayer on the wall surface, which helps in inhibiting further deposition of particles by unfavourable physicochemical interactions with the suspended particles.76,80
 | ||
| Fig. 4 (a) Schematic illustration of particle stabilisation helping in fouling resistance. (b) Schematic of repellence characteristics of SLIPS antifouling coatings (tethered perfluorocarbon in the porous surface and liquid perfluorocarbon forming a stable film on top) (reproduced from ref. 81 with permission from Springer, © 2014). | ||
In addition to tuning particle surface chemistry, surface modification of the reactor wall can prevent fouling. Amorphous silica coating,82 silanisation74 and lubricant-infused slippery polymer83,84 based surface modification has been used for antifouling surfaces. These surface treatments are very different in terms of involved chemistry, but the concept is similar, i.e., increase the surface energy required for particles or reagents to deposit on the reactor wall. Examples of commercially available amorphous silica coating via chemical vapour deposition on metal surfaces are SilkoNert® or Dursan® (SilcoTek, USA). These coatings are corrosion resistant and promote high oleo and hydrophobicity (contact angle of water >100°), which can resist fouling caused by bacterial biofilm formation;82 they were successfully used for a thermal decomposition flow synthesis of iron oxide nanoparticles.85 Silane treatment of reactor wall surfaces have been reported to generate hydrophobic surfaces. This reduces the wetting of wall surfaces and the adhesion of gold nanoparticles or their nuclei to the wall surfaces, suppressing the wall deposition and reactor fouling.74
Slippery liquid infused porous surfaces (SLIPS) technology has been developed to make low friction flow channels with omniphobic slippery surfaces to prevent wall adhesion of particles (including blood corpuscles and bacterial biofilms).81,86 For SLIPS flow reactors, the inner wall of plastic tubing such as polydimethylsiloxane, poly(methyl methacrylate), polysulfone and polyvinyl chloride, is first transformed into porous or roughened surfaces by casting, surface deposition or etching techniques.87 This can also be achieved by covalently binding a flexible molecular perfluorocarbon layer, or tethered perfluorocarbon. Then, different liquid perfluorocarbon lubricants, such as perfluorodecalin and perfluoropolyethers are used to infiltrate the surface.81,86 This results in the formation of a stably immobilised, molecularly smooth, liquid overlayer, which prevents adhesion to the underlying wall surface (see Fig. 4b).
Synthesis of nanoparticles in single phase flow results in fouling or clogging of the reactor in many cases, but several examples exist in single phase flow reactors showing stable operation without clogging. This is certainly inherent to the synthetic procedure, especially pH values, solvents used, and additives, which explains why some protocols are particular prominent in flow, such as thermal decomposition synthesis of iron oxide nanoparticles in polyols.88–90 However, the fouling prevention mechanisms in such examples are not evident and require further investigation.
Reactors featuring high shear or external forces
Constrictive fouling of reactors can lead to reactor clogging, thereby rendering the flow reactors inoperable. In such situations constrictive fouling or at least reactor clogging, can be minimised and even prevented by usage of high shear and external forces.High shear forces (and the induced lift particles experience) are important to prevent sedimentation and particle accumulation in flow systems handling microparticles. Hence, flow reactors and operations providing high shear are likewise important when nanoparticles are not yet stabilised and large agglomerates form. In addition, the forces high shear rates induce can detach material accumulated at the reactor wall, which is why they can reduce constrictive fouling and prevent clogging (but not surface fouling entirely). This was shown for example for iron oxide nanoparticle synthesis in capillary based systems,64 and in a chip reactor where a minimum flow rate was required to avoid clogging.90 With increasing flowrates, the high shear between reacting liquid and reactor wall minimises the possibility of surface interactions, which reduces the extent of nanoparticle deposition on the walls and surface fouling.74,91 The high flow rates required for sufficient shear in straight channels as well as fast mixing,92 however, are not always practical. Reactors with active mixing systems such as the spinning disk reactors (see Fig. 5a) or continuous stirred tank reactors (CSTRs, see section “Reactors with low surface to volume ratio”) can provide high shear rates for clogging free operation at all flow rates. For nanoparticle synthesis, the precursor solution is commonly introduced in the centre of a rotating disc, forming a thin film on the disc surface under the influence of high centrifugal force. In addition to the large interfacial area and shear between the liquid and disc surface, the centrifugal force forms waves and ripples on the film and generates smaller eddies throughout, enhancing gas–liquid mass transfer (for open systems) and mixing.93–95 Increasing disc speeds and flowrates and using grooved surfaces on the disc intensifies micro-mixing and allows near ideal plug flow in the travelling film of liquid along the disc surface, providing flexibility to manipulate the fabrication of nanoparticles.96,97 Syntheses of iron oxide, titanium dioxide, silver, curcumin and chitosan nanoparticles have been demonstrated in spinning disc reactors, with no clogging reported.96,98–102
 | ||
| Fig. 5 High shear and ultrasound assisted continuous flow reactors for NP synthesis. (a) Spinning disk reactor for magnesium hydroxide and oxide NPs (reproduced from ref. 103 with permission from the American Chemical Society, © 2007), (b) Ultrasound-assisted capillary microreactor (reproduced from ref. 104 with permission from Elsevier, © 2009). (c and d) Microreactors directly coupled with piezoelectric transducer to prevent clogging (reproduced from ref. 105 and 106 with permission from Elsevier, © 2011 & 2019). | ||
An alternative to high shear forces is to incorporate external forces such as ultrasonication with the reactor (see Fig. 5b–d).107–110 Immersing flow reactors in commercial ultrasonic baths (see Fig. 5b) is commonly used for transporting ultrasonic energy into microreactors. Coupling piezoelectric transducers (see Fig. 5c and d) directly to a microreactor chip surface has been reported to be more energy efficient.105 Ultrasound at low frequencies generates transient cavitation bubbles which oscillate and collapse, and can break-up agglomerates and formed constrictions, thereby preventing clogging.110,111 At high frequencies, acoustophoretic effects have been used to focus (micro) particles to the channel centre to prevent sedimentation, which causes clogging.112
Also worth highlighting is the potential of microwave reactor systems to mitigate fouling for temperature induced nanoparticle syntheses. Conventional heating systems increase the reactor wall temperature first, making it the reactor hotspot and hence, a preferable location for reactions to occur which increases the likelihood of fouling. On the contrary, microwave systems heat the reactor volume homogeneously, which can mitigate fouling, especially for temperature sensitive syntheses.113,114
Though it is evident that high shear rate and external force systems can reduce constrictive fouling and mitigate clogging, they should not be considered as fouling-free and may suffer from scalability and operating in limited range of flow parameters such as high flow rates, low residence times (e.g., usually ≪1 min in spinning disc reactors), or high pressure.
Hydrodynamic focusing reactors
Hydrodynamic focusing allows using a single phase system to mix reagents, while keeping reactions distant from the reactor wall.115 In a typical hydrodynamic flow focusing device, a core stream is directed in the centre of a flow channel through hydrodynamic forces. These forces are exerted to the core stream by fully miscible lateral/sheath streams that come in direct contact with it by using contactors of specialised geometry. Fig. 6 shows a summary of flow focusing devices. | ||
| Fig. 6 Hydrodynamic focusing reactors for NP synthesis. (a) On-chip 2D flow focusing device (reproduced from ref. 116 with permission from the American Physical Society, © 1998). (b) On-chip 3D flow focusing device used for PLGA–PEG NP synthesis and demonstration of 3D focusing advantage (reproduced from ref. 119 with permission from Wiley, © 2011). (c) On-chip 3D flow focusing with high aspect ratio lateral sheath stream channels, used for iron oxide NP synthesis (reproduced from ref. 120 with permission from the American Chemical Society, © 2018). (d) Co-axial capillary configuration for 3D flow focusing used for titanium dioxide NP synthesis (reproduced from ref. 121 with permission from Elsevier, © 2004). (e) Ultra small co-axial flow device made by 2-photon polymerisation 3D printing used for lipid NP production (reproduced from ref. 36 with permission from the Royal Society of Chemistry, © 2021). (f) Co-axial capillary devices in series for multi-step silica coated iron oxide NP synthesis (reproduced from ref. 122 with permission from Wiley, © 2009). (g) Fouling at the confluence point of a co-axial capillary device during silver NP synthesis (reproduced from ref. 35 with permission from the Royal Society of Chemistry, © 2015). (h) On-chip device with two sequential 3D flow focusing steps to prevent fouling at the confluence point during iron oxide NP synthesis, by introducing an inert separating stream (reproduced from ref. 79 with permission from the Royal Society of Chemistry, © 2022). | ||
When nanoparticle formation takes place in the central core stream, there is no interaction with the channel walls, thus preventing fouling. In addition, hydrodynamic focusing offers enhanced and controllable mass transfer (since by squeezing the core stream, the diffusion length is effectively decreased), and reduces the residence time distribution of the reactive stream, since the velocity profile near the centre of the channel where the core stream is confined is more uniform compared to the parabolic velocity profile in the whole flow channel.115
In hydrodynamic focusing configurations, mixing takes place via inter-diffusion of reactants into and out of the core stream. This diffusional mixing is greatly enhanced by the reduction of the core stream width (focusing) which is affected by the core/sheath flowrate and viscosity ratio. Mixing in this type of reactor is especially effective when the reactants in the lateral streams are used in excess and the process is driven by diffusion from the sheath into a very thin core stream.
Flow reactor designs that enable hydrodynamic focusing can be as simple as a cross junction in a microfluidic chip (on-chip flow focusing) with two lateral sheath streams confining the core channel into a thin liquid sheet116 (see Fig. 6a). These, 2D flow focusing devices have been widely used for the production of polymeric nanoparticles.117,118 Due to their simplicity, these reactor designs cannot prevent contact of the core stream with the top and bottom of the channel, eventually leading to fouling.119
Various reactor designs have been reported to isolate the core stream from all directions (3D flow focusing), completely preventing contact of the core stream with the flow channel walls and thus, avoid fouling during nanoparticle synthesis. An on-chip device that used additional sheath streams for vertical focusing prior to a cross junction (see Fig. 6b), completely isolated the core stream from the walls.119,123 In other approaches, 3D flow focusing was achieved by using chip devices comprising of multiple layers that create high aspect ratio channels for the lateral sheath streams. In these devices, the lateral sheath stream channels are wider (see Fig. 6c) allowing the sheath streams to engulf the core stream.79,120,124 CFD simulations have been shown to be a simple but valuable tool when designing custom geometries to enable 3D flow focusing.79,119 On-chip 3D hydrodynamic focusing devices have been used in many occasions for the production of nanoparticles including polymeric119,123 and inorganic materials.79,120,124 The advantage in fouling resistance over 2D hydrodynamic focusing devices is illustrated in Fig. 6b where fouling at the top and bottom walls is only observed in the 2D version of the device, shortly after mixing of the reactants.
Alternatively to on-chip devices, 3D flow focusing has been achieved by aligning capillaries in co-axial configuration where the outer stream sheaths a core stream (see Fig. 6d). Co-axial flow focusing devices have been used to synthesise titania nanoparticles121 and silver and gold nanoparticles with various stabilisers.35,125 More recently, a microfluidic device manufactured via 2-photon polymerisation utilised a co-axial mixer to mitigate fouling during the production of lipid nanoparticles36 as shown in Fig. 6e. Finally, it has been demonstrated that multiple co-axial flow devices can be used in series continuously producing nanoparticles in a multistep process, while avoiding particle wall interactions in all steps.122Fig. 6f shows this configuration for the continuous production of silica coated iron oxide nanoparticles.
Despite their appealing properties, flow focusing devices face challenges in their successful implementation in continuous nanoparticle synthesis. Uncontrolled reaction–diffusion can take place near the confluence point, which in rapid reactions has been reported to cause fouling at the entry point of the core stream as shown in Fig. 6g,35 especially after long operation times or under high solid loads. This can be overcome with the introduction of a separating stream that acts as a diffusion barrier near the confluence point, preventing reaction close to the wall of the mixing point. The principle has been proven effective for fouling prevention during nanoparticle synthesis in a multilayer annular device,126 and in a triple stream on-chip 3D flow focusing device (see Fig. 6h).79
It must be noted that while flow focusing devices can be very effective for nanoparticle synthesis, there are several issues to be considered before their application. The diffusion based mixing due to laminar flow can be slow unless the core stream is sufficiently squeezed or the dimensions of the device are greatly reduced which leads to either the need of adjusting synthetic protocols for highly unequal flowrates, or greatly reducing the throughput. In addition, mixing in laminar flow can lead to local concentration profiles which may affect the synthetic process.127 Finally, while hydrodynamic focusing can mitigate fouling during the initial steps and for limited channel lengths, nanoparticle syntheses often require multiple steps and reaction times exceeding the residence times in single devices. If the reaction is slower than the nanoparticles diffusion through the sheath stream towards the wall, fouling may still occur. Thus, the use of hydrodynamic focusing reactors is usually reserved for fast reactions while for slower, or multiple step reactions, flow focusing reactors can be used to prevent fouling during the initial reaction stages.
Reactors with low surface to volume ratio
In the processes/reactors described so far, constrictive fouling cannot be neglected due to the dimensional similarity of depositions with the characteristic reactor dimensions (e.g., channel diameter), which are kept small to enhance transport rates. In conventional batch processes, fouling reports are rare, mostly due to the orders of magnitude lower surface to volume ratios. Hence, precursor decomposition and nanoparticle formation and growth occur mostly in the bulk solution, away from the walls, and the synthetic conditions do not change much if the reactor walls foul. The continuous process analogues of low surface-volume ratio processes are larger scale (>1 cm diameter) plug flow reactors (PFR) and continuous stirred tank reactors (CSTR). Apart from having a low surface-volume ratio, the active stirring in CSTRs provides control over shear, reactant mixing rate and prevents settling of eventually formed larger particles, which makes them well suited for solids handling. When assembling multiple CSTRs in series, the residence time and the residence time distribution, both important parameters for nanoparticle synthesis, can be optimised.While versatile and relatively easy to implement, due to the low surface to volume ratio, CSTRs suffer from slow (relatively to micro-processes) heat transfer rates and poorly mixed areas (dead volumes) which may limit their effectiveness in syntheses where the control of reaction conditions is of paramount importance. Recently developed miniaturised CSTR technology,128,129 (reducing the reactor volume to <5 mL) was shown to drastically improve transport properties, while retaining fouling resistance. Miniaturisation increases the surface to volume ratio of CSTR systems to levels closer to millifluidic devices, making it easier to achieve good mixing and improve heat transfer. In addition, miniaturisation enables compact designs that minimise interconnections between the CSTRs and thus reduces dead volumes and improves fouling resistance, since the interconnections are the most fouling-prone parts of CSTR systems. Fig. 7a, shows a representation of surface to volume ratios for different reactor scales (calculated for cylindrical tank reactors and tubing). In terms of fouling, bigger is better, but the opposite holds true for heat and mass transfer and residence time control. CSTR systems can operate over a wide range of scales making them a flexible tool to operate at the scale the synthesis requires. Nevertheless, CSTR systems are still underused for continuous nanoparticle production.
 | ||
| Fig. 7 CSTR systems for NP synthesis. (a) Comparison of length-scale and surface to volume ratio of various types of reactors. (b) Lab scale CSTR cascade for silver nanoparticle production (reproduced from ref. 16 with permission from Wiley, © 2018). (c) Larger scale system for iron oxide nanoparticle production (reproduced from ref. 130 with permission from Elsevier, © 2020). (d) Miniaturised CSTR cascade for functionalised silica nanoparticle synthesis (reproduced from ref. 128 with permission from the American Chemical Society, © 2020). (e) High temperature miniaturised CSTR cascade for core shell quantum dot synthesis (reproduced from ref. 129 with permission of the Royal Society of Chemistry, © 2021). | ||
Lab scale serial CSTRs have been successfully employed for nanoparticle synthesis. Various configurations of 50 mL and 100 mL CSTRs in series (see Fig. 7b) have been used for the production of silver nanoparticles16 while a cascade of 1 and 0.5 L CSTRs followed by two 3 L CSTRs (see Fig. 7c) has been used for iron oxide nanoparticle synthesis via a partial oxidation precipitation synthesis which benefitted from the residence time control offered by the CSTR cascade.130 Following the miniaturisation trend, an 8-CSTR cascade of 0.4 mL per tank made via 3D printing (see Fig. 7d) was used to produce silica nanoparticles functionalised with polyethyleneimine (PEI) and capped with δ-gluconolactone via multiple single phase reagent addition steps,128 and a miniaturised (1.3 mL) CSTR cascade designed to operate at high temperatures (see Fig. 7e) was used to produce core shell quantum dots.129
Despite the benefits of miniaturisation, it has not yet been established how mixing of miniaturised CSTRs compares to properly designed microfluidic/millifluidic flow reactors. Hence, while they are suitable for reactions with moderate or slow kinetics, further investigations are required for fast reactions. In addition, (closed) micro-CSTRs are susceptible to the accumulation of gas bubbles, similar to micro- and millifluidic devices. Operation of micro-CSTRs in vertical orientation, albeit complicated to achieve, has been shown to mitigate gas accumulation.128
Jet reactors
Jet reactors are alternative reactor designs to typical tubular flow reactors, which are less confined in nature. In jet reactors, which are typically used for fast reactions and where particles form rapidly, mixing occurs away from the reactor walls, which helps to mitigate fouling. Their fouling resistance (for fast reactions) and excellent mixing characteristics have led to their recent emergence in continuous micro/nanoparticle synthesis.131,132 In a typical jet reactor at least two liquid jet streams are introduced at high velocities in a mixing zone, where their kinetic energy is converted into chaotic motion through impinging or redirecting the flow in a course of a small volume.133 The impinged liquid jets mix rapidly35,134 (within 0.2–10 ms), making jet reactors ideal candidates when the timescales for nanoparticle formation are ≪1 s.For the purpose of this review, jet reactors are split into categories depending on the impingement mixing zone.133 The two main categories are confined and free impinging jet reactors, which may exhibit different characteristics both in hydrodynamics, mixing and their fouling resistance behaviour. In confined jet reactors, liquid streams enter a mixing chamber filled with the reactive mixture at high velocities, forming jets that introduce energy into the system and induce rapid mixing. In contrast, when a mixing chamber is filled with gas (e.g., atmospheric air) or a mixing chamber is absent, liquid streams introduced from the inlets at sufficiently high velocities form free jets that impinge head-on to form a liquid sheet, subsequently disintegrating into ligaments and droplets (see Fig. 8i). These configurations are known as free impinging jet reactors (FIJR).135 It should be noted that in the literature, the term “confined impinging jet” has been used to describe free impinging jet reactor configurations where a chamber containing the mixing zone limits the expansion of the liquid sheet.
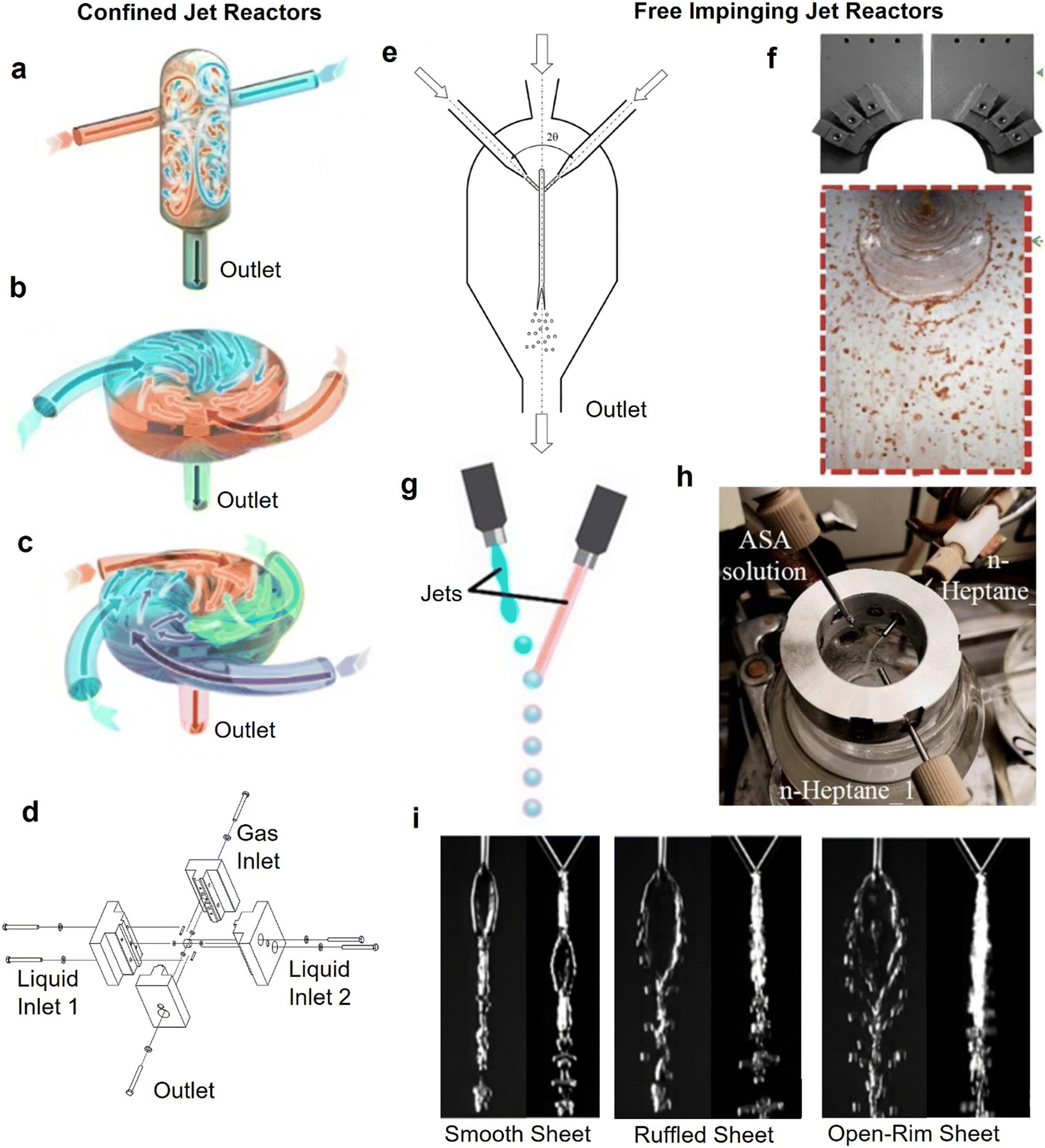 | ||
| Fig. 8 Jet reactors for NP synthesis. (a) Conventional confined impinging jet reactor for flash nanoprecipitation and (b and c) multiple inlet vortex mixers (reproduced from ref. 137 with permission from Elsevier, © 2021). (d) Micro-jet reactor for synthesis of cadmium sulfide nanoparticles (reproduced from ref. 144 with permission from Wiley, © 2019). (e) Free impinging jet reactor enclosed in a mixing chamber for synthesis of lanthanum orthophosphate nanoparticles (reproduced from ref. 145 with permission from Springer, © 2021). (f) Free impinging jet reactor (reproduced from ref. 146 with permission from Elsevier, © 2020). (g) In-air droplet microfluidic reactor (reproduced from ref. 147 with permission from AAAS, © 2018). (h) Three-stream impinging jet reactor for antisolvent crystallisation (reproduced from ref. 131 with permission from Elsevier, © 2021). (i) Flow patterns observed in free impinging jet geometries for increasing flowrates, pictures show side view and front view (reproduced from ref. 148 with permission from Springer, © 2016). | ||
Confined jet reactors
In confined jet reactors (CJR), two or more liquid jets impinge in a liquid-filled mixing chamber surrounded by walls from all adjacent sides. The liquid jets travel through the reactive liquid phase, and upon impingement, the energy is dissipated via creation of eddies in the confined mixing chamber.133,136CJRs' fouling resistance stems from the large volume of the mixing chamber relative to the intensive mixing zone where nanoparticles are rapidly produced, and by the relatively high shear due to the increased flow velocities required for efficient mixing. The use of a CJR can offer effective solution to prevent fouling for very fast nanoparticle formation processes (compared to the residence time in the mixing chamber). This makes them a valuable tool in flow synthesis of nanoparticles involving fast kinetics, which is indicated by dedicated reviews on usage of CJRs for flash nanoprecipitation.65,137
One of the first applications of CJRs in material synthesis, involved two liquid jets (see Fig. 8a), providing rapid mixing in a cylindrical chamber to prepare nanoparticles via flash nanoprecipitation.138 Though the initial CJR geometry developed by Prud'homme and co-workers was widely used, mixing quality of such reactor design is reported to be susceptible to flow imbalance.136 If the flow rates of the two liquid jets are different, the impingement point deviates from the centre, which results in more spatially segregated and heterogeneous mixture of the two liquid streams. The requirement of identical flowrates of the two liquid jets for efficient mixing, makes it challenging to implement the CJR geometry in chemistries requiring different flowrate of the reactant streams. Multiple inlet vortex CJR geometries with two, or four inlet streams mixing in perpendicular fashion, tangentially to the mixing chamber139,140 (see Fig. 8b and c) enables mixing of streams of unequal flowrates, while retaining the ability of CJRs to provide rapid micromixing (in the range of milliseconds for Reynolds number greater than 1600).141 The momentum from each stream contributes independently to drive micromixing in the mixing chamber, enabling to have one or more streams at high flow rate and another stream at a lower flow rate and still get good micromixing. Such multi-vortex CJRs have been demonstrated for continuous flash nanoprecipitation of curcumin without any issues of fouling and clogging.142 A similar design was used by Wojtalik et al.139 but with two inlet channels arranged tangentially to the outlet channel to develop a two inlet vortex CJR for synthesis of molybdenum disulphide nanoparticles. A dead volume-free reactor design was recently introduced by drilling small channels (0.1–1 mm) into a solid cube, creating jet nozzles and a very small mixing chamber (see Fig. 8d).143,144 Small nozzle diameters allowed for higher velocity jets to mix efficiently in the small mixing chamber and a third inert gas phase was used from the top to flush the reactor to avoid clogging.
If nanoparticle formation is not completed before the reactive streams come in contact with the reactor wall, the CJR resembles a standard single-phase system in terms of the fouling characteristics. Hence, the formation kinetics of the nanoparticles, and the CJR dimensions and design are key to avoid fouling of the walls, jet orifices, as well as of the outlet of the mixing chamber.
Free impinging jet reactors
Similar to confined jet reactors, free impinging jet reactors provide intense mixing due to rapid energy dissipation during the impingement,148–150 and confine the mixing zone mid-air (see Fig. 8f), hence limiting the interaction with the walls during and after mixing. These characteristics result in an excellent system to prevent fouling for nanoparticle syntheses involving rapid precursor decomposition and particle formation steps taking place (or being completed) in the confined mixing zone.A FIJR enclosed in a mixing chamber (see Fig. 8e) has been used for the production of a plethora of mineral based nanoparticles such as bismuth orthoferrite,132 lanthanum orthophosphate,145 and titanium dioxide,151 which required a rapid mixing step. An open air FIJR has been used to produce MgO nanocrystals to prevent clogging problems associated with the sol–gel production method.152 FIJRs have been implemented in silver nanoparticle production via rapid synthetic routes where the rapid mixing offered by the system led to improved product quality.148,153 It has been demonstrated that the shape, size and thickness of the liquid sheet formed in the impingement plane, (which are affected by the total flowrate of the jets and the impingement angle), are important parameters in the synthesis, defining the mixing time, and thus the product quality. With increasing jet Weber number different flow regimes such as smooth sheet, ruffled sheet and open rim sheet have been observed after the impingement, as shown in Fig. 8i, which drastically affect the mixing characteristics. For example, in an open-rim sheet (obtained at high Weber numbers), reactants in the detached ligaments and droplets are poorly mixed.148 Knowledge of the flow pattern is especially important for free impinging jets inside a mixing chamber, as the edges of the liquid sheet or detached droplets and ligaments that touch the walls of the mixing chamber, can be a source of fouling accumulation.
Despite its advantages and its non-fouling nature, the application of FIJR in nanoparticle synthesis has been limited. The main disadvantage is the lack of control of reaction residence time due to the reactor only consisting of a small mixing zone before the solution is collected (which means that for successful use of this reactor, the reaction must be complete in the liquid sheet i.e. almost instantaneously). This is further aggravated by the finding that optimal mixing only occurs only at a specific operational window of Weber number,148 preventing the use of flowrate as a means of adjusting residence time. Thus, FIJRs are difficult to implement for processes involving multiple steps such as growth or particle stabilisation. In addition, FIJRs are difficult to accommodate unequal flowrates (which may be a necessity in nanoparticle synthesis), further decreasing their applicability. These disadvantages could potentially be mitigated by drawing inspiration from crystallisation processes. Triple impinging jets have been demonstrated for accommodating unequal flowrates, while coupling a FIJR with a stirred vessel can allow some control of the residence time (see Fig. 8h). Finally, an alternative operation-mode of impinging jets is to manipulate liquid jets with vibrating piezoelectric elements in order to produce droplets, which then merge in air to form monodisperse droplets (see Fig. 8g).147 Such operation can also potentially be used for fouling free nanoparticle synthesis, specifically for fast kinetics while allowing increased control (e.g., by modulating the rate of droplet impingement).
Multiphase reactors
Multiphase flow reactors have a long history for nanoparticle synthesis, most commonly using segmented flow, where one immiscible phase gets dispersed (dispersed phase) in another one (continuous phase). Segmented flow can improve mixing throughout the synthesis, provide a plug flow like residence time distribution and compartmentalise the phases into small volumes, making it popular for high-throughput screening. Fouling can be prevented when the continuous phase obstructs the dispersed phase, preventing the nanoparticle synthesis domain from contacting the reactor wall. Furthermore, segmented flow can prevent the sedimentation of larger particles, such as agglomerates, forming when nanoparticle stabilisation is insufficient or incomplete.Therefore, multiphase reactors offer solutions where nanoparticle syntheses require low flow rates and/or long residence times, mixing throughout the synthesis (improved compared to single phase equivalent), and where stabilisation is a concern. This makes them an important flow chemistry tool, which is reflected by the many dedicated reviews on multiphase flow reactors for nanoparticle synthesis.154–158 Most commonly used are two-phase segmented flow reactors, not least, due to their simplicity in design and operation.
Two-phase segmented flow reactors
Gas–liquid segmentation yields many of the advantages described and has been used successfully for nanoparticle flow syntheses,159–162 also at large scale (∼l per h scale),154,163 and using the dispersed gas phase as reagent164–167 or for controlled vaporisation from the liquid phase.168 Gas–liquid segmentation, however, has the liquid phase as the continuous phase. Hence, the reactive (liquid) phase remains in contact with the wall (see Fig. 9a, top) and there is no protective film between the reacting liquid and the wall, preventing fouling.169 Exceptions of non-fouling gas–liquid multiphase systems are the discussed free impinging jets, levitating droplets, e.g., as used for dynamic particle characterisation in solution (at synchrotron facilities170,171 or in rare cases via aerosols.172,173 | ||
| Fig. 9 Two-phase segmented flow reactors for NP synthesis. (a) Flow pattern in gas–liquid (top; grey liquid phase contacts the wall, see red dotted line) and liquid–liquid (bottom; grey dispersed liquid phase not contacting the wall) flow (reproduced from ref. 169 with permission from Elsevier, © 2012). (b) Reagent mixing before segmentation.178 (c) Reagent mixing during segmentation using chip (top) and capillary (middle & bottom) systems (reproduced from ref. 179, 175 and 176 with permission from Wiley, © 2015, and the Royal Society of Chemistry, © 2012 & 2014). (d) Reagent mixing after segmentation merging droplets (top; Qx, Qy are reagent solution flow rates) via electrical coalescence (bottom; electrodes in black) (reproduced from ref. 180 with permission from Wiley, © 2008). | ||
To prevent fouling via segmentation, the synthesis must happen in the dispersed phase, which is possible using liquid–liquid segmented flow (see Fig. 9a, bottom). Therefore, liquid–liquid segmentation is well-established for non-fouling nanoparticle synthesis in droplet (dispersed phase is spherical) or slug (dispersed phase extends to the reactor wall without making contact) flow systems, facilitating long term operation but also larger scale production, e.g., at the 1–10 g per h scale.174
From a reactor engineering perspective, it is not complicated to establish liquid–liquid segmentation. Off-the-shelf components such as standard microfluidic chips or Y- and T-connectors (made of material with sufficient wettability with the continuous phase) are usually sufficient. What is challenging, however, is to add reagents to already formed droplets or slugs. As a result, reagent addition/mixing as specified by the synthetic protocol is usually completed prior to segmentation. Therefore, liquid–liquid segmentation is applied where reagent solutions (e.g., precursor and reducing agent) can be mixed before segmentation (see Fig. 9b) and the reaction/nanoparticle particle formation is not fast enough to cause fouling before then. This is for example the case when precursor decomposition requires elevated temperatures and a heating step follows segmentation. If the reaction/nanoparticle particle formation kinetics are fast and fouling is of concern as soon as the reagent solutions mix, the mixing and segmentation step can be combined. This requires more intricate reactor designs to ensure that the reactive solutions are being sufficiently engulfed by the continuous phase when coming in contact during droplet or slug formation. It can be achieved using customised droplet chip systems feeding solutions directly where segmentation occurs with or without additional separating streams, or capillary-based systems allowing segments to form distant from the reactor wall (see Fig. 9c).175,176 The merging of reagent droplets (containing different solutions) after segmentation can be achieved, for example by electrically controlled droplet coalescence (see Fig. 9d) or hydrodynamic coupling.177 Alternative ways to initiate nanoparticle formation after segmentation are reagent supply through transfer from the continuous phase or via radiation (e.g., UV photons). Examples of non-fouling nanoparticle syntheses using such liquid–liquid segmented flow reactor concepts are provided in Table 2. Disadvantages originating from the dispersion in a continuous liquid phase are discussed at the end of the multiphase reactor section.
| Type | Nanoparticle (NP) synthesis | Ref. |
|---|---|---|
| a Perfluorinated continuous phase. | ||
| Reagent mixing before segmentation | Gold and silver NPs, mixing precursor solution and reducing agent solution prior segmentation via T-connectors; heating post segmentation | 178, 181–183 |
| Seeded growth of gold NPs, mixing seed solution with new precursor solution prior simultaneous mixing with hydrogen peroxide and segmentation in standard cross connector; heating post segmentation | 184 | |
| Fe-metal–organic framework NPs, mixing precursor (DMF–water solution) with ligand (DMF solution) prior segmentation via Y-connectors; heating post segmentation | 185 | |
| Metal and metal oxide NPs, thermal decomposition synthesis in polyol dispersed in n-dodecane after mixing precursors and reactants/additive solutions in micro-mixer prior to segmentation; heating up to 180 °C post segmentation | 174 | |
| Metal halide perovskite quantum dot NPs with tuned bandgaps through reacting a cesium lead bromide solution with zinc iodine/chloride/bromide, oleic acid and oleylamine. Reagent concentrations were set mixing six reagent solutions via standard cross connectors and passive mixers before segmentation in a standard T connectora | 186 | |
| Reagent mixing during segmentation | Seeded growth of gold NPs, mixing three streams (precursor, reducing agent/stabiliser and seed solution) when forming droplets using a droplet chip reactor | 176, 187 |
| Silver NPs, mixing the precursor and the reducing agent/stabiliser solution during segmentation via 4-way connector/micromixer; heating during and post segmentation | 169 | |
| Rhodium and rhodium–silver NPs, thermal decomposition/reduction synthesis merging precursors and additive streams (in ethylene glycol) just before segmentation in 3D printed droplet generator;a heating up to 120 °C post segmentation | 188 | |
| Iron oxide NPs via co-precipitation, mixing the ferric and ferrous iron precursor solution with the base solution (NH4OH) and preventing premature mixing before segmentation is completed using a separating water stream in a droplet chip reactor or droplet formation distant from the wall in a capillary-based system with two symmetric silica capillaries meeting at a 90° angle in the centre of the host channel feeding in the continuous phase | 175, 189, 190 | |
| Gold NPs, mixing precursors and polyvinyl pyrrolidone solution with ascorbic acid solution and segmentation in standard cross connector or counter (continuous phase)-current flow focusing reactor | 191, 192 | |
| Palladium NPs, mixing three aqueous reactive solutions (I precursor, II capping agent and stabilisers, III reducing agent) introduced via silica capillaries with side-by-side outlets forming droplets distant from the wall in a capillary-based system; heating post segmentation | 193 | |
| Palladium NPs, mixing palladium precursor solution with ascorbic acid and additive solution, where droplets form using a droplet chip reactor | 194 | |
| Lead sulfide/selenide NPs, mixing lead precursor solution (octadecene and oleic acid mixture) with silicon or selenium precursor solution (e.g., in octadecene) during segmentation in standard cross connector (the central stream was the continuous phase);a heating up to ∼150 °C post segmentation | 12, 179 | |
| Cesium lead halide quantum dot NPs, mixing a cesium precursor solution with a lead halide precursor solution (octadecene and oleic acid mixture) during segmentation in standard cross connector (the central stream was the continuous phase);a heating up to ∼230 °C post segmentation | 195 | |
| Cadmium sulfide, cadmium selenide NPs, mixing cadmium precursor solution with the sulphur precursor solution, while preventing premature mixing before segmentation using a droplet chip reactor with a separating water stream | 196 | |
| Cadmium selenide quantum dot NPs, thermal decomposition synthesis mixing cadmium and selenium precursors solutions (both in octadecene) via two symmetric silica capillaries meeting at an 90° angle in the centre of the host channel feeding in the continuous phasea (heating >220 °C post segmentation) and titanium oxide and silver NPs, mixing aqueous titanium or silver precursor solution with base or reducing agent solution using the same reactor design | 197 | |
| Reagent mixing after segmentation | Iron oxide NPs via co-precipitation, merging droplets of the ferric and ferrous iron precursor solution and the base solution (electrically controlled droplet coalescence, and via hydrodynamic coupling) in droplet chip reactors | 180, 198 |
| Cadmium sulphide NPs, merging droplets of the cadmium and sulphur precursor solutions (passive droplet fusion via constrained channel) in droplet chip reactor | 177 | |
| Seeded growth of branched gold NPs, mixing precursor and additives when forming initial droplets prior to addition of seed solution to droplets (electrically controlled feed addition via integrated electrodes) using droplet chip reactor | 199 | |
| Reagent supply after segmentation | Gold nanoclusters, segmenting precursor solution in continuous phase (carbon monoxide saturated heptane) providing reducing agent via transfer from continuous to dispersed phase | 200 |
| Radiation induced reaction after segmentation | Gold NPs, mixing precursor solution and mild reducing agent solution prior segmentation via T-connectors; UV reduction of precursor post segmentation | 201 |
| Palladium, platinum, rhodium and iridium NPs, mixing precursors with photo-initiator and segmenting fluid in cross connector; UV induced reduction of precursor post segmentationa | 202 | |
Although this variety of liquid–liquid segmented flow reactors does offer some flexibility, they have mostly been used for simple synthetic protocols with a single reagent addition step. Hence, translating batch protocols with staggered reagent addition steps into flow remains challenging using liquid–liquid flow reactors, which is why the more complex but more versatile multiple-phase (≥3) reactor systems are gaining interest.
Multiple-phase segmented flow reactors
Although more complex due to the introduction of an additional phase, multiple-phase segmentation such as gas–liquid–liquid flow, can overcome the limitation of single reagent addition step. This was first demonstrated by Nightingale et al. showing how gas–liquid–liquid flow makes it possible to repeatedly add controlled quantities of reagent solution to already formed droplets.203 Using a customised PTFE element for (gas–liquid–liquid) droplet generation and fused silica tubing piercing a capillary for subsequent reagent addition, they showed how vital the gas phase is. The gas phase introduced during droplet formation spaced the droplets uniformly (also referred to as self-synchronisation) and suppressed new droplet formation when feeding new reagent solution (of the same phase as the droplets), hence, allowing to feed solution to the already formed droplets only (see Fig. 10a). The same work demonstrated the versatility of gas–liquid–liquid flow for non-fouling multistep nanoparticle syntheses showing a five-stage quantum dot synthesis (1× initial nanoparticle formation, 4× precursor solution addition after segmentation) at low flow rates (<200 μL min−1). Soon after, gas–liquid–liquid segmentation was demonstrated also at higher flow rates for palladium nanoparticle synthesis (∼5 mL min−1 yielding ∼10 L d−1, see Fig. 10b) using a reactor made from off-the-shelf parts.18 | ||
| Fig. 10 Gas–liquid–liquid segmented flow reactors for NP synthesis. (a) Initial demonstration of multi-step reagent addition into slugs using capillary-based system (reproduced from ref. 203 with permission from Springer, © 2014). (b) Litres per day production of palladium NPs via sodium borohydride reduction of precursor in capillary reactor made from off-the-shelf parts (reproduced from ref. 18 with permission from the Royal Society of Chemistry, © 2017). (c) Quantum dot NP synthesis using customised 4 + 1 way connector for segmentation (two reagents were mixed just before segmentation) reproduced from ref. 204 with permission from Wiley, © 2021. (d) Four-phase segmented flow (gas–liquid–liquid–liquid) for automatically conducted ligand exchange of cadmium selenide quantum dots (reproduced from ref. 206 with permission from the Royal Society of Chemistry, © 2021). (e) Modular three-phase segmented flow reactor platform with custom elements for droplet generation and reagent addition into droplets (shown in images 1–4) used to synthesise iron oxide NPs (reproduced from ref. 37 with permission from the Royal Society of Chemistry, © 2023). | ||
More recently, gas–liquid–liquid segmentation was used for a two stage lead halide perovskite quantum dot nanoparticle synthesis operated autonomously via robotic experimentation to tune particle/optical properties.204 The capillary-based reactor comprised a 4 + 1 way connector for segmentation (two reagents were mixed just before segmentation) which was customised to minimise precursor contact time before segmentation, with the continuous liquid phase inlet immediately followed by introduction of the gas phase (see Fig. 10c) and a standard T-connector for the subsequent reagent addition step.204 A similar gas–liquid–liquid segmented flow reactor design was used for lead halide perovskite nanoparticle synthesis.205 Gas–liquid–liquid–liquid (i.e., quaternary segmentation, see Fig. 10d) flow reactors were also used successfully by the same group to process reactive phase systems for continuous ligand exchange of quantum dots using commercially available crosses, connectors and tubing only.206
Non-fouling gas–liquid–liquid flow reactors proved their ability to produce nanomaterials at small and large scales and versatility for multistep nanoparticle syntheses. Nevertheless, examples are scarce indicating reactor design and operation challenges for gas–liquid–liquid systems. These may include the relatively high pressure drop for long reactors or high flow rates, the uniform multiple-phase segmentation (most studies used customised droplet generators) requiring pumps providing consistent feed rates, complications due to the compressible gas phase (e.g., its expansion with heating or with pressure fluctuations), or the robust addition of reagent solution into droplets over a broad flow rate regime and the continuous liquid phase required (all gas–liquid–liquid reactors for nanoparticle synthesis used perfluorinated oils). To address these challenges a modular “Lego like” flow reactor platform designed specifically for gas–liquid–liquid segmented systems was presented recently (see Fig. 10e).37 Its reactor elements were machined from highly hydrophobic plastic for robust segmentation, the droplet generator had a movable nozzle to control the droplet spacing and all elements were made transparent for visual inspection. The platform was showcased for a multistep iron oxide nanoparticle synthesis involving two reagent addition steps and a heating step (to 70 °C) using heptane as continuous liquid phase.
Liquid–liquid or gas–liquid–liquid flow reactors often appear as the obvious choice to prevent fouling for nanoparticle flow syntheses. The consequences of dispersing the reactive liquid phase, in addition to the additional separation step required, need to be considered carefully. Solutes that can participate in the nanoparticle formation process, can enter but also exit the dispersed phase through the large interfacial area with the continuous phase. In addition, nanoparticles with higher affinity to the continuous phase might accumulate at this interphase or transfer completely to the continuous phase.207 It is therefore not surprising, that nanoparticle synthesis in segmented flow reactors is sensitive to the hydrodynamics within the dispersed phase as well as the droplet/slug size, velocity, or spacing (which is also important to avoid coalescence).169,208–210
Fouling monitoring and detection
Once a flow reactor clogs, it can be very difficult to remove the constrictions. Dissolving the constrictions can be challenging as they are stalled in the processed solution, making it hard to replace the solvents in their surroundings, i.e., a simple rinse is not possible. Higher pressures (if possible) and ultra-sonication to break up constrictions or temperature treatments (if the constriction's solubility varies with temperature) do not guarantee success. Detecting fouling early is therefore not only important to avoid any interference with the synthetic conditions, it could also save a “reactor's life”.Many fouling monitoring techniques rely on pressure measurements to detect pressure drop increase due to constrictive fouling. Sensitive and/or multiple pressure probes are recommended, as flow reactors (and especially microreactors) have an inherently high pressure drop and relative changes due to thin films of deposit are not easily detected. A continuous increase in pressure indicates progressive fouling, whereas a sudden increase indicates a rapidly occurring or growing constriction, for example due the onset of crystallisation (or solidification) fouling, local deposition of agglomerated particles or complete channel blockage in multi (parallel) channel systems.211,212 Irregular but reoccurring pressure fluctuations can indicate the emergence of constrictions and their subsequent break up due to the higher pressure. An addition or alternative to fouling monitoring via pressure measurements are temperature measurements where heating is involved, e.g., at the outlet or via thermal imaging of the whole system, as well as flow rate measurements in multi (parallel) channel systems.212,213
Detecting surface fouling, local or traversed, with these techniques is challenging, as while the synthetic conditions might be affected, the operation conditions remain mostly unaffected. Hence, alternative options need to be considered. Optical inspections by eye or any imaging (or spectroscopic) technique remain a valuable tool to detect surface fouling during or post synthesis in transparent systems, such as reactors made of glass or transparent plastic. For non-transparent reactors, e.g. made of metal or silicon, imaging techniques based on X-ray or near-mid infrared radiation could aid fouling identification.214,215
Where imaging is difficult, post synthesis reactor weighing or cleaning can be used to assess fouling. When using cleaning, the reactor should be rinsed first with a solvent (e.g. the solvent used for the synthesis) that removes all process solution, before injecting a cleaning solution that dissolves possible accumulations at the wall. This could be for example aqua regia for noble metal nanoparticle synthesis or hydrochloric acid for iron or iron oxide nanoparticle synthesis. Changes of the cleaning solution effluent, e.g., increased conductivity or coloration quantified via UV-vis, can indicate residuals post synthesis, hence fouling during the synthesis.85
The best way to demonstrate that a flow reactor operates truly fouling free and that the nanoparticle quality does not change over time are long term operation studies. This proof can be provided through sample collection during the operation and post-synthesis characterisation of the nanoparticles including the yield/particle concentration. Real-time nanoparticle characterisation (to be used in-line, on-line or at-line) would be more practical from a process control perspective. The most valuable information for nanoparticle syntheses, however, is hardly accessible in real time as measurements of size and shape, particle concentration, and functionalisation are often restricted to post-synthesis characterisation only. Exceptions are quantum dots or plasmonic nanoparticles where fluorescence or UV-vis spectroscopy can provide valuable information of the synthesised nanoparticles including their concentration, hence the occurrence of fouling.178 Also indirect fouling detection through (close to) real-time measurements of changes in the nanoparticle solution's electrical or optical properties (e.g., conductivity, obscuration, refractive index, etc.) or a reduced precursor conversion, can be enabled via process analytical technology that is well established for fine chemical production in flow reactors.216–218
Conclusion and perspectives
Flow reactors are beyond doubt part of the journey to develop and produce new nanomaterials for current and future applications. Their characteristics giving advantages over their batch equivalent, however, make them more prone to fouling. This problem is well recognised but requires more attention.Many studies on nanoparticle synthesis via flow chemistry are motivated by scalable production through long-term operation. Investigations of nanoparticle property changes over operation time (minutes, hours, days), however, are scarce and samples are commonly taken just (and only) after steady state operation is reached. This makes it challenging to evaluate the true potential of many flow syntheses reported.
This review discussed the several flow reactor designs that have been successfully used to mitigate fouling. The diversity of reactors discussed shows that there is no single non-fouling flow reactor suitable for nanomaterial synthesis at all relevant synthetic protocols, but there exists a well-equipped flow chemistry toolbox with reactors and reactor materials to choose from.
Where possible, flow reactor design should follow initial studies (in batch or flow) to understand the time scales of the synthesis, the particle formation steps involved, and the sensitivity in terms of process parameters (e.g., mixing time, heating rate and temperature, pH, concentration etc.) to understand what is critical to the synthesis, as well as long term operation studies to screen reactor materials and synthetic conditions. The final reactor design should be tailored to each nanoparticle synthesis to meet the critical process parameters and product specifications, while preventing fouling to ensure operational stability. Not necessarily a single reactor, but a combination of multiple flow reactors (e.g., for different stages of nanoparticle formation), may be most suited to synthesise nanoparticles of the required quality without fouling. Reactor fouling needs to be considered at the early design stages and developing flow syntheses “worrying about the problem later” should be avoided.
In line with the discussion on nanoparticle synthesis induced fouling and the discussed reactor technologies, we suggest the following simplified guidelines (visualised in Fig. 11) to design non-fouling flow systems.
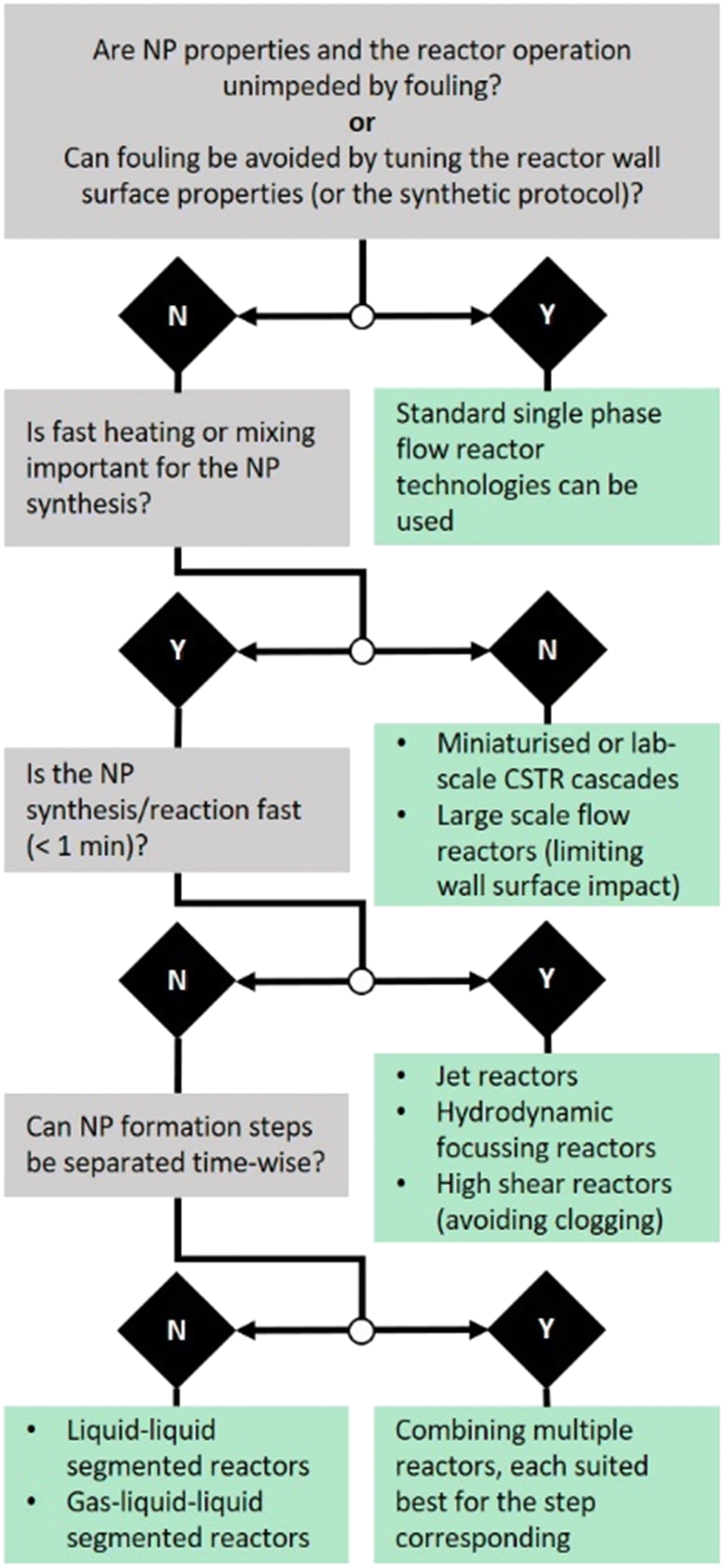 | ||
| Fig. 11 Decision tree of guidelines to design non-fouling flow reactors. | ||
1. Are the reactor operation or the nanoparticle properties unimpeded by fouling? Can fouling be avoided by selecting a suitable reactor wall material considering its (macroscopic) zeta potential and wettability with the solid phase formed?
If the answer to one of the two questions is yes, “standard” single phase reactor systems can be considered.
2. Is fast mixing or fast heating important for any of the nanoparticle formation steps such as precursor decomposition and particle formation?
If yes, and reaction times are short, high shear reactors avoiding clogging as well as flow focusing or jet reactors seem sensible. If yes, but longer reaction time is required, liquid–liquid segmentation can be considered. Liquid–liquid segmentation is a convenient choice where fast heating is required and can provide fast mixing too (usually slower than for the high shear reactors discussed), e.g., before or during segmentation. Here, reaction time refers to the entire synthesis (if completed in the reactor) or the reaction step(s) performed in the reactor.
If no, miniaturised or lab scale CSTR cascades can be used or other larger scale flow reactors.
3. Can the particle formation steps be separated time-wise, with different critical process parameters for each step? If yes, the flow reactor could comprise multiple elements for the steps involved. Examples are:
A: impinging jet reactor for mixing sensitive and fouling prone precursor decomposition & particle formation, followed by a CSTR cascade system for particle growth requiring long residence times.
B: single phase reactor systems for precursor mixing and liquid–liquid segmentation before nanoparticle formation (e.g., when initiated via heating or UV radiation).
C: flow focusing reactors for precursor decomposition and particle formation followed by simple single phase reactors, e.g., using coiled flow inverters of suitable wall material, for particle growth and stabilisation.
Designing non-fouling flow reactors is possible but not trivial. The way to tame flow chemistry's “demon of reactor fouling” is guided by the synthesis considered and its particle formation kinetics. The powerful tools available range from high-shear systems, specialised reactor wall materials, CSTR cascades, flow focusing reactors and jet reactors, to multiphase systems such as liquid–liquid and recently established versatile gas–liquid–liquid flow reactors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the EPSRC UK for financial support (EP/M015157/1, EP/V050796/1).References
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- V. F. Cardoso, A. Francesko, C. Ribeiro, M. Bañobre-López, P. Martins and S. Lanceros-Mendez, Adv. Healthcare Mater., 2018, 7, 1700845 CrossRef PubMed.
- M. Iranmanesh and J. Hulliger, Chem. Soc. Rev., 2017, 46, 5925–5934 RSC.
- V. Montes-Garcia, M. A. Squillaci, M. Diez-Castellnou, Q. K. Ong, F. Stellacci and P. Samorì, Chem. Soc. Rev., 2021, 50, 1269–1304 RSC.
- H. Wei Tan, J. An, C. Kai Chua, T. Tran, H. W. Tan, J. An, C. K. Chua and T. Tran, Adv. Electron. Mater., 2019, 5, 1800831 CrossRef.
- V. Sebastian, M. Arruebo and J. Santamaria, Small, 2014, 10, 835–853 CrossRef CAS PubMed.
- R. Bawa, G. F. Audette and B. E. Reese, Handbook of clinical nanomedicine: Law, business, regulation, safety, and risk, Pan Stanford Publishing Pte. Ltd., Boca Raton, 2016 Search PubMed.
- T. I. Ramos, C. A. Villacis-Aguirre, K. V. López-Aguilar, L. Santiago Padilla, C. Altamirano, J. R. Toledo and N. Santiago Vispo, Pharmaceutics, 2022, 14, 247 CrossRef CAS PubMed.
- X. Z. Lin, A. D. Terepka and H. Yang, Nano Lett., 2004, 4, 2227–2232 CrossRef CAS.
- V. Sebastián and K. F. Jensen, Nanoscale, 2016, 8, 15288–15295 RSC.
- I. Lignos, L. Protesescu, S. Stavrakis, L. Piveteau, M. J. Speirs, M. A. Loi, M. V. Kovalenko and A. J. deMello, Chem. Mater., 2014, 26, 2975–2982 CrossRef CAS.
- A. A. Volk, R. W. Epps and M. Abolhasani, Adv. Mater., 2021, 33, 2004495 CrossRef CAS PubMed.
- R. W. Epps, A. A. Volk, K. G. Reyes and M. Abolhasani, Chem. Sci., 2021, 12, 6025–6036 RSC.
- H. Tao, T. Wu, S. Kheiri, M. Aldeghi, A. Aspuru-Guzik and E. Kumacheva, Adv. Funct. Mater., 2021, 31, 2106725 CrossRef CAS.
- J. B. Deshpande and A. A. Kulkarni, Chem. Eng. Technol., 2018, 41, 157–167 CrossRef CAS.
- M. O. Besenhard, L. Panariello, C. Kiefer, A. P. LaGrow, L. Storozhuk, F. Perton, S. Begin, D. Mertz, N. T. K. Thanh and A. Gavriilidis, Nanoscale, 2021, 13, 8795–8805 RSC.
- W. K. Wong, S. K. Yap, Y. C. Lim, S. A. Khan, F. Pelletier and E. C. Corbos, React. Chem. Eng., 2017, 2, 636–641 RSC.
- S. Bomhard, J. Schramm, R. Bleul, R. Thiermann, P. Höbel, U. Krtschil, P. Löb and M. Maskos, Chem. Eng. Technol., 2019, 42, 2085–2094 CrossRef.
- O. Długosz and M. Banach, React. Chem. Eng., 2020, 5, 1619–1641 RSC.
- J. Ahn, J. Ko, S. Lee, J. Yu, Y. T. Kim and N. L. Jeon, Adv. Drug Delivery Rev., 2018, 128, 29–53 CrossRef CAS PubMed.
- Z. Liu, J. Zhu, C. Peng, T. Wakihara and T. Okubo, React. Chem. Eng., 2019, 4, 1699–1720 RSC.
- J. Ma, S. M. Y. Lee, C. Yi and C. W. Li, Lab Chip, 2017, 17, 209–226 RSC.
- J. Sui, J. Yan, D. Liu, K. Wang and G. Luo, Small, 2020, 16, 1902828 CrossRef CAS PubMed.
- P. Shrimal, G. Jadeja and S. Patel, Chem. Eng. Res. Des., 2020, 153, 728–756 CrossRef CAS.
- J. Tao, S. F. Chow and Y. Zheng, Acta Pharm. Sin. B, 2019, 9, 4–18 CrossRef PubMed.
- M. Maeki, N. Kimura, Y. Sato, H. Harashima and M. Tokeshi, Adv. Drug Delivery Rev., 2018, 128, 84–100 CrossRef CAS PubMed.
- M. J. W. Evers, J. A. Kulkarni, R. van der Meel, P. R. Cullis, P. Vader and R. M. Schiffelers, Small Methods, 2018, 2, 1700375 CrossRef.
- J. Dong, J. Lau, S. A. Svoronos and B. M. Moudgil, Kona Powder Part. J., 2022, 39, 262–269 CrossRef.
- Chemistry of Nanomaterials - a Practical Guide, ed. C. Kumar, De Gruyter, Berlin, Boston, 2020 Search PubMed.
- A. V. Nikam, B. L. V. Prasad and A. A. Kulkarni, CrystEngComm, 2018, 20, 5091–5107 RSC.
- P. Zardi, T. Carofiglio and M. Maggini, Chem. – Eur. J., 2021, 28, 1–16 Search PubMed.
- R. Abedini-Nassab, M. Pouryosef Miandoab and M. Şaşmaz, Micromachines, 2021, 12, 768 CrossRef PubMed.
- N. Ajinkya, X. Yu, P. Kaithal, H. Luo, P. Somani and S. Ramakrishna, Materials, 2020, 13, 4644 CrossRef CAS PubMed.
- R. Baber, L. Mazzei, N. T. K. Thanh and A. Gavriilidis, RSC Adv., 2015, 5, 95585–95591 RSC.
- P. Erfle, J. Riewe, H. Bunjes and A. Dietzel, Lab Chip, 2021, 21, 2178–2193 RSC.
- M. O. Besenhard, S. Pal, L. Storozhuk, S. Dawes, N. T. K. Thanh, L. Norfolk, S. Staniland and A. Gavriilidis, Lab Chip, 2023, 23, 115–124 RSC.
- J. Dong, S. A. Svoronos, J. C. Lau and B. Moudgil, Chem. Eng. Sci., 2022, 249, 117293 CrossRef CAS.
- H. Huang, H. Du Toit, M. O. Besenhard, S. Ben-Jaber, P. Dobson, I. Parkin and A. Gavriilidis, Chem. Eng. Sci., 2018, 198, 422–430 CrossRef.
- A. Majumder and Z. K. Nagy, Cryst. Growth Des., 2015, 15, 1129–1140 CrossRef CAS.
- L. Schnöing, W. Augustin and S. Scholl, Food Bioprod. Process., 2020, 121, 1–19 CrossRef.
- E. Tummons, Q. Han, H. J. Tanudjaja, C. A. Hejase, J. W. Chew and V. V. Tarabara, Sep. Purif. Technol., 2020, 248, 116919 CrossRef CAS.
- M. Schoenitz, L. Grundemann, W. Augustin and S. Scholl, Chem. Commun., 2015, 51, 8213–8228 RSC.
- N. Epstein, Heat Transfer Eng., 1983, 4, 43–56 CrossRef CAS.
- A. A. Lapkin, K. Loponov, G. Tomaiuolo and S. Guido, in Sustainable Flow Chemistry: Methods and Applications, ed. L. Vaccaro, Wiley-VCH, Weinheim, 2017, pp. 277–308 Search PubMed.
- A. Marin, H. Lhuissier, M. Rossi and C. J. Kähler, Phys. Rev. E, 2018, 97, 021102 CrossRef CAS PubMed.
- G. C. Agbangla, É. Climent and P. Bacchin, Comput. Fluids, 2014, 94, 69–83 CrossRef.
- Y. Chen, J. C. Sabio and R. L. Hartman, J. Flow Chem., 2015, 5, 166–171 CrossRef.
- L. Sicignano, G. Tomaiuolo, A. Perazzo, S. P. Nolan, P. L. Maffettone and S. Guido, Chem. Eng. J., 2018, 341, 639–647 CrossRef CAS.
- R. L. Hartman, J. R. Naber, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Org. Process Res. Dev., 2010, 14, 1347–1357 CrossRef CAS.
- A. Lassenberger, T. A. Grünewald, P. D. J. van Oostrum, H. Rennhofer, H. Amenitsch, R. Zirbs, H. C. Lichtenegger and E. Reimhult, Chem. Mater., 2017, 29, 4511–4522 CrossRef CAS PubMed.
- P. Guardia, J. Pérez-Juste, A. Labarta, X. Batlle and L. M. Liz-Marzán, Chem. Commun., 2010, 46, 6108–6110 RSC.
- K. J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267–276 RSC.
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, ACS Nano, 2015, 9, 7052–7071 CrossRef CAS PubMed.
- P. Georgiev, A. Bojinova, B. Kostova, D. Momekova, T. Bjornholm and K. Balashev, Colloids Surf., A, 2013, 434, 154–163 CrossRef CAS.
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- E. E. Finney and R. G. Finke, J. Colloid Interface Sci., 2008, 317, 351–374 CrossRef CAS PubMed.
- J. Polte, X. Tuaev, M. Wuithschick, A. Fischer, A. F. Thuenemann, K. Rademann, R. Kraehnert and F. Emmerling, ACS Nano, 2012, 6, 5791–5802 CrossRef CAS PubMed.
- K. Georgieva, D. J. Dijkstra, H. Fricke and N. Willenbacher, J. Colloid Interface Sci., 2010, 352, 265–277 CrossRef CAS PubMed.
- M. Wuithschick, S. Witte, F. Kettemann, K. Rademann and J. Polte, Phys. Chem. Chem. Phys., 2015, 17, 19895–19900 RSC.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- G. Cotin, C. Kiefer, F. Perton, D. Ihiawakrim, C. Blanco-Andujar, S. Moldovan, C. Lefevre, O. Ersen, B. Pichon, D. Mertz and S. Bégin-Colin, Nanomaterials, 2018, 8, 881 CrossRef PubMed.
- C. Wei and Q. Liu, CrystEngComm, 2017, 19, 3254–3262 RSC.
- M. O. Besenhard, A. P. LaGrow, A. Hodzic, M. Kriechbaum, L. Panariello, G. Bais, K. Loizou, S. Damilos, M. Margarida Cruz, N. T. K. Thanh and A. Gavriilidis, Chem. Eng. J., 2020, 399, 125740 CrossRef CAS.
- W. S. Saad and R. K. Prud'Homme, Nano Today, 2016, 11, 212–227 CrossRef CAS.
- V. Ball, Front. Bioeng. Biotechnol., 2018, 6, 109 CrossRef PubMed.
- H. Okudera and A. Hozumi, Thin Solid Films, 2003, 434, 62–68 CrossRef CAS.
- A. H. Bari, R. B. Jundale and A. A. Kulkarni, Chem. Eng. J., 2020, 398, 125427 CrossRef CAS.
- J. Polte, R. Erler, A. F. Thünemann, F. Emmerling and R. Kraehnert, Chem. Commun., 2010, 46, 9209 RSC.
- H. Gavilán, E. H. Sánchez, M. E. F. Brollo, L. Asín, K. K. Moerner, C. Frandsen, F. J. Lázaro, C. J. Serna, S. Veintemillas-Verdaguer, M. P. Morales and L. Gutiérrez, ACS Omega, 2017, 2, 7172–7184 CrossRef PubMed.
- L. Storozhuk, M. O. Besenhard, S. Mourdikoudis, A. P. LaGrow, M. R. Lees, L. D. Tung, A. Gavriilidis and N. T. K. Thanh, ACS Appl. Mater. Interfaces, 2021, 13, 45870–45880 CrossRef CAS PubMed.
- C. Henry, J. P. Minier and G. Lefèvre, Adv. Colloid Interface Sci., 2012, 185–186, 34–76 CrossRef CAS PubMed.
- C. M. Cejas, F. Monti, M. Truchet, J. P. Burnouf and P. Tabeling, Langmuir, 2017, 33, 6471–6480 CrossRef CAS PubMed.
- J. Wagner and J. M. Köhler, Nano Lett., 2005, 5, 685–691 CrossRef CAS PubMed.
- T. Luxbacher, The Zeta Potential for Solid Surface Analysis - A practical guide to streaming potential measurement | Web-Books in the Austria-Forum, Anton Paar, 2014 Search PubMed.
- J. L. Perry and S. G. Kandlikar, Microfluid. Nanofluid., 2008, 5, 357–371 CrossRef.
- I. Chowdhury and S. L. Walker, J. Colloid Interface Sci., 2012, 369, 16–22 CrossRef CAS PubMed.
- N. Sen, R. Chakravarty, K. K. Singh, S. Chakraborty, L. Panicker and K. T. Shenoy, Chem. Eng. Process., 2022, 109036 CrossRef CAS.
- G. Gkogkos, M. O. Besenhard, L. Storozhuk, N. Thi Kim Thanh and A. Gavriilidis, Chem. Eng. Sci., 2022, 251, 117481 CrossRef CAS.
- S. G. Yiantsios and A. J. Karabelas, Int. J. Multiphase Flow, 1998, 24, 283–293 CrossRef CAS.
- D. C. Leslie, A. Waterhouse, J. B. Berthet, T. M. Valentin, A. L. Watters, A. Jain, P. Kim, B. D. Hatton, A. Nedder, K. Donovan, E. H. Super, C. Howell, C. P. Johnson, T. L. Vu, D. E. Bolgen, S. Rifai, A. R. Hansen, M. Aizenberg, M. Super, J. Aizenberg and D. E. Ingber, Nat. Biotechnol., 2014, 32, 1134–1140 CrossRef CAS PubMed.
- T. Gu, A. Meesrisom, Y. Luo, Q. N. Dinh, S. Lin, M. Yang, A. Sharma, R. Tang, J. Zhang, Z. Jia, P. D. Millner, A. J. Pearlstein and B. Zhang, Food Control, 2021, 130, 108275 CrossRef CAS.
- C. Howell, A. Grinthal, S. Sunny, M. Aizenberg and J. Aizenberg, Adv. Mater., 2018, 30, 1802724 CrossRef PubMed.
- H. Geng and S. K. Cho, Lab Chip, 2019, 19, 2275–2283 RSC.
- M. O. Besenhard, A. P. Lagrow, S. Famiani, M. Pucciarelli, P. Lettieri, N. T. K. Thanh and A. Gavriilidis, React. Chem. Eng., 2020, 5, 1474–1483 RSC.
- C. Howell, T. L. Vu, C. P. Johnson, X. Hou, O. Ahanotu, J. Alvarenga, D. C. Leslie, O. Uzun, A. Waterhouse, P. Kim, M. Super, M. Aizenberg, D. E. Ingber and J. Aizenberg, Chem. Mater., 2015, 27, 1792–1800 CrossRef CAS.
- A. K. Epstein, T. S. Wong, R. A. Belisle, E. M. Boggs and J. Aizenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13182–13187 CrossRef CAS PubMed.
- N. Akkurt, C. L. Altan and M. F. Sarac, J. Supercond. Novel Magn., 2022, 35, 615–623 CrossRef CAS.
- E. Bertuit, S. Neveu and A. Abou-Hassan, Nanomaterials, 2021, 12, 119 CrossRef PubMed.
- L. Uson, M. Arruebo, V. Sebastian and J. Santamaria, Chem. Eng. J., 2018, 340, 66–72 CrossRef CAS.
- S. Yang, T. Zhang, L. Zhang, S. Wang, Z. Yang and B. Ding, Colloids Surf., A, 2007, 296, 37–44 CrossRef CAS.
- L. Falk and J.-M. Commenge, Chem. Eng. Sci., 2010, 65, 405–411 CrossRef CAS.
- M. Meeuwse, J. van der Schaaf, B. F. M. Kuster and J. C. Schouten, Chem. Eng. Sci., 2010, 65, 466–471 CrossRef CAS.
- B. de Caprariis, M. Di Rita, M. Stoller, N. Verdone and A. Chianese, Chem. Eng. Sci., 2012, 76, 73–80 CrossRef CAS.
- X. Sen Wu, Y. Luo, G. W. Chu, Y. C. Xu, L. Sang, B. C. Sun and J. F. Chen, Ind. Eng. Chem. Res., 2018, 57, 7692–7699 CrossRef.
- S. Mohammadi, A. Harvey and K. V. K. Boodhoo, Chem. Eng. J., 2014, 258, 171–184 CrossRef CAS.
- X. Chen, N. M. Smith, K. S. Iyer and C. L. Raston, Chem. Soc. Rev., 2014, 43, 1387–1399 RSC.
- F. Haseidl, N. C. Jacobsen and K.-O. Hinrichsen, Chem. Ing. Tech., 2013, 85, 540–549 CrossRef CAS.
- W. H. Khan and V. K. Rathod, Chem. Eng. Process., 2014, 80, 1–10 CrossRef CAS.
- G. Vilardi, M. Stoller, L. Di Palma, K. Boodhoo and N. Verdone, Chem. Eng. Process., 2019, 146, 107683 CrossRef CAS.
- J. W. Loh, J. Schneider, M. Carter, M. Saunders and L. Y. Lim, J. Pharm. Sci., 2010, 99, 4326–4336 CrossRef CAS PubMed.
- S. D. Pask, Z. Cai, H. Mack, L. Marc and O. Nuyken, Macromol. React. Eng., 2013, 7, 98–106 CrossRef CAS.
- C. Y. Tai, C. Te Tai, M. H. Chang and H. S. Liu, Ind. Eng. Chem. Res., 2007, 46, 5536–5541 CrossRef CAS.
- S. Aljbour, H. Yamada and T. Tagawa, Chem. Eng. Process., 2009, 48, 1167–1172 CrossRef CAS.
- S. Kuhn, T. Noël, L. Gu, P. L. Heider and K. F. Jensen, Lab Chip, 2011, 11, 2488–2492 RSC.
- C. Delacour, C. Lutz and S. Kuhn, Ultrason. Sonochem., 2019, 55, 67–74 CrossRef CAS PubMed.
- V. Sebastián, N. Zaborenko, L. Gu and K. F. Jensen, Cryst. Growth Des., 2017, 17, 2700–2710 CrossRef.
- F. Castro, S. Kuhn, K. Jensen, A. Ferreira, F. Rocha, A. Vicente and J. A. Teixeira, Chem. Eng. Sci., 2013, 100, 352–359 CrossRef CAS.
- F. Castro, S. Kuhn, K. Jensen, A. Ferreira, F. Rocha, A. Vicente and J. A. Teixeira, Chem. Eng. J., 2013, 215–216, 979–987 CrossRef CAS.
- Z. Dong, C. Delacour, K. M. Carogher, A. P. Udepurkar and S. Kuhn, Materials, 2020, 13, 344 CrossRef CAS PubMed.
- D. F. Rivas and S. Kuhn, in Sonochemistry: From Basic Principles to Innovative Applications, Springer, Cham, 2017, pp. 225–254 Search PubMed.
- Z. Dong, D. Fernandez Rivas and S. Kuhn, Lab Chip, 2019, 19, 316–327 RSC.
- R. Manno, V. Sebastian, R. Mallada and J. Santamaria, Ind. Eng. Chem. Res., 2019, 58, 12711–12711 CrossRef.
- S. Horikoshi, T. Sumi and N. Serpone, Chem. Eng. Process., 2013, 73, 59–66 CrossRef CAS.
- M. Lu, A. Ozcelik, C. L. Grigsby, Y. Zhao, F. Guo, K. W. Leong and T. J. Huang, Nano Today, 2016, 11, 778–792 CrossRef CAS PubMed.
- J. B. Knight, A. Vishwanath, J. P. Brody and R. H. Austin, Phys. Rev. Lett., 1998, 80, 3863–3866 CrossRef CAS.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906–2912 CrossRef CAS PubMed.
- T. Baby, Y. Liu, A. P. J. Middelberg and C. X. Zhao, Chem. Eng. Sci., 2017, 169, 128–139 CrossRef CAS.
- M. Rhee, P. M. Valencia, M. I. Rodriguez, R. Langer, O. C. Farokhzad and R. Karnik, Adv. Mater., 2011, 23, H79–H83 CrossRef CAS PubMed.
- J. Bemetz, A. Wegemann, K. Saatchi, A. Haase, U. O. Häfeli, R. Niessner, B. Gleich and M. Seidel, Anal. Chem., 2018, 90, 9975–9982 CrossRef CAS PubMed.
- M. Takagi, T. Maki, M. Miyahara and K. Mae, Chem. Eng. J., 2004, 101, 269–276 CrossRef CAS.
- A. Abou-Hassan, R. Bazzi and V. Cabuil, Angew. Chem., 2009, 121, 7316–7319 CrossRef.
- J. M. Lim, N. Bertrand, P. M. Valencia, M. Rhee, R. Langer, S. Jon, O. C. Farokhzad and R. Karnik, Nanomedicine, 2014, 10, 401–409 CrossRef CAS PubMed.
- Y. Wang and M. Seidel, Microfluid. Nanofluid., 2021, 25, 1–10 CrossRef.
- R. Baber, L. Mazzei, N. T. K. Thanh and A. Gavriilidis, Nanoscale, 2017, 9, 14149–14161 RSC.
- H. Nagasawa and K. Mae, Ind. Eng. Chem. Res., 2006, 45, 2179–2186 CrossRef CAS.
- A. Abou-Hassan, J. F. Dufrêchfer, O. Sandre, G. Mériguet, O. Bernard and V. Cabuil, J. Phys. Chem. C, 2009, 113, 18097–18105 CrossRef CAS.
- I. Lignos, H. Ow, J. P. Lopez, D. McCollum, H. Zhang, J. Imbrogno, Y. Shen, S. Chang, W. Wang and K. F. Jensen, ACS Appl. Mater. Interfaces, 2020, 12, 6699–6706 CrossRef CAS PubMed.
- I. Lignos, Y. Mo, L. Carayannopoulos, M. Ginterseder, M. G. Bawendi and K. F. Jensen, React. Chem. Eng., 2021, 6, 459–464 RSC.
- T. Asimakidou, A. Makridis, S. Veintemillas-Verdaguer, M. P. Morales, I. Kellartzis, M. Mitrakas, G. Vourlias, M. Angelakeris and K. Simeonidis, Chem. Eng. J., 2020, 393, 124593 CrossRef CAS.
- K. Tacsi, Á. Joó, É. Pusztai, A. Domokos, Z. K. Nagy, G. Marosi and H. Pataki, Chem. Eng. Process., 2021, 165, 108446 CrossRef CAS.
- O. V. Proskurina, R. S. Abiev, D. P. Danilovich, V. V. Panchuk, V. G. Semenov, V. N. Nevedomsky and V. V. Gusarov, Chem. Eng. Process., 2019, 143, 107598 CrossRef CAS.
- B. K. Johnson and R. K. Prud'homme, AIChE J., 2003, 49, 2264–2282 CrossRef CAS.
- L. Chen, H. Zeng, Y. Guo, X. Yang and B. Chen, Chem. Eng. Process., 2022, 177, 108991 CrossRef CAS.
- V. Hessel, H. Löwe and F. Schönfeld, Chem. Eng. Sci., 2005, 60, 2479–2501 CrossRef CAS.
- C. P. Fonte, M. A. Sultan, R. J. Santos, M. M. Dias and J. C. B. Lopes, Chem. Eng. J., 2015, 260, 316–330 CrossRef CAS.
- H. Hu, C. Yang, M. Li, D. Shao, H. Q. Mao and K. W. Leong, Mater. Today, 2021, 42, 99–116 CrossRef CAS PubMed.
- B. K. Johnson and R. K. Prud'homme, Aust. J. Chem., 2003, 56, 1021–1024 CrossRef CAS.
- M. Wojtalik, Z. Bojarska and Ł. Makowski, J. Solid State Chem., 2020, 285, 121254 CrossRef CAS.
- C. E. Markwalter and R. K. Prud'homme, J. Pharm. Sci., 2018, 107, 2465–2471 CrossRef CAS PubMed.
- Y. Liu, C. Cheng, Y. Liu, R. K. Prud'homme and R. O. Fox, Chem. Eng. Sci., 2008, 63, 2829–2842 CrossRef CAS.
- S. F. Chow, C. C. Sun and A. H. L. Chow, Eur. J. Pharm. Biopharm., 2014, 88, 462–471 CrossRef CAS PubMed.
- P. Ranadive, A. Parulkar and N. A. Brunelli, React. Chem. Eng., 2019, 4, 1779–1789 RSC.
- J. Hiemer, A. Clausing, T. Schwarz and K. Stöwe, Chem. Eng. Technol., 2019, 42, 2018–2027 CrossRef CAS.
- R. S. Abiev, O. V. Proskurina, M. O. Enikeeva and V. V. Gusarov, Theor. Found. Chem. Eng., 2021, 55, 12–29 CrossRef CAS.
- M. Hafezi, M. Mozaffarian, M. Jafarikojour, M. Mohseni and B. Dabir, J. Photochem. Photobiol., A, 2020, 389, 112198 CrossRef CAS.
- C. W. Visser, T. Kamperman, L. P. Karbaat, D. Lohse and M. Karperien, Sci. Adv., 2018, 4, eaao1175 CrossRef PubMed.
- R. Baber, L. Mazzei, N. T. K. Thanh and A. Gavriilidis, J. Flow Chem., 2016, 6, 268–278 CrossRef CAS.
- S. Pal, K. Madane, M. Mane and A. A. Kulkarni, Ind. Eng. Chem. Res., 2021, 60, 969–979 CrossRef CAS.
- R. S. Abiev and A. A. Sirotkin, Fluids, 2020, 5, 179 CrossRef CAS.
- A. V. Zdravkov, Y. S. Kudryashova and R. S. Abiev, Russ. J. Gen. Chem., 2020, 90, 1677–1680 CrossRef CAS.
- D. V. R. Kumar, B. L. V. Prasad and A. A. Kulkarni, Ind. Eng. Chem. Res., 2013, 52, 17376–17382 CrossRef CAS.
- K. Sahoo and S. Kumar, Chem. Eng. Process., 2021, 165, 108439 CrossRef CAS.
- A. M. Nightingale, J. H. Bannock, S. H. Krishnadasan, F. T. F. O'Mahony, S. A. Haque, J. Sloan, C. Drury, R. McIntyre and J. C. deMello, J. Mater. Chem. A, 2013, 1, 4067–4076 RSC.
- L. J. Pan, J. W. Tu, H. T. Ma, Y. J. Yang, Z. Q. Tian, D. W. Pang and Z. L. Zhang, Lab Chip, 2018, 18, 41–56 RSC.
- Y. Ding, P. D. Howes and A. J. deMello, Anal. Chem., 2020, 92, 132–149 CrossRef CAS PubMed.
- Y. He, K.-J. Kim and C. Chang, Nanomaterials, 2020, 10, 1421 CrossRef CAS PubMed.
- Y. Gao, B. Pinho and L. Torrente-Murciano, Curr. Opin. Chem. Eng., 2020, 29, 26–33 CrossRef.
- V. Sebastian Cabeza, S. Kuhn, A. A. Kulkarni and K. F. Jensen, Langmuir, 2012, 28, 7007–7013 CrossRef CAS PubMed.
- J. Mahin and L. Torrente-Murciano, Chem. Eng. J., 2020, 396, 125299 CrossRef CAS.
- G. H. Albuquerque, R. C. Fitzmorris, M. Ahmadi, N. Wannenmacher, P. K. Thallapally, B. P. McGrail and G. S. Herman, CrystEngComm, 2015, 17, 5502–5510 RSC.
- R. W. Epps, K. C. Felton, C. W. Coley and M. Abolhasani, Lab Chip, 2017, 17, 4040–4047 RSC.
- L. Wang, L. R. Karadaghi, R. L. Brutchey and N. Malmstadt, Chem. Commun., 2020, 56, 3745–3748 RSC.
- H. Huang, H. Du Toit, S. Ben-Jaber, G. Wu, L. Panariello, N. T. K. Thanh, I. P. Parkin and A. Gavriilidis, React. Chem. Eng., 2019, 4, 884–890 RSC.
- A. Larrea, V. Sebastian, A. Ibarra, M. Arruebo and J. Santamaria, Chem. Mater., 2015, 27, 4254–4260 CrossRef CAS PubMed.
- V. Sebastian, S. Basak and K. F. Jensen, AIChE J., 2016, 62, 373–380 CrossRef CAS.
- K. J. Hartlieb, M. Saunders, R. J. J. Jachuck and C. L. Raston, Green Chem., 2010, 12, 1012–1017 RSC.
- C. Zeng, C. Wang, F. Wang, Y. Zhang and L. Zhang, Chem. Eng. J., 2012, 204–205, 48–53 CrossRef.
- D. V. Ravi Kumar, B. L. V. Prasad and A. A. Kulkarni, Chem. Eng. J., 2012, 192, 357–368 CrossRef CAS.
- A. M. Seddon, S. J. Richardson, K. Rastogi, T. S. Plivelic, A. M. Squires and C. Pfrang, J. Phys. Chem. Lett., 2016, 7, 1341–1345 CrossRef CAS PubMed.
- M. Agthe, T. S. Plivelic, A. Labrador, L. Bergström and G. Salazar-Alvarez, Nano Lett., 2016, 16, 6838–6843 CrossRef CAS PubMed.
- J. Feng, G. Biskos and A. Schmidt-Ott, Sci. Rep., 2015, 5, 1–9 Search PubMed.
- P. Maguire, D. Rutherford, M. Macias-Montero, C. Mahony, C. Kelsey, M. Tweedie, F. Pérez-Martin, H. McQuaid, D. Diver and D. Mariotti, Nano Lett., 2017, 17, 1336–1343 CrossRef CAS PubMed.
- A. Testino, F. Pilger, M. Lucchini, J. Quinsaat, C. Stähli and P. Bowen, Molecules, 2015, 20, 10566–10581 CrossRef CAS PubMed.
- K. Kumar, A. M. Nightingale, S. H. Krishnadasan, N. Kamaly, M. Wylenzinska-Arridge, K. Zeissler, W. R. Branford, E. Ware, A. J. deMello and J. C. deMello, J. Mater. Chem., 2012, 22, 4704 RSC.
- S. Duraiswamy and S. A. Khan, Part. Part. Syst. Charact., 2014, 31, 429–432 CrossRef CAS.
- L. H. Hung, K. M. Choi, W. Y. Tseng, Y. C. Tan, K. J. Shea and A. P. Lee, Lab Chip, 2006, 6, 174–178 RSC.
- S. Damilos, I. Alissandratos, L. Panariello, A. N. P. Radhakrishnan, E. Cao, G. Wu, M. O. Besenhard, A. A. Kulkarni, C. Makatsoris and A. Gavriilidis, J. Flow Chem., 2021, 11, 553–567 CrossRef CAS.
- I. Lignos, S. Stavrakis, A. Kilaj and A. J. deMello, Small, 2015, 11, 4009–4017 CrossRef CAS PubMed.
- L. Frenz, A. El Harrak, M. Pauly, S. Bégin Colin, A. D. Griffiths and J. C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS PubMed.
- A. Knauer, A. Thete, S. Li, H. Romanus, A. Csáki, W. Fritzsche and J. M. Köhler, Chem. Eng. J., 2011, 166, 1164–1169 CrossRef CAS.
- L. Panariello, S. Damilos, H. Du Toit, G. Wu, A. N. P. Radhakrishnan, I. P. Parkin and A. Gavriilidis, React. Chem. Eng., 2020, 5, 663–676 RSC.
- G. Ochoa-Vazquez, B. Kharisov, A. Arizmendi-Morquecho, A. Cario, C. Aymonier, S. Marre and I. Lopez, IEEE Trans. Nanobioscience, 2022, 21, 135–140 Search PubMed.
- L. Panariello, K. Chuen To, Z. Khan, G. Wu, G. Gkogkos, S. Damilos, I. P. Parkin and A. Gavriilidis, Chem. Eng. J., 2021, 423, 129069 CrossRef CAS.
- L. Paseta, B. Seoane, D. Julve, V. Sebastián, C. Téllez and J. Coronas, ACS Appl. Mater. Interfaces, 2013, 5, 9405–9410 CrossRef CAS PubMed.
- R. W. Epps, M. S. Bowen, A. A. Volk, K. Abdel-Latif, S. Han, K. G. Reyes, A. Amassian and M. Abolhasani, Adv. Mater., 2020, 32, 2001626 CrossRef CAS PubMed.
- S. Duraiswamy and S. A. Khan, Small, 2009, 5, 2828–2834 CrossRef CAS PubMed.
- P. Kunal, E. J. Roberts, C. T. Riche, K. Jarvis, N. Malmstadt, R. L. Brutchey and S. M. Humphrey, Chem. Mater., 2017, 29, 4341–4350 CrossRef CAS.
- O. Kašpar, A. H. Koyuncu, A. Hubatová-Vacková, M. Balouch and V. Tokárová, RSC Adv., 2020, 10, 15179–15189 RSC.
- C. D. Ahrberg, J. W. Choi and B. G. Chung, Beilstein J. Nanotechnol., 2018, 9, 2413–2420 CrossRef CAS PubMed.
- J. Yue, F. H. Falke, J. C. Schouten and T. A. Nijhuis, Lab Chip, 2013, 13, 4855–4863 RSC.
- M. V. Bandulasena, G. T. Vladisavljević and B. Benyahia, Chem. Eng. Sci., 2019, 195, 657–664 CrossRef CAS.
- Y. H. Kim, L. Zhang, T. Yu, M. Jin, D. Qin and Y. Xia, Small, 2013, 9, 3462–3467 CrossRef CAS PubMed.
- G. Gaikwad, P. Bangde, K. Rane, J. Stenberg, L. Borde, S. Bhagwat, P. Dandekar and R. Jain, Microfluid. Nanofluid., 2021, 25, 27 CrossRef CAS.
- I. Lignos, S. Stavrakis, G. Nedelcu, L. Protesescu, A. J. deMello and M. V. Kovalenko, Nano Lett., 2016, 16, 1869–1877 CrossRef CAS PubMed.
- I. Shestopalov, J. D. Tice and R. F. Ismagilov, Lab Chip, 2004, 4, 316–321 RSC.
- A. M. Nightingale, S. H. Krishnadasan, D. Berhanu, X. Niu, C. Drury, R. McIntyre, E. Valsami-Jones and J. C. deMello, Lab Chip, 2011, 11, 1221–1227 RSC.
- L. Zou, B. Huang, X. Zheng, H. Pan, Q. Zhang, W. Xie, Z. Zhao and X. Li, Mater. Chem. Phys., 2022, 276, 125384 CrossRef CAS.
- S. Abalde-Cela, P. Taladriz-Blanco, M. G. De Oliveira and C. Abell, Sci. Rep., 2018, 8, 2440 CrossRef PubMed.
- H. Huang, G. B. Hwang, G. Wu, K. Karu, H. Du Toit, H. Wu, J. Callison, I. P. Parkin and A. Gavriilidis, Chem. Eng. J., 2020, 383, 123176 CrossRef CAS.
- H. Du Toit, T. J. Macdonald, H. Huang, I. P. Parkin and A. Gavriilidis, RSC Adv., 2017, 7, 9632–9638 RSC.
- L. Hafermann and J. M. Köhler, Chem. Eng. Technol., 2015, 38, 1138–1143 CrossRef CAS.
- A. M. Nightingale, T. W. Phillips, J. H. Bannock and J. C. deMello, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- K. Abdel-Latif, R. W. Epps, F. Bateni, S. Han, K. G. Reyes and M. Abolhasani, Adv. Intell. Syst., 2021, 3, 2000245 CrossRef.
- F. Bateni, R. W. Epps, K. Antami, R. Dargis, J. A. Bennett, K. G. Reyes and M. Abolhasani, Adv. Intell. Syst., 2022, 4, 2200017 CrossRef.
- A. A. Volk, R. W. Epps, D. Yonemoto, F. N. Castellano and M. Abolhasani, React. Chem. Eng., 2021, 6, 1367–1375 RSC.
- L. Zhang, Y. Wang, L. Tong and Y. Xia, Nano Lett., 2014, 14, 4189–4194 CrossRef CAS PubMed.
- A. A. Kulkarni and V. Sebastian Cabeza, Langmuir, 2017, 33, 14315–14324 CrossRef CAS PubMed.
- C. B. Kerr, R. W. Epps and M. Abolhasani, Lab Chip, 2019, 19, 2107–2113 RSC.
- N. Sen, V. Koli, K. K. Singh, L. Panicker, R. Sirsam, S. Mukhopadhyay and K. T. Shenoy, Chem. Eng. Process., 2018, 125, 197–206 CrossRef CAS.
- S. Pal and A. A. Kulkarni, Chem. Eng. Sci., 2016, 153, 344–353 CrossRef CAS.
- C. Spiegel, M. Kraut, G. Rabsch, C. Küsters, W. Augustin and S. Scholl, Chem. Eng. Technol., 2019, 42, 2067–2075 CrossRef CAS.
- Y. Tanaka, O. Tonomura, K. Isozaki and S. Hasebe, Chem. Eng. J., 2011, 167, 483–489 CrossRef CAS.
- A. Aliseda and T. J. Heindel, Annu. Rev. Fluid Mech., 2021, 53, 543–567 CrossRef.
- A. Perro, G. Lebourdon, S. Henry, S. Lecomte, L. Servant and S. Marre, React. Chem. Eng., 2016, 1, 577–594 RSC.
- J. Li, H. Šimek, D. Ilioae, N. Jung, S. Bräse, H. Zappe, R. Dittmeyer and B. P. Ladewig, React. Chem. Eng., 2021, 6, 1497–1507 RSC.
- M. Rodriguez-Zubiri and F. X. Felpin, Org. Process Res. Dev., 2022, 26, 1766–1793 CrossRef CAS.
- M. A. Morin, W. Zhang, D. Mallik and M. G. Organ, Angew. Chem., 2021, 133, 20774–20794 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |