
Development of a high surface area Cu electrocatalyst for effective nitrous oxide reduction reaction†
Siraphat
Nilvichean
a,
Kornkamon
Meesombad
 b,
Teera
Butburee
b,
Pongkarn
Chakthranont
*b and
Rungthiwa
Methaapanon
*a
b,
Teera
Butburee
b,
Pongkarn
Chakthranont
*b and
Rungthiwa
Methaapanon
*a
aCenter of Excellence in Particle and Material Processing Technology, Department of Chemical Engineering, Chulalongkorn University, Bangkok, 10330 Thailand. E-mail: rungthiwa.m@chula.ac.th
bNational Nanotechnology Center (NANOTEC), National Science and Technology Development Agency (NSTDA), Pathum Thani, 12120 Thailand. E-mail: pongkarn.cha@nanotec.or.th
First published on 22nd September 2022
Abstract
Electrochemical reduction of nitrous oxide (N2ORR) into benign nitrogen (N2) under mild reaction conditions is one approach for reducing emissions of this long-lived anthropogenic greenhouse gas and their environmental impact. In this work, high surface area Cu was investigated as a non-noble N2ORR catalyst alternative to conventional noble metals such as Pd, Pt, and Ir. Nanostructured Cu2O was electrodeposited onto a Cu substrate and electrochemically reduced to metallic Cu prior to the reaction. When compared to a flat Cu electrode, the high surface area Cu results in a 130 mV improved onset potential and 3.5 times higher N2 partial current density. The specific activity of the high surface area Cu indicates that the improved activity is due to the increased electrochemically active surface area. However, the mass transport of reactants becomes limiting at a very large surface area. Peak activity is observed on a Cu electrode with a surface area that is ∼30 times the geometric surface area. The stability test shows that the electrode can maintain its high surface area and catalytic activity for at least 7 hours, demonstrating that high surface area Cu is a viable alternative to noble metal N2ORR catalysts, outperforming other non-noble catalysts previously reported.
1. Introduction
Although nitrous oxide (N2O) is less well-known as a greenhouse gas (GHG), it has a 310-fold greater global warming potential than carbon dioxide (CO2) due to its long lifetime of 116 ± 9 years.1,2 Moreover, approximately 10% of N2O in the stratosphere can be photolyzed to nitrogen oxides (NOx), which can rapidly degrade ozone molecules,3 making it the most significant ozone depleting substance emitted today.4 N2O is a part of the nitrogen cycle and is emitted into the atmosphere by both natural (oceans, soils, lightning, etc.), and anthropogenic sources such as agriculture, transportation, industry, and waste management sectors.5 In addition, N2O is a common anesthetic in surgical operations, where the majority of the gas is not metabolized by patients. Due to the lack of regulation and cost-effective solutions, highly concentrated and localized N2O medical gas waste is routinely released into the atmosphere without scrubbing, accounting for roughly 1% of anthropogenic N2O emissions.6 The atmospheric concentration of N2O has continuously increased at a rate of 2% per decade since the pre-industrial era, reaching 335 ppb in 2022.5,7–9 It is therefore critical to develop measures to mitigate the impact of N2O by reducing emissions from all possible sources.A potential approach for N2O abatement is by thermocatalytically or electrocatalytically converting the gas, either directly from the emitting sources or the air, into benign nitrogen. N2O decomposition via thermochemical reaction at high temperature is a mature process and several N2O abatement units are commercially available for medical gas waste treatment, especially in Japan and Europe. For example, the Anesclean® unit is currently used in Japanese hospitals,10 and the Medclair® destruction unit11 from the Swedish Medtech organization and the EXCIDIO® catalytic destruction facility12 from Linde Healthcare are employed in Scandinavia. These thermochemical systems are effective in removing 99% of N2O from the gas feed; nevertheless, they require high temperature ranging from 300–500 °C and often uses expensive precious metal catalysts such as Rh,13 resulting in high energy consumption and high costs.14 Furthermore, the high temperature could also lead to toxic by-products such as NO and NO2.15 As a result, more cost-effective N2O abatement processes are required for the technology to be extensively implemented around the world.
N2O decomposition via an electrochemical route has the potential to be less energy intensive than the thermal process since it can be operated even at ambient temperature. Additionally, by coupling the reaction with renewable electricity, the processes can be carbon negative. It has been demonstrated that with the right electrocatalysts, an active and selective N2O reduction reaction (N2ORR) can be achieved. In most of the previous works, noble metal catalysts such as platinum (Pt),16 palladium (Pd),17,18 and iridium (Ir)19 have been utilized as N2ORR catalysts. Among them, Pd offers the highest N2ORR activity and a high Faradaic efficiency toward N2 formation.20 Various Pd-based catalysts have been investigated for N2ORR such as bimetallic catalysts15 and core–shell structures.21 While these Pd-based catalysts have the earliest onset potential of any catalyst now available,15,21,22 their exorbitant cost makes large-scale production of this technology challenging.
Based on the catalyst screening study by Kudo et al.,20 among non-noble metals, copper (Cu) is the most attractive alternative as it offers high activity and high selectivity at a relatively low overpotential, on par with noble metals like gold and silver and only slightly inferior to Pd. Additionally, Cu is low-cost, abundantly available, and non-toxic. The overall N2ORR reaction and the reaction steps involved on the Cu surface can be written as follows:
| N2O → N2O* N2O adsorption step |
| N2O* → N2 + O* N2O dissociation step |
| O* + H2O + 2e− → 2OH− O* reduction step |
Theoretical calculation suggested that the N2O dissociation energy barrier on the Cu surface is relatively low.23 However, previous experimental studies utilizing Cu show that there is still significant room for improvement when compared to the performance of noble metal alternatives. As the electrochemical reactions occur exclusively on the surface of the electrode, increasing the electrochemically active surface area (ECSA) of the catalyst can effectively increase the overall activity. Although several prior studies sought to boost the electrochemical activity of other N2ORR catalysts in this manner, the specific activities of the high surface area catalysts were typically not investigated, making it difficult to understand the origin of enhanced activity. In addition, comparing N2ORR performances between studies is challenging since there was no standardized procedure, i.e., different cell designs and electrolyte compositions were employed. Most importantly, the Faradaic efficiency that indicates the selectivity of the process toward N2 formation was rarely reported (see Table S1† for a comparison of reported N2ORR activities).

In this study, a high surface area Cu electrocatalyst was fabricated via electrodeposition of cubic Cu2O nanoparticles on a Cu substrate. The effects of electrodeposition parameters temperature and time were investigated and optimized to maximize the surface area and catalytic activity towards N2ORR. The performances based on the increased surface area, including the onset potential, current density, and Faradaic efficiency based on detectable gas products, were reported. The kinetics of the process was also investigated using electrochemical impedance spectroscopy (EIS) to shed light on the origin of enhanced activity. The optimized high surface area electrode exhibited an improved onset potential, surpassing other Cu-based N2ORR catalysts previously reported,21,22,24–26 an increased partial current density towards N2, and excellent stability. The performance exhibited by this high surface area Cu electrode affirms its potential as a N2ORR catalyst alternative to noble metals.
2. Materials and methods
2.1 Catalyst preparation and characterization
High surface area Cu electrodes were prepared by electrodepositing nanostructured Cu2O onto Cu foil substrates. First, a Cu foil (4.2 cm × 2.8 cm, thickness 0.254 mm, 99.9%, Alfa Aesar) was cleaned by sonication in acetone, isopropanol, and Millipore water (18.2 MΩ cm) for 30 min each. Each foil was then electropolished for 2 min in 85% phosphoric acid at 1.5 V vs. Ag/AgCl, using a Pt foil counter electrode (99.99%, Alfa Aesar) and a Ag/AgCl reference electrode to achieve a mirror-finished surface. All electrochemical experiments were controlled using a PARSTAT MC potentiostat (Ametek, PMC-2000A). After being rinsed with water and dried under a nitrogen flow, the polished foil was used as a substrate for the electrodeposition of Cu2O. The electrolytic solution was composed of 0.2 M CuSO4·5H2O (analytical grade, Ajax Finechem) and 3 M lactic acid (90%, Sigma-Aldrich) in Millipore water. The pH of the solution was adjusted to 12.5 with NaOH.27 Lactic acid helps stabilize Cu ions under alkaline conditions, which facilitates the formation of Cu+.27,28 The electrodeposition was performed in a beaker without stirring, at a fixed cathodic current density of −1.7 mA cm−2. The deposition temperature was controlled using a water bath. The deposition temperature (25 °C, 40 °C, and 50 °C) and time (2–40 min) were investigated to maximize the surface area and electrocatalytic activity. Each deposition condition was repeated at least 3 times to ensure the repeatability of the experiment.The surface morphology of the electropolished Cu foil and electrodeposited Cu2O electrodes was characterized by scanning electron microscopy (SEM, Hitachi, SU5000). The thickness of the electrodeposited Cu2O layer was assessed using a stylus profiler (Bruker, DektakXT). The particle size distribution was obtained by measuring the edge-to-edge lengths of at least 20 random particles in the SEM images via ImageJ software. The lattice fringes of the electrodeposited Cu2O were imaged by high-resolution transmission electron microscopy (HRTEM, JEOL, JEM-2100). The surface of the flat electropolished Cu electrode was characterized by atomic force microscopy (AFM, Hitachi, 5500M). The roughness and surface area of the AFM data were analyzed via Gwyddion software. The crystalline features of the particles were identified by grazing incident X-ray diffraction (GI-XRD, Rigaku TTRAX III) analysis at an incident angle of 0.4°, a scan rate of 2.5° min−1, and a sampling step of 0.02°. Diffraction patterns were measured over the 2θ range of 10–80°. The crystallite size (Dp) was also calculated from Scherrer's equation 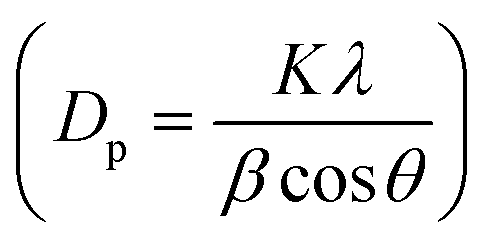 , where K is 0.94 for spherical crystallites with a cubic symmetry, β is the full width at half maximum (FWHM) of the peak at 2θ = 36.5°, and λ is the X-ray wavelength of 1.54178 Å. The X-ray penetration depth was calculated by AbsorbDX software using a mass attenuation coefficient of 47.23 cm2 g−1, 50% porosity, and 99% penetration. The oxidation states of the catalysts were evaluated by X-ray photoelectron spectroscopy (XPS, Shimadzu Kratos, AXIS Supra+). The XPS peak fittings were performed in CasaXPS software, referencing the binding energy of the C 1s peak at 284.8 eV. The XPS regions of Cu 2p3/2 and Cu 2p1/2 were fitted using Shirley-type backgrounds. As the oxidation states of Cu are difficult to differentiate from the Cu 2p spectrum only, Cu LMM Auger scans were performed to identify the Cu, Cu2O, and CuO species. The compositions of the electrode before and after 7 h stability testing were characterized using a micro-energy dispersive X-ray fluorescence spectrometer (μ-EDXR, EDAX Orbis PC, Rh target).
, where K is 0.94 for spherical crystallites with a cubic symmetry, β is the full width at half maximum (FWHM) of the peak at 2θ = 36.5°, and λ is the X-ray wavelength of 1.54178 Å. The X-ray penetration depth was calculated by AbsorbDX software using a mass attenuation coefficient of 47.23 cm2 g−1, 50% porosity, and 99% penetration. The oxidation states of the catalysts were evaluated by X-ray photoelectron spectroscopy (XPS, Shimadzu Kratos, AXIS Supra+). The XPS peak fittings were performed in CasaXPS software, referencing the binding energy of the C 1s peak at 284.8 eV. The XPS regions of Cu 2p3/2 and Cu 2p1/2 were fitted using Shirley-type backgrounds. As the oxidation states of Cu are difficult to differentiate from the Cu 2p spectrum only, Cu LMM Auger scans were performed to identify the Cu, Cu2O, and CuO species. The compositions of the electrode before and after 7 h stability testing were characterized using a micro-energy dispersive X-ray fluorescence spectrometer (μ-EDXR, EDAX Orbis PC, Rh target).
2.2 Electrochemical experiment
All electrochemical reactions were conducted in a custom-made electrochemical compression cell (Fig. 1). The cathode compartment was separated from the anode compartment by an anionic exchange membrane (Sustainion™ X37-50 RT, Dioxide Material). The cell consists of a Cu working electrode, a leakless Ag/AgCl reference electrode (eDAQ), and a Pt foil counter electrode (99.9%, Alfa Aesar). The geometric surface area of the sample was 6 cm2. Each compartment was filled with 8 mL of 0.1 M KOH (99.99%, Sigma Aldrich) electrolyte. N2O (99.8%) or Ar (99.999%) gases were fed into the bottom of both compartments via gas dispersion tubes which generated small gas bubbles in the electrolyte. The gas flow rate was controlled by mass flow controllers (Bronkhorst, El-flow) to be 20 mL min−1 unless otherwise specified. The cathode gas was N2O for the N2ORR experiment and Ar for the hydrogen evolution reaction (HER) experiment, while the anode gas was always Ar.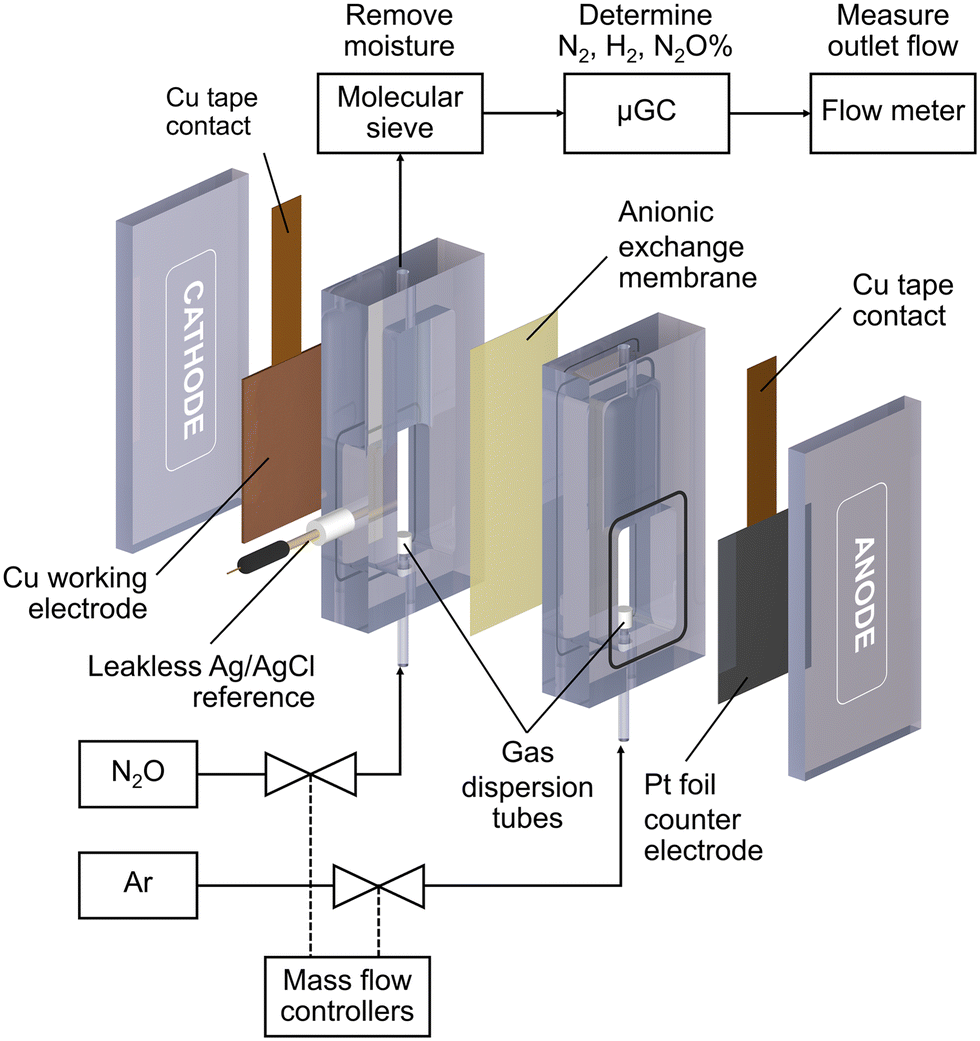 | ||
| Fig. 1 A schematic drawing of the electrochemical cell setup for N2ORR experiments. | ||
Each Cu electrode was subjected to 5 cyclic voltammograms (CV) from 0 V to −2.0 V vs. Ag/AgCl at 10 mV s−1 under Ar to measure the background HER activity and under N2O for the N2ORR activity survey. The N2ORR experiment was performed under chronoamperometry mode at −1.0, −1.2, −1.4, −1.6, and −1.8 V vs. Ag/AgCl for 2400 s each. Prior to each chronoamperometry experiment, the Cu catalyst was electrochemically reduced to metallic Cu via linear sweep voltammetry from 0 to −1.8 V vs. Ag/AgCl at 50 mV s−1. For ease of comparison with other studies, all the electrochemical data were converted from Ag/AgCl to the reversible hydrogen electrode (RHE) according to this equation:
| E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.1967 + 0.059 (pHsolution) |
Before and after the N2ORR experiment, CVs of the electrochemically reduced Cu electrodes were measured in a non-Faradaic region between 0.7 and 0.8 V vs. Ag/AgCl at 7 different scan rates ranging from 5 mV s−1 to 100 mV s−1. The double-layer capacitance (Cdl) was determined from the slope of the capacitive current densities  at 0.75 V vs. Ag/AgCl plotted against the scan rates. The surface area discussed in this study is represented by the roughness factor, which is defined as follows:
at 0.75 V vs. Ag/AgCl plotted against the scan rates. The surface area discussed in this study is represented by the roughness factor, which is defined as follows:
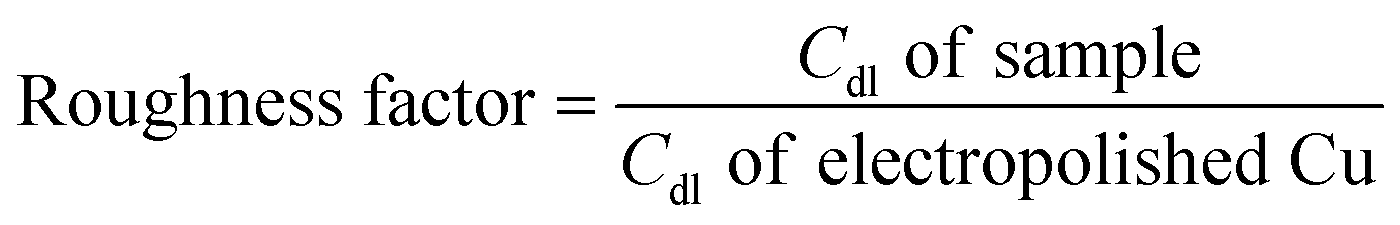
The stability of the high surface area Cu electrode was assessed by performing a prolonged chronoamperometry experiment at −0.44 V vs. RHE (−1.4 V vs. Ag/AgCl) under a 20 mL min−1 N2O feed. The gas product was collected once every hour throughout the 7 h experiment.
2.3 Product measurement
The reduction gas products evolved from the cathodic compartment during chronoamperometry experiments were analyzed using an online micro-gas chromatograph (μGC, Varian CP-4900) equipped with a Molsieve 5A column at 140 °C for H2 and N2 detection and a Porapak Q column at 70 °C for N2O detection, using Ar and He as the reference gases for the thermal conductivity detectors (TCD), respectively. The gas product collection began after the reduction reaction had started for 2 min and continued sampling every 5 min until the end of the experiment, resulting in 8–10 injections per potential. The N2, H2, and N2O concentrations (mol%) were calibrated using a standard mixed gas to quantify the product concentrations from the GC peak areas. The outlet gas flow rate was also measured using a digital film flow meter (Horiba SF-1U), which allowed us to correctly quantify the total amount of gas products. The Faradaic efficiency (FE) of N2 and H2 were calculated according to the following equation:where n is the number of electrons required to produce N2 or H2 (n = 2 for both products), F is Faraday's constant (96
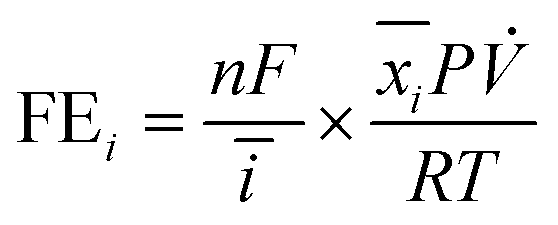
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), ī (A) is the average current passed during the chronoamperometry experiment,
485 C mol−1), ī (A) is the average current passed during the chronoamperometry experiment, ![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) i is the average mole fraction of each product, P is the pressure of our experiment (1 atm),
i is the average mole fraction of each product, P is the pressure of our experiment (1 atm), ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) is the outlet gas flow rate measured using a digital flow meter (L s−1), T is the temperature (298.15 K), and R is the gas constant (0.082057 L atm K−1 mol−1).
is the outlet gas flow rate measured using a digital flow meter (L s−1), T is the temperature (298.15 K), and R is the gas constant (0.082057 L atm K−1 mol−1).
3. Results and discussion
3.1 Synthesis and characterization of electrodeposited Cu2O
Electrodeposition is a scalable and highly tunable method suitable for synthesizing high surface area Cu-based electrocatalysts.29 In this work, the deposition parameters of Cu2O, including temperature and time, were examined and optimized for maximum electrochemical N2ORR activity. The effects of these deposition parameters on the morphology of Cu2O were investigated to identify the conditions that result in a catalyst with the largest surface area and, more importantly, the highest N2ORR activity.The morphologies of the electropolished Cu foil (Cu-Epolish) and Cu2O electrodeposited at 25 °C (controlled room temperature), 40 °C, and 50 °C for 20 min at −1.7 mA cm−2 were studied. Fig. 2 shows SEM images of the as-deposited Cu2O electrodes, the double-layer capacitances (Cdl) determined from the non-Faradaic region of the electrochemically reduced electrodes, and their corresponding roughness factors. All the as-deposited Cu2O electrodes have cubic structures, as commonly acknowledged to be enclosed by (100) facets,30,31 which is the most stable surface of Cu2O.30 The HRTEM image of the Cu2O structure electrodeposited at 40 °C (Fig. S1†) also exhibited clear lattice fringes with an interval of 0.2 nm, consistent with the lattice spacing between (200) planes of cubic Cu2O.32 Cu2O structures deposited at 25 °C and 40 °C show similar multiple-scale clusters of small cubic structures. The individual cube and the clusters of Cu deposited at 40 °C appeared slightly larger. The cubic structures of Cu2O deposited at 50 °C, on the other hand, grew substantially larger, and the morphology changed from clusters of small cubes to an isolated large cubic architecture.
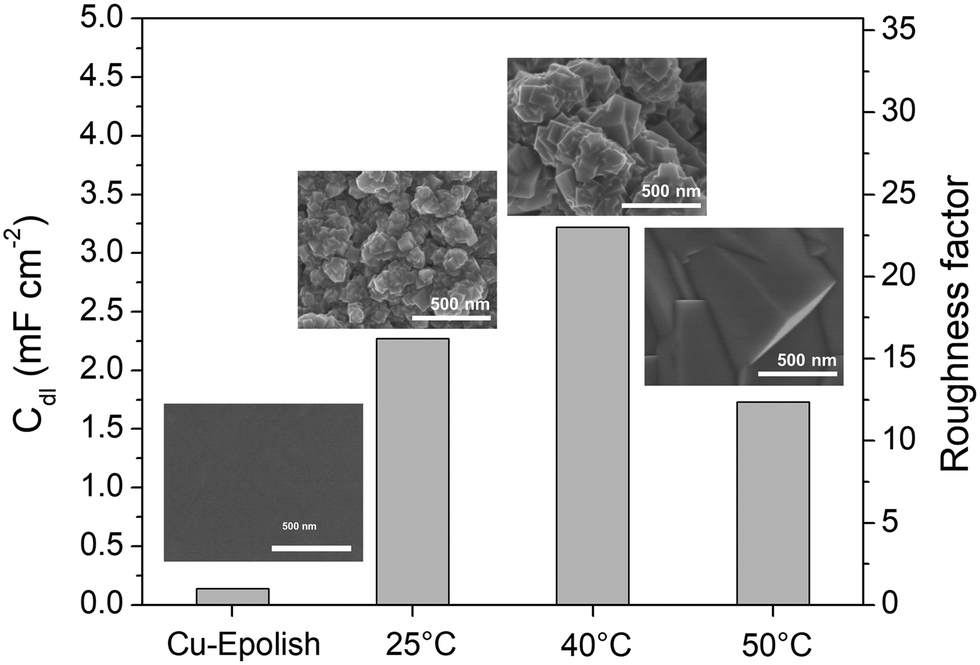 | ||
| Fig. 2 SEM images of the electropolished Cu foil (Cu-Epolish) and Cu2O electrodes deposited at −1.7 mA cm−2 for 20 min at 25 °C, 40 °C, and 50 °C, as well as their double-layer capacitances (Cdl) and roughness factors. | ||
The overall roughness of the surface, calculated by normalizing the Cdl of the electrochemically reduced Cu2O electrodes with that of the electropolished Cu, is the determining factor for the relative surface area. The roughness factors of the nanostructured Cu electrodes deposited at 25 °C, 40 °C, and 50 °C were 16.2, 23.0, and 12.4, respectively. Compared to the significantly flat surface of electropolished Cu (surface ratio = 1.0006 from AFM; see Fig. S2†), the electrochemically active surface areas (ECSA) of these samples are approximately 16.2, 23.0, and 12.4 times that of the geometric area, respectively. Despite its larger particle sizes, the nanostructured Cu electrode deposited at 40 °C exhibits a higher surface area than the one deposited at 25 °C potentially due to the thicker Cu2O film or a more complete coating (see the photo of the electrode surface in Fig. S3†) as a result of a higher deposition rate at the higher temperature. Increasing the temperature further to 50 °C resulted in a significant reduction in surface area, which is expected from the larger grain size observed in the SEM image. As explained by previous studies, the higher deposition temperature leads to an increased growth rate but a reduced nucleation rate, resulting in lower density and larger crystals.33 This hypothesis aligns with the trend in the deposition potential (Fig. S3d†). The deposition potentials at 25 °C and 40 °C were around 0.4 and 0.41 V vs. Ag/AgCl, respectively, while the deposition potential at 50 °C shifted by 100 mV to around 0.51 V vs. Ag/AgCl. The significant shift in the deposition potential at 50 °C can change the local concentration of Cu2+ at the electrode surface, which affects the balance between the nucleation and growth rates, leading to a different morphology. Based on the surface area and the even distribution of particles, the deposition at 40 °C was investigated further for the optimization of deposition time.
Electrodeposition of Cu2O at 40 °C was conducted at varying deposition times and the SEM images are shown in Fig. 3b–f. These images show that the uniform Cu2O film did not form since the beginning – there appeared to be some nucleation period. The 2 min deposition exhibits a low coverage and a small particle size. The particle size grew larger, and the surface coverage increased with longer electrodeposition time. After 10 min, the electrode surface appeared fully covered by Cu2O nanocubes. The Cu2O particle size, as measured from edge-to-edge lengths of cubic particles observed in the SEM images (Fig. 3g and S4a†), grew during the initial deposition and started plateauing at approximately 20 min, implying the balance between nucleation and growth. The longer deposition time led to the creation of new cubes of roughly the same size. The nucleation of the new cubic structures preferred to form atop the formerly formed cluster rather than laterally, creating clusters of merged nanocubes. Interestingly, the roughness factor, calculated from the post-reduced Cdl shown in Fig. S5,† continued to increase almost linearly at ∼1 fold min−1 even after the complete surface coverage (Fig. 3h) and the average particle size leveling off. The film thickness as measured by a stylus profiler also progressed in the same manner with a growth rate of ∼0.1 μm min−1 (Fig. 3h and S4b†). This suggests that new accessible surfaces were created during film growth.
 | ||
| Fig. 3 SEM images of (a) electropolished Cu and Cu2O deposited at varying times: (b) 2 min, (c) 10 min, (d) 20 min, (e) 30 min, and (f) 40 min at a bath temperature of 40 °C. (g) Averaged particle sizes obtained from SEM images and crystallite sizes calculated from Scherrer's equation and (h) thicknesses of the as-deposited Cu2O films and roughness factors of the electrochemically reduced Cu2O at varying deposition times. (i) GIXRD patterns of the as-deposited Cu2O catalysts. | ||
To confirm the state of Cu formation, the crystallographic structures of the electropolished Cu foil and electrodeposited Cu2O at varying electrodeposition times were investigated by grazing incident X-ray diffraction (GIXRD). Fig. 3i shows the GIXRD patterns of the electropolished Cu foil and the electrodeposited Cu2O at varying deposition times. The peaks at 2θ = 43.4°, 50.5°, and 74.2° found in the electropolished Cu can be attributed to metallic Cu, while the peaks at 2θ = 29.6°, 36.5°, 42.4°, 61.5°, and 73.7° in the electrodeposited electrodes belong to Cu2O. Although the cubic structures of Cu2O were discernible from the SEM images through the apparent right-angle corners which indicates the exposure of Cu2O (100) facets, the diffraction pattern resembled the powder diffraction (JCPDS: 71-3645) as these nanocubes organized on the surface at random orientation. The Cu2O-related peaks could be observed after 2 min of deposition and the size of the peak increased with the deposition time. The peaks correlated to metallic Cu also subsided with a longer deposition time. At 20 min of deposition time, the metallic Cu-related peaks could no longer be detected, implying complete coverage of a thick Cu2O film on the Cu substrate. The thickness of the film under this condition was approximately 2.6 μm. The disappearance of the metallic Cu peaks was consistent with the 2.2 μm penetration depth of GIXRD through the porous Cu2O layer (assuming 50% porosity). The crystallite size as estimated from Scherrer's equation established the same trend as the SEM image where the size reached a plateau after 20 min of deposition (Fig. 3g). However, the crystallite size appeared to be significantly smaller than the particle size measured from SEM, albeit the implication of single crystal particles from the apparent cubes. Since the distribution of particle size was broad, the size disparity between the particle (from SEM) and the crystallite (from XRD) could be from the biased particle sampling from the SEM image toward the larger particles, as they are more clearly visible. In addition, the XRD tends to underestimate the size of large crystallites, especially under the grazing incident geometry where there was considerable dispersion of the sample-to-detector distance.34
3.2 Electroreduction of N2O
N2ORR experiments were carried out at ambient temperature (25 °C) and pressure. The electrodes were cycled at a scan rate of 10 mV s−1 in 0.1 M KOH electrolyte purged with 20 mL min−1 Ar or N2O at a potential between 0.5 and −1.0 V vs. RHE. The cathodic scans of the 3rd CV scans of all Cu electrodes measured under Ar (open symbols) and N2O (filled symbols) are shown in Fig. 4a. Within the potential window of −0.3 to 0.3 V vs. RHE, N2ORR is more favorable than the hydrogen evolution reaction (HER) as the current density under the N2O feed exhibits an earlier onset potential compared to the current density under Ar. All current–voltage curves collected under N2O consist of 2 potential regimes: a kinetic-controlled regime at the lower overpotential, where the current density exponentially rises with applied overpotential, and a mass transport-controlled regime at the higher overpotential, where the current density reaches a limit governed by the diffusion of N2O to the catalyst surface.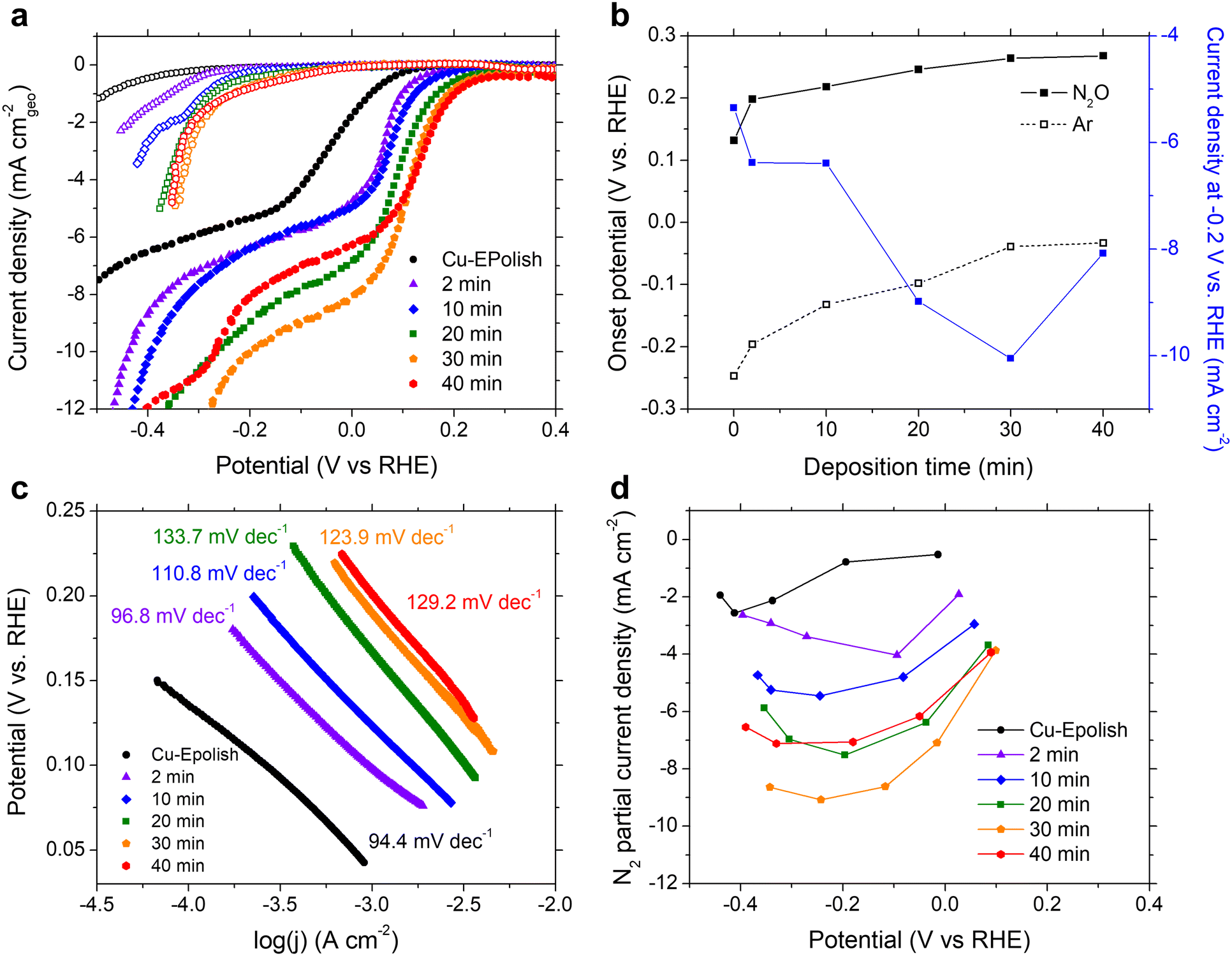 | ||
| Fig. 4 (a) Current–voltage curves of all the Cu catalysts. Filled symbols: under continuous N2O bubbling, open symbols: under continuous Ar bubbling, scan rate: 10 mV s−1, and electrolyte: 0.1 M KOH. (b) Onset potentials (at −0.1 mA cm−2) of the electrodes measured under N2O and Ar and current density at −0.2 V vs. RHE. (c) Tafel slopes of the Cu electrodes extracted from the CV measured under a 20 mL min−1 N2O gas flow rate. (d) Partial current density of N2. | ||
In the kinetic-controlled regime, the onset potential under N2O (defined here at −0.1 mA cm−2) of the electropolished Cu electrode was 0.13 vs. RHE, +0.38 V shifted from the onset potential under Ar (Fig. 4a). The high surface area Cu electrodes prepared by electrodeposition exhibit increasingly more anodic onset potentials for both N2ORR and HER with longer deposition time. However, the onset potential for N2ORR reaches a maximum at around 0.27 V vs. RHE for the Cu-40 min electrode, exhibiting a +0.30 V shift over its HER onset (Fig. 4b). Hence, the high surface area Cu electrode offers a 130 mV reduction in the N2ORR onset potential compared to the flat Cu. Fig. 4c shows that the Tafel slopes extracted from the kinetic-controlled regime slightly increase with the surface area, ranging from 94 mV dec−1 for electropolished Cu to ∼130 mV dec−1 for high surface area Cu electrodes with long deposition times. This suggests that the higher surface area resulted in slightly slower kinetics. In the mass transport-controlled regime, all electrodes exhibit a maximum current density ranging from −5 to −10 mA cm−2. The diffusion-limited current density of N2ORR (measured at −0.2 V vs. RHE) increases with the surface area of the electrode until Cu-30 min, which achieves the highest current density of −10 mA cm−2, a 2-fold improvement of the flat Cu. Interestingly, despite having the largest roughness factor, the limiting current density of Cu-40 min was lower than those of Cu-30 min and Cu-20 min (Fig. 4b).
The concentration of reduction products N2 and H2 were detected by online micro-gas chromatography (μGC) during the N2ORR experiment. Together with the measured outlet flow rate, the Faradaic efficiencies of N2 and H2 produced at varying applied potentials were calculated and the values are shown in Fig. S6.† The sum of the N2 and H2 Faradaic efficiencies of all the catalysts were close to unity, confirming that these gases were the two main products that evolved within the investigated potential window. The partial current density of N2 formation catalyzed by Cu shown in Fig. 4d exhibits a volcano trend where at a low overpotential, the N2 partial current density generally increases with the applied potential, until a maximum is reached, and then the current decreases with the applied potential at a high overpotential. This is because at a lower overpotential, between 0 and 0.2 V vs. RHE, N2ORR is the only process happening in the solution, and increasing the applied potential drives the rate of N2ORR. At a higher cathodic potential, HER contributes progressively to the process and the higher overall currents arise from the concomitant N2 and H2 production. In the regime that HER was expected, the longer deposition time slightly improved the N2 Faradaic efficiency. Among all the samples, the Cu-30 min electrode achieved the highest N2 partial current density of −9.1 mA cm−2, which is on-par with that of Pd,15 and a 3.5-fold improvement over the flat Cu electrode.
We learned that Cu-30 min exhibits the highest N2 partial current density at all the applied potentials, despite its lower roughness factor of ∼30 compared to Cu-40 min of ∼40 (Fig. 4d). To further understand the origin of the current limitation of these high surface area Cu electrodes, the current density of each electrode was normalized by its roughness factor to yield a specific activity based on the electrochemically active surface area (ECSA), as shown in Fig. 5a. When accounting for the surface area, all the high surface area Cu electrodes exhibit similar onset potentials to the electropolished Cu. Furthermore, the kinetic-controlled activity of the high surface area Cu electrodes seems to match well with that of the electropolished Cu. The major difference lies in the transition potential from the kinetic-controlled to the mass transport-controlled regime. The electrode with a larger surface area results in an earlier transition to the mass transport-controlled regime and a lower maximum specific current density. This phenomenon was investigated further by electrochemical impedance spectroscopy (EIS). Fig. 5b shows the Nyquist impedance spectra of the electropolished Cu and Cu-30 min samples at −0.18, −0.29, and −0.39 V vs. RHE. All the spectra exhibit a single semicircle that can be modeled by a simple Randles circuit (shown in Fig. 5b), which is composed of electrolyte resistance (Rs) in series with a parallel circuit of a constant phase element related to the double-layer capacitance (Qdl) and charge transfer resistance (Rct) related to the reduction reaction. The extracted double layer capacitance (Cdl) and charge transfer resistance (Rct) are shown in Fig. 5c (see Table S1† for the fitting values and the method of Cdl calculation). It was found that at the same potential, both samples display the same charge transfer resistances, but Cu-30 min has a substantially higher Cdl. This result confirms that the enhanced activity indeed arises from the higher number of active sites, while the specific activity per site remains constant. Additionally, for Cu-30 min at −0.18 V vs. RHE, the impedance at low frequency deviates from a typical Randles circuit and exhibits a 45° angle, which is a characteristic of a Warburg element associated with a mass transfer effect. This finding aligns with our observation from the specific activity plot (Fig. 5a) and suggests that the larger surface area experiences a higher mass transport limitation at the active sites. Hence, when the deposition time is too long, the extra surface area is no longer beneficial to the N2ORR activity because mass transport becomes a dominating process.
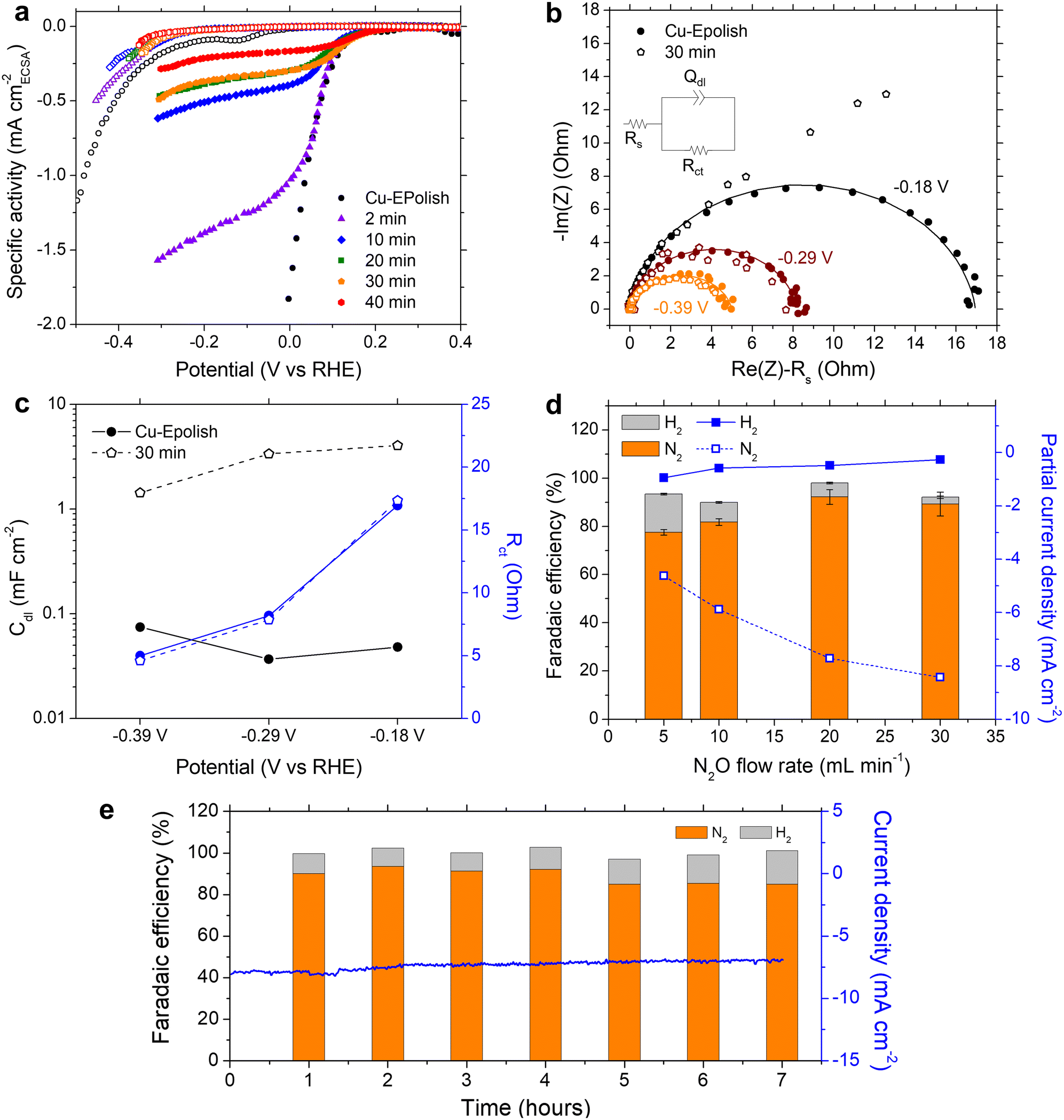 | ||
| Fig. 5 (a) Specific N2ORR activity normalized by ECSA. (b) Nyquist plots of Cu-Epolish and Cu-30 min measured at −0.18, −0.29, and −0.39 V vs. RHE (the electrolyte resistance, Rs, was subtracted) and (c) the Cdl and Rct parameters extracted from the EIS spectra. (d) The effect of the N2O flow rate on the Faradaic efficiency and partial current density of Cu-30 min at 0.12 V vs. RHE. (e) N2ORR stability test measurements using the Cu-30 min electrode at a constant N2O flow rate of 20 mL min−1 at 0.12 V vs. RHE. | ||
The mass transport limitation on the electrode performance was studied further by varying the N2O gas flow rate during the N2ORR experiment of Cu-30 min. The increased N2O flow rate enhanced the overall current density (Fig. S7†) and the Faradaic efficiency, leading to a higher N2 partial current density and a lower H2 partial current density (Fig. 5d). This result implies that the reaction at a low flow rate was indeed limited by the mass transport of N2O to the active sites. The increased selectivity, i.e., the increase in N2 product ratio over all the products, with higher flow rates demonstrates that the competing HER process diminished when sufficient N2O was supplied to the catalytic sites. The trend is indicative that our nanostructured Cu catalyst has high selectivity towards N2ORR over HER, and the overall N2ORR activity can be improved further if the mass transport of N2O can be enhanced. Increasing the N2O flow rate in our electrochemical cell further may result in a slightly higher N2ORR rate; nevertheless, the system will still be limited by the low N2O solubility in water of only 24.3 mM. Based on our limiting current density (jlim) of electropolished Cu and the diffusion of N2O in water of 1.8 × 10−9 m2 s−1,22 the diffusion layer (δ) of our system was as large as ∼150 μm  .35 Hence, to overcome the mass transport limitation, the electrochemical cell must be redesigned to increase the N2O concentration at the surface of the Cu catalyst; for example, by improving the fluid hydrodynamics to increase mixing,36 increasing the cell pressure,37 modifying the electrolyte,25 or changing the cell configuration to a gas diffusion electrode.38,39
.35 Hence, to overcome the mass transport limitation, the electrochemical cell must be redesigned to increase the N2O concentration at the surface of the Cu catalyst; for example, by improving the fluid hydrodynamics to increase mixing,36 increasing the cell pressure,37 modifying the electrolyte,25 or changing the cell configuration to a gas diffusion electrode.38,39
Despite the mass transport limitation of N2O, the high surface area Cu-30 min electrode was proven to be a robust N2ORR catalyst as the overall current density remained stable throughout 7 h under 0.12 V vs. RHE and a 20 mL min−1 N2O flow rate, as shown in Fig. 5e. Over the course of the experiment, the Faradaic efficiency of H2 slightly increased from ∼10% in the first 4 hours to ∼13% in the last 3 hours. XRF analysis of the post-stability testing sample suggests that there was a trace of Pt contamination on the surface of Cu, which was not present prior to the 7 h stability testing (Fig. S8†). This is likely due to the redeposition of the Pt ion, which was dissolved out from the anode under prolonged oxidation in water, similar to previous reports.40 As Pt is highly selective toward HER over N2ORR, this Pt contamination on the cathode may contribute to the slight increase in the Faradaic efficiency towards H2.
3.3 Post-reaction characterization
To investigate the chemical and structural changes of the high surface area Cu catalyst after N2ORR, three stages of the electrodes (electropolished Cu substrate, as-deposited Cu2O, and high surface area Cu after N2ORR) were examined by XPS, XRD, and SEM. Both core and Auger spectra (Cu LMM) were measured in XPS to accurately identify Cu0, Cu+, and Cu2+ species. This is because the two oxidation states are hardly distinguishable using XPS spectra alone as the Cu 2p peaks of metallic Cu and Cu2O have very similar binding energies and the satellite peaks of Cu+ are rather weak.As shown in Fig. 6a and b, the electropolished Cu substrate was composed of metallic Cu0 and Cu2+. The binding energies of the main Cu 2p3/2 and Cu 2p1/2 peaks are centered at approximately 932.9 and 952.8 eV, respectively. Together with the kinetic energy split from the Auger spectra, the main Cu peaks were assigned to Cu0, which is expected from the Cu foil substrate. The secondary Cu 2p3/2 peak at 935.3 eV is coupled with the satellite peaks at 941.3 eV and 943.8 eV, suggesting Cu2+ species.35 Cu2+ species were likely derived from post-process oxidation as the external surface of Cu is known to be easily oxidized under ambient conditions. On the as-deposited electrode, the peak shift to lower kinetic energy and the extinction of the kinetic energy peak split from the Auger spectra (Fig. 6b) affirm the existence of Cu2O and the lack of metallic Cu on the surface of the as-deposited electrode.41,42 A small amount of CuO (about 4.5%) likely caused by air exposure42 existed on the surface, as a minor Cu 2p3/2 peak was detected at 953.3 eV. After the N2ORR process, the Cu 2p and Cu LMM spectra resembled those of the electropolished Cu, comprising mainly metallic Cu and CuO. The results suggest that during the N2ORR process, Cu was essentially in the metallic form. A similar conclusion can also be drawn from the XRD patterns in Fig. 6c, which clearly shows that the as-deposited Cu2O phase was completely reduced to metallic Cu. The peaks at 43.4°, 50.5°, and 74.2° corresponded to the (111), (200), and (220) planes of powder diffraction of Cu nanoparticles (JCPDS 04-0836). The intensity ratios of these peaks are different from those of the Cu foil, which exhibits a preferred orientation in the (200) plane, suggesting that the metallic Cu indeed originated from the randomly oriented electrodeposited Cu2O. This discovery aligns well with the N2ORR activity, as the electropolished Cu and the high surface area Cu, derived from Cu2O, exhibit a similar kinetic-controlled specific activity, suggesting a similar nature of the active sites. The SEM images in Fig. S9† show that after N2ORR testing, the reduced Cu-30 min electrode still displayed a high surface area, but the cubic nanostructures had lost their definition and become rounded. This surface restructuring under reaction conditions is a common phenomenon that can be influenced by both physical and chemical driving forces.43 The dynamic surface reconfiguration could be induced by temporary oxidation of surface copper by nitrous oxide, followed by reduction under reaction conditions.44 This rounding effect was more prominent after 7 h of stability testing, which might result in a lower surface area and a lower current density over time. Nevertheless, the stability results implied that the electrode remained porous and with a high surface area despite the loss of definitive facet structures.
 | ||
| Fig. 6 (a) XPS spectra (Cu 2p3/2 and Cu 2p1/2 core peaks), (b) Cu LMM Auger spectra, and (c) GIXRD patterns of the Cu-Epolish, the as-prepared Cu2O electrodeposited for 30 min, and Cu-30 min after N2ORR. | ||
4. Conclusions
Cu was utilized as a non-noble electrocatalyst in the electrochemical reduction of N2O to N2. The active surface area of the catalyst was enhanced by electrodepositing nanostructured Cu2O onto a Cu substrate. At the optimal deposition conditions of −1.7 mA cm−2, 30 min, and 40 °C, the electrochemically reduced Cu2O demonstrated a surface area ∼30-fold that of electropolished Cu. The Cu catalyst was selective to N2ORR, providing more than 90% Faradaic efficiency toward N2 formation. Compared to a flat Cu electrode, the optimized nanostructured Cu electrode improved the N2ORR onset potential by 130 mV and increased the N2 partial current density 3.5 fold. Increasing the surface area of Cu further resulted in a decreased performance due to mass transport limitation. With a comparable kinetically regulated specific activity to the flat Cu electrode, the improved performance of the nanostructured electrode was attributed to the increased number of active sites. The high surface area Cu catalyst was robust and remained highly stable over the course of the 7 hour stability testing. This study demonstrates that high surface area Cu is a promising N2ORR catalyst substitute for noble metals.Author contributions
SN performed all the experiments and wrote the first draft. KM and TB helped with characterization. PC and RM conceptualized the work and finalized the manuscript. All the authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nanotechnology Center (NANOTEC), Thailand [Grant Number P1752588] and the Thailand Science Research and Innovation Fund, Chulalongkorn University [Grant number CU_FRB65_hea (75)_169_21_35]. The authors would like to acknowledge the NSTDA Characterization and Testing Service Center (NCTC) for the characterization tools, as well as Dr. Kasempong Srisawad and Dr. Chakrit Sriprachuabwong for their assistance with the stylus profiler.References
- J. T. Houghton, et al., Climate change 2001: the scientific basis: contribution of Working Group I to the third assessment report of the Intergovernmental Panel on Climate Change, 2001, Cambridge university press Search PubMed.
- M. J. Prather, et al., Measuring and modeling the lifetime of nitrous oxide including its variability, J. Geophys. Res.: Atmos., 2015, 120(11), 5693–5705 CrossRef CAS PubMed.
- R. W. Portmann, J. S. Daniel and A. R. Ravishankara, Stratospheric ozone depletion due to nitrous oxide: influences of other gases, Philos. Trans. R. Soc., B, 2012, 367(1593), 1256–1264 CrossRef CAS PubMed.
- A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century, Science, 2009, 326(5949), 123–125 CrossRef CAS PubMed.
- H. Tian, et al., A comprehensive quantification of global nitrous oxide sources and sinks, Nature, 2020, 586(7828), 248–256 CrossRef CAS PubMed.
- M. Charlesworth and F. Swinton, Anaesthetic gases, climate change, and sustainable practice, Lancet Planet. Health, 2017, 1(6), e216–e217 CrossRef PubMed.
- U. M. Skiba and R. M. Rees, Nitrous oxide, climate change and agriculture, CAB Rev., 2014, 9, 010 Search PubMed.
- P. Arias, et al., Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Technical Summary, 2021 Search PubMed.
- E. Dlugokencky, Global N2O Monthly Means, NOAA/GML 2022 [cited 2022 9/6/2022]; Available from: https://gml.noaa.gov/ccgg/trends_n2o/ Search PubMed.
- S. Yamauchi, et al., Removal of sevoflurane and nitrous oxide from waste anesthetic gases by using Anesclean®, the system for treating waste anesthetic gases, Masui, 2010, 59(7), 930–934 Search PubMed.
- K. Tjus, Test of MDU nitrous oxide from Medclair. 2020, Medclair: IVL Swedish Environmental Research Institute, 2020 Search PubMed.
- L.-H. Norway, EXCIDIO – Nitrous Oxide Converter, 2022, Available from: https://www.linde-healthcare.no/en/products_services_ren/products_services_care_area/hospital_clinical_care/excidio/index.html Search PubMed.
- M. Ek and K. Tjus, Decreased emission of nitrous oxide from delivery wards—case study in Sweden, Mitig. Adapt. Strateg. Glob. Chang., 2008, 13(8), 809–818 CrossRef.
- M. Konsolakis, Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects, ACS Catal., 2015, 5(11), 6397–6421 CrossRef CAS.
- S. Baek, et al., Pd–Cu alloy catalyst synthesized by citric acid-assisted galvanic displacement reaction for N2O reduction, J. Appl. Electrochem., 2020, 50(4), 395–405 CrossRef CAS.
- N. Konishi, K. Hara and T. Sakata, Electrochemical Reduction of N2O on Gas-Diffusion Electrodes, Bull. Chem. Soc. Jpn., 1996, 69, 2159–2162 CrossRef CAS.
- Y. Yoshida, et al., Catalytic reduction of nitrous oxide with atomic hydrogen permeating through palladized Pd sheet electrodes, Electrochim. Acta, 1999, 44(20), 3585–3587 CrossRef CAS.
- B. Wang, Electrocatalytic Properties of Nitrous Oxide and Its Voltammetric Detection at Palladium Electrodeposited on a Glassy Carbon Electrode, Anal. Chem., 1998, 21812187 Search PubMed.
- R. Gómez and M. J. Weaver, Reduction of Nitrous Oxide on Iridium Single-Crystal Electrodes, Langmuir, 2002, 18(11), 4426–4432 CrossRef.
- A. Kudo and A. Mine, Electrocatalysis for N2O reduction on metal electrodes, J. Electroanal. Chem., 1996, 408(1), 267–269 CrossRef.
- K. Kim, et al., Systematic Approach to Designing a Highly Efficient Core–Shell Electrocatalyst for N2O Reduction, ACS Catal., 2021, 11(24), 15089–15097 CrossRef CAS.
- A. Aziznia, et al., Electroreduction of nitrous oxide on platinum and palladium: Toward selective catalysts for methanol–nitrous oxide mixed-reactant fuel cells, Electrochim. Acta, 2011, 56(14), 5238–5244 CrossRef CAS.
- K. Kim, et al., Catalytic decomposition of N2O on PdxCuy alloy catalysts: A density functional theory study, Appl. Surf. Sci., 2020, 510, 145349 CrossRef CAS.
- A. Kudo and A. Mine, Electrocatalytic reduction of nitrous oxide on metal and oxide electrodes in aqueous solution, Appl. Surf. Sci., 1997, 121–122, 538–542 CrossRef.
- O. S. Kwon, et al., Optimization of Solution Condition for an Effective Electrochemical Reduction of N2O, Electroanalysis, 2019, 31(4), 739–745 CrossRef CAS.
- K. H. Kim, et al., Porous indium electrode with large surface area for effective electroreduction of N2O, Electrochem. Commun., 2016, 62, 13–16 CrossRef CAS.
- J. Kaur, et al., All-oxide solar cells based on electrodeposited Cu2O absorber and atomic layer deposited ZnMgO on precious-metal-free electrode, Sol. Energy Mater. Sol. Cells, 2017, 161, 449–459 CrossRef CAS.
- Y. Yang, Y. Li and M. Pritzker, Control of Cu2O Film Morphology Using Potentiostatic Pulsed Electrodeposition, Electrochim. Acta, 2016, 213, 225–235 CrossRef CAS.
- I. S. Brandt, et al., Electrodeposition of Cu2O: growth, properties, and applications, J. Solid State Electrochem., 2017, 21(7), 1999–2020 CrossRef CAS.
- Y. Shang and L. Guo, Facet-Controlled Synthetic Strategy of Cu2O-Based Crystals for Catalysis and Sensing, Adv. Sci., 2015, 2(10), 1500140 CrossRef PubMed.
- Y. Su, et al., Controlling Surface Termination and Facet Orientation in Cu2O Nanoparticles for High Photocatalytic Activity: A Combined Experimental and Density Functional Theory Study, ACS Appl. Mater. Interfaces, 2017, 9(9), 8100–8106 CrossRef CAS PubMed.
- D. Zhao, et al., Notable in situ surface transformation of Cu2O nanomaterials leads to dramatic activity enhancement for CO oxidation, RSC Adv., 2017, 7(60), 37596–37603 RSC.
- M.-C. Huang, et al., Temperature dependence on p-Cu2O thin film electrochemically deposited onto copper substrate, Appl. Surf. Sci., 2014, 301, 369–377 CrossRef CAS.
- W. L. Tan, Y.-B. Cheng and C. R. McNeill, Direct assessment of structural order and evidence for stacking faults in layered hybrid perovskite films from X-ray scattering measurements, J. Mater. Chem. A, 2020, 8(25), 12790–12798 RSC.
- A. Filzwieser, K. Hein and G. Mori, Current density limitation and diffusion boundary layer calculation using CFD method, JOM, 2002, 54(4), 28–31 CrossRef CAS.
- S. Baek, et al., N2O dissolution with Couette-Taylor flow mixer for the effective electrochemical N2O reduction reaction, Chem. Eng. J., 2018, 335, 915–920 CrossRef CAS.
- A. D. King and C. Coan, Solubility of water in compressed carbon dioxide, nitrous oxide, and ethane. Evidence for hydration of carbon dioxide and nitrous oxide in the gas phase, J. Am. Chem. Soc., 1971, 93, 1857–1862 CrossRef CAS.
- N. Furuya and H. Yoshiba, Electroreduction of nitrous oxide to nitrogen using a gas-diffusion electrode loaded with Pt catalyst, J. Electroanal. Chem. Interfacial Electrochem., 1991, 303(1), 271–275 CrossRef CAS.
- T. N. Nguyen and C.-T. Dinh, Gas diffusion electrode design for electrochemical carbon dioxide reduction, Chem. Soc. Rev., 2020, 49(21), 7488–7504 RSC.
- M. Tian, et al., Influence of the Working and Counter Electrode Surface Area Ratios on the Dissolution of Platinum under Electrochemical Conditions, ACS Catal., 2016, 6(8), 5108–5116 CrossRef CAS.
- Y. Gao, et al., Three-dimensional porous Cu@Cu2O aerogels for direct voltammetric sensing of glucose, Microchim. Acta, 2019, 186(3), 192 CrossRef PubMed.
- C. C. Chusuei, M. A. Brookshier and D. W. Goodman, Correlation of Relative X-ray Photoelectron Spectroscopy Shake-up Intensity with CuO Particle Size, Langmuir, 1999, 15(8), 2806–2808 CrossRef CAS.
- Z. Weng, et al., Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction, Nat. Commun., 2018, 9(1), 415 CrossRef PubMed.
- X. He, et al., Controllable in Situ Surface Restructuring of Cu Catalysts and Remarkable Enhancement of Their Catalytic Activity, ACS Catal., 2019, 9(3), 2213–2221 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2re00249c |
| This journal is © The Royal Society of Chemistry 2023 |