
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntermolecular exo-selective Diels–Alder reaction catalysed by dual-functional Brønsted acid: conformational restriction of transition states by hydrogen bonds as the key interaction†
Taishi Nakanishi and
Masahiro Terada *
*
Department of Chemistry, Graduate School of Science, Tohoku University, Aramaki, Aoba-ku, Sendai, Miyagi 980-8578, Japan. E-mail: mterada@tohoku.ac.jp
First published on 12th December 2023
Abstract
An exo-selective Diels–Alder (exo-DA) reaction in which the formed diastereomer is different from that formed in the conventional endo-selective Diels–Alder (endo-DA) reaction was developed, which involves a dual-functional Brønsted acid as a catalyst and not only a dienophile (vinylquinoline) but also an acyclic diene (dienylcarbamate) having a sterically less demanding substituent. Factors necessary for achieving the exo-DA reaction were extracted through an exhaustive computational search of the corresponding transition states, in which the relative orientation of the dienophile and the acyclic diene is firmly defined by hydrogen bonding interactions with a dual-functional Brønsted acid catalyst. It was experimentally verified that the combined use of the dual-functional acid catalyst, such as phosphoric acid, and the conformationally restricted diene (dienylcarbamate), which was realized by the introduction of a substituent at the 2-position of the diene unit, is the key to achieving the exo-DA reaction. A catalytic enantioselective exo-DA reaction was also attempted by using a chiral phosphoric acid catalyst, which gave rise to the corresponding exo-adduct with fairly good enantioselectivity.
Introduction
The Diels–Alder (DA) reaction is one of the most fundamental and versatile reactions for the synthesis of six-membered cyclic compounds and is widely used in total synthesis and other applications, whether in an intra- or intermolecular fashion (Fig. 1).1 In general, intermolecular normal-electron-demand DA reactions proceed in an endo-selective manner.2,3 The secondary orbital interaction has long been considered a major contributing factor to the endo selectivity, although dipoles, magnetic properties, and steric effects have also been found to play an important role; this issue is still being actively debated.4 On the other hand, the development of the exo-selective DA (exo-DA) reaction, in which the formed diastereomer is different from that formed by the conventional endo-selective DA (endo-DA) reaction, is an uphill battle for synthetic organic chemists, and considerable effort has been devoted to its development because the comprehensive synthesis of stereoisomers is achieved using versatile DA reactions.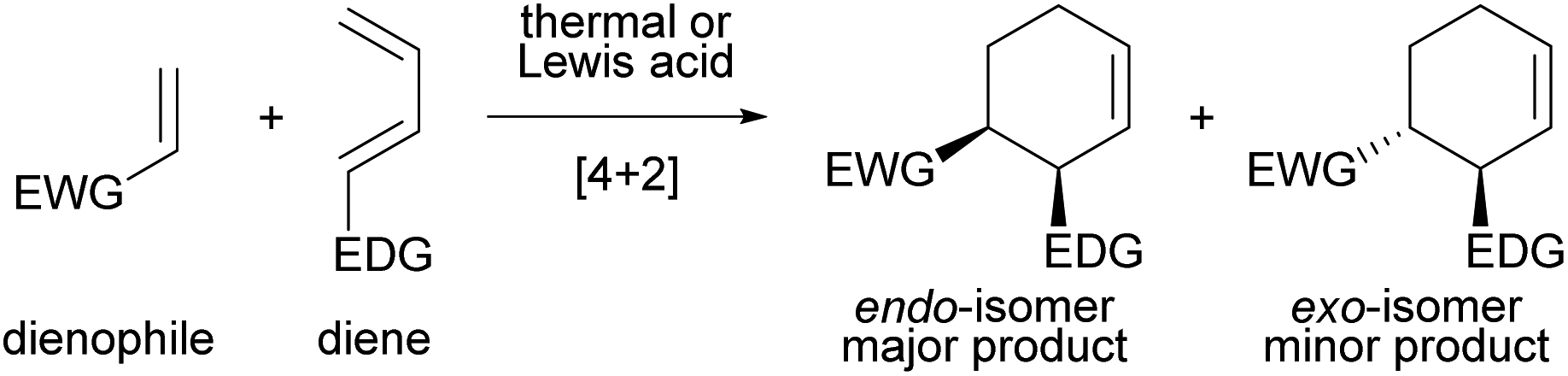 | ||
| Fig. 1 Intermolecular normal-electron-demand Diels–Alder reaction, giving endo-isomer as the major product. | ||
exo-DA reactions are generally achieved by using bulky catalysts or bulky dienes and dienophiles to restrict the directions of approach of these components in the transition state (TS), in which the endo-TS is destabilized by steric repulsion, resulting in a kinetically unfavourable pathway. In principle, dienes bearing either a cyclic structure5 or a bulky substituent6–8 are often used. The exo-DA reactions have also been established by designing dienophiles including (i) dienophiles anchored in the s-cis conformation,4a,9 (ii) bulky organometallic dienophiles (and dienes),10 and (iii) acrolein derivatives having substituents at the α-position.11 Other exo-selective methods originating from the use of sterically bulky Lewis acid catalysts12 and supramolecular catalysts13 have also been reported. However, these prior examples are essentially limited to reactions of cyclopentadiene because the use of cyclopentadiene tends to facilitate the exo-selective reaction. In contrast, the exo-DA reaction using acyclic dienes is largely unexplored, and there are only a few examples of exo-DA reactions achieved by using acyclic dienes, which proceed through weak non-covalent interactions, such as hydrogen bonding, rather than the steric repulsion.14–17
In the exo-DA reaction catalysed by bulky B(C6F5)3,14a detailed mechanistic studies have revealed that specific C–H⋯F interactions are the key to stabilizing the TS structure in the exo-reaction pathway.14b Recently, computational studies predicted an exo-DA reaction when the directions of approach of a diene and a dienophile were restricted by hydrogen bonding interactions,15 although it has yet to be proven experimentally. In 1998, Wilson et al. reported an asymmetric exo-DA reaction that uses an antibody as a catalyst.16 The DA reaction proceeded in an exo-selective manner without taking advantage of the steric effect of the substrates themselves. In this particular catalytic system, the relative orientation of the diene and the dienophile was controlled by the multiple hydrogen bonding interactions between the antibody catalyst and the substrates. However, the hydrogen bonding interaction approach has several limitations compared with the controlling method based on the steric repulsive effect.
Brønsted acids such as phosphoric acid and carboxylic acid are expected to exhibit dual function even though they are monofunctional acid catalysts because these molecules have both acidic and basic sites in the sole functional unit. Indeed, the dual-functional phosphoric acid unit has been effectively utilized in catalytic enantioselective reactions18 in which a nucleophile and an electrophile are easily captured owing to the dual-functional nature of the acid catalyst through hydrogen bonding interactions, and hence the reaction proceeds in an enantioselective manner under a chiral environment created by a chiral phosphoric acid catalyst. In this context, we hypothesized that if the relative orientation of the diene and the dienophile in TSs of the DA reaction could be restricted by the hydrogen bonds formed between the dual-functional acid catalyst19 and these substrates, the exo-DA reaction would be realized without having to use a bulky catalyst or the introduction of sterically demanding substituents to the substrates (Fig. 2).
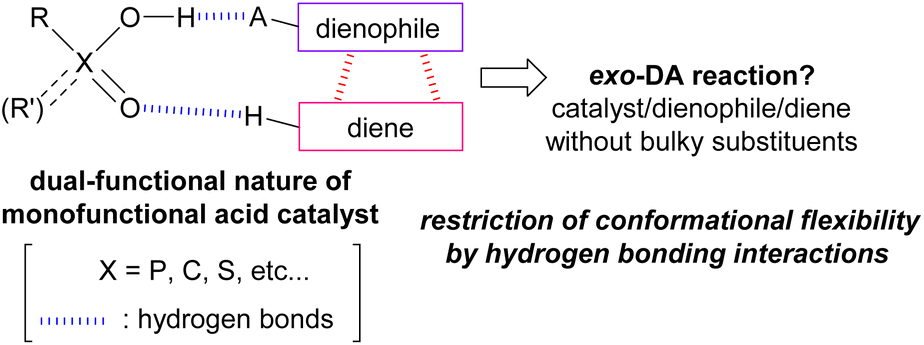 | ||
| Fig. 2 Working hypothesis. | ||
To verify this hypothesis, we devised a model reaction, that is, the DA reaction of vinylquinolines 1 with dienylcarbamates 2 catalysed by phosphoric acid, which we had previously reported as an endo-selective reaction.20 Computational analysis of this reaction revealed that the dual-functional nature of the phosphoric acid catalyst strictly restricts the relative orientation of the diene and the dienophile. Extraction of factors necessary for the exo-selectivity from this reaction system by computational studies and their experimental verification are expected to offer new insights into the control of the endo-/exo-selectivity in the DA reactions. Indeed, we developed an exo-DA reaction by designing the substrates and the catalytic system based on an exhaustive search for TSs using DFT calculations (Scheme 1). Herein we report new knowledge to realizing the exo-DA reaction, that is, not only regulating the conformational flexibility of the diene but also employing the dual-functional acid catalyst to restrict the relative orientation of the diene and the dienophile by virtue of the hydrogen bonding interaction.
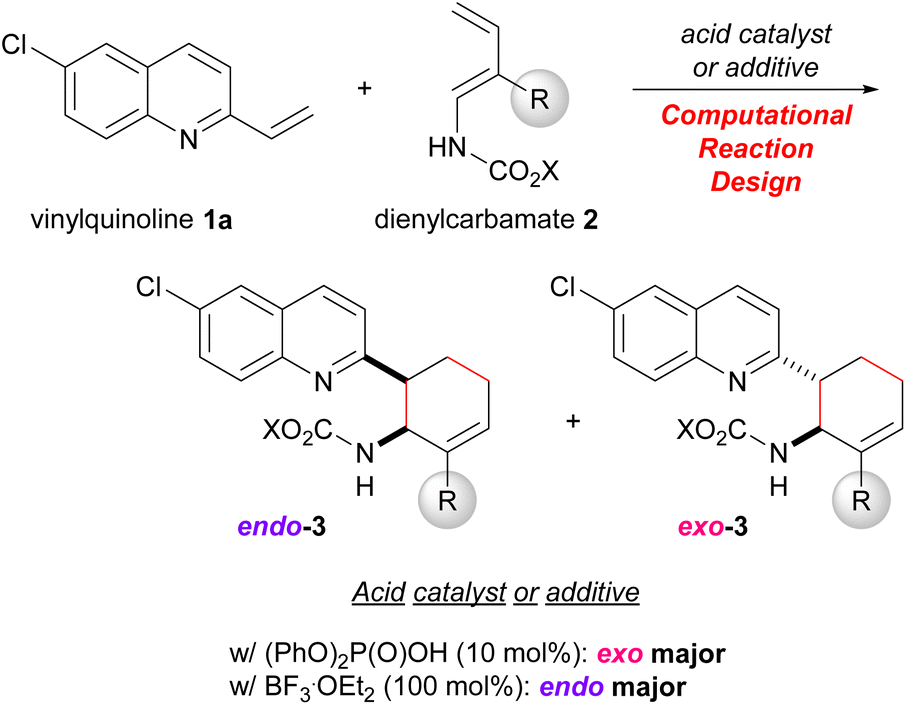 | ||
| Scheme 1 The developed exo-DA reaction that uses a dual-functional acid catalyst. | ||
Results and discussion
Initially, model calculations were performed to construct an evaluation system for endo-/exo-selectivity. The calculations were conducted in the gas phase at B3LYP-D3/6-31g(d) level of theory.‡21,22 In the model calculations, vinylquinoline 1b and dienylcarbamate 2a were employed and all possible conformations, relative orientations, and enantiotopic faces of these substrates were considered because of using chiral phosphoric acid (R)-4 as the model catalyst (Fig. 3). For vinylquinoline 1b, there are two types of conformational isomers, s-trans and s-cis. For dienylcarbamate 2a, four types of conformational isomers s-cis–cis, s-cis–trans, s-trans–cis, and s-trans–trans, in which the first and second s-cis/s-trans indicate the geometry of C1–N1−C2−C3 and O1–C1−N1−C2 respectively, were considered. In addition, four types of TSs, R-endo, S-endo, R-exo, and S-exo, originating from the relative orientation (endo and exo) and the enantiotopic faces (R and S) of dienylcarbamate 2a, were generated. Totally, the existence of 2 × 4 × 4 = 32 TS structures was assumed.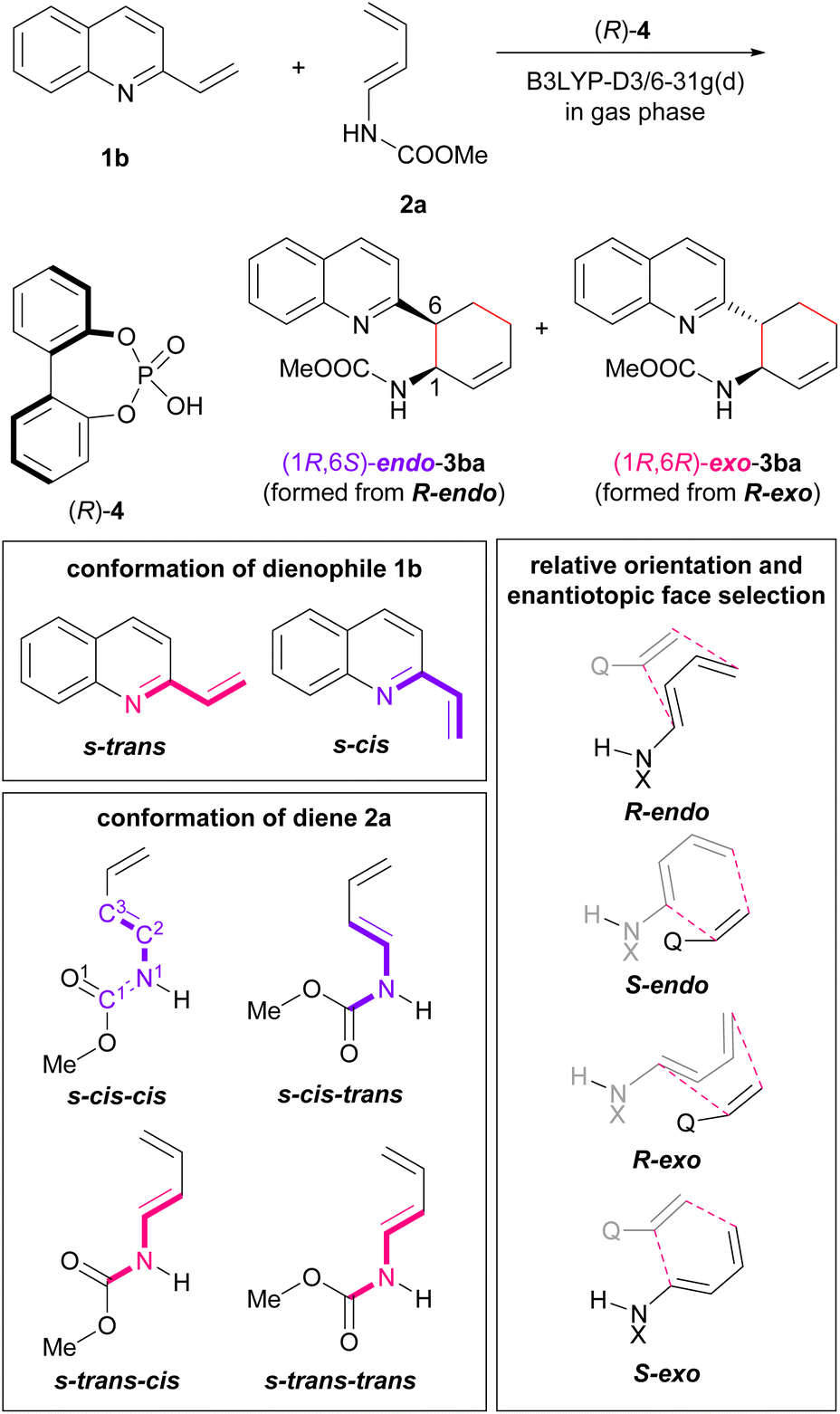 | ||
| Fig. 3 Method for model calculation. | ||
The calculated TSs and the relative free energies ΔΔG‡ of these TSs are summarized in Table 1. All TSs of the model system indicate that the relative free energies ΔΔG‡ of R-endo and R-exo TSs are comparable to those of the corresponding S-endo and S-exo TSs. Because vinylquinoline 1b is activated through protonation at the nitrogen atom by phosphoric acid, an N–H⋯O hydrogen bond is formed in all TSs (Fig. 4; see ESI† for 3D structures of all TSs). Focusing on the relationship between the conformation of vinylquinoline 1b and the free energy of TSs, overall, the s-trans conformation of vinylquinoline 1b is more energetically favourable than the s-cis conformation. As expected, the basic site, namely, the phosphoryl oxygen, of the acid catalyst captures the dienylcarbamate through the hydrogen bond formed with the carbamate N–H proton, and hence, TSs are stabilized by this hydrogen bonding interaction. In contrast, when the carbamate N–H proton is structurally unable to interact with the phosphoryl oxygen of the catalyst, TSs are markedly destabilized. Indeed, when vinylquinoline 1b formed the s-cis conformation, most of the corresponding TSs did not form a hydrogen bond and thus were unstable (see ESI† for 3D structures). At the most stable TS in the model calculation, a small difference in free energy (ΔΔG‡ = 0.4 kcal mol−1) was observed between endo-TS [TS-Cc(R-en)] and exo-TS [TS-Tc(R-ex)]. However, more importantly, exo-TS slightly predominated despite the fact that the DA reaction generally proceeds in an endo-selective manner. It is considered that this small energy gap originated from the hydrogen bonding interaction between the phosphoric acid and both substrates, as shown in Fig. 4.
| 1b | 2a | TSa | ΔΔG‡ b (kcal mol−1) | TS |
|---|---|---|---|---|
| a Transition states originate from the relative orientations (endo and exo), and enantiotopic faces (R and S) are generated.b Relative free energies of R-endo and R-exo TSs are tabulated, and the corresponding relative free energies of S-endo and S-exo TSs are shown in parentheses.c The corresponding S-exo TS could not be optimized. | ||||
| s-trans | cis–cis | R-endo | 0.4 (0.5) | TS-Cc(R-en) |
| R-exo | 13.7 (15.3) | — | ||
| cis–trans | R-endo | 3.8 (4.1) | TS-Ct(R-en) | |
| R-exo | 16.5 (−)c | — | ||
| trans–cis | R-endo | 11.2 (11.7) | — | |
| R-exo | 0.0 (1.1) | TS-Tc(R-ex) | ||
| trans–trans | R-endo | 9.5 (9.9) | — | |
| R-exo | 2.3 (3.5) | TS-Tt(R-ex) | ||
| s-cis | cis–cis | R-endo | 11.2 (14.3) | — |
| R-exo | 8.5 (7.5) | — | ||
| cis–trans | R-endo | 9.1 (11.6) | — | |
| R-exo | 10.9 (10.2) | — | ||
| trans–cis | R-endo | 5.7 (5.6) | — | |
| R-exo | 12.4 (11.0) | — | ||
| trans–trans | R-endo | 10.3 (10.9) | — | |
| R-exo | 9.4 (7.3) | — | ||
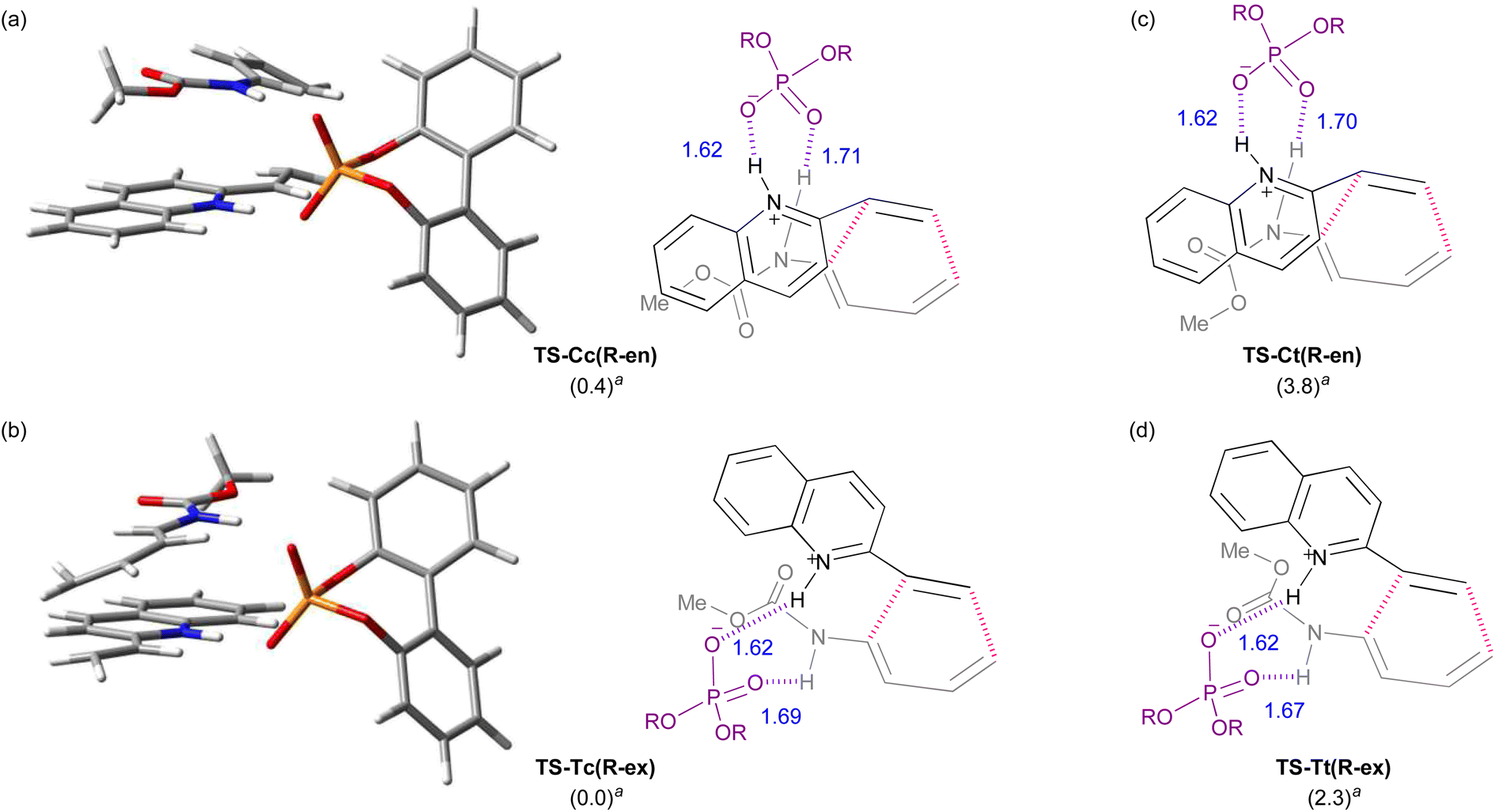 | ||
| Fig. 4 3D structures and schematic models of the most stable endo- and exo-TSs: (a) endo-TS: TS-Cc(R-en). (b) exo-TS: TS-Tc(R-ex). Schematic models of the second most stable endo- and exo-TSs: (c) endo-TS: TS-Ct(R-en). (d) exo-TS: TS-Tt(R-ex). aRelative free energy (kcal mol−1). | ||
Furthermore, among the R-endo and R-exo TSs calculated, it was found that the four TSs are relatively stable, their energies being within 5.0 kcal mol−1 from the energy of the most stable TS [TS-Tc(R-ex)] (Table 1 and Fig. 4). Detailed analysis of these four TSs revealed that the geometry around C1–N1−C2−C3 of dienylcarbamate 2a was different between the endo- and exo-TSs: dienylcarbamate 2a had an s-cis conformation in the endo-TS (Fig. 4a and c) and, in contrast, an s-trans conformation in the exo-TS (Fig. 4b and d). These results suggest that if the s-cis conformation of dienylcarbamate is selectively destabilized, the reaction will proceed preferentially from the s-trans conformation, giving rise to the exo-adduct in a stereoselective manner. In order to establish the exo-DA reaction, we proposed a molecular design in which a substituent is introduced at the 2-position of the diene unit (in the previous original numbering: C3) to destabilize the s-cis conformation selectively with little detrimental effect on the s-trans conformation (Fig. 5). On the basis of this proposal, we designed six dienylcarbamates 2b–g having a substituent at the 2-position and calculated the difference in free energy between endo- and exo-TS in the model system. All newly designed substrates 2b–g except 2f (R = SiMe3) were modified without having to introduce sterically demanding substituents.
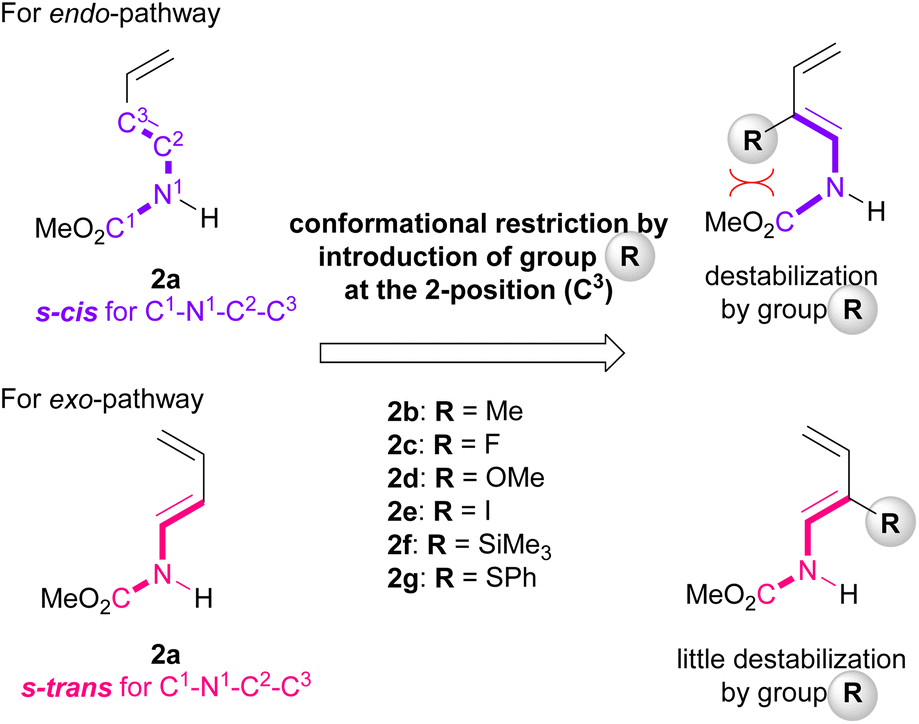 | ||
| Fig. 5 Concept of newly designed substrates for exo-DA reaction. | ||
DFT calculations were performed using newly designed dienylcarbamates 2b–g having a substituent at the 2-position, vinylquinoline 1b, and phosphoric acid (R)-4. The relative free energies of selected substrates 2b, 2c, and 2g are summarized in Table 2 (all TS energies of the model system using substrates 2b–g including S-endo and S-exo TSs are shown in ESI†). It became clear from the selected examples that while certain reactions through the S-enantiotopic face of dienylcarbamate 2 (S-endo) and its corresponding R-counterpart gave similar ΔΔG‡ values, the majority of cases showed that the reaction through the R-enantiotopic face had a smaller ΔΔG‡ value than the reaction through its S-counterpart. More importantly, as shown in Table 2 and all TSs in ESI,† the TSs giving the exo-adduct, namely, TSR-Tc(R-ex) (TSR: R = substituent introduced at the 2-position), are the most energetically favourable for newly designed dienylcarbamates 2b–g. Noteworthy is the fact that dienylcarbamate 2c having a small substituent such as fluorine at the 2-position undergoes the exo-DA reaction in a highly selective manner (calculated exo/endo = >20/1).
| R | TS | ΔΔG‡ a (kcal mol−1) |
|---|---|---|
| a Relative free energies of R-endo and R-exo TSs are shown, and the corresponding relative free energies of S-endo and S-exo TSs are partially shown in parentheses.b ΔΔG‡ for TSMe-Cc(S-en).c ΔΔG‡ for TSMe-Ct(S-en).d ΔΔG‡ for TSF-Cc(S-en). | ||
| Me (2b) | TSMe-Cc(R-en) | 5.3 (5.2)b |
| TSMe-Ct(R-en) | 9.4 (9.3)c | |
| TSMe-Tc(R-ex) | 0.0 | |
| TSMe-Tt(R-ex) | 2.7 | |
| F (2c) | TSF-Cc(R-en) | 2.7 (2.6)d |
| TSF-Ct(R-en) | 5.3 | |
| TSF-Tc(R-ex) | 0.0 | |
| TSF-Tt(R-ex) | 2.9 | |
| SPh (2g) | TSSPh-Cc(R-en) | 6.4 |
| TSSPh-Ct(R-en) | 9.5 | |
| TSSPh-Tc(R-ex) | 0.0 | |
| TSSPh-Tt(R-ex) | 2.4 | |
The 3D TSs and the schematic models derived from the calculations using dienylcarbamate 2b (R = Me) are shown as a representative (Fig. 6). In the endo-TS, the introduction of the R group at the 2-position (in the previous original numbering: C3) resulted in a marked steric repulsion between substituent R and the carbamate unit because C1–N1−C2−C3 of dienylcarbamate 2b took an s-cis conformation. As predicted, this steric congestion resulted in a twisted conformation of the dienylcarbamates, in which the carbamate unit markedly deviated from the coplanarity with the diene unit by around 30° (Fig. 6a and c). In contrast, in the exo-TS, the introduction of the R group caused little steric repulsion because C1–N1−C2−C3 took an s-trans conformation (Fig. 6b and d). Hence the corresponding dihedral angle was expected to be close to 180°. Indeed, the dihedral angle was around 170° and the coplanarity between the diene and carbamate units was almost maintained. As described above, the proposal, “the introduction of substituent R at the 2-position destabilizes the s-cis conformation, pushing up the energy level of the endo-TS”, was confirmed by the DFT calculations.
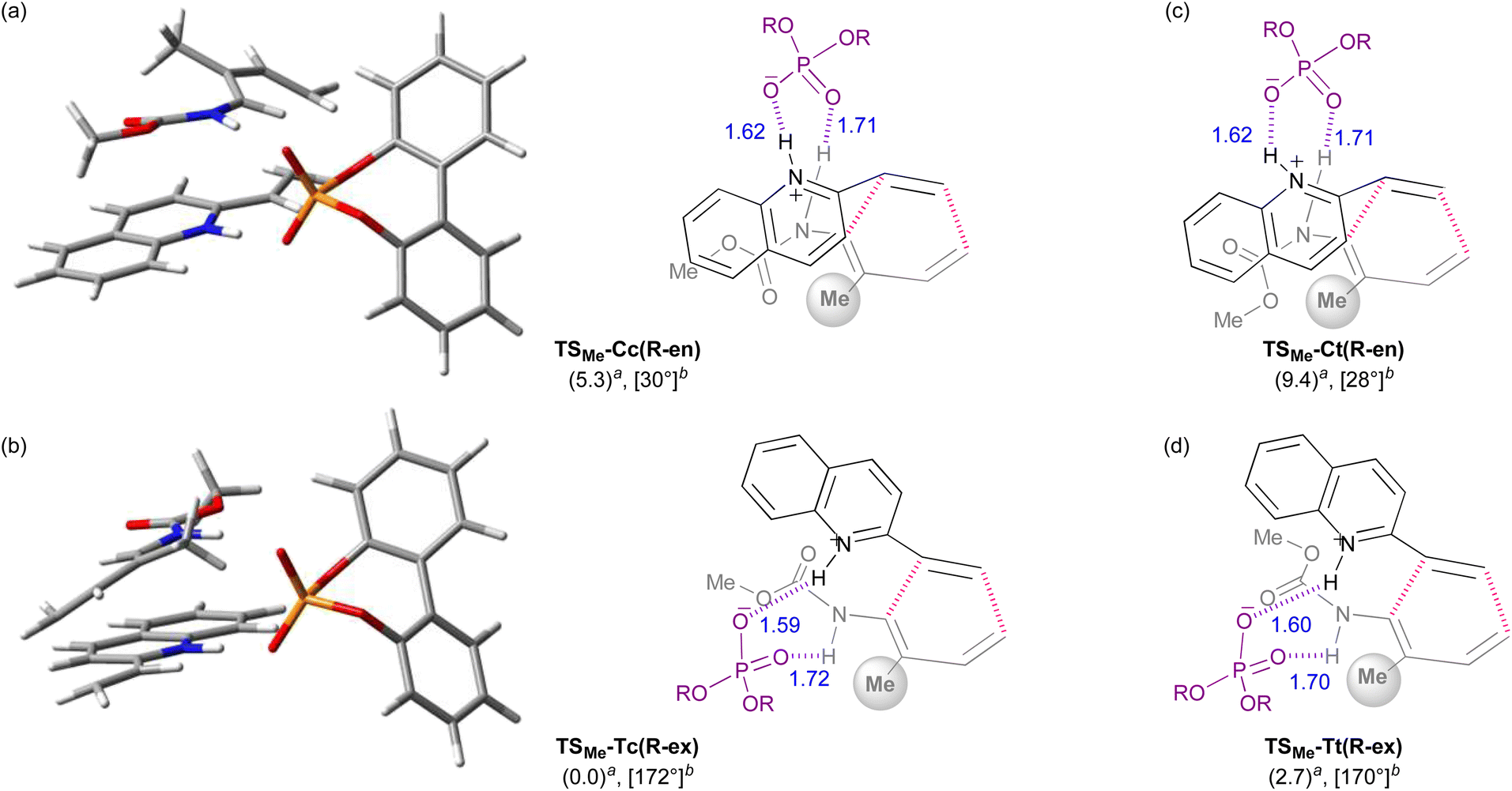 | ||
| Fig. 6 DFT calculations of the transition states in the reaction of dienylcarbamate 2b (R = Me) with vinylquinoline 1b catalysed by phosphoric acid (R)-4. 3D structures and schematic models of the most stable endo- and exo-TSs: (a) endo-TS: TSMe-Cc(R-en). (b) Exo-TS: TSMe-Tc(R-ex). Schematic models of the second most stable endo- and exo-TSs: (c) endo-TS: TSMe-Ct(R-en). (d) Exo-TS: TSMe-Tt(R-ex). aRelative free energy (kcal mol−1). bDihedral angle of C1–N1−C2−C3 for dienylcarbamate 2b (R = Me). | ||
Our greatest concern was whether the exo-stereochemical outcome predicted by the DFT calculations could be verified experimentally. A newly designed substrate was investigated using dienylcarbamate 2b having a methyl group at the 2-position (R = Me) (Table 3). Furthermore, in order to confirm the importance of the formation of hydrogen bonds between the catalyst/additive and both substrates, vinylquinoline 1a and dienylcarbamate 2b, we employed different acid catalyst/additive to investigate endo-/exo-selectivity in the intended DA reaction. As predicted, the use of diphenyl phosphate [(PhO)2P(O)OH] as the acid catalyst facilitated the formation of an exo-adduct in a highly selective manner (entry 1).§ In contrast, endo-selectivity was predominant when the reaction was performed using Lewis acids such as boron trifluoride diethyl ether complex (BF3·OEt2)23 and tris(pentafluorophenyl)borane [B(C6F5)3] (entries 2 and 3). The predominant formation of the endo-adduct was also observed under thermal reaction conditions (without using an acid catalyst/additive) (entry 4) as a common stereochemical outcome in the intermolecular normal-electron-demand DA reaction. Furthermore, trichloroacetic acid and p-toluene sulfonic acid were considered promising candidates for the dual-functional Brønsted acid catalyst similar to phosphoric acid. In fact, under the influence of these acid catalysts, the exo-adduct was obtained as the major diastereomer, albeit with a slightly low selectivity (entries 5 and 6). These results indicate that the type of acid catalyst/additive has a significant impact on the endo-/exo-selectivity. As hypothesized for the present reaction system, the dual-functional nature of the acid catalyst is essential for the exo-DA reaction. Even if the s-cis conformation of dienylcarbamate is effectively destabilized by the steric effect of the substituent (R = Me in Fig. 6), no exo-selectivity would be achieved when no hydrogen bonds are formed between the acid catalyst and both substrates. The relative stereochemistry of 3ab was determined by comparing the 1H NMR coupling constants of the endo- and exo-products7a,8b (see ESI† for details).
|
||||
|---|---|---|---|---|
| Entry | Catalyst/additive (mol%) | Time (h) | Yieldb (%) | Endo/exoc |
| a Unless otherwise noted, all reactions were carried out using 0.1 mmol of 1a, 0.12 mmol of 2b, MS 5A (50 mg), and catalyst/additive in toluene (1 mL) at room temperature.b Isolated yields.c Determined by 1H NMR measurement of the crude mixture.d Without MS 5A. | ||||
| 1 | (PhO)2P(O)OH (10) | 48 | 70 | 3/97 |
| 2d | BF3·OEt2 (100) | 24 | 43 | 81/19 |
| 3 | B(C6F5)3 (10) | 48 | 50 | 82/18 |
| 4 | None, 100 °C | 48 | 13 | 62/38 |
| 5 | Cl3CCO2H (10) | 48 | 75 | 15/85 |
| 6 | TsOH·H2O (10) | 48 | 52 | 25/75 |
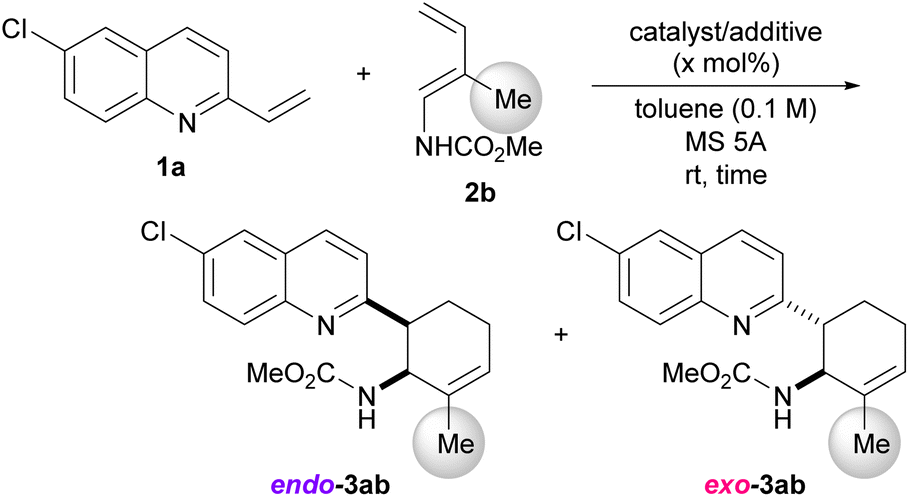
Next, using a series of dienylcarbamates 2, we investigated the substituent effect on the endo-/exo-selectivity (Table 4). In the case of simple dienylcarbamate 2a, instead of 2b having a methyl substituent, either diphenyl phosphate [(PhO)2P(O)OH] (condition A) or boron trifluoride diethyl ether complex (BF3·OEt2) (condition B) resulted in the formation of an endo- adduct as the major diastereomer (entries 1 and 2). Similarly, under the same reaction conditions, dienylcarbamate 2h having a methyl group at the 3-position underwent the reaction with low to moderate endo-selectivity (entries 3 and 4). These results suggest that the conformational restriction of dienylcarbamate by the introduction of a substituent at the 2-position is the key to achieving an exo-selective reaction, as predicted by the DFT calculations (Table 2 and Fig. 5). Dienylcarbamate having a methyl substituent at the 2-position was further modified by changing the carbamate unit to the Boc group from methoxycarbonyl (entries 5 and 6). exo-Selectivity was observed when (PhO)2P(O)OH was used as the acid catalyst (entry 5), whereas the endo-adduct was produced when BF3·OEt2 was used (entry 6). Even when the substituent at the 2-position was replaced by a thiophenyl group instead of a methyl group, (PhO)2P(O)OH facilitated the reaction in an exo-selective manner (entry 7). Again, the use of BF3·OEt2 resulted in the endo-selectivity in the same reaction (entry 8). It should be emphasized that the combined use of the dual-functional acid catalyst and the conformationally restricted dienylcarbamate is crucial to accomplishing the DA reaction in an exo-selective manner.
|
|||||
|---|---|---|---|---|---|
| Entry | Cond.b | 2 | 3 | Yieldc (%) | Endo/exod |
| a Unless otherwise noted, all reactions were carried out using 0.1 mmol of 1a, 0.12 mmol of 2, and MS 5A (50 mg) in toluene (1 mL) at room temperature.b Condition A: 10 mol% (PhO)2P(O)OH was used as an acid catalyst. Condition B: 100 mol% BF3·OEt2 was used as an acid additive.c Isolated yields.d Determined by 1H NMR measurement of the crude mixture. | |||||
| 1 | A | 2a | 3aa | 68 | 72/28 |
| 2 | B | 60 | 83/17 | ||
| 3 | A | 2h | 3ah | 80 | 60/40 |
| 4 | B | 21 | 71/29 | ||
| 5 | A | 2i | 3ai | 45 | 25/75 |
| 6 | B | 50 | 80/20 | ||
| 7 | A | 2g | 3ag | 24 | 26/74 |
| 8 | B | 28 | 69/31 | ||
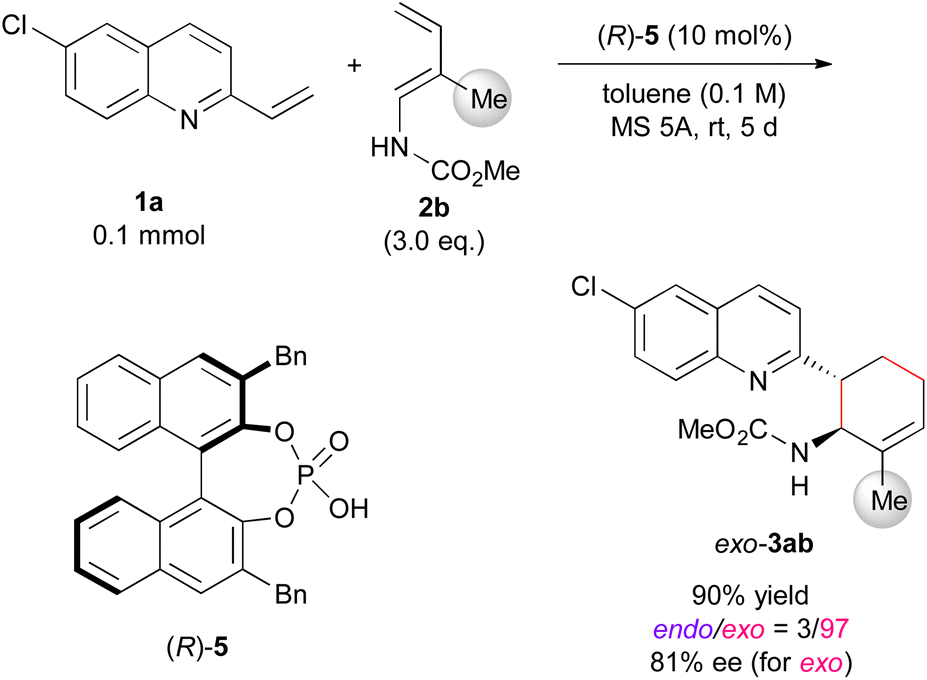
Finally, we attempted to expand the developed exo-DA reaction to an enantioselective variant using chiral phosphoric acid (Scheme 2).24 In the reaction of 1a with 2b, the use of (R)-5 having benzyl groups at the 3,3′-positions resulted in exo-DA product 3ab in good yield with high exo-selectivity and fairly good enantioselectivity¶ (see ESI† for catalyst screening). The number of examples of enantioselective exo-DA reactions using acyclic dienes under the influence of chiral catalyst6a,7a,12j,16,25 is far fewer than those of endo-DA reactions. Hence, the present approach provides valuable insights into the development of novel catalytic enantioselective exo-DA reactions using acyclic dienes.
 | ||
| Scheme 2 Initial attempt at enantioselective exo-DA reaction. | ||
Conclusions
We have demonstrated a novel approach to establish an exo-DA reaction of an acyclic diene without introducing sterically demanding substituents into either a catalyst, a dienophile (vinylquinoline), or an acyclic diene (dienylcarbamate). Factors necessary for achieving the exo-DA reaction, in which the relative orientation of the dienophile and the acyclic diene is firmly defined by hydrogen bonding interactions with a dual-functional Brønsted acid catalyst, were extracted by thorough computational analysis of the corresponding transition states and verified experimentally. As predicted by DFT calculations, conformational restriction of the diene (dienylcarbamate) by the introduction of a substituent at the 2-position of the diene unit is the key to achieving the DA reaction in an exo-selective manner. Thus, the effective utilization of the hydrogen bonding interaction realized by a combination of the dual-functional acid catalyst and the conformationally restricted diene (dienylcarbamate) is crucial to achieving the exo-DA reaction. The present approach is expected to give new insights into the control of endo-/exo-selectivity in the DA reactions and contribute to the development of stereodivergent methods for the DA reactions. Further studies along this line are in due course in our laboratory.Data availability
The exploratory investigation, experimental procedures, computational data, and characterization data are available.Author contributions
T. N. contributed conceptualization, design of the work, data curation, formal analysis, investigation (experimental and theoretical studies), and writing – original draft. M. T. contributed conceptualization, project administration, writing – review & editing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computation was performed using Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C127). This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (JP17H06447) from MEXT, Japan (M. T.), a Grant-in-Aid for Challenging Research (Exploratory) (JP22K19018) from JSPS (M. T.), and a Grant-in-Aid for Young Scientists (JP21J14551) from JSPS, Japan (T. N.).Notes and references
- (a) K. C. Nicolau, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef; (b) M. Gregoritza and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 97, 438–453 CrossRef CAS PubMed; (c) M. M. Heravi, T. Ahmadi, M. Ghavidel, B. Heidari and H. Hamidi, RSC Adv., 2015, 5, 101999–102705 RSC.
- K. Alder and G. Stein, Angew. Chem., 1937, 50, 510–519 CrossRef CAS.
- (a) R. Hoffmann and R. B. Woodward, J. Am. Chem. Soc., 1965, 87, 4388–4389 CrossRef CAS; (b) R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Academic Press, New York, 1970 Search PubMed.
- (a) W. R. Roush and B. B. Brown, J. Org. Chem., 1992, 57, 3380–3387 CrossRef CAS; (b) J. I. García, J. A. Mayoral and L. Salvatella, Acc. Chem. Res., 2000, 33, 658–664 CrossRef PubMed; (c) A. Arrieta, F. P. Cossío and B. Lecea, J. Org. Chem., 2001, 66, 6178–6180 CrossRef CAS PubMed; (d) J. I. García, J. A. Mayoral and L. Salvatella, Eur. J. Org. Chem., 2005, 85–90 CrossRef; (e) C. S. Wannere, A. Paul, R. Herges, K. N. Houk, H. F. Schaefer III and P. von Rague Schleyer, J. Comput. Chem., 2007, 28, 344–361 CrossRef CAS PubMed; (f) I. Fernandez and F. M. Bickelhaupt, J. Comput. Chem., 2014, 35, 371–376 CrossRef CAS PubMed; (g) W. J. Lording, T. Fallon, M. S. Sherburn and M. N. Paddon-Row, Chem. Sci., 2020, 11, 11915–11926 RSC.
- If one alkene is embedded in a congested ring structure, exo-selectivity is often expressed, see: (a) T. Yoon, S. J. Danishefsky and S. A. de Gala, Angew. Chem., Int. Ed. Engl., 1994, 33, 853–855 CrossRef; (b) J. D. Ha, C. H. Kang, K. A. Belmore and J. K. Cha, J. Org. Chem., 1998, 63, 3810–3811 CrossRef CAS; (c) M. Ge, B. M. Stoltz and E. J. Corey, Org. Lett., 2000, 2, 1927–1929 CrossRef CAS PubMed.
- (a) H. Usuda, A. Kuramochi, M. Kanai and M. Shibasaki, Org. Lett., 2004, 6, 4387–4390 CrossRef CAS PubMed; (b) G. M. Sammis, E. M. Flamme, H. Xie, D. M. Ho and E. Sorensen, J. Am. Chem. Soc., 2005, 127, 8612–8613 CrossRef CAS PubMed; (c) Y. Zeng and J. Aube, J. Am. Chem. Soc., 2005, 127, 15712–15713 CrossRef CAS PubMed.
- (a) Z. Liu, X. Lin, N. Yang, Z. Su, C. Hu, P. Xiao, Y. He and Z. Song, J. Am. Chem. Soc., 2016, 138, 1877–1883 CrossRef CAS PubMed; (b) G.-M. Ho, C.-J. Huang, E. Y.-T. Li, S.-K. Hsu, T. Wu, M. M. L. Zulueta, K. B. Wu and S.-C. Hung, Sci. Rep., 2016, 6, 35147 CrossRef PubMed; (c) J. Wang, Z. Liu, J. Li, Z. Song, C. Hu and Z. Su, J. Org. Chem., 2019, 84, 3940–3952 CrossRef CAS PubMed; (d) C.-J. Huang and E. Y. Li, RSC Adv., 2019, 9, 7246–7250 RSC.
- (a) Y.-H. Lam, C. Bobbio, I. R. Cooper and V. A. A. Gouverneur, Angew. Chem., Int. Ed., 2007, 46, 5106–5110 CrossRef CAS PubMed; (b) Y.-H. Lam, P. H.-Y. Cheong, J. M. B. Mata, S. J. Stanway, V. Gouverneur and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 1947–1957 CrossRef CAS PubMed.
- (a) W. R. Roush, A. P. Essenfeld, J. S. Warmus and B. B. Brown, Tetrahedron Lett., 1989, 30, 7305–7308 CrossRef CAS; (b) K. Takeda, I. Imaoka and E. Yoshii, Tetrahedron, 1994, 50, 10839–10848 CrossRef CAS; (c) W. R. Roush, M. L. Reilly, K. Koyama and B. B. Brown, J. Org. Chem., 1997, 62, 8708–8721 CrossRef CAS; (d) W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1998, 120, 7411–7419 CrossRef CAS; (e) W. R. Roush, D. A. Barda, C. Limberakis and R. K. Kunz, Tetrahedron, 2002, 58, 6433–6454 CrossRef CAS; (f) W. R. Roush, C. Limberakis, R. K. Kunz and D. A. Barda, Org. Lett., 2002, 4, 1543–1546 CrossRef CAS PubMed; (g) J. Qi and W. R. Roush, Org. Lett., 2006, 8, 2795–2798 CrossRef CAS PubMed.
- (a) S. R. Gilbertson, X. Zhao, D. P. Dawson and K. L. Marshall, J. Am. Chem. Soc., 1993, 115, 8517–8518 CrossRef CAS; (b) M. W. Wright, T. L. Smalley, M. E. Welker and A. L. Rheingold, J. Am. Chem. Soc., 1994, 116, 6777–6791 CrossRef CAS; (c) B. M. Richardson and M. E. Welker, J. Org. Chem., 1997, 62, 1299–1304 CrossRef CAS; (d) T. S. Powers, W. Jiang, J. Su, W. D. Wulff, B. E. Waltermire and A. L. Rheingold, J. Am. Chem. Soc., 1997, 119, 6438–6439 CrossRef CAS; (e) J. Barluenga, R. M. Canteli, J. Florez, S. García-Granda, A. Gutierrez-Rodríguez and E. Martín, J. Am. Chem. Soc., 1998, 120, 2514–2522 CrossRef CAS.
- The reaction of α-substituted acrolein with cyclopentadiene is well known for its tendency to develop exo selectivity. see selected examples: (a) E. J. Corey and T. P. Loh, J. Am. Chem. Soc., 1991, 113, 8966–8967 CrossRef CAS; (b) E. P. Kündig, B. Bourdin and G. Bernardinelli, Angew. Chem., Int. Ed. Engl., 1994, 33, 1856–1858 CrossRef; (c) Y. Hayashi, J. J. Rohde and E. J. Corey, J. Am. Chem. Soc., 1996, 118, 5502–5503 CrossRef CAS; (d) K. Ishihara, H. Kurihara, M. Matsumoto and H. Yamamoto, J. Am. Chem. Soc., 1998, 120, 6920–6930 CrossRef CAS; (e) Y.-C. Teo and T.-P. Loh, Org. Lett., 2005, 7, 2539–2541 CrossRef CAS PubMed; (f) F. Fu, Y.-C. Teo and T.-P. Loh, Org. Lett., 2006, 8, 5999–6001 CrossRef CAS PubMed.
- (a) K. Maruoka, H. Imoto and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 12115–12116 CrossRef CAS; (b) E. P. Kündig, C. M. Saudan, V. Alezra, F. Viton and G. Bernardinelli, Angew. Chem., Int. Ed., 2001, 40, 4481–4485 CrossRef; (c) K. Ishihara and K. Nakano, J. Am. Chem. Soc., 2005, 127, 10504–10505 CrossRef CAS PubMed; (d) T. Kano, Y. Tanaka and K. Maruoka, Org. Lett., 2006, 8, 2687–2689 CrossRef CAS PubMed; (e) H. Gotoh and Y. Hayashi, Org. Lett., 2007, 9, 2859–2862 CrossRef CAS PubMed; (f) Y. Hayashi, S. Samanta, H. Gotoh and H. Ishikawa, Angew. Chem., Int. Ed., 2008, 47, 6634–6637 CrossRef CAS PubMed; (g) M. Bakos, Z. Dobi, D. Fegyverneki, Á. Gyömöre, I. Fernández and T. Soós, ACS Sustainable Chem. Eng., 2018, 6, 10869–10875 CrossRef CAS; (h) M. Hou, M. Xu, B. Yang, H. He and S. Gao, Org. Lett., 2021, 23, 7487–7491 CrossRef CAS PubMed; (i) B. P. Bondzić, K. Daskalakis, T. Taniguchi, K. Monde and Y. Hayashi, Org. Lett., 2022, 24, 7455–7460 CrossRef PubMed; (j) Y.-H. Li, S.-L. Zhang, Y. Lu, B. Xiao, T.-Y. Sun, Q.-Q. Xu, J.-H. Chen and Z. Yang, Angew. Chem., Int. Ed., 2023, 62, e202303075 CrossRef CAS PubMed.
- (a) A. Sakakura, K. Suzuki and K. Ishihara, Adv. Synth. Catal., 2006, 348, 2457–2465 CrossRef CAS; (b) M. Hatano, T. Mizuno, A. Izumiseki, R. Usami, T. Asai, M. Akakura and K. Ishihara, Angew. Chem., Int. Ed., 2011, 50, 12189–12192 CrossRef CAS PubMed; (c) M. Hatano and K. Ishihara, Chem. Commun., 2012, 48, 4273–4283 RSC; (d) K. Matsui, K. Toh, M. Hatano and K. Ishihara, Org. Lett., 2022, 24, 6483–6488 CrossRef CAS PubMed.
- (a) J.-H. Zhou, B. Jiang, F.-F. Meng, Y.-H. Xu and T.-P. Loh, Org. Lett., 2015, 17, 4432–4435 CrossRef CAS PubMed; (b) D. Yepes, P. Pérez, P. Jaque and I. Fernández, Org. Chem. Front., 2017, 4, 1390–1399 RSC.
- D. Taherinia, M. M. Mahmoodi and A. Fattahi, New J. Chem., 2021, 45, 16760–16772 RSC.
- A. Heine, E. A. Stura, J. T. Yli-Kauhaluoma, C. Gao, Q. Deng, B. R. Beno, K. N. Houk, K. D. Janda and I. A. Wilson, Science, 1998, 279, 1934–1940 CrossRef CAS PubMed.
- D. Taherinia and A. Fattahi, Sci. Rep., 2022, 12, 22225 CrossRef PubMed.
- (a) Y. Toda, T. Korenaga, R. Obayashi, J. Kikuchi and M. Terada, Chem. Sci., 2021, 12, 10306–10312 RSC; (b) J. P. Reid and J. M. Goodman, J. Am. Chem. Soc., 2016, 138, 7910–7917 CrossRef CAS PubMed; (c) P. A. Champagne and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 12356–12359 CrossRef CAS PubMed; (d) C. Liu, M. Besora and F. Maseras, Chem. –Asian J., 2016, 11, 411–416 CrossRef CAS PubMed; (e) F. Li, T. Korenaga, T. Nakanishi, J. Kikuchi and M. Terada, J. Am. Chem. Soc., 2018, 140, 2629–2642 CrossRef CAS PubMed; (f) K. Kanomata, Y. Nagasawa, M. Yamanaka, Y. Shibata, F. Egawa, J. Kikuchi and M. Terada, Chem.–Eur. J., 2020, 26, 3364–3372 CrossRef CAS PubMed.
- (a) J. Pous, T. Courant, G. Bernadat, B. I. Iorga, F. Blanchard and G. Masson, J. Am. Chem. Soc., 2015, 137, 11950–11953 CrossRef CAS PubMed; (b) N. Momiyama, H. Tabuse, H. Noda, M. Yamanaka, T. Fujinami, K. Yamanishi, A. Izumiseki, K. Funayama, F. Egawa, S. Okada, H. Adachi and M. Terada, J. Am. Chem. Soc., 2016, 138, 11353–11359 CrossRef CAS PubMed; (c) T. Varlet, C. Gelis, P. Retailleau, G. Bernadat, L. Neuville and G. Masson, Angew. Chem., Int. Ed., 2020, 59, 8491–8496 CrossRef CAS PubMed.
- T. Nakanishi and M. Terada, Chem. Sci., 2023, 14, 5712–5721 RSC.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) S. Portela and I. Fernández, J. Org. Chem., 2022, 87, 9307–9315 CrossRef CAS PubMed; (b) A. E. Davis, J. M. Lowe and M. K. Hilinski, Chem. Sci., 2021, 12, 15947–15952 RSC.
- Chiral phosphoric acid catalysts: (a) N. Momiyama, H. Tabuse and M. Terada, J. Am. Chem. Soc., 2009, 131, 12882–12883 CrossRef CAS PubMed; (b) Y.-Y. Wang, K. Kanomata, T. Korenaga and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 927–931 CrossRef CAS PubMed; (c) Y. Ota, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2018, 57, 13917–13921 CrossRef CAS PubMed; (d) S. Umemiya and M. Terada, Org. Lett., 2021, 23, 3767–3771 CrossRef CAS PubMed; (e) T. Uchikura, N. Kamiyama, T. Mouri and T. Akiyama, ACS Catal., 2022, 12, 5209–5216 CrossRef CAS; (f) T. Uchikura, K. Aruga, R. Suzuki and T. Akiyama, Org. Lett., 2022, 24, 4699–4703 CrossRef CAS PubMed . For seminal studies of chiral phosphoric acid catalysts, see:; (g) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (h) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- Y. Sudo, D. Shirasaki, S. Harada and A. Nishida, J. Am. Chem. Soc., 2008, 130, 12588–12589 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07688a |
| ‡ Since the highest priority was to grasp the overall feature of the endo-/exo-selectivity of the present DA reaction, we selected B3LYP-D3, which has a relatively low computational load. In our previous work, we thoroughly searched several functions of the DFT calculation and confirmed that B3LYP-D3 is one of the reliable ones for the transition state analysis in the present reaction system (see, ref. 20). |
| § The DA reaction of parent 2-vinylquinolines (1b) with 2b using (PhO)2P(O)OH as the catalyst (condition A shown in Table 4) gave the exo-adduct as the major stereoisomer (69% combined yield of endo/exo-isomers) with exactly the same stereoselectivity (endo/exo = 3/97) as the DA reaction of chloro-substituted 1a with 2b (Table 3, entry 1). |
| ¶ The absolute configuration of exo-3ab has not been determined yet. |
| This journal is © The Royal Society of Chemistry 2023 |