
C–H activation and subsequent C–C bond formation in rigid alkenes catalyzed by Ru(III) metallates†
S.
Dharani
a,
G.
Kalaiarasi
 a,
Vincent M.
Lynch
b,
K.
Srinivasan
c and
R.
Prabhakaran
*a
a,
Vincent M.
Lynch
b,
K.
Srinivasan
c and
R.
Prabhakaran
*a
aDepartment of Chemistry, Bharathiar University, Coimbatore 641046, India. E-mail: rpnchemist@gmail.com
bDepartment of Chemistry, University of Texas, Austin, TX 78712-1224, USA
cDepartment of Physics, Bharathiar University, Coimbatore 641046, India
First published on 19th October 2022
Abstract
Pyrazolone derived Ru(III) complexes were synthesized and comprehensively characterized by spectral techniques. The true nature of the complexes was revealed by X-ray crystallography in which the ligand coordinated in a monobasic bidentate fashion. The catalytic efficiency of the complexes was analyzed in the C–H activation reaction of α,β-unsaturated carbonyl compounds. The reactions proceeded well in ethanol with potassium hydroxide as a base. In addition to Heck type coupling at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, ethanol mediated ethoxylation of the C–Cl bond at the C2 position of the quinoline moiety was witnessed and validated by establishing the structure of a representative intermediate through X-ray diffraction analysis. All the catalytic products were characterized by 1H and 13C NMR analysis and representatives by mass spectral studies.
C bond, ethanol mediated ethoxylation of the C–Cl bond at the C2 position of the quinoline moiety was witnessed and validated by establishing the structure of a representative intermediate through X-ray diffraction analysis. All the catalytic products were characterized by 1H and 13C NMR analysis and representatives by mass spectral studies.
Introduction
Transition metal catalyzed C–H activation reactions have taken a front lead in recent years for the construction of C–C and C–heteroatom bonds. Activation of a C–H bond minimizes the number of synthetic steps, and in most cases, it is accompanied by cyclization with nearby atoms or groups, accounting for tandem reactions.1,2 This breakthrough research has been currently involved in developing eco-friendly synthetic conditions for cost-effective and sustainable chemistry.3,4 Transition metal complexes especially those of palladium and rhodium were found to be crucial for C–H activation while promoting C–C bond formation under mild reaction conditions.5–11 Simultaneously, efforts have been taken towards the catalytic activation and conversion of C–H bonds by using other metal catalysts like ruthenium, nickel and iridium.12–15 Ruthenium catalysts are attractive owing to their stability in air and moisture and easy insertion into C–H bonds to form a cyclometalated species.12 More stable Ru(II) catalysts prepared in a facile way were intensively employed in such reactions.16–18 An early example of C–H activation with aryl halides was reported by Oi et al. by using a Ru(II) catalyst {[Ru(p-cymene)Cl2]2}.19 With [Ru(p-cymene)Cl2]2 as a catalyst, synthesis of many organic compounds such as maleimides,20 homoisoflavonoids, flavones,21 isoquinolines,22 and isoindolinones,23,24 has been accomplished. Similar to [Ru(p-cymene)Cl2]2, other ruthenium(II) precursors like RuCl3,25 Ru3(CO)12,26,27 [RuCl2(PPh3)3],28,29 and [RuCl2(cod)]n30 have also been applied. Most of these reactions generate the catalyst in situ by reaction with a ligand, viz., triphenylphosphine, (adamantyl)2P(O)H, etc.18,31–33 C–H activations catalyzed by ruthenium based catalytic systems can be categorized into various types, viz., i) arylation with aryl halides, boranes and silanes, ii) ortho-alkylation of arenes with haloalkanes and amides, iii) ortho-alkenylation of arenes with vinyl acetates and boronic acids and iv) ortho-ruthenation enabled remote C–H functionalization in which meta- and para-functionalized products are obtained.34 Further, it has been evidenced that directing groups like amide, carbonyl, ester, hydroxyl, silane, and phosphate play an important role in C–H activation reactions.35 In arene substrates, these groups mostly direct the incoming moiety to their ortho position.36 With carbonyl as the directing group, a pioneer work demonstrated a highly regioselective o-alkylation of acetophenone derivatives with alkenes by using a low valent ruthenium complex as a catalyst.15 This landmark contribution paved the way for various C–H alkylations and arylations. Ogiwara et al. arylated the ortho position of acyl derivatives with boronic acid esters using the catalysts [RuHCl(CO)(PAr3)3] and [RuH2(CO)(PAr3)3].37 With [RuCl2(PPh3)3] as a catalyst, ortho C–H arylation of 2-aroylbenzofurans with boronic acid has been carried out.38 [Ru(p-cymene)Cl2]2 mediated C–H alkylation reactions of acetophenone derivatives with maleimides and alkenes were reported by Bettadapur et al.,39 and Pan et al.40 Similar reports of alkylation for heterocyclic compounds using [RuCl2(PPh3)3] were made by Ramana and his co-workers.41,42 Though Ru(II) complexes have been applied for C–C bond constructions, their applications in Heck type coupling reactions are very limited. A report on Ru(II) catalyzed olefination (with and without a carbonyl group) in the presence of sodium acetate was made by Na et al.43 Munoz-Molina et al. later reported a similar Heck reaction for alkenylation of alkyl bromides with a ruthenium(II) cyclopentadienyl complex in the presence of N,N-diisopropylethylamine.44 All these reactions indicated the importance of ruthenium catalyzed C–H activation reactions.Pyrazolones, a well-known class of nitrogen compounds, are present in nature in the form of various alkaloids. Synthetically developed pyrazole and pyrazolone derivatives like celecoxib (a COX-2 inhibitor), remogliflozin etabonate (a type 2 diabetes drug), trocoxil (a veterinary drug), phenazone (antipyretic and analgesic), metamizole (antipyretic), and edaravone (a neuroprotective agent) have found medicinal applications. In addition, they were reported to inhibit human chk1 kinase, HIV integrase and p38. They are also used as synthons for producing other pyrazolone derivatives.45 Because of their pharmaceutical significance, several organocatalytic and transition metal catalytic strategies have been developed. Coordination chemistry makes use of pyrazolones as ligands, whose denticity can be enhanced by functionalizing the C3 or C4 position for generating new sites, facilitating effective chelation with the metal. As catalysts, pyrazolone derived complexes have been applied in polymerization, the Hantzsch reaction and oxidation reactions.46
A review of the literature finds only less availability of a predefined ruthenium(III) catalyst derived from a pyrazolone based amide ligand and its application in C–H activation reactions involving Heck type coupling has not been explored much. Hence, we have developed ruthenium(III) complexes of 4-(2-hydroxybenzyl)-3-methyl-5-oxo-2,5-dihydro pyrazole-1-carbothioic acid methylamide and 4-(2-hydroxybenzyl)-3-methyl-5-oxo-2,5-dihydro pyrazole-1-carbothioic acid ethylamide, which were applied as catalysts in the C–H activation of α,β-unsaturated carbonyl compounds.
Results and discussion
Ru(III) complexes (RuL1 and RuL2) were synthesized from the reaction of 4-(2-hydroxybenzyl)-3-methyl-5-oxo-2,5-dihydropyrazole-1-carbothioic acid methylamide (HL1)47 and 4-(2-hydroxybenzyl)-3-methyl-5-oxo-2,5-dihydropyrazole-1-carbothioic acid ethylamide (HL2)47 with [Ru(acac)2(CH3CN)2] in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloroform/methanol medium under reflux (Scheme 1). These complexes have shown complete dissolution in chloroform, dichloromethane, methanol, ethanol, acetonitrile, benzene, toluene, DMF and DMSO and were insoluble in water.
:1 chloroform/methanol medium under reflux (Scheme 1). These complexes have shown complete dissolution in chloroform, dichloromethane, methanol, ethanol, acetonitrile, benzene, toluene, DMF and DMSO and were insoluble in water.
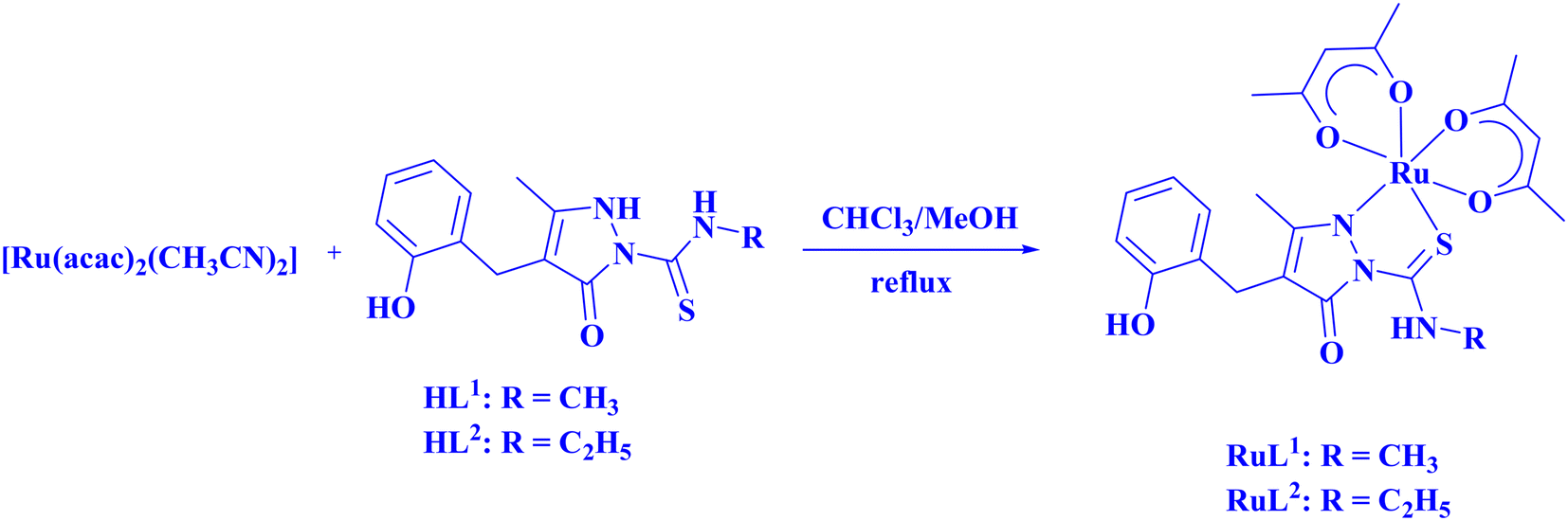 | ||
| Scheme 1 Synthesis of Ru(III) complexes RuL1 and RuL2. | ||
Infrared spectroscopy
In the IR spectra, the ligands HL1 and HL2 displayed bands corresponding to the presence of –OH, –NH, CO and CS groups. The IR spectra of the Ru(III) complexes RuL1 and RuL2 were compared with those of their parent ligands. The band due to –NH in the ligands at 3218 cm−1 (HL1) and 3170 cm−1 (HL2) disappeared in the complexes suggesting the involvement of nitrogen with the ruthenium ion after deprotonation. The CO group of the pyrazolone ring showed stretching at 1627 cm−1 (HL1) and 1614 cm−1 (HL2) in the ligands, which have experienced a slight downfield shift in the complexes (RuL1 1619 cm−1 and RuL2 1603 cm−1) indicating the non-involvement of pyrazolone oxygen in the coordination. The band corresponding to CS in the ligands at 844 cm−1 (HL1) and 833 cm−1 (HL2) shifted to lower frequency values of around 800 cm−1 (RuL1) and 802 cm−1 (RuL2), suggesting the coordination of the sulfur atom in the thione form.48 In addition, a band at 1517 (RuL1) and 1519 cm−1 (RuL2) in the complexes corresponded to the mixed mode of vibration due to the stretching of CO and CC groups in the coordinated acetylacetone.49
Electronic spectroscopy
The electronic spectra of the ligands and complexes were recorded in chloroform (Fig. S1†). The ligand HL1 exhibited a single sharp absorption band at 272 nm assignable to π–π* transition and HL2 exhibited three absorption bands centered at 234, 271 and 349 nm corresponding to π–π* and n–π* transitions.48,50 The ground state of the ruthenium(III) ion is 2T2g (t2g5 configuration) and the excited states related to the t2g4eg1 configuration are 2A2g and 2A1g. Hence, from the ground state to excited states, two transitions are possible. The complex RuL1 showed three main bands at 279, 389 and 649 nm and RuL2 exhibited three bands at 278, 390 and 647 nm. The bands at around 278–279 nm were assigned to intra ligand transitions, the bands at 389–390 nm were due to metal to ligand charge transfer transition (MLCT) and the bands at 647–649 nm were attributed to the transition between metal ‘d’ orbitals (d–d transition).1H NMR spectroscopy
The 1H NMR spectra of the ligands HL1 and HL2 are shown in Fig. S2 and S3.† The hydroxyl proton of the ligands resonated as a doublet at δ 11.11 (HL1) and as a singlet at 11.38 ppm (HL2), the –NH proton of the pyrazolone ring as a singlet at δ 8.99 (HL1) and 9.43 ppm (HL2) and the terminal –NH proton as a doublet at δ 7.34 (HL1) and as a triplet at around 6.67–6.71 ppm (HL2). Methyl protons of the ring resonated as a singlet at δ 3.21 ppm for HL1 and 2.16 ppm for HL2. The –NHCH3 protons of the ligand HL1 were observed at δ 2.30 ppm as a doublet and the –NHCH2 of HL2 as a singlet at 3.45 ppm. Aromatic protons were observed in the region δ 6.77–7.54 ppm. The unresolved signals in the 1H NMR spectra clearly revealed the paramagnetic nature of the complexes.EPR spectroscopy
The paramagnetic nature of the complexes RuL1 and RuL2 was ascertained from their X-band EPR spectra recorded at room temperature in the solid state (Fig. S4†). The spectra clearly indicated the anisotropy of the complexes. The complexes RuL1 and RuL2 exhibited axial distortion in an octahedral geometry with two different ‘g’ values g‖ and g⊥ as given in Table S1.† The average g value gav was calculated from the equation gav = (g‖ + 2 × g⊥)/3. The obtained ‘g’ values gx = gy ≠ gz indicated tetragonal distortion in an octahedral field.X-ray crystallography
The ligand HL2 (Fig. 1) and the complexes RuL1 (Fig. 2) and RuL2 (Fig. 3) were structurally characterized by single crystal X-ray diffraction analysis. The ligand HL2 crystallized in an orthorhombic crystal system with the space group Pbca. Crystallographic data and the respective bond lengths and angles are given in Tables 1 and 2. The complexes RuL1 and RuL2 were found to be in a triclinic crystal system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. In both complexes, the ligands behaved in a mononegative bidentate manner by the coordination of a nitrogen atom of the pyrazolone ring and the thione sulfur atom to the ruthenium ion with Ru(1)–N(1) and Ru(1)–S(1) bond distances of 2.011 Å (RuL1), 2.000 Å (RuL2), 2.329 Å (RuL1), and 2.337 Å (RuL2), respectively. Two acetylacetonate units coordinated to the ruthenium ion through two oxygen atoms from each unit with Ru–O bond distances of Ru(1)–O(3) 2.012 Å (RuL1) 2.023 Å (RuL2), Ru(1)–O(4) 2.065 Å (RuL1) 2.018 Å (RuL2), Ru(1)–O(5) 2.022 Å (RuL1) 2.061 Å (RuL2), and Ru(1)–O(6) 2.025 Å (RuL1) 2.016 Å (RuL2). The bite angles N(1)–Ru(1)–S(1) were found to be 84° (RuL1) and 83° (RuL2). The trans angles N(1)–Ru(1)–O(6) (175°), S(1)–Ru(1)–O(4) (177°) and O(3)–Ru(1)–O(5) (178°) of the complex RuL1 and O(3)–Ru(1)–O(6) (179°), O(5)–Ru(1)–S(1) (178°) and N(1)–Ru(1)–O(4) (175°) of the complex RuL2 (Table 2) are less than 180° indicating severe distortion in the octahedral geometry. The torsion angles of the complexes RuL1 and RuL2 were 1.32° and 1.35°, respectively. The observed bond distances and bond angles are comparable to the reported values.51–53
space group. In both complexes, the ligands behaved in a mononegative bidentate manner by the coordination of a nitrogen atom of the pyrazolone ring and the thione sulfur atom to the ruthenium ion with Ru(1)–N(1) and Ru(1)–S(1) bond distances of 2.011 Å (RuL1), 2.000 Å (RuL2), 2.329 Å (RuL1), and 2.337 Å (RuL2), respectively. Two acetylacetonate units coordinated to the ruthenium ion through two oxygen atoms from each unit with Ru–O bond distances of Ru(1)–O(3) 2.012 Å (RuL1) 2.023 Å (RuL2), Ru(1)–O(4) 2.065 Å (RuL1) 2.018 Å (RuL2), Ru(1)–O(5) 2.022 Å (RuL1) 2.061 Å (RuL2), and Ru(1)–O(6) 2.025 Å (RuL1) 2.016 Å (RuL2). The bite angles N(1)–Ru(1)–S(1) were found to be 84° (RuL1) and 83° (RuL2). The trans angles N(1)–Ru(1)–O(6) (175°), S(1)–Ru(1)–O(4) (177°) and O(3)–Ru(1)–O(5) (178°) of the complex RuL1 and O(3)–Ru(1)–O(6) (179°), O(5)–Ru(1)–S(1) (178°) and N(1)–Ru(1)–O(4) (175°) of the complex RuL2 (Table 2) are less than 180° indicating severe distortion in the octahedral geometry. The torsion angles of the complexes RuL1 and RuL2 were 1.32° and 1.35°, respectively. The observed bond distances and bond angles are comparable to the reported values.51–53
 | ||
| Fig. 1 ORTEP diagram of the ligand HL2 (thermal ellipsoids at a 50% probability level). | ||
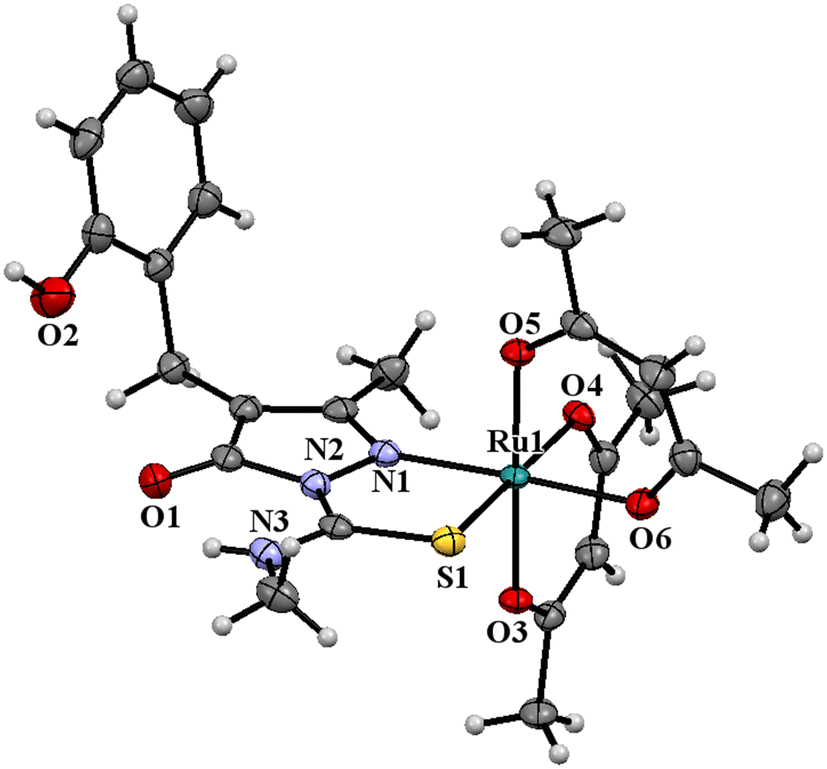 | ||
| Fig. 2 ORTEP diagram of the Ru(III) complex RuL1 (thermal ellipsoids at a 50% probability level). | ||
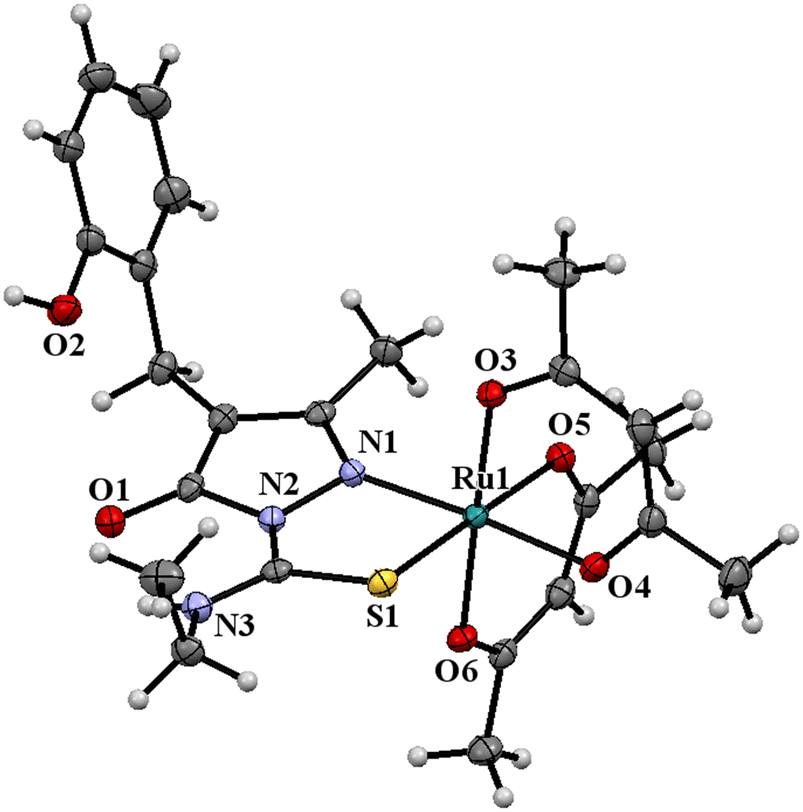 | ||
| Fig. 3 ORTEP diagram of the Ru(III) complex RuL2 (thermal ellipsoids at a 50% probability level). | ||
| HL 2 | RuL 1 | RuL 2 | |
|---|---|---|---|
| Empirical formula | C14H17N3O2S | C23H28N3O6RuS | C24H30N3O6RuS |
| Formula weight | 291.36 | 575.61 | 589.64 |
| Temperature | 100(2) K | 100(2) K | 100(2) K |
| Wavelength | 0.7107 Å | 1.5418 Å | 1.5418 Å |
| Crystal system | Orthorhombic | Triclinic | Triclinic |
| Space group | Pbca |
P |
P |
| Unit cell dimensions | |||
| a | 13.042(3) Å | 9.247(4) Å | 8.641(2) Å |
| b | 10.705(2) Å | 11.966(4) Å | 12.432(5) Å |
| c | 20.581(5) Å | 11.968(5) Å | 13.006(7) Å |
| α | 90° | 63.486(4)° | 63.222(5)° |
| β | 90° | 87.750(3)° | 82.336(3)° |
| γ | 90° | 86.958(3)° | 86.701(3)° |
| Volume | 2873.3(10) Å3 | 1183.2(9) Å3 | 1236.3(10) Å3 |
| Z | 8 | 2 | 2 |
| Density | 1.347 Mg m−3 | 1.616 Mg m−3 | 1.584 Mg m−3 |
| Absorption coefficient | 0.230 mm−1 | 6.575 mm−1 | 6.307 mm−1 |
| F(000) | 1232 | 590 | 606 |
| θ range for data collection | 2.521 to 28.287° | 4.094 to 72.914° | 3.963 to 73.132° |
| Limiting indices | −17 ≤ h ≤ 17, −13 ≤ k ≤ 14, −27 ≤ l ≤ 27 | −11 ≤ h ≤ 9, −14 ≤ k ≤ 14, −14 ≤ l ≤ 14 | −10 ≤ h ≤ 10, −12 ≤ k ≤ 15, −16 ≤ l ≤ 16 |
| Reflections collected | 50085 |
13457 |
14034 |
| Independent reflections | 3574 [R(int) = 0.0758] | 4639 [R(int) = 0.0224] | 4852 [R(int) = 0.0290] |
| Absorption correction | Semi-empirical from equivalents | Gaussian and multi scan | Gaussian and multi scan |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3574/288/239 | 4639/0/317 | 4852/0/326 |
| Goodness-of-fit on F2 | 1.034 | 1.038 | 1.037 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0438, wR2 = 0.1091 | R 1 = 0.0266, wR2 = 0.0687 | R 1 = 0.0227, wR2 = 0.0578 |
| R indices (all data) | R 1 = 0.0708, wR2 = 0.1209 | R 1 = 0.0273, wR2 = 0.0693 | R 1 = 0.0243, wR2 = 0.0590 |
| Bond lengths | HL 2 | RuL 1 | RuL 2 |
|---|---|---|---|
| O(1)–C(1) | 1.259(02) | — | — |
| N(1)–C(1) | 1.406(02) | — | — |
| N(1)–N(2) | 1.370(02) | 1.406(03) | 1.405(02) |
| S(1)–C(4) | 1.648(04) | — | — |
| N(3)–C(4) | 1.328(05) | — | — |
| Ru(1)–N(1) | — | 2.011(18) | 2.000(15) |
| Ru(1)–S(1) | — | 2.329(06) | 2.337(05) |
| Ru(1)–O(3) | — | 2.012(15) | 2.023(13) |
| Ru(1)–O(4) | — | 2.065(15) | 2.018(13) |
| Ru(1)–O(5) | — | 2.022(15) | 2.061(13) |
| Ru(1)–O(6) | — | 2.025(16) | 2.016(13) |
| Bond angles | |||
|---|---|---|---|
| N(1)–Ru(1)–S(1) | — | 84.61(5) | 83.32(4) |
| S(1)–Ru(1)–O(3) | — | 89.81(5) | 93.31(4) |
| O(3)–Ru(1)–O(6) | — | 88.37(6) | 179.05(5) |
| O(6)–Ru(1)–O(4) | — | 86.82(7) | 88.26(5) |
| O(4)–Ru(1)–O(5) | — | 87.65(6) | 86.43(5) |
| O(5)–Ru(1)–N(1) | — | 89.86(7) | 98.06(6) |
| O(3)–Ru(1)–O(4) | — | 91.98(6) | 90.87(5) |
| O(5)–Ru(1)–S(1) | — | 90.52(5) | 178.55(4) |
| O(5)–Ru(1)–O(6) | — | 90.29(6) | 93.34(5) |
| O(6)–Ru(1)–S(1) | — | 91.06(5) | 87.12(4) |
| N(1)–Ru(1)–O(3) | — | 91.51(7) | 90.75(6) |
| N(1)–Ru(1)–O(4) | — | 97.51(7) | 175.32(5) |
| N(1)–Ru(1)–O(6) | — | 175.67(7) | 90.14(6) |
| S(1)–Ru(1)–O(4) | — | 177.19(4) | 92.21(4) |
| O(3)–Ru(1)–O(5) | — | 178.62(6) | 86.22(5) |
Hydrogen bonding interactions
The ligand HL2 exhibited two intermolecular hydrogen bonding interactions – one between H(23)O of one molecule and O(1) of another molecule and vice versa and another between H(21)N of one molecule and O(2) of an adjacent molecule and vice versa and one intramolecular hydrogen bonding interaction between H(3)N of one molecule and O(1) of an adjacent molecule and vice versa (Fig. S5, Table S2†). In the complexes RuL1 and RuL2, two molecules are interconnected through one intramolecular (between N(3)–H(3) and O(1)) hydrogen bonding interaction and one intermolecular (between O(2)–H(2) and O(1)) hydrogen bonding interaction (Fig. S6 and S7 and Table S2†). These interactions in the complexes resulted in a pseudo binuclear structure.Catalytic application in C–H activation of α,β-unsaturated carbonyl compounds
The applications of Ru(II) precatalysts in C–H activation reactions are numerous but those of Ru(III) complexes are rare. Hence, the synthesized Ru(III) complexes RuL1 and RuL2 were applied as catalysts in the C–H activation reactions of α,β-unsaturated carbonyl compounds. For optimizing the reaction conditions, the chalcone derived from 2-chloro-3-formylbenzo quinoline and acetyl ferrocene was reacted with bromobenzene in various solvents with potassium hydroxide as the base under reflux with the complex RuL1 as the catalyst (Table 3). Optimization of solvents indicated that the reaction was very specific as it proceeded only in ethanol (Table 3, entry 1) and not in other solvents (Table 3, entries 2–11). The scheme given in Table 3 shows the expected product. However, the characterization of the obtained product indicated that in addition to Heck type coupling at the CC bond, the C–Cl bond of the quinoline ring has been activated by ethanol, i.e., solvent mediated ethoxylation of the C–Cl bond. When the polarities of methanol, ethanol and isopropanol are compared, ethanol is found to be more polar than isopropanol but less polar than methanol. Maybe this reaction occurs neither in a more polar nor in a less polar solvent than ethanol. We are uncertain in offering any concrete reason for why the reaction occurred only in ethanol and not in methanol or isopropanol at this stage.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Base | Temperature (°C) | Time (h) | Yielda (%) |
| Reaction conditions: rigid alkene (1 mmol), bromobenzene (1 mmol), base (2 mmol), and Ru(III) catalyst (0.005 mmol). a Isolated yield. rt – room temperature. NR – no reaction. | ||||||
| 1 | EtOH | RuL 1 | KOH | Reflux | 3 | 84 |
| 2 | MeOH | RuL 1 | KOH | Reflux | 3 | NR |
| 3 | CH3CN | RuL 1 | KOH | Reflux | 3 | NR |
| 4 | DMF | RuL 1 | KOH | Reflux | 3 | NR |
| 5 | DMSO | RuL 1 | KOH | Reflux | 3 | NR |
| 6 | CHCl3 | RuL 1 | KOH | Reflux | 3 | NR |
| 7 | DCM | RuL 1 | KOH | Reflux | 3 | NR |
| 8 | Benzene | RuL 1 | KOH | Reflux | 3 | NR |
| 9 | Toluene | RuL 1 | KOH | Reflux | 3 | NR |
| 10 | 2-Methoxyethanol | RuL 1 | KOH | Reflux | 3 | NR |
| 11 | Isopropanol | RuL 1 | KOH | Reflux | 3 | NR |
| 12 | EtOH | RuL 2 | KOH | Reflux | 3 | 91 |
| 13 | EtOH | RuL 2 | KOH | 60 | 3 | 67 |
| 14 | EtOH | RuL 2 | KOH | 50 | 3 | 42 |
| 15 | EtOH | RuL 2 | KOH | rt | 3 | 36 |

The application of the complex RuL2 enhanced the yield of the product to 91% (Table 3, entry 12) and hence further reactions were carried out with RuL2 as the catalyst. The reactions kept at 60 °C, 50 °C and room temperature resulted in only a lower yield (Table 3, entries 13–15). Variation of the base and the catalyst loading (Table 4) suggested 2 mmol of potassium hydroxide to be the better base with 0.005 mmol of the catalyst RuL2 for better progress of the reaction (Table 4, entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base (mmol) | Catalyst (mmol) | Temperature | Time (h) | Yielda (%) |
| Reaction conditions: rigid alkene (1 mmol), bromobenzene (1 mmol), base (as specified), RuL2 (as specified). a Isolated yield. NR – no reaction. | |||||
| 1 | KOH (2) | 0.005 | Reflux | 3 | 91 |
| 2 | NaOH (2) | 0.005 | Reflux | 3 | 78 |
| 3 | K2CO3 (2) | 0.005 | Reflux | 3 | 43 |
| 4 | Na2CO3 (2) | 0.005 | Reflux | 3 | 36 |
| 5 | Cs2CO3 (2) | 0.005 | Reflux | 3 | 42 |
| 6 | Et3N (2) | 0.005 | Reflux | 3 | 59 |
| 7 | KOH (2) | No catalyst | Reflux | 3 | NR |
| 8 | No base | 0.005 | Reflux | 3 | NR |
| 9 | KOH (1.5) | 0.005 | Reflux | 3 | 84 |
| 10 | KOH (1) | 0.005 | Reflux | 3 | 63 |
| 11 | KOH (2) | 0.0025 | Reflux | 3 | 72 |
| 12 | KOH (2) | 0.00125 | Reflux | 3 | 37 |
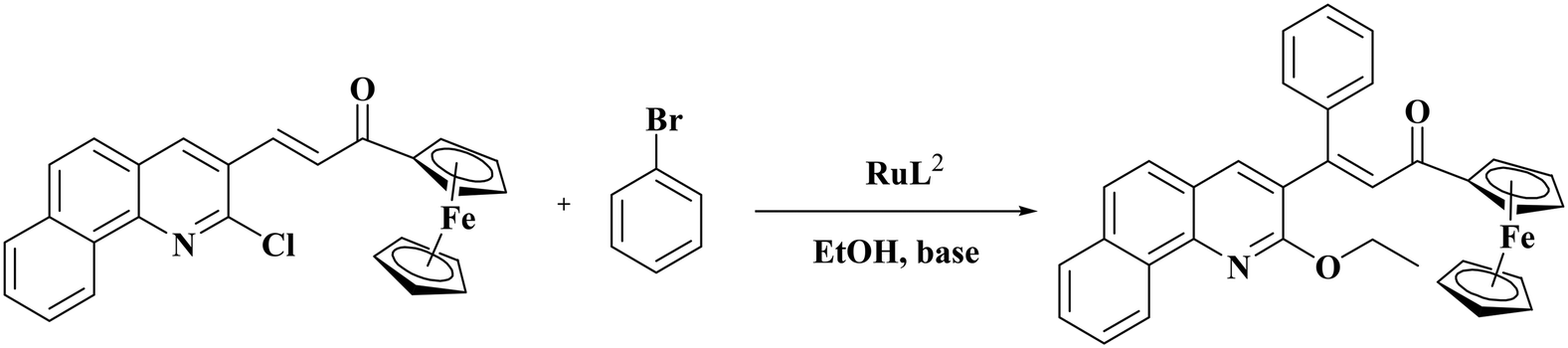
Extension of the reaction scope was done with chalcones specifically derived from quinolines and acetyl ferrocenes (Table 5). In a particular series with respect to the ketonic part in chalcone, benzoquinoline derived chalcones gave a comparatively higher yield than others. The 1H and 13C NMR spectra of the products are shown in Fig. S8 and S39.† Mass spectra of representative products were recorded as supporting evidence and are provided in Fig. S40–S47.† In order to check the utility of the reaction, three different aryl bromides were taken for coupling with the quinolinyl chalcone. Their NMR spectra are presented in Fig. S48–S50.†
|
|
||
|---|---|---|
| Reaction conditions: rigid alkene (1 mmol), bromobenzene (1 mmol), base (2 mmol), and 6B (0.005 mmol). a Isolated yield. | ||
 A1, 91%,a 3 h
A1, 91%,a 3 h |
 A2, 84%,a 3 h
A2, 84%,a 3 h |
 A3, 85%,a 3 h
A3, 85%,a 3 h |
 A4, 88%,a 3 h
A4, 88%,a 3 h |
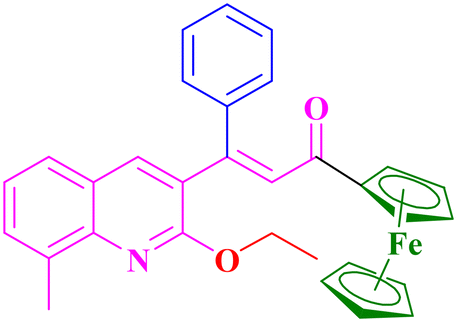 A5, 87%,a 3 h
A5, 87%,a 3 h |
 A6, 82%,a 3 h
A6, 82%,a 3 h |
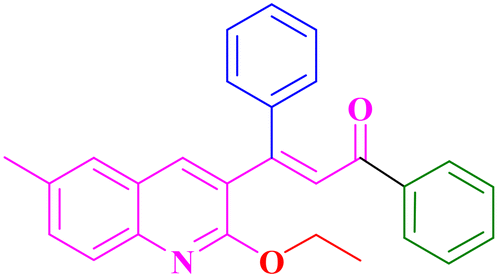 A7, 82%,a 3 h
A7, 82%,a 3 h |
 A8, 74%,a 3 h
A8, 74%,a 3 h |
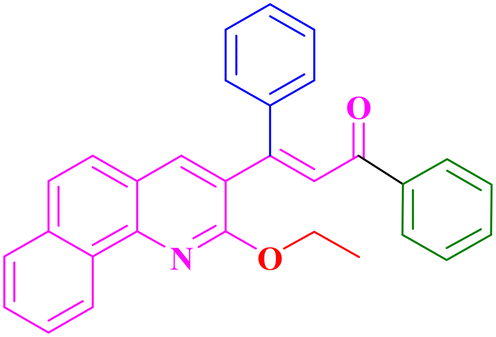 A9, 86%,a 3 h
A9, 86%,a 3 h |
 A10, 80%,a 3 h
A10, 80%,a 3 h |
 A11, 82%,a 3 h
A11, 82%,a 3 h |
 A12, 83%,a 3 h
A12, 83%,a 3 h |
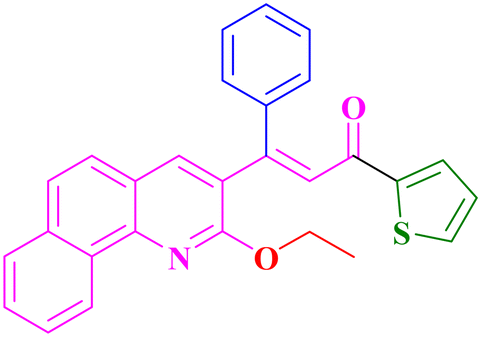 A13, 86%,a 3 h
A13, 86%,a 3 h |
 A14, 79%,a 3 h
A14, 79%,a 3 h |
 A15, 81%,a 3 h
A15, 81%,a 3 h |
 A16, 84%,a 3 h
A16, 84%,a 3 h |
 A17, 81%,a 3 h
A17, 81%,a 3 h |
 A18, 84%,a 3 h
A18, 84%,a 3 h |
 A19, 89%,a 3 h
A19, 89%,a 3 h |
||

The formation of the intermediate ethoxy quinolinyl chalcone was validated by carrying out a representative reaction without the addition of bromobenzene; the compound was isolated through column chromatography and its true nature was established by X-ray diffraction analysis. The crystallographic data are presented in Table S3.† From the ORTEP diagram (Fig. 4), it is clear that the C–Cl bond of the quinoline moiety has been activated by ethanol and resulted in an ethoxylated intermediate. From the above observation, it is inferred that during the product formation, ethoxylation has taken place prior to Heck coupling of the chalcone.
 | ||
| Fig. 4 ORTEP diagram of the A12 ethoxy intermediate (thermal ellipsoids at a 50% probability level). | ||
Through the isolation of the ethoxy intermediate, the occurrence of ethoxylation prior to Heck coupling was confirmed. This indicated that the presence of chlorine somehow hindered the Heck coupling because of the possible resonance due to conjugation. The catalyst regenerated at the end of the overall reaction as presented in the scheme under Table 5 completely matched with the virgin catalyst, showing its chemical identity. In addition, no spot for the ligand was observed in TLC both during the progress and at the end of the reaction. Based on these observations, a mechanistic catalytic pathway has been suggested starting with ethoxylation and proceeding to Heck coupling (Scheme 2).
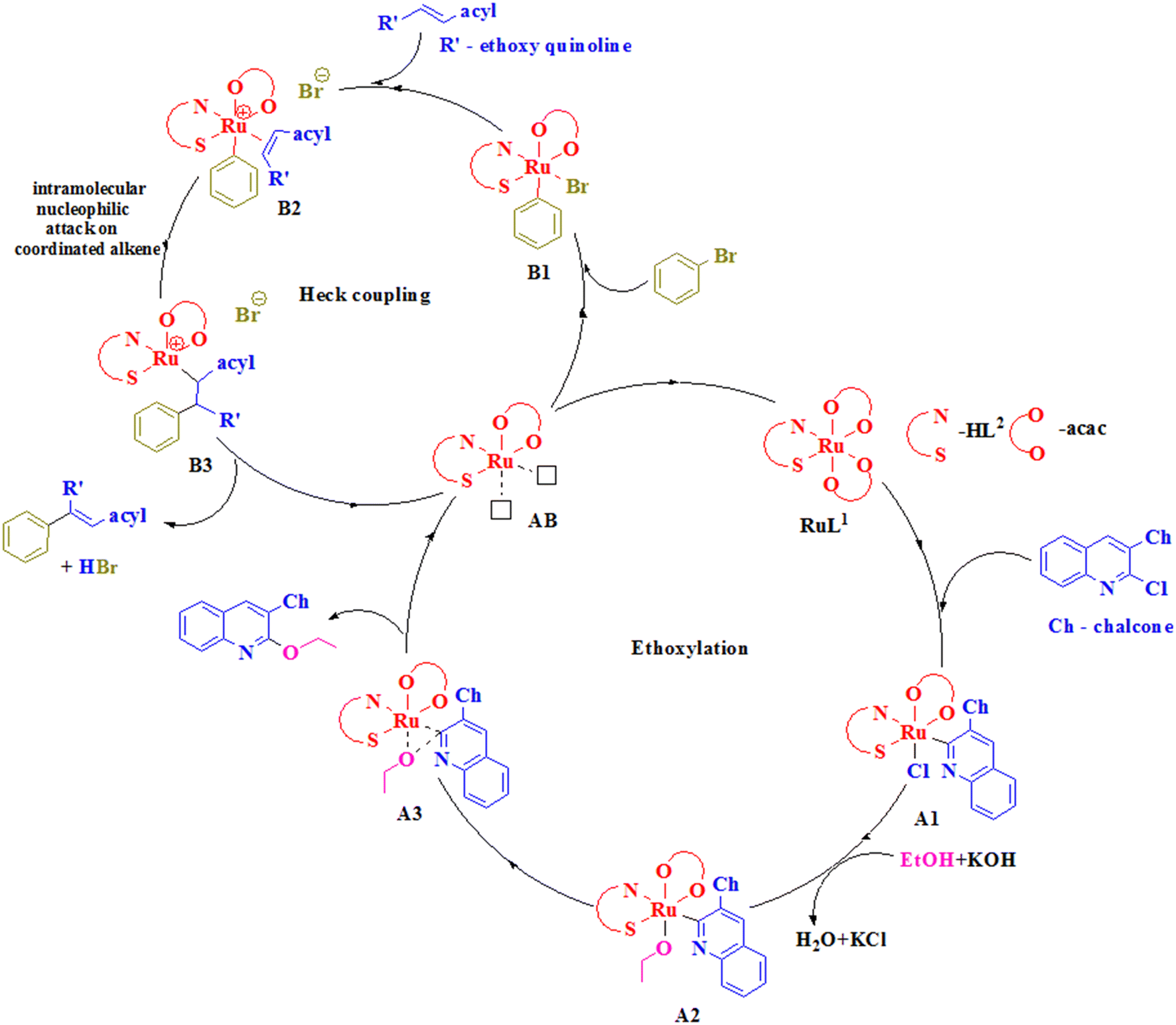 | ||
| Scheme 2 Plausible mechanism for the formation of the arylated quinolinyl ethoxy chalcone. | ||
The mechanism started with the oxidative addition of the quinolinyl chalcone to the catalyst RuL2 to generate a Ru(IV) species A1. This species, upon reaction with the ethoxide ion obtained by the reaction of ethanol with potassium hydroxide, exchanged with the chloride ion and resulted in an intermediate A2. A2 formed A3, which underwent reductive elimination to give the ethoxylated quinoline chalcone, and the species AB formed was involved in Heck coupling.54 Initially, oxidative addition of bromobenzene produced B1, which interacted with the ethoxy quinoline chalcone and formed B2. In B2, intramolecular nucleophilic attack on the coordinated alkene led to generation of the intermediate B3 which underwent Ru–C bond activation to form a coupled product by reductive elimination.
A review of the literature indicated no Ru(III) catalyzed reaction similar to the one presented in the current work, i.e., inclusion of quinoline C–Cl bond alkoxylation followed by Heck type coupling at the alkenyl C–H of an α,β-unsaturated ketone. In addition, there are only limited reports on Ru(II) catalyzed Heck coupling reactions. Hence, a comparison of the catalyst activities was made only for Heck coupling (Table 6). It indicated that our complex was able to catalyze the coupling reaction at a relatively lower temperature compared to the others.
Conclusion
Two Ru(III) complexes RuL1 and RuL2 of pyrazolone thioamide ligands have been synthesized and characterized by using IR, UV-visible, 1H NMR, EPR spectroscopy and X-ray crystallographic analysis. The ligands behaved in a monobasic bidentate manner while coordinating to the ruthenium ion by using their pyrazolone nitrogen and thione sulfur atoms. The coordination of two acetylacetonate units satisfied the other coordination sites. The complexes were tested as catalysts for the C–H activation reaction of α,β-unsaturated carbonyl compounds. Optimization of reaction conditions indicated ethanol and potassium hydroxide to be the better solvent and base, respectively. Upon reaction with bromobenzene, Heck type coupling occurred at the CC bond. Further, ethanol mediated ethoxylation at the C2 position of the quinoline moiety took place through C–Cl bond activation. This was validated by solving the structure of an isolated ethoxy intermediate. The products were characterized by 1H and 13C NMR spectroscopy and the mass spectra were recorded for representative products.
Experimental section
Synthesis of the Ru(III) complex RuL1
A methanolic solution (10 cm3) of 4-(2-hydroxy-benzyl)-3-methyl-5-oxo-2,5-dihydro-pyrazole-1-carbothioic acid methylamide HL1 (100 mg, 36 mmol) was added dropwise to [Ru(acac)2(CH3CN)2] (139 mg, 36 mmol) in chloroform (10 cm3) and refluxed for an hour. The reaction mixture was dried, subjected to thin layer chromatography and purified through column chromatography to obtain a green product. The product obtained was recrystallized from chloroform/methanol (4:1) to yield single crystals suitable for X-ray crystallography.
Synthesis of the Ru(III) complex RuL2
A methanolic solution (10 cm3) of 4-(2-hydroxy-benzyl)-3-methyl-5-oxo-2,5-dihydro-pyrazole-1-carbothioic acid ethylamide HL2 (100 mg, 36 mmol) was added dropwise to [Ru(acac)2(CH3CN)2] (132 mg, 36 mmol) in chloroform (10 cm3) and refluxed for an hour. The reaction mixture was dried, subjected to thin layer chromatography and purified through column chromatography to obtain a green product. The product obtained was recrystallized from chloroform/methanol (4:1) to yield single crystals suitable for X-ray crystallography.
O), 844 (νCS). UV-vis (DMSO), λmax (ε): 246 (42132) nm (dm3 mol−1 cm−1) (π–π* transition), 276 (74647) nm (dm3 mol−1 cm−1) (π–π* transition). 1H NMR (CDCl3, δ ppm, J Hz): δ 11.11 (d, J = 21.6 Hz, 1H, –OH), δ 8.99 (s, 1H, –NHring), δ 7.34 (d, J = 6 Hz, 1H, C6–H), δ 7.09 (d, J = 4.4 Hz, 1H, C5–H), δ 6.89 (d, J = 7.6 Hz, 2H, C3–H & C4–H), δ 6.77–6.80 (t, J = 7.2 Hz, 1H, –NH), δ 3.53 (s, 2H, –CH2), δ 3.20 (s, 3H, –CH3ring), δ 2.31 (s, 3H, –NHCH3![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ).
O), 833 (νCS). UV-vis (DMSO), λmax (ε): 234 (24354) nm (dm3 mol−1 cm−1) (π–π* transition), 271 (62934) nm (dm3 mol−1 cm−1) (π–π* transition), 349 (8933) nm (dm3 mol−1 cm−1) (n–π* transition). 1H NMR (DMSO-d6, δ ppm, J Hz): δ 11.38 (s, 1H, –OH), δ 9.43 (s, 1H, –NHring), δ 7.54 (d, J = 8 Hz, 2H, C3–H), δ 7.36–7.40 (t, J = 8 Hz, 2H, C4–H), δ 7.11–7.14 (t, J = 7.2 Hz, 1H, C5–H), δ 6.97–7.04 (m, 2H, –CH2), δ 6.77 (d, J = 7.6 Hz, 1H, C6–H), δ 6.67–6.71 (t, J = 7.6 Hz, 1H, –NH), δ 3.45 (s, 2H, –NHCH2
).
O), 833 (νCS). UV-vis (DMSO), λmax (ε): 234 (24354) nm (dm3 mol−1 cm−1) (π–π* transition), 271 (62934) nm (dm3 mol−1 cm−1) (π–π* transition), 349 (8933) nm (dm3 mol−1 cm−1) (n–π* transition). 1H NMR (DMSO-d6, δ ppm, J Hz): δ 11.38 (s, 1H, –OH), δ 9.43 (s, 1H, –NHring), δ 7.54 (d, J = 8 Hz, 2H, C3–H), δ 7.36–7.40 (t, J = 8 Hz, 2H, C4–H), δ 7.11–7.14 (t, J = 7.2 Hz, 1H, C5–H), δ 6.97–7.04 (m, 2H, –CH2), δ 6.77 (d, J = 7.6 Hz, 1H, C6–H), δ 6.67–6.71 (t, J = 7.6 Hz, 1H, –NH), δ 3.45 (s, 2H, –NHCH2![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ), δ 2.16 (s, 3H, –CH3ring), δ 1.99 (s, 3H, CH2CH3). Single crystals suitable for X-ray diffraction were obtained by recrystallization in a chloroform and methanol (4:1 v/v) mixture.
O 1619, νCS 800, νCO (acetylacetone) 1517. UV-vis (DMSO), λmax, nm (ε, dm3 mol−1 cm−1) 279 (65297) (intra ligand transition), 389 (27028) (MLCT), 649 (19459) (d–d transition). EPR (X-band, solid state): g‖ = 2.664, g⊥ = 2.131, gav = 2.302. Single crystals suitable for X-ray diffraction were obtained by recrystallization in a chloroform and methanol (4:1 v/v) mixture.
O 1603, νCS 802, νCO (acetylacetone) 1519. UV-vis (DMSO), λmax, nm (ε, dm3 mol−1 cm−1) 278 (69330) (intra ligand transition), 390 (25461) (MLCT), 647 (15445) (d–d transition). EPR (X-band, solid state): g‖ = 2.181, g⊥ = 1.996, gav = 2.057. Single crystals suitable for X-ray diffraction were obtained by recrystallization in a chloroform and methanol (4:1 v/v) mixture.
), δ 2.16 (s, 3H, –CH3ring), δ 1.99 (s, 3H, CH2CH3). Single crystals suitable for X-ray diffraction were obtained by recrystallization in a chloroform and methanol (4:1 v/v) mixture.
O 1619, νCS 800, νCO (acetylacetone) 1517. UV-vis (DMSO), λmax, nm (ε, dm3 mol−1 cm−1) 279 (65297) (intra ligand transition), 389 (27028) (MLCT), 649 (19459) (d–d transition). EPR (X-band, solid state): g‖ = 2.664, g⊥ = 2.131, gav = 2.302. Single crystals suitable for X-ray diffraction were obtained by recrystallization in a chloroform and methanol (4:1 v/v) mixture.
O 1603, νCS 802, νCO (acetylacetone) 1519. UV-vis (DMSO), λmax, nm (ε, dm3 mol−1 cm−1) 278 (69330) (intra ligand transition), 390 (25461) (MLCT), 647 (15445) (d–d transition). EPR (X-band, solid state): g‖ = 2.181, g⊥ = 1.996, gav = 2.057. Single crystals suitable for X-ray diffraction were obtained by recrystallization in a chloroform and methanol (4:1 v/v) mixture.
Crystal structure determination
Single crystals of the ligand HL2, the complexes RuL1 and RuL2 and the intermediate A12 were grown as clear, colorless (HL2), green (RuL1 and RuL2) and yellow (A12 intermediate) prisms by slow evaporation from chloroform/methanol medium. The data were collected at −173 °C using a Nonius Kappa CCD diffractometer with a Bruker AXS Apex II detector and a graphite monochromator with MoKα radiation (λ = 0.71073 Å) for HL2 and the A12 intermediate and CuKα radiation (λ = 1.54184 Å) for the complexes RuL1 and RuL2. Reduced temperatures were maintained by use of an Oxford Cryosystems 700 low-temperature device. Data reduction was performed using SAINT V8.27B.55 The structure was solved by direct methods using SHELXT56 and refined by full-matrix least-squares on F2 with anisotropic displacement parameters for t non-H atoms using SHELXL-2016/6.57 Structure analysis was aided by the use of the programs PLATON,58 OLEX2,59 and WinGX.60Procedure for C–H activation of α,β-unsaturated carbonyl compounds
The α,β-unsaturated carbonyl compound (1 mmol), bromobenzene (1 mmol), potassium hydroxide (2 mmol) and the catalyst (0.005 mmol) were dissolved in 10 cm3 of ethanol and refluxed for 3 h. The solution was cooled and worked up with ethyl acetate and water, and the organic layer was collected, passed through anhydrous Na2SO4, filtered and dried. The product was isolated by using column chromatography with petroleum ether (60–80 °C) and ethyl acetate as eluents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author S. D. greatly acknowledges UGC, New Delhi, India for UGC-BSR fellowship (F.25-1/2014-15(BSR)/7-26/2007(BSR) dated 05.11.2015). The authors S. D. and R. P. acknowledge DST-PURSE-Phase II, Bharathiar University for the NMR instrumentation facility.References
- M. L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901 RSC.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879 RSC.
- C. J. Clarke, W. C. Tu, O. Levers, A. Brohl and J. P. Hallett, Chem. Rev., 2018, 118, 747 CrossRef CAS PubMed.
- C. Arroniz, A. Ironmonger, G. Rassias and I. Larrosa, Org. Lett., 2013, 15, 910 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed.
- H. Zhang, H. Y. Wang, Y. Luo, C. Chen, Y. Cao, P. Chen, Y. L. Guo, Y. Lan and G. Liu, ACS Catal., 2018, 8, 2173 CrossRef CAS.
- Y. Li, S. Liu, Z. Qi, X. Qi, X. Li and Y. Lan, Chem. – Eur. J., 2015, 21, 10131 CrossRef CAS PubMed.
- X. Qin, X. Li, Q. Huang, H. Liu, D. Wu, Q. Guo, J. Lan, R. Wang and J. You, Angew. Chem., Int. Ed., 2015, 54, 7167 CrossRef CAS PubMed.
- S. Yu, Y. Li, L. Kong, X. Zhou, G. Tang, Y. Lan and X. Li, ACS Catal., 2016, 6, 7744 CrossRef CAS.
- S. Yu, G. Tang, Y. Li, X. Zhou, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2016, 55, 8696 CrossRef CAS PubMed.
- P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721 CrossRef CAS.
- L. Huang, D. Hackenberger and L. J. Goossen, Angew. Chem., Int. Ed., 2015, 54, 12607 CrossRef CAS PubMed.
- D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715 CrossRef CAS PubMed.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS.
- A. Bris, I. Turel and J. Roithova, Eur. J. Org. Chem., 2018, 2018, 6107 CrossRef CAS.
- G. Duarah, P. P. Kaishap, T. Begum and S. Gogoi, Adv. Synth. Catal., 2019, 361, 654 CrossRef CAS.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Org. Lett., 2001, 3, 2579 CrossRef CAS PubMed.
- Y. Zhou, H. Liang, Y. Sheng, S. Wang, Y. Gao, L. Zhan, Z. Zheng, M. Yang, G. Liang, J. Zhou, J. Deng and Z. Song, J. Org. Chem., 2020, 85, 9230 CrossRef CAS PubMed.
- G. C. E. Raja, J. Y. Ryu, J. Lee and S. Lee, Org. Lett., 2017, 19, 6606 CrossRef CAS PubMed.
- N. Sharma, V. Bahadur, U. K. Sharma, D. Saha, Z. Li, Y. Kumar, J. Colaers, B. K. Singh and E. V. Van der Eycken, Adv. Synth. Catal., 2018, 360, 3083 CrossRef CAS.
- Y. C. Yuan, C. Bruneau, T. Roisnel and R. G. Doria, Org. Biomol. Chem., 2019, 17, 7517 RSC.
- X. Q. Hu, Y. X. Hou, Z. K. Liu and Y. Gao, Org. Chem. Front., 2021, 8, 915 RSC.
- N. Luo and Z. K. Yu, Chem. – Eur. J., 2010, 16, 787 CrossRef CAS.
- C. S. Yi, S. Y. Yun and I. A. Guzei, J. Am. Chem. Soc., 2005, 127, 5782 CrossRef CAS PubMed.
- C. S. Yi and S. Y. Yun, J. Am. Chem. Soc., 2005, 127, 17000 CrossRef CAS PubMed.
- T. Kochi, S. Urano, H. Seki, E. Mizushima, M. Sato and F. Kakiuchi, J. Am. Chem. Soc., 2009, 131, 2792 CrossRef CAS PubMed.
- T. Kochi, A. Tazawa, K. Honda and F. Kakiuchi, Chem. Lett., 2011, 40, 1018 CrossRef CAS.
- S. Oi, H. Sato, S. Sugawara and Y. Inoue, Org. Lett., 2008, 10, 1823 CrossRef CAS PubMed.
- S. Oi, Y. Tanaka and Y. Inoue, Organometallics, 2006, 25, 4773 CrossRef CAS.
- L. Ackermann, Org. Lett., 2005, 7, 3123 CrossRef CAS PubMed.
- P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2010, 49, 6629 CrossRef CAS PubMed.
- C. Shan, L. Zhu, L. B. Qu, R. Bai and Y. Lan, Chem. Soc. Rev., 2018, 47, 7552 RSC.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- L. Ping, D. S. Chung, J. Bouffard and S. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC.
- Y. Ogiwara, M. Miyake, T. Kochi and F. Kakiuchi, Organometallics, 2017, 36, 159–164 CrossRef CAS.
- D. J. Paymode and C. V. Ramana, J. Org. Chem., 2015, 80, 11551–11558 CrossRef CAS PubMed.
- K. R. Bettadapur, V. Lanke and K. R. Prabhu, Org. Lett., 2015, 17, 4408–4411 CrossRef CAS PubMed.
- S. Pan, H. Jiang, Y. Zhang, D. Chen and Z. Zhang, Synlett, 2016, 1997–2002 CAS.
- Y. Kommagalla, K. Srinivas and C. V. Ramana, Chem. – Eur. J., 2014, 20, 7884–7889 CrossRef CAS.
- K. Srinivas, Y. Dangat, Y. Kommagalla, K. Vanka and C. V. Ramana, Chem. – Eur. J., 2017, 23, 7570–7581 CrossRef CAS PubMed.
- Y. Na, S. Park, S. B. Han, H. Han, S. Ko and S. Chang, J. Am. Chem. Soc., 2004, 126, 250–258 CrossRef CAS.
- J. M. Munoz-Molina and P. J. Perez, J. Org. Chem., 2019, 84, 8289–8296 CrossRef CAS.
- P. Chauhan, S. Mahajan and D. Enders, Chem. Commun., 2015, 51, 12890–12907 RSC.
- F. Marchetti, C. Pettinari, C. D. Nicola, A. Tombesi and R. Pettinari, Coord. Chem. Rev., 2019, 401, 213069 CrossRef CAS.
- R. Prabhakaran and S. Dharani, Indian Pat., 202241023805, 2022 Search PubMed.
- G. Kalaiarasi, S. Rex Jeya Rajkumar, S. Dharani, J. G. Malecki and R. Prabhakaran, RSC Adv., 2018, 8, 1539 RSC.
- R. Karvembu and K. Natarajan, Polyhedron, 2002, 21, 219 CrossRef CAS.
- P. Kalaivani, R. Prabhakaran, M. V. Kaveri, R. Huang, R. J. Staples and K. Natarajan, Inorg. Chim. Acta, 2013, 405, 415 CrossRef CAS.
- D. A. Hey, M. J. Sauer, P. J. Fischer, E. M. H. J. Esslinger, F. E. Kuhn and W. Baratta, ChemCatChem, 2020, 12, 3537 CrossRef CAS.
- S. Chatterjee, S. Mandal, S. Samanta and S. Goswami, Dalton Trans., 2012, 41, 7057 RSC.
- D. Das, B. Sarkar, D. Kumbhakar, T. K. Mondal, S. M. Mobin, J. Fiedler, F. A. Urbanos, R. Jimenez-Aparicio, W. Kaim and G. K. Lahiri, Chem. – Eur. J., 2011, 17, 11030 CrossRef CAS.
- S. Nandhini, S. Dharani, C. Elamathi, F. Dallemer and R. Prabhakaran, Appl. Organomet. Chem., 2021, 35, e6436 CrossRef CAS.
- Bruker, SAINT V8.34A, Bruker AXS Inc, Madison, WI, 2013 Search PubMed.
- G. M. Sheldrick and SHELXT, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 91, 9 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00317a |
| This journal is © The Royal Society of Chemistry 2023 |