Probing the effects of fructose concentration on the evolution of humins during fructose dehydration†
Received
12th August 2022
, Accepted 10th October 2022
First published on 11th October 2022
Abstract
5-Hydroxymethylfurfural (HMF), considered as a “sleeping giant” of sustainable chemistry, is generally produced by fructose dehydration. Till now, high HMF yields have been achieved, whereas large-scale production of HMF is hampered by the formation of undesired humins, especially at higher fructose concentrations (>10 wt%). In this work, we report the effects of fructose concentration (4.5–360.0 wt%) on the evolution pathways of humins during the H2SO4-catalyzed dehydration of fructose in water. We show that both etherification–dehydration–condensation and degradative condensation of fructose and/or HMF are involved in the formation of humins, wherein the increase of fructose concentration promotes the former path and inhibits the latter one because of the promotional effect on the formation of difructose anhydride (DFA) species. The progressive dehydrations and condensations of DFAs under experimental conditions lead to humins, but the reversible hydrolysis of DFAs to fructose favors the HMF formation. Further, we demonstrate that the addition of a typical polar aprotic solvent such as tetrahydrofuran (THF) or 1,4-dioxane (DIO) to water as a co-solvent could stabilize the DFA species and increase the HMF yield by more than 10% in the conversion of high-concentration fructose (72.0 wt%). This understanding provides an indispensable insight on factors influencing humin formation for future advances on HMF biorefineries.
Introduction
The effective transformation of readily available biomass, a naturally renewable source of carbon, to useful chemicals and liquid fuels could settle the problems due to shortage of fossil resources and increasing carbon emissions in the world.1–3 5-Hydroxymethylfurfural (HMF), known as a “sleeping giant” of sustainable chemistry, is a representative biomass-derived chemical.4–7 It has been considered as one of the “Top 10 + 4” platform chemicals as additions to the original list from the U.S. Department of Energy (DOE).8 So far, the synthesis of HMF via dehydration of a hexose (typically fructose) has attracted great attention,9–14 wherein remarkable progress has been made to improve the yield and selectivity to HMF by developing effective solvent and catalyst systems. For example, with the catalysis of a sulfonated carbonaceous catalyst, an excellent yield (98.0%) of HMF was achieved from fructose (5 wt%) in a mixed solvent of tetrahydrofuran (THF) and dimethyl sulfoxide (DMSO).15 However, when the fructose concentration was increased to 20 wt%, the HMF yield was significantly reduced to 53.2%. In fact, the high HMF yield and selectivity were not compatible with the high fructose concentration in most of the existing catalytic systems.16,17 High HMF yields (>90%) were normally achieved when the concentration of fructose was extremely low (1–5 wt%).15,18,19 Several studies have been attempted to improve the HMF yield in the conversion of high-concentration fructose. For example, the use of a macroporous sulfonated styrene–divinylbenzene resin (DR-2030) catalyst in DMSO20 or betaine-based solid acid catalyst in biphasic solvent with a deep eutectic solvent as an additive21 led to higher HMF yields (>85%) from fructose (30 wt%), wherein DMSO or ChCl played important roles in promoting fructose dehydration and stabilizing HMF, although the high boiling point solvents were hard to recycle. Besides, the conversion of high-concentration fructose (20–80 wt%) in ionic liquids (ILs) also led to satisfactory HMF yields (>60%),22–25 but always suffered from poor mass transfer due to the high viscosity of ILs. Efficient systems for converting high-concentration fructose to HMF remains a challenge. As early as 1986, the phenomenon of a decrease in HMF selectivity with an increase of fructose concentration was observed by van Dam et al.,26 which was preliminarily attributed to the formation of humins via the irreversible polymerization of fructose during fructose dehydration. Till now, the formation of humins has become the main bottleneck that tremendously limits the economic feasibility of HMF biorefineries.11 Therefore, it is of great significance to study the pathway and influencing factors of humin formation in fructose dehydration, especially at high fructose concentrations.
Depending on the reaction conditions, fructose-to-HMF dehydration might generate humins via both the self-/cross-condensation and degradative condensation of fructose/HMF.27–35 Recent studies showed that both the type of catalyst and the composition of solvent would affect the evolution pathway of humins.36 To be specific, kinetic studies revealed that a Brønsted acid catalyst favored both the self-/cross-condensation of fructose/HMF and the degradative condensation of fructose to humins, whereas a Lewis acid catalyst promoted the self-condensation of fructose and degradative condensation of HMF to humins.33,37 Our previous work showed that the solvent composition influenced the humin formation by affecting the distribution of fructose tautomers in reaction systems, wherein the predominant existence of α-fructofuranose, α/β-fructopyranose, and chain fructose led to the degradative condensation of fructose to humins.32 However, considering the possible influence of fructose concentration, the formation of humins was briefly ascribed to the increasing collision probability of reactive species responsible for the self- and/or cross-polymerizations.15,38,39 Nowadays, limited insight has been given to understand the reaction chemistry for humin formation at high fructose concentrations due to the lack of product analysis, which hampers the development of effective reaction systems for advancing HMF biorefineries.
In this work, we report the effects of fructose concentration on the evolution of humins during acid-catalyzed fructose dehydration in water. H2SO4 was selected as the catalyst because it acted as a representative and effective Brønsted acid catalyst in fructose dehydration. Considering that a higher acid concentration (pH < 0.6) and reaction temperature (>180 °C) would aggravate the humin formation whereas a lower acid concentration (pH > 1.6) and reaction temperature (<120 °C) would slow down the fructose dehydration,40 a moderate acid concentration (pH = 1.0) and reaction temperature (140 °C) were chosen. Besides, the fructose concentration ranged from 4.5 to 360.0 wt%, aiming to provide a universal understanding on its influence on the product distributions and humin formations. The structures of humins and possible interactions between fructose molecules in aqueous solution were analyzed by electrospray ionization mass spectrometry (ESI-MS), attenuated total reflection-infrared (ATR-IR) spectroscopy, and Fourier transform infrared (FT-IR) spectroscopy. The evolution of humins was isotopically traced by 13C nuclear magnetic resonance (NMR) spectroscopy.
Experimental
Materials
D-Fructose (>99.0%), HMF (>95.0%), and levulinic acid (LA, >97.0%) were purchased from TCI Reagent Factory. Isotopically labeled D-fructose (D-[1-13C]-fructose and D-[2-13C]-fructose) were purchased from Shanghai ZZBIO CO. Ltd. Formic acid (FA, 98%) was purchased from J&K Scientific Ltd. Furfural (FF, 99%) and D2O (99.9%) were purchased from Adamas-β. Sulfuric acid (96–98%) was purchased from Chengdu Jinshan Chemical Reagent Factory. 3-(Trimethylsilyl)propionic acid sodium salt (TPS, 98%) was purchased from Cambridge Isotope Laboratories Inc. All chemicals were used as received. Ultra-pure water (resistance >18.2 mΩ cm−1) was used in all experiments.
Catalytic experiment
The H2SO4-catalyzed dehydration of fructose was carried out in a 15 mL thick-walled pressure tube (Synthware Corporation). In a typical synthesis experiment, fructose (4.5–360.0 wt%) was dissolved in 1 mL H2O containing 0.05 M H2SO4, followed by purging with high purity nitrogen for 3 min to remove the air. The mixture was heated for a given time (5–150 min) at 140 °C with magnetic stirring. After the reaction, the mixture was allowed to cool to room temperature, followed by dilution with water and filtration before analysis. By using THF or 1,4-dioxane (DIO) as a co-solvent, the total volume of mixed solvents remained constant at 1 mL whereas the volume ratio of THF or DIO was 30% or 70%.
Product analysis
The products from fructose dehydration were analyzed quantitatively by high performance liquid chromatography (HPLC, Waters 1525-1487) equipped with an Aminex column (HPX-87 column, Bio-Rad, 50 °C), a refractive index detector (2414), and a PDA detector (2998). The mobile phase was a 5.0 mM aqueous H2SO4 solution at a flow rate of 0.6 mL min−1. Fructose was detected using the RI detector; HMF, FA, LA, and FF were detected using a UV detector at a wavelength of 210 nm. The conversion of fructose and the yields of products were determined by comparison with external standard curves obtained using authentic samples at different concentrations. The conversion of fructose and yields of products were defined as eqn (1) and (2), respectively.| | | Conversion of fructose [mol%] = (moles of fructose reacted)/(moles of starting fructose) × 100% | (1) |
| | | Yield of products [mol%] = (moles of products formed)/(moles of starting fructose) × 100% | (2) |
The actual carbon balance (ACB) was defined as the molar ratio between the output of carbon and input of carbon (eqn (3)), wherein the output of carbon including small-molecular products and unreacted substrate was quantitatively detected by HPLC. Then, the unbalanced carbon (100%-ACB) was indicative of the actual carbon loss during the fructose-to-HMF conversion, including the losses from dehydration condensations and degradative condensations from fructose and/or HMF.35 Herein, the degradative condensations of fructose and/or HMF generated stoichiometric amounts of FA and undetected C5 fragments. To consider the carbon loss from the degradation of fructose and/or HMF, the theoretical carbon balance (TCB) was assumptively calculated by the ratio of the sum of the output of carbon and carbon in degraded fructose/HMF to the input of carbon (eqn (4)). Therefore, the difference between TCB and ACB represented the carbon loss from degradative condensation of fructose and/or HMF.32
| | | ACB [mol%] = (output of carbon)/(input of carbon) × 100% | (3) |
| | | TCB [mol%] = (output of carbon + carbon in degraded fructose or HMF)/(input of carbon) × 100% | (4) |
ESI-MS spectra
ESI-MS (LCMS-IT-TOF, Shimadzu) spectra were used to analyze the possible chemical structure of the reaction mixture before and after the reaction. The operating parameters were as follows: ionization voltage, 4.5 kV; interface temperature, 250 °C; nebulizer gas flow, 90 L h−1 (N2); detector voltage, 1.60 kV; continuum mode. Aqueous fructose solutions with various concentrations before the reaction were detected in the range of m/z = 204–500 to avoid the appearance of a strong signal assigned to [Fru + Na]+, whereas the reaction mixtures quenched at various reaction times were detected in the range of m/z = 50–500.
ATR-IR spectra
ATR-IR spectra were used to explore the possible interactions between fructose molecules in aqueous solution at room temperature. A real-time online reaction analysis system for ReactIR iC10 and a mercury cadmium telluride (MCT) detector were used. The wavenumber range was 650–4000 cm−1, and the resolution was 4 cm−1. The spectra were acquired with a single scanning time of 1 min with a scanning frequency of 256 times per min.
FT-IR spectra
FT-IR spectra of the solid humins were acquired using a Fourier transform infrared spectrometer (INVENIO-R, Bruker) in the wavenumber range of 400–4000 cm−1 with a spectral resolution of 4 cm−1. Before characterization, the solid humins were washed with water until the filtrate became transparent and dried at 80 °C overnight.
Isotope-labelling 13C NMR spectra
In the isotopic tracing experiment, one third of fructose was replaced with D-[1-13C]-fructose or D-[2-13C]-fructose as an isotopic tracer. The reaction mixture after reacting fructose (72.0 wt%) at 140 °C for 0.5 h was characterized using a 13C NMR spectrometer (Bruker AVANCE-II 600 MHz) operating at 150.9 MHz using a 5 mm broad-band probe, at 25 ± 0.5 °C. 0.5 mL of reaction mixture was mixed with 0.2 mL D2O and 5 mg TPS in an NMR tube, wherein TPS was used as the internal standard to calibrate the chemical shift.41 The NMR parameters were as follows: pulprog, zgig30; pulse width, 10 μs; acquisition time, 9.04 μs; 256 scans.
Results and discussion
Effect of the initial fructose concentration on the product distributions
The product distributions after the H2SO4-catalyzed fructose dehydration at 140 °C are shown in Table 1. By increasing the fructose concentration from 4.5 wt% to 360.0 wt%, the conversion of fructose at a constant reaction time was increased monotonously, whereas the yield of HMF remained almost unchanged. This variation was indicative of the aggravation of the side reactions in the conversion of high-concentration fructose. FA, LA, and a small amount of FF were detected as the side products. Among others, the formation of LA indicated the occurrence of HMF rehydration, accompanied by the formation of FA with a stoichiometric equimolar amount.33,37 However, the stoichiometric excess formation of FA relative to LA suggested the occurrence of degradative condensation of fructose and/or HMF, which was thought to be one of the paths for humin formation in fructose-to-HMF dehydration.34,42 Assuming that all of the carbons in fructose were lost via the condensation of degraded fructose and/or HMF, the theoretical carbon balance (TCB) calculated by the sum of the output of carbon and carbon in degraded fructose/HMF divided by the input of carbon should reach 100%. However, the far lower TCB indicated the occurrence of other condensation paths responsible for humin formation (i.e., the path of dehydration–condensation of fructose to humins).43 Therefore, the carbon lost from the dehydration–condensation path could be evaluated by 100% − TCB, whereas the carbon lost from the degradative condensation path could be quantified by the difference between TCB and the actual carbon balance (ACB) after the reaction. As seen from Table 1, the carbon lost from the dehydration–condensation path was more serious than that from the degradative condensation path with increasing initial fructose concentration, thereby demonstrating that the use of high-concentration fructose favored the humin formation via the path of dehydration–condensation of fructose and/or HMF rather than degradative condensation.
Table 1 Product distributions of fructose dehydration at different initial fructose concentrationsa
| Entry |
Fructose conc.b/wt% |
Conv./% |
Yield/% |
TCBc/% |
ACBd/% |
(100%-TCB)/% |
(TCB-ACB)/% |
| HMF |
FA |
LA |
FA–LA |
FF |
|
Reaction conditions: 1 mL H2O, 0.05 M H2SO4, 140 °C, 0.5 h, under a N2 atmosphere.
Fructose concentration = (mass of fructose/mass of H2O) × 100%.
TCB [mol%] = (output of carbon + carbon in degraded fructose)/(input of carbon) × 100%.
ACB [mol%] = (output of carbon)/(input of carbon) × 100%.
|
| 1 |
4.5 |
24.4 |
11.8 |
1.2 |
0.6 |
0.6 |
0.1 |
88.7 |
88.2 |
11.7 |
0.5 |
| 2 |
18.0 |
30.6 |
9.6 |
2.1 |
0.3 |
1.8 |
0.1 |
81.1 |
79.6 |
18.9 |
1.5 |
| 3 |
72.0 |
39.5 |
10.5 |
4.0 |
0.7 |
3.3 |
0.1 |
75.0 |
72.3 |
25.0 |
2.8 |
| 4 |
180.0 |
56.5 |
9.0 |
7.7 |
0.4 |
7.3 |
0.1 |
60.2 |
54.1 |
39.8 |
6.1 |
| 5 |
270.0 |
63.7 |
9.9 |
11.5 |
0.7 |
10.8 |
0.2 |
57.8 |
48.8 |
42.2 |
9.0 |
| 6 |
360.0 |
68.1 |
10.3 |
14.2 |
0.8 |
13.4 |
0.2 |
56.6 |
45.4 |
43.4 |
11.2 |
Identification of the humins
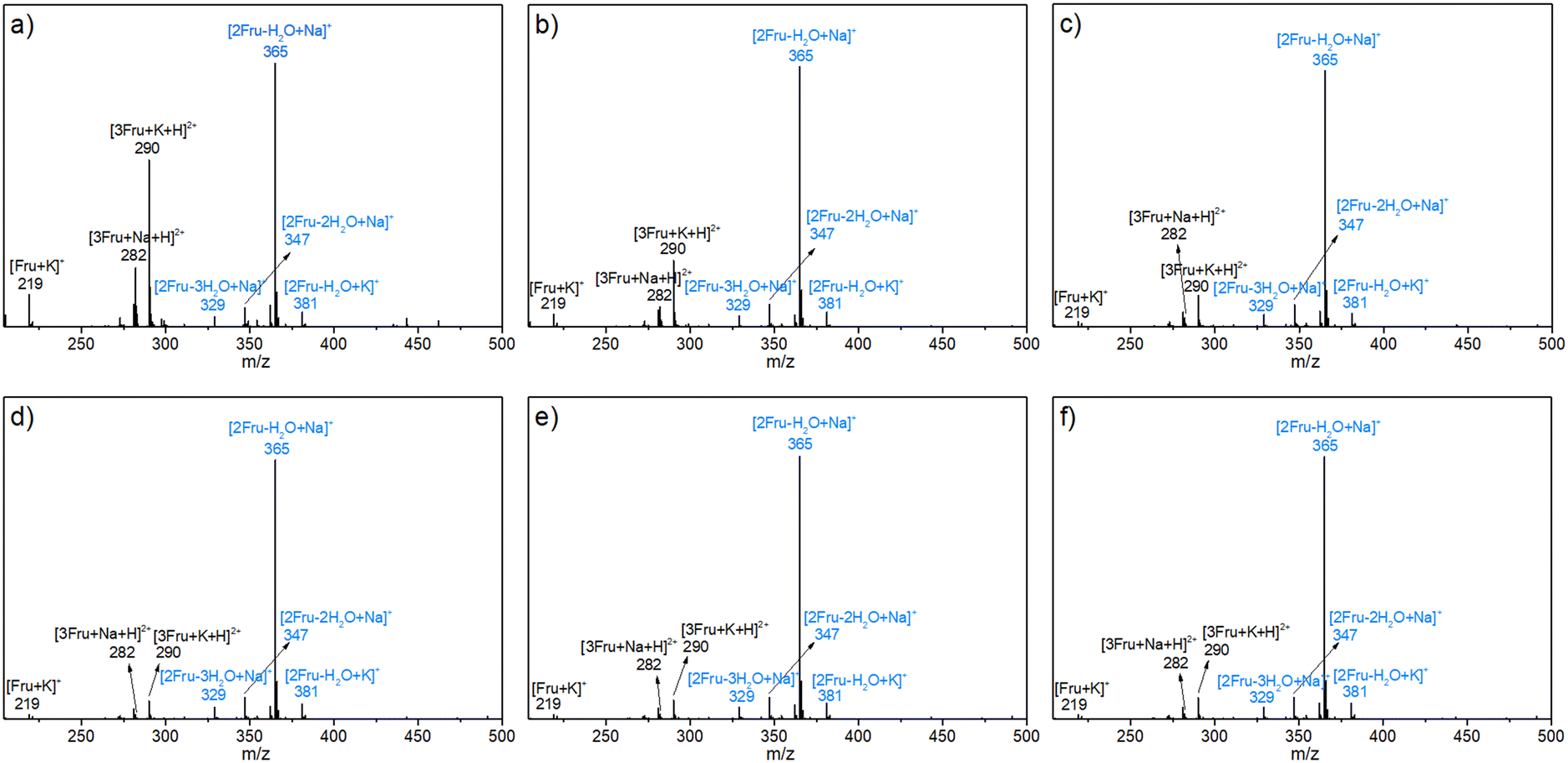
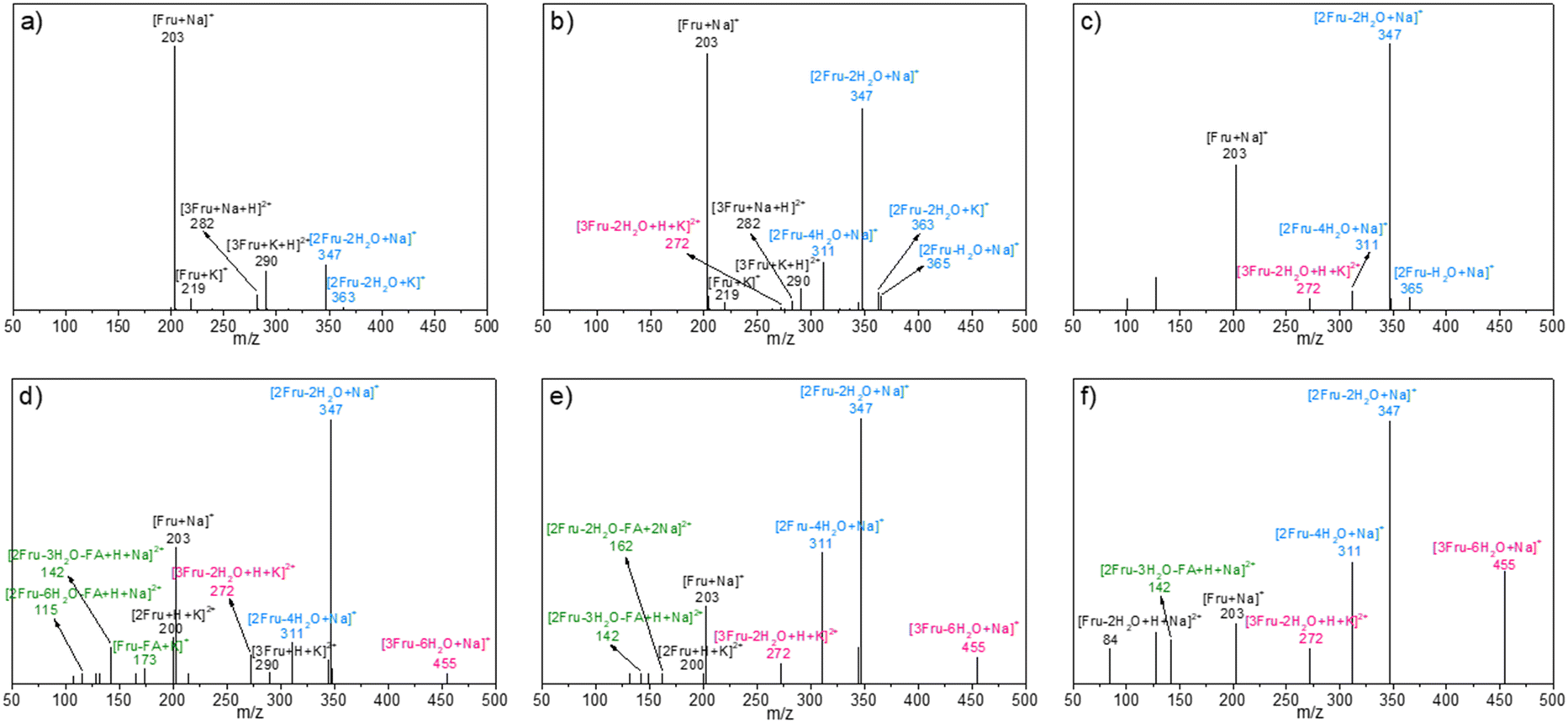
The possible structures of the reaction mixture before and after the reaction were analyzed using the ESI-MS spectra, wherein Na+ and K+ were derived from the ionization of sodium salts and potassium salts in water. As shown in Fig. 1a, in addition to the fructose monomer, low-concentration fructose (4.5 wt%) existed predominantly as a difructose (i.e., [2Fru–H2O]) species at room temperature by detecting the ionic peaks of [2Fru–H2O + Na]+ and [2Fru–H2O + K]+ with m/z values of 365 and 381, respectively. This observation revealed that fructose underwent intermolecular etherification to form dimerized fructose at room temperature. Besides, it was found that the relative intensity of this dimer was increased with the rise of initial fructose concentration to 360.0 wt% (Fig. 1b–f). That is, the intermolecular etherification of fructose became serious in high-concentration fructose solutions. In addition, [2Fru–2H2O + Na]+ (m/z = 347) and [2Fru–3H2O + Na]+ (m/z = 329) species were also detected, which indicated that difructose could undergo intramolecular dehydration at room temperature. To rule out the possibility of dehydration during the detection process of ESI-MS, sucrose and cellobiose were selected as contrast samples under the same detection conditions (Fig. S1†).44 The results showed that no dehydrated products from the above-mentioned disaccharides were detected during the detection process, thereby illustrating the reliability of the ESI-MS spectra in the present work.
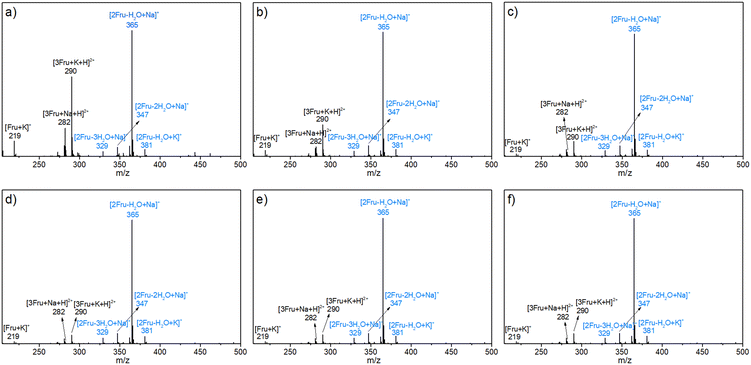 |
| | Fig. 1 ESI-MS spectra of the aqueous fructose solutions with various concentrations at room temperature. a) 4.5 wt%, b) 18.0 wt%, c) 72.0 wt%, d) 180.0 wt%, e) 270.0 wt%, and f) 360.0 wt%. | |
After the reaction at 140 °C for 0.5 h, the signals assigned to [2Fru–2H2O + Na]+ species became more and more dominant with increasing initial fructose concentration, accompanied with the decrease in intensity of signals assigned to the fructose monomer (Fig. 2). These results revealed that the fructose monomer might be consumed by successive conversion to difructose via intermolecular dehydration, followed by being converted to difructose anhydrides (DFAs) via intramolecular dehydration under experimental conditions. These DFAs were once thought to be reversible intermediates to fructose and HMF.45–48 However, the deep dehydration of DFAs was observed by detecting the [2Fru–4H2O + Na]+ species with m/z = 311 after converting higher-concentration fructose (18.0 wt%). Besides, the intensity of this [2Fru–4H2O + Na]+ species in ESI-MS spectra was found to be increased with increasing initial fructose concentration to 360.0 wt%. Notably, some trimers (i.e., [3Fru–2H2O + H + K]2+ or [2Fru + HMF + H2O + H + K]2+, m/z = 272; [3Fru–6H2O + H + K]2+, [2Fru + HMF–3H2O + H + K]2+, or [Fru + 2HMF + H + K]2+, m/z = 455) were also detected after the conversion of fructose with initial concentrations of 18.0–360.0 wt%, which demonstrated the further condensation of the difructose species with fructose/HMF and the progressive intramolecular dehydration during the conversion of high-concentration fructose. Obviously, this etherification–dehydration–condensation of fructose and/or HMF during fructose-to-HMF dehydration represented one of the typical paths responsible for humin formation, which became more serious at higher fructose concentrations. This conclusion was consistent with that deduced from the TCB (vide supra). Besides, FA-related species [Fru–FA + K]+ (m/z = 173), [2Fru–2H2O–FA + 2Na]2+ (or [Fru–FA + HMF + H2O + 2Na]2+) (m/z = 162), [2Fru–3H2O–FA + H + Na]2+ (or [Fru–FA + HMF + H + Na]2+) (m/z = 142), and [2Fru–6H2O–FA + H + Na]2+(or [Fru–FA–3H2O + HMF + H + Na]2+, [2HMF–FA + H + Na]2+) (m/z = 115) were also detected in the reaction mixture after reaction of high-concentration fructose (180–360 wt%), which confirmed the occurrence of degradative condensation of fructose and/or HMF to humins. However, these species were less obvious in the reaction mixture at relatively lower fructose concentration (<72.0 wt%), possibly due to the lower occurrence of degradative condensation at low fructose conversions (Table 1).
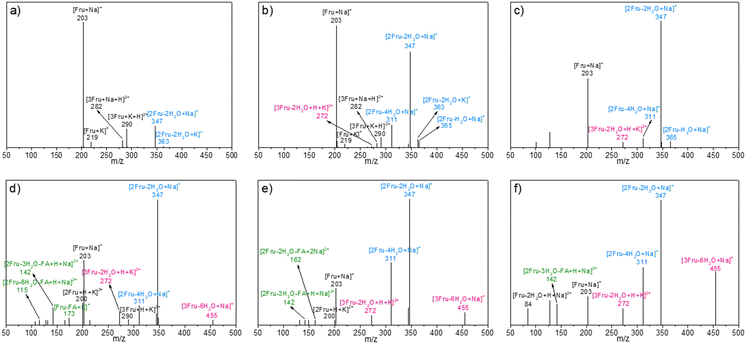 |
| | Fig. 2 ESI-MS spectra of the reaction mixture after reacting fructose with various initial concentrations at 140 °C for 0.5 h. a) 4.5 wt%, b) 18.0 wt%, c) 72.0 wt%, d) 180.0 wt%, e) 270.0 wt%, and f) 360.0 wt%. | |
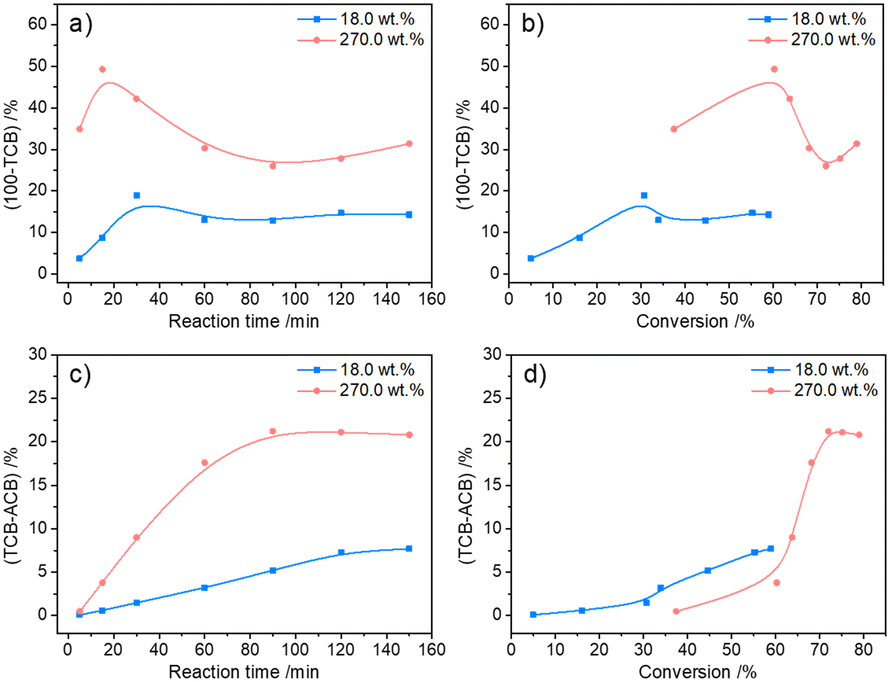
Evolution of the humins with time
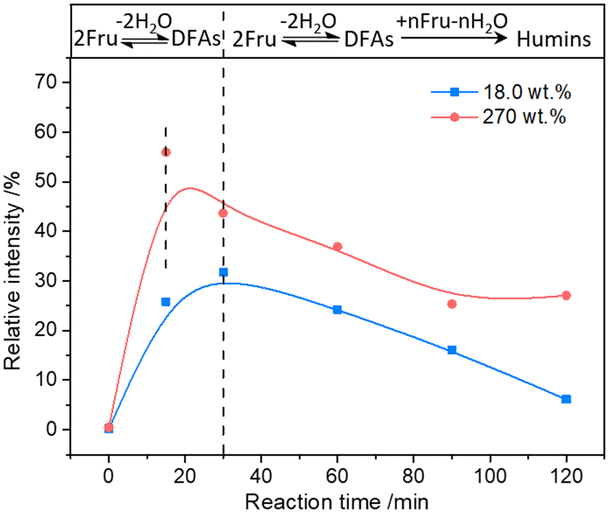
The possible evolution of humins during fructose-to-HMF dehydration was explored by analyzing the product variations with time, wherein fructose with relatively lower (18.0 wt%) and higher (270.0 wt%) initial concentrations was used as the substrate. As shown in Fig. 3a and b, a higher carbon loss from the etherification–dehydration–condensation of fructose and/or HMF was found in the conversion of high-concentration fructose rather than low-concentration fructose, which confirmed the concentration-aggravated self-condensation of fructose and/or HMF to humins via an etherification–dehydration–condensation path. Besides, the observed increase of carbon loss at the initial reaction stage (18.0 wt%, within 15 min; 270.0 wt%, within 30 min) in both systems was possibly ascribed to the formation of DFAs, whereas the subsequent decrease of carbon loss was reasonably attributed to the reversible hydrolysis of DFAs to fructose. As proof, the relative intensities of the peaks assigned to DFAs to the total intensity of all peaks in the ESI-MS spectra (Fig. S2 and S3†)44 of the reaction mixture provided semiquantitative information on the DFAs, which illustrated the increase of DFA content within 15 min and 30 min, respectively, in converting high- or low-concentration fructose, followed by the decrease of content with prolonged reaction time (Fig. 4). Therefore, DFAs were proved to be protective intermediates for HMF formation. However, when the fructose conversion exceeded 70% in converting high-concentration fructose, the carbon loss was enhanced again (Fig. 3b), thereby demonstrating the exacerbation of the dehydration–condensation of fructose and/or HMF in the later reaction stage. Meanwhile, the ESI-MS spectra (Fig. S2 and S3†)44 of the reaction mixture revealed the formation of [2Fru–nH2O] (n = 4–7) and [3Fru–nH2O] (n = 2–9) species, and the relative intensities of which were increased with prolonged reaction time. For this reason, the DFAs were deemed to act also as unwanted intermediates responsible for the etherification–dehydration–condensation to humins. In this case, the facile formation of humins via the etherification–dehydration–condensation path in converting high-concentration fructose could be interpreted by the favorable formation of DFA intermediates under the experimental conditions.
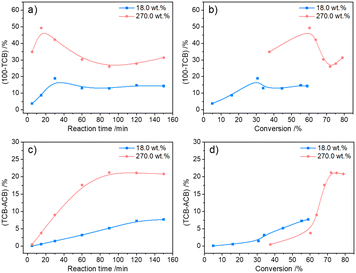 |
| | Fig. 3 The variation of carbon loss via dehydration–condensation or degradative condensation in fructose-to-HMF dehydration. a) 100% − TCB as a function of reaction time, b) 100% − TCB as a function of fructose conversion, c) TCB − ACB as a function of reaction time, and d) TCB − ACB as a function of fructose conversion. Reaction conditions: 1 mL of H2O, 0.05 M of H2SO4, 140 °C, under a N2 atmosphere. TCB [mol%] = (output of carbon + carbon in degraded fructose or HMF)/(input of carbon) × 100%. ACB [mol%] = (output of carbon)/(input of carbon) × 100%. | |
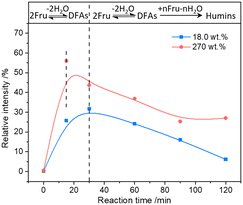 |
| | Fig. 4 The relative intensity variation of the peak assigned to the [2Fru–2H2O + Na]+ species to the total intensity of all peaks in the ESI-MS spectra of the reaction mixture with time. | |
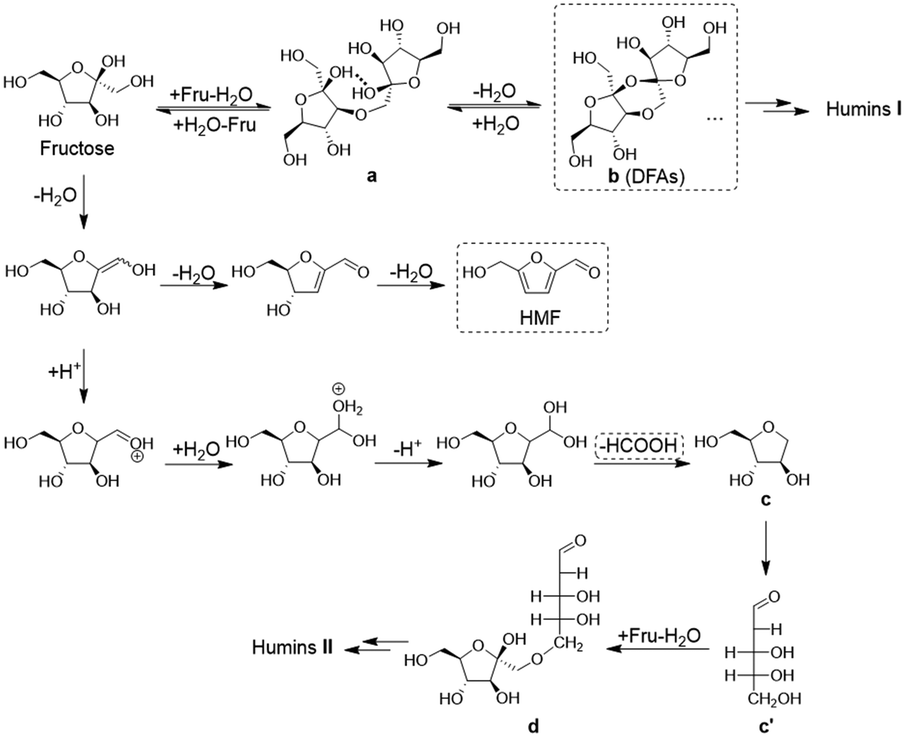
Considering the path of degradative condensation of fructose and/or HMF to humins, the results in Fig. 3c showed that the carbon loss in converting both high- and low-concentration fructose increased with the reaction time. However, the carbon loss in converting high-concentration fructose was generally lower at a constant fructose conversion (≤60%), which illustrated that the degradative condensation of fructose and/or HMF was alleviated at the initial reaction stage. The isotopically traced 13C NMR spectra (Fig. S4†)44 revealed that the degradation of fructose to FA occurred via the C1–C2 cleavage of fructose because the characteristic peak of FA (173.3 ppm) was only detected in the reaction mixture from the reaction of 1-13C labeled fructose. After the degradation, the unstable C5 intermediates (c/c′ in Scheme 1) would further condense with fructose molecules to form a C11 species (d in Scheme 1), which was detected in the 13C NMR spectra of the reaction mixture from the reaction of 2-13C labeled fructose by detecting a peak at 192.3 ppm assigned to [2Fru–H2O–FA].
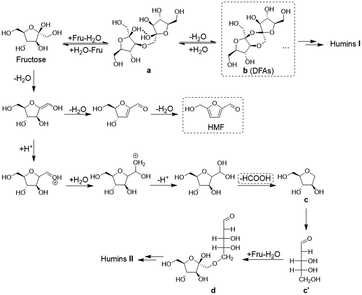 |
| | Scheme 1 Evolution pathways to HMF and humins during fructose conversion. | |
Obviously, the etherification–dehydration–condensation and the degradative condensation of fructose and/or HMF to humins were competitive at the initial stage of fructose conversion, where the reversible formation of DFAs at high fructose concentration favored the former path and suppressed the latter one. In addition, after reacting fructose for a long reaction time of 2 h, some solid humins were collected and characterized using FT-IR spectra (Fig. S5†).44 The characteristic FT-IR bands centered at 1611 and 1019 cm−1, respectively, were assigned to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O–C for furan rings, which indicated the existence of furan rings in solid humins. Moreover, the strong stretching vibrations of C(H)O at 1665 cm−1 further confirmed the involvement of HMF in solid humins. Therefore, HMF was unstable under the experimental conditions, and would undergo self- or cross-condensation to form soluble and even insoluble humins (i.e., solid humins). In addition, the increase of the relative intensity of the peak at 1611 cm−1 in the FT-IR spectra with increasing initial fructose concentration demonstrated the aggravation of humin formation from HMF.
C and C–O–C for furan rings, which indicated the existence of furan rings in solid humins. Moreover, the strong stretching vibrations of C(H)O at 1665 cm−1 further confirmed the involvement of HMF in solid humins. Therefore, HMF was unstable under the experimental conditions, and would undergo self- or cross-condensation to form soluble and even insoluble humins (i.e., solid humins). In addition, the increase of the relative intensity of the peak at 1611 cm−1 in the FT-IR spectra with increasing initial fructose concentration demonstrated the aggravation of humin formation from HMF.
Possible factors influencing the evolution pathways of humins
We are interested in the factors influencing the evolution pathways of humins during the fructose-to-HMF dehydration. Firstly, the pH values of the aqueous fructose solutions with different concentrations were measured at room temperature, wherein a decrease of pH value from 6.99 to 6.56 was observed with increasing fructose concentration from 4.5 wt% to 360.0 wt% (Table S1†).44 This result was indicative of the increase of H+ concentration owing to the increase of hydroxy groups capable of being ionized in water. On the one hand, the enhanced Brønsted acidity of the aqueous fructose solution favored the fructose-to-HMF dehydration, thereby accelerating the fructose conversion. On the other hand, the enhanced Brønsted acidity should also be responsible for the promotional effect on the etherification–dehydration–condensation of fructose and/or HMF to humins at the initial reaction stage of fructose conversion because this path was also promoted by a Brønsted acid.33,37
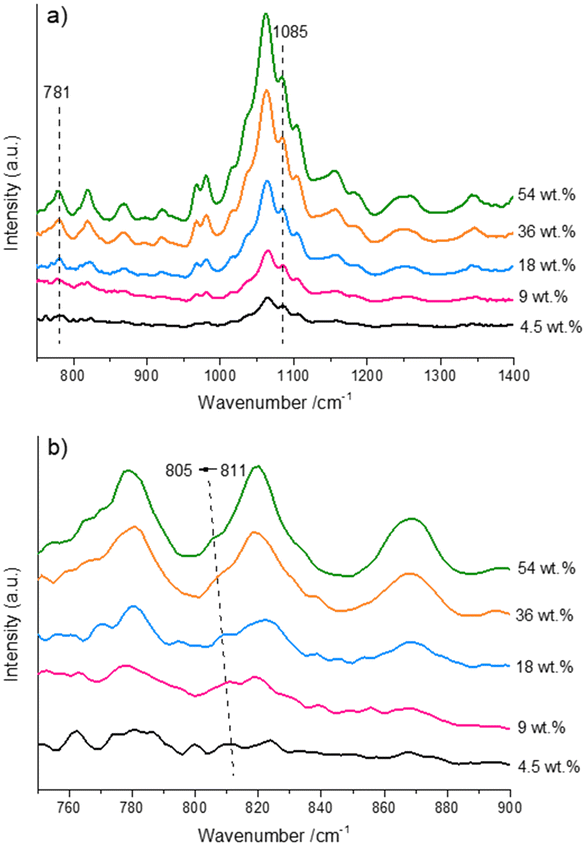
In addition, the possible interactions between fructose molecules at room temperature were explored using the ATR-IR spectra of the aqueous fructose solutions (Fig. 5). As seen from Fig. 5a, the intensity of the characteristic signals (Table S2†)44 assigned to fructose was increased with increasing fructose concentration. Notably, a band centered at 811 cm−1 was observed (Fig. 5b), which was attributed to the O–C2–C3 stretching vibration of β-β-3,1′-difructofuranose (a in Scheme 1). Hence, we inferred that some of difructose species existed as β-β-3,1′-difructofuranose in solutions at room temperature. It was observed that when the initial fructose concentration was increased from 4.5 wt% to 54.0 wt%, the band assigned to the difructose was redshifted from 811 cm−1 to 805 cm−1, which suggested the strengthening of the hydrogen-bonding involving the C2–OH of β-β-3,1′-difructofuranose. This result might be ascribed to the enhanced collision between the hydroxyl groups in difructose and other fructose molecules induced by the increase of fructose concentration. Then, the strengthening of this hydrogen bond in difructose would promote further intramolecular etherification to form DFAs (b in Scheme 1) under the role of the increased Brønsted acid. This result well explained the concentration-aggravated formation of DFA species under reaction conditions.
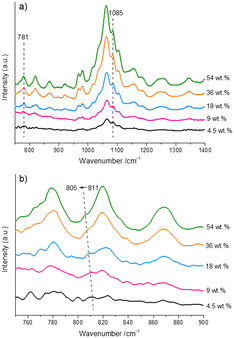 |
| | Fig. 5 ATR-IR spectra of the aqueous fructose solutions with different concentrations at room temperature. a) The wavenumber range is 750–1400 cm−1. b) Local magnification of the region with wavenumbers of 750–900 cm−1. | |
Putting together, the evolution pathways of humin formation during fructose conversion were concluded as shown in Scheme 1. At room temperature, fructose underwent intermolecular etherification to form difructose (a) in aqueous solution, which became serious when the fructose concentration became higher. At the initial stage of the fructose-to-HMF dehydration, the intramolecular dehydration of difructose led to the generation of DFAs (b), which acted as both protective intermediates to fructose/HMF and unwanted intermediates to humins I formed via the path of etherification–dehydration–condensation. As protective intermediates, the large amount of DFAs generated in the conversion of high-concentration fructose also alleviated the degradative condensation of fructose to humins II at the initial reaction stage. Thus, the reversible formation of DFAs at high fructose concentration aggravated the etherification–dehydration–condensation of fructose and/or HMF to form humins I, and at the same time, competed with the degradative condensation of fructose and/or HMF to humins II.
Stabilization of DFAs for accelerating fructose-to-HMF dehydration at high fructose concentration
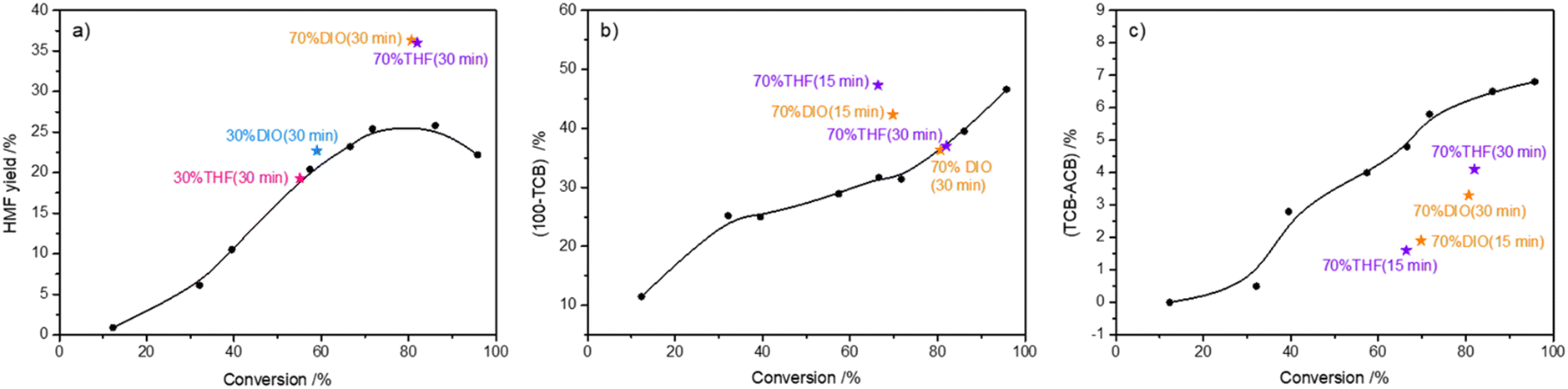
In the literature,32,48–51 the solvent composition was reported to significantly influence the selectivity to HMF in fructose-to-HMF dehydration. In this part, the possible roles of typical polar aprotic solvents (i.e., THF, DIO) in the conversion of high-concentration fructose to HMF were investigated by reacting fructose (72.0 wt%) at 140 °C for 30 min. As shown in Fig. 6a, the addition of THF or DIO with a volume ratio of 30% as the cosolvent did not affect the yield of HMF significantly when compared with the reaction performed in pure water. However, when the volume ratio was increased to 70%, the HMF yield was significantly improved by more than 10%. Nevertheless, the carbon loss from the etherification–dehydration–condensation of fructose and/or HMF did not change (Fig. 6b), whereas the carbon loss from the degradative condensation of fructose and/or HMF was significantly lowered (Fig. 6c). In light of these results, it was assumed that the addition of THF or DIO as the co-solvent with a volume ratio of 70% promoted the fructose-to-HMF dehydration by inhibiting humin formation. However, it was observed that the carbon loss from etherification–dehydration–condensation of fructose and/or HMF at a shorter reaction time (15 min) was larger (Fig. 6b), which suggested that the as-formed intermediates at this reaction time would reversibly transform back to fructose, and finally HMF. The ESI-MS spectra (Fig. S6b and c†)44 of the reaction mixture at that reaction time revealed the predominant existence of DFAs, thereby highlighting the stabilization of DFAs by adding THF or DIO as a co-solvent to water. Meanwhile, the relative intensities of the dehydrated species assigned to the [2Fru–6H2O + K + H]2+ (m/z = 146) and [3Fru–9H2O + K + H]2+ (m/z = 209) species were significantly weaker in the THF–H2O or DIO–H2O co-solvent than those in pure H2O (Fig. S6a†),44 which illustrated the relieving of DFAs from further intramolecular dehydration and intermolecular condensation by the presence of the co-solvent. When the reaction time was increased to 30 min, the consumption of DFAs was observed (Fig. S6d and e†),44 which suggested the positive transition of DFAs to fructose and finally HMF. As a result, the degradative condensation of fructose and/or HMF was alleviated by adding the co-solvents (Fig. 6c). Therefore, the stabilization of DFAs by the presence of typical polar aprotic solvents during the conversion of high-concentration fructose contributed to a higher HMF yield by alleviating the formation of humins via both paths of etherification–dehydration–condensation and degradative condensation of fructose/HMF.
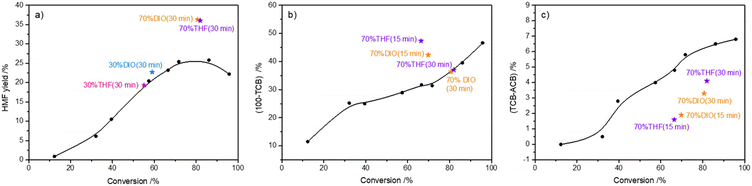 |
| | Fig. 6 a) HMF yield, b) 100 − TCB, and c) TCB − ACB as a function of fructose conversion. Black dots (•) represent experiments performed in H2O for a reaction time of 5–180 min. Pink pentagrams ( ) represent experiments performed in THF–H2O solvent (V/V = 3/7) for 30 min. Blue pentagrams ( ) represent experiments performed in THF–H2O solvent (V/V = 3/7) for 30 min. Blue pentagrams ( ) represent experiments performed in DIO–H2O solvent (V/V = 3/7) for 30 min. Purple pentagrams ( ) represent experiments performed in DIO–H2O solvent (V/V = 3/7) for 30 min. Purple pentagrams ( ) represent experiments performed in THF–H2O solvent (V/V = 7/3) for 15 min and 30 min. Orange pentagrams ( ) represent experiments performed in THF–H2O solvent (V/V = 7/3) for 15 min and 30 min. Orange pentagrams ( ) represent experiments performed in DIO–H2O solvent (V/V = 7/3) for 15 min and 30 min. All reactions were carried out with 72.0 wt% fructose and 0.05 M H2SO4 in 1 mL solvent at 140 °C under a N2 atmosphere. ) represent experiments performed in DIO–H2O solvent (V/V = 7/3) for 15 min and 30 min. All reactions were carried out with 72.0 wt% fructose and 0.05 M H2SO4 in 1 mL solvent at 140 °C under a N2 atmosphere. | |
Conclusions
In this work, the effects of fructose concentration (4.5–360.0 wt%) in aqueous fructose solution on the dehydration of fructose were studied. A universal concentration-induced deterioration of humin formation was demonstrated. We revealed that the strengthened H-bonds between fructose molecules and the increased Brønsted acidity in the high-concentration fructose solution promoted the formation of difructose at room temperature, which underwent intramolecular dehydration to form DFAs under reaction conditions. Herein, DFAs acted as protective intermediates for the reversible generation of fructose/HMF and inhibition of the degradative condensation to humins. Meanwhile, DFAs were also capable of undergoing further intramolecular dehydration and intermolecular condensations to form humins, which represented a main path for the evolution of humins in high-concentration fructose. Besides, the stabilization of DFAs by adding some typical polar aprotic solvents (e.g., THF, DIO) as a co-solvent of water would increase the HMF yield by more than 10% through the mitigation of both etherification–dehydration–condensation and degradative condensation of fructose/HMF to humins in the conversion of high-concentration fructose (72.0 wt%). These deep insights on the concentration-aggravated humin formation and feasible strategy for mitigating humin formation provide an indispensable understanding on the reaction chemistry of HMF biorefineries.
Author contributions
Yexin Hu: data acquirement and analysis, and writing – original draft. Hui Li and Ping Hu: ATR-IR and 13C NMR experiments. Linzhen Li, Di Wu and Zhidan Xue: data discussion. Liangfang Zhu and Changwei Hu: conceptualization, funding acquisition, project administration, supervision, and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 21875149), the 111 Project (B17030), and the Basal Research Fund of the Central University.
Notes and references
- J. R. Rostrup-Nielsen, Science, 2005, 308, 1421–1422 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- S. Gajula and C. R. K. Reddy, Biofuels, Bioprod. Biorefin., 2021, 15, 1221–1232 CrossRef CAS.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119–231 RSC.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976–2982 CrossRef CAS.
- Z. Xue, M.-G. Ma, Z. Li and T. Mu, RSC Adv., 2016, 6, 98874–98892 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- D. Hu, M. Zhang, H. Xu, Y. Wang and K. Yan, Renewable Sustainable Energy Rev., 2021, 147, 11253 CrossRef.
- J. Slak, B. Pomeroy, A. Kostyniuk, M. Grilc and B. Likozar, Chem. Eng. J., 2022, 429, 132325 CrossRef CAS.
- C. Thoma, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544–3564 CrossRef CAS PubMed.
- I. K. M. Yu and D. C. W. Tsang, Bioresour. Technol., 2017, 238, 716–732 CrossRef CAS PubMed.
- X. Zou, C. Zhu, Q. Wang and G. Yang, Biofuels, Bioprod. Biorefin., 2019, 13, 153–173 CrossRef CAS.
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS PubMed.
- J. Wang, J. Ren, X. Liu, G. Lu and Y. Wang, AIChE J., 2013, 59, 2558–2566 CrossRef CAS.
- C. Carlini, P. Patrono, A. M. R. Galletti and G. Sbrana, Appl. Catal., A, 2004, 275, 111–118 CrossRef CAS.
- J. Wang, W. Xu, J. Ren, X. Liu, G. Lu and Y. Wang, Green Chem., 2011, 13, 2678–2681 RSC.
- J. R. Bernardo, M. C. Oliveira and A. C. Fernandes, Mol. Catal., 2019, 465, 87–94 CrossRef CAS.
- W. Sebati, S. S. Ray and R. Moutloali, Catalysts, 2019, 9, 656 CrossRef CAS.
- S.-H. Pyo, M. Sayed and R. Hatti-Kaul, Org. Process Res. Dev., 2019, 23, 952–960 CrossRef CAS.
- Y. Feng, M. Li, Z. Gao, X. Zhang, X. Zeng, Y. Sun, X. Tang, T. Lei and L. Lin, ChemSusChem, 2019, 12, 495–502 CrossRef CAS.
- G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345–9348 CrossRef CAS.
- S. Marullo, C. Rizzo and F. D'Anna, Front. Chem., 2019, 7, 134 CrossRef PubMed.
- Y. Nie, Q. Hou, C. Bai, H. Qian, X. Bai and M. Ju, J. Cleaner Prod., 2020, 274, 123023 CrossRef CAS.
- Q. Hou, W. Li, M. Zhen, L. Liu, Y. Chen, Q. Yang, F. Huang, S. Zhang and M. Ju, RSC Adv., 2017, 7, 47288–47296 RSC.
- H. E. van Dam, A. P. G. Kieboom and H. Vanbekkum, Starch-Starke, 1986, 38, 95–101 CrossRef CAS.
- D. Jung, P. Körner and A. Kruse, Biomass Convers. Biorefin., 2019, 11, 1155–1170 CrossRef.
- B. A. Fachri, R. M. Abdilla, H. H. v. d. Bovenkamp, C. B. Rasrendra and H. J. Heeres, ACS Sustainable Chem. Eng., 2015, 3, 3024–3034 CrossRef CAS.
- Z. Cheng, J. L. Everhart, G. Tsilomelekis, V. Nikolakis, B. Saha and D. G. Vlachos, Green Chem., 2018, 20, 997–1006 RSC.
- Y. Shao, W. Lu, Y. Meng, D. Zhou, Y. Zhou, D. Shen and Y. Long, Sci. Total Environ., 2021, 755, 142499 CrossRef CAS PubMed.
- T. D. Swift, C. Bagia, V. Choudhary, G. Peklaris, V. Nikolakis and D. G. Vlachos, ACS Catal., 2014, 4, 259–267 CrossRef CAS.
- X. Fu, Y. Hu, Y. Zhang, Y. Zhang, D. Tang, L. Zhu and C. Hu, ChemSusChem, 2020, 13, 501–512 CrossRef CAS.
- J. Tang, L. Zhu, X. Fu, J. Dai, X. Guo and C. Hu, ACS Catal., 2016, 7, 256–266 CrossRef.
- T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS.
- X. Fu, Y. Hu, P. Hu, H. Li, S. Xu, L. Zhu and C. Hu, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.09.012.
- L. Zhu, X. Fu, Y. Hu and C. Hu, ChemSusChem, 2020, 13, 4812–4832 CrossRef CAS PubMed.
- T. Dallas Swift, H. Nguyen, A. Anderko, V. Nikolakis and D. G. Vlachos, Green Chem., 2015, 17, 4725–4735 RSC.
- B. F. M. Kuster, Starch-Starke, 1990, 42, 314–321 CrossRef CAS.
- Z. Cao, M. Li, Y. Chen, T. Shen, C. Tang, C. Zhu and H. Ying, Appl. Catal., A, 2019, 569, 93–100 CrossRef CAS.
- X. Zhang, K. Wilson and A. F. Lee, Chem. Rev., 2016, 116, 12328–12368 CrossRef CAS.
- T. Barclay, M. Ginic-Markovic, M. R. Johnston, P. Cooper and N. Petrovsky, Carbohydr. Res., 2012, 347, 136–141 CrossRef CAS PubMed.
- W. Guo, Z. Zhang, J. Hacking, H. J. Heeres and J. Yue, Chem. Eng. J., 2021, 409, 128182 CrossRef CAS.
- Y. Hu, Y. Zhang, X. Fu, D. Tang, H. Li, P. Hu, L. Zhu and C. Hu, Ind. Eng. Chem. Res., 2022, 61, 5786–5796 CrossRef CAS.
- See the ESI† for details.
- Y. Huang, P.-Y. Chao, T.-Y. Cheng, Y. Ho, C.-T. Lin, H.-Y. Hsu, J.-J. Wong and T.-C. Tsai, Chem. Eng. J., 2016, 283, 778–788 CrossRef CAS.
- T.-Y. Cheng, P.-Y. Chao, Y.-H. Huang, C.-C. Li, H.-Y. Hsu, Y.-S. Chao and T.-C. Tsai, Microporous Mesoporous Mater., 2016, 233, 148–153 CrossRef CAS.
- E. Suarez-Pereira, E. M. Rubio, S. Pilard, C. Ortiz Mellet and J. M. Garcia Fernandez, J. Agric. Food Chem., 2010, 58, 1777–1787 CrossRef CAS PubMed.
- G. S. Svenningsen, R. Kumar, C. E. Wyman and P. Christopher, ACS Catal., 2018, 8, 5591–5600 CrossRef CAS.
- L. Shuai and J. Luterbacher, ChemSusChem, 2016, 9, 133–155 CrossRef CAS PubMed.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, P. Bai, K. Ma, M. Neurock and J. A. Dumesic, Nat. Catal., 2018, 1, 199–207 CrossRef CAS.
- H. Kimura, M. Nakahara and N. Matubayasi, J. Phys. Chem. A, 2013, 117, 2102–2113 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Changwei
Hu
* and
Changwei
Hu
 ) represent experiments performed in THF–H2O solvent (V/V = 3/7) for 30 min. Blue pentagrams (
) represent experiments performed in THF–H2O solvent (V/V = 3/7) for 30 min. Blue pentagrams ( ) represent experiments performed in DIO–H2O solvent (V/V = 3/7) for 30 min. Purple pentagrams (
) represent experiments performed in DIO–H2O solvent (V/V = 3/7) for 30 min. Purple pentagrams ( ) represent experiments performed in THF–H2O solvent (V/V = 7/3) for 15 min and 30 min. Orange pentagrams (
) represent experiments performed in THF–H2O solvent (V/V = 7/3) for 15 min and 30 min. Orange pentagrams ( ) represent experiments performed in DIO–H2O solvent (V/V = 7/3) for 15 min and 30 min. All reactions were carried out with 72.0 wt% fructose and 0.05 M H2SO4 in 1 mL solvent at 140 °C under a N2 atmosphere.
) represent experiments performed in DIO–H2O solvent (V/V = 7/3) for 15 min and 30 min. All reactions were carried out with 72.0 wt% fructose and 0.05 M H2SO4 in 1 mL solvent at 140 °C under a N2 atmosphere.