
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo-dimensional covalent organic frameworks made of triquinoxalinylene derivatives are promising anodes for high-performance lithium and sodium ion batteries†
Tianze Xu,
Youchao Yang,
Tianyang Liu* and
Yu Jing *
*
Jiangsu Co-Innovation Centre of Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China. E-mail: liutianyang@njfu.edu.cn; yujing@njfu.edu.cn
First published on 28th November 2023
Abstract
Searching for electrode materials with good electrical conductivity, fast charge/discharge rates and high storage capacity is essential for the development of high-performance metal ion batteries. Here, by performing first principles calculations, we have explored the feasibility of using two dimensional (2D) covalent organic frameworks (COFs) constructed by tri-quinazoline, triquinoxalinylene and benzoquinone, and tribenzoquinoxaline-5,10-dione and benzoquinone (BQ2), as electrode materials for lithium and sodium ion batteries. All the designed 2D COFs show good structure stability and are semiconductors with a band gap of 1.63–2.93 eV because of the high electron conjugation of the skeletons. The pyrazine N and carbonyl groups are revealed to be the active sites to combine Li/Na, while the Li-/Na-binding strength can be highly enhanced when the pyrazine N and the carbonyl group are located in adjacent sites. The designed 2D COFs show a low Li and Na diffusion barrier in the range of 0.28–0.56 eV to guarantee high rate performance for LIBs/SIBs. With abundant redox active sites, 2D BQ2-COF shows a high theoretical capacity of 1030 mA h g−1 with an average open circuit voltage of 0.80 and 0.67 V for LIBs and SIBs, respectively, which is comparable to that of the most advanced inorganic anode materials. Composed of only light elements, the designed 2D COFs are predicted to be promising anode materials with high energy density, good conductivity and high-rate performance for sustainable LIBs and SIBs.
1. Introduction
The large consumption of fossil fuels has caused a growing threat of the energy crisis and environmental pollution. Solving these problems depends on the large-scale utilization of clean and renewable resources,1–3 such as solar, wind and tidal energy. However, these sustainable energy sources are intermittent in nature, thus require the application of efficient energy storage systems.4–6 Lithium-ion batteries (LIBs) are the most widely applied energy storage devices, because of their high energy density, large output power and good safety.7–10 However, since the widespread use of LIBs has caused increasing concerns of lithium depletion, sodium-ion batteries (SIBs) have been developed as possible alternatives to LIBs because of the high abundance of sodium on the earth. Generally, LIBs and SIBs work based on similar electrochemical principles, and their electrochemical performance is mainly determined by the electrode materials. Currently, the most widely studied electrode materials are inorganic materials,11–14 which usually contain toxic and expensive transition metals, thus show restrained applications in LIBs and SIBs. Moreover, since the radius of Na is larger than that of Li, many anode materials of LIBs cannot be applied to SIBs. The exploration of environmentally benign and sustainable electrode materials is essential for the long-term development of LIBs and SIBs.Composed of only light elements, organic electrode materials show a low environmental footprint, high structure diversity and high design flexibility, thus are promising candidates to replace traditional inorganic electrode materials.15,16 Remarkably, the energy storage process of organic electrode materials is completed by redox reaction with Li or Na, thus is not limited by the radius of metal ions. Many organic electrode materials such as carbonyl and imine compounds can be widely found in nature or can be produced from biomass raw materials, thus are sustainable electrode materials for LIBs and SIBs and have gained wide research interest.17,18 However, organic electrodes based on small organic molecules suffer from poor stability and low capacity retention because of their high solubility in electrolyte and low electrical conductivity. The development of effective strategies to significantly reduce the solubility of organic molecules and enhance their conductivity is essential to obtaining high-performance organic electrode materials.19–22
Polymerization of small redox-active organic molecules into periodic covalent organic frameworks (COFs) can significantly reduce their solubility in electrolytes and enhance the electron conjugation in long-range skeletons, contributing to improved stability and electrical conductivity for LIBs and SIBs. Especially, the fast development of two dimensional (2D) materials in the past decade23 has significantly motivated the exploration of 2D electrode materials for LIBs and SIBs.24–27 The high specific surface area and large open surface space are intrinsically beneficial for the fast transfer of electrons and metal ions in electrolyte. Remarkably, featuring pore structures, 2D COFs can provide ordered open channels to further facilitate ion transport for fast charge and discharge processes.28,29 Therefore, coupling small organic molecules into 2D polymers or COFs is of great significance for the construction of high-performance OEMs.30 Since many amine and carbonyl compounds have been demonstrated to be promising electrode materials for LIBs and SIBs,31–35 the construction of 2D conjugated COFs using these molecules as building monomers is an attractive strategy to obtain high performance electrode materials. However, it requires the molecules to be with high symmetry and that they can be connected in two dimensions via available experimental methods. Recently, tri-quinazoline (3Q), triquinoxalinylene (TQBQ) and tribenzoquinoxaline-5,10-dione (3BQ)36 have been demonstrated to be effective organic electrodes for LIBs and SIBs because of the high redox activity of the carbonyl and pyrazine N sites.37 Remarkably, the D3h symmetry of these molecules allows the construction of corresponding 2D COFs with a honeycomb lattice (Fig. 1), which can be experimentally realized by simple condensation reactions. For example, Shi et al. demonstrated that TQBQ-COF can be synthesized by a triple condensation reaction between tetraminophenone and cyclohexanehexaone, and show a high specific capacity of 452.0 mA h g−1 and good cycling stability for sodium batteries.31,36 However, the energy storage process of these molecules based 2D COFs need to be clearly understood and it is intriguing to figure out the structure–property relationship of multiple carbonyls and pyrazine N sites for the electrochemical performance of 2D COFs for LIBs and SIBs.
 | ||
| Fig. 1 Top and side views of a structurally optimized 2D COFs of 3Q (a), TQBQ (b), 3BQ (c) and BQ2 (d) in a 2 × 2 × 1 supercell. The vacuum layer was set to be 15 Å. | ||
Via first principles calculations, we here explored the potential of 2D COFs made of 3Q, TQBQ, 3BQ and benzoquinone (BQ2) molecules as electrode materials for LIBs and SIBs. Note that 2D-3Q and 2D-TQBQ have been experimentally realized,31,61 while 2D-3BQ and 2D-BQ2 have never been reported before. It was found that all the examined 2D COFs show good structural stability and are semiconductors with a band gap of 1.63, 2.78, 2.93, and 1.96 eV for 2D-3Q, -TQBQ, -3BQ and -BQ2, respectively. The influences of multiple carbonyl and pyridine functional groups on the electrochemical performance of 2D COFs were systematically studied by estimating the combination of Li/Na on different surface sites and their diffusion within the pores and across the 2D frameworks. The results show that the bridge site of two pyridine N is the most preferred site for the combination of Li/Na and all examined 2D COFs show a low diffusion barrier for Li/Na ions. By estimating the theoretical capacity and open-circuit voltage, we found that all the examined 2D COFs show high capacities of 515.4–1030.1 mA h g−1 and low average OCV for Li and Na storage, respectively, and are promising anode materials for LIB and SIB batteries.
2. Computational methods
Geometric and electronic properties of 2D COFs before and after Li/Na combination were studied by performing first principles calculations as implemented in Vienna Ab initio Simulation Package (VASP).38,39 Projector-augmented wave method40 was employed to describe ion–electron interactions. The exchange–correlation was treated with the generalized gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE) functional.41 To avoid the interaction between neighboring images, the vacuum layer was set to 15 Å along the c direction. The DFT-D3 method with the Grimme zero damping correction was adopted in all calculations to describe weak interactions.42 The cutoff energy was set to be 420 eV for plane-wave basis set and the convergence criterion (Ecc) were set to be 1 × 10−5 eV in energy and 0.01 eV Å−1 for Hellmann–Feynman forces, respectively. For all calculation steps, the Monkhorst–Pack k-point grid of the Brillouin zone was sampled as 3 × 3 × 1.43Ab initio molecular dynamics (AIMD) simulations were performed to evaluate the thermal dynamic stability of the examined 2D COFs. A 2 × 2 supercell consisting of more than 200 atoms was used in the AIMD simulation with the lattice parameters of 33.21, 33.25, 50.37, 49.03 Å for 2D 3Q-COF, TQBQ-COF, 3BQ-COF, BQ2-COF, respectively. The temperature was controlled by using the Nosé–Hoover method,44 which was set to 500 K and lasted for 5 ps with a time step of 0.5 fs. The Heyd–Scuseria–Ernzerhof hybrid functional (HSE06) was adopted to describe the band structures of target 2D COFs. Finally, the diffusion paths of Li/Na atoms on the surface of 2D COFs were estimated by employing the climbing image nudged elastic band (CI-NEB) method as compiled in VASP, the spring constant value was set for −5 eV Å−2.45
The formation energy (Ef) of 2D COF monolayer was calculated by the following equation
| Ef = E2DCOF − nEA − mEB − xEH2O | (1) |
The combination energy (Ec) of Li or Na was defined as
| Ec = E(2DCOF+M) − E2DCOF − EM | (2) |
| Eave = [E(2DCOF+nM) − E2DCOF+(n−1)M − xEM]/x | (3) |
To explicitly understand the interaction between the adsorbed Li/Na and the 2D COFs, the charge density difference (Δρ) is calculated by the following equation
| Δρ = ρ2DCOF+M − ρ2DCOF − ρM | (4) |
The theoretical storage capacity (TSC) can be defined as
| TSC = xF/M2DCOF | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1) and M2DCOF is the molecular weight of the 2D COFs monolayer.
485 C mol−1) and M2DCOF is the molecular weight of the 2D COFs monolayer.
The average open-circuit voltage (OCV) was estimated based on the change of Gibbs free energy by neglecting the effect of volume and entropy at room temperature, as described by
| OCV ≈ [E2DCOF+Mx1 − E2DCOF+Mx2 + (x2 − x1)EM]/(x2 − x1)e | (6) |
3. Results and discussion
3.1 Geometry and electronic properties of 2D COFs of 3Q, TQBQ, 3BQ and BQ2
The optimized monolayer of 2D 3Q-COF, 2D TQBQ-COF, 2D 3BQ-COF and 2D BQ2-COF are shown in Fig. 1. It can be found that all four structures show a planar configuration with a 2D honeycomb lattice. For 2D COFs of 3Q, TQBQ and 3BQ, there are six pairs of pyrazine N atoms in each unit cell as active centers, which can be prepared by using building blocks of tri-quinazoline, triquinoxalinylene and tribenzoquinoxaline-5,10-dione, respectively. While 2D BQ2-COF contains twelve pairs of pyridinic N in the unit cell. For 2D COFs of 3Q, TQBQ and 3BQ, there are three pairs of carbonyl functional groups, while 2D BQ2-COF consists of six pairs of carbonyls. According to previous studies,31,32 these functional groups will serve as redox active sites for the combination of Li/Na. The optimized lattice parameters of 2D 3Q-COF, TQBQ-COF, 3BQ-COF, BQ2-COF are 16.50, 16.63, 25.18, 24.52 Å, respectively. The pore diameter is 11.46, 11.16, 20.20, 19.05 Å for 2D 3Q-COF, TQBQ-COF, 3BQ-COF, BQ2-COF, respectively. The porous structures and large pore diameters will be beneficial for the fast migration of Li and Na.The thermodynamic stability of these structures were verified by performing AIMD simulations. As shown in Fig. S1,† the 2D monolayers remain stable without significant deformations and distortions after 5 ps at 500 K, which is much higher than the room temperature, indicating the good thermodynamic stability of the examined 2D COFs. Moreover, the distorted structures of the examined 2D COFs can recover to the original planar configuration after geometry optimization, demonstrating their structure robustness. Since 2D COFs can be realized by a triple condensation reaction, to evaluate the synthesis feasibility of 2D 3Q-, TQBQ-, 3BQ- and BQ2-COFs, we estimated their formation energy based on eqn (1). It was found that the formation energy of these four 2D COFs from the corresponding building monomers is −7.1, −3.12, −1.91, −0.35 eV, respectively. This result indicates that the proposed 2D 3BQ- and BQ2-COFs is experimentally realizable. Furthermore, the synthesis of these two structures could be realized by using different methods and building blocks.
The electronic properties of these four 2D COFs were then explored. It is well known that PBE functional usually underestimates the band gaps of 2D materials. Thus, we depicted the band structure of target monolayers at the HSE06 level. As shown in Fig. 2, all the examined structures are semiconductors with a band gap of 1.63, 2.78, 2.93, 1.96 eV, respectively. The small band gap of 2D 3Q-COF can be ascribed to the high electron delocalization along the 2D skeleton. Especially, the electron in the pz orbital of sp2 hybridized N can conjugate effectively with the π electrons of connected benzene rings, forming an extended π-conjugation, as demonstrated by the charge density distribution of CBM and VBM (Fig. 2a).33,36 However, for the other three 2D COFs, the electron conjugation along the 2D network is blocked by the appearance of carbonyls because of the high electronegativity of O. As illustrated by the charge density distribution analysis, there is negligible contribution from carbonyl groups to the CBM of 2D TQBQ-COF, 3BQ-COF and VBM of BQ2-COF, respectively. As a result, the CB of 2D TQBQ-COF, 3BQ-COF and VB of BQ2-COF are less dispersive comparing with the conduction and valence bands of 2D 3Q-COF, forming flat bands with localized charges. By comparing the band structure and charge density distribution of BQ2- and TQBQ-COFs, it can be found that the π conjugation of the 2D COFs can be enhanced by increasing concentration of pyridic N, resulting in a narrowed band gap of BQ2-COF.
 | ||
| Fig. 2 Band structures of 2D COFs of 3Q (a), TQBQ (b), 3BQ (c) and BQ2 (d) calculated at the HSE06 level, respectively, and the charge density distribution for corresponding CBM and VBM. The Fermi level is set to 0 eV and indicated by the dashed red line in the band structure profile, and the isosurface for the orbitals is set to 0.00127 e Å−3. | ||
3.2 Adsorption of single Li/Na on 2D COFs
The feasibility of using these structures as lithium/sodium electrode materials is first examined by estimating the adsorption of a single Li/Na atom on all possible adsorption sites (Fig. 3), including the site above the central benzene ring (HB), above the pyrazine ring (HPy), above the core benzene ring by sharing C atoms with 3 pairs of pyrazine rings (HPyB), on the bridge of two adjacent pyrazine N atoms (SPy), on the bridge of a pyrazine N atom and a carbonyl group (SPC) and above the benzene ring which contains the carbonyl groups (HCB). The most favorable adsorption sites were determined by comparing the adsorption energies of different positions (Table S1†). According to eqn (2), the more negative the value of the adsorption energy, the stronger the interaction between 2D COFs and Li/Na. It can be found that SPy is the optimal adsorption site for Li or Na for all four structures, which is consistent with the findings of previous studies.15,31,36 Meanwhile, the appearance of carbonyl functional groups at the adjacent site of pyrazine N can significantly increase the adsorption energy of metal atoms. The enhanced Li/Na adsorption at the Spy site can be ascribed to the fact that the oxygen atom is more electronegative than the hydrogen atom at the same position. According to the Bader charge analysis, there is about 0.88/0.86|e| charge transferred from the adsorbed Li/Na to the neighboring pyridine N in 2D 3Q-COF, for the other three 2D COFs, there is about 0.91/0.88, 0.90/0.87, and 0.90/0.89|e| transferred from the adsorbed Li/Na to the neighboring pyridine N and carbonyl groups, respectively, indicating that the adsorbed metal atoms transfer a pronouncedly amount of charge and are heavily ionized. As shown in Fig. 4 and S3,† the electrons mainly accumulate between Li/Na and neighboring O and N atoms at the SPy site owing to the strong interaction between O, N and Li/Na. | ||
| Fig. 3 Schematics for the adsorption of Li/Na on the HB, HPy, HPyB, SPy, HCB and SPC sites of (a–d) 2D 3Q-COF, (e–h) 2D TQBQ-COF, (i–m) 2D 3BQ-COF and (n–r) 2D BQ2-COF from top and side views. The white, purple, pink, gray, blue and red balls represent H, Li/Na, C, N and O atoms, respectively. | ||
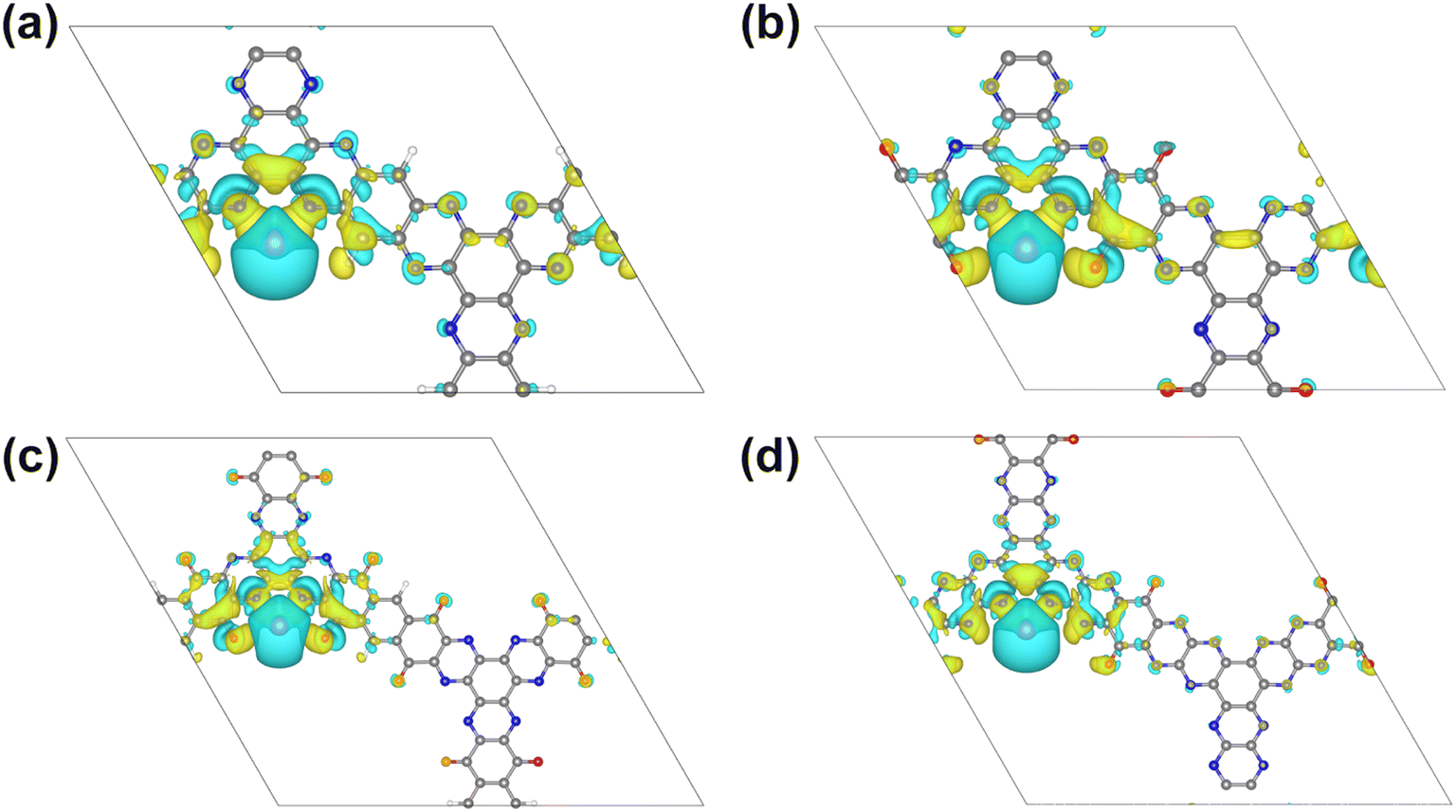 | ||
| Fig. 4 The differential charge distributions of 2D COFs of 3Q (a), TQBQ (b), 3BQ (c) and BQ2 (d) with Li adsorption at the SPy site, respectively. The white, purple, gray, blue and red balls represent H, Li, C, N and O atoms, respectively. The isosurface is set to 0.0013 e Å−3. The yellow and blue regions denote the accumulation and depletion of charge, respectively. | ||
The electronic structure of 2D COFs after Li/Na adsorption at the optimal SPy site was then plotted in Fig. 2 and S3.† It can be found that there is band dispersion crossing the Fermi level after Li or Na adsorption and the DOS diagram show that there is an obvious hybridization between the 2p orbitals of C, N, O atoms and the s orbital of Li or Na. Therefore, the examined 2D COFs show a transition from semiconductor to metals after Li/Na adsorption, indicating improved electrical conductivity during discharging process, which is beneficial for their application as electrode materials for lithium and sodium ion batteries.
3.3 Diffusion of Li/Na atoms on 2D COFs
The charge/discharge rate performance was estimated by ascertaining the migration of Li and Na on the surface of these 2D COFs. As the large pore size ensures sufficient contact between the electrode material and the electrolyte, the metal ions can diffuse rapidly to the redox active center across the pore channels, which is beneficial for rapid charging/discharging process.46,47 However, the diffusion of metal atoms on the surface of skeleton was also simulated to better understand the kinetics of charging/discharging process. Because of the significant adsorption energy difference between the SPy site and the other benzene ring sites, (Table S1†) the diffusion barrier of a single Li for all three paths on the examined systems are rather large (Fig. S3†). Thus, the migration of Li atoms on lithiated structures were then examined by establishing a model with seven adsorbed Li/Na and the favorable energy migration paths and corresponding diffusion barrier can be accordingly determined.48 As shown in Fig. 5 and S4,† for all models, two SPy sites located in the same chemical environment were set as the initial and final states, respectively. Therefore, the possible migration paths of Li/Na atoms between the two SPy sites through the HPyB site (SPy → HPyB → SPy), the HPy site (SPy → HPy → SPy), the HCB site (SPy → HCB → SPy) and the HB site (SPy → HB → SPy) were examined. The pathway through the HB site has a high migration energy barrier due to the affect of carbonyl group on part of the structure with the Li/Na atom. For 2D TQBQ and BQ2 COF, Li/Na can diffuse faster through the core benzene ring along path-I by showing a smaller diffusion barrier of 0.48/0.35, and 0.34/0.36 eV, respectively. For 2D 3Q and 3BQ COF, Li atom diffuse faster along path-II (3Q COF) and path-I (3BQ COF) with a barrier of 0.28 and 0.56 eV, respectively, while the optimal diffusion path for Na is just the reverse with a diffusion barrier of 0.38 and 0.55 eV along path-I (3Q COF) and II (3BQ COF), respectively. Generally, the diffusion of Li is more efficient than that of Na. Although the optimal migration paths are different, these 2D COFs have comparable or even lower diffusion barriers in range of 0.28–0.56 eV compared to other 2D materials,49–55 indicating their good rate performance as electrode materials. | ||
| Fig. 5 The possible diffusion paths and energy barrier of Li atom on 2D COFs of 3Q (a), TQBQ (b), 3BQ (c) and BQ2 (d), respectively. | ||
3.4 TSC and average OCV of 2D COFs
Finally, we investigated the theoretical storage capacity of 2D COFs and the relative average open-circuit voltage (OCV), which represent the two most important indicators to determine the performance of electrode materials. The maximum theoretical storage capacity was estimated by considering all possible adsorption sites from both sides of the monolayer.56–58 For these four structures, the most favorable SPy sites were first occupied by Li/Na from both sides, the average adsorption energy was calculated to be negative, indicating that additional Li/Na adsorption on the sub-optimal sites could be possible. As a result, 2D 3Q-COF, TQBQ-COF, 3BQ-COF and BQ2-COF show a maximum theoretical capacity of 602.27, 515.40, 792.93, 1030.08 mA h g−1 (Fig. 6 and S5†), respectively. The predicted capacity of 2D 3Q-COF and TQBQ-COF agrees well with the experimentally reported results,31,61 demonstrating the reliability of our calculations. It can be found that although the introduction of carbonyl functional group can increase the redox active sites, it does not significantly improve the maximum storage capacity. This is because the increase of carbonyl group is accompanied by the addition of benzene rings which do not participate in the electrochemical reaction, resulting in increased molar mass of the examined structure. The skeleton of 2D COFs was distorted to different extent after accommodating Li/Na at the maximum capacity, but recovered to the initial planar configuration after removing the adsorbed Li/Na followed with re-optimization. This result indicates that these structures possess high structure robustness and good cycling stability when used as electrode materials. Remarkably, as demonstrated in Fig. 7a, the theoretical capacities of the examined 2D COFs are much higher than most of other 2D inorganic electrode materials.56–65 Among them, 2D BQ2-COF shows the highest theoretical capacity of 1030.08 mA h g−1 for both LIBs and SIBs, due to the high concentration of redox-active functional groups (pyrazine and carbonyl). | ||
| Fig. 6 (a–d) Top and side views of the optimized configuration of 2D COFs of 3Q (a), TQBQ (b), 3BQ (c) and BQ2 (d) with maximum adsorbed Li atoms. | ||
 | ||
| Fig. 7 (a) The maximum capacity of 2D 3Q-COF, 2D TQBQ-COF, 2D 3BQ-COF and 2D BQ2-COF for LIBs/SIBs in comparison with that of other 2D anode materials. (b) Theoretical storage capacity (TSC) and average open-circuit voltage (OCV) for 2D 3Q-COF, 2D TQBQ-COF, 2D 3BQ-COF and 2D BQ2-COF, respectively. | ||
The average OCV of 2D COFs was then estimated to evaluate their potential to be used as cathode or anode for MIBs. Compared with 2D BQ2-COF, 2D 3Q-COF, 2D TQBQ-COF and 2D 3BQ-COF only show one voltage plateau (Fig. S6†) because all adsorption sites are in the same chemical environment. As depicted in Fig. 7b and S6,† when 2D 3Q-COF, 2D TQBQ-COF, 2D 3BQ-COF, 2D BQ2-COF are occupied by Li/Na to reach the maximum theoretical capacity, the average OCVs is calculated to be 0.94/0.64, 1.89/1.58, 1.13/0.92, and 0.74/0.67 V for LIBs/SIBs, respectively (Table S2†). The above results indicated that these four 2D COFs are promising organic materials with high theoretical capacity to be used as anode materials for LIBs/SIBs.
4. Conclusions
In summary, we have comprehensively investigated the adsorption and diffusion behaviors of Li/Na on four 2D COFs by performing first principles calculations and studied their feasibility as electrode materials for LIBs or SIBs. All examined 2D COFs are semiconductors with a band gap of 1.63–2.93 eV. The incorporation of N can enhance π electron conjugation along the 2D network. It was found that the side pyrazine is the optimal site to accommodate Li or Na, and all structures exhibit a transition from semiconductor to metal after the adsorption of metal atoms. The target 2D COFs show a low diffusion barrier, indicating their good rate performance for rapid charge and discharge process. The theoretical capacity of 2D BQ2-COF is predicted to be as high as 1030 mA h g−1, while the low average OCVs renders it an appropriate anode materials for LIBs and SIBs. Since 2D 3Q-COF66 and 2D TQBQ-COF31,67 have been realized by a triple condensation reaction between hexaketocyclohexane and 1,2,4,5-benzenetetramine/tetraminophenone (TABQ) and cyclohexanehexaone, respectively, 2D 3BQ-COF and 2D BQ2-COF can be expected to be obtained using similar methods in real practice. Our investigation reveals the relationship between composition, structure and electronic properties of 2D COFs and provide new insights for the rational design of promising organic electrode materials with desired electrochemical performance for LIBs and SIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the financial support in China by National Natural Science Foundation of China (no. 22223204, 21903046).Notes and references
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- Z. Li, A. Siddiqi, L. D. Anadon and V. Narayanamurti, Renewable Sustainable Energy Rev., 2018, 82, 3833–3847 CrossRef.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590–593 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- A. Konarov, J. H. Jo, J. U. Choi, Z. Bakenov, H. Yashiro, J. Kim and S.-T. Myung, Nano Energy, 2019, 59, 197–206 CrossRef CAS.
- M. Zhou, F. Lu, X. Shen, W. Xia, H. He and X. Zeng, J. Mater. Chem. A, 2015, 3, 21201–21210 RSC.
- H. Yu and H. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268–1280 CrossRef CAS PubMed.
- P. He, H. Yu, D. Li and H. Zhou, J. Mater. Chem., 2012, 22, 3680–3695 RSC.
- Z. Lei, Q. Yang, Y. Xu, S. Guo, W. Sun, H. Liu, L.-P. Lv, Y. Zhang and Y. Wang, Nat. Commun., 2018, 9, 576 CrossRef PubMed.
- A. Jaffe, A. S. Valdes and H. I. Karunadasa, Chem. Mater., 2015, 27, 3568–3571 CrossRef CAS.
- X. Zhu and Y. Jing, J. Power Sources, 2022, 531, 231291 CrossRef CAS.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef CAS PubMed.
- H. Chen, M. Armand, M. Courty, M. Jiang, C. P. Grey, F. Dolhem, J.-M. Tarascon and P. Poizot, J. Am. Chem. Soc., 2009, 131, 8984–8988 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed.
- J. Zhou, L. Wang, M. Yang, J. Wu, F. Chen, W. Huang, N. Han, H. Ye, F. Zhao, Y. Li and Y. Li, Adv. Mater., 2017, 29, 1702061 CrossRef PubMed.
- X. Cai, W. Yi, J. Chen, L. Lu, B. Sun, Y. Ni, S. A. T. Redfern, H. Wang, Z. Chen and Y. Chen, J. Mater. Chem. A, 2022, 10, 6551–6559 RSC.
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef PubMed.
- H. Chen, X. Suo, Z. Yang and S. Dai, Adv. Mater., 2022, 34, 2107947 CrossRef CAS PubMed.
- Y. An, S. Tan, Y. Liu, K. Zhu, L. Hu, Y. Rong and Q. An, Energy Storage Mater., 2021, 41, 354–379 CrossRef.
- M.-S. Wu, N. T. H. Luu, T.-H. Chen, H. Lyu, T.-W. Huang, S. Dai, X.-G. Sun, A. S. Ivanov, J.-C. Lee, I. Popovs and W. Kaveevivitchai, Adv. Energy Mater., 2021, 11, 2100330 CrossRef CAS.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 178 CrossRef CAS PubMed.
- M. Wu, Y. Zhao, B. Sun, Z. Sun, C. Li, Y. Han, L. Xu, Z. Ge, Y. Ren, M. Zhang, Q. Zhang, Y. Lu, W. Wang, Y. Ma and Y. Chen, Nano Energy, 2020, 70, 104498 CrossRef CAS.
- S.-B. Xia, T. Liu, W.-J. Huang, H.-B. Suo, F.-X. Cheng, H. Guo and J.-J. Liu, J. Energy Chem., 2020, 51, 303–311 CrossRef.
- S. Kandambeth, J. Jia, H. Wu, V. S. Kale, P. T. Parvatkar, J. Czaban-Jóźwiak, S. Zhou, X. Xu, Z. O. Ameur, E. Abou-Hamad, A.-H. Emwas, O. Shekhah, H. N. Alshareef and M. Eddaoudi, Adv. Energy Mater., 2020, 10, 2001673 CrossRef CAS.
- Y. Zhang, Z. Sun, X. Kong, Y. Lin and W. Huang, J. Mater. Chem. A, 2021, 9, 26208–26215 RSC.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- D. J. Chadi, Phys. Rev. B: Solid State, 1977, 16, 1746–1747 CrossRef.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Kandambeth, V. S. Kale, O. Shekhah, H. N. Alshareef and M. Eddaoudi, Adv. Funct. Mater., 2022, 12, 2100177 CAS.
- L. Zhai, G. Li, X. Yang, S. Park, D. Han, L. Mi, Y. Wang, Z. Li and S.-Y. Lee, Adv. Funct. Mater., 2022, 32, 2108798 CrossRef CAS.
- D. Wu, B. Yang, H. Chen and E. Ruckenstein, Energy Storage Mater., 2019, 16, 574–580 CrossRef.
- X. Zhang, J. Hu, Y. Cheng, H. Y. Yang, Y. Yao and S. A. Yang, Nanoscale, 2016, 8, 15340–15347 RSC.
- L. Xiong, J. Hu, S. Yu, M. Wu, B. Xu and C. Ouyang, Phys. Chem. Chem. Phys., 2019, 21, 7053–7060 RSC.
- B. Mortazavi, O. Rahaman, S. Ahzi and T. Rabczuk, Appl. Mater. Today, 2017, 8, 60–67 CrossRef.
- M. Mortazavi, C. Wang, J. Deng, V. B. Shenoy and N. V. Medhekar, J. Power Sources, 2014, 268, 279–286 CrossRef CAS.
- Y. Zhang, Z.-F. Wu, P.-F. Gao, S.-L. Zhang and Y.-H. Wen, ACS Appl. Mater. Interfaces, 2016, 8, 22175–22181 CrossRef CAS PubMed.
- J. Liu, M. Qiao, X. Zhu, Y. Jing and Y. Li, RSC Adv., 2019, 9, 15536–15541 RSC.
- B. Xiao, Y.-c. Li, X.-f. Yu and J.-b. Cheng, ACS Appl. Mater. Interfaces, 2016, 8, 35342–35352 CrossRef CAS PubMed.
- Y. Jing, J. Liu, Z. Zhou, J. Zhang and Y. Li, J. Phys. Chem. C, 2019, 123, 26803–26811 CrossRef CAS.
- D.-X. Song, L. Xie, Y.-F. Zhang, Y. Lu, M. An, W.-G. Ma and X. Zhang, ACS Appl. Energy Mater., 2020, 3, 7699–7709 CrossRef CAS.
- Y. Yang, W. Geng, T. Xu and Y. Jing, 2D Mater., 2022, 9, 034003 CrossRef.
- D. Er, J. Li, M. Naguib, Y. Gogotsi and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 11173–11179 CrossRef CAS PubMed.
- S. Qi, F. Li, J. Wang, Y. Qu, Y. Yang, W. Li and M. Zhao, Carbon, 2019, 141, 444–450 CrossRef CAS.
- S. Banerjee, S. Neihsial and S. K. Pati, J. Mater. Chem. A, 2016, 4, 5517–5527 RSC.
- Q. Sun, Y. Dai, Y. Ma, T. Jing, W. Wei and B. Huang, J. Phys. Chem. Lett., 2016, 7, 937–943 CrossRef CAS PubMed.
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Phys. Chem. Chem. Phys., 2015, 17, 13921–13928 RSC.
- C. Tang, S. Wang, K. Zhang and C. Cheng, Electrochim. Acta, 2021, 388, 138641 CrossRef CAS.
- Y. Jing, Z. Zhou, C. R. Cabrera and Z. Chen, J. Phys. Chem. C, 2013, 117, 25409–25413 CrossRef CAS.
- M. K. Shehab, K. S. Weeraratne, T. Huang, K. U. Lao and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2021, 13, 15083–15091 CrossRef CAS PubMed.
- W. Zhou, H. Shen, H. Xie, Y. Shen, W. Kang, Q. Wang, Y. Kawazoe and P. Jena, J. Phys. Chem. Lett., 2021, 12, 12142–12149 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07655e |
| This journal is © The Royal Society of Chemistry 2023 |