
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine catalyzed synthesis of imidazo[1,2-a]pyrazine and imidazo[1,2-a]pyridine derivatives and their anticancer activity†
Rajavenkatesh Krishnamoorthy *a and
Parthiban Anaikutti*b
*a and
Parthiban Anaikutti*b
aOrganic and Bioorganic Chemistry Laboratory, CSIR-Central Leather Research Institute (CSIR-CLRI), Adyar, Chennai, Tamil Nadu-600020, India. E-mail: krajavenkateshcmp5@gmail.com; rajavenkateshk2@gmail.com
bCentre for GMP, National Institute of Pharmaceutical Education and Research (NIPER-G), Guwahati, Assam-781101, India. E-mail: parthichem84@gmail.com; parthiban@niperguwahati.in; Tel: +91 9843080794
First published on 13th December 2023
Abstract
An efficient iodine-catalyzed method for synthesizing imidazo[1,2-a]pyrazines and imidazo[1,2-a]pyridines via one-pot three-component condensations has been reported. The product, generated in situ by the reaction between an aryl aldehyde and 2-aminopyridine or 2-aminopyrazine, undergoes [4 + 1] cycloaddition with tert-butyl isocyanide, affording the corresponding imidazopyrazine and imidazopyridine derivatives in good yields. The photophysical properties of these new fluorescent derivatives are also presented. The anti-cancer activities of the synthesized compounds (10a–m) and (12a–l) were evaluated against four cancer cells (Hep-2, HepG2, MCF-7, A375) and the normal Vero cell. Significant anti-cancer activities were observed and compared with the standard drug Doxorubicin. In vitro experimental results revealed that compound 12b is a promising lead with IC50 values of 11 μM, 13 μM, 11 μM, 11 μM, and 91 μM against Hep-2, HepG2, MCF-7, A375, and Vero, respectively.
Introduction
With improper and uncontrolled cell division, cancer encompasses over 100 diseases affecting the body extensively. Globally, cancer ranks as the second leading cause of mortality, accounting for 1 in 6 deaths in 2018, totaling 9.6 million deaths.1 Diverse malignancies impact both genders. Current anti-cancer medications exhibit distinct modes of action, leading to varying effects on cancer types and healthy cells.2 Moreover, limited evidence distinguishes malignant cells biochemically from normal ones, impeding targeted treatments.3 A critical concern is cancer's resistance to multi-drug chemotherapy, coupled with poor cure rates, severe side effects, and patient harm. Thus, vital for cancer treatment, the development of superior, more efficient, and less harmful chemotherapeutic drugs remains imperative.4Multicomponent reactions (MCRs) offer simple and atom-economical methods for synthesizing complex molecular structures with a wide range of biological activities.5 Consequently, product purification resulting from MCRs becomes simpler, as almost all starting materials are incorporated into the formed product. Additional advantages of MCRs, such as being less time-consuming and facilitating diversity-oriented synthesis with good yields, have captured the attention of chemists, particularly for the synthesis of biologically active complex molecular structures. Therefore, the development of environmentally friendly and efficient MCRs is acknowledged as a critical topic in green chemistry.
Nitrogen-containing heterocyclic compounds have attracted significant interest due to their broad range of applications, especially in biomedicine. Within this context, isocyanide-based MCRs like Ugi-3CRs, Ugi-4CRs, and Passerini reactions have been employed to synthesize structurally diverse small molecule libraries.6 Among the wide spectrum of nitrogen heterocyclic compounds, imidazo[1,2-a]pyrazine and pyridines hold importance as a class of bridgehead heterocycles. Notable drug candidates within this class include zolpidem (1), alpidem (2), saripidem (3), olprinone (4), zolimidine (5), and ND-009628 (6) (Fig. 1). These compounds exhibit various pharmacological properties, such as sedative-hypnotics as well as antibacterial,7 antifungal,8 antiviral9–12 and anti-inflammatory13,14 activities. The inhibitory effects of imidazo[1,2-a]pyrazines against insulin-like growth factor-I receptor (IGF-IR),15 p13K,16 aurora kinase,17 and tyrosine kinase EphB4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 have garnered substantial attention. Recently, Li et al. research groups reported imidazo[1,2-a]pyrazines and its analogs shows inhibition of SARS-CoV and SARS-CoV-2 main protease with excellent activity with IC50 values as low as 21 nM.19 Consequently, numerous attempts have been made to synthesize these compounds.20,21
18 have garnered substantial attention. Recently, Li et al. research groups reported imidazo[1,2-a]pyrazines and its analogs shows inhibition of SARS-CoV and SARS-CoV-2 main protease with excellent activity with IC50 values as low as 21 nM.19 Consequently, numerous attempts have been made to synthesize these compounds.20,21
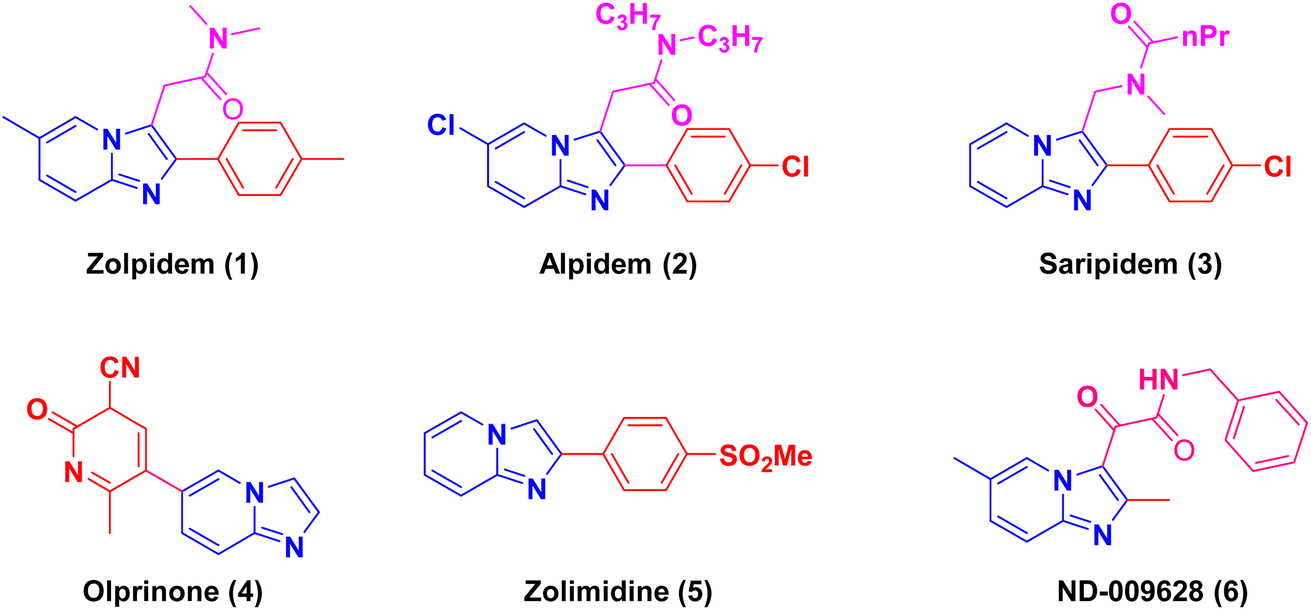 | ||
| Fig. 1 Examples of anticancer/antiviral/antibacterial activities of imidazo[1,2-a]pyridine molecules. | ||
Previously reported methods for synthesizing imidazo derivatives using Lewis acid catalysts like ZnCl2,22 Sc(OTf)3,23 MgCl2,24 and ZrCl425 suffer from long reaction times and low yields. As a result, the development of new methodologies offering cost-effective and eco-friendly procedures with high yields becomes both challenging and imperative. Within this context, we focused on a new methodology that utilizes tert-butyl isocyanide-based Ugi-type multicomponent reactions for the efficient preparation of a series of heterocyclic fluorophores with good yields. Iodine serves as a catalyst in the synthesis of imidazo[1,2-a]pyrazines and imidazo[1,2-a]pyridines, making it a viable choice in MCRs due to its low cost, easy availability, and benign nature.26 The three-component condensation involving tert-butyl isocyanide, an aryl aldehyde, and 2-aminopyridine or 2-aminopyrazine at room temperature leads to the formation of imidazo[1,2-a]pyrazine and imidazo[1,2-a]pyridine derivatives. Herein, we present this straightforward one-pot synthesis of imidazo[1,2-a]pyrazine and pyridine derivatives. The advantage of tert-butyl isocyanide-based multicomponent reactions at room temperature is emphasized. To the best of our knowledge, this is the first report where tert-butyl isocyanide, an aryl aldehyde, and 2-aminopyrazine or 2-aminopyridine, in the presence of iodine as a catalyst in ethanol, result in highly functionalized imidazo[1,2-a]pyrazine and pyridine derivatives with excellent yields. Henceforth in continuation of our efforts in medicinal chemistry research27 and considering the biological importance of the imidazo[1,2-a]pyrazine and imidazo[1,2-a]pyridine derivatives,28 the present work aimed to design, synthesis and biological activity of imidazo[1,2-a]pyrazine and imidazo[1,2-a]pyridine derivatives by in vitro anticancer studies.
Results and discussion
Chemistry
The low yield of the desired compounds in isocyanide-based multicomponent reactions is due to the decomposition of acid-sensitive 2-morpholinoethyl, p-methoxyphenyl, trimethylsilyl cyanides and tert-butyl isocyanide (at higher temperature).29 Thus, a pilot reaction involving 2-aminopyrazine 7, 4-nitrobenzaldehyde 8, and tert-butyl isocyanide 9 was conducted at room temperature for approximately 1 hour in both polar and non-polar solvents, employing 5 mol% of various Lewis acids such as ceric ammonium nitrate (CAN), SnCl4, SnCl2, InCl3, FeCl3, PTSA·H2O, and I2 as catalysts to evaluate their efficiency in terms of reaction yield and time (Scheme 1, Table 1). Initially, the reaction was carried out with different catalysts (entries 1–5; Table 1), resulting in moderate yields and however, product has to be isolate in conventional workup and column chromatography. The reaction with ferric chloride (FeCl3) (entry 6; Table 1) yielded poor results. On the other hand, the presence of 5 mol% of I2 (entry 7; Table 1) as a catalyst in ethanol solvent provided excellent yields of imidazo[1,2-a]pyrazines with high purity. The reaction was also tested in various solvents such as MeOH, H2O, ACN, DCM (entries 8–11: Table 1), and the non-polar solvent toluene (entry 12), yielding moderate to low yields. Among the catalysts tested, iodine proved to be cost-effective and eco-friendly for the synthesis of desired product 10a (Table 1). Further experimentation with 10 mol% iodine did not significantly improve the overall yield. The reaction did not proceed in the absence of iodine in ethanol (entry 13). Based on optimization results, the iodine–ethanol condition was found to be ideal for this three-component reaction, providing an excellent yield. Notably, under the iodine–ethanol condition, the product precipitated in the reaction vessel, simplifying purification through straightforward filtration. Hence, no additional purification techniques were required to obtain the pure desired product 10a. | ||
| Scheme 1 Schematic diagram of formation of imidazo[1,2-a]pyrazines. | ||
| Entry | Catalyst (5 mol%) | Solvent | Time (h) | Yieldb (%) |
|---|---|---|---|---|
| a The reaction was performed using 1.0 mmol each of 4-nitrobenzaldehyde, 2-aminopyrazine and tert-butyl isocyanide at room temperature.b Isolated yield. | ||||
| 1 | CAN | EtOH | 20 | 54 |
| 2 | SnCl4 | EtOH | 24 | 58 |
| 3 | SnCl2 | EtOH | 24 | 48 |
| 4 | InCl3 | EtOH | 24 | 55 |
| 5 | PTSA·H2O | EtOH | 24 | 75 |
| 6 | FeCl3 | EtOH | 24 | 25 |
| 7 | I2 | EtOH | 5 | 98 |
| 8 | I2 | MeOH | 15 | 85 |
| 9 | I2 | Water | 24 | 80 |
| 10 | I2 | ACN | 30 | 57 |
| 11 | I2 | DCM | 30 | 51 |
| 12 | I2 | Toluene | 30 | 25 |
| 13 | No catalyst | EtOH | 24 | — |
The desired product 10a was characterized by IR, 1H NMR, 13C NMR and ESI-HRMS analysis. FT-IR spectra analysis given that the N–H stretching of amine functional group was appeared strong peak at 3350 cm−1 and the UV spectra analysis gave broad absorbance spectrum at 368 nm to confirm that the amine group present in the molecule. The 1H NMR spectra of 10a revealed that NH proton was appeared at 3.08 ppm as singlet and the t-butyl group was observed at 1.09 as singlet. The aromatic pyrazine hydrogen was located at δ 9.03 as a singlet and the remaining two hydrogens were appeared at δ 8.32–8.30 ppm as doublets in pyrazine ring. The four aromatic hydrogens were located in multiplets at 8.30–7.91 ppm (ESI Fig. 1†). The 13C NMR spectrum of 10a displayed 12 distinct signals, including two aliphatic regions one at 30 ppm that proved t-butyl CH3 carbon and another carbon at 57 ppm that confirmed t-butyl quaternary carbon. Total of ten carbons with five CH and five quaternary carbons were appeared in aromatic regions at δ 147.21 to 116.30 ppm (ESI Fig. 2†). The purified imidazo[1,2-a]pyrazines 10a subjected to ESI-HRMS to identify the mass of product and mass calculated for C16H17N5O2:ESI-HRMS = 312.1460 which was found to be 312.1465 (M + H)+. After optimizing the reaction conditions, we synthesized different imidazo[1,2-a]pyrazine derivatives (10a–m) using tert-butyl isocyanide 9 and appropriate aryl aldehydes 8. In almost all the cases, the reaction proceeded well and the formations of the cyclized products were in moderate to good yields (ESI Fig. 1–24†). To further expand the scope of this methodology and the catalytic role of iodine, cyclohexyl isocyanide is also used for the synthesis of the corresponding imidazo[1,2-a]pyrazine derivatives (10a–m) (Scheme 2) in good yields.
 | ||
| Scheme 2 Library synthesis of imidazo[1,2-a]pyrazines derivatives (10a–m). | ||
To broaden the scope of this room temperature protocol, the one-pot reaction involving 2-aminopyridine 11, tert-butyl isocyanide 9 and different aryl aldehyde 8 was carried out. Iodine effectively catalyzed the cyclo-condensation of different aldehydes with 2-aminopyridine and tert-butyl isocyanide resulting in the corresponding 3-aminoimidazo[1,2-a]pyridines (12a–l) (Scheme 3) in good yields (ESI Fig. 25–50†). The HPLC spectrum analysis of the most potent compounds (10b and 12b) were carried out on reverse phase C18 column using CH3CN:H2O (50:50, 1:1) as a mobile phase and we obtained more than 98% purity of the samples (ESI Fig. 51 and 52†).
 | ||
| Scheme 3 Library synthesis of imidazo[1,2-a]pyridines derivatives (12a–l). | ||
After optimizing the reaction conditions, a plausible mechanism is presented in Scheme 4. In the initial step, condensation of 2-aminopyrazine 7 or 2-amino pyridine 11 with an aryl aldehyde 8 yields the formation of the imine ion A. This ion is subsequently activated by the Lewis acid iodine, facilitating the nucleophilic addition of tert-butyl isocyanide 9. This addition leads to the creation of the intermediate iminium ion B. The nucleophilic addition of tert-butyl isocyanide 9 to intermediate B undergoes [4 + 1] cycloaddition generates intermediate C. Finally, intramolecular interaction involving the nitrogen of the pyrazine or pyridine ring in intermediate C propels the entire reaction towards the formation of cyclized products imidazopyrazine 10 and imidazopyridine 12, as anticipated in Scheme 4.
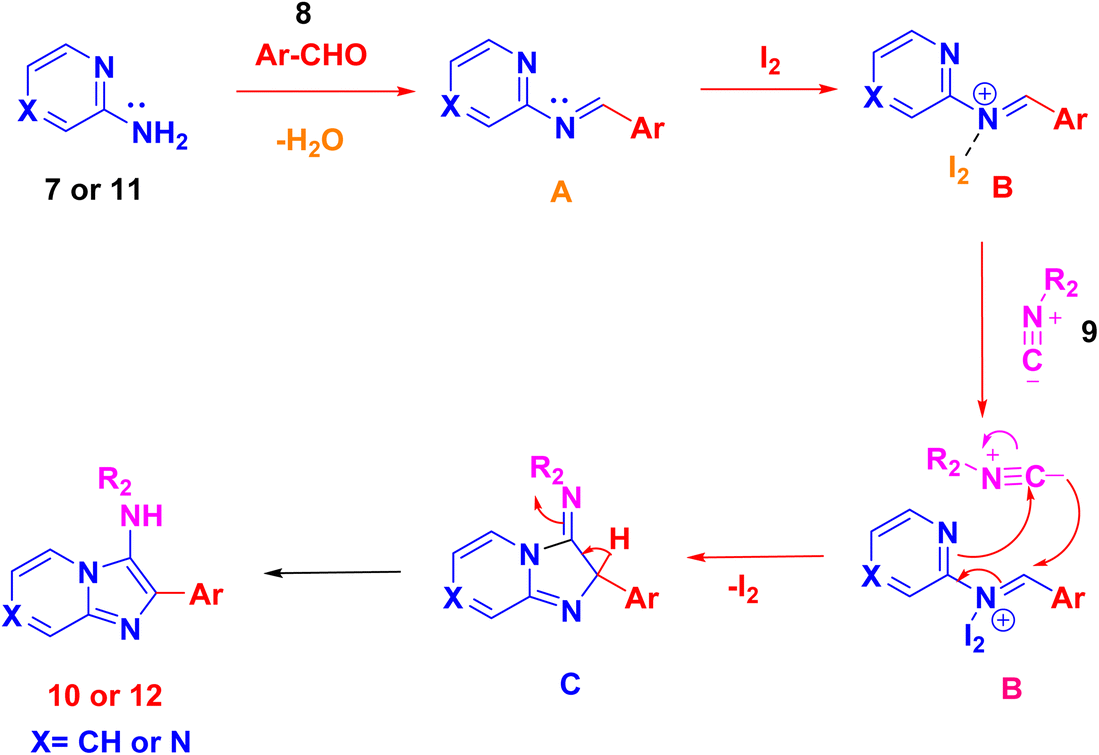 | ||
| Scheme 4 Plausible mechanism for formation of compounds (10 and 12). | ||
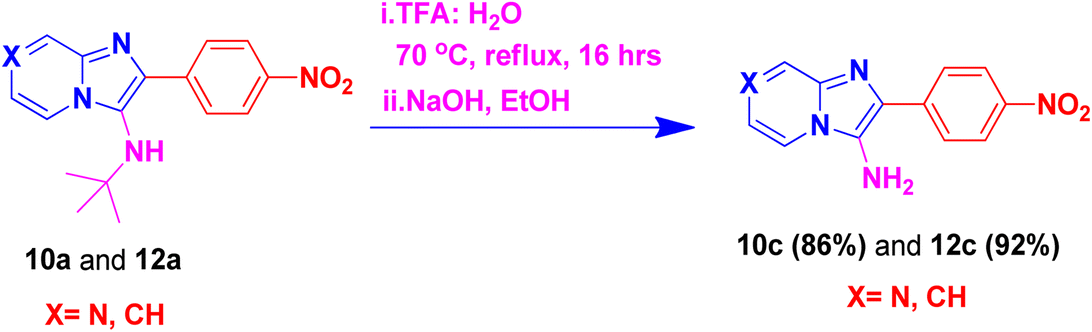
By using this methodology, we have synthesized nitro substituted imidazo[1,2-a]pyrazines (10a, 10c, 10e, 10j and 10l) and imidazo[1,2-a]pyridine derivatives (12a, 12c, 12e, 12g and 12k). According to the literature, the nitro group containing molecules act as both a pharmacophore as well as toxicophore and making it even much more fascinating for medicinal chemists.30 In most cases, the nitro group containing compounds were showed lesser anticancer activity and also affect fluorescent properties. In order to increase anticancer as well as fluorescent properties,31 we have carried out reduction process with those compounds using H2N–NH2, Pd/C afforded amine containing imidazo[1,2-a]pyrazines (10b, 10d, 10f, 10k and 10m) and imidazo[1,2-a]pyridine derivatives (12b, 12d, 12f, 12h and 12l) with good yields. Using TFA:H2O (9:1) and followed by adjust pH using NaOH with EtOH (to avoid TFA-amine formation) compounds 10a, 10b, 12a and 12b having the NH-attachment of t-butyl group was deprotected and gives 10c, 10d, 12c and 12d compounds.32 The general procedure for synthesizing derivatives (10b–d) and (12b–d) is presented in Scheme 5.
 | ||
| Scheme 5 General schematic line synthesis of compounds (10b–d) and (12b–d). | ||
Considering the significance of fluorescent molecules acting as chemosensors and biosensors.33–35 The absorption and fluorescence characteristics of all compounds were investigated in media of different polarity. Among the derivatives, 10b, 10i, 12b and 12i exhibited more favorable fluorescence emission spectra compared to the other compounds. Consequently, these specific compounds were chosen for further biological evaluation based on their recorded fluorescence spectra. Among the solvents tested, acetonitrile displayed the maximum blue shift in emission (Fig. 2). Therefore, acetonitrile was selected as the solvent for measuring the fluorescence characteristics of imidazo[1,2-a]pyrazine and pyridine derivatives 10b, 10i, 12b, and 12i. The absorbance and fluorescence characteristics are significantly influenced by the nature of functional groups (Fig. 2) present at the 2-position in the aryl ring of the imidazo[1,2-a]pyrazine and pyridine system. From this data, it can be observed that compound 10i (which has an additional nitrogen atom in the pyrazine ring compared to 12i's pyridine ring) exhibited the maximum emission at ∼850 nm, surpassing other derivatives such as 10b, 12b, and 12i.
 | ||
| Fig. 2 Fluorescence spectra of imidazo[1,2-a]pyrazine and pyridine derivatives 10b, 10i, 12b and 12i (1 μM) in acetonitrile. | ||
Anticancer activity
The in vitro anticancer properties of all synthesized imidazo[1,2-a]pyrazine (10a–m) imidazo[1,2-a]pyridine (12a–l) compounds were screened against four cancer cell lines such as laryngeal carcinoma cell line (Hep-2), hepatocellular carcinoma cell line (HepG2), human skin cancer cell line (A375) and breast cancer cell line (MCF-7) cell lines and the cytotoxicity to the non-cancerous normal cell (Vero) by MTT assay.36 The compounds were examined for in vitro cytotoxicity utilizing a panel of cancer cell lines and a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) reduction test for 48 hours. These findings prove the synthesized compounds have anti-proliferative properties. Among other compounds, 2-(4-aminophenyl)-N-(tert-butyl)imidazo[1,2-a]pyridin-3-amine (12b) was found to be most potent compound, with IC50 values of 11 μM (Hep-2), 13 μM (HepG2), 11 μM (MCF-7), 11 μM (A375) and 91 μM (Vero) cells lines, respectively. These values are almost comparable to the inhibitory value of the standard drug, Doxorubicin IC50 values of 10 μM (Hep-2), 1.5 μM (HepG2), 0.85 μM (MCF-7), 5.16 μM (A375) and 14 μM (Vero) cells lines (Table 2). From the interpretation of results, the structure–activity relationship (SAR) has been drawn and graphically represented in Fig. 4. It is evident that the central core, imidazo[1,2-a]pyrazine and pyridine is an essential requirement for displaying the anticancer activity. The first imidazo[1,2-a]pyrazine series (10a–m) involves the intact imidazopyrazine skeleton scaffold with variations of electron-donating functional amine groups viz. are NH2, cyclohexylamine, and tert-butylamine on the 3rd position of imidazo[1,2-a]pyrazine molecules. Among these molecules, the compound 10b has shown good IC50 at a μM concentration against all the cell lines 20 μM (Hep-2), 18 μM (HepG2), 21 μM (MCF-7), 16 μM (A375), 76 μM (Vero) respectively due to increasing order electron-donating nature of tert-butylamine when compared than cyclohexylamine 10k and parent amine 10d containing imidazo[1,2-a]pyrazine compounds. Similarly, 10i has retained decent activity against two cell lines with IC50 values of 17 μM (MCF-7) and 16 μM (A375), respectively, because of electron-donating groups such as tert-butylamine at 3rd and N,N-dimethyl aniline attached at 2nd positions of imidazo[1,2-a]pyrazine. Due to electron-donating nature of tert-butylamine at the 3rd position and amine located at the steric ortho position, the resulting molecule 10f was showed moderate activity with values of 25 μM (Hep-2), 20 μM (HepG2), 26 μM (MCF-7), 20 μM (A375) and 85 μM (Vero) respectively. Similar to the functional group differences, the compounds obtained from the structural dissimilarities on the 2nd position of imidazo[1,2-a]pyrazine molecules could not help to increase the activity. For example, the electron-withdrawing group nitro-substituted compounds 10e and 10l at meta positions of phenyl rings showed lesser anti-cancer activities and lost two folds its activity than nitro group substituted at para position compounds such as 10a, 10c and 10j owing to steric hindrance between the nitro group and imidazopyrazine ring. The compounds 10g and 10m showed significant inhibition against all the cells due to amine substitution at ortho position of aryl ring in imidazopyrazine ring. The second category imidazo[1,2-a]pyridine compounds (12a–l) involves both variations at the substitutions on the 3rd position and 2nd position of imidazo[1,2-a]pyridine and showed better anticancer activities similar like imidazo[1,2-a]pyrazine series (10a–m) owing to electron donating (amines) as well as electron withdrawing groups (NO2). It is interesting to notice that imidazo[1,2-a]pyridine compounds (12a–l) is found to have more significant anticancer activities than imidazo[1,2-a]pyrazine series (10a–m) with the removal of one nitrogen atom at 7th position of imidazole ring. Among both series (10a–m) and (12a–l), compound 12b, tertiary butylamine group at 2nd position and phenylamine group at 3rd position showed more substantial anticancer effects with IC50 values at 11 μM, 13 μM, 11 μM, 11 μM and 91 μM concentrations against Hep-2, HepG2, MCF-7, A375, and Vero cell lines respectively. Similarly, 12f, 12i, and 12l have displayed better inhibition against the Hep-2, HepG2, MCF-7, A375, and Vero respectively, because of electron donating groups present at 3rd and 2nd position of imidazopyridine ring. The electron withdrawing group bearing compounds 12c, 12e, and 12g exhibited lower inhibition IC50 values than electron donating containing compounds 12i, 12j, and 12l. The compound 12b more preferentially binds to cancer cells' outer membranes via electrostatic interfaces and kills the cancer cells from their IC50 value Table 2. Therefore, the cytotoxicity of cell viability demonstrates that all compounds (10b, 10i, 12b, and 12i) could be utilized to target kill cancer cells while being non-toxic to non-cancerous cells (Fig. 3).| S. no. | Compounds | In vitro studies | ||||
|---|---|---|---|---|---|---|
| Anticancer (IC50)b (μM) | ||||||
| Hep-2 | HepG2 | MCF-7 | A375 | Vero | ||
| a The cytotoxic effect of compounds against human tumor cell lines was determined by a rapid colorimetric assay, using MTT (5 mg ml−1) and compared with untreated controls.b Results are the average of three independent experiments. Hep2-laryngeal carcinoma; HepG2-hepatocellular carcinoma; MCF-7-breast carcinoma; A375-human skin cancer; non-cancerous normal Vero cell. Full detailed experimental procedure has been given in experimental section. | ||||||
| 1 | 10a | 50 ± 3.42 | 40 ± 2.52 | >100 | 36 ± 3.1 | 80 ± 5.72 |
| 2 | 10b | 20 ± 1.64 | 18 ± 1.32 | 21 ± 1.64 | 16 ± 0.96 | 76 ± 40 |
| 3 | 10c | 45 ± 1.6 | 30 ± 1.43 | 40 ± 3.8 | 24 ± 2.48 | 80 ± 6.72 |
| 4 | 10d | 38 ± 2.2 | 34 ± 2.64 | 30 ± 3.32 | 25 ± 1.45 | 96 ± 14 |
| 5 | 10e | 80 ± 1.37 | 40 ± 2.52 | 100 ± 8.64 | 34 ± 3.2 | >100 |
| 6 | 10f | 25 ± 5.2 | 20 ± 4.64 | 26 ± 2.38 | 20 ± 1.57 | 85 ± 0.22 |
| 7 | 10g | 22 ± 2.15 | 25 ± 4.54 | 30 ± 1.24 | 20 ± 5.8 | 78 ± 5.0 |
| 8 | 10h | 45 ± 0.5 | 34 ± 5.5 | 50 ± 6.41 | 30 ± 2.84 | >100 |
| 9 | 10i | 28 ± 3.1 | 26 ± 0.9 | 17 ± 0.52 | 16 ± 1.20 | 91 ± 38 |
| 10 | 10j | 44 ± 6.4 | 40 ± 1.4 | 24 ± 0.15 | 25 ± 5.41 | 64 ± 64 |
| 11 | 10k | 34 ± 4.56 | 28 ± 2.48 | 65 ± 2.41 | 30 ± 1.5 | 75 ± 2.8 |
| 12 | 10l | 64 ± 2.45 | 32 ± 4.64 | 80 ± 4.53 | 27 ± 5.0 | 67 ± 4.54 |
| 13 | 10m | 28 ± 4.28 | 20 ± 2.15 | 44 ± 2.68 | 23 ± 1.5 | 75 ± 2.8 |
| 14 | 12a | 32 ± 5.5 | 30 ± 2.5 | 55 ± 5.25 | 45 ± 2.5 | 85 ± 14 |
| 15 | 12b | 11 ± 0.8 | 13 ± 1.2 | 11 ± 0.5 | 11 ± 0.07 | 91 ± 45 |
| 16 | 12c | 35 ± 2.4 | 48 ± 4.8 | >100 | 50 ± 1.8 | 74 ± 58 |
| 17 | 12d | 20 ± 5.8 | 35 ± 2.5 | 50 ± 1.82 | 38 ± 2.4 | 88 ± 85 |
| 18 | 12e | 55 ± 0.4 | 62 ± 4.8 | >100 | >100 | 78 ± 28 |
| 19 | 12f | 28 ± 5.0 | 32 ± 0.1 | 24 ± 5.8 | 28 ± 1.5 | 90 ± 58 |
| 20 | 12g | 50 ± 5.2 | >100 | >100 | 62 ± 2.4 | 65 ± 14 |
| 21 | 12h | 44 ± 1.4 | 54 ± 1.5 | 61 ± 2.4 | 55 ± 1.5 | 74 ± 50 |
| 22 | 12i | 20 ± 2.7 | 19 ± 1.3 | 17 ± 0.92 | 12 ± 0.7 | 87 ± 50 |
| 23 | 12j | 45 ± 5.0 | 35 ± 4.8 | 30 ± 5.0 | 25 ± 1.4 | 70 ± 12 |
| 24 | 12k | 30 ± 8.0 | 40 ± 1.5 | 52 ± 0.8 | 41 ± 5.0 | 85 ± 02 |
| 25 | 12l | 20 ± 5.2 | 25 ± 0.2 | 35 ± 1.5 | 30 ± 0.1 | 90 ± 10 |
| 26 | Doxorubicin | 10 ± 0.5 | 1.5 ± 0.09 | 0.85 ± 0.02 | 5.1 ± 0.12 | 14 ± 10 |
 | ||
| Fig. 3 Cell viability assay (up to 100 μM): normal Vero cell and cancer cells (Hep-2, HepG2, A375, and MCF-7) showing no cytotoxic effects against normal cells. Scale bar = 20 micrometer. | ||
 | ||
| Fig. 4 The anticancer SAR studies of imidazo[1,2-a]pyrazine (10a–m) and imidazo[1,2-a]pyridine (12a–l) derivatives. | ||
Conclusions
In conclusion, we have successfully developed an iodine-catalyzed tert-butyl isocyanide based three-component condensation at room temperature for the first time. Use of iodine as a catalyst offers a cost-effective method for the synthesis of imidazo[1,2-a]pyrazine (10a–m) and imidazo[1,2-a]pyridine (12a–l) derivatives. Compared to tert-butyl isocyanide based three-component condensations at higher temperatures, the present methodology offers good yields at room temperature. Simple workup and short reaction time are the other desirable features of this methodology. The compounds 10b, 10i, 12b and 12i showed better fluorescence emission properties compared to other derivatives. The fluorescent compounds 10b, 10i, 12b and 12i also show significant anticancer activity against cancer cells. Interestingly, compound 12b was displayed promising more anticancer activities against the four cancer cells and normal cell, namely, Hep-2, HepG2, MCF-7, A375, and Vero and equated with positive control, Doxorubicin. The compound stability during in vivo animal experiments would be major concern when these molecules are considered to deliver small drug molecules. These are the limitations of the present study, and the same aspects are explored currently.Experimental section
General information
Ethanol (AR grade) and double distilled water were used in all experiments. All the materials for synthesis were purchased from commercial suppliers and used without further purification. Absorption spectra were recorded on SPECORD 200 PLUS and Cary 50 Bio UV-visible spectrophotometer. Fluorescence measurements were performed on a HITACHI F-4500 fluorescence spectrophotometer (Excitation wavelength 355 nm; Slit width 5 nm). NMR spectra were recorded using a Bruker Advance 400 MHz spectrometer operated at 400 MHz. ESI-HRMS spectrum was obtained on a Thermo exactive PE Sciex API3000 mass spectrometer. Further dilutions all measurements were carried out at room temperature. We have prepared 1 μM solutions for the experiments. Stock solutions (1 M) were prepared in de-ionized water. Melting points were determined in open glass capillary with a Stuart scientific SMP3 apparatus and are uncorrected. All the compounds were checked by FT-IR, 1H and 13C NMR and mass spectrometry (Thermo Finningan LCQ-deca XP-plus) equipped with an ESI source and an ion trap detector. Chemical shifts are reported in part per million (ppm). Thin layer chromatography (TLC) was carried out on 5Y 20 cm plates with a layer thickness of 0.25 mm (Merck Silica gel 60 F254).Materials and methods
:H2O (50:50, 1:1) as an elutant to check the purity of the samples.000 cells per well and incubated at 37 °C, 5% CO2, 95% air and relative humidity (100%) for 48 h prior to addition of compounds. After 48 hours, compounds at various concentrations were added and incubated at 37 °C, 5% CO2, 95% air and relative humidity (100%) for 48 hours. Triplicate was maintained and the medium containing without the sample were served as control. After 48 hours, 50 μL of MTT (5 mg ml−1) in triple distilled water was added to each well and incubated at 37 °C for 4 hours. The medium with MTT was then flicked off and the formed formazan crystals were solubilised in 100 μL of DMSO and then measured the absorbance at 570 nm using micro plate reader. The percentage cell inhibition was determined using the following formula and summarized in Table 2.| Cell inhibition (%) = 100 − absorbance (sample)/absorbance (control) × 100 |




N-(tert-Butyl)-2-(4-nitrophenyl)imidazo[1,2-a]pyrazin-3-amine (10a).
Orange solid (3.6 g, 98%); melting point 138–140 °C; FT-IR (KBr, cm−1): ν/cm 3326 (NH); 1H NMR (400 MHz, CDCl3; Me4Si) δH 9.03 (s, 1H, H-8, Py), 8.31 (d, J = 10.1 Hz, 4H, H-5 & H-6, Py, H-12 & H-12′, Ph), 8.14 (d, J = 4.4 Hz, 1H, H-11, Ph), 7.91 (d, J = 4.5 Hz, 1H, H-11′, Ph), 3.22 (brs, 1H, NH), 1.12 (s, 9H, Me). 13C NMR (100 MHz, CDCl3) δC 147.21 (C-13, C), 143.97 (C-8, CH), 140.82 (C-10, C), 139.57 (C-9, C), 137.59 (C-3, C), 129.29 (C-2, C), 128.71 (C-6, CH), 126.09 (C-11 & 11′, CH), 123.71 (C-12 & 12′, CH), 116.30 (C-5, CH), 57.30 (C-14, t-BuC), 30.50 (Me). ESI-HRMS: C16H17N5O2 requires 312.1460 (M + H); found 312.1465 and λmax (EtOH)/nm 360.

2-(4-Aminophenyl)-N-(tert-butyl)imidazo[1,2-a]pyrazin-3-amine (10b).
Yellowish green solid (3.0 g, 81%); HPLC purity 98%; melting point 165–167 °C; FT-IR (KBr, cm−1): ν/cm 3326 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.80 (dd, J = 16.1, 1.3 Hz, 1H, H-8, Py), 8.41–8.29 (m, 2H, H-6 & H-5, Py), 7.99 (d, J = 8.7 Hz, 1H, H-11, Ph), 7.85 (d, J = 8.6 Hz, 1H, H-11′, Ph), 7.78 (dd, J = 9.4, 4.6 Hz, 1H, H-12, Ph), 6.87 (d, J = 8.7 Hz, 1H, H-12′, Ph), 6.59 (d, J = 8.6 Hz, 2H, NH2), 4.61 (d, J = 28.0 Hz, 1H, NH), 0.97 (s, 9H, Me). 13C NMR (100 MHz, DMSO-d6) δC 152.18 (C-13, C), 148.95 (C-8, CH), 142.31(C-9, C), 142.24 (C-3, C), 141.91 (C-2, C), 129.30 (C-6, CH), 128.77 (C-11, CH), 125.81 (C-10, C), 122.29 (C-5, CH), 117.26 (C-12, CH), 56.51(C, t-Bu), 30.51 (Me). ESI-HRMS: C16H19N5 requires 282.1719 (M + H); found 282.1718 and λmax (EtOH)/nm 362.

2-(4-Nitrophenyl)imidazo[1,2-a]pyrazin-3-amine (10c).
Light brown solid (3.2 g, 86%); melting point 152–155 °C; FT-IR (KBr, cm-1): ν/cm 3330 (NH); 1H NMR (400 MHz, DMSO-d6) δH 9.40 (d, J = 1.4 Hz, 1H, H-8, Py), 8.85 (d, J = 1.4 Hz, 1H, H-5, Py), 8.58–8.48 (m, 1H, H-6, Py), 8.42–8.14 (m, 4H, Ph), 6.30 (s, 2H, NH2). 13C NMR (100 MHz, DMSO-d6) δC 145.56 (C-13, C), 143.67 (C-8, CH) 143.18 (C-10, C), 141.49 (C-9, C), 134.82 (C-3, C), 131.79 (C-2, C), 128.27 (C-6, CH), 126.91 (C-11 & 11′, CH), 125.43 (C-12 & 12′, CH), 123.96 (C-5, CH). ESI-HRMS: C12H9N5O2 requires 256.0834 (M + H); found 256.0830 and λmax (EtOH)/nm 425.

2-(4-Aminophenyl)imidazo[1,2-a]pyrazin-3-amine (10d).
Dark brown solid (2.9 g, 91%); melting point 174–177 °C; FT-IR (KBr, cm−1): ν/cm 3319 (NH); 1H NMR (400 MHz, DMSO-d6) δ 9.15 (s, 1H, H-8, Py), 8.73 (d, J = 8 Hz, 1H, H-6, Py), 8.11 (d, J = 8.2 Hz, 2H, H-12 & 12′, Ph), 7.80 (d, J = 3.8 Hz, 1H, H-5, Py), 7.57 (d, J = 8.4 Hz, 2H, H-11 & 11′, Ph), 7.57 (d, J = 8.4 Hz, 2H, NH2), 7.43 (d, J = 2.9 Hz, 2H, NH2); 13C NMR (100 MHz, DMSO-d6) δ 137.46 (C-13, C), 136.03 (C-8, CH), 133.52 (C-9, C), 132.20 (C-3, C), 132.10 (C-2, C), 131.45 (C-6, CH), 128.92 (C-11 & 11′, CH), 124.19 (C-10, C), 118.12 (C-5, CH), 116.76 (C-12 & 12′, CH). ESI-HRMS: C12H11N5 requires 226.1093 (M + H); found 226.1098 and λmax (EtOH)/nm 376.

N-(tert-Butyl)-2-(2-nitrophenyl)imidazo[1,2-a]pyrazin-3-amine (10e).
Pale yellow solid (3.5 g, 93%); melting point 156–159 °C; FT-IR (KBr, cm−1): ν/cm 3261 (NH); 1H NMR (400 MHz, CDCl3) δH 9.01 (d, J = 1.5 Hz, 1H, H-8, Py), 8.11 (dd, J = 4.6, 1.5 Hz, 1H, H-5, Py), 7.98 (dd, J = 8.1, 1.1 Hz, 1H, H-6, Py), 7.90 (d, J = 4.6 Hz, 1H, H-11, Ph), 7.79 (dd, J = 7.7, 1.4 Hz, 1H, H-12, Ph), 7.70 (td, J = 7.6, 1.3 Hz, 1H, H-12′, Ph), 7.56 (ddd, J = 8.1, 7.5, 1.5 Hz, 1H, H-13, Ph), 2.83 (sbr, 1H, NH), 0.97 (s, 9H, Me). 13C NMR (100 MHz, CDCl3) δC 149.35 (C-11, C), 143.86 (C-8, CH), 139.48 (C-9, C), 138.96 (C-3, C), 137.58 (C-12′, CH), 132.77 (C-2, C), 132.76 (C-11′, CH), 129.29 (C-6, CH), 129.18 (C-13, CH), 126.50 (C-12, CH), 126.12 (C-5, CH), 116.21 (C-10, C), 56.12 (t-Bu C), 30.10 (Me). ESI-HRMS: C16H17N5O2 312.1460 (M + H); found 312.1462 and λmax (EtOH)/nm 360.

2-(2-Aminophenyl)-N-(tert-Butyl)imidazo[1,2-a]pyrazin-3-amine (10f).
Greenish yellow solid (3.2 g, 90%); melting point 167–169 °C; FT-IR (KBr, cm−1): ν/cm 3360 (NH); 1H NMR (400 MHz, CDCl3) δH 8.97 (d, J = 1.4 Hz, 1H, H-8, Py), 8.16 (dd, J = 4.6, 1.4 Hz, 1H, H-5, Py), 7.85 (d, J = 4.6 Hz, 1H, H-6, Py), 7.47 (dd, J = 7.6, 1.5 Hz, 1H, H-11′, Ph), 7.27 (s, 1H, H-13, Ph), 7.19 (td, J = 8.0, 1.5 Hz, 1H, H-12, Ph), 6.82 (dd, J = 8.0, 0.8 Hz, 2H, H-12′, NH2), 0.97 (d, J = 3.1 Hz, 9H, Me), 13C NMR (101 MHz, CDCl3) δC 144.72 (C-11, C), 143.93 (C-8, CH), 137.42 (C-9, C), 132.70 (C-3, C), 130.43 (C-2, C), 129.47 (C-6, CH), 128.82 (C-5, CH), 126.26 (C-13, CH), 124.13 (C-11′, CH), 118.72 (C-12′, CH), 116.68 (C-12, CH), 116.29 (C-10, C), 57.13 (t-BuC), 29.38 (Me). ESI-HRMS: C16H19N5 requires 282.1719 (M + H); found 282.1715 and λmax (EtOH)/nm 362.

2-(2-Aminophenyl)imidazo[1,2-a]pyrazin-3-amine (10g).
Dark brown solid (2.7 g, 84%); melting point 176–179 °C; FT-IR (KBr, cm−1): ν/cm 3299 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.79 (d, J = 9.9 Hz, 1H, H-11′, Ph), 8.34 (t, J = 5.5 Hz, 1H, H-8, Py), 7.90 (dd, J = 20.5, 7.9 Hz, 1H, H-5, Py), 7.75(dd, J = 4.6, 2.2 Hz, 1H, H-6, Py), 7.57 (t, J = 8.2 Hz, 1H, H-13, Ph), 6.79 (d, J = 7.9 Hz, 1H, H-12, Ph), 6.65 (t, J = 7.4 Hz, 1H, H-12′, Ph), 6.04 (s, 2H, NH2), 5.69 (s, 2H, NH2). 13C NMR (101 MHz, DMSO-d6) δC 149.23 (C-11, C), 146.91 (C-8, CH), 143.01 (C-9, C), 141.75 (C-3, C), 132.80 (C-2, C), 131.33 (C-6, CH), 129.19 (C-13, CH), 128.77 (C-11′, CH), 128.54 (C-5, CH), 128.39 (C-12, CH), 124.71 (C-12′, CH), 116.46 (C-10, C). ESI-HRMS: C12H11N5 requires 226.1093 (M + H); found 226.1095 and λmax (EtOH)/nm 384.

N-(tert-Butyl)-2-(4-(dimethylamino)phenyl)imidazo[1,2-a]pyrazin-3-amine (10h).
Yellow solid (3.3 g, 90%); melting point 205–207 °C; FT-IR (KBr, cm−1):ν/cm 3257 (NH); 1H NMR (400 MHz, CDCl3) δH 8.95 (d, J = 6.9 Hz, 1H, H-8, Py), 8.11 (dd, J = 4.0, 4.0 Hz, 1H, H-6, Py), 7.82–7.81 (m, 3H, H-5, Py & H-11, H-11′, Ph), 6.80 (d, J = 9.0 Hz, 2H, H-12 & 12′, Ph), 3.02 (s, 6H, N(Me)2), 2.07 (br s, 1H, NH), 1.08 (s, 9H, Me). 13C NMR (101 MHz, CDCl3) δC 150.23 (C-13, C), 142.29 (C-8, CH), 142.61 (C-9, C), 137.18 (C-3, C), 129.02 (C-2, C), 128.68 (C-6, CH), 123.99 (C-11 & 11′, CH), 121.92 (C-10, C), 116.2 (C-5, CH), 112.03 (C-12 & 12′, CH), 56.85 (t-BuC), 40.36 (N(Me)2), 30.42 (Me). ESI-HRMS: C16H23N5 requires 310.2032 (M + H); found 310.2036 and λmax (EtOH)/nm 370.

2-(4-(Dimethylamino)phenyl)imidazo[1,2-a]pyrazin-3-amine (10i).
Pale Yellow solid (2.9 g, 87%); melting point 210–213 °C; FT-IR (KBr, cm−1): ν/cm 3312 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.88 (s, 1H, H-8, Py), 8.33 (dd, J = 17.5, 5.9 Hz, 1H, H-5, Py), 7.88 (d, J = 4.5 Hz, 3H, H-6, Py & H-11 & H-11′, Ph), 7.74–7.72 (m, 2H, H-12 & H-12′, Ph), 6.83 (d, J = 8.6 Hz, 2H, NH2), 2.98 (s, 6H, N(Me)2); 13C NMR (101 MHz, DMSO-d6) δC 151.16 (C-13, C), 140.34 (C-8. CH), 134.74 (C-9, C), 132.37 (C-3, C), 129.85 (C-2, C), 128.78 (C-6, CH), 119.46 (C-11 & C-11′, CH), 119.29 (C-10, C), 112.47 (C-12 & 12′, CH), 39.36 (N(Me)2); ESI-HRMS: C14H15N5 requires 254.1406 (M + H); found 254.1408 and λmax (EtOH)/nm 368.

N-Cyclohexyl-2-(4-nitrophenyl)imidazo[1,2-a]pyrazin-3-amine (10j).
Yellowish brown solid (3.6 g, 97%); melting point 154–156 °C; FT-IR (KBr, cm−1): ν/cm 3249 (NH); 1H NMR (400 MHz, CDCl3) δH 9.02 (s, 1H, H-8, Py), 8.32 (s, 4H, H-5 & 6, Py, H-12 & H-12′, Ph), 7.94 (dd, J = 38.3, 4.0 Hz, 2H, H-11 & H-11′, Ph), 3.21 (br s, 1H, NH), 3.01 (s, 1H, aliphatic CH), 1.81–1.60 (m, 5H, CH2), 1.30–1.12 (m, 5H, CH2). 13C NMR (101 MHz, CDCl3) δC 147.16 (C-13, C), 144.15 (C-10, C), 140.12 (C-8, CH), 137.12 (C-3, C), 136.48 (C-2, C), 129.40 (C-6, CH), 127.68 (C-11, CH), 127.54 (C-11′, CH), 123.98 (C-12 & 12′, CH), 115.51 (C-5, CH), 57.22 (aliphatic CH), 34.43 (CH2), 25.47 (CH2), 24.78 (CH2). ESI-HRMS: C18H19N5O2 requires 338.1617 (M + H); found 338.1615 and λmax (EtOH)/nm 364.

2-(4-Aminophenyl)-N-cyclohexylimidazo[1,2-a]pyrazin-3-amine (10k).
Yellowish brown solid (3.4 g, 94%); melting point 172–175 °C; FT-IR (KBr, cm−1): ν/cm 3249 (NH); 1H NMR (400 MHz, CDCl3) δH 8.93 (d, J = 1.4 Hz, 1H, H-8, Py), 7.98 (dd, J = 4.6, 1.4 Hz, 1H, H-5, Py), 7.82 (dd, J = 5.6, 2.9 Hz, 3H, H-6, Py, H-11 & H-11′, Ph), 6.80–6.66 (m, 2H, H-12 & H-12′, Ph), 6.50 (s, 2H, NH2), 3.21 (br s, 1H, NH), 3.01 (s, 1H, aliphatic CH), 1.75 (dd, J = 42.2, 7.9 Hz, 6H, CH2), 1.28–1.06 (m, 4H, CH2). 13C NMR (101 MHz, CDCl3) δC 146.165 (C-13, C), 144.25 (C-8, CH), 141.16 (C-3, C), 136.61 (C-2, C), 128.79 (C-6, CH), 128.49 (C-11 & C-13′, CH), 125.39 (C-10, C), 123.75 (C-5, CH), 115.51 (C-12 & C-12′, CH), 56.77 (aliphatic CH), 34.28 (CH2), 29.69 (CH2), 25.63 (CH2), 24.79 (CH2). ESI-HRMS: C18H21N5 requires 308.1875 (M + H); found 308.1878 and λmax (EtOH)/nm 390.

2-(2-Nitrophenyl)imidazo[1,2-a]pyrazin-3-amine (10l).
Dark brown solid (3.2 g, 86%); melting point 159–161 °C; FT-IR (KBr, cm−1): ν/cm 3208 (NH); 1H NMR (400 MHz, CDCl3) 1H NMR (400 MHz, CDCl3) δH 8.77 (s, 1H, H-8, Py), 8.10 (s, 2H, H-5 & 6, Py), 7.79 (dd, J = 67.9, 41.3 Hz, 4H, H-11, H-12, H-12′ & H-13, Ph), 3.69 (br s, 1H, NH), 2.72 (s, 1H, aliphatic CH), 1.68–1.38 (m, 6H, CH2), 1.19–0.71 (m, 4H, CH2). 13C NMR (101 MHz, CDCl3) δC 148.19 (C-11, C), 136.98 (C-8, CH), 134.11 (C-9, C), 133.62 (C-3, C), 132.37 (C-12′, CH), 131.38 (C-2, C), 127.86 (C-11′, CH), 125.39 (C-6, CH), 125.14 (C-13, CH), 122.65 (C-10, C), 121.46 (C-12, CH), 116.72 (C-5, CH), 56.43 (aliphatic CH), 33.52(CH2), 25.26 (CH2), 24.48 (CH2). ESI-HRMS: C16H17N5O2 requires 338.1617 (M + H); found 338.1614 and λmax (EtOH)/nm 390.

2-(2-Aminophenyl)-N-cyclohexylimidazo[1,2-a]pyrazin-3-amine (10m).
Dark brown solid (2.6 g, 80%); melting point 178–181 °C; FT-IR (KBr, cm−1): ν/cm 3299 (NH); 1H NMR (400 MHz, CDCl3) δH 8.12 (dd, J = 6.8, 1.2 Hz, 1H, H-11′, Ph), 7.56–7.49 (m, 2H, H-8 & H-5, Py), 7.31 (d, J = 2.4 Hz, 1H, H-6, Py), 7.19–7.12 (m, 2H, H-13 & H-12, Ph), 6.86–6.77 (m, 3H, H-12′, Ph, NH2), 3.69 (br s, NH), 2.12 (s, 1H, aliphatic CH), 1.65 (dd, J = 71.4, 23.8 Hz, 6H, CH2), 1.12 (d, J = 7.3 Hz, 4H, CH2). 13C NMR (101 MHz, CDCl3) δC 145.14 (C-11, C), 141.15 (C-8, CH), 135.01 (C-9, C), 129.59 (C-3, C), 128.57 (C-2, C), 125.78 (C-6, CH), 123.72 (C-13, CH), 122.53 (C-11′, CH), 119.25 (C-5, CH), 118.18 (C-12, CH), 116.88 (C-12′, CH), 116.51 (C-10, C), 56.01 (aliphatic CH), 33.91 (CH2), 25.64 (CH2), 24.73 (CH2). ESI-HRMS: C18H21N5 requires 308.1875 (M + H); found 308.1870 and λmax (EtOH)/nm 384.

N-(tert-Butyl)-2-(4-nitrophenyl)imidazo[1,2-a]pyridin-3-amine (12a).
Yellowish brown solid (3.5 g, 93%); melting point 140–142 °C; FT-IR (KBr, cm−1): ν/cm 3350 (NH); 1H NMR (400 MHz, CDCl3) δH 8.25 (s, 4H, H-12 & H-12′, H-11 & H-12′, Ph), 8.18 (d, J = 6.9 Hz, 1H, H-5, Py), 7.55 (d, J = 9.0 Hz, 1H, H-8, Py), 7.23–7.15 (m, 1H, H-7, Py), 6.82 (t, J = 6.8 Hz, 1H, H-6, Py), 3.08 (br s, 1H, NH), 1.09 (s, 9H, Me). 13C NMR (101 MHz, CDCl3) δC 146.62 (C-13, C), 142.54 (C-9, C), 141.99 (C-10, C), 137.08 (C-3, C), 128.39 (C-2, C), 125.01 (C-7, CH), 123.54 (C-5, CH), 123.41 (C-11 & C-11′, CH), 117.70 (C-12 & C-12′, CH), 114.56 (112.02, C-8, CH), (C-6, CH), 56.79 (aliphatic CH), 30.48 (Me). ESI-HRMS: C16H17N5O2 requires 311.1508 (M + H); found 311.1500 and λmax (EtOH)/nm 368.

2-(4-Aminophenyl)-N-(tert-butyl)imidazo[1,2-a]pyridin-3-amine (12b).
Pale yellowish brown solid (3.2 g, 91%); HPLC purity 98%; melting point 160–163 °C; FT-IR (KBr, cm−1): ν/cm 3258 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.42 (d, J = 6.9 Hz, 1H, H-5, Py), 7.73 (dd, J = 7.7, 1.3 Hz, 2H, H-11 & H-11′, Ph), 7.47 (d, J = 9.1 Hz, 1H, H-8, Py), 7.24–7.16 (m, 1H, H-7, Py), 7.09–6.72 (m, 3H, H-12 & H-12′, Ph, H-6, Py) 3.26 (brs, 3H, NH, NH2), 0.93 (s, 9H, Me). 13C NMR (101 MHz, DMSO-d6) δC 146.86 (C-13, C), 141.08 (C-9, C), 130.90 (C-3, C), 128.50 (C-2, C), 124.18 (C-11 & C-11′, CH), 124.12 (C-7, CH), 124.09 (C-5, CH), 119.78 (C-12 & C-12′, CH), 116.77 (C-6, CH), 116.20 (C-8, CH), 56.04 (aliphatic CH), 30.31 (Me). ESI-HRMS: C17H20N4 requires 281.1766 (M + H); found 281.1760 and λmax (EtOH)/nm 360.

2-(4-Nitrophenyl)imidazo[1,2-a]pyridin-3-amine (12c).
Light brown solid (3.2 g, 92%); melting point 154–156 °C; FT-IR (KBr, cm−1): ν/cm 3208 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.86 (d, J = 29.0 Hz, 1H, H-5, Py), 8.22 (d, J = 7.3 Hz, 1H, H-12, Ph), 8.00 (t, J = 11.1 Hz, 1H, H-12′, Ph), 7.96–7.85 (m, 2H, H-11 & H-11′, Ph), 7.69 (d, J = 7.6 Hz, 1H, H-8, Py), 7.47 (t, J = 9.2 Hz, 2H, H-7 & H-6, Py), 6.57 (s, 2H, NH2). 13C NMR (101 MHz, DMSO-d6) δC 157.71 (C-13, C), 157.33 (C-9, C), 147.22 (C-10, C), 143.21 (C-3, C), 139.36 (C-2, C), 127.85 (C-12 & C-12′, CH), 127.59 (C-7, CH), 124.48 (C-5, CH), 117.52 (C-11 & C-11′, CH), 114.75 (C-6, CH), 114.15 (C-8, CH), ESI-HRMS: C13H10N4O2 requires 255.0800 (M + H); found 255.0807 and λmax (EtOH)/nm 332.

2-(4-Aminophenyl)imidazo[1,2-a]pyridin-3-amine (12d).
Dark brown solid (2.8 g, 87%); melting point 170–172 °C; FT-IR (KBr, cm−1): ν/cm 3400 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.86 (d, J = 29.0 Hz, 1H, H-8, Py), 8.22 (d, J = 7.3 Hz, 1H, H-5, Py), 8.00 (t, J = 11.1 Hz, 2H, H-6 & H-7, Py), 7.96–7.85 (m, 2H, H-11 & H-11′, Ph), 7.69 (d, J = 7.6 Hz, 2H, H-12 & H-12′, Ph), 7.01 (t, J = 9.2 Hz, 2H, NH2), 6.99–6.57 (d, J = 8.4 Hz, 2H, NH2). 13C NMR (101 MHz, DMSO-d6) δC 144.46 (C-13, C), 136.11 (C-9, C), 135.72 (C-3, C), 135.27 (C-2, C), 131.72 (C-11 & C-11′, CH), 128.13 (C-7, CH), 125.11 (C-7, CH), 116.51 (C-5, CH), 113.92(C-10, C), 112.52 (C-12 & C-12′, CH), 112.05 (C-6, CH). ESI-HRMS: C13H12N4 requires 225.1140 (M + H); found 225.1146 and λmax (EtOH)/nm 314.

N-(tert-Butyl)-2-(2-nitrophenyl)imidazo[1,2-a]pyridin-3-amine (12e).
Dark Reddish brown solid (3.5 g, 94%); melting point 156–159 °C; FT-IR (KBr, cm−1): ν/cm 3249 (NH); 1H NMR (400 MHz, CDCl3) δH 8.19 (d, J = 6.9 Hz, 1H, H-5, Py), 7.91 (dd, J = 8.1, 0.7 Hz, 1H, H-11′, Ph), 7.81 (dd, J = 7.7, 1.4 Hz, 1H, H-12, Ph), 7.66 (t, J = 7.6 Hz, 1H, C-12′, Ph), 7.57–7.47 (m, 2H, H-13 & H-8, Py), 7.21–7.13 (m, 1H, H-7, Py), 6.81 (t, J = 6.8 Hz, 1H, H-6, Py), 2.74 (br s, 1H, NH), 0.92 (s, 9H, Me). 13C NMR (101 MHz, CDCl3) δC 149.47 (C-11, C), 142.42 (C-9, C), 136.17 (C-3, C), 132.83 (C-2, C), 132.43 (C-12′, CH), 130.27 (C-11′, CH), 128.40 (C-13, CH), 124.68 (C-10, C), 124.27 (C-7, CH), 123.34 (C-5, CH), 117.70 (C-8, CH), 111.80 (C-6, CH), 55.63 (t-Bu C), 30.05 (Me). ESI-HRMS: C17H18N4O2 requires 311.1508 (M + H); found 311.1505 and λmax (EtOH)/nm 396.

2-(2-Aminophenyl)-N-(tert-butyl)imidazo[1,2-a]pyridin-3-amine (12f).
Pale brown solid (2.8 g, 80%); melting point 176–179 °C; FT-IR (KBr, cm−1): ν/cm 3258 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.42 (d, J = 6.9 Hz, 1H, H-11′, Ph), 7.73 (dd, J = 7.7, 1.3 Hz, 1H, H-5, Py), 7.47 (d, J = 9.1 Hz, 1H, H-8, Py), 7.24–7.16 (m, 1H, H-13, Ph), 6.90 (d, J = 6.6 Hz, 2H, H-7 & H-6, Py), 6.74 (d, J = 7.4 Hz, 1H, H-12′, Ph), 6.61 (d, J = 6.2 Hz, 1H, H-12, Ph), 4.60 (s, 2H, NH2), 3.25(s br NH), 0.93 (s, 9H, Me). 13C NMR (101 MHz, DMSO-d6) δC 146.86 (C-11, C), 141.08 (C-9, C), 130.90 (C-3, C), 128.50 (C-2, C), 124.18 (C-5, CH), 124.12 (C-7, CH), 124.09 (C-5, CH), 119.78 (C-11′, CH), 116.77 (C-12 & C-12′, CH), 116.20 (C-6, CH), 116.10 (C-8, CH), 111.46 (C-10, C), 56.04 (t-Bu C), 32.01 (Me). ESI-HRMS: C17H20N4 requires 281.1760 (M + H); found 281.1764 and λmax (EtOH)/nm 362.

2-(2-Nitrophenyl)imidazo[1,2-a]pyridin-3-amine (12g).
Dark brown solid (3.0 g, 85%); melting point 164–167 °C; FT-IR (KBr, cm−1): ν/cm 3330 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.42 (d, J = 6.9 Hz, 1H, H-5, Py), 7.73 (dd, J = 7.7, 1.3 Hz, 1H, H-11′, Ph), 7.47 (d, J = 9.1 Hz, 1H, H-12, Ph), 7.21 (dd, J = 16.9, 2.4 Hz, 2H, H-12′ & H-13, Ph), 7.01 (d, J = 7.0 Hz, 1H, H-8, Py), 6.90 (d, J = 6.6 Hz, 1H, H-7, Py), 6.74 (d, J = 7.4 Hz, 1H, H-6, Py), 6.61 (br s, 1H, NH2). 13C NMR (101 MHz, DMSO-d6) δC 148.10 (C-11′, C), 135.34 (C-9, C), 134.72 (C-3, C), 134.14 (C-2, C), 131.53 (C-12′, CH), 131.38 (C-11′, CH), 130.35 (C-13, CH), 125.98 (C-7, CH), 125.17 (C-5, CH), 122.50 (C-10, C), 116.31 (C-12, CH), 112.43 (C-8, CH), 111.39 (C-6, CH). ESI-HRMS: C13H10N4O2 requires 255.0882 (M + H); found 255.0888 and λmax (EtOH)/nm 363.

2-(2-Aminophenyl)imidazo[1,2-a]pyridin-3-amine (12h).
Dark reddish brown solid (2.5 g, 83%); melting point 179–181 °C; FT-IR (KBr, cm−1): ν/cm 3336 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.50 (d, J = 6.8 Hz, 1H, C-11′, Ph), 8.23 (d, J = 7.1 Hz, 1H, H-5, Py), 7.94 (d, J = 9.0 Hz, 1H, H-8, Py), 7.80 (d, J = 7.4 Hz, 1H, H-13, Ph), 7.61 (s, 1H, H-7, Py), 7.38–7.31 (m, 2H, H-12′ & H-12, Ph), 7.17 (dd, J = 17.8, 9.3 Hz, 1H, H-6, Py), 7.07–6.99 (m, 2H, NH2), 3.40 (s, NH2). 13C NMR (101 MHz, DMSO-d6) δC 143.93 (C-11′, C), 141.94 (C-9, C), 130.35 (C-3, C), 129.84 (C-2, C), 125.88 (C-13, CH), 125.55 (C-5, CH), 125.32 (C-7, CH), 124.47 (C-11′, CH), 122.48 (C-12, CH), 120.93 (C-12′, CH), 120.54 (C-6, CH), 115.20 (C-8, CH), 110.56 (C-10, C). ESI-HRMS: C13H12N4 requires 225.1140 (M + H); found 225.1145 and λmax (EtOH)/nm 366.

N-(tert-Butyl)-2-(4-(dimethylamino)phenyl)imidazo[1,2-a]pyridin-3-amine (12i).
White solid (3.4 g, 92%); melting point 206–208 °C; FT-IR (KBr, cm−1): ν/cm 3280 (NH); 1H NMR (400 MHz, CDCl3) δH 8.23 (d, J = 6.9 Hz, 1H, H-5, Py), 7.87–7.81 (m, 2H, H-11 & H-11′, Ph), 7.56 (d, J = 9.0 Hz, 1H, H-7, Py), 7.13 (ddd, J = 8.9, 6.7, 1.3 Hz, 1H, H-8, Py), 6.83–6.74 (m, 3H, H-6, Py, H-12 & H-12′, Ph), 3.41 (br s, 1H, NH), 3.02 (s, 6H, N(Me)2), 1.08 (s, 9H, Me). 13C NMR (101 MHz, CDCl3) δC 149.87 (C-13, C), 141.67 (C-9, C), 139.52 (C-3, C), 128.96 (C-2, C), 123.91 (C-11 & C-11′, CH), 123.38 (C-5, CH), 122.25 (C-7, CH), 116.72 (C-10, C), 112.15 (C-6, CH), 111.21 (C-8, CH), (C-12 & C-12′, CH), 56.37 (t-Bu C), 40.52 (N(Me)2), 30.43 (Me). ESI-HRMS: C19H24N4 requires 309.2079 (M + H); found 309.2070 and λmax (EtOH)/nm 358.

2-(4-(Dimethylamino)phenyl)imidazo[1,2-a]pyridin-3-amine (12j).
Pale white solid (3.0 g, 88%); melting point 214–217 °C; FT-IR (KBr, cm−1): ν/cm 3385 (NH); 1H NMR (400 MHz, DMSO-d6) δH 8.18 (d, J = 6.7 Hz, 1H, H-5, Py), 7.75 (d, J = 8.7 Hz, 2H, H-11 & H-11′, Ph), 7.61 (d, J = 9.0 Hz, 1H, H-8, Py), 7.39–7.31 (m, 1H, H-7, Py), 6.99 (t, J = 6.6 Hz, 1H, H-6, Py), 6.81 (d, J = 8.8 Hz, 2H, H-12 & H-12′, Ph), 3.45 (br s, NH2), 2.95 (s, 6H, N(Me)2). 13C NMR (101 MHz, DMSO-d6) δC 157.75 (C-13, C), 157.11 (C-9, C), 150.62 (C-3, C), 141.78 (C-2, C), 127.89 (C-11 & C-11′, CH), 125.88 (C-5, CH), 123.90 (C-7, CH), 120.66 (C-10, C), 116.87(C-8, CH), 112.88 (C-6, CH), 112.54 (C-12 & C-12′, CH), 31.04 (N(Me)2). ESI-HRMS: C15H16N4 requires 253.1453 (M + H); found 253.1456 and λmax (EtOH)/nm 358.

N-Cyclohexyl-2-(4-nitrophenyl)imidazo[1,2-a]pyridin-3-amine (12k).
Orange solid (3.3 g, 90%); melting point 237–240 °C; FT-IR (KBr, cm−1): ν/cm 3290 (NH); 1H NMR (400 MHz, CDCl3) δH 8.32 (dd, J = 9.1, 4.2 Hz, 1H, H-5, Py), 8.05 (d, J = 6.9 Hz, 2H, H-12 & H-12′, Ph), 7.54 (d, J = 9.1 Hz, 2H, H-11 & H-11′, Ph), 7.22–7.14 (m, 2H, H-8 & H-7, Py), 6.83 (d, J = 6.7 Hz, 1H, H-6, Py), 3.11 (s br, 1H, NH), 2.97 (td, J = 10.0, 5.6 Hz, 1H, aliphatic CH), 2.16 (s, 2H, CH2), 1.88–1.54 (m, 4H, CH2), 1.22 (dt, J = 18.4, 9.5 Hz, 4H, CH2). 13C NMR (101 MHz, CDCl3) δC 146.50 (C-13, C), 142.09 (C-9, C), 141.19 (C-10, C), 134.41 (C-3, C), 127.23 (C-2, C), 126.55 (C-11 & C-11′, CH), 124.86 (C-5, CH), 123.78 (C-7, CH), 122.70 (C-12 & C-12′, CH), 117.85 (C-6, CH), 112.23 (C-8, CH), 57.08 (aliphatic CH), 34.30 (CH2), 25.61 (CH2), 24.82 (CH2). ESI-HRMS: C19H20N4O2 requires 337.1665 (M + H); found 337.1668 and λmax (EtOH)/nm 370.

2-(4-Aminophenyl)-N-cyclohexylimidazo[1,2-a]pyridin-3-amine (12l).
Reddish brown solid (2.8 g, 84%); melting point 248–251 °C; FT-IR (KBr, cm−1): ν/cm 3350 (NH); 1H NMR (400 MHz, CDCl3) δH 8.09 (d, J = 6.7 Hz, 1H, H-5, Py), 7.92 (d, J = 8.3 Hz, 1H, H-8, Py), 7.84 (d, J = 8.3 Hz, 2H, H-11 & H-12′, Ph), 7.50 (d, J = 8.8 Hz, 1H, H-7, Py), 7.08 (d, J = 8.1 Hz, 1H, H-6, Py), 6.77 (d, J = 7.1 Hz, 2H, H-11 & H-11′, Ph), 3.81 (sbr, 2H, NH2), 3.10 (dd, J = 13.6, 4.3, 1H, NH), 3.07–2.95 (m, 1H, aliphatic CH), 1.71 (dd, J = 71.0, 25.7 Hz, 5H), 1.19 (dt = J 18.9, 9.0 Hz, 5H, CH2) 13C NMR (101 MHz, CDCl3) δC 145.68 (C-13, C), 141.33 (C-9, C), 137.17 (C-3, C), 128.19 (C-2, C), 127.75 (C-11 & C-11′, CH), 124.96 (C-10, C), 123.45 (C-5, CH), 122.80 (C-7, CH), 115.10 (C-6, CH), 114.06 (C-8, CH), 111.24 (C-12 & C-12′, CH), 56.82 (aliphatic CH), 34.15 (CH2), 25.76 (CH2), 24.82 (CH2). ESI-HRMS: C19H22N4 requires 307.1923 (M + H); found 307.1928 and λmax (EtOH)/nm 348.

Author contributions
A. P.: supervision, formal analysis, original draft-review & editing. R. K.: conceptualization, methodology, formal analysis, writing.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
The authors heartily thank to Dr S. Thennarasu, Chief Scientist and Head, CSIR-Central Leather Research Institute (CSIR-CLRI), Adyar, Chennai, Tamilnadu-600020, India for providing research support and Central Instrumentation Facilities (CIF) for characterizing the compounds. We heartfelt thank to Prof. USN Murty, Director of the National Institute of Pharmaceutical Education and Research Guwahati (NIPER-G) for excellent support. Authors also thankful to Department of Biotechnology under the Centre of GMP extraction facility for necessary support.References
- C. B. Blackadar, World J. Clin. Oncol., 2016, 7, 54–86 CrossRef PubMed.
- P. S. Roy and B. J. Saikia, Indian J. Cancer, 2016, 53, 441–442 CrossRef CAS PubMed.
- W. Report, WHO Cancer Report, 2019 Search PubMed.
- S. Srivastava, E. J. Koay, A. D. Borowsky, A. M. Marzo, S. Ghosh, P. D. Wagner and B. S. Kramer, Nat. Rev. Cancer, 2019, 19, 349–358 CrossRef CAS PubMed.
- (a) A. Domling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed; (b) H. S. P. Rao and A. Parthiban, Org. Biomol. Chem., 2014, 12, 6223–6238 RSC; (c) A. Parthiban and P. Makam, RSC Adv., 2022, 12, 29253–29290 RSC.
- (a) Y. Tanaka, T. Hasui and M. Suginome, Org. Lett., 2007, 9, 4407–4410 CrossRef CAS PubMed; (b) L. EI Kaim, L. Grimaud and J. Oble, Angew. Chem., Int. Ed., 2005, 44, 7961–7964 CrossRef PubMed; (c) P. R. Andreana, C. C. Liu and S. L. Schreiber, Org. Lett., 2004, 6, 4231–4233 CrossRef CAS PubMed.
- (a) N. M. Shukla, D. B. Salunke, E. Yoo, C. A. Mutz, R. Balakrishna and S. A. Davi, Bioorg. Med. Chem., 2012, 20, 5850–5863 CrossRef CAS PubMed; (b) M. H. Fischer and A. Lusi, J. Med. Chem., 1972, 15, 982–985 CrossRef PubMed.
- M. H. Fischer and A. Lusi, J. Med. Chem., 1972, 15, 982–985 CrossRef PubMed.
- K. S. Gudmundsson, J. D. Williams, J. C. Drach and L. B. Townsend, J. Med. Chem., 2003, 46, 1449–1455 CrossRef CAS PubMed.
- C. Hamdouchi, J. D. Blas, M. Del Prado, J. Gruber, B. A. Heinz and L. Vance, J. Med. Chem., 1999, 42, 50–53 CrossRef CAS PubMed.
- M. Lhassani, O. Chavignon, J. M. Chezal, J. C. Teulade, J. P. Chapat, R. Snoeck, G. Andrei, J. Balzarini, E. D. Clercq and A. Gueiffer, Eur. J. Med. Chem., 1999, 34, 271–275 CrossRef CAS.
- J. B. Veron, H. Allouchi, C. Enguedhar Gueiffier, R. Snoeck, G. Andrei, E. D. Clercq and A. Gueiffier, Bioorg. Med. Chem., 2008, 16, 9536–9545 CrossRef CAS PubMed.
- (a) L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305–312 CrossRef CAS PubMed; (b) Y. K. M. Flores, M. E. C. Aldrete, H. S. Zamora, J. C. Basurto and M. Camargo, Med. Chem. Res., 2012, 21, 775–782 CrossRef.
- M. G. Rimoli, L. Avallone, P. Caprariis, E. Luraschi, W. Filippelli, L. Berrino and F. Rossi, Eur. J. Med. Chem., 1997, 32, 195–203 CrossRef CAS.
- M. J. Mulvihill, J. Qun Sheng, H. R. Coate, A. Cooke, H. Dong, L. Feng, K. Foreman, R. F. Maryland, A. Honda, G. Mak, K. M. Mulvihill, A. I. Nigro, M. O. Connor, C. Pirrit, A. G. Steinig, S. Kam, K. M. Stolz, S. Yingchuan, P. A. R. Tavares, Y. Yao and N. W. Gibson, Bioorg. Med. Chem., 2008, 16, 1359–1375 CrossRef CAS PubMed.
- S. M. Gonzalez, A. I. Hernandez, C. Varela, R. A. Sonsoles, R. M. Alvarez, A. B. Garcia, M. Lorenzo, M. Rivero, J. Oyarzabal, R. Obdulia, J. R. Bischoff, M. Albarran, A. Cebria, P. Alfonso, W. Link, J. Fominaya and J. Pastor, Bioorg. Med. Chem. Lett., 2012, 22, 1874–1878 CrossRef PubMed.
- D. B. Belanger, M. J. Williams, P. J. Curran, A. K. Mandal, Z. Meng, M. P. Rainka, T. Yu, N. Y. Shih, M. A. Siddiqui, M. Liu, S. Tevar, S. Lee, L. Lianzhu, K. Gray, B. Yareko, J. Jones, E. B. Smith, D. B. Prelusky and A. D. Basso, Bioorg. Med. Chem. Lett., 2010, 20, 6739–6743 CrossRef CAS PubMed.
- S. A. Mitchell, M. D. Danca, P. A. Blomgren, J. W. Darrow, K. S. Currie, J. E. Kropf, S. H. Lee, S. L. Gallion, J. M. Xiong, D. A. Pippin, R. W. Desimone, D. R. Brittelli, D. C. Eustice, A. Bourret, M. Hill-Dezewi, P. M. Maciejewski and L. L. Elkin, Bioorg. Med. Chem. Lett., 2009, 19, 6991–6995 CrossRef CAS PubMed.
- X. Li and Y. Song, Eur. J. Med. Chem., 2023, 260, 115772 CrossRef CAS PubMed.
- (a) L. Yimin and W. Zhang, QSAR Comb. Sci., 2004, 23, 827–835 CrossRef; (b) A. Shaabani, E. Soleimani and J. Moghimi Rad, Synth. Commun., 2008, 38, 1090–1095 CrossRef CAS; (c) M. I. Sarah, T. Heather and W. Mark, Tetrahedron Lett., 2003, 44, 4369–4371 CrossRef.
- (a) S. K. Guchhait, C. Madaan and B. S. Thakkar, Synthesis, 2009, 3293–3300 CrossRef CAS; (b) S. Guchhait and C. Madaan, Synlett, 2009, 4, 628–632 CrossRef; (c) S. K. Guchhait and C. Madaan, Tetrahedron Lett., 2011, 52, 56–58 CrossRef CAS.
- (a) L. R. Amanda, M. Pulane and J. P. Christopher, Tetrahedron Lett., 2007, 48, 4079–4082 CrossRef; (b) M. Krasavin, S. Tsirulnikov, M. Nikulnikov, Y. Sandulenko and K. Bukhryakov, Tetrahedron Lett., 2008, 49, 7318–7321 CrossRef CAS.
- C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635–3638 CrossRef CAS.
- L. R. Odell, M. Nilson, J. Gising, O. Lagerlund, D. Muthas, A. Nordqvist, A. Karlen and M. Larhed, Bioorg. Med. Chem. Lett., 2009, 19, 4790–4793 CrossRef CAS PubMed.
- (a) Y. M. Ren, C. Cai and R. C. Yang, RSC Adv., 2013, 3, 7182–7204 RSC; (b) A. T. Khan, M. M. Khan and K. K. R. Bannuru, Tetrahedron, 2010, 66, 7762–7772 CrossRef CAS; (c) A. T. Khan, A. Ghosh and M. M. Khan, Tetrahedron Lett., 2012, 53, 2622–2626 CrossRef CAS.
- (a) Z. T Bhutia, P. C. Panjikar, S. Iyer, A. Chatterjee and M. Banerjee, ACS Omega, 2020, 22, 13333–13343 CrossRef PubMed; (b) U. Tekale Sunil, S. Kauthale Sushma, A. Dake Satish, R. Sarda Swapnil and P. Pawar Rajendra, Curr. Org. Chem., 2012, 16, 1385–2728 Search PubMed; (c) L. Royer, S. K. De and R. A. Gibbs, Tetrahedron Lett., 2005, 46, 4595–4597 CrossRef CAS; (d) J. Q. Wang, Z. Y. Zuo and W. He, Catalysts, 2022, 12, 821 CrossRef; (e) S. S. Wu, C. T. Feng, D. Hu, Y. K. Huang, Z. Li, Z. G. Luo and S. T. Ma, Org. Biomol. Chem., 2017, 15, 1680–1685 RSC; (f) Y. Z. Yan, Y. Xu, B. Niu, H. F. Xie and Y. Q. Liu, J. Org. Chem., 2015, 80, 5581–5587 CrossRef CAS PubMed.
- (a) A. Parthiban, R. Sivasankar, B. Rajdev, R. N. Asha, T. C. Jeyakumar, R. Periakaruppan and V. G. M. Naidu, J. Mol. Struct., 2022, 1270, 133885 CrossRef CAS; (b) A. Parthiban and P. Makam, Bioorg. Chem., 2020, 105, 104379 CrossRef PubMed; (c) P. Anaikutti, M. Selvaraj, J. Prabhakaran, T. Pooventhiran, T. C. Jeyakumar, R. Thomas and P. Makam, J. Mol. Struct., 2022, 1266, 133464 CrossRef CAS; (d) A. Parthiban, J. Muthukumaran, A. Moushumi, S. Jayachandran, R. Krishna and H. S. P. Rao, Med. Chem. Res, 2014, 23, 642–659 CrossRef CAS; (e) A. Parthiban, M. Kumaravel, J. Muthukumaran, R. Rukkumani, R. Krishna and H. S. P. Rao, Med. Chem. Res., 2015, 24, 1226–1240 CrossRef CAS; (f) A. Parthiban, M. Kumaravel, J. Muthukumaran, R. Rukkumani, R. Krishna and H. S. P. Rao, Med. Chem. Res., 2016, 25, 1308–1315 CrossRef CAS; (g) A. Parthiban, J. Muthukumaran, A. Manhas, K. Srivastava, R. Krishna and H. S. P. Rao, Bioorg. Med. Chem. Lett., 2015, 25, 4657–4663 CrossRef CAS PubMed.
- G. S. Mani, S. P. Shaik, Y. Tangella, S. Bale, C. Godugu and A. Kamal, Org. Biomol. Chem., 2017, 15, 6780 RSC.
- M. Umkehrer, G. Ross, N. Jager, C. Burdack, J. Kolb, H. Hu, M. Alvim Gaston and C. Hulme, Tetrahedron Lett., 2007, 48, 2213–2216 CrossRef CAS.
- G. Chauvière, B. Bouteille, B. Enanga de, C. Albuquerque, S. L. Croft, M. Dumas and J. Périé, J. Med. Chem., 2003, 46, 427–440 CrossRef PubMed.
- P. Fernando da Silva Santos-Júnior, L. Rocha Silva, L. José Quintans-Júnior and E. Ferreira da Silva-Júnior, Bioorg. Med. Chem. Lett., 2022, 75, 128930 CrossRef PubMed.
- (a) B. F. Lundt, N. L. Johansen, A. Vølund and J. Markussen, Int. J. Pept. Protein Res., 1978, 12, 258–268 CrossRef CAS PubMed; (b) V. Evans, M. F. Mahon and R. L. Webster, Tetrahedron, 2014, 70, 7593–7597 CrossRef CAS; (c) J. D. Catt and W. L. Matier, J. Org. Chem., 1974, 39, 566–568 CrossRef CAS; (d) P. Pollock, K. P. Cole, S. W. Roberts and M. Faul, Org. Synth., 2012, 89, 537–548 CrossRef CAS.
- (a) N. R. Chereddy, K. Suman, P. S. Korrapati, S. Thennarasu and A. B. Mandal, Dyes Pigm., 2012, 95, 606–613 CrossRef CAS; (b) N. R. Chereddy, S. Thennarasu and A. B. Mandal, Dalton Trans., 2012, 41, 11753–11759 RSC.
- (a) W. Jiasheng, L. Weimin, G. Jiechao, Z. Hongyan and W. Pengfei, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC; (b) G. Aragay, J. Pons and A. Merkoc-I, Chem. Rev., 2011, 111, 3433 CrossRef CAS PubMed.
- G. H. Bae, S. Kim, N. K. Lee, A. Dagar, J. H. Lee, J. Lee and I. Kim, RSC Adv., 2020, 10, 7265–7288 RSC.
- (a) R. Krishnamoorthy, M. Singh, A. Parthiban, P. L. Edwin, S. Dhanasekaran and T. Sathiah, Bioorg. Chem., 2023, 134, 106434 CrossRef CAS PubMed; (b) S. Pajaniradje, M. K. Kumar, R. Radhakrishanan, S. A. Sufi, S. Subramaniam, P. Anaikutti, H. S. P. Rao and R. Rajagopalan, Lett. Drug Des. Discovery, 2020, 17, 1146–1154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The 1H NMR, 13C NMR data of all compounds are given. See DOI: https://doi.org/10.1039/d3ra07842f |
| This journal is © The Royal Society of Chemistry 2023 |