
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a flow process for an easy and fast access to 2-pyrone derivatives†
Grazia Isa C.
Righetti
,
Francesca
Tentori
,
Elisabetta
Brenna
 and
Cristian
Gambarotti
*
and
Cristian
Gambarotti
*
Department of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milano, Italy. E-mail: cristian.gambarotti@polimi.it
First published on 13th October 2022
Abstract
2-Pyrones are compounds widely present in nature and they represent interesting building blocks both in medicinal and synthetic chemistry. Due to their peculiar pharmacological activity and structure, they have attracted much attention during the last decades and several protocols for their synthesis have been developed. In this work we propose the synthesis of bio-sourced 2-pyrones, exploiting continuous-flow conditions for an easy, sustainable and fast access to these important molecules.
Introduction
Nowadays, in the pressing need to find more sustainable chemical processes to reduce our carbon footprints, valorisation of biomass has gained much attention, not only as a renewable feedstock for the production of energy and biofuels, but also as a starting material for the production of high value-added bio-based chemicals that, currently, are mainly derived from petrochemical routes.1,2 For this reason, the use of biomass for the synthesis of valuable bio-sourced molecules has a central role in the achievement of a circular and green economy.3Among all, carbohydrates represent one of the major fractions of biomass and for this reason they are good candidates as sustainable raw materials to be applied in organic synthesis.4
Pyrones represent a family of six-membered cyclic unsaturated molecules containing an oxygen atom and a carbonyl group. The 2-pyrone motif is widely present in many naturally occurring products derived from plants, animals, marine organisms, bacteria and insects, where they are often involved in defence mechanisms.5 It has already been proven that these compounds exhibit some peculiar pharmacological behaviour, and besides it has been observed that even a small change in the substituents on the pyrone ring often leads to a different biological effect. Molecules containing 2-pyrones have also shown potential activity against tuberculosis6–8 and HIV.9–11
Their peculiar chemical structure, which combines a diene with an aromatic-like ring,12 makes 2-pyrone motifs interesting also from the synthetic chemistry point of view13,14 especially those to be employed in Diels–Alder reactions.15
In this work, we propose a two-step synthesis of 3-hydroxy-2H-pyran-2-one starting from galactaric acid under continuous-flow conditions. Nowadays flow chemistry is widely considered a powerful tool for the development of more efficient and sustainable processes for the preparation of high-value added products.16–19 In particular, the reduced dimensions of continuous-flow reactors cause a significant enhancement of mass and heat transfer phenomena with an improvement in terms of yields20–22 and selectivity23–26 as in the case of the synthetic process of 2-pyrones herein described.
Results and discussion
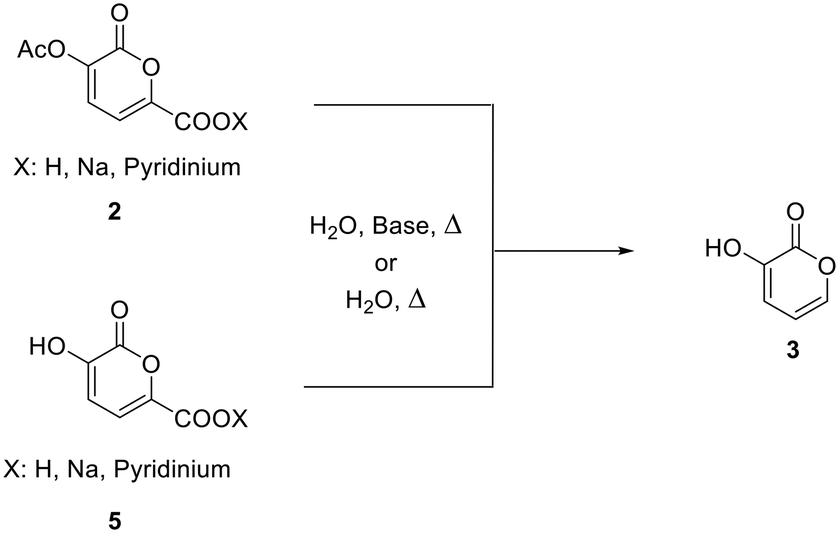
In a recently published paper27 it was demonstrated how galactaric acid (or mucic acid, 1, Scheme 1) can be efficiently employed as a renewable starting material for the synthesis of 2-pyrone derivatives. In particular, we focused our attention on 3-hydroxy-2-pyrone (3) which is a very interesting and high-priced building block for the synthesis of poly-functionalized aromatic products. Therefore, we envisaged the possibility of developing a new protocol for a more efficient synthesis, with a further improvement in terms of yields by means of a continuous flow reactor.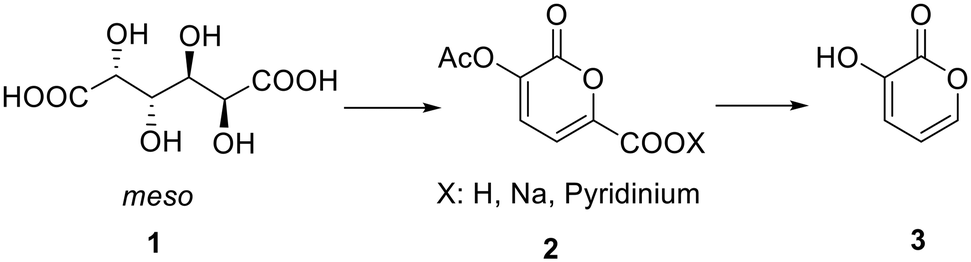 | ||
| Scheme 1 General reaction pathway from galactaric acid (1) to 3-hydroxy-2-pyrone (3). | ||
Based on the previous findings, the development of this new process was divided into 3 steps: (a) modification of the reaction conditions for the synthesis of 2-pyrone 2 in batch, (b) development of a new decarboxylation protocol, and (c) the transfer of the processes from a batch to a continuous flow reactor.
Synthesis of pyrone 2 from galactaric acid 1 – batch conditions
The first step is the conversion of galactaric acid to pyrone 2 (Scheme 2). As already known, this can be obtained in one step by reacting galactaric acid in acetic anhydride in the presence of a base. As is known, galactaric acid is highly insoluble in most solvents and in order to be carried out in a continuous flow reactor, the reaction system needs to be homogeneous. For this reason, we studied the dissolution of acid 1 in a suitable solvent, e.g. DMSO, prior to the treatment with the Ac2O/base. When heated in DMSO, acid 1 affords the corresponding galactaro lactone which is completely soluble in this solvent and more reactive than its precursor in the following cyclization.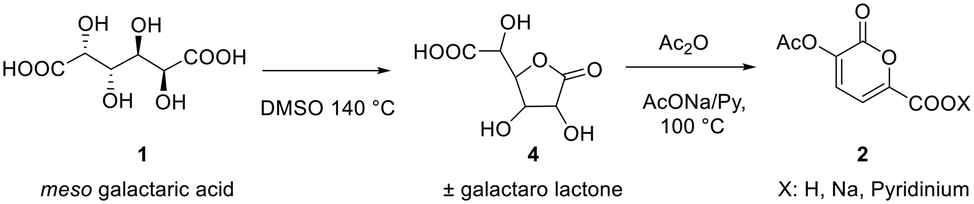 | ||
| Scheme 2 Steps for the synthesis of 2-pyrone in flow. | ||
The conditions for the conversion of galactaric acid into the corresponding lactone and then into pyrone 2 were carefully investigated under batch conditions. The first experiment with pyridine afforded a yield of 68% after 3 h at 100 °C in the presence of 1.2 equivalents of the base and 6 mL of acetic anhydride (entry 1, Table 1). With the same quantity of the base and at the same temperature, a more diluted system (entry 2) seemed to slightly favour the reaction outcome. The influence of DMSO on the reaction was also tested using the minimum quantity of solvent needed to dissolve mucic acid (0.75 mL for 500 mg, entry 3); in this case a yield of 77% in the final product was obtained. Given that acetic acid is the co-product of the reaction, sodium acetate was also tested as a base, obtaining an 85% yield for the desired product (entry 4). It is noteworthy how the use of pyridine allowed obtaining a homogeneous solution for our protocol, while the same was not true for sodium acetate.
In the presence of both bases, the final product crystallized in the reaction flask after solvent removal.
Synthesis of pyrone 2 from galactaric acid (1) – continuous flow conditions
To implement the reaction under continuous-flow conditions the preparation of lactone 4 was needed to obtain a homogeneous system. Once the lactone was obtained, this was cooled to room temperature and sodium acetate was added to the solution which was then injected into a 10 mL PFA tubular reactor kept at 100 °C. The conversion was evaluated at different flow rate values. 3-Acetoxy-2-oxo-2H-pyran-6-carboxylic pyrone 2 was obtained in 52% yield with a flow rate of 650 μL min−1 (corresponding to a residence time of 15 min), while a 76% yield was obtained (Table 2, entries 1–3) with a flow rate of 150 μL min−1 (residence time of 67 min). As a comparison, 3 hours were required to reach the same yield in the batch reaction (Table 1).To compare batch and continuous-flow reactions, a definition for the yield, which is generally adopted, takes into account the amount of product formed per unit of time and volume, and it is called the “space–time yield” (STY). For this reaction we compared entry 4 (Table 1) for the batch configuration and entry 3 (Table 2) for the flow configuration and STY values were calculated as follows:
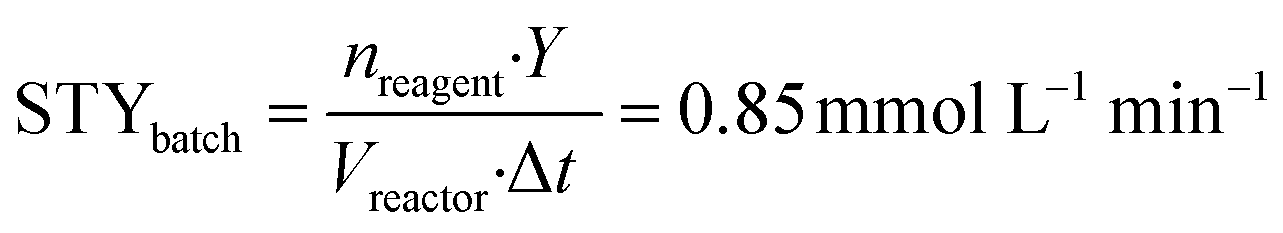
nreagent = galactaric acid (1) amount (mmol); Y = analytical yield of pyrone 2 by 1H NMR; Vreactor = reactor volume (mL); Δt = reaction time (for batch); τ = residence time (for flow).
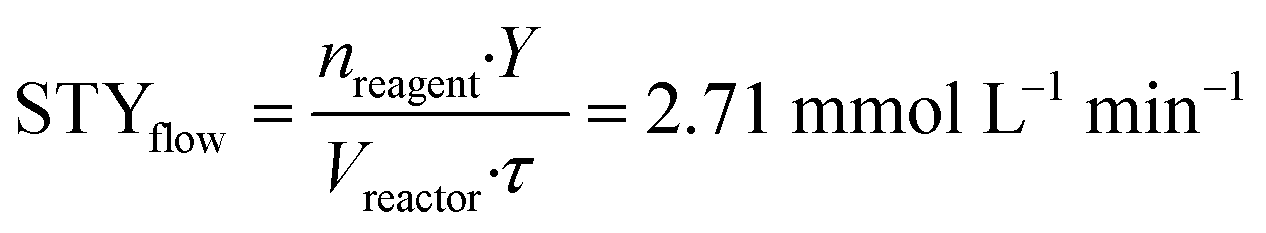
Observing the results, it can be stated that the synthesis of pyrone 2 performed in a continuous-flow reactor determined an approximately three-fold higher STY with respect to the batch process. This improvement can be ascribed to the shorter residence time achieved with the continuous-flow configuration, caused by the intrinsically high transport properties of small-scaled reactors.
Decarboxylation of pyrone 2 to 3 – batch conditions
Very recently, a simple protocol for the decarboxylation of 3-hydroxy-2-oxo-2H-pyran-6-carboxylic acid to obtain 3-hydroxy-2-pyrone 3 has been published.28 According to this article, the decarboxylation reaction proceeds efficiently when the pyrone is refluxed in acid aqueous media. Based on this finding, the possibility of performing the decarboxylation reaction directly on pyrones 2 and 5 by transferring the protocol to a continuous flow system has been investigated. Since pyrones 2 and 5 are easily obtained as sodium or pyridinium salts, the protocol has been investigated in the presence of bases. Firstly, the decarboxylation reaction was performed on substrate 2 in the presence of a base (Table 3). Experiments performed in the presence of sodium hydroxide at 80–100 °C showed substantial decomposition of the substrate, while only traces of 3-hydroxy-2-pyrone were found after 24 h at 50 °C (Scheme 3).| Entry | [Subs.] (M) | Base, eq. | T (°C) | t (h) | Product percentagea (%) |
|---|---|---|---|---|---|
| a Analytical measurement. b Degradation. | |||||
| 1 | 2, 1 | TEA, 1 | 50 | 24 | Tr. |
| 2 | 2, 0.2 | AcONa, 0.2 | 100 | 24 | Degr. |
| 3 | 2, 0.2 | AcONa, 0.2 | 50 | 24 | Traces |
| 4 | 2, 0.2 | AcONa, 0.2 | 80 | 24 | 80b |
| 5 | 2, 1 (pyridinium) | AcONa, 0.2 | 80 | 6 | 77 |
| 6 | 2, 1 (sodium salt) | AcONa, 0.2 | 80 | 6 | 67 |
| 7 | 2, 1.5 | AcONa, 0.1 | 80 | 3 | 68 |
| 8 | 2, 1.5 | AcONa, 0.1 | 90 | 3 | 74 |
| 9 | 5, 0.2 | AcONa, 1 | 100 | 24 | 50b |
| 10 | 5, 0.2 | AcONa, 1 | 80 | 24 | 98 |
| 11 | 5, 1 | TEA, 1 | 50 | 24 | Tr. |
 | ||
| Scheme 3 Decarboxylation of 2 or 5 to obtain 3. | ||
An attempt of decarboxylation was also performed in the presence of TEA at 50 °C and only traces of the product were found (entry 1). Given that acetic acid is also the co-product of this second step, decarboxylation was also tried in the presence of sodium acetate as a base. When the substrate is heated at 100 °C for 24 hours, degradation of the pyrone ring also occurs (entry 2). Only traces of the decarboxylation product were present when the same reaction was performed at 50 °C (entry 3), while 80% of the reaction product along with degradation was obtained when the reaction was performed at 80 °C (entry 4).
In light of these results, the reaction was performed under milder conditions and for a shorter time to preserve the 2-pyrone ring. The reaction was carried out at 80 °C for 6 hours, obtaining a 77% yield starting from the pyridinium salt (entry 5) and a 67% yield starting from the sodium salt (entry 6). These results highlighted how the nature of the counterion of the carboxylic group does not influence the outcome of the reaction; for this reason, the decarboxylation protocols were carried out using the sodium salt since it is highly soluble in water with respect to the pyridinium salt.
A more concentrated system with a catalytic amount of base (0.1 eq.) led to a 68% yield in 3 hours at 80 °C, while at 90 °C the yield increased to 74% (entries 7 and 8).
Since decarboxylation of pyrone 2 usually occurred together with some degradation, the same reaction was performed on the deprotected pyrone 5. It was confirmed that heating of the substrate for a prolonged time in the presence of a base causes the degradation of the pyrone ring both at 100 °C and 80 °C. On the other hand, when sodium acetate was used in an equimolar amount and the system was heated for 24 h at 80 °C, an analytical yield of 98% of 3-hydroxy-2-pyrone was obtained (entry 10).
Given that the 2-pyrone ring gave rise to degradation when heated in the presence of a base, it was decided to test the reactivity of the latter toward decarboxylation in water.
Surprisingly, when pyrone 5 was heated in water at 100 °C for 24 h, a 41% yield of the product was obtained (entry 1, Table 4). In this case, the 1H-NMR spectrum of the reaction mixture did not show any signal due to the degradation derivatives of the pyrone moiety. In light of this result, 2 was also subjected to the same conditions.
It was found that the decarboxylation in water is faster for 2 than for 5, giving an analytical yield of 97% in 24 h (entry 2).
The role of temperature in the process was also tested (entries 2–4). The overall yield increased with an increase in temperature. In fact, when the reaction was performed at 130 °C, the analytical yield was 63% in 6 h (entry 3). It is noteworthy that the substrate could not be heated for a prolonged time at 130 °C as degradation of the 2-pyrone ring took place. To obtain a similar yield at a lower temperature and in order to prevent substrate degradation, 24 h of heating was necessary (entry 4).
A comparison between the experiments performed at different concentration values (entries 4–7) clearly indicates that this latter variable does not play a key role in the overall final yield. In fact, in any case high and very similar analytical yields (92–99%) were obtained when the substrate was heated at 100 °C for 24 h.
The final product could be easily isolated from the reaction mixture by extraction with ethyl acetate.
Decarboxylation of pyrone 2 to 3 – continuous flow
As the decarboxylation in water gave high yields with substrate 2, it was decided to use it as the reactant, avoiding therefore the isolation of 5 for the flow step.The concentration of the substrate (0.5 M or 0.2 M) was dictated by the solubility of the substrate in water (Table 5). In fact, a more concentrated solution did not start from a homogeneous reaction mixture.
Since temperature was found to be a critical parameter, we tested the continuous-flow conditions at 120 °C and 130 °C.
In the first experiment, continuous-flow decarboxylation of pyrone 2 was conducted at 130 °C, with a flow rate of 100 μL min−1 (corresponding to a contact time of 100 min, entry 1) in a 10 mL coil reactor kept at 130 °C and at 4 bar by means of a BPR. The reaction resulted in complete 2 conversion but with a rather low product yield (29%) by 1H-NMR analysis. Actually, this unsatisfactory result could be ascribed to the much more efficient heat transfer of the flow system that caused rapid degradation of the starting compound. In fact, by decreasing the temperature to 120 °C (while keeping the same residence time and pressure) the yield increased to 71%.
In this case also, a comparison between the batch and flow-modes can be achieved exploiting the definition of the space–time yield. Comparing entry 7 (Table 4) for batch and entry 2 (Table 5) for flow, we obtained the following STY values:
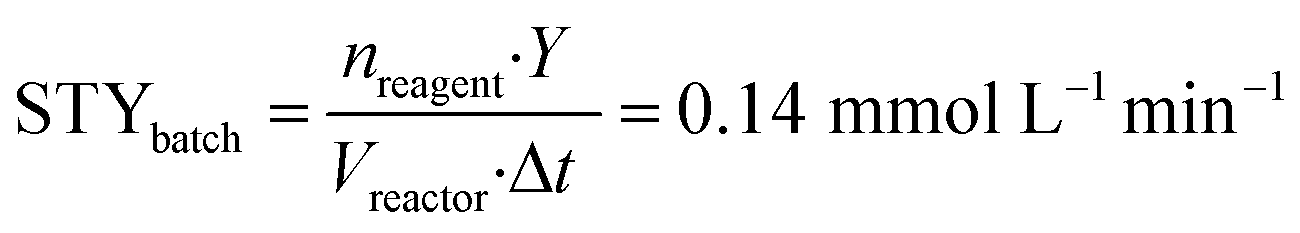
nreagent = pyrone 2 amount (mmol); Y = analytical yield of pyrone 3 by 1HNMR; Vreactor = reactor volume (mL); Δt = reaction time (for batch); τ = residence time (for flow).
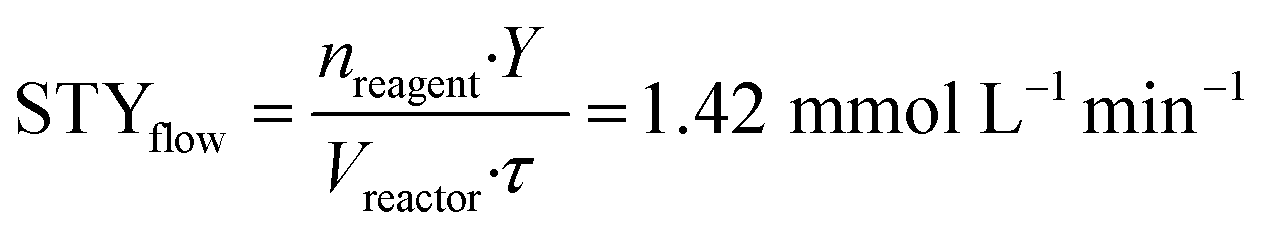
In this second reaction, as for the previous one, performances of flow operation are higher than the batch one: decarboxylation under continuous-flow conditions shows a ten-fold higher STY than the batch configuration, for the same reasons reported before. Therefore, the increased performances in flow mode for both steps demonstrate that the flow configuration can significantly contribute to a rapid, efficient synthesis of 2-pyrone derivatives.
The complete flow diagram of the synthesis of pyrone 3 starting from galactaric acid (1) is reported in Fig. 1.
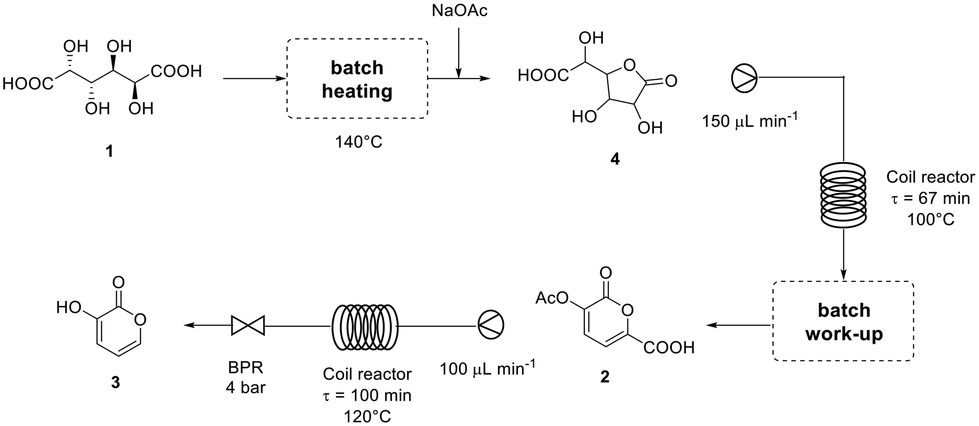 | ||
| Fig. 1 Flow diagram of the synthesis of 3-hydroxy-2-pyrone (3) from galactaric acid (1) developed in this work. | ||
Experimental
All reagents and solvents were purchased from Merck (Merck Life Science s.p.a., Milan, Italy) and used without any further purification. The 1H-NMR spectra were recorded on a Bruker AV 400 MHz instrument, equipped with a 5 mm multinuclear probe, and 32 scans were acquired with an acquiring time of 3 seconds for each spectrum. The 1H-NMR analysis was performed in the presence of an external standard: 1,4-dinitro benzene for DMSO-d6 and trioxane for D2O. The continuous flow reactions were performed using an E-Series Integrated Flow Chemistry system from Vapourtec (Alfatech s.p.a., Genoa, Italy) equipped with a 10 mL PFA coiled tubular reactor (1.0 mm i.d. × 12.75 m length). The coil reactor was kept under controlled pressure using back pressure regulators (BPRs) from IDEX Health & Science L.L.C. (Microcolumn s.r.l., Desio, Italy).Synthesis of pyrone 2 (as sodium salt) from galactaric acid with sodium acetate – batch conditions
Galactaric acid (1) (500 mg. 2.38 mmol) is placed in a 2 neck round bottom flask equipped with a thermometer and a magnetic stirrer. DMSO (1 mL) is added, and the system is heated to 140 °C (internal temperature). This is left under heating until complete dissolution of mucic acid and formation of a light brown solution. The solution is brought from 140 °C to 100 °C. 197.0 mg sodium acetate (2.38 mmol, 1 eq.) is added together with acetic anhydride (12 mL). The system is left under stirring for 3 hours, and then the solution is brought back to room temperature. The solvent is evaporated under reduced pressure as much as possible and the reaction flask is kept in a fridge overnight. Pyrone 2 sodium salt crystallizes in the flask as a colourless crystal (88% isolated yield) together with 2 molecules of acetic acid. 1H-NMR (400 MHz, DMSO-d6) δ = 7.45 (d, 1H, J = 7.2 Hz), 6.95 (d, 1H, J = 7.2 Hz), 2.27 (s, 3H), 1.91 (s, 6H) ppm; 13C-NMR (100.6 MHz, DMSO-d6) δ = 174.0, 169.1, 162.6, 158.4, 156.4, 137.9, 133.5, 106.6, 22.9, 21.2 ppm.Synthesis of pyrone 2 (as pyridinium salt) from galactaric acid with pyridine – batch conditions
Galactaric acid (1) (500 mg. 2.38 mmol) is placed in a 2 neck round bottom flask equipped with a thermometer and a magnetic stirrer. DMSO (1 mL) is added, and the system is heated to 140 °C (internal temperature). This is left under heating until complete dissolution of mucic acid and formation of a light brown solution. The solution is brought from 140 °C to 100 °C. Pyridine (230 μL) and acetic anhydride (12 mL) are added to the reaction flask. The system is left under stirring for 3 hours, and then the solution is brought back to room temperature. The solvent is evaporated under reduced pressure as much as possible and the reaction flask is kept in a fridge overnight. Pyrone 2 (pyridinium salt) crystallizes in the flask as a colourless crystal (64% isolated yield) together with 2 molecules of acetic acid. 1H-NMR (400 MHz, DMSO-d6) δ = 7.40 (d, 1H, J = 7.2 Hz), 6.80 (d, 1H, J = 7.2 Hz), 2.26 (s, 3H), 1.90 (s, 6H) ppm.General procedure for the decarboxylation of pyrone 2 (sodium salt) to 3 in water – batch conditions
Pyrone 2 (sodium salt) is placed in a round bottom reaction flask and dissolved in 10 mL of distilled water (see Table 4 for the concentrations). The light yellow solution is then heated to 100 °C for 24 hours. After this time, a sample is withdrawn to calculate the analytical yield of the final product. 3-Hydroxy-2-pyrone is then extracted with ethyl acetate (4 × 25 mL). The solvent is removed under reduced pressure and the final product is obtained as a light brown oil together with some colourless crystals. The batch is then further purified at the vacuum pump to obtain 3 as a white solid (76% isolated yield). 1H-NMR (400 MHz, D2O) δ = 7.29 (dd, 1H, J = 5.1 Hz, J = 1.5 Hz), 6.78 (dd, 1H, J = 7.2 Hz, J = 1.5 Hz), 6.34 (dd, 1H, J = 7.2 Hz, J = 5.1 Hz) ppm; 1H-NMR (400 MHz, CDCl3) δ = 7.14 (dd, 1H, J = 5.1 Hz, J = 1.6 Hz), 6.66 (dd, 1H, J = 7.1 Hz, J = 1.6 Hz), 6.20 (dd, 1H, J = 7.1 Hz, J = 5.1 Hz) ppm; 13H-NMR (100.6 MHz, CDCl3) δ = 162.0, 142.8, 142.5, 114.8, 107.5 ppm.Synthesis of pyrone 2 (sodium salt) from galactaric acid (1) – continuous flow conditions
Galactaric acid (1) (500 mg, 2.38 mmol) is placed in a 2 neck round bottom flask then DMSO (1 mL) is added. The system is heated to 140 °C and left under stirring until complete dissolution of 1. This is cooled to room temperature. Sodium acetate (1.2 eq.) is dissolved in 200 μL of water and added to the lactone solution. The system is kept at 50 °C under stirring and pumped, at a flow rate of 150 μL min−1, in a 10 mL PFA tubular coil kept at 100 °C (residence time of 67 minutes). The mixture collected at the outlet was analysed by 1H-NMR in DMSO-d6.Decarboxylation of pyrone 2 (sodium salt) to 3 – continuous flow conditions
10 mL of a 0.2 M solution of pyrone 2 was prepared. The solution was pumped at a flow rate of 100 μL min−1 to a 10 mL PFA heated tubular coil kept at 120 °C corresponding to a residence time of 100 min. The coil reactor was kept at a constant pressure of 4 bar using a back pressure regulator. The mixture collected at the outlet was analysed by 1H-NMR in D2O.Conclusions
In order to allow easy and fast access to bio-sourced 2-pyrone derivatives, the synthesis protocols for these molecules were optimized in order to be transferred to a continuous flow system. A new, green and easy decarboxylation protocol to obtain the expensive 3-hydroxy-2-pyrone was also developed. All the protocols were successfully transferred to a continuous flow system. This latter allowed us to obtain 2-pyrones in high yield and in a much shorter time with respect to a batch reaction (66 minutes vs. 3 hours for 3-acetoxy-2-oxo-2H-pyran-6-carboxylic acid and 33 minutes vs. 6/24 hours for 3-hydroxy-2-pyrone).Author contributions
All the authors contributed equally in the research activity.Conflicts of interest
“There are no conflicts to declare”.Notes and references
- R. Sakuta and N. Nakamura, Int. J. Mol. Sci., 2019, 20, 1 CrossRef PubMed.
- S. Rautiainen, P. Lehtinen, J. Chen, M. Vehkamäki, K. Niemelä, M. Leskelä and T. Repo, RSC Adv., 2015, 5, 19502 RSC.
- P. Gallezot, Green Chem., 2007, 9, 295 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- G. P. Mcglacken and I. J. S. Fairlamb, Nat. Prod. Rep., 2005, 22, 369 RSC.
- G. Appendino, E. Mercalli, N. Fuzzati, L. Arnoldi, M. Stavri, S. Gibbons, M. Ballero and A. Maxia, J. Nat. Prod., 2004, 67, 2108 CrossRef CAS.
- K. Mitra, A. Chadha and M. Doble, Eur. J. Pharm. Sci., 2019, 135, 103 CrossRef CAS PubMed.
- R. Mata, I. Morales, O. Pérez, I. Rivero-Cruz, L. Acevedo, I. Enriquez-Mendoza, R. Bye, S. Franzblau and B. Timmermann, J. Nat. Prod., 2004, 67, 1961 CrossRef CAS PubMed.
- M. He, N. Yang, C. Sun, X. Yao and M. Yang, Med. Chem. Res., 2011, 20, 200 CrossRef CAS.
- Y. S. Lee, S. N. Kima, Y. S. Lee, J. Y. Lee, C. Y. Lee, H. S. Kim and H. Park, Arch. Pharm. Pharm. Med. Chem., 2000, 333, 319 CrossRef CAS.
- S. Hagen, J. V. N. Vara Prasad and B. D. Tait, Adv. Med. Chem., 2000, 5, 159 CAS.
- C. W. Bird, Tetrahedron, 1986, 42, 89 CrossRef CAS.
- J. J. Lee and G. A. Kraus, Green Chem., 2014, 16, 2111 RSC.
- G. A. Kraus, G. R. Pollock, C. L. Beck, K. Palmer and A. H. Winter, RSC Adv., 2013, 3, 12721 RSC.
- C. Gambarotti, M. Lauria, G. I. C. Righetti, G. Leonardi, R. Sebastiano, A. Citterio and A. Truscello, ACS Sustainable Chem. Eng., 2020, 8, 11152 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796 CrossRef CAS PubMed.
- K. F. Jense, AIChE J., 2017, 63, 858 CrossRef.
- M. Baumann and I. R. Baxendale, J. Org. Chem., 2015, 1, 1194 Search PubMed.
- I. R. Baxendale, L. Brocken and C. J. Mallia, Green Process. Synth., 2013, 2, 211 CAS.
- V. Hessel, Chem. Eng. Technol., 2009, 32, 1655 CrossRef CAS.
- V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746 CrossRef CAS PubMed.
- K. G. Hugentobler, M. Rasparini, L. A. Thompson, K. E. Jolley, A. J. Blacker and N. J. Turner, Org. Process Res. Dev., 2017, 21, 195 CrossRef CAS.
- J. M. Muñoz, J. Alcázar, A. de la Hoz and A. Díaz-Ortiz, Eur. J. Org. Chem., 2012, 2, 260 CrossRef.
- J.-I. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896 RSC.
- D. Cantillo and C. O. Kappe, React. Chem. Eng., 2017, 2, 7 RSC.
- P. Žnidaršič-Plazl, Curr. Opin. Green Sustainable Chem., 2021, 31, 100 Search PubMed.
- G. Leonardi, J. Li, G. I. C. Righetti, A. Truscello, C. Gambarotti, G. Terraneo, A. Citterio and R. Sebastiano, Eur. J. Org. Chem., 2020, 2, 241 CrossRef.
- G. Leonardi, A. Truscello, G. G. Mondrone and R. Sebastiano, Results Chem., 2022, 4, 100280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00312k |
| This journal is © The Royal Society of Chemistry 2023 |