
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, docking, ADMET and anticancer evaluations of N-alkyl substituted iodoquinazoline derivatives as dual VEGFR-2 and EGFR inhibitors†
Marwa Alsulaimanya,
Khaled El-Adl *bc,
Ahmed K. B. Aljohanid,
Hussam Y. Alharbie,
Omar M. Alatawif,
Majed S. Aljohanie,
Ahmed El-morsyg,
Sara A. Almadanih,
Abdulrahman A. Alsimareei,
Samir A. Salamaj,
Doaa E. Keshekkl and
Abeer A. Mohamedcm
*bc,
Ahmed K. B. Aljohanid,
Hussam Y. Alharbie,
Omar M. Alatawif,
Majed S. Aljohanie,
Ahmed El-morsyg,
Sara A. Almadanih,
Abdulrahman A. Alsimareei,
Samir A. Salamaj,
Doaa E. Keshekkl and
Abeer A. Mohamedcm
aDepartment of Pharmacognosy & Pharmaceutical Chemistry, College of Pharmacy, Taibah University, Medina, 42353, Saudi Arabia
bPharmaceutical Medicinal Chemistry and Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Nasr City 11884, Cairo, Egypt. E-mail: eladlkhaled74@yahoo.com; eladlkhaled74@azhar.edu.eg; khaled.eladl@hu.edu.eg
cChemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt
dPharmacognosy and Pharmaceutical Chemistry Department, College of Pharmacy, Taibah University, Al-Madinah Al-Munawarah 41477, Saudi Arabia
eDepartment of Chemistry, Faculty of Science, Taibah University, Yanbu, Saudi Arabia
fDepartment of Chemistry, Faculty of Science, University of Tabuk, Tabuk, 47512, Saudi Arabia
gPharmaceutical Chemistry Department, College of Pharmacy, The Islamic University, Najaf, Iraq
hDepartment of Pharmacology and Toxicology, College of Pharmacy, Taibah University, Medina 42353, Saudi Arabia
iDepartment of Basic Science (Chemistry), College of Science and Humanities, Shaqra University, Afif, Saudi Arabia
jDivision of Biochemistry, Department of Pharmacology, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944, Kingdom of Saudi Arabia
kDepartment of Biology, Jumum College University, Umm Al-Qura University, P.O Box7388, Makkah 21955, Sudia Arabia
lAgriculture Genetic Engineering Research Institute (AGERI), Agriculture Research Centre, Giza, Egypt
mEgyptian Drug Authority (EDA), 51 Wezaret El-Zeraa St, Dokki, Giza, A. R., Egypt
First published on 13th December 2023
Abstract
Fifteen new 1-alkyl-6-iodoquinazoline derivatives 5a–d to 9a–e were designed and synthesized and their anticancer activities were evaluated against HepG2, MCF-7, HCT116 and A549 cancer cell lines via dual targeting of EGFR and VEGFR-2. The newly synthesized compounds were designed based on the structure requirements of the target receptors and were confirmed using spectral data. Compound 9c showed the highest anticancer activities with EC50 = 5.00, 6.00, 5.17 and 5.25 μM against HepG2, MCF-7, HCT116 and A549 cell lines correspondingly. Moreover, compounds 5d, 8b, 9a, 9b, 9d, and 9e exhibited very good anticancer effects against the tested cancer cell lines. The highly effective seven derivatives 5d, 8b, 9a–e were examined against VERO normal cell lines to estimate their cytotoxic capabilities. Compounds 9c, 9b, 9d, 9a, 9e and 5d excellently inhibited VEGFR-2 activity with IC50 = 0.85, 0.90, 0.90, 1.00, 1.20 and 1.25 μM respectively. Moreover, compounds 9c, 9d, 9e, 5d, 8b and 9b excellently inhibited EGFRT790M activity with IC50 = 0.22, 0.26, 0.30, 0.40, 0.45 and 0.50 μM respectively. Also, compounds 9c, 9d and 9e excellently inhibited EGFRWT activity with IC50 = 0.15, 0.20 and 0.25 μM respectively. As planned, compound 9c showed excellent dual EGFR/VEGFR-2 inhibitory activities. Consonantly, ADMET study was calculated in silico for the supreme three worthwhile compounds 9b, 9c and 9e in contrast to sorafenib and erlotinib as reference drugs. The obtained results concluded that, our compounds might be useful as prototype for design, optimization, adaptation and investigation to have more powerful and selective dual VEGFR-2/EGFRT790M inhibitors with higher antitumor activity.
1. Introduction
The prevention of tumor progression has involved blocking angiogenesis as a prevalent strategy. Vascular endothelial growth factor receptor (VEGFR) shows a fundamental role in VEGF-induced response regulation in endothelial cells and increased microvascular permeability in solid tumors.1 VEGFRs bind to VEGF and other related ligands, starting a cascade of signals leading to the proliferation and endothelial cell migration and new blood vessel development.2 Also, in both normal and abnormal angiogenesis, VEGFR is a main component, making it a target for the development of new cancer treatments.3 VEGFR has three subtypes, which are VEGFR-1, VEGFR-2 and VEGFR-3. The most important for angiogenesis is the VEGFR-2.4 In various cancers such as breast, lung, liver and colorectal cancer, VEGFR-2 is overexpressed. So, blocking VEGFR-2 with suitable tyrosine kinase inhibitors is the most significant target in anti-angiogenic therapy.5,6Existing research is being conducted to improve new strategies targeting signaling pathways.7,8 Tyrosine kinases modulate the growth factors signaling so they are considered as a significant target in cancer therapy. One of the most popular targets in anticancer therapy is protein tyrosine kinases (PTKs).9 Targeted therapy showed fewer toxic side effects compared to chemotherapy as they aimed at cancer-specific molecules and signaling pathways.10
VEGFR-2 inhibitors can be divided into three classes: class I, which binds to the ATP binding region (hinge region). Class II binds to the ATP binding site and extends into the adjacent hydrophobic back pocket, showing higher affinity and selectivity compared to class I; and class III blocks the receptor through hydrophobic interactions in the allosteric hydrophobic back pocket. Due to their prolonged TK suppression, researchers are actively seeking to develop new type II and III VEGFR-2 inhibitors.11–13
There are several quinazoline derivatives were reported as antitumor agents targeting VEGFR-2 kinase inhibitors.14,15 Alternatively, several potential anticancer agents were designed with numerous quinazoline scaffolds.16–18 Also, secondary resistance to approved VEGFR-2 inhibitors after the initial course of treatment highlights the need for novel therapeutic alternatives.19,20
On the other hand, VEGF and epidermal growth factor (EGF) act through similar signaling pathways and can act independently of each other in cancer development and resistance to treatment.21 EGFR (epidermal growth factor receptor) is the most common important PTK in cellular proliferation, differentiation and signaling pathways.22 Multitarget agents are involved in signaling pathways preventing cancer progression by targeting different biological receptors resulting in reduced toxicity synergistic effects.23 Thus, the simultaneous VEGFR and EGFR signaling pathways blockade proved to be an attractive approach to cancer treatment.14 Research suggests that tumors with EGFR mutations are more reliant on VEGF than those with the normal EGFR gene.24 As a result, targeting both VEGF and EGFR simultaneously is considered a rational approach for treating EGFR-mutant non-small cell lung cancer (NSCLC) treatment.25
EGFR mutation is a common mechanism of resistance to tyrosine kinase inhibitors (TKIs) involved in cancer progression.26 In 90% of EGFR-mutant NSCLC cases, an exon 19 deletion or exon 21 L858R mutations occur, both of which render tumors responsive to EGFR TKIs.27,28 Also T790M, known as Thr790Met, is a gatekeeper mutation of EGFR. The mutation substitutes a threonine (T) with a methionine (M) at position 790 of exon 20. The EGFRT790M mutation restores the affinity of the kinase for ATP to wild-type levels, and therefore at cellular ATP levels, the potency of the TKI is reduced.29 In metastatic cases, EGFR TKIs are considered first-line therapy due to their excellent tolerability and progression-free survival (PFS).30,31 Despite being initially effective, resistance to EGFR TKI therapy consequently occurs. The survival rate for individuals with metastatic lung cancer caused by a mutant EGFR gene is only 15% over five years, highlighting the need for new and improved therapeutic approaches. One possible addition to EGFR inhibition in non-small cell lung cancer is targeting the VEGF pathway.32,33 The suppression of the VEGF signaling pathway is a crucial step in cancer treatment.34 Lately, there is considerable attention paid to designating dual TKIs that can prevent cancer development.35,36
Numerous FDA approved quinazoline based small molecule inhibitors display clinical importance via targeting either VEGFR-2 or EGFR signaling pathways and inhibiting tumor progression.37,38
Quinazoline is a good scaffold for drug discovery. In addition to anticancer activities targeting different biological targets, they also show good potential as antiviral39 and antibacterial40 drug discovery.
In this research we decide to synthesize new iodoquinazolines derivatives as anticancer agents due to iodine's high atomic number, and ease of attachment to organic compounds, and it has also found favour as a non-toxic radiocontrast material. Because of the specificity of its uptake by the human body, radioactive isotopes of iodine can also be used to treat thyroid cancer. Moreover, there are many approved drugs containing iodo substitutions e.g. 2-iodo-4′-methoxychalcone,41 amiodarone and iodochlorhydroxyquinoline (Clioquinol) (Fig. 1). On the other hand, there are many quinazoline derivatives targeting either EGFR and/or VEGFR-2 signaling pathways such as Gefitinib, Erlotinib, Vandetanib, Lapatinib, WHI-P180 and Afatinib38 (Fig. 1).
 | ||
| Fig. 1 Examples of drugs containing iodo substitutions and some quinazoline multitargeted TKIs. | ||
The hybridization strategy aimed at combining the EGFR, VEGFR inhibition and anticancer activity of quinazolines with other privileged pharmacophores possessing antitumor potency could enhance the activity of the compounds.2,42 In this work, we designed some new iodoquinazolinones as potential VEGFR-2 and EGFR inhibitors by keeping the acetamide linker attached to the N-3 of the iodoquinazoline ring and modification of the distal side chains to be aliphatic, aromatic and/or heterocyclic rings.
The current investigation is an extension to our preceding work12,43–51 that reveals the efficacy of the hybridization strategy between iodoquinazolinone and different moieties as EGFR, VEGFR-2 inhibitors and anticancer agents. We designed and synthesized a series of new 6-iodoquinazolinone bearing different moieties with acetamide linker as dual EGFR-mutant/VEGFR-2 inhibitors for the treatment of various cancer types, as liver (HepG2), breast (MCF-7), colorectal (HCT-116) and NSCLC (A549). In silico molecular modeling study was performed for our compounds as dual VEGFR-2/EGFR inhibitors to explore their binding mode in the enzymes active sites or predict their ADMET properties.
1.1. Rationale and structure-based design
Our derivatives were designed based on the pharmacophoric features of both EGFR and VEGFR-2 inhibitors. We synthesized a series of new iodoquinazoline derivatives inspired by the bioisosteric modifications of VEGFR-2 inhibitors (sorafenib and sunitinib) at four different positions: (1) a flat hetero-aromatic ring that occupies the ATP binding domain, (2) a central hydrophobic spacer, (3) a linker with a functional group that has both H-bond acceptor and donor properties to bind with key amino acids (Glu885 and Asp1046), and (4) a terminal hydrophobic moiety that fits into the allosteric hydrophobic pocket (Fig. 2).52,53 | ||
| Fig. 2 Basic structural features of VEGFR-2 inhibitors. | ||
Also, the iodoquinazolinone derivatives 5a–d to 9a–e possess the essential pharmacophoric features of EGFR-TKIs. These derivatives are based on structural modifications of erlotinib at four different positions: (1) a terminal hydrophobic head that fits into hydrophobic region I, (2) a flat hetero-aromatic system that occupies the adenine binding pocket, (3) HBD and/or HBA (NH as HBD) that forms important hydrogen bond interactions with amino acid residues in the linker region, and (4) a hydrophobic tail that fits into hydrophobic region II (Fig. 3).54
 | ||
| Fig. 3 Basic structural features of EGFR inhibitors. | ||
2. Results and discussion
2.1. Chemistry
The synthetic strategy for preparation of the target compounds 5a–d to 9a–e is depicted in Scheme 1 and 2. Synthesis was initiated by reaction of chloroacetyl chloride with the appropriate amine Ia–c to afford the corresponding chloroacetamide IIa–c.55,56 The chloroactamide IIa underwent reaction with the appropriate methyl and/or ethylamine according to the mixed anhydride procedure56 to afford the corresponding derivatives IIIa,b respectively. On the other hand anthranilic acid (1) reacted with iodine to afford 5-iodoanthranilic acid (2) which underwent alkylation with ethyl iodide and/or propyl iodide to give the corresponding N-alkyl derivatives (3a,b).57 Fusion of 5-iodo-N-alkylanthranilic acid (3a,b) with urea afforded the corresponding 6-iodo-1-alkylquinazoline-2,4(1H,3H)-dione (4a,b).57 In situ reaction of 6-iodo-1-alkylquinazoline-2,4(1H,3H)-dione (4a,b) with the appropriate chloroacetamide IIb,c afforded the corresponding ester derivatives 5a–d. Also reaction of 6-iodo-1-alkylquinazoline-2,4(1H,3H)-dione (4a,b) with the appropriate chloroacetamide IIIa,b to get the corresponding amide derivatives 6a,b (Scheme 1). Reaction of the appropriate ethyl ester 5b,d with hydrazine hydrate resulted in the corresponding hydrazide derivative 7a,b which underwent cyclization with carbon disulfide giving the corresponding 5-sulfanyloxadiazole 8a,b respectively. On the other hand, condensation of the appropriate acid hydrazide 7a,b with the appropriate benzaldehyde afforded the corresponding Schiff's bases 9a–e respectively (Scheme 2). | ||
| Scheme 1 Synthetic route for preparation of the target compounds 5a–d and 6a,b. | ||
 | ||
| Scheme 2 Synthetic route for preparation of the target compounds 7a,b to 9a–e. | ||
2.2. Docking studies
For molecular docking investigations, Molsoft software was used. The PDB IDs for VEGFR-2 (PDB ID 4ASD)48,58 and EGFRT790M (PDB ID 3W2O)55,56 were used in each experiment, respectively.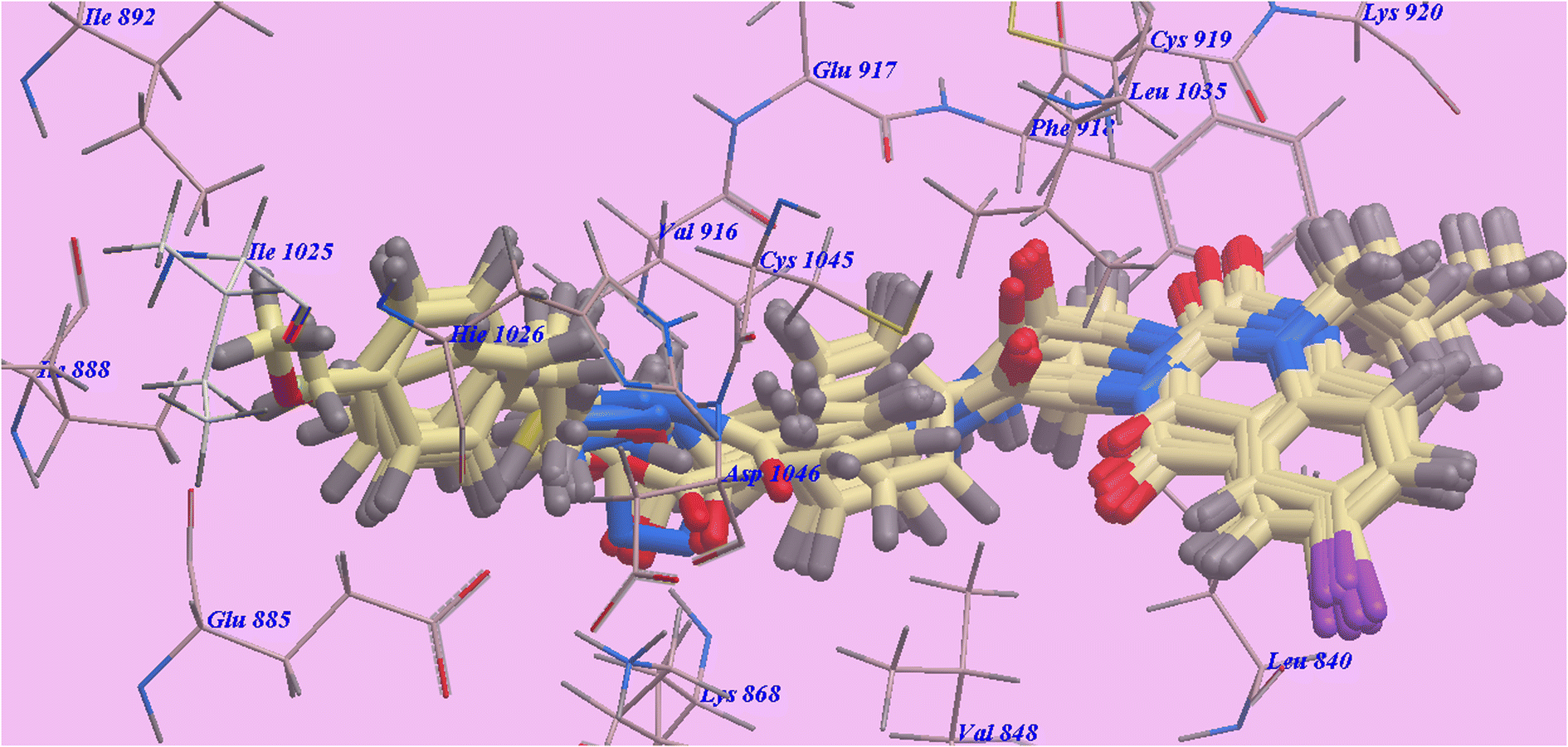 | ||
| Fig. 4 Compounds superimposed inside the active site of 4ASD. | ||
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 5a | −86.20 | 8a | −88.77 |
| 5b | −90.99 | 8b | −93.50 |
| 5c | −87.37 | 9a | −102.82 |
| 5d | −95.01 | 9b | −107.14 |
| 6a | −91.18 | 9c | −110.10 |
| 6b | −88.47 | 9d | −108.45 |
| 7a | −79.59 | 9e | −102.12 |
| 7b | −83.13 | Sorafenib | −99.50 |
The Sorafenib-suggested binding mechanism produced 5 H-bonds and an energy value of −99.50 kcal mol−1. It generated 2 hydrogen bonds with Cys919 (2.51 Å and 2.10 Å), two with Glu885 (1.77 Å and 2.75 Å), and one with Asp1046 (1.50 Å). The pocket created by Cys919, Phe918, Leu1035, Lys920, Glu917, Val848 and Leu840 occupied by the N-methylpicolinamide group. Additionally, the hydrophobic groove formed by Cys1045, Leu1035, Thr916, Lys868, and Val848 occupied by the central phenyl linker. Additionally, Cys1045, Asp1046, Hie1026, Ile888, Ile892 and Glu885 formed a hydrophobic channel that occupied by the terminal 3-trifluromethyl-4-chlorophenyl group (Fig. 5). The urea spacer had a crucial part in the binding of the VEGFR-2 enzyme, but it also played a crucial role in the high binding affinity of Sorafenib. These results encourage us to experiment with various linkers in order to obtain efficient VEGFR-2 inhibitors.
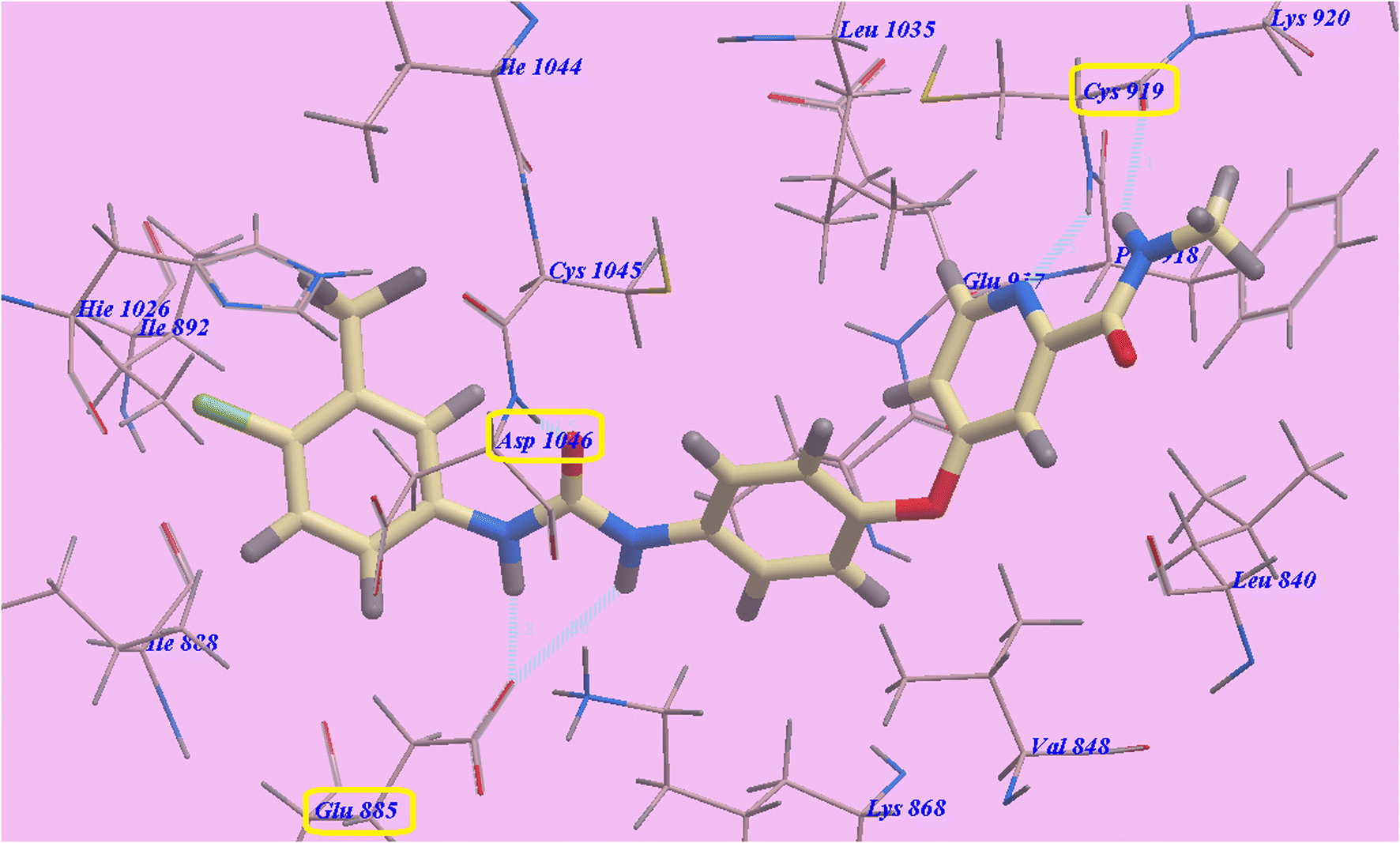 | ||
| Fig. 5 Sorafenib predicted mechanism of binding with 4ASD. Dotted lines represent atoms that are H-bonded. | ||
Compound 9c predicted binding mode is nearly identical to that of Sorafenib, which showed an affinity value of −110.10 kcal mol−1 and 6 H-bonds. 1-Ethyl-6-iodoquinazoline ring formed one H-bond with Cys919 (2.33 Å). The acetamide linker also formed another H-bond with Cys919 (2.96 Å). The acid hydrazide linker formed three H-bonds with Asp1046 (2.50 Å) and Glu885 (2.15 Å and 2.21 Å), while the 4-methoxy group at the side chain formed one H-bond with Ile1025 (2.98 Å). The pocket formed by Leu1035, Lys920, Cys919, Phe918, Glu917, Val848 and Leu840 occupied by the Iodoquinazoline scaffold. Additionally, the hydrophobic channel created by Hie1026, Ile1025, Ile892, Ile888 and Glu885 occupied by the distal 4-methoxyphenyl moiety (Fig. 6).
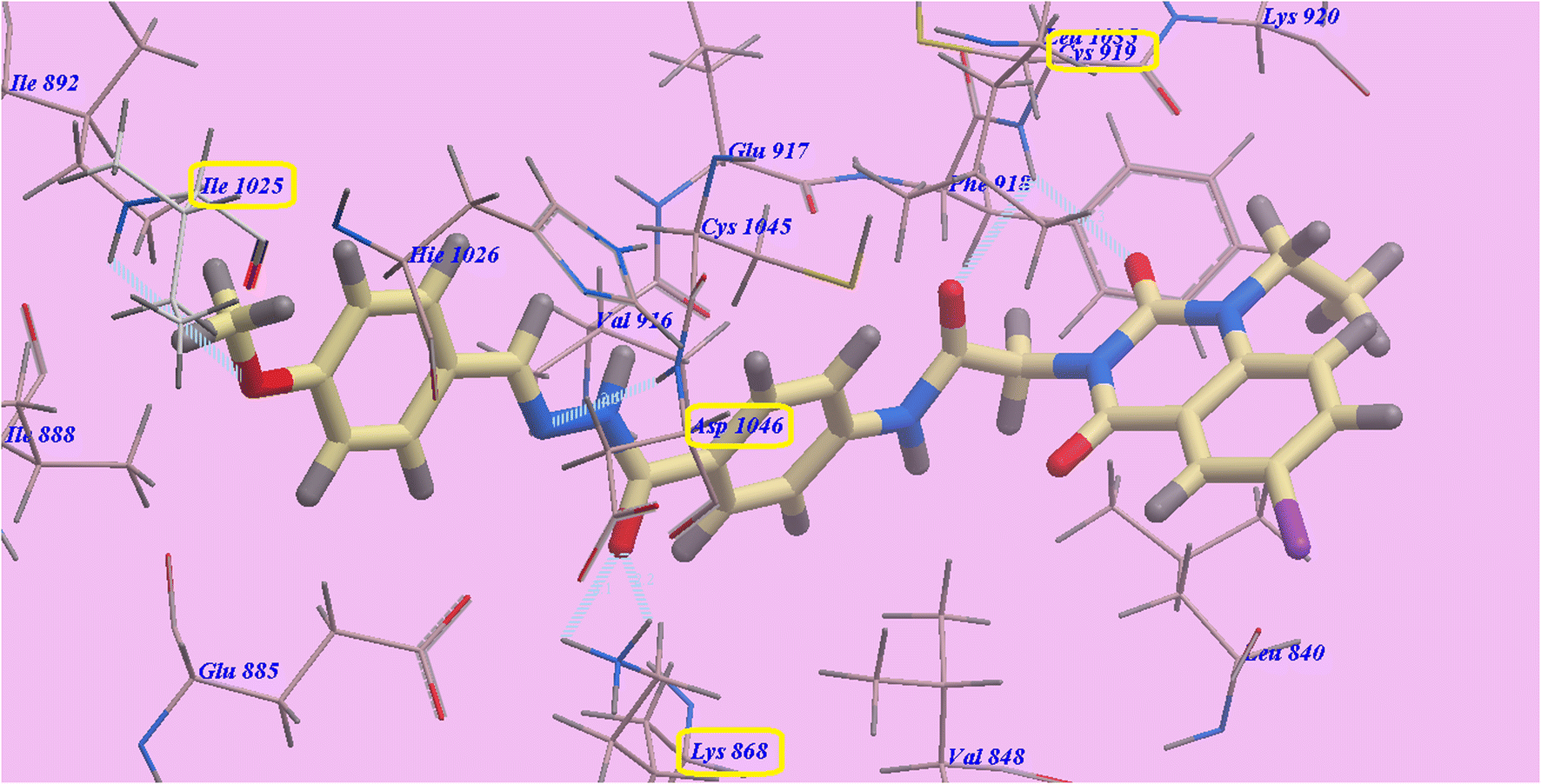 | ||
| Fig. 6 9c predicted binding mode with 4ASD. | ||
Compound 9d predicted binding mode is nearly identical to that of Sorafenib and 9c, which showed an affinity value of −108.45 kcal mol−1 and 5 H-bonds. One H-bond was created with the 6-iodo-1-propylquinazoline ring and Cys919 (2.70 Å). The acetamide linker also formed another H-bond with Cys919 (2.97 Å). The carbonyl of hydrazide linker made two H-bonds with Lys868 (2.02 Å and 2.63 Å), and its NH group designed a third hydrogen bond with Asp1046 (2.43 Å) (Fig. 7). Moreover, compound 9b suggested binding mode has five H-bonds and an affinity value of −107.14 kcal mol−1, which is almost identical to that of compound 9c. It formed five H-bonds with Cys919 (2.74 Å and 2.98 Å), Lys868 (2.00 Å and 2.80 Å) and Asp1046 (2.28 Å) (Fig. 8).
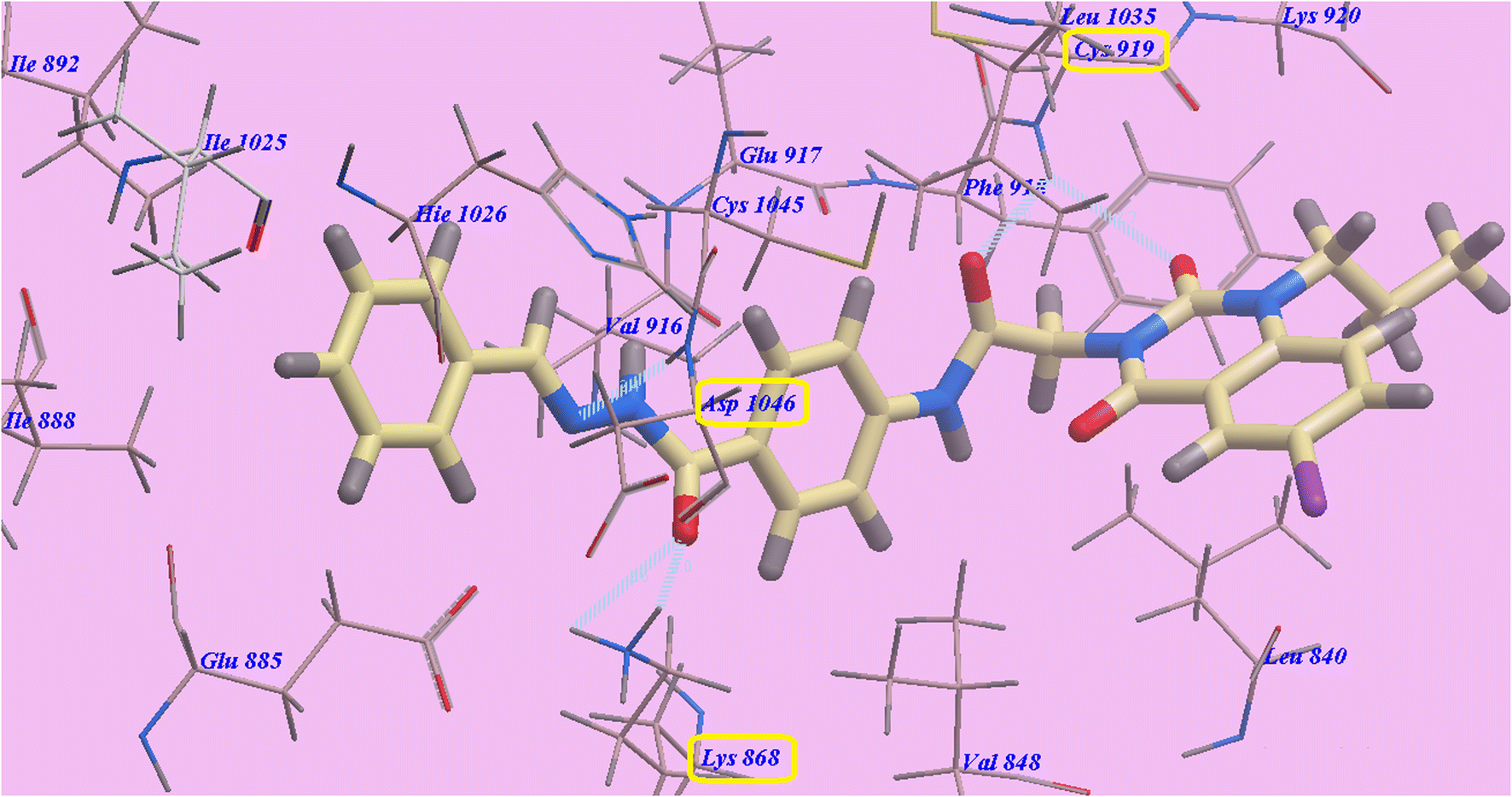 | ||
| Fig. 7 Docking results for 9d with 4ASD. | ||
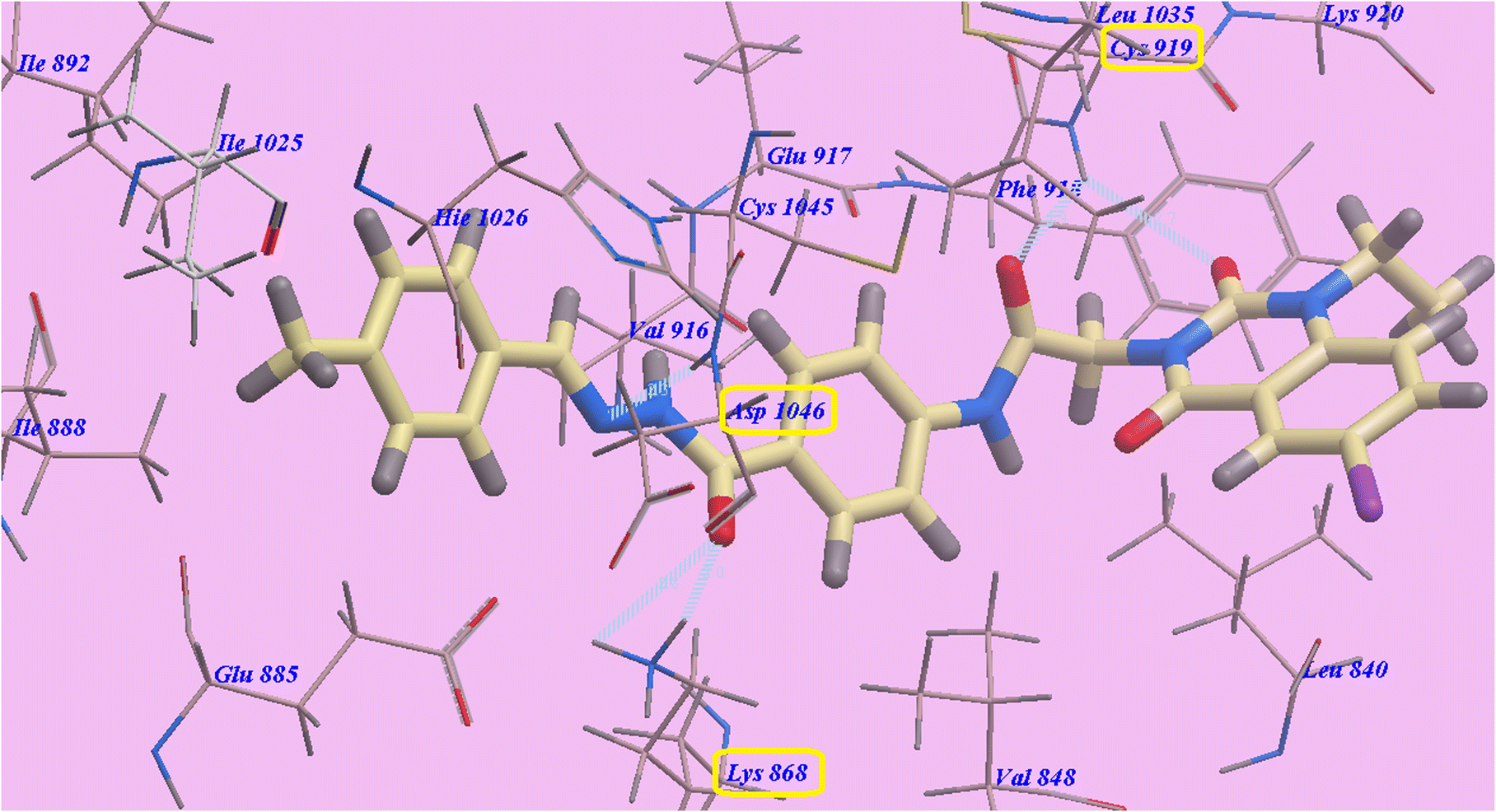 | ||
| Fig. 8 Docking results for 9b with 4ASD. | ||
The obtained binding moods in docking results (Table 1) led us to the conclusion that the used linkers shared the same groove as the urea linker of Sorafenib and performed the same function, both of which are necessary for greater affinity towards the VEGFR-2 enzyme. The establishment of an H-bond with cysteine 919 and the emergence of hydrophobic interactions may have contributed to the heterocyclic N-alkyl-6-iodoquinazoline scaffold's higher affinity towards the VEGFR-2 enzyme.
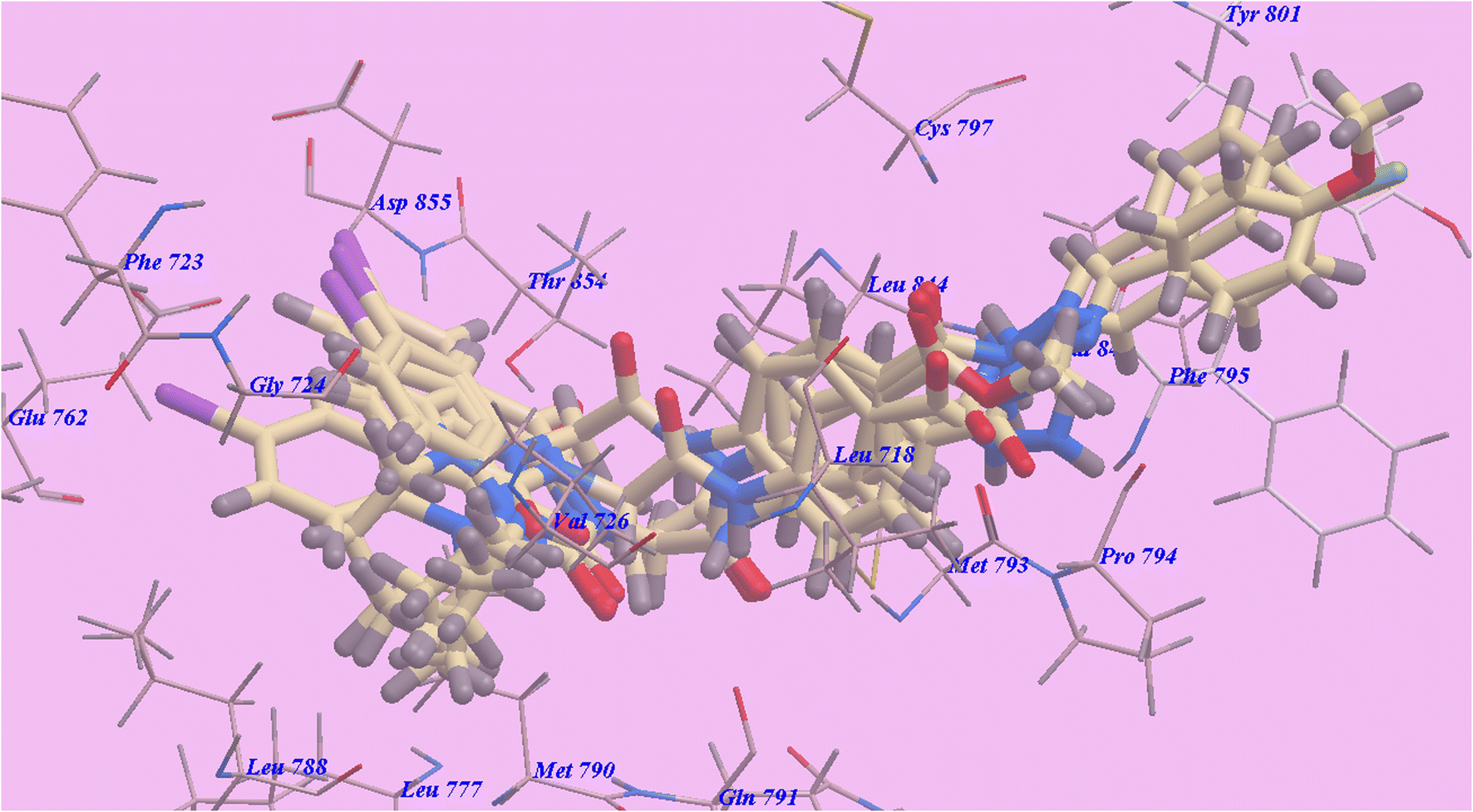 | ||
| Fig. 9 Certain docked compounds superimposed inside the binding pocket of 3W2O. | ||
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 5a | −82.03 | 8a | −80.89 |
| 5b | −83.02 | 8b | −85.85 |
| 5c | −80.71 | 9a | −83.59 |
| 5d | −86.58 | 9b | −84.20 |
| 6a | −81.91 | 9c | −90.95 |
| 6b | −83.76 | 9d | −89.30 |
| 7a | −73.37 | 9e | −88.10 |
| 7b | −69.77 | Erlotinib | −82.77 |
Erlotinib's hypothesized binding mode revealed an binding energy value of −82.77 kcal mol−1 and four H-bonds, one of the two 2-methoxyethoxy groups established one H-bond with the residing amino acid Cys797 (2.05 Å), The creation of two H bonds with Val726 (2.97 Å) and Met793 (1.82 Å) and stabilized the quinazoline moiety. The NH spacer and Thr854 (2.99 Å) established one H-bond. The hydrophobic region I, which formed by Val726, Asp855, Thr854, Glu791, Met790, Leu777, Glu762, Ile759, and Phe723, was occupied by the 3-ethynylphenyl head. Additionally, the 2-methoxyethoxy tail occupied the hydrophobic area II created by Val845, Leu844, Pro794, Met793, and Leu718 (Fig. 10).
 | ||
| Fig. 10 Docking results for Erlotinib with 3W2O, H-bonded atoms are specified by dotted lines. | ||
Compound 9c predicted binding mode is nearly identical to that of erlotinib, which displayed an affinity value of −90.95 kcal mol−1 and 6 H-bonds. Iodoquinazoline scaffold made two hydrogen bonds with Thr854 (2.22 Å) and Met790 (2.96 Å), while acetamide linker formed one hydrogen bond with Met793 (2.27 Å). Moreover, the hydrazide linker formed three H-bonds with Met793 (2.96 Å), Pro794 (2.77 Å) and Phe795 (2.95 Å). Moreover, the 4-methoxy group formed H-bond with Tyr801 (2.98 Å). The hydrophobic region II, which created by Val845, Leu844, Cys797, Phe795, Pro794, Met793, Leu718 and Tyr801, was occupied by the distal 4-methoxyphenyl side chain and the acid hydrazide linker. The hydrophobic region I, which was created by Asp855, Thr854, Met790, Leu788 and Gly724 was occupied by iodoquinazoline scaffold (Fig. 11).
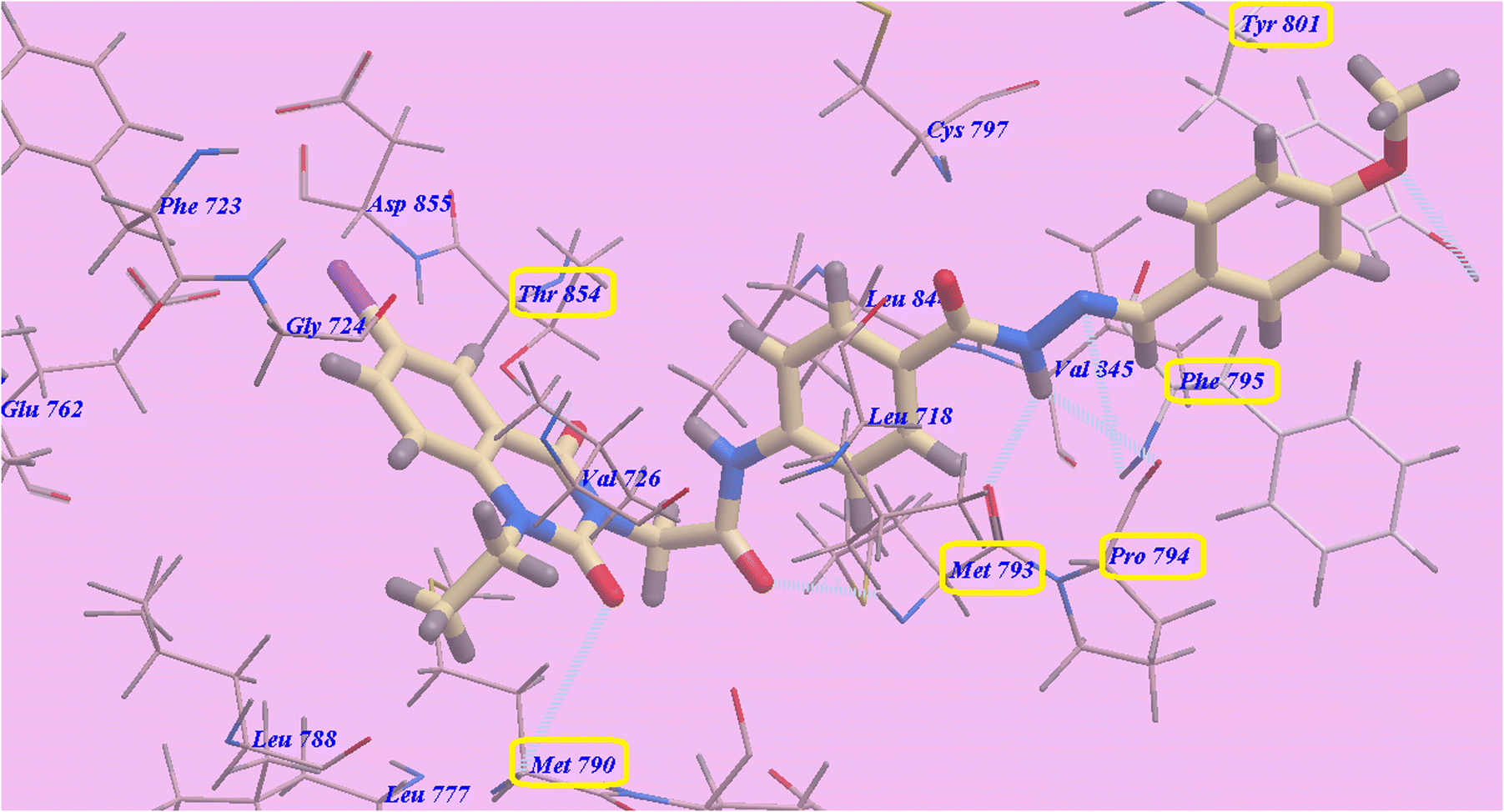 | ||
| Fig. 11 Predicted binding mode for 9c with 3W2O. | ||
Compounds 9d and 9e have suggested binding mechanisms that are nearly identical to those of erlotinib and 9c, with affinities of −89.30 kcal mol−1 (formed five H-bonds) (Fig. 12) and −88.10 kcal mol−1 (formed 5 H-bonds) (Fig. 13), respectively.
 | ||
| Fig. 12 Docking results for compound 9d with 3W2O. | ||
 | ||
| Fig. 13 Docking results for compound 9e with 3W2O. | ||
2.3. Biological evaluation
| Comp. | EC50 (μM)a | CC50 (μM)b | IC50 (μM)c | |||||
|---|---|---|---|---|---|---|---|---|
| HepG2 | MCF-7 | HCT116 | A549 | VERO | VEGFR-2 | EGFRT790M | EGFRWT | |
| a EC50 (50% effective concentration).b CC50 (50% cytotoxic concentration).c IC50 (50% inhibitory concentration).d NT: not tested compounds. | ||||||||
| 5a | 12.53 ± 0.7 | 11.60 ± 0.7 | 11.44 ± 0.7 | 8.45 ± 0.7 | NTd | 2.00 ± 0.25 | 0.86 ± 0.50 | NTd |
| 5b | 7.59 ± 0.7 | 7.40 ± 0.7 | 6.55 ± 0.7 | 6.78 ± 0.7 | NTd | 1.50 ± 0.10 | 0.80 ± 0.50 | NTd |
| 5c | 7.75 ± 0.7 | 7.96 ± 0.7 | 6.93 ± 0.7 | 6.73 ± 0.7 | NTd | 1.50 ± 0.50 | 0.92 ± 0.50 | NTd |
| 5d | 7.50 ± 0.7 | 7.37 ± 0.7 | 6.52 ± 0.7 | 6.68 ± 0.7 | 46.60 ± 0.50 | 1.25 ± 0.10 | 0.40 ± 0.35 | 0.32 ± 0.03 |
| 6a | 7.80 ± 0.8 | 8.50 ± 0.8 | 7.60 ± 0.8 | 7.95 ± 0.8 | NTd | 1.40 ± 0.20 | 0.90 ± 0.50 | NTd |
| 6b | 8.50 ± 0.8 | 8.15 ± 0.8 | 8.35 ± 0.8 | 6.70 ± 0.8 | NTd | 1.65 ± 0.10 | 0.60 ± 0.35 | NTd |
| 7a | 16.00 ± 1.5 | 15.50 ± 1.5 | 13.25 ± 1.5 | 13.66 ± 1.5 | NTd | 2.50 ± 0.25 | 1.50 ± 0.50 | NT |
| 7b | 15.50 ± 1.5 | 13.90 ± 1.5 | 15.90 ± 1.5 | 15.10 ± 1.5 | NTd | 2.25 ± 0.25 | 2.05 ± 0.50 | NTd |
| 8a | 7.55 ± 0.9 | 8.25 ± 0.9 | 7.25 ± 0.9 | 10.10 ± 0.9 | NTd | 1.50 ± 0.10 | 1.00 ± 0.50 | NTd |
| 8b | 7.50 ± 0.7 | 7.45 ± 0.7 | 6.75 ± 0.7 | 6.70 ± 0.5 | 50.75 ± 0.50 | 1.38 ± 0.10 | 0.45 ± 0.35 | 0.38 ± 0.03 |
| 9a | 6.80 ± 0.7 | 7.50 ± 0.7 | 6.29 ± 0.7 | 6.75 ± 0.7 | 45.50 ± 0.50 | 1.00 ± 0.10 | 0.75 ± 0.50 | NTd |
| 9b | 6.20 ± 0.7 | 6.70 ± 0.6 | 5.93 ± 0.6 | 6.75 ± 0.6 | 49.25 ± 0.50 | 0.90 ± 0.10 | 0.50 ± 0.35 | 0.35 ± 0.03 |
| 9c | 5.00 ± 0.5 | 6.00 ± 0.5 | 5.17 ± 0.5 | 5.25 ± 0.5 | 55.00 ± 0.50 | 0.85 ± 0.10 | 0.22 ± 0.30 | 0.15 ± 0.03 |
| 9d | 5.65 ± 0.5 | 6.45 ± 0.5 | 6.50 ± 0.5 | 5.48 ± 0.5 | 50.39 ± 0.50 | 0.90 ± 0.10 | 0.26 ± 0.30 | 0.20 ± 0.03 |
| 9e | 7.36 ± 0.7 | 7.30 ± 0.7 | 6.70 ± 0.7 | 6.30 ± 0.7 | 46.00 ± 0.50 | 1.20 ± 0.10 | 0.30 ± 0.30 | 0.25 ± 0.03 |
| Sorafenib | 4.00 ± 0.33 | 5.05 ± 0.50 | 5.58 ± 0.55 | 4.04 ± 0.33 | NTd | 0.84 ± 0.04 | NTd | NTd |
| Erlotinib | 7.73 ± 0.67 | 13.91 ± 1.3 | 8.20 ± 0.34 | 5.49 ± 0.45 | NTd | NTd | 0.24 ± 0.22 | 0.18 ± 0.02 |
Regarding HepG2, compounds 5b–d, 6a–b, 8a–b and 9a,b,d,e exhibited very good anticancer effects with EC50 ranging from 5.65 to 8.50 μM. Derivative 5a with EC50 = 12.53 μM, showed good cytotoxicity. On the other hand, derivatives 7a and 7b exhibited moderate anticancer effects with EC50 = 16.00 and 15.50 μM respectively.
Concerning MCF-7, derivatives 5b–d, 6a–b, 8a–b and 9a,b,d,e exhibited very good anticancer effects with EC50 ranging from 6.45 to 8.50 μM. Derivatives 5a and 7b with EC50 = 11.60 and 13.90 μM respectively, showed good cytotoxicity. On the other hand, derivative 7a exhibited moderate anticancer effects with EC50 = 15.50 μM.
Derivatives 5b–d, 6a–b, 8a–b and 9a,b,d,e exhibited very good anticancer effects with EC50 ranging from 5.93 to 8.35 μM against HCT-116. Derivatives 5a and 7a with EC50 = 11.44 and 13.25 μM respectively, showed good cytotoxicity. On the other hand, derivative 7b exhibited moderate anticancer effects with EC50 = 15.90 μM.
Derivative 5a–d, 6a–b, 8b and 9a,b,d,e exhibited very good anticancer effects with EC50 ranging from 5.48 to 8.45 μM against A549. Derivatives 7a and 8a with EC50 = 13.66 and 10.10 μM, showed good cytotoxicity. On the other hand, derivative 7b presented moderate anticancer effect with EC50 = 15.10 μM.
2.4. Selectivity index (SI)
Finally, the highly effective six derivatives 5d, 8b and 9a–e were examined against VERO normal cell lines to estimate their cytotoxic capabilities. All conclusions revealed that each discovered compound possessed low toxicity regarding VERO normal cells with 50% cytotoxic concentration (CC50) extending from 45.50 to 55.00 μM. One criterion for the anticancer drug to be good it should not affect the non-cancer cells. A molecule could be considered as highly selective if it presented SI value ≥5. A molecule with moderate selectivity presents SI value >2 while low selectivity is considered if the SI is lower than 2.62 In this study, derivatives 5d, 8b, 9a, 9b, 9c, 9d and 9e are correspondingly 6.21, 6.77, 6.69, 7.94, 11.00, 8.92 and 6.25 folds more toxic respecting HepG2 than normal VERO cells. Uniformly, derivatives 5d, 8b, 9a, 9b, 9c, 9d and 9e are consequently 6.32, 6.81, 6.07, 7.35, 9.17, 7.81 and 6.30 folds toxic in MCF-7 than in normal VERO cells. Furthermore, structures 5d, 8b, 9a, 9b, 9c, 9d and 9e are respectively 7.15, 7.52, 7.23, 8.31, 10.64, 7.75 and 6.87 folds grander lethalness in HCT-116 than in ordinary VERO cells. Furthermore, products 5d, 8b, 9a, 9b, 9c, 9d and 9e are respectively 6.98, 7.57, 6.74, 7.30, 10.48, 9.20 and 7.30 folds toxicity in A549 than in ordinary VERO cells. All derivatives exhibited high selectivity against the tested cancer cell lines where SI values >5.Compounds 9c, 9b, 9d, 9a, 9e and 5d excellently inhibited VEGFR-2 activity with IC50 = 0.85, 0.90, 0.90, 1.00, 1.20 and 1.25 μM respectively. Candidates 5b, 5c, 6a, 6b, 8a and 8b significantly inhibited VEGFR-2 at IC50 range of 1.38–1.65 μM. Alternatively, compounds 5a, 7a and 7b moderately inhibited VEGFR-2 at IC50 = 2.00, 2.50 and 2.25 μM, respectively.
Based on the structure of the synthesized derivatives and the data in Table 3, we can divide the tested compounds into three groups. Compounds 5a–d, 6a,b and 7a,b which have ester, amide and hydrazide moieties respectively as side chains, belong to the first category. Generally in this group esters exhibited higher anticancer activities than amides and hydrazides respectively.
Compound 5d with the more hydrophobic 1-propyl and ethyl ester side chain exhibited higher anticancer activities than compound 5b with 1-ethyl and ethyl ester side chain, compound 5c with 1-propyl and methyl ester side chain and 5a with 1-ethyl and methyl ester side chain against the four tested cell lines except A549 where the order of activity is 5d > 5c > 5b > 5a. Moreover, the ester derivatives 5d–c showed higher activities than the amides 6a and 6b against HepG2, MCF-7 and HCT-116, but in case of A549 the order of activity is 5d > 6b > 5c > 5b > 6a. In addition, the amide 6a with 1-ethyl and ethyl amide side chain displayed higher activities than the amide 6b with 1-propyl and methyl amide side chain against HepG2 and HCT-116 but 6b showed higher activities than 6a against MCF-7 and A549. The hydrazide derivatives 7a and 7b displayed the lowest anticancer activities along this series. The hydrazide 7b with 1-propyl substitution exhibited higher activities than 7a with 1-ethyl against HepG2 and MCF-7 cell lines but in case of HCT-116 and A549 cell lines 7a showed higher activities than 7b.
Compounds 8a and 8b in the second group have a distal hydrophobic heteroaromatic oxadiazole moiety. Compound 8b with 1-propyl and oxadiazol-5-thiol side chain groups showed stronger effects than compound 8a with 1-ethyl and oxadiazol-5-thiol side chain groups against the four HepG2, MCF-7, HCT-116 and A549 cell lines. This finding indicated that the long chain alkyl group at position-1 increases the anticancer activities.
The third group consists of compounds 9a–e with distal hydrophobic Schiff's bases side chains. Compound 9c with 1-ethyl and highly electron donating methoxy group at position-4 of the phenyl side chain showed strongest activities against the four cell lines along this group. Derivative 9d with 1-propyl and unsubstituted phenyl side chain showed higher activities than compound 9b 1-ethyl and 4-methylphenyl side chain and 9a with 1-ethyl and unsubstituted phenyl side chain against the four cell lines except HCT-116 where 9b > 9a > 9d. Moreover, derivatives 9b demonstrated higher action than 9a against HepG2, MCF-7, and HCT-116 but showed the same activity against A549. Finally, derivative 9d with 1-propyl and unsubstituted phenyl side chain showed higher activities than compound 9e with 1-propyl and the electron withdrawing 4-chlorophenyl side chain against the four tested cell lines. These findings enable us to conclude that substitution with electron donating groups at para-position showed higher activities than electron withdrawing ones.
2.5. ADMET; in silico studies profile
In silico report of the highly active derivatives 9b, 9c and 9e was conducted for their physicochemical character evaluation and the proposed ADMET profile. It was predicted using pkCSM descriptor algorithm procedures66 and matched to the rule of five described by Lipinski.67 Good absorption properties were expected for the molecules that accomplish at least three rules: (i) no more than five hydrogen bond donors, (ii) no more than 10 hydrogen bond acceptors, (iii) molecular weight less than 500, (iv) not more than 5 for log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P. In the current work, the standard anticancer agent sorafenib violate the logP rule while our new compounds 9b, 9c and 9e violate the molecular weight rule but erlotinib doesn't violate any rule.
P. In the current work, the standard anticancer agent sorafenib violate the logP rule while our new compounds 9b, 9c and 9e violate the molecular weight rule but erlotinib doesn't violate any rule.
As a result of obtaining data from Table 4, we can assume that compounds 9b, 9c and 9e have very good GIT absorption in human (82.751 to 88.2) which indicates easier to cross different biological membranes.68 So, they may show a significantly high bioavailability through GIT. Concerning CNS penetrability, our prepared compounds can reach CNS (CNS permeability values −2.266 to −2.535), comparable to that of sorafenib (−2.007) and erlotinib (−3.216).
| Parameter | 9b | 9c | 9e | Sorafenib | Erlotinib |
|---|---|---|---|---|---|
| Physicochemical properties | |||||
| Molecular weight | 609.424 | 625.423 | 643.869 | 464.831 | 393.443 |
| LogP |
3.49872 | 3.1989 | 4.2338 | 5.5497 | 3.4051 |
| Rotatable bonds | 7 | 8 | 8 | 5 | 10 |
| Acceptors | 7 | 8 | 7 | 4 | 7 |
| Donors | 2 | 2 | 2 | 3 | 1 |
| Surface area | 225.514 | 230.628 | 235.817 | 185.111 | 169.532 |
|
|||||
| Absorption | |||||
| Water solubility | −4.304 | −4.504 | −4.457 | −4.822 | −4.736 |
| Caco2 permeability | 0.384 | 0.399 | 0.421 | 0.689 | 1.431 |
| Human intest. absorption | 86.879 | 82.751 | 88.2 | 89.043 | 94.58 |
| Skin permeability | −2.75 | −2.748 | −2.743 | −2.767 | −2.741 |
| Substrate for P-glycoprotein | + | + | + | + | − |
| Inhibitor of P-glycoprotein I | + | + | + | + | + |
| Inhibitor of P-glycoprotein II | + | + | + | + | + |
|
|||||
| Distribution | |||||
| VDss (human) | −0.388 | −0.353 | −0.326 | −0.29 | 0.199 |
| Human unbound fraction | 0.101 | 0.092 | 0.086 | 0.065 | 0.059 |
| Permeability throughout BBB | −0.7 | −0.912 | −0.878 | −1.684 | −0.745 |
| Permeability to CNS | −2.286 | −2.535 | −2.266 | −2.007 | −3.216 |
|
|||||
| Metabolism | |||||
| CYP2D6 substrate | − | − | − | − | − |
| CYP3A4 substrate | + | + | + | + | + |
| Inhibition of CYP1A2 | + | − | + | + | + |
| Inhibition of CYP2C19 | + | + | + | + | + |
| Inhibition of CYP2C9 | + | + | + | + | + |
| Inhibition of CYP2D6 | − | − | − | − | − |
| Inhibition of CYP3A4 | + | + | + | + | + |
|
|||||
| Excretion | |||||
| Clearance | −0.817 | −0.838 | −0.858 | −0.219 | 0.702 |
| Renal OCT2 substrate | − | − | − | − | − |
|
|||||
| Toxicity | |||||
| AMES toxicity | − | − | − | − | − |
| Human max. tolerated dose | 0.421 | 0.421 | 0.533 | 0.549 | 0.839 |
| Inhibitor of hERG I | − | − | − | − | − |
| hERG II inhibitor | + | + | + | + | + |
| Acute toxic activity (LD50) | 2.439 | 2.468 | 2.346 | 2.538 | 2.393 |
| Chronic toxic activity (LOAEL) | 1.873 | 1.74 | 1.925 | 1.198 | 1.37 |
| Hepatotoxic effect | + | + | + | + | + |
| Skin sensitization | − | − | − | − | − |
| T. Pyriformis toxicity | 0.361 | 0.333 | 0.343 | 0.383 | 0.309 |
| Minnow toxic activity | 0.849 | 0.526 | 0.171 | 0.189 | −0.1 |
It is well known that CYP3A4, the major drug-metabolizing enzyme, could be inhibited by sorafenib, erlotinib and also our derivatives 9b, 9c and 9e. Elimination was expected depending on the total clearance which is a considerable factor in deciding dose intervals. The data showed that erlotinib confirmed higher clearance rates compared with sorafenib and our new compounds which demonstrated low clearance values. Thus, erlotinib could be eliminated faster, and as a result, supposed to have shorter dosing intervals. Unlike erlotinib, the prepared compounds exhibited slowly clearance rate, which signifies a long duration of action and extended dosing intervals. Toxicity is the final ADMET profile studied factor. Like presented in Table 4, sorafenib, erlotinib, and the novel compounds 9b, 9c and 9e shared the drawback of unwanted hepatotoxic actions. Erlotinib, sorafenib, 9b, 9c and 9e demonstrated high maximum tolerated dose. These involve the advantage of the broad therapeutic index of erlotinib, sorafenib, and our derivatives respectively. Lastly, the oral chronic toxic doses of the novel compounds 9b, 9c and 9e are higher than that of sorafenib and erlotinib.
3. Conclusion
Fifteen new 1-alkyl-6-iodoquinazoline derivatives bearing different aliphatic, aromatic and/or heteroaromatic moieties were designed and synthesized as dual EGFRT790M/VEGFR-2 inhibitors. The cytotoxic activities of the new compounds were screened in vitro against HepG2, MCF-7, HCT116 and A549 using an MTT assay. Their ability to bind with both EGFR and VEGFR-2 receptors was examined by molecular modeling. Compound 9c showed the highest anticancer activities with EC50 = 5.00, 6.00, 5.17 and 5.25 μM against HepG2, MCF-7, HCT116 and A549 cell lines correspondingly. Despite it showed lower activities than sorafenib (EC50 = 4.00, 5.05, 5.58 and 4.04 μM) versus HepG2, MCF-7 and A549 contrarily subjacent performance against HCT116, it demonstrated higher actions than erlotinib (EC50 = 7.73, 13.91, 8.20 and 5.49 μM), against the four tested cell lines. Moreover, compounds 5d, 8b, 9a, 9b, 9d, and 9e exhibited very good anticancer effects against the tested cancer cell lines. The highly effective seven derivatives 5d, 8b, 9a, 9b, 9c, 9d and 9e were examined against VERO normal cell lines to estimate their cytotoxic capabilities. All conclusions revealed that each discovered compound possessed low toxicity regarding VERO normal cells with CC50 extending from 45.50 to 55.00 μM. Additionally, all derivatives were evaluated for their ability as dual inhibitors of VEGFR-2 and EGFRT790M in comparing to sorafenib and erlotinib as standards with IC50 = 0.84 and 0.24 respectively. Moreover, compounds 9c, 9d, 9e, 5d, 8b and 9b excellently inhibited EGFRT790M activity with IC50 = 0.22, 0.26, 0.30, 0.40, 0.45 and 0.50 μM respectively. Also, compounds 9c, 9d and 9e excellently inhibited EGFRWT activity with IC50 = 0.15, 0.20 and 0.25 μM respectively in comparing to erlotinib (IC50 = 0.18 μM). As planned, compound 9c showed excellent dual EGFR/VEGFR-2 inhibitory activities. Consonantly, ADMET study was calculated in silico for the supreme three worthwhile compounds 9b, 9c and 9e in contrasting to sorafenib and erlotinib as reference drugs.4. Experimental
4.1. Chemistry
All melting points were carried out by open capillary method on a Gallen kamp Melting point apparatus at faculty of pharmacy Al-Azhar University and were uncorrected. The infrared spectra were recorded on pye Unicam SP 1000 IR spectrophotometer at Microanalytical Unit, Faculty of Pharmacy, Cairo University using potassium bromide disc technique. Proton magnetic resonance 1HNMR spectra were recorded on a Bruker 400 MHZ-NMR spectrophotometer at Microanalytical Center, Ain Shams University, and/or a Mercury 300 MHZ-NMR spectrophotometer Faculty of Science, Cairo University. 13CNMR spectra were recorded on a Bruker 100 MHZ-NMR spectrophotometer at Microanalytical Unit, Faculty of pharmacy, Cairo University. TMS was used as internal standard and chemical shifts were measured in d scale (ppm). The mass spectra were carried out on Direct Probe Controller Inlet part to Single Quadropole mass analyzer in Thermo Scientific GCMS model ISQ LT using Thermo X-Calibur software at the Regional Center for Mycology and Biotechnology, Al-Azhar University. Elemental analyses (C, H, N) were performed on a CHN analyzer at Regional Center for Mycology and Biotechnology, Al-Azhar University. All compounds were within ±0.4 of the theoretical values. The reactions were monitored by thin-layer chromatography (TLC) using TLC sheets precoated with UV fluorescent silica gel Merck 60 F254 plates and were visualized using UV lamp and different solvents as mobile phases.Alkyl 4-(2-chloroacetamido)benzoate (IIb,c), 4-(2-chloroacetamido)-N-alkylbenzamide (IIIa,b), 5-iodoanthranilic acid (2), 5-iodo-N-alkylanthranilic acid (3a,b), and 6-iodo-1-alkylquinazoline-2,4(1H,3H)-dione (4a,b), were synthesized according to the reported procedures.55–57
4.1.1.1. General method. A mixture of equimolar quantities of 6-iodo-1-ethylquinazoline-2,4(1H,3H)-dione (4a) and/or 6-iodo-1-propylquinazoline-2,4(1H,3H)-dione (4b) (0.01 mole) and the appropriate alkyl 4-(2-chloroacetamido)benzoate IIb,c (0.01 mole) in dry acetone (50 ml) was heated under reflux for 10 h in the presence of K2CO3 (1.38 g, 0.01 mole). The reaction mixture was filtered off while hot. The reaction mixture was allowed to attain room temperature and the precipitated solids were filtered, dried and crystallized from ethanol to give the corresponding target compounds, 5a–d respectively.
4.1.1.2. Methyl 4-{2-[1-ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (5a). Yield, 74%; m.p. 250–2 °C; IRνmax (cm−1): 3484 (NH), 3053 (C–H aromatic), 2917 (C–H aliphatic), 1752, 1718, 1701, 1685 (4C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1HNMR (400 MHz, DMSO-d6): 1.23 (t, 3H, NCH2CH3), 3.82 (s, 3H, COOCH3), 4.14–4.21 (q, 2H, NCH2CH3), 4.80 (s, 2H, CH2CO), 7.31–8.10 (m, 7H, 7Ar–H), 10.63 (s, 1H, NH, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 10.1, 36.5, 46.4, 50.4, 86.9, 115.8, 118.2, 121.4 (2), 127.2, 127.9 (2), 136.8, 140.0, 145.4, 150.3, 155.3, 158.3, 163.4, 168.4; MS (m/z): 507 (M+, 17.09%), 420 (80.04%), 294 (100% base), 268 (69.10%), 144 (70.06%); Anal. Calcd. For C20H18IN3O5 (m.w. 507.28): C, 47.35; H, 3.58; N, 8.28. Found: C, 47.30; H, 3.66; N, 8.33.
O); 1HNMR (400 MHz, DMSO-d6): 1.23 (t, 3H, NCH2CH3), 3.82 (s, 3H, COOCH3), 4.14–4.21 (q, 2H, NCH2CH3), 4.80 (s, 2H, CH2CO), 7.31–8.10 (m, 7H, 7Ar–H), 10.63 (s, 1H, NH, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 10.1, 36.5, 46.4, 50.4, 86.9, 115.8, 118.2, 121.4 (2), 127.2, 127.9 (2), 136.8, 140.0, 145.4, 150.3, 155.3, 158.3, 163.4, 168.4; MS (m/z): 507 (M+, 17.09%), 420 (80.04%), 294 (100% base), 268 (69.10%), 144 (70.06%); Anal. Calcd. For C20H18IN3O5 (m.w. 507.28): C, 47.35; H, 3.58; N, 8.28. Found: C, 47.30; H, 3.66; N, 8.33.
4.1.1.3. Ethyl 4-{2-[1-ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (5b). Yield, 75%; m.p. 257–9 °C; IRνmax (cm−1): 3481 (NH), 3046 (C–H aromatic), 2969 (C–H aliphatic), 1725, 1672, 1653 (4C
O); 1HNMR (400 MHz, DMSO-d6): 1.21–1.39 (2t, 6H, 2 CH2CH3), 4.16–4.19 (q, 2H, NCH2CH3), 4.27–4.29 (q, 2H, OCH2CH3), 4.80 (s, 2H, –CH2CO), 7.32–8.10 (m, 7H, 7Ar–H), 10.62 (s, 1H, NH, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 14.3, 21.5, 33.9, 42.6, 62.2, 89.3, 119.6, 130.3 (2), 130.5, 130.8 (2), 134.7, 141.9, 147.7, 150.7, 154.8, 158.3, 165.4, 167.2, 167.4; Anal. Calcd. For C21H20IN3O5 (m.w. 521.31): C, 48.38; H, 3.87; N, 8.06. Found: C, 48.45; H, 3.75; N, 7.98.
4.1.1.4. Methyl 4-{2-[6-iodo-2,4-dioxo-1-propyl-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (5c). Yield, 74%; m.p. 250–2 °C; IRνmax (cm−1): 3310 (NH), 3070 (C–H aromatic), 2922 (C–H aliphatic), 1722, 1697, 1666, 1650 (4C
O); 1HNMR (400 MHz, DMSO-d6): 1.29–1.34 (t, 3H, CH2CH2CH3), 3.57 (s, 3H, COOCH3), 3.79–3.83 (m, 2H, CH2CH2CH3), 4.26–4.31 (t, 2H, CH2CH2CH3), 4.82 (s, 2H, CH2CO), 7.34–8.09 (m, 7H, 7Ar–H), 10.62 (s, 1H, NH, D2O exchangeable); MS (m/z): 521 (M+, 35.30%), 498 (48.96%), 421 (100% base), 338 (79.89%), 235 (78.10%), 221 (85.87%); Anal. Calcd. For C21H20IN3O5 (m.w. 521.31): C, 48.38; H, 3.87; N, 8.06. Found: C, 48.50; H, 3.80; N, 7.94.
4.1.1.5. Ethyl 4-{2-[6-iodo-2,4-dioxo-1-propyl-1,4-dihydroquinazolin-3(2H)-yl]acetamido}benzoate (5d). Yield, 75%; m.p. 260-2 °C; IRνmax (cm−1): 3318 (NH), 3058 (C–H aromatic), 2925 (C–H aliphatic), 1733, 1686, 1671 (4C
O); 1HNMR (400 MHz, DMSO-d6): 0.92–0.97 (t, 3H, CH2CH2CH3), 1.29–1.33 (t, 3H, OCH2CH3), 1.63–1.70 (m, 2H, CH2CH2CH3), 4.07–4.12 (t, 2H, CH2CH2CH3), 4.25–4.31 (q, 2H, OCH2CH3), 4.80 (s, 2H, CH2CO), 7.31–8.04 (m, 7H, 7Ar–H), 10.62 (s, 1H, NH, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 9.4, 14.2, 23.1, 46.3, 56.3, 89.9, 112.5, 113.5, 114.4, 120.9 (2), 130.4 (2), 137.7, 141.4, 145.0, 150.4, 153.4, 154.8, 156.4, 164.3, 168.1; Anal. Calcd. For C22H22IN3O5 (m.w. 535.34): C, 49.36; H, 4.14; N, 7.85. Found: C, 49.53; H, 4.06; N, 8.02.
4.1.2.1. General method. A mixture of equimolar quantities of 6-iodo-1-alkylquinazoline-2,4(1H,3H)-dione (4a,b) (0.01 mole) and the appropriate 4-(2-chloroacetamido)-N-substitutedbenzamide (IIIa,b) (0.01 mole) in dry acetone (50 ml) was heated under reflux for 12 h in the presence of K2CO3 (1.38 g, 0.01 mole). The reaction mixture was filtered off while hot. The reaction mixture was concentrated and the afforded precipitated solids were filtered, dried and crystallized from ethanol to give the corresponding target compounds 6a,b.
4.1.2.2. 4-{2-[1-Ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamido}-N-ethylbenzamide (6a). Yield, 75%; m.p. 273–5 °C; IRνmax (cm−1): 3315, 3283 (2NH), 3064 (C–H aromatic), 2922 (C–H aliphatic), 1703, 1659, 1632 (4C
O); 1HNMR (400 MHz, DMSO-d6): 1.21–125 (t, 3H, NHCH2CH3), 1.30–1.33 (t, 3H, NCH2CH3), 4.16–4.24 (q, 2H, NHCH2CH3), 4.27–4.31 (q, 2H, NCH2CH3), 4.81 (s, 2H, CH2CO), 7.32–8.10 (m, 7H, 7Ar–H), 9.73 (s, 1H, NHCH2CH3, D2O exchangeable), 10.63 (s, 1H, CONH, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 9.1, 13.7, 32.8, 43.7, 55.4, 91.9, 113.9, 119.53, 120.1 (2), 121.1, 126.8, 127.1 (2), 129.9, 132.9, 134.5, 138.9, 141.5, 166.2, 168.0; Anal. Calcd. For C21H21IN4O4 (m.w. 520.33): C, 48.48; H, 4.07; N, 10.77. Found: C, 48.40; H, 3.95; N, 10.80.
4.1.2.3. 4-{2-[6-Iodo-2,4-dioxo-1-propyl-1,4-dihydroquinazolin-3(2H)-yl]acetamido}-N-methylbenzamide (6b). Yield, 71%; m.p. 276–8 °C; IRνmax (cm−1): 3285 (2NH), 3062 (C–H aromatic), 2907 (C–H aliphatic), 1734, 1686, 1667 (4C
O); 1HNMR (400 MHz, DMSO-d6): 0.91–0.96 (t, 3H, CH2CH2CH3), 1.59–1.66 (m, 2H, CH2CH2CH3), 3.83 (s, 3H, COOCH3), 3.96–4.02 (t, 2H, CH2CH2CH3), 4.30 (s, 2H, CH2CO), 7.23–8.02 (m, 7H, 7Ar–H), 10.60 (s, 1H, NHCH3, D2O exchangeable), 11.50 (s, 1H, CONH, D2O exchangeable); MS (m/z): 522 (M++2, 1.87%), 521 (M++1, 13.78%), 520 (M+, 26.11%), 480 (53.91%), 352 (100% base), 293 (52.06%), 157 (70.02%); Anal. Calcd. For C21H21IN4O4 (m.w. 520.33): C, 48.48; H, 4.07; N, 10.77. Found: C, 48.58; H, 4.15; N, 10.59.
4.1.3.1. General method. Hydrazine hydrate 100% (0.5 ml, 0.01 mol) was added dropwise to a stirred solution of the appropriate ethyl ester 5b and/or 5d (0.002 mol) in absolute ethanol (20 ml). The mixture was stirred well and heated at 70 °C for 6 h. The reaction mixture was concentrated, cooled and the crude product was filtered and recrystallized from ethanol to afford the corresponding hydrazide derivatives 7a,b respectively.
4.1.3.2. 2-(1-Ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl)-N-(4-(hydrazinecarbonyl)phenyl)-acetamide (7a). Yield, 75%; m.p. 300–2 °C; IRνmax (cm−1): 3250, 3190 (2NH & NH2), 3036 (C–H aromatic), 2927 (C–H aliphatic), 1710, 1685 (4C
O); 1HNMR (400 MHz, DMSO-d6): 1.23 (t, 3H, NCH2CH3), 4.16–4.19 (q, 2H, NCH2CH3), 4.42 (s, 2H, NH2), 4.80 (s, 2H, CH2CO), 7.31–8.10 (m, 7H, 7Ar–H), 9.63 (s, 1H, NHNH2, D2O exchangeable), 10.48 (s, 1H, NHPh, D2O exchangeable); Anal. Calcd. For C19H18IN5O4 (m.w. 507.29): C, 44.99; H, 3.58; N, 13.81. Found: C, 45.12; H, 3.65; N, 13.70.
4.1.3.3. N-[4-(hydrazinecarbonyl)phenyl]-2-(6-iodo-2,4-dioxo-1-propyl-1,4-dihydroquinazolin-3(2H)-yl)acetamide (7b). Yield, 70%; m.p. 306-8 °C; IRνmax (cm−1): 3436, 3234, 3179 (2NH & NH2), 3038 (C–H aromatic), 2915 (C–H aliphatic), 1685, 1670 (4C
O); 13CNMR (100 MHz, DMSO-d6): 10.7, 22.9, 32.8, 46.6, 55.4, 90.7, 118.1, 126.7 (2), 127.4 (2), 135.5, 140.0, 143.1, 147.4, 152.3, 156.6, 161.7, 166.7, 171.6; Anal. Calcd. For C20H20IN5O4 (m.w. 521.32): C, 46.08; H, 3.87; N, 13.43. Found: C, 45.90; H, 3.96; N, 13.37.
4.1.4.1. 2-[1-Ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]-N-[4-(5-mercapto-1,3,4-oxadiazol-2-yl)phenyl]acetamide (8a). Yield, 74%; m.p. 283–5 °C; IRνmax (cm−1): 3301, 3192 (2NH), 3036 (C–H aromatic), 2919 (C–H aliphatic), 1689, 1665, 1643 (3C
O), 1263 (CS); 1HNMR (400 MHz, DMSO-d6): 1.21–1.26 (t, 3H, CH2CH3), 4.17–4.19 (q, 2H, CH2CH3), 4.82 (s, 2H, COCH2), 7.32–8.11 (m, 7H, 7Ar–H), 10.66 (s, 1H, NH, D2O exchangeable), 14.59 (s, 1H, SH, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 12.3, 34.9, 52.1, 91.6, 114.8, 116.9, 122.8, 123.6, 124.6 (2), 125.4, 128.9 (2), 129.4, 139.5, 148.5, 153.1, 157.4, 163.7, 170.1; Anal. Calcd. for C20H16IN5O4S (m.w. 549.34): C, 43.73; H, 2.94; N, 12.75; Found: C, 43.90; H, 3.06; N, 12.65.
4.1.4.2. 2-[6-Iodo-2,4-dioxo-1-propyl-1,4-dihydroquinazolin-3(2H)-yl]-N-[4-(5-mercapto-1,3,4-oxadiazol-2-yl)phenyl]acetamide (8b). Yield, 68%; m.p. 287–9 °C; IRνmax (cm−1): 3447, 3189 (2NH), 3050 (C–H aromatic), 2967 (C–H aliphatic), 1692, 1653 (3C
O), 1262 (CS); 1HNMR (400 MHz, DMSO-d6): 0.92–0.97 (t, 3H, CH2CH2CH3), 1.63–1.70 (m, 2H, CH2CH2CH3), 4.07–4.12 (t, 2H, CH2CH2CH3), 4.81 (s, 2H, COCH2), 7.31–8.09 (m, 7H, 7Ar–H), 10.66 (s, 1H, NH, D2O exchangeable), 14.59 (s, 1H, SH, D2O exchangeable); MS (m/z): 565 (M++2, 3.96%), 564 (M++1, 9.96%), 520 (M+, 60.00%), 443 (85.24%), 334 (80.01%), 328 (100% base), 233 (79.97%); Anal. Calcd. for C21H18IN5O4S (m.w. 563.37): C, 44.77; H, 3.22; N, 12.43. Found: C, 44.84; H, 3.11; N, 12.56.
4.1.5.1. General method. Equimolar amounts of appropriate hydrazide 7a and/or 7b (0.002 mol) and the appropriate aromatic aldehyde namely benzaldehyde, 2-chlorobenzaldehyde, 4-chlorobenzaldehyde, 4-methylbanzaldehyde and 4-methoxybanzaldehyde (0.002 mol) were refluxed in gl. acetic acid (25 ml) for 4 hours and the reaction was followed up by TLC. After completion of the reaction, the mixture was cooled, poured into water, filtered and crystallized from ethanol to afford the corresponding target compounds 9a–e respectively.
4.1.5.2. N-[4-(2-benzylidenehydrazine-1-carbonyl)phenyl]-2-[1-ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]acetamide (9a). Yield, 72%; m.p. 290–2 °C; IRνmax (cm−1): 3262 (2NH), 3050 (C–H aromatic), 2950 (C–H aliphatic), 1722, 1697, 1666, 1652 (4C
O); 1HNMR (400 MHz, DMSO-d6): 1.24–1.31 (t, 3H, CH2CH3), 4.18–4.20 (q, 2H, CH2CH3), 4.82 (s, 2H, COCH2), 7.32–8.12 (m, 12H, 12Ar–H), 8.45 (s, 1H, –NCH), 10.68 (s, 1H, NHCO, D2O exchangeable), 11.75 (s, 1H, N–NHCO, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 13.4, 34.9, 56.6, 91.7, 114.7, 116.8, 121.8 (2), 122.4, 123.6, 124.3, 124.5 (2), 125.6 (2), 128.9, 129.4, 129.5 (2), 140.0, 148.5, 152.3, 156.4, 160.0, 164.6, 168.7; Anal. Calcd. for C26H22IN5O4 (m.w. 595.40): C, 52.45; H, 3.72; N, 11.76. Found: C, 52.35; H, 3.78; N, 11.70.
4.1.5.3. 2-[1-Ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]-N-{4-[2-(4-methylbenzylidene)hydrazine-1-carbonyl]phenyl}acetamide (9b). Yield, 72%; m.p. 297–9 °C; IRνmax (cm−1): 3313, 3255 (2NH), 3033 (C–H aromatic), 2922 (C–H aliphatic), 1700, 1675 (4C
O); 1HNMR (400 MHz, DMSO-d6): 1.22–1.26 (t, 3H, CH2CH3), 2.34 (s, 3H, CH3), 4.17–4.20 (q, 2H, CH2CH3), 4.82 (s, 2H, COCH2), 7.25–8.11 (m, 11H, 11Ar–H), 8.42 (s, 1H, –NCH), 10.57 (s, 1H, NHCO, D2O exchangeable), 11.67 (s, 1H, N–NHCO, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 13.4, 21.8, 56.6, 87.4, 114.6 (2), 116.6 (2), 118.8, 121.8 (2), 123.9, 124.3, 125.6, 128.9, 129.3 (2), 129.5, 140.0, 143.3, 145.7, 147.7, 153.7, 156.6, 159.4, 160.9, 170.1; Anal. Calcd. for C27H24IN5O4 (m.w. 609.42): C, 53.21; H, 3.97; N, 11.49. Found: C, 53.14; H, 4.15; N, 11.65.
4.1.5.4. 2-[1-Ethyl-6-iodo-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]-N-{4-[2-(4-methoxybenzylidene)-hydrazine-1-carbonyl]phenyl}acetamide (9c). Yield, 70%; m.p. 302–4 °C; IRνmax (cm−1): 3262, 3184 (2NH), 3036 (C–H aromatic), 2932 (C–H aliphatic), 1733, 1665, 1641 (4C
O); 1HNMR (400 MHz, DMSO-d6): 1.22–1.27 (t, 3H, CH2CH3), 3.81 (s, 3H, OCH3), 4.18–4.20 (q, 2H, CH2CH3), 4.82 (s, 2H, COCH2), 7.27–8.11 (m, 11H, 11Ar–H), 8.42 (s, 1H, –NCH), 10.58 (s, 1H, NHCO, D2O exchangeable), 11.75 (s, 1H, N–NHCO, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 14.7, 33.8, 50.6, 62.0, 87.4, 99.3, 113.6 (2), 115.9, 117.1, 123.7 (2), 131.2 (2), 134.9 (2), 136.5, 137.7, 138.8, 150.0, 151.0, 154.8, 158.4, 162.9, 165.6, 169.9, 178.5; MS (m/z): 626 (M+ + 1, 41.76%), 625 (M+, 65.91%), 558 (74.18%), 353 (100% base), 334 (81.89%), 289 (74.26%); Anal. Calcd. for C27H24IN5O5 (m.w. 625.42): C, 51.85; H, 3.87; N, 11.20. Found: C, 52.00; H, 3.58; N, 11.40.
4.1.5.5. N-[4-(2-benzylidenehydrazine-1-carbonyl)phenyl]-2-[6-iodo-2,4-dioxo-1-propyl-1,4-dihydroquinazolin-3(2H)-yl]acetamide (9d). Yield, 65%; m.p. 307–9 °C; IRνmax (cm−1): 3447, 3283 (2NH), 3063 (C–H aromatic), 2979 (C–H aliphatic), 1692, 1658, 1653 (4C
O); 1HNMR (400 MHz, DMSO-d6): 0.93–1.02 (t, 3H, CH2CH2CH3), 1.64–1.71 (m, 2H, CH2CH2CH3), 4.08–4.13 (t, 2H, CH2CH2CH3), 4.81 (s, 2H, COCH2), 7.32–8.11 (m, 12H, 12Ar–H), 8.45 (s, 1H, –NCH), 10.57 (s, 1H, NHCOCH2, D2O exchangeable), 11.74 (s, 1H, N–NHCO, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 11.8, 21.7, 47.3, 56.8, 89.9, 112.8 (2), 116.3, 124.5 (2), 127.8, 129.1 (2), 130.5 (2), 131.3, 138.3, 138.9, 146.6, 150.6, 153.7, 157.4, 157.9, 161.5, 165.3, 168.7, 171.6; Anal. Calcd. for C27H24IN5O4 (m.w. 609.42): C, 53.21; H, 3.97; N, 11.49. Found: C, 53.30; H, 4.05; N, 11.42.
4.1.5.6. N-{4-[2-(4-chlorobenzylidene)hydrazine-1-carbonyl]phenyl}-2-{6-iodo-2,4-dioxo-1-propyl-1,4-dihydroquinazolin-3(2H)-yl}acetamide (9e). Yield, 70%; m.p. >310 °C; IRνmax (cm−1): 3316, 3280 (2NH), 3073 (C–H aromatic), 2950 (C–H aliphatic), 1701, 1659 (4C
O); 1HNMR (400 MHz, DMSO-d6): 0.93–0.98 (t, 3H, CH2CH2CH3), 1.64–1.69 (m, 2H, CH2CH2CH3), 4.08–4.13 (t, 2H, CH2CH2CH3), 4.82 (s, 2H, COCH2), 7.32–8.11 (m, 11H, 11Ar–H), 8.86 (s, 1H, –NCH), 10.59 (s, 1H, NHCOCH2, D2O exchangeable), 11.98 (s, 1H, N–NHCO, D2O exchangeable); 13CNMR (100 MHz, DMSO-d6): 10.9, 21.3, 46.8, 54.9, 96.8, 113.9 (2), 118.9, 122.8 (2), 123.6 (2), 124.4, 125.6, 129.7, 129.9 (2), 131.8, 142.7, 143.5, 148.8, 152.5, 153.2, 160.0, 169.4; MS (m/z): 645 (M++2, 14.69%), 643 (M+, 44.72%), 580 (58.22%), 492 (60.98%), 471 (100% base), 367 (46.13%), 87 (82.00%); Anal. Calcd. for C25H19ClIN5O4 (m.w. 643.87): C, 50.37; H, 3.60; N, 10.88. Found: C, 50.22; H, 3.74; N, 11.08.
4.2. Docking studies
Both VEGFR-2 (PDB ID 4ASD)48,58 and EGFRT790M (PDB ID 3W2O)55,56 were used by Molsoft program to carry out docking studies.4.3. Biological testing
Author contributions
K. El-Adl, Samir A. Salama, Abeer Mohamed, Ahmed Aljohani, Ahmed El-morsy, Doaa Keshek and Marwa Alsulaimany were responsible for the conception and rational design of the work. K. El-Adl, Abeer Mohamed, Ahmed Aljohani, Omar Alatawi, Hussam Alharbi, Majed Aljohani and Marwa Alsulaimany were responsible for the data collection and synthesis of the new compounds. K. El-Adl performed the molecular docking study. Hussam Alharbi, Majed Aljohani, Ahmed El-morsy, Ahmed Aljohani, Marwa Alsulaimany and K. El-Adl were responsible for spectral data analysis. Doaa Keshek, Sara Almadani, Abdulrahman Alsimaree, Samir A. Salama and K. El-Adl conducted the cytotoxicity assay. Sara Almadani, Abdulrahman Alsimaree, Samir A. Salama and K. El-Adl conducted the in silico pharmacokinetic study. All authors discussed the results and contributed to the writing and revision of the original manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We would like to acknowledge the Deanship of Scientific Research, Taif University for funding this work.References
- M. Shibuya, N. Ito and L. Claesson-Welsh, Structure and Function of Vascular Endothelial Growth Factor Receptor-1 And-2, Vascular Growth Factors and Angiogenesis, 1999, pp. 59–83 Search PubMed.
- A. A. Shah, M. A. Kamal and S. Akhtar, Tumor angiogenesis and VEGFR-2: mechanism, pathways and current biological therapeutic interventions, Curr. Drug Metab., 2021, 22, 50–59 CAS.
- J. S. Wey, O. Stoeltzing and L. M. Ellis, Vascular endothelial growth factor receptors: expression and function in solid tumors, Clin. Adv. Hematol. Oncol., 2004, 2, 37–45 Search PubMed.
- M. M. Ghorab, M. S. Alsaid, A. M. Soliman and F. A. Ragab, VEGFR-2 inhibitors and apoptosis inducers: synthesis and molecular design of new benzo [g] quinazolin bearing benzenesulfonamide moiety, J. Enzyme Inhib. Med. Chem., 2017, 32, 893–907 CrossRef CAS PubMed.
- Y.-S. Ng, D. Krilleke and D. T. Shima, VEGF function in vascular pathogenesis, Exp. Cell Res., 2006, 312, 527–537 CrossRef CAS PubMed.
- K. M. Cook and W. D. Figg, Angiogenesis inhibitors: current strategies and future prospects, Ca-Cancer J. Clin., 2010, 60, 222–243 CrossRef PubMed.
- Q. Liu, S. Yu, W. Zhao, S. Qin, Q. Chu and K. Wu, EGFR-TKIs resistance via EGFR-independent signaling pathways, Mol, Cancer, 2018, 17, 1–9 Search PubMed.
- P. Dent, D. T. Curiel, P. B. Fisher and S. Grant, Synergistic combinations of signaling pathway inhibitors: mechanisms for improved cancer therapy, Drug Resistance Updates, 2009, 12, 65–73 CrossRef CAS PubMed.
- S. S. Zahran, F. A. Ragab, M. G. El-Gazzar, A. M. Soliman, W. R. Mahmoud and M. M. Ghorab, Antiproliferative, antiangiogenic and apoptotic effect of new hybrids of quinazoline-4 (3H)-ones and sulfachloropyridazine, Eur. J. Med. Chem., 2023, 245, 114912 CrossRef CAS PubMed.
- A. Arora and E. M. Scholar, Role of tyrosine kinase inhibitors in cancer therapy, J. Pharmacol. Exp. Ther., 2005, 315, 971–979 CrossRef CAS PubMed.
- P. Traxler and P. Furet, Strategies toward the design of novel and selective protein tyrosine kinase inhibitors, Pharmacol. Ther., 1999, 82, 195–206 CrossRef CAS PubMed.
- A. G. A. El-Helby, H. Sakr, I. H. Eissa, H. Abulkhair, A. A. Al-Karmalawy and K. El-Adl, Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors, Arch. Pharm., 2019, 352, 1900113 CrossRef CAS PubMed.
- M. A. Abdelgawad, K. El-Adl, S. S. El-Hddad, M. M. Elhady, N. M. Saleh, M. M. Khalifa, F. Khedr, M. Alswah, A. A. Nayl and M. M. Ghoneim, Design, molecular docking, synthesis, anticancer and anti-hyperglycemic assessments of thiazolidine-2, 4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists, Pharmaceuticals, 2022, 15, 226 CrossRef CAS PubMed.
- H.-Q. Zhang, F.-H. Gong, J.-Q. Ye, C. Zhang, X.-H. Yue, C.-G. Li, Y.-G. Xu and L.-P. Sun, Design and discovery of 4-anilinoquinazoline-urea derivatives as dual TK inhibitors of EGFR and VEGFR-2, Eur. J. Med. Chem., 2017, 125, 245–254 CrossRef CAS PubMed.
- R. Newton, K. A. Bowler, E. M. Burns, P. J. Chapman, E. E. Fairweather, S. J. Fritzl, K. M. Goldberg, N. M. Hamilton, S. V. Holt and G. V. Hopkins, The discovery of 2-substituted phenol quinazolines as potent RET kinase inhibitors with improved KDR selectivity, Eur. J. Med. Chem., 2016, 112, 20–32 CrossRef CAS PubMed.
- M. M. Ghorab, M. S. Alsaid and A. M. Soliman, Dual EGFR/HER2 inhibitors and apoptosis inducers: New benzo [g] quinazoline derivatives bearing benzenesulfonamide as anticancer and radiosensitizers, Bioorg. Chem., 2018, 80, 611–620 CrossRef CAS PubMed.
- S. R. A. E. Hadi, D. S. Lasheen, D. H. Soliman, E. Z. Elrazaz and K. A. M. Abouzid, Scaffold hopping and redesign approaches for quinazoline based urea derivatives as potent VEGFR-2 inhibitors, Bioorg. Chem., 2020, 101, 103961, DOI:10.1016/j.bioorg.2020.103961.
- H. A. Allam, E. E. Aly, A. K. B. A. W. Farouk, A. M. El Kerdawy, E. Rashwan and S. E. S. Abbass, Design and Synthesis of some new 2,4,6-trisubstituted quinazoline EGFR inhibitors as targeted anticancer agents, Bioorg. Chem., 2020, 98, 103726, DOI:10.1016/j.bioorg.2020.103726.
- Y. Cao, J. Arbiser, R. J. D'Amato, P. A. D'Amore, D. E. Ingber, R. Kerbel, M. Klagsbrun, S. Lim, M. A. Moses and B. Zetter, Forty-year journey of angiogenesis translational research, Sci. Transl. Med., 2011, 3, 114rv113 Search PubMed.
- F. Khedr, M.-K. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Phthalazine-based VEGFR-2 inhibitors: Rationale, design, synthesis, in silico, ADMET profile, docking, and anticancer evaluations, Arch. Pharm., 2021, e2100201, DOI:10.1002/ardp.202100201.
- D. Panigraphy, S. Huang, M. W. Kieran and A. Kaipainen, PPARγ as a therapeutic target for tumor angiogenesis and metastasis, Cancer Biol. Ther., 2005, 4, 687–693 CrossRef PubMed.
- A. Gschwind, O. M. Fischer and A. Ullrich, The discovery of receptor tyrosine kinases: targets for cancer therapy, Nat. Rev. Cancer, 2004, 4, 361–370 CrossRef CAS PubMed.
- S.-N. Li and H.-Q. Li, Epidermal growth factor receptor inhibitors: a patent review (2010–present), Expert Opin. Ther. Pat., 2014, 24, 309–321 CrossRef CAS PubMed.
- M. R. Raspollini, F. Castiglione, F. Garbini, A. Villanucci, G. Amunni, G. Baroni, V. Boddi and G. L. Taddei, Correlation of epidermal growth factor receptor expression with tumor microdensity vessels and with vascular endothelial growth factor expression in ovarian carcinoma, Int. J. Surg. Pathol., 2005, 13, 135–142 CrossRef CAS PubMed.
- D. Frezzetti, M. Gallo, M. R. Maiello, A. D'Alessio, C. Esposito, N. Chicchinelli, N. Normanno and A. De Luca, VEGF as a potential target in lung cancer, Expert Opin. Ther. Targets, 2017, 21, 959–966 CrossRef CAS PubMed.
- A. Ayati, S. Moghimi, S. Salarinejad, M. Safavi, B. Pouramiri and A. Foroumadi, A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy, Bioorg. Chem., 2020, 99, 103811 CrossRef CAS PubMed.
- N. Wagner and K.-D. Wagner, PPARs and angiogenesis—Implications in pathology, Int. J. Mol. Sci., 2020, 21, 5723 CrossRef CAS PubMed.
- E. Fröhlich and R. Wahl, Chemotherapy and chemoprevention by thiazolidinediones, BioMed Res. Int., 2015, 2015, 845340 Search PubMed.
- T. Stockley, C. A. Souza, P. K. Cheema, B. Melosky, S. Kamel-Reid, M. S. Tsao, A. Spatz and A. Karsan, Evidence-based best practices for EGFR T790M testing in lung cancer in Canada, Curr. Oncol., 2018, 25(2), 163–169, DOI:10.3747/co.25.4044.
- X. Nan, C. Xie, X. Yu and J. Liu, EGFR TKI as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer, Oncotarget, 2017, 8, 75712 CrossRef PubMed.
- E. R. Haspinger, F. Agustoni, V. Torri, F. Gelsomino, M. Platania, N. Zilembo, R. Gallucci, M. C. Garassino and M. Cinquini, Is there evidence for different effects among EGFR-TKIs? Systematic review and meta-analysis of EGFR tyrosine kinase inhibitors (TKIs) versus chemotherapy as first-line treatment for patients harboring EGFR mutations, Crit. Rev. Oncol. Hematol., 2015, 94, 213–227 CrossRef PubMed.
- L. A. Byers and J. V. Heymach, Dual targeting of the vascular endothelial growth factor and epidermal growth factor receptor pathways: rationale and clinical applications for non-small-cell lung cancer, Clin. Lung Cancer, 2007, 8, S79–S85 CrossRef CAS PubMed.
- L. Hu, M. Fan, S. Shi, X. Song, F. Wang, H. He and B. Qi, Dual target inhibitors based on EGFR: Promising anticancer agents for the treatment of cancers (2017-), Eur. J. Med. Chem., 2022, 227, 113963 CrossRef CAS PubMed.
- D. Panigrahy, S. Singer, L. Q. Shen, C. E. Butterfield, D. A. Freedman, E. J. Chen, M. A. Moses, S. Kilroy, S. Duensing and C. Fletcher, PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis, J. Clin. Invest., 2002, 110, 923–932 CrossRef CAS PubMed.
- C. L. Arteaga, Epidermal growth factor receptor dependence in human tumors: more than just expression?, Oncologist, 2002, 7, 31–39 CrossRef CAS PubMed.
- S. Wilhelm, C. Carter, M. Lynch, T. Lowinger, J. Dumas, R. A. Smith, B. Schwartz, R. Simantov and S. Kelley, Discovery and development of sorafenib: a multikinase inhibitor for treating cancer, Nat. Rev. Drug Discovery, 2006, 5, 835–844 CrossRef CAS PubMed.
- H. A. Yu and W. Pao, Afatinib—new therapy option for EGFR-mutant lung cancer, Nat. Rev. Clin. Oncol., 2013, 10, 551–552 CrossRef CAS PubMed.
- M. A. Abdullaziz, H. T. Abdel-Mohsen, A. M. El Kerdawy, F. A. Ragab, M. M. Ali, S. M. Abu-Bakr, A. S. Girgis and H. I. El Diwani, Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors, Eur. J. Med. Chem., 2017, 136, 315–329 CrossRef CAS PubMed.
- M. Ashraf-Uz-Zaman, X. Li, Y. Yao, C. B. Mishra, B. K. Moku and Y. Song, Quinazolinone Compounds Have Potent Antiviral Activity against Zika and Dengue Virus, J. Med. Chem., 2023, 66(15), 10746–10760, DOI:10.1021/acs.jmedchem.3c00924.
- R. Bouley, D. Ding, Z. Peng, M. Bastian, E. Lastochkin, W. Song, M. A. Suckow, V. A. Schroeder, W. R. Wolter, S. Mobashery and M. Chang, Structure–activity relationship for the 4(3h)-quinazolinone antibacterials, J. Med. Chem., 2016, 59(10), 5011–5021, DOI:10.1021/acs.jmedchem.6b00372.
- Y. T. Tseng, Y. H. Tsai, F. Fülöp, F. R. Chang and Y. C. Lo, 2-Iodo-4'-Methoxychalcone Attenuates Methylglyoxal-Induced Neurotoxicity by Activation of GLP-1 Receptor and Enhancement of Neurotrophic Signal, Antioxidant Defense and Glyoxalase Pathway, Molecules, 2019, 24(12), 2249, DOI:10.3390/molecules24122249.
- R. Ghomashi, S. Ghomashi, H. Aghaei and A. R. Massah, Recent Advances in Biological Active Sulfonamide based Hybrid Compounds Part A: Two-Component Sulfonamide Hybrids, Curr. Med. Chem., 2023, 30(37), 4181–4255, DOI:10.2174/0929867330666221128142730..
- A. G. A. El-Helby, R. R. Ayyad, H. Sakr, K. El-Adl, M. M. Ali and F. Khedr, design, synthesis, molecular docking, and anticancer activity of phthalazine derivatives as VEGFR-2 inhibitors, Arch. Pharm., 2017, 350, 1700240 CrossRef PubMed.
- M. M. Ghorab, A. M. Soliman, K. El-Adl and N. S. Hanafy, New quinazoline sulfonamide derivatives as potential anticancer agents: Identifying a promising hit with dual EGFR/VEGFR-2 inhibitory and radiosensitizing activity, Bioorg. Chem., 2023, 140, 106791, DOI:10.1016/j.bioorg.2023.106791.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem, S. I. Eissa, H. Elkady and I. H. Eissa, Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents, Bioorg. Chem., 2020, 104, 104218, DOI:10.1016/j.bioorg.2020.104218.
- N. M. Saleh, M. S. A. El-Gaby, K. El-Adl and N. E. A. Abd El-Sattar, Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors, Bioorg. Chem., 2020, 104, 104350, DOI:10.1016/j.bioorg.2020.104350.
- A. E. Abdallah, I. H. Eissa, A. B. M. Mehany, H. Sakr, A. Atwa, K. El-Adl and M. A. El-Zahabi, Immunomodulatory quinazoline-based thalidomide analogs: Design, synthesis, apoptosis and anticancer evaluations, J. Mol. Struct., 2023, 1281, 135164, DOI:10.1016/j.molstruc.2023.135164.
- N. S. Hanafy, N. A. A. M. Aziz, S. S. A. El-Hddad, M. A. Abdelgawad, M. M. Ghoneim, A. F. Dawood, S. Mohamady, K. El-Adl and S. Ahmed, Design, synthesis, and docking of novel thiazolidine-2,4-dione multitarget scaffold as new approach for cancer treatment, Arch. Pharm., 2023, e2300137, DOI:10.1002/ardp.202300137.
- D. Adel, K. El-Adl, T. Nasr, T. M. Sakr and W. Zaghary, Pyrazolo[3,4-d]pyrimidine derivatives as EGFRT790M and VEGFR-2 dual TK inhibitors: Design, synthesis, molecular docking, ADMET profile and anticancer evaluations, J. Mol. Struct., 2023, 1291, 136047, DOI:10.1016/j.molstruc.2023.136047.
- H. Elkady, K. El-Adl, H. Sakr, A. S. Abdelraheem, S. I. Eissa and M. A. El-Zahabi, Novel promising benzoxazole/benzothiazole-derived immunomodulatory agents: Design, synthesis, anticancer evaluation, and in silico ADMET analysis, Arch. Pharm., 2023, e2300097, DOI:10.1002/ardp.202300097.
- M. A. El-Zahabi, H. Elkady, H. Sakr, A. S. Abdelraheem, S. I. Eissa and K. El-Adl, Design, synthesis, anticancer evaluation, in silico docking and ADMET analysis of novel indole-based thalidomide analogs as promising immunomodulatory agents, J. Biomol. Struct. Dyn., 2023, 1–18, DOI:10.1080/07391102.2023.2187217.
- K. Lee, K.-W. Jeong, Y. Lee, J. Y. Song, M. S. Kim, G. S. Lee and Y. Kim, Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors, Eur. J. Med. Chem., 2010, 45, 5420–5427 CrossRef CAS PubMed.
- V. A. Machado, D. Peixoto, R. Costa, H. J. Froufe, R. C. Calhelha, R. M. Abreu, I. C. Ferreira, R. Soares and M.-J. R. Queiroz, Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno [3, 2-b] pyridin-7-ylthio) phenyl] ureas: Discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors, Bioorg. Med. Chem., 2015, 23, 6497–6509 CrossRef CAS PubMed.
- Y. Liu and N. S. Gray, Rational design of inhibitors that bind to inactive kinase conformations, Nat. Chem. Biol., 2006, 2, 358–364 CrossRef CAS PubMed.
- N. A. A. M. Aziz, R. F. George, K. El-Adl and W. R. Mahmoud, Design, synthesis, in silico docking, ADMET and anticancer evaluations of thiazolidine-2,4-diones bearing heterocyclic rings as dual VEGFR-2/EGFRT790M tyrosine kinase inhibitors, RSC Adv., 2022, 12(20), 12913–12931, 10.1039/D2RA01119K.
- N. A. A. M. Aziz, R. F. George, K. El-Adl and W. R. Mahmoud, Exploration of thiazolidine-2,4-diones as tyrosine kinase inhibitors: Design, synthesis, ADMET, docking, and antiproliferative evaluations, Arch. Pharm., 2023, 356(3), e2200465, DOI:10.1002/ardp.202200465.
- K. El-Adl, A.-G. A. El-Helby, H. Sakr and S. S. A. El-Hddad, Design, synthesis, molecular docking, and anticancer evaluations of 1-benzylquinazoline-2,4(1H,3H)-dione bearing different moieties as VEGFR-2 inhibitors, Arch. Pharm., 2020, 353(8), e2000068, DOI:10.1002/ardp.202000068.
- S. A. Elmetwally, K. F. Saied, I. H. Eissa and E. B. Elkaeed, Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers, Bioorg. Chem., 2019, 88, 102944, DOI:10.1016/j.bioorg.2019.102944.
- D. A. Scudiero, R. H. Shoemaker, K. D. Paull, A. Monks, S. Tierney, T. H. Nofziger, M. J. Currens, D. Seniff and M. R. Boyd, Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines, Cancer Res., 1988, 48, 4827–4833 CAS.
- A. Huether, M. Höpfner, A. P. Sutter, D. Schuppan and H. Scherübl, Erlotinib induces cell cycle arrest and apoptosis in hepatocellular cancer cells and enhances chemosensitivity towards cytostatics, J. Hepatol., 2005, 43, 661–669 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS PubMed.
- N. E. A. Abd El-Sattar, K. El-Adl, M. A. El-Hashash, S. A. Salama and M. M. Elhady, Design, synthesis, molecular docking and in silico ADMET profile of pyrano[2,3-d]pyrimidine derivatives as antimicrobial and anticancer agents, Bioorg. Chem., 2021, 115, 105186, DOI:10.1016/j.bioorg.2021.105186.
- Y. Jia, C. M. Quinn, A. I. Gagnon and R. Talanian, Homogeneous time-resolved fluorescence and its applications for kinase assays in drug discovery, Anal. Biochem., 2006, 356(2), 273–281 CrossRef CAS PubMed.
- S. Sogabe, Y. Kawakita, S. Igaki, H. Iwata, H. Miki, D. R. Cary, T. Takagi, S. Takagi, Y. Ohta and T. Ishikawa, Structure-based approach for the discovery of pyrrolo[3, 2-d]pyrimidine-based EGFR T790M/L858R mutant inhibitors, ACS Med. Chem. Lett., 2013, 4, 201–205 CrossRef CAS PubMed.
- S. A. Elmetwally, K. F. Saied, I. H. Eissa and E. B. Elkaeed, Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers, Bioorg. Chem., 2019, 88, 102944, DOI:10.1016/j.bioorg.2019.102944.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures, J. Med. Chem., 2015, 58, 4066, DOI:10.1021/acs.jmedchem.5b00104.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17 CrossRef.
- A. Beig, R. Agbaria and A. Dahan, Oral delivery of lipophilic drugs: the tradeoff between solubility increase and permeability decrease when using cyclodextrin-based formulations, PLoS One, 2013, 8, e68237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07700d |
| This journal is © The Royal Society of Chemistry 2023 |