
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical photocatalytic and sonophotocatalytic reduction of nitroarenes in water under blue LED irradiation using β-CD modified TiO2 as a green nest photocatalyst†
Leila Ghasemia,
Ayub Ahmadia,
Raheleh Abedinia,
Foad Kazemi *ab and
Babak Kaboudina
*ab and
Babak Kaboudina
aDepartment of Chemistry, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, 45137-66731, Iran. E-mail: kazemi_f@iasbs.ac.ir
bCenter of Climate Change and Global Warming, Institute for Advanced Studies in Basic Sciences (IASBS), Zanjan, Iran
First published on 28th November 2023
Abstract
Photocatalysis using natural photosynthesis is a green technology that is gaining popularity in a number of industries due to its potential for environmental applications and the use of solar energy. Focus is being placed on using inexpensive materials and light-emitting diodes (LEDs) of various wavelengths in photocatalytic reactions in order to improve the performance of solar-driven photocatalysts at a lower cost. In this study, a scalable, highly efficient photocatalytic and sonophotocatalytic method was investigated for the reduction of nitro-compounds by a water/titania/β-cyclodextrin system under sunlight and blue LED irradiation, using sodium sulfide as a sacrificial electron donor. β-Cyclodextrin, chemically bound to TiO2 nanoparticles as an encapsulating agent, hosted nitro compounds in aqueous media and formed an inclusion complex. In addition, this method was used to successfully carry out one-pot reduction-amidation of nitroarene compounds in the presence of acetic anhydride. Interestingly, it was found that ultrasound has a synergistic effect on photocatalytic reduction and considerably reduces the duration time. In this regard, a fast, practical sonophotocatalytic reduction of nitroarenes was carried out in an ultrasound bath.
1 Introduction
Nowadays, photocatalysis technology is inspired by natural photosynthesis1,2 as a green technology due to its high potential for solar energy utilization and storage. As a result, it has received increasing research interest in many fields, such as energy, health, environment, pollution control, and value-added chemical synthesis.3 To improve the performance of solar-driven photocatalysts, using low-cost materials and processing technologies for sunlight harvesting achieves significant higher solar conversion performance at a lower cost.4–6Recently, light-emitting diodes (LEDs) emitting light of different wavelengths (infrared, visible, or near-ultraviolet) have been used as an adjustable tool in photocatalytic reactions. Among photocatalysts, titanium dioxide (TiO2) in different types and forms is used primarily as a photocatalyst due to its unique properties, such as low cost, physical and chemical stability, wide availability, environmentally friendly nature, nontoxicity, and high reactivity, which have made it widely used.7 However, a substrate's adsorption on a catalyst's surface blocks active sites. It makes them unavailable for other substrate molecules, which prolongs the reaction time, leads to wasted energy, and often produces by-products, consequently reducing catalytic efficiency. Therefore, designing an equipped system that continuously cleans the catalyst's surface during photocatalytic operation is necessary to overcome this limitation. In this regard, sonophotocatalysis, as a green-hybrid strategy, is a promising method to create innovativeness for the photocatalysis technique.
Sonophotocatalysis involves using ultrasound (US), ultraviolet (UV), or visible light on semiconductors. Researchers have been paying attention to ultrasound irradiation due to its advantages, such as safety, cleanliness, not needing additional chemicals, and simplicity of operation, as a source of green energy.8,9
When ultrasound waves pass through an aqueous solution, they produce alternating adiabatic compression and rarefaction, leading to acoustic cavitation. The reduction in pressure is created in the rarefaction part of the ultrasonic waveform microbubbles. The size of these microbubbles grows over several cycles until they become very unstable and collapse. The microbubbles in the solution create localized supercritical conditions with extremely high temperatures and pressures. These supercritical conditions, created by ultrasonic waves, help improve catalyst performance by increasing the mass transfer rate, continuous cleaning, and increasing the surface area (due to fragmentation and separation) of the catalysts.10 Interestingly, sonophotocatalysis is performed in water, which is a green, available, and cheap solvent compared to other organic solvents. In addition to producing active species and radicals, it creates a synergistic effect between sonolysis and photocatalysis that can increase the reaction rate.11–16
However, sonophotocatalysis, through advanced oxidation processes (AOPs), has been most widely used in environmental remediation, especially waste-water treatment.17–22
There is less attention in the chemical synthesis field. Dimitrios A. Giannakoudakis introduced sonophotocatalysis as a hybrid process intensification method (HPIM) and used it successfully for selective partial photooxidation of benzyl alcohol to benzaldehyde.23 In this work, in continuing our interest in using TiO2@β-CD as a ‘green nest’ photocatalyst system, we report the practical photocatalytic reduction of nitroarenes and, more interestingly, the synergistic effect of ultrasound on the reaction under sunlight and blue LED irradiation, using sodium sulfide as a sacrificial electron donor. Additionally, we investigate the one-pot photocatalytic synthesis of amides.
2 Results and discussion
Water has attracted much attention in organic synthesis compared to other organic solvents because it is a green, available, and, more importantly, cheap and safe solvent. With this in mind, we chose water as the reaction medium. In order to improve the interaction of the nitro compound with active sites of TiO2 in aqueous media, we used β-cyclodextrin (β-CD) as a green nest, according to the previous work of our group, that 0.1 mmol of Nitro Compound was reduced, in the presence of 0.1 mmol of β-CD, 30 mg of titania (P25), and oxalic acid (15 mg) as a reducing agent. It should be mentioned that the reduction was carried out in the presence of sunlight.24 To improve and develop our previous report, in this work, we adjusted the reaction condition in order to use light-emitting diodes (LEDs) for the practical scale of starting materials. In this regard, sodium sulfide was used as a reducing agent in photocatalytic reactions for the first time. Initial experiments on nitrobenzene as a model compound were conducted to study the optimal reaction conditions. Initially, 1 mmol of nitrobenzene was added to 10 mL of water, 30 mg of commercial TiO2 (P25), and 0.15 g of β-CD were added to the mixture. Then, three mmol of sodium sulfide was added to the reaction mixture. The reaction mixture was first deoxygenated with Ar gas and sealed with a septum. The flask was irradiated under stirring with sunlight and a blue LED. To demonstrate that β-CD, as a host molecule, improves the efficiency of the reaction in an aqueous medium, we investigated the presence and absence of β-CD in the reaction.Without β-CD, the photoreduction did not work with an excellent yield (Table 1, entries 12 and 19). The amounts of nitrobenzene (0.5, 1, 1.2, 1.3, and 1.4 mmol) were optimized, and the best conversion was obtained for 1.2 mmol of nitrobenzene after irradiation for 5 hours (Table 1, entry 3). To improve the performance of the photocatalysis technique, we investigated the effect of TiO2-P25 and irradiation time on the conversion of nitrobenzene under the above conditions. Interestingly, the best results were obtained when 7.5 mg of TiO2 was used for 1.2 mmol of nitrobenzene, 15 mg of β-cyclodextrin, 3.6 mmol of sodium sulfide, 10 mL of water, and blue LED (Table 1, entry 8).
| Entry | Nitrobenzene (M) | Na2S (M) | β-CD (g) | TiO2 (mg) | Yielda (%) | Time (h) |
|---|---|---|---|---|---|---|
| a GC yield and under blue LED.b Under 395 nm LED.c 365 nm LED.d Green LED.e Sunlight intensity between 1020–1070 Lux.f Sonophotoreduction method.g Sonication.h Photoreduction method. | ||||||
| 1 | 0.5 | 1.5 | 0.15 | 30 | 98 | 5 |
| 2 | 1 | 3 | 0.15 | 30 | 98 | 5 |
| 3 | 1.2 | 3.6 | 0.15 | 30 | 98 | 5 |
| 4 | 1.3 | 3.9 | 0.15 | 30 | 85 | 5 |
| 5 | 1.4 | 4.2 | 0.15 | 30 | 65 | 5 |
| 6 | 1.2 | 3.6 | 0.15 | 20 | 98 | 5 |
| 7 | 1.2 | 3.6 | 0.15 | 10 | 98 | 5 |
| 8 | 1.2 | 3.6 | 0.15 | 7.5 | 98 | 5 |
| 9 | 1.2 | 3.6 | 0.15 | 5 | 90 | 5 |
| 10 | 1.2 | 3.6 | 0.15 | 4 | 87 | 5 |
| 11 | 1.2 | 3.6 | 0.15 | 3.5 | 77 | 5 |
| 12 | 1.2 | 3.6 | 0 | 7.5 | 54 | 5 |
| 13 | 1.2 | 3.6 | 0.05 | 7.5 | 68 | 5 |
| 14 | 1.2 | 3.6 | 0.1 | 7.5 | 81 | 5 |
| 15b | 1.2 | 3.6 | 0.15 | 7.5 | 86 | 7 |
| 16c | 1.2 | 3.6 | 0.15 | 7.5 | 69 | 24 |
| 17d | 1.2 | 3.6 | 0.15 | 7.5 | 98 | 3 |
| 18e | 1.2 | 3.6 | 0.15 | 7.5 | 98 | 2 |
| 19f | 1.2 | 3.6 | 0 | 7.5 | 0 | 2 |
| 20g | 1.2 | 3.6 | 0.15 | 7.5 | 0 | 2 |
| 21h | 1.2 | 3.6 | 0.15 | 7.5 | 0 | 2 |
In addition to the blue LED, we examined the reaction in the presence of violet and green LEDs and sunlight. Nitroarene compounds were reduced with excellent efficiency in the presence of blue LED as well sunlight (Table 1, entries 8, 18). In the next step, we evaluated the effect of sacrificial agents on the reduction of nitrobenzene by investigating their presence and absence. In the absence of any sacrificial agents, the reaction did not proceed. Among the sacrificial agents tested, sodium sulfide showed the best performance with a 98% yield and was selected as an excellent reducing agent (Table S1,† entry 6).
Then, the optimal conditions were applied to other nitro-aromatic compounds with electron-donating and electron-withdrawing groups, and the corresponding amines were obtained with good to excellent yields (Table 2). In both photoreduction methods, we found that nitrobenzenes containing electron-withdrawing groups were readily converted to the corresponding aromatic amines with high yields (Table 2, entries 4, 6, 8, 9, and 10).
| Entry | Nitro | Amine |
|
|
|
|---|---|---|---|---|---|
| a Reaction condition: TiO2-P25 (7.5 mg), β-cyclodextrin (0.15 g), nitro compound (1.2 mmol), irradiation with sunlight (1020–1070 Lux), Na2S (3.6 mmol).b Irradiation with blue LED (10 W), Na2S (3.6 mmol).c Irradiation with blue LED (10 W), Na2S (3.6 mmol), sonication time 2 h.d Large-scale reaction condition: TiO2-P25 (22 mg), nitro compound (6 mmol), irradiation with blue LED (10 W), Na2S (18 mmol), sonication time 10 h. | |||||
| 1 | 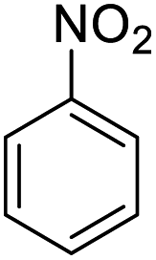 |
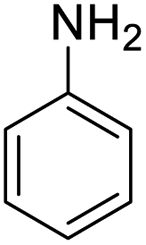 |
98 (3 h) | 100 (5 h) | 100 |
| 2 | 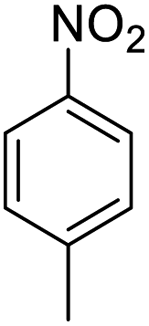 |
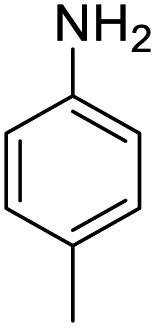 |
94 (4 h) | 97 (15 h) | 98 |
| 3 | 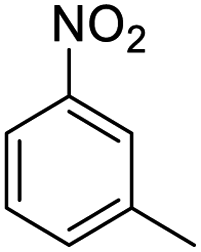 |
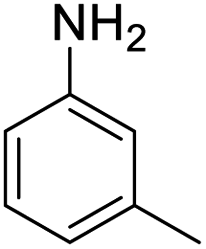 |
96 (3.5 h) | 95 (15 h) | 98 |
| 4 | 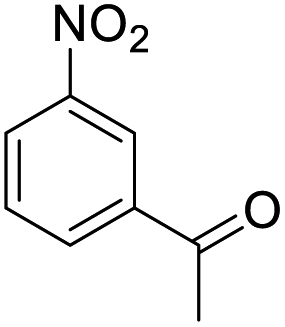 |
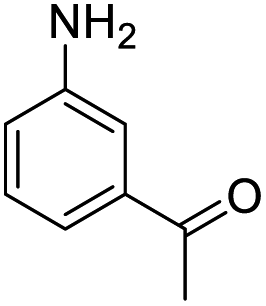 |
94 (3.5 h) | 96 (5 h) | 75 |
| 5 | 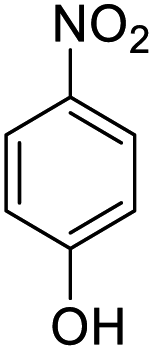 |
 |
15 (3 h) | 10 (24 h) | 14 |
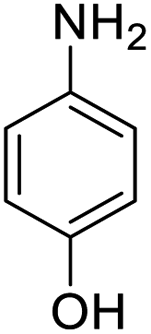
| 6 |  |
 |
100 (3 h) | 98 (15 h) | 98 |
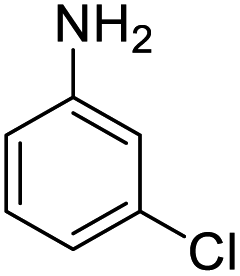
| 7 | 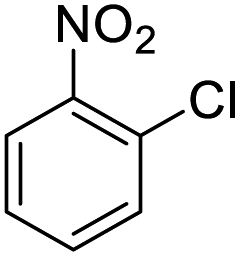 |
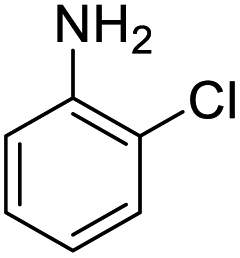 |
48 (3 h) | 25 (15 h) | 52 |
| 8 | 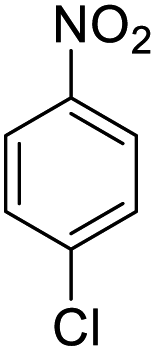 |
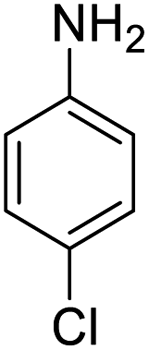 |
95 (3 h) | 97 (15 h) | 95 |
| 9 | 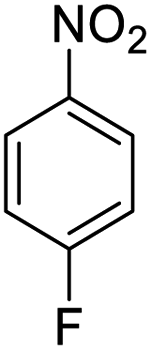 |
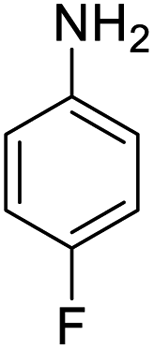 |
98 (3 h) | 94 (5 h) | 80 |
| 10 | 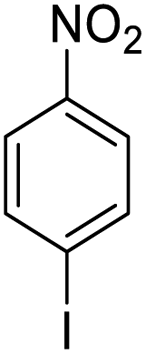 |
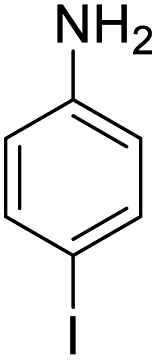 |
98 (3 h) | 97 (5 h) | 99 |
| 11 |  |
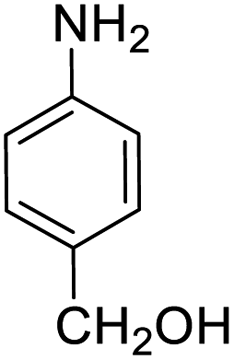 |
95 (3 h) | 96 (5 h) | 100 |
| 12 | 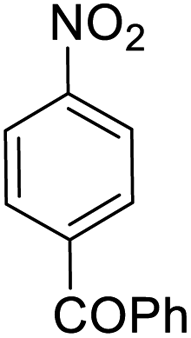 |
 |
80 (3 h) | 80 (5 h) | 75 |
| 13 | 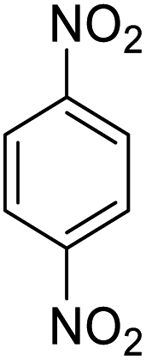 |
 |
100 (3.5 h) | 98 (14 h) | 100 |
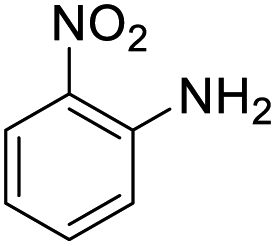
| 14 | 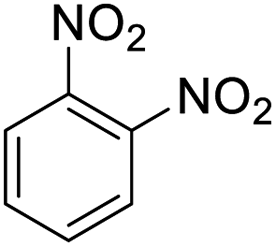 |
 |
98 (3 h) | 99 (5 h) | 85 |
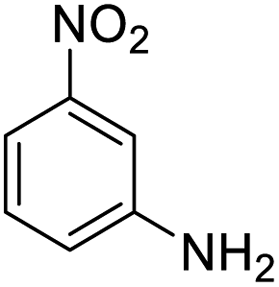
| 15 | 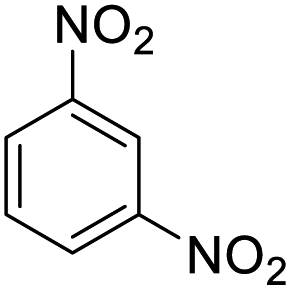 |
 |
98 (3 h) | 76 (5 h) | 100 |
| 16 | 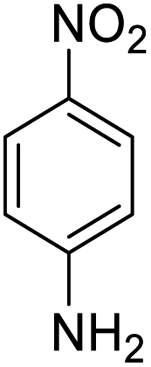 |
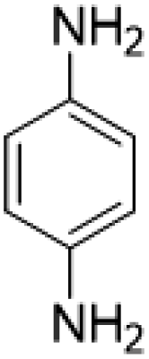 |
15 (3.5 h) | 5 (22 h) | 7 |
| 17 | 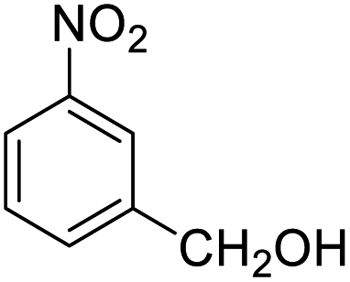 |
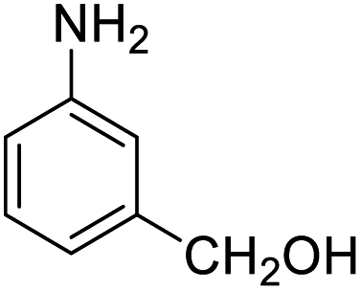 |
93 (3 h) | 94 (5 h) | 98 |
| 18d | 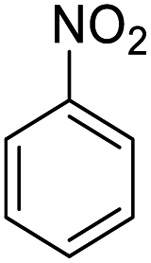 |
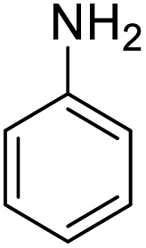 |
— | — | 85 (10 h) |



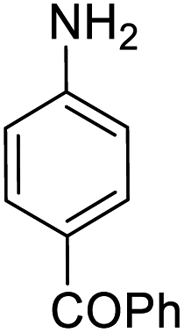
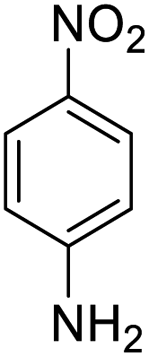
It appears that the solubility of nitrobenzenes in the reaction medium is a crucial factor for the progress of the reaction. Therefore, 4-nitrobenzophenone, despite having an electron-withdrawing group, had a lower reaction rate than other compounds due to its poor solubility and only converted to 4-aminobenzophenone with a yield of 80% (Table 2, entry 12). The remarkable thing about our green system is its ability to selectively reduce nitro compounds in the presence of other reducible functional groups such as ketones and halides. Moreover, no unwanted reduction in the carbonyl group was observed when reducing nitro-aromatic compounds in the presence of carbonyl groups (Table 2, entry 12). Additionally, we obtained the chemoselective reduction of 1,2-dinitrobenzene to 2-nitroaniline (Table 2, entry 14). Reusability is a significant parameter for evaluating the performance of a photocatalyst. To assess this, we tested the activity of the TiO2-P25 catalyst five times in the presence of sunlight and blue LED. The catalyst system performed very well during the five runs in the nitrobenzene reduction reaction. TEM images and XRD analysis did not show any differences in the lattice structure in of β-CD/TiO2 after five times (Fig. 1, S4, and S5†).
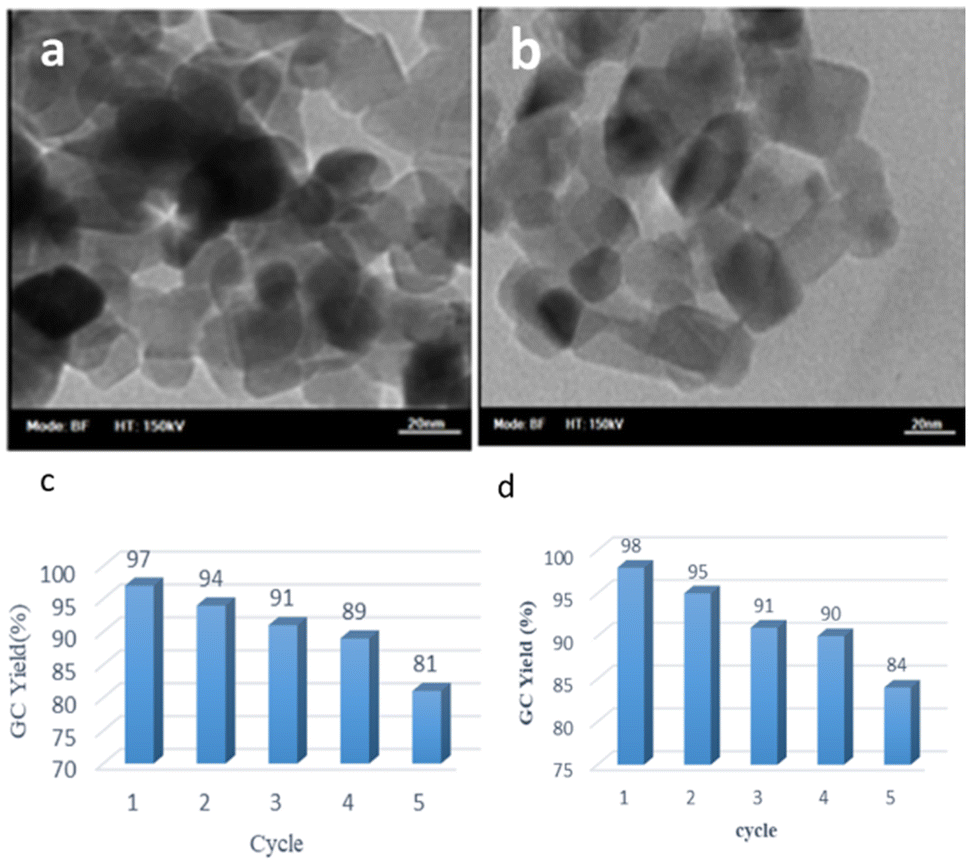 | ||
| Fig. 1 TEM image of (a) reused β-CD-TiO2 (b) β-CD-TiO2, (c) reusability of the catalyst in the photoreduction of nitrobenzene under blue LED irradiation, (d) under sunlight irradiation. | ||
The presence of β-CD in the titania surface was confirmed with various characterizations (Fig. S1–S3†). In the FTIR spectrum of TiO2-β-CD, the intensity of the band at 3400 cm−1 is increased in comparison with P25, that could be due to the presence of the O–H groups in the β-CD. The peak located at 1676 cm−1 corresponded to the stretching mode of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Also, the band at 1414 cm−1 attributed to the O–H in-plane bending, while the antisymmetric C–O–C stretch is located at 1156 cm−1. The C–O stretching vibrations mode is clear at 1030 cm−1.25 Considering the UV-visible diffuse reflectance spectroscopy, the β-CD-TiO2 displayed a higher absorption in the visible light region in comparison with TiO2.26
C bonds. Also, the band at 1414 cm−1 attributed to the O–H in-plane bending, while the antisymmetric C–O–C stretch is located at 1156 cm−1. The C–O stretching vibrations mode is clear at 1030 cm−1.25 Considering the UV-visible diffuse reflectance spectroscopy, the β-CD-TiO2 displayed a higher absorption in the visible light region in comparison with TiO2.26
We further investigated TiO2-P25 and the recycled TiO2-P25 catalyst using transmission electron microscopy (TEM) analysis. We observed no changes in the size and shape of the nanoparticles, and the size of the nanoparticles was found to be in the range of 20–30 nanometers (Fig. 1). In continuation, we used this green photocatalysis method for the one-pot amidation of nitroarene compounds in the presence of acetic anhydride in water (Fig. 2).
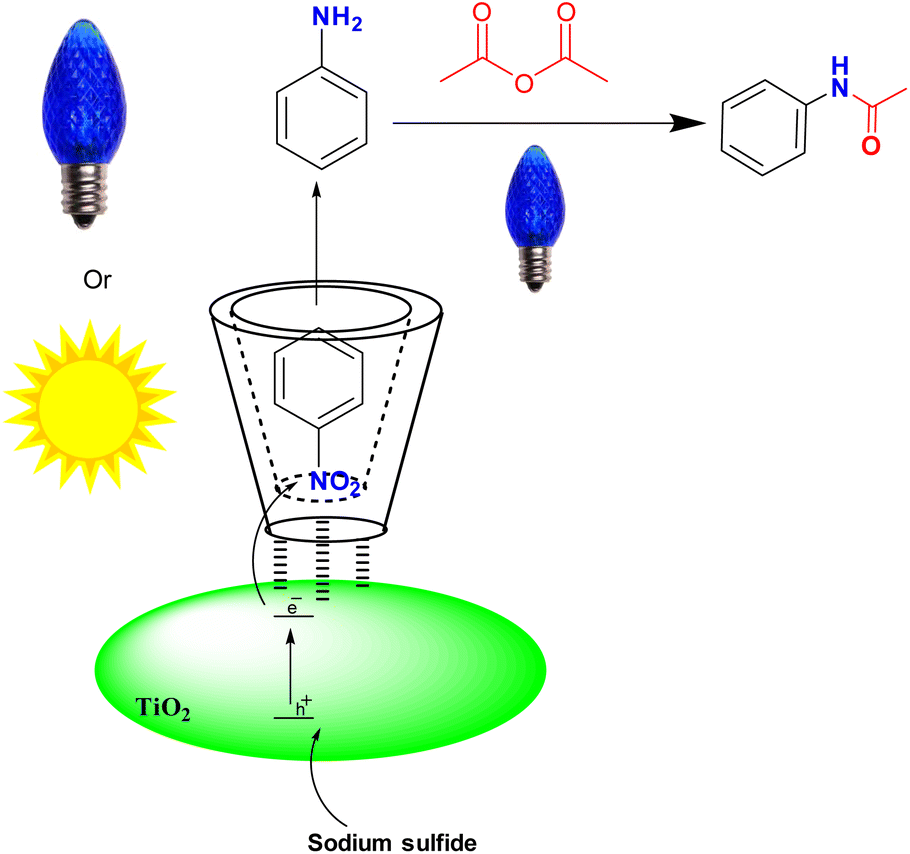 | ||
| Fig. 2 One-pot N-acetylation of the nitro compounds using β-CD-TiO2 under blue LED irradiation in water. | ||
Under the optimized conditions for nitro compound reduction with blue LED, we optimized the amount of acetic anhydride to 3.6 mmol, which resulted in a yield of 97% (Table S2,† entry 3). However, this reaction did not work under sunlight (Table S2,† entry 4).
The NMR spectrum of the obtained product confirmed the formation of the acetanilide compound (see ESI†). We then tested the reaction for preparing acetamides from other nitro compounds (Table 3).
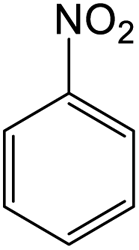
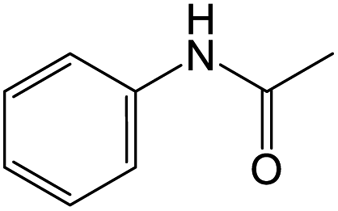
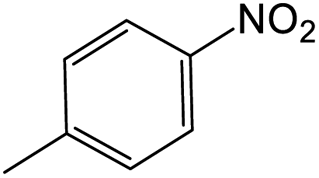
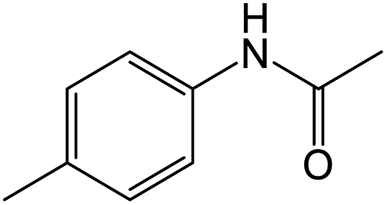
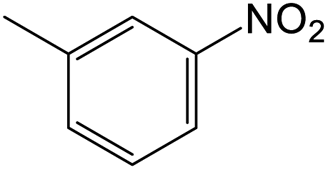
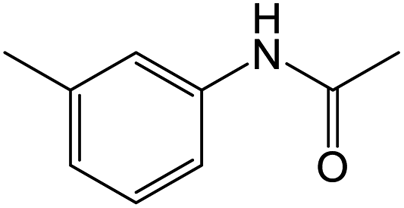
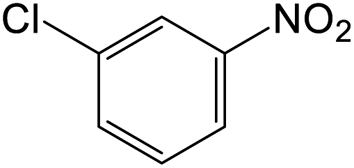
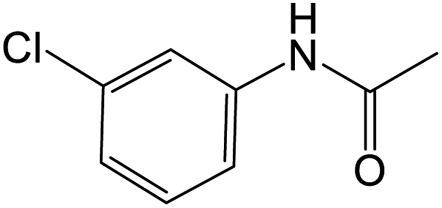
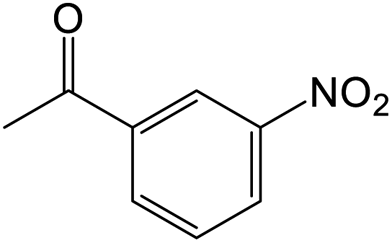
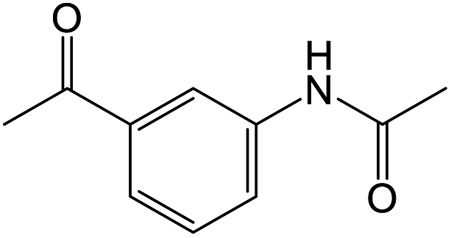
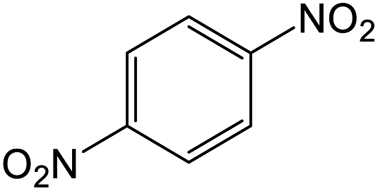
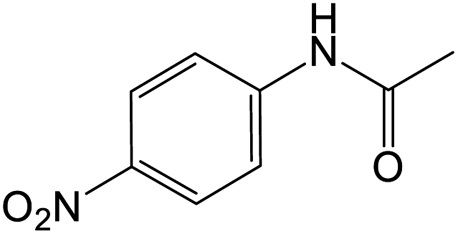
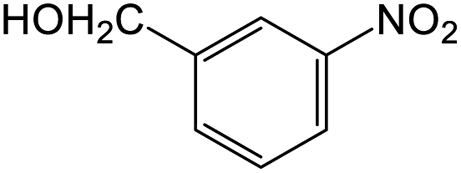
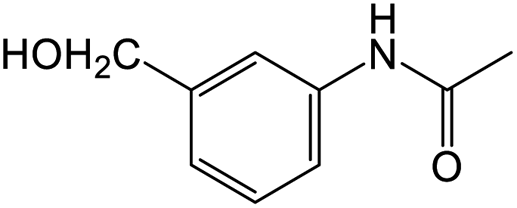
The results showed that different nitroaromatics types and various functional groups were selectively converted to their corresponding amide compounds in water. Therefore, we propose that due to their intrinsic physicochemical properties, the nitro compounds are reduced in the hydrophobic cavity of β-CD, and the resulting amine rapidly reacts in the presence of acetic anhydride.
Due to the cleaning and highly active dispersing properties of sonication, we investigated the synergetic effect of ultrasound on the photocatalytic reduction of nitroarenes. Interestingly, when we carried out the optimized photocatalytic reaction under commercial ultrasound, we observed a significant decrease in the reaction time and a faster reduction of nitrobenzene.
In the sonophotocatalytic method, the flask was first placed in the ultrasound bath and then exposed to radiation with a blue LED (Fig. S5†). The reaction time in the photocatalytic condition under LED irradiation varied from 5 to 24 hours. However, interestingly, all the nitro compounds were reduced within only 2 hours (Table 2).
For Example, the reduction time of 4-nitrotoluene decreased from 15 to 2 hours under sonophotocatalytic conditions compared to photocatalytic conditions (Table 2, entry 2). This can be attributed to the efficient dispersion of the catalyst and starting materials and the surface refreshing of the catalyst with a jet effect.
In summary, the Scheme used to compare present work compared to our previous work (Fig. 3):
 | ||
| Fig. 3 Reduction of nitro compound. | ||
Comparison of the present and our previous report.
2.1 Reaction condition our previous work24
Nitro benzene (0.1 mmol), TiO2-P25 (30 mg), β-cyclodextrin (15 mg), water, oxalic acid (15 mg), sun light, 3 h.2.2 Reaction condition present work
As it can be found, using Na2S instead of oxalic acid resulted in successful photoreduction under visible LED, with increasing amounts of nitro compounds from 0.1 to 1.2 mmol and decreasing amounts of catalyst from 30 mg (for 0.1 mmol nitrobenzene) to 7.5 mg (for 1.2 mmol nitrobenzene). The sonophotocatalytic condition also showed a significant increase in the reaction rate, and all the compounds were reduced within 2 hours.
3 Conclusions
The photocatalytic and sonophotocatalytic methods can be used as green protocols to reduce nitro compounds in aqueous media in the presence of sodium sulfide as a sacrificial reagent. Both methods have successfully reduced Nitro compounds containing electron-donating and electron-withdrawing groups.In this method, nitro compounds are solubilized in water through encapsulation in the inner β-CD cavities and then reduced by TiO2 nanoparticles attached to host–guest complexes in the presence of sodium sulfide. Compared to our previous work,24 this green protocol consumes less catalyst. It reduces light scattering, allowing for the reduction of larger amounts of nitro compounds from 0.1 mmol to 1.2 mmol in aqueous environments. Although the photocatalyst technique is effective for the reduction of nitro compounds, a remarkable achievement of this study is that we were able to increase the initial amount of nitro from 0.1 mmol to 1.2 mmol in the aqueous environment with a shorter period of time using the sonophotocatalysis method, which continuously cleans the catalyst surface with ultrasound waves.
In conclusion, this method has excellent reusability and is suitable for reducing nitroarenes on a large scale using many initial materials. In addition, we successfully performed one-pot N-acetylation through the photocatalysis technique using acetic anhydride in an aqueous medium with in situ prepared amines and converted them to the corresponding amides.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Institute for Advanced Studies in Basic Sciences (IASBS) for financial support in this work. We would like to extend our sincere thanks to Dr Zahra Zand for her helpful comments in this work.Notes and references
- J. Lv, J. Xie, A. G. A Mohamed, X. Zhang, Y. Feng, L. Jiao, E. Zhou, D. Yuan and Y. Wang, Nat. Rev. Chem, 2023, 7(2), 91, DOI:10.1038/s41570-022-00448-9.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501, 10.1039/C3CS60405E.
- (a) K. Q. Lu, Y. H. Li, F. Zhang, M. Y. Qi, X. Chen and Z. R. Tang, Nat. Commun., 2020, 11, 5181, DOI:10.1038/s41467-020-18944-1; (b) Y. Vyas, P. Chundawat, D. Dharmendra, P. Chaubisa, M. Kumar, P. B. Punjabi and C. Ameta, Nanoscale Adv., 2023, 5, 4833–4851, 10.1039/D3NA00268C; (c) Y. Vyas, p. Chundawat, D. Dharmendra, P. B. Punjabi and C. Ameta, Int. J. Hydrogen Energy, 2021, 46, 37208–37241, DOI:10.1016/j.ijhydene.2021.09.004.
- (a) H. A. Atwater and P. Albert, Nat. Mater., 2010, 9, 205, DOI:10.1038/nmat2629; (b) Y. Vyas, S. Gupta, P. Punjabi and C. Ameta, ChemistrySelect, 2022, 7, e202201099, DOI:10.1002/slct.202201099.
- (a) G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. Van Grondelle, Nat. Chem., 2011, 3, 763, DOI:10.1038/nchem.1145; (b) Y. Vyas, P. Chundawat, p. Dharmendra, P. B. Punjabi and C. Ameta, ChemistrySelect, 2021, 6, 8566–8580, DOI:10.1002/slct.202102156.
- (a) J. A. Schuller, E. S. Barnard, W. S. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193, DOI:10.1038/nmat2630; (b) Y. Vyas, P. Chundawat, D. Dharmendra, P. B. Punjabi and C. Ameta, Int. J. Hydrogen Energy, 2021, 46, 37208–37241, DOI:10.1016/j.ijhydene.2021.09.004.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 719, 4545, DOI:10.1021/jp0273934.
- A. Gedanken, Ultrason. Sonochem., 2004, 11, 47, DOI:10.1016/j.ultsonch.2004.01.037.
- J. Madhavan, P. S. S. Kumar, S. Anandan, F. Grieser and M. Ashokkumar, J. Hazard. Mater., 2010, 177, 944, DOI:10.1016/j.jhazmat.2010.01.009.
- P. R. Gogate and A. B. Pandit, AIChE J., 2004, 50, 1051, DOI:10.1002/aic.10079.
- M. Mrowetz, E. Selli and C. Pirola, Ultrason. Sonochem., 2003, 10, 247, DOI:10.1016/S1350-4177(00)00030-4.
- S. Matzusawa, J. Tanaka, S. Sato and T. Ibusuki, J. Photochem. Photobiol., A, 2002, 149, 183, DOI:10.1016/S1010-6030(02)00004-7.
- L. Davydov, E. Reddy, P. France and P. Smirniotis, Appl. Catal., B, 2001, 32, 95, DOI:10.1016/S0926-3373(01)00126-6.
- Y. Kado, M. Atobe and T. Nonaka, Ultrason. Sonochem., 2001, 8, 69, DOI:10.1016/S1350-4177(00)00072-9.
- E. Selli, Phys. Chem. Chem. Phys., 2002, 4, 6123, 10.1039/B205881B.
- C. G. Joseph, G. L. Puma, A. Bono and D. Krishnaiah, Ultrason. Sonochem., 2009, 16, 583, DOI:10.1016/j.ultsonch.2009.02.002.
- N. Talebian, M. R. Nilforoushan and F. J. Mogaddas, Ceram. Int., 2013, 39, 4913, DOI:10.1016/j.ceramint.2012.11.085.
- L. Liang, Y. Tursun, A. Nulahong, T. Dilinuer, A. Tunishaguli, G. Gao, A. Abulikemu and K. Okitsu, Ultrason. Sonochem., 2017, 39, 93, DOI:10.1016/j.ultsonch.2017.03.054.
- V. Vinesh, A. R. M. Shaheer and B. Neppolian, Ultrason. Sonochem., 2018, 50, 302, DOI:10.1016/j.ultsonch.2018.09.030.
- S. Sunasee, K. T. Wong, G. Lee, S. Pichiah, S. Ibrahim, C. Park, N. C. Kim, Y. Yoon and M. Jang, Environ. Sci. Pollut. Res., 2017, 24, 15488, DOI:10.1007/s11356-017-9124-0.
- Z. Yan, L. Zhang, Z. Zhao, H. Qi, Y. Li and D. Cang, Ultrason. Sonochem., 2018, 47, 133, DOI:10.1016/j.ultsonch.2018.03.020.
- S. Kucukcongar, A. G. J. Alwindawi, M. Turkyilmaz and I. Ozaytekin, Water, Air, Soil Pollut., 2023, 234, 367, DOI:10.1007/s11270-023-06136-8.
- D. A. Giannakoudakis, D. Łomot and J. C. Colmenares, Green Chem., 2020, 22, 4896, 10.1039/d0gc00329h.
- M. Abdollahi Kakroudi, F. Kazemi and B. Kaboudin, RSC Adv., 2014, 4, 52762, 10.1039/C4RA08059A.
- B. Casu, M. Reggiani, G. Gallo and A. Vigevani, Tetrahedron, 1968, 2, 803–821, DOI:10.1016/0040-4020(68)88030-5.S.
- T. Tachikawa, S. Tojo, M. Fujitsuka and T. Majima, Chem. – Eur. J., 2006, 12, 7585–7594, DOI:10.1002/chem.200600097.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06530h |
| This journal is © The Royal Society of Chemistry 2023 |