
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective total synthesis of (3Z)- and (3E)-elatenynes†
Soo Yeon Kwak‡
 a,
Iljin Shin‡a,
Hongjun Janga,
Youngjik Parka,
Seongju Lima,
Dongjoo Leea,
Hyoungsu Kim*a and
Deukjoon Kimb
a,
Iljin Shin‡a,
Hongjun Janga,
Youngjik Parka,
Seongju Lima,
Dongjoo Leea,
Hyoungsu Kim*a and
Deukjoon Kimb
aCollege of Pharmacy and Research Institute of Pharmaceutical Science and Technology (RIPST), Ajou University, Suwon 16499, Korea. E-mail: hkimajou@ajou.ac.kr
bCollege of Pharmacy, Seoul National University, Seoul 08826, Korea
First published on 11th December 2023
Abstract
We describe here the highly stereoselective total synthesis of the Laurencia C15 acetogenins (3Z)- and (3E)-elatenynes having a 7,12-dibromo-6,9-cis-10,13-cis adjacent bis-tetrahydrofuran (THF) core. The present synthesis features a highly stereoselective, protecting group-dependent, chelate-controlled intramolecular amide enolate alkylation (IAEA) for the synthesis of key intermediate 7-hydroxy-6,7-cis-6,9-cis-THF intermediate 10, deployment of the sequential ate complex (n-BuLi/DIBAL-H) reduction/Keck allylation/cross metathesis (CM) protocol for the stereoselective introduction of the C(10)–C(15) unit, a sequential Sharpless asymmetric dihydroxylation (SAD)/intramolecular Williamson etherification for the construction of the 10,13-cis-THF ring, and a modified Nakata chloromethanesulfonate-mediated SN2 displacement for the 7,12-dibromo functionality. Furthermore, our strategy based on chelate-controlled IAEA methodology would provide access to any member of the C15 adjacent bis-THF acetogenin class.
Marine algae produce a diverse set of oxacyclic C15 acetogenins, among which some, as shown in Fig. 1, have a 2,2′-bifuranyl (adjacent bis-THF) core structure.1 (3Z)-Elatenyne (1a) was first isolated from the marine algae Laurencia elata by Hall and Reiss in 1986,2a and Erickson reported isolating (3E)-elatenyne (1b) from the marine alga Laurencia majuscule in 1989.2b Later, 1a was re-isolated from Laurencia decumbens by Wang in 2007 (ref. 2c) and from Laurencia elata by Urban in 2011.2d The isolation of several closely related Laurencia C15 acetogenins has been reported, including notoryne (2),3 chloroenyne (3) from L. majuscule,4 laurendecumenyne B (4),5 and laurefurenynes A (5a) and B (5b).6 It is worth mentioning at this point that the structures depicted in Fig. 1 have been revised or confirmed by total synthesis.2h,3c–e,6b,c
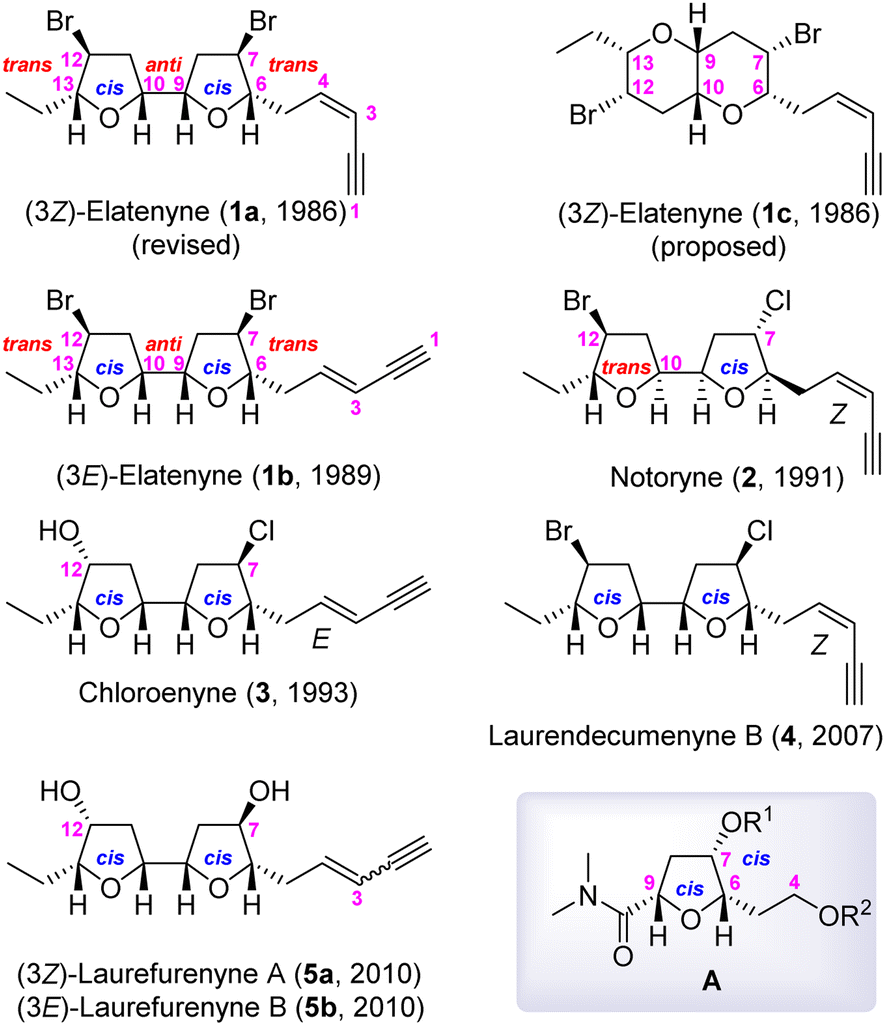 | ||
| Fig. 1 Laurencia adjacent bis-tetrahydrofuranoid natural products. | ||
Based on extensive 1H and 13C NMR spectroscopic analyses, the structure of (3Z)-elatenyne (1a) was initially proposed by Hall and Reiss to have a pyrano[3,2-b]pyran core (fused bis-THP), as depicted in 1c.2a However, the 1c structure was shown by Burton, et al., to be incorrect through the total synthesis thereof.2e,f The Burton and the Goodman groups collaborated to predict the correct 2,2′-bifuranyl skeleton (adjacent bis-THF) structure and relative stereochemistry of 1a through comparison of the 13C NMR chemical shifts of 1a with the Boltzmann-weighted GIAO 13C NMR chemical shifts determined through DFT methods.2g Later, a collaborative effort by the Kim and Burton groups achieved the total synthesis of 1a and ent-1a utilizing a modular and biomimetic approach, respectively.2h Despite the collaborative effort, the unequivocal assignment of the absolute stereochemistry of 1a was still not possible. Eventually, Urban and Fujita confirmed the absolute stereochemistry of 1a as that shown in Fig. 1 using the crystalline sponge method.2i
The structural features of these C15 adjacent bis-THF acetogenins have received considerable attention from organic chemistry community, culminating to several total syntheses: the modular synthesis on the basis of analysis 13C NMR chemical shifts,2h,e,3e,6b biomimetic approach,2e,h,3c,e the cyclization of chlorohydrin derived from anti-aldol reaction,6c the Sharpless asymmetric dihydroxylation (SAD)/Williamson cyclization sequence,4b and the bromo-etherification.3d
Based on the insights garnered from our highly stereoselective syntheses of oxylipids7a and asimitrin,7b we formulated a synthetic strategy which provides access to any member of this C15 adjacent bis-THF acetogenin class through a highly stereoselective construction of the 2,5-disubstituted-3-oxygenated tetrahydrofuran moiety A (Fig. 1) via intramolecular amide enolate alkylation (IAEA).7 In addition, our strategy utilizes Marshall's protocol [cross metathesis (CM)/SAD/Williamson cyclization]8 for the efficient construction of 2nd THF skeleton in the adjacent bis-THF unit.
To demonstrate the synthetic potential of this strategy, we describe herein the asymmetric total synthesis of (3Z)-elatenyne (1a) and (3E)-elatenyne (1b) featuring a highly stereoselective and chelate-controlled IAEA for constructing key intermediate 7-hydroxy-6,7-cis-6,9-cis-THF 10. This is followed by a sequential ate complex (n-BuLi/DIBAL-H) reduction/Keck allylation/cross metathesis (CM) protocol for stereoselective introduction of the C(10)–C(15) unit.
As shown in our retrosynthetic plan (Scheme 1), we envisioned that respective total syntheses of (3Z)-elatenyne (1a) and (3E)-elatenyne (1b) could be readily accomplished through stereoselective incorporation of the (Z)- and (E)-enyne units into 7,12-dibromo-adjacent bis-THF 6. This intermediate could be accessed by bis-bromination of the adjacent 7,12-dihydroxy-bis-THF 7, which in turn could be constructed from the tetrahydrofuranyl syn-diol 8 through an intramolecular Williamson etherification. We planned to synthesize 8 from homoallylic alcohol 9 by employing cross metathesis (CM) and Sharpless asymmetric dihydroxylation (SAD) as key steps. By this route, (10S)-9,10-syn homoallylic alcohol 9 could be stereoselectively prepared through application of ate complex (n-BuLi/DIBAL-H) reduction/Keck allylation protocols to yield α-alkoxy amide 10 (vide infra). Based on our previous work,7 we were confident that key 6,7-cis-6,9-cis-THF intermediate 10 could be accessed by subjecting 6,7-syn-ω-bromo-α-alkoxy amide 11 to our stereoselective chelate-controlled IAEA reaction. Finally, we imagined that IAEA substrate 11 could be prepared in a straightforward manner from the known 6,7-syn epoxy alcohol 12.
 | ||
| Scheme 1 Retrosynthetic plan. | ||
Our synthesis began with the preparation of IAEA substrate 11, as outlined in Scheme 2. Thus, known epoxy alcohol 12 (ref. 9) was subjected to O-alkylation with N,N-dimethyl chloroacetamide to afford the desired epoxy α-alkoxy amide 13 in 94% yield. The regioselective opening of the terminal epoxide 13 was achieved through the action of (n-Bu)4NBr in the presence of Mg(NO3)2·6H2O to furnish the 6,7-syn-bromoamide 14 with an excellent 96% yield.10 Protection of the hydroxyl group in 14 as the PMB ether with 4-methoxybenzyl 2,2,2-trichloroacetimidate in the presence of a catalytic amount of p-toluenesulfonic acid (PTSA)11 gave rise to key IAEA substrate 11 in good yield (88%).
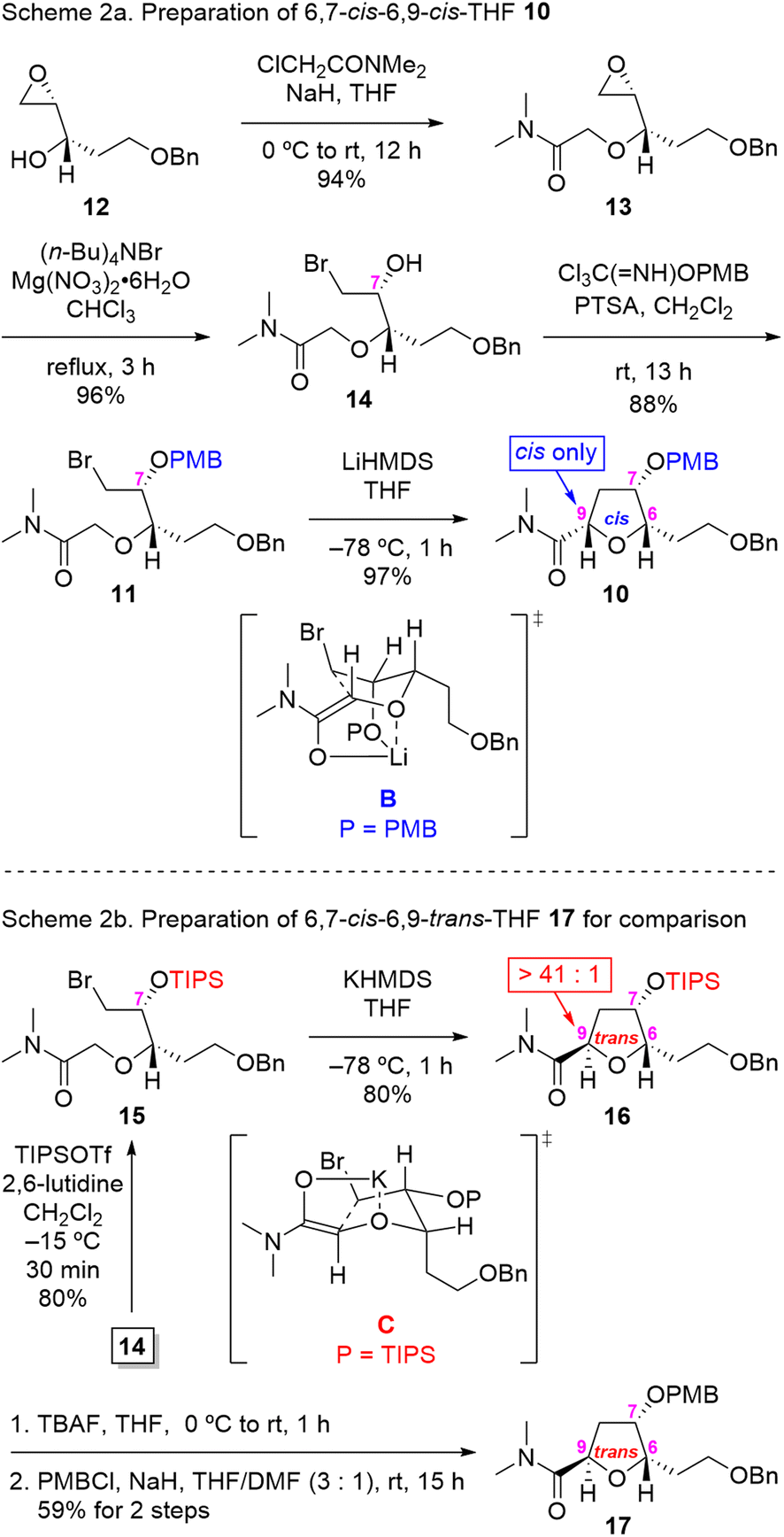 | ||
| Scheme 2 Stereoselective synthesis of 6,9-cis-THF 10 and 6,9-trans-THF 17 via IAEA. | ||
With IAEA substrate 11 in hand, we proceeded to address the pivotal stereoselective IAEA reaction of PMB-protected bromo α-alkoxy amide 11 for the construction of key intermediate 10. Treatment of 11 with LiHMDS in THF at −78 °C for 1 h led to the desired 6,7-cis-6,9-cis-THF 10 in 97% yield as a single stereoisomer (by 1H NMR analysis, see ESI† for details), presumably via chelated transition state geometry B. The NOE interaction between protons on [C(6) and C(7)] and [C(6) and C(9)] in 10 was supportive of the assigned cis relative stereochemistry.
To establish the diastereoselectivity of the IAEA reaction in a rigorous manner, we decided to synthesize the corresponding 6,9-trans isomer 17 for comparison purposes as shown at the bottom of Scheme 2. To this end, subjection of TIPS-protected bromo α-alkoxy amide 15 (prepared by TIPS protection of alcohol 14) to KHMDS in THF at −78 °C for 1 h gave rise to the desired 6,9-trans-THF 16 in 80% yield as the major isomer (dr > 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by 1H NMR analysis), presumably via transition state C. Deprotection of the TIPS protecting group in 16 by exposure to TBAF and subsequent protection of the resultant alcohol as the PMB ether provided the 6,9-trans-THF 17 in 59% yield (two steps).
:1 by 1H NMR analysis), presumably via transition state C. Deprotection of the TIPS protecting group in 16 by exposure to TBAF and subsequent protection of the resultant alcohol as the PMB ether provided the 6,9-trans-THF 17 in 59% yield (two steps).
Having accomplished a highly stereoselective synthesis of the desired 6,9-cis-THF 10, we turned our attention to the construction of the crucial adjacent bis-THF 21 as shown in Scheme 3. This requires the stereoselective synthesis of 9,10-syn homoallylic alcohol 9 from α-alkoxy amide 10 through application of our direct ketone synthesis/L-Selectride protocol.7 Thus, the Grignard reaction of 10 with CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2MgBr, and the subsequent L-Selectride reduction of the resulting ketone 18, afforded the desired 9,10-syn-homoallylic alcohol 9 in moderate yield (59% for two steps) and good selectivity (dr = 8:1 by 1H NMR analysis). In an alternative approach, 10 was reduced using the ate complex derived from n-BuLi and DIBAL-H12 and subjected to Keck allylation13 to afford homoallylic alcohol 9 in improved yield (75% for two steps) and improved selectivity (dr = 46:1 by 1H NMR analysis)14 CM reaction of the alcohol 9 with cis-3-hexene in the presence of Grubbs second-generation catalyst [G-II, (H2IMes)(Cy3P)Cl2RuCHPh]15 afforded alkene 20 as an inseparable mixture of stereoisomers (95% total yield, E/Z = 6:1 by 1H NMR analysis). Tosylation of alkene 20 (E/Z = 6:1) and subsequent AD-mix β-mediated SAD reaction16 of the resulting tosylate afforded the pure syn-diol 8 in 57% overall yield from 9 (three steps) after separation. Internal Williamson cyclization of 8 in refluxing pyridine or NaH in THF/DMF (3:2) furnished the desired adjacent bis-THF 21 in 86% or 84% yield, respectively.
CHCH2MgBr, and the subsequent L-Selectride reduction of the resulting ketone 18, afforded the desired 9,10-syn-homoallylic alcohol 9 in moderate yield (59% for two steps) and good selectivity (dr = 8:1 by 1H NMR analysis). In an alternative approach, 10 was reduced using the ate complex derived from n-BuLi and DIBAL-H12 and subjected to Keck allylation13 to afford homoallylic alcohol 9 in improved yield (75% for two steps) and improved selectivity (dr = 46:1 by 1H NMR analysis)14 CM reaction of the alcohol 9 with cis-3-hexene in the presence of Grubbs second-generation catalyst [G-II, (H2IMes)(Cy3P)Cl2RuCHPh]15 afforded alkene 20 as an inseparable mixture of stereoisomers (95% total yield, E/Z = 6:1 by 1H NMR analysis). Tosylation of alkene 20 (E/Z = 6:1) and subsequent AD-mix β-mediated SAD reaction16 of the resulting tosylate afforded the pure syn-diol 8 in 57% overall yield from 9 (three steps) after separation. Internal Williamson cyclization of 8 in refluxing pyridine or NaH in THF/DMF (3:2) furnished the desired adjacent bis-THF 21 in 86% or 84% yield, respectively.
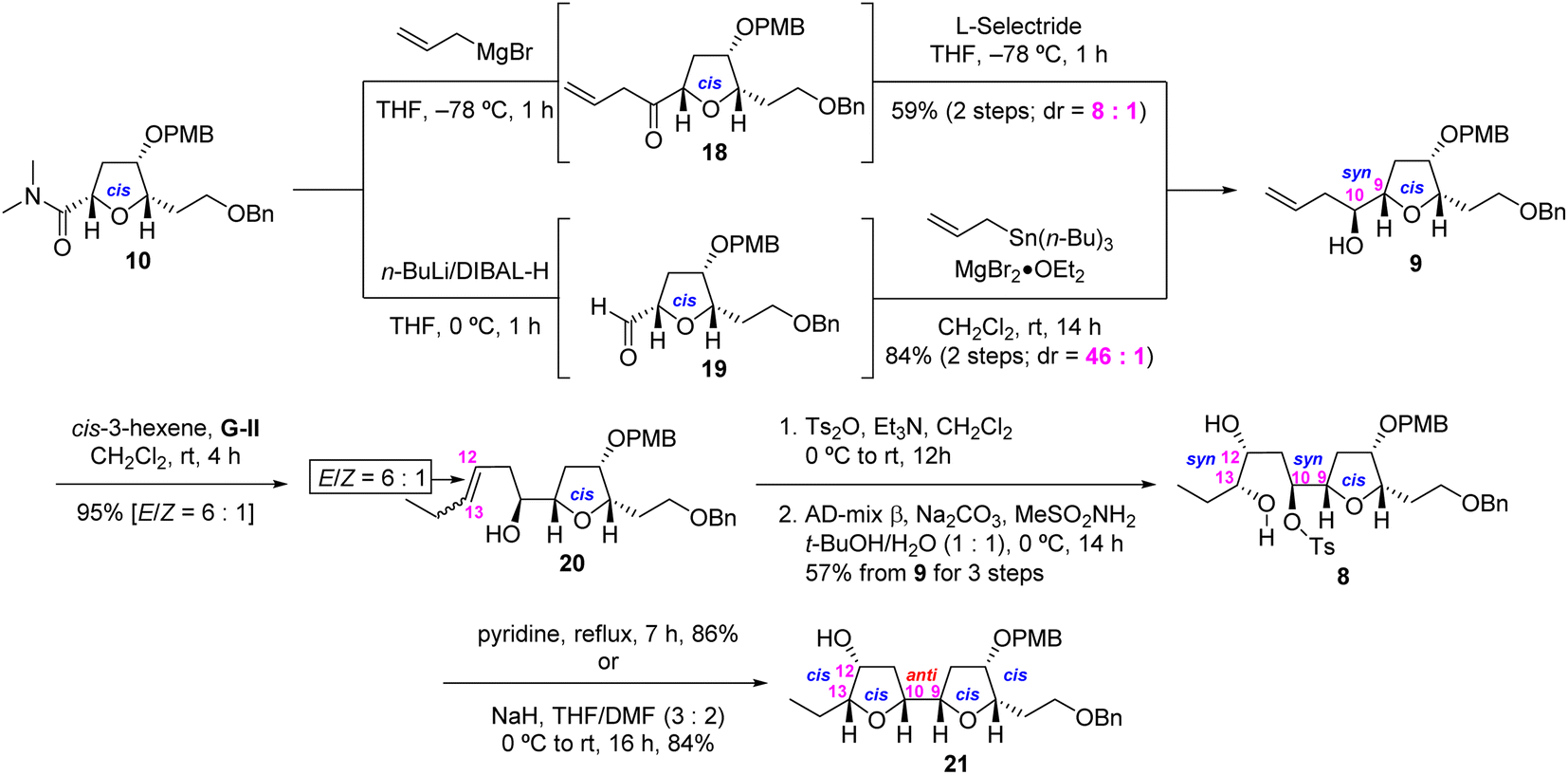 | ||
| Scheme 3 Construction of 7,12-dihydroxy adjacent bis-THF 21. | ||
Having acquired adjacent bis-THF 21, we proceeded to introduce the bis-bromide functionality to both the C(7) and C(12) positions utilizing the two-step modified Nakata chloromethanesulfonate-mediated SN2 displacement protocol2h,7b,17 (Scheme 4). To this end, treatment of bis secondary alcohol 7, obtained from 21 after PMB deprotection (92%), with chloromethanesulfonyl chloride (McCl) in the presence of 2,6-lutidine and subsequent exposure of the resulting sulfonate to (n-Bu)4NBr in refluxing THF furnished the desired 7,12-dibromo-bis-THF 6 in an overall yield of 63% from 21 in two steps. It is of note that the two-step Nakata protocol was superior to Hooz bromination in term of yield and purification in our hands [69% vs. 58%; see ESI† for details].18
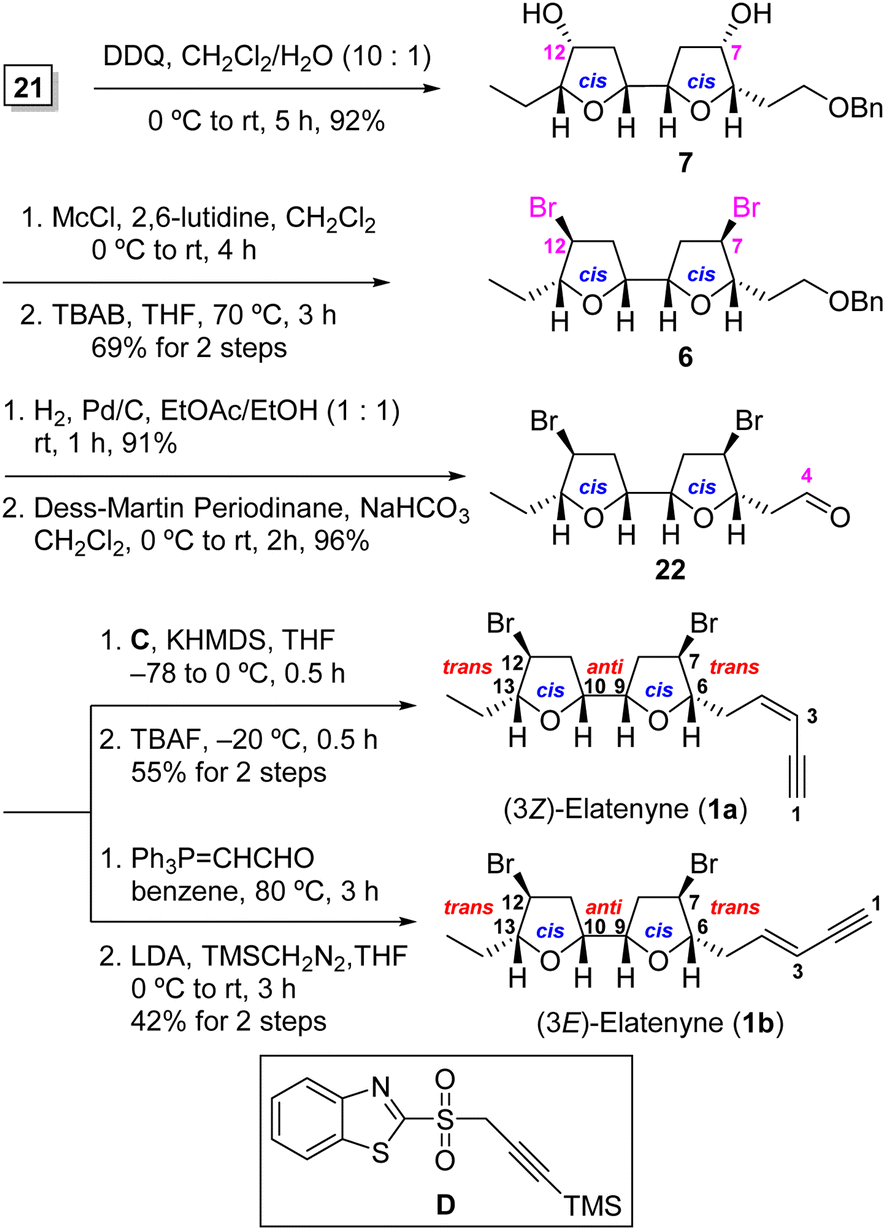 | ||
| Scheme 4 Completion of total synthesis of 1a and 1b. | ||
Having successfully installed both the C(7) and C(12) bromide atoms in 1a and 1b, the remaining task was attaching the C(4) enyne appendages. Catalytic hydrogenolysis of benzyl ether 6, followed by Dess–Martin oxidation19 of the resultant primary alcohol gave rise to aldehyde 22. The stereoselective Julia–Kocienski olefination20 of aldehyde 22 with benzothiazole sulfone C by treatment with KHMDS in THF at –78 to 0 °C for 0.5 h gave rise to the (3Z)-TMS-enyne (Z/E = 31:1 by 1H NMR analysis), which was desilylated with TBAF to afford (3Z)-elatenyne (1a) in 55% overall yield for the two steps from 22. For the second target, Wittig olefination of aldehyde 22 with Ph3PCHCHO [(triphenylphosphoranylidene)acetaldehyde] gave exclusively the (E)-α,β-unsaturated aldehyde, which was then subjected to the condition of Colvin–Ohira homologation21 using trimethylsilyldiazomethane and LDA to afford (3E)-elatenyne (1b) in 42% overall yield for two steps. The spectral characteristics of our synthetic material 1a and 1b were in good agreement with those reported for both the natural and synthetic (3Z)-2a,d,h and (3E)2b,h-elatenynes, respectively.
Conclusions
In summary, we have accomplished the total synthesis of both (3Z)-elatenyne (1a) and (3E)-elatenyne (1b), featuring the protecting group-dependent chelate-controlled IAEA methodology for a highly stereoselective construction of key intermediate 6,7-cis-6,9-cis-THF 10. Other key features of the synthesis include the sequential ate complex reduction/Keck allylation for stereoselective establishment of 9,10-syn configuration, the CM/SAD/Williamson cyclization sequence for the efficient construction of the bis-THF moiety, and the chloromethanesulfonate-mediated SN2 displacement for installation of the 7,12-dibromo functionality. Application of our strategy on the basis of chelate-controlled IAEA and the Marshall's protocol to the synthesis of other members of the adjacent C15 bis-THF acetogenin class in Fig. 1 is currently under investigation in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF-2020R1A2C2010329) and a grant (21153MFDS602 & 21163MFDS369) from the Ministry of Food and Drug Safety.Notes and references
- (a) B.-G. Wang, J. B. Gloer, N.-Y. Ji and J.-C. Zhao, Halogenated Organic Molecules of Rhodomelaceae Origin: Chemistry and Biology, Chem. Rev., 2013, 113, 3632–3685 CrossRef CAS PubMed; (b) T. Wanke, A. C. Philippus, G. A. Zatelli, L. F. O. Vieira, C. Lhullier and M. Falkenberg, C15 Acetogenins from the Laurencia Complex: 50 Years of Research-An Overview, Rev. Bras. Farmacogn., 2015, 25, 569–587 CrossRef CAS; (c) Z. F. Zhou, M. Menna, Y.-S. Cai and Y. W. Guo, Polyacetylenes of Marine Origin: Chemistry and Bioactivity, Chem. Rev., 2015, 115, 1543–1596 CrossRef CAS PubMed; (d) M. Harizani, E. Ioannou and V. Roussis, The Laurencia paradox: An endless source of chemodiversity, in Progress in the Chemistry of Organic Natural Products, ed. A. D. Kinghorn, H. Galk, S. Gibbons and J. Kobayashi, Springer, Berlin, 2016, vol. 102, pp. 91–252 Search PubMed.
- (a) J. G. Hall and J. A. Reiss, Elatenyne – a Pyrano[3,2-B]Pyranyl Vinyl Acetylene from the Red Alga Laurencia elata, Aust. J. Chem., 1986, 39, 1401–1409 CrossRef CAS; (b) The absolute configuration of natural (E)-elatenyne was not determined because no optical rotation data were reported in the original isolation paper; see, K. Kim, M. R. Brennan and K. L. Erickson, Lauroxolanes from the marine alga Laurencia majuscule, Tetrahedron Lett., 1989, 30, 1757–1760 CrossRef; (c) N. Y. Ji, X. M. Li, K. Li and B. G. Wang, Laurendecumallenes A–B and Laurendecumenynes A–B, Halogenated Nonterpenoid C15-Acetogenins from the Marine Red Alga Laurencia decumbens, J. Nat. Prod., 2007, 70, 1499–1502 CrossRef CAS PubMed; (d) D. A. Dias and S. Urban, Phytochemical studies of the southern Australian marine alga, Laurencia elata, Phytochemistry, 2011, 72, 2081–2089 CrossRef CAS PubMed; (e) H. M. Sheldrake, C. Jamieson and J. W. Burton, The Changing Faces of Halogenated Marine Natural Products: Total Synthesis of the Reported Structures of Elatenyne and an Enyne from Laurencia majuscule, Angew. Chem., Int. Ed., 2006, 45, 7199–7202 CrossRef CAS; (f) H. M. Sheldrake, C. Jamieson, S. I. Pascu and J. W. Burton, Synthesis of the Originally Proposed Structures of Elatenyne and an Enyne from Laurencia Majuscule, Org. Biomol. Chem., 2009, 7, 238–252 RSC; (g) S. G. Smith, R. S. Paton, J. W. Burton and J. M. Goodman, Stereostructure Assignment of Flexible Five-Membered Rings by GIAO 13C NMR Calculations: Prediction of the Stereochemistry of Elatenyne, J. Org. Chem., 2008, 73, 4053–4062 CrossRef CAS; (h) B. S. Dyson, J. W. Burton, T. I. Sohn, B. Kim, H. Bae and D. Kim, Total Synthesis and Structure Confirmation of Elatenyne: Success of Computational Methods for NMR Prediction with Highly Flexible Diastereomers, J. Am. Chem. Soc., 2012, 134, 11781–11790 CrossRef CAS; (i) S. Urban, R. Brkljača, M. Hoshino, S. Lee and M. Fujita, Determination of the Absolute Configuration of the Pseudo- Symmetric Natural Product Elatenyne by the Crystalline Sponge Method, Angew. Chem., Int. Ed., 2016, 55, 2678–2682 CrossRef CAS PubMed.
- (a) For the isolation and structure determination of notoryne see: H. Kikuchi, T. Suzuki, E. Kurosawa and M. Suzuki, The Structure of Notoryne, a Halogenated C15 Nonterpinoid with a Novel Carbon Skeleton from the Red Alga Laurencia Nipponica Yamada, Bull. Chem. Soc. Jpn., 1991, 64, 1763–1775 CrossRef CAS; (b) 3(E)-Notoryne has also been reported as a natural product, see: A. Fukuzawa, M. Aye, M. Nakamura, M. Tanura and A. Murai, Structure elucidation of laureoxanyne, a new nonisoprenoid C-15-enyne, using lactoperoxidase, Tetrahedron Lett., 1990, 31, 4895–4898 CrossRef CAS ; For total synthesis of 3, see:; (c) M. Lee, First Asymmetric Total Synthesis of (Z)-and (E)-Notorynes, M.S. thesis, Seoul National University, Seoul, Korea, 2009 Search PubMed; (d) S. Senapati, S. Das and C. V. Ramana, Total Synthesis of Notoryne, J. Org. Chem., 2018, 83, 12863–12868 CrossRef CAS; (e) E. D. Shepherd, B. S. Dyson, W. E. Hak, Q. N. N. Nguyen, M. Lee, M. J. Kim, T. I. Sohn, D. Kim, J. W. Burton and R. S. Paton, Structure Determination of a Chloroenyne from Laurencia majuscula Using Computational Methods and Total Synthesis, J. Org. Chem., 2019, 84, 4971–4991 CrossRef CAS.
- (a) A. D. Wright, G. M. Konig, R. Denys and O. Sticher, Seven New Metabolites From The Marine Red Alga Laurencia Majuscula, J. Nat. Prod., 1993, 56, 394–401 CrossRef CAS; (b) For total synthesis of 3, see: S. Senapati, N. A. Unmesh, M. N. Shet, I. Ahmad, N. Ajikumar and C. V. Ramana, Unified Approach for the Total Synthesis of Bis-THF C15 Acetogenins: A Chloroenyne from Laurencia majuscula, Laurendecumenyne B and Laurefurenynes A/B, Synthesis, 2021, 53, 2903–2910 CrossRef CAS ; and also see: ref. 3e..
- (a) For isolation of 4, see: ref. 2c;; (b) For total synthesis of 4, see: ref. 2h and 4b..
- (a) W. M. Abdel-Mageed, R. Ebel, F. A. Valeriote and M. Jaspars, Laurefurenynes A–F, new Cyclic Ether Acetogenins from a Marine Red Alga, Laurencia sp, Tetrahedron, 2010, 66, 2855–2862 CrossRef CAS ; For total synthesis of 5a and 5b, see:; (b) D. J. Shepherd, P. A. Broadwith, B. S. Dyson, R. S. Paton and J. W. Burton, Structure Reassignment of Laurefurenynes A and B by Computation and Total Synthesis, Chem. – Eur. J., 2013, 19, 12644–12648 CrossRef CAS; (c) M. T. Holmes and R. A. Britton, Total Synthesis and Structural Revision of Laurefurenynes A and B, Chem. – Eur. J., 2013, 19, 12649–12652 CrossRef CAS PubMed.
- (a) H. Jang, I. Shin, D. Lee, H. Kim and D. Kim, Stereoselective Substrate-Controlled Asymmetric Syntheses of both 2,5-cis- and 2,5-trans-Tetrahydrofuranoid Oxylipids: Stereodivergent Intramolecular Amide Enolate Alkylation, Angew. Chem., Int. Ed., 2016, 55, 6497–6501 CrossRef CAS PubMed; (b) S. Y. Kwak, Y. Park, S. Lim, H. Jang, D. Lee, H. Kim and D. Kim, Total Synthesis and Structure Confirmation of (–)-Asimitrin, a C37 Annonaceous Acetogenin with a Hydroxylated Adjacent Bis-Tetrahydrofuran Core, Org. Lett., 2023, 25, 6659–6664 CrossRef CAS; (c) I. Shin, D. Lee and H. Kim, Substrate-Controlled Asymmetric Total Synthesis and Structure Revision of (–)-Bisezakyne A, Org. Lett., 2016, 18, 4420–4423 CrossRef CAS PubMed; (d) I. Shin, H. Jang, S. Y. Kwak, Y. Park, D. Lee, H. Kim and D. Kim, Highly Stereodivergent Construction of C2-Symmetric cis,cis- and trans,trans-2,6-dioxabicyclo[3.3.0]octane Framework by Double Intramolecular Amide Enolate Alkylation: Total Synthesis of (+)-Laurenidificin and (+)-Aplysiallene, Org. Lett., 2022, 24, 8780–8785 CrossRef CAS.
- J. A. Marshall and J. J. Sabatini, Synthesis of cis- and trans-2,5-Disubstituted Tetrahydrofurans by a Tandem Dihydroxylation-SN2 Cyclization Sequence, Org. Lett., 2005, 7, 4819–4822 CrossRef CAS PubMed.
- (a) H. Lee, H. Kim, S. Baek, S. Kim and D. Kim, Total Synthesis and Determination of the Absolute Configuration of (+)-Neoisoprelaurefucin, Tetrahedron Lett., 2003, 44, 6609–6612 CrossRef CAS; (b) The ee value of known epoxy alcohol 12 was determined as >86.9% by analysis of the 1H NMR spectrum of the corresponding Mosher esters, see the ESI† for the details..
- Y.-G. Suh, B.-A. Koo, J.-A. Ko and Y.-S. Cho, A Facile and Highly Regioselective of Epoxides to Bromohydrins Using Tetrabutylammonium Bromide and Magnesium Nitrate, Chem. Lett., 1993, 22, 1907–1910 CrossRef.
- T. Iversen and D. R. Bundle, Benzyl trichloroacetimidate, a versatile reagent for acid-catalysed benzylation of hydroxy-groups, J. Chem. Soc. Chem. Commun., 1981, 1240–1241 RSC.
- S. Kim and K. H. Ahn, Ate complex from diisobutylaluminum hydride and n-butyllithium as a powerful and selective reducing agent for the reduction of selected organic compounds containing various functional groups, J. Org. Chem., 1984, 49, 1717–1724 CrossRef CAS.
- G. E. Keck and E. P. Boden, Stereocontrolled additions of allyltri(n-butyl)stannane to a-hydroxyaldehyde derivatives. A useful route to monoprotected erythro or threo diols, Tetrahedron Lett., 1984, 25, 265–268 CrossRef CAS.
- The stereochemistry of C(10) in 9 was confirmed unambiguously utilizing Mosher ester analysis. (a) I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, High-Field FT NMR Application of Mosher's Method. The Absolute Configuration of Marine Terpenoids, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS; (b) J. A. Dale and H. S. Mosher, Nuclear Magnetic Resonance Enantiomer Regents. Configurational Correlations via Nuclear Magnetic Resonance Chemi-cal Shifts of Diastereomeric Mandelate, O-Methylmandelate, and α-Methoxy-α-trifluoromethylphenylacetate (MTPA) Esters, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS.
- A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, A General Model for Selectivity in Olefin Cross Metathesis, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Catalytic Asymmetric Dihydroxylation, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- T. Shimizu, S. Hiranuma and T. Nakata, Efficient Method for Inversion of Secondary Alcohols by Reaction of Chloromethanesulfonates with Cesium Acetate, Tetrahedron Lett., 1996, 37, 6145–6148 CrossRef CAS.
- The five-step bromination of 21 utilizing modified Nakata chloromethanesulfonate-mediated SN2 displacement afforded bis-bromide 6 in 41% yield over five steps, see ESI† for details.
- D. B. Dess and J. C. Martin, A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species, J. Am. Chem. Soc., 1991, 113, 7277–7287 CrossRef CAS.
- (a) C. Bonini, L. Chiummiento and V. Videtta, Direct Preparation of Z-1,3-Enyne Systems with a TMS-Propargylic Sulfone: Application of a One-Pot Julia Olefination, Synlett, 2006, 2079–2082 CrossRef CAS; (b) G. Kim, T. I. Sohn, D. Kim and R. S. Paton, Asymmetric Total Synthesis of (+)-Bermudenynol, a C15 Laurencia Metabolite with a Vinyl Chloride Containing Oxocene Skeleton, through Intramolecular Amide Enolate Alkylation, Angew. Chem., Int. Ed., 2014, 53, 272–276 CrossRef CAS PubMed.
- (a) E. W. Colvin and B. J. Hamill, One-Step Conversion of Carbonyl Compounds into Acetylenes, J. Chem. Soc., Chem. Commun., 1973, 151–152 RSC; (b) E. W. Colvin and B. J. Hamill, A Simple Procedure for the Elaboration of Carbonyl Compounds into Homologous Alkynes, J. Chem. Soc., Perkin Trans. 1, 1977, 869–874 RSC; (c) S. Ohira, K. Okai and T. Moritani, Generation of Alkylidenecarbenes by the Alkenation of Carbonyl Compounds with Lithiotrimethylsilyldiazomethane, J. Chem. Soc., Chem. Commun., 1992, 721–722 RSC; (d) K. Miwa, T. Aoyama and T. Shioiri, Extension of the Colvin rearrangement using trimethylsilyldiazomethane. A new synthesis of alkynes, Synlett, 1994, 107–108 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07741a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |