
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and vectorial functionalisation of pyrazolo[3,4-c]pyridines†
Elizabeth V. Bedwell a,
Flavio da Silva Emeryb,
Giuliano C. Clososkic and
Patrick G. Steel*a
a,
Flavio da Silva Emeryb,
Giuliano C. Clososkic and
Patrick G. Steel*a
aDepartment of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK. E-mail: p.g.steel@durham.ac.uk
bDepartamento de Ciências Farmacêuticas, Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brazil
cDepartamento de Ciências Biomoleculares, Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brazil
First published on 23rd November 2023
Abstract
Heterocycles are a cornerstone of fragment-based drug discovery (FBDD) due to their prevalence in biologically active compounds. However, novel heterocyclic fragments are only valuable if they can be suitably elaborated to compliment a chosen target protein. Here we describe the synthesis of 5-halo-1H-pyrazolo[3,4-c]pyridine scaffolds and demonstrate how these compounds can be selectively elaborated along multiple growth-vectors. Specifically, N-1 and N-2 are accessed through protection-group and N-alkylation reactions; C-3 through tandem borylation and Suzuki–Miyaura cross-coupling reactions; C-5 through Pd-catalysed Buchwald–Hartwig amination; and C-7 through selective metalation with TMPMgCl.LiCl followed by reaction with electrophiles or transmetalation to ZnCl2 and Negishi cross-coupling. Linking multiple functionalisation strategies emulates a hit-to-lead pathway and demonstrates the utility of pyrazolo[3,4-c]pyridines to FBDD.
Introduction
Through the identification of weakly binding small-molecules (fragments) and their subsequent elaboration into larger more potent lead compounds, fragment-based drug discovery (FBDD) has become a major strategy for the development of new drug leads.1,2 Heterocyclic compounds represent attractive starting points for FBDD due to an ability to engage with the target protein through a wide variety of intermolecular interactions, coupled with the potential to optimise drug-like properties (lipophilicity, hydrogen bonding capacity, and polarity) through modification of substitution patterns. Given the requirements to compliment a protein's 3D-structure, fragment elaboration requires multiple positions, also called growth-vectors, to be decorated with different motifs to introduce new, or improve existing, binding interactions with the target. It is therefore important to be able to functionalise a given fragment along the available growth-vectors in a chemically diverse yet directionally controlled manner.Whilst FBDD has been successfully applied to several studies, this has largely been achieved using a relatively limited set of heteroaromatic scaffolds.3 A computational enumeration of the underutilized areas of chemical space was conducted to address this deficiency, introducing a set of novel heterocyclic motifs, the “heteroaromatic rings of the future”, as valuable inputs to inspire new drug candidates.4 This stimulated a number of reports describing initial access to these structures.5 However, further functionalisation or introduction of alternative substituent patterns often requires developing challenging de novo syntheses. The complexity of this synthesis limits the structural diversity of compounds that can be rapidly generated for testing and so creates a bottle-neck in FBDD programs.3 Consequently, promising fragment hits may be down-prioritised due to supposed synthetic intractability.6
To address these issues and increase efficiency of hit-to-lead elaboration of heterocyclic fragments, we have initiated a programme to explore late-stage functionalisation of heterocyclic cores that can be applied in a sequential and vectorially diverse manner. In this report we describe the application of complementary C–H activation methods to the vectorial elaboration of a 1H-pyrazolo[3,4-c]pyridine 1 scaffold (Fig. 1) as a representative example of this fragment set.
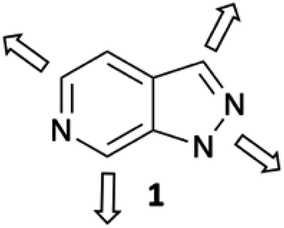 | ||
| Fig. 1 1H-Pyrazolo[3,4-c]pyridine 1; the heterocyclic scaffold chosen for elaboration by vectorial functionalisation along the 4 major growth vectors. | ||
Results
Access to the 5-methoxy-pyrazolo[3,4-c]pyridine was recently reported by Silva Júnior et al. in an adaptation of the procedure first reported by Chapman and Hurst based on the classical Huisgen indazole synthesis.7,8 In further optimisation, introduction of dichloroethane (DCE) as a co-solvent enhanced scalability and enabled isolation of the 1-(5-halo-pyrazolo[3,4-c]pyridine-1-yl)ethan-1-ones 3 without need for purification (Scheme 1). Following simple deacetylation of 3 with NaOMe/MeOH, the desired 5-halo-1H-pyrazolo[3,4-c]pyridines 4 were isolated in excellent overall yield. With the scaffold in hand, the initial objective was to develop selective N-protection sequences. Using precedents based on standard indazole chemistry and tailoring reaction conditions enabled functionalisation of either N-1 or N-2.9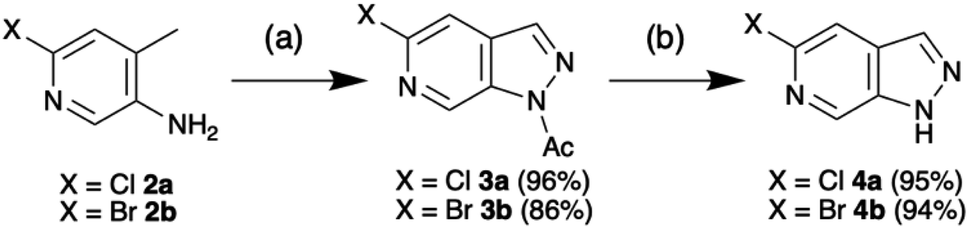 | ||
| Scheme 1 Synthesis of 5-chloro-1H-pyrazolo[3,4-c]pyridine 4a and 5-bromo-1H-pyrazolo[3,4-c]pyridine 4b. Conditions: (a) NaNO2, Ac2O, DCE, rt – 90 °C, 20 h, (b) MeOH, NaOMe, rt, 1 h. | ||
Mesylation (Ms) selectively afforded the N-1 protected product 5a in 92% yield (Table 1, row 1). However, this group proved less useful than hoped due to the tendency for migration to C-3 as exploited by Silva Júnior et al.10–12 Tetrahydropyran (THP) protection of the bromo-scaffold 4b was similarly straightforward as the N-1 isomer 6a and N-2 isomer 6b could be produced selectively based on reaction time (Table 1, rows 2–3) with longer reaction times favouring the thermodynamically more stable N-1 protected product.9 Surprisingly, the chloro-analogues 7 could not be produced selectively without significant impact on the yield. Fortunately, the N-1 and N-2 protected products, 7a and 7b, could be readily separated by chromatography (Table 1, row 4). Complete selectivity in the introduction of a trimethylsilylethoxymethyl (SEM) group was also challenging. However, by careful choice of base, either NaH or N,N-dicyclohexylmethylamine, predominant formation of either the N-1 or N-2 isomer could be achieved with the organic base favouring the formation of the N-2 products, 8b and 9b (Table 1, rows 5–8).
|
|||||
|---|---|---|---|---|---|
| Entry | X = | Reaction conditions | R = | PG1 (yield) | PG2 (yield) |
| 1 | Cl | MsCl, NaH, THF, 0 °C-rt, 2 h | -Ms | 5a (92%) | 5b (0%) |
| 2 | Br | DHP, pTsOH, DCM, rt, 2 h | -THP | 6a (6%) | 6b (75%) |
| 3 | Br | DHP, pTsOH, DCM, rt, 22 h | -THP | 6a (82%) | 6b (0%) |
| 4 | Cl | DHP, pTsOH, DCM, rt, 4 h | -THP | 7a (14%) | 7b (66%) |
| 5 | Br | SEMCl, Cy2MeN, THF, 0 °C-rt, 18 h | -SEM | 8a (18%) | 8b (32%) |
| 6 | Br | SEMCl, NaH, THF, 0 °C-rt, 6 h | -SEM | 8a (47%) | 8b (26%) |
| 7 | Cl | SEMCl, NaH, THF, 0 °C-rt, 6 h | -SEM | 9a (45%) | 9b (29%) |
| 8 | Cl | SEMCl, Cy2MeN, THF, 0 °C-rt, 18 h | -SEM | 9a (21%) | 9b (44%) |
| 9 | Br | MeI, NaH, 0 °C-rt, 1 h | -Me | 10a (36%) | 10b (50%) |
| 10 | Cl | MeI, NaH, 0 °C-rt, 1 h | -Me | 11a (31%) | 11b (42%) |
| 11 | Cl | PrI, NaH, 0 °C-rt, 24 h | -Pr | 12a (27%) | 12b (35%) |
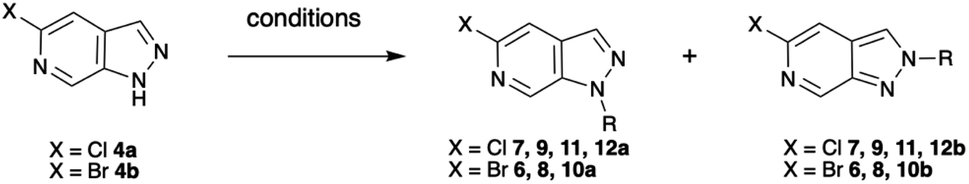
Finally, simple alkylation afforded the corresponding N-alkylated species as mixtures that could be separated by column chromatography (Table 1, rows 9–11).
As a first vector of functionalisation, we opted to explore Buchwald–Hartwig amination at C-5 (Scheme 2). A vast array of conditions exists in the literature varying base, ligand, solvent, and reaction time depending on the substrate.13 Following precedents established by Slade,9 the combination of Pd2(dba)3, rac-BINAP, and NaOtBu in THF was successfully applied to reactions of 6b with primary, secondary, and aromatic amines generating products in yields of 62–75%. These conditions were also explored for various protection group strategies which revealed the reaction of 8a as the most successful affording 18 in 97% yield.
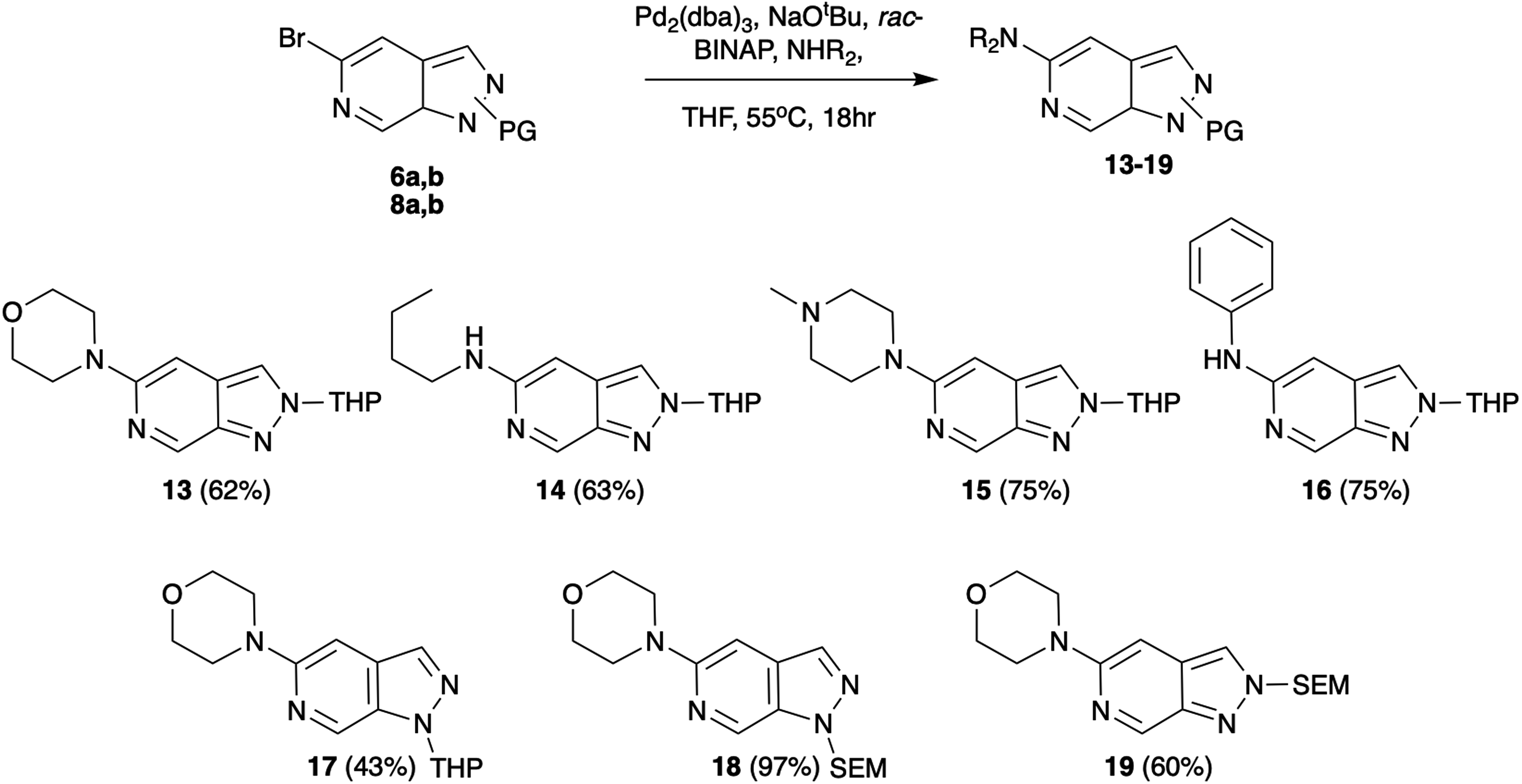 | ||
| Scheme 2 Pd-catalysed Buchwald–Hartwig amination examples. | ||
Selecting C-3 as the next target vector, we again took precedent from indazole chemistry in which iridium catalysed C–H borylation has been shown to occur regioselectively in this position. Consistent with the literature reports, using conditions based on those optimised by Sadler et al.14,15 both 9a and 9b underwent efficient borylation at the C-3 position. The utility of these boronate esters was demonstrated through in situ Suzuki–Miyaura cross-coupling. Whilst Suzuki–Miyaura cross-coupling of the 9a substrate proceeded readily under standard conditions of Cs2CO3 and Pd(dppf)Cl2 in DMAc with yields of 47–60% (Scheme 3), rapid protodeborylation was a barrier to product generation for reaction of 9b. This was addressed by use of a CuCl additive to increase the rate of transmetalation via formation of an intermediary Cu species.16 Under these modified conditions, the desired C-3 functionalised products could be obtained in moderate to good overall yields (31–48%, Scheme 3).
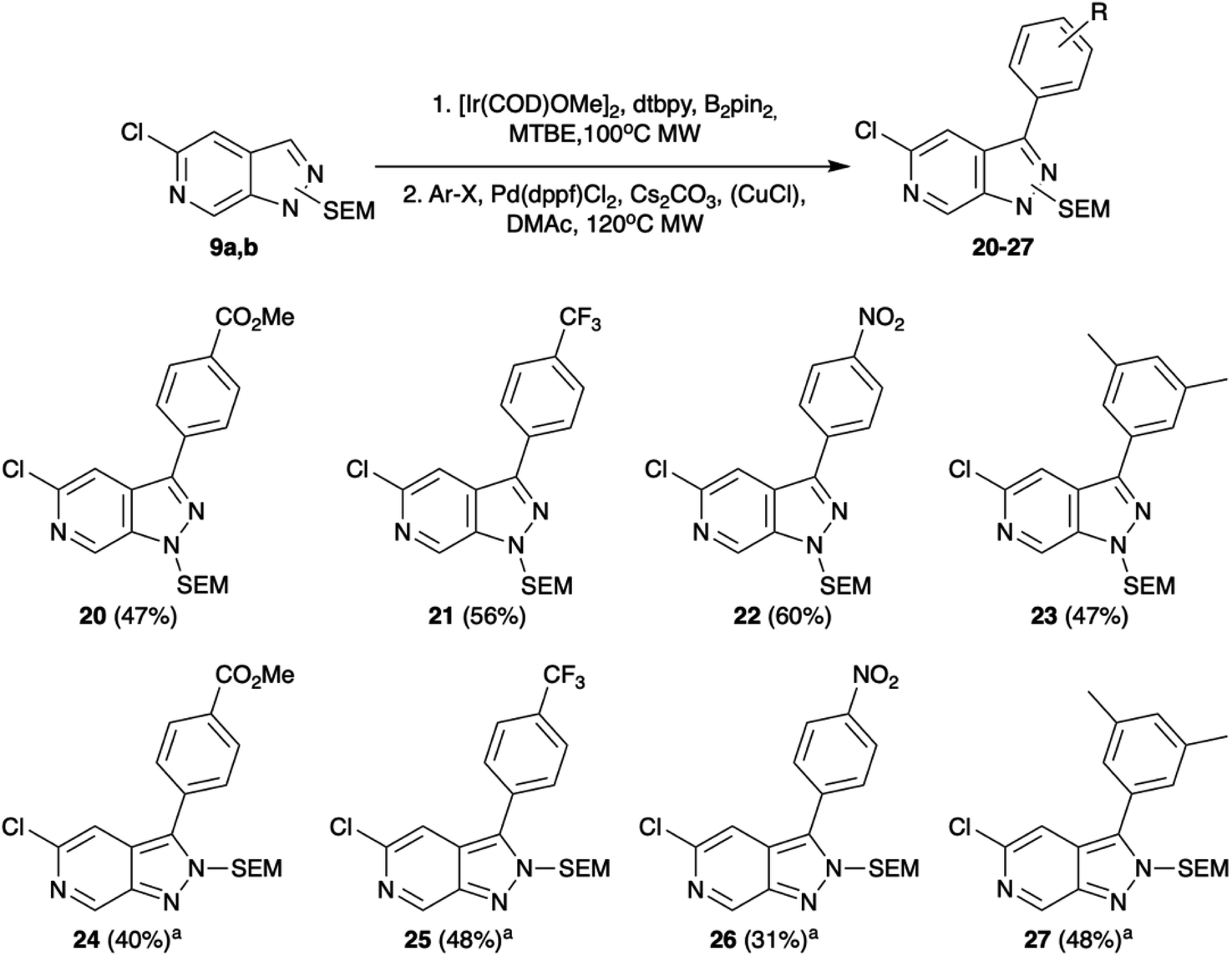 | ||
| Scheme 3 Tandem C–H borylation and Suzuki–Miyaura cross-coupling of 9. Conditions: (1) [Ir(COD)OMe]2, dtbpy, B2pin2, MTBE, 100 °C, MW, (2). Ar-X, Pd(dppf)Cl2, Cs2CO3, DMAc, 120 °C, MW. aAdditional CuCl. | ||
Having established methodologies to functionalise N-1, C-3, and C-5, we then turned to the remaining vector of C-7. Initial attempts to exploit the azinyl nature of C-7 and couple the heterocycle with nbutyl-lithium led to nucleophilic addition at the C-7 position, albeit in very low yields. Consequently, we turned to more selective metalation chemistries with the mixed magnesium-lithium TMP (2,2,4,4-tetramethylpiperidine) bases.
Regioselective metalation by TMP-metal bases has been previously reported for a broad range of aromatic and heteroaromatic compounds.17–19 When applied to the N-1 SEM protected analogue 9a this afforded the desired metalation at C-7. The efficiency of the magnesiation step proved to be highly temperature dependent with optimal conversion, following trapping with iodine, being obtained at −40 °C (Table S1, ESI†). To demonstrate the broad scope of this chemistry, the intermediate organomagnesium species were treated with a variety of electrophiles including aldehydes, diphenyl disulfide, and DMF for yields of 48–66%. Alternatively, transmetalation with ZnCl2 afforded a range of arylated products via Negishi cross-coupling chemistry with yields of 71–83% (Scheme 4). Interestingly, TMPMgCl.LiCl treatment of the N-2 SEM-protected compound 13b led to metalation at C-3 instead of C-7 (Scheme 5). However, this reaction was inefficient and produced a complex mixture of products.
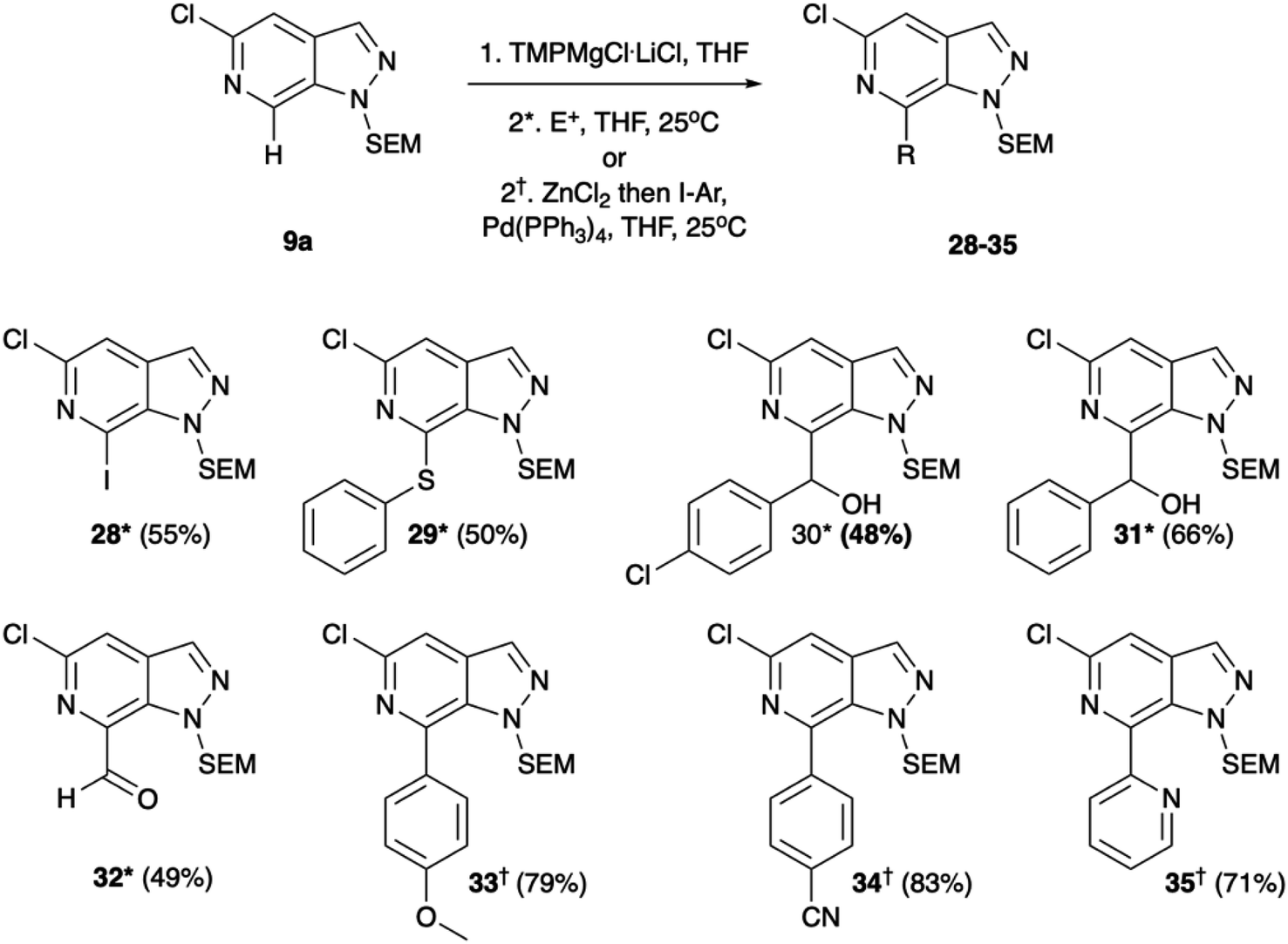 | ||
| Scheme 4 Metalation with TMPMgCl.LiCl reaction with electrophiles and transmetalation with ZnCl2 for Negishi cross-coupling. Conditions*: (1) TMPMgCl·LiCl, THF, −40 °C, (2) E+, THF, 25 °C or conditions†: (1). TMPMgCl.LiCl, THF, −40 °C, (2) ZnCl2 then I–Ar, Pd(PPh3)4, THF, 25 °C. | ||
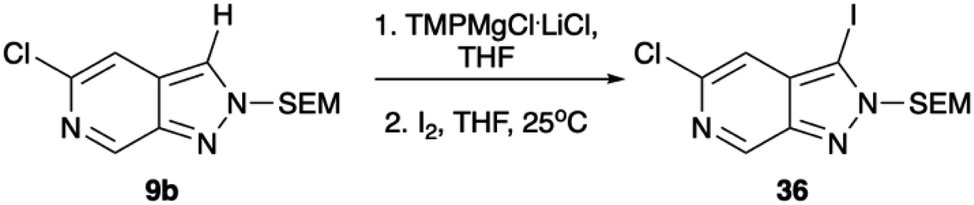 | ||
| Scheme 5 Metalation with TMPMgCl.LiCl reaction with iodine. Conditions: (1) TMPMgCl.LiCl, THF, −78 °C, 30 min, (2) I2, THF, 25 °C, 1 h. | ||
As the C-3 vector had already been thoroughly explored through Ir-catalysed borylation, this route was not pursued further.
While individual elaboration along each growth vector is a valuable tool to explore fragment space, it is likely that an FBDD optimisation may require multiple vectors of growth. As a final aspect we, therefore, sought to demonstrate how these functionalisation strategies could be combined to emulate a hit-to-lead pathway common to medicinal chemistry development.
In the first sequence, a combination of C-3 borylation, Suzuki–Miyaura cross-coupling and a tandem N-methylation/SEM-deprotection was explored (Scheme 6A). In the case of N-2 SEM-protected compound 25, reaction with [Me3O][BF4] (trimethyloxonium tetrafluoroborate) saw efficient formation of the N1-methylated product 37. In the case of the N-1 SEM-protected isomers, methylation of N-2 occurred readily with [Me3O][BF4] to afford the methyl salts 38 and 39.
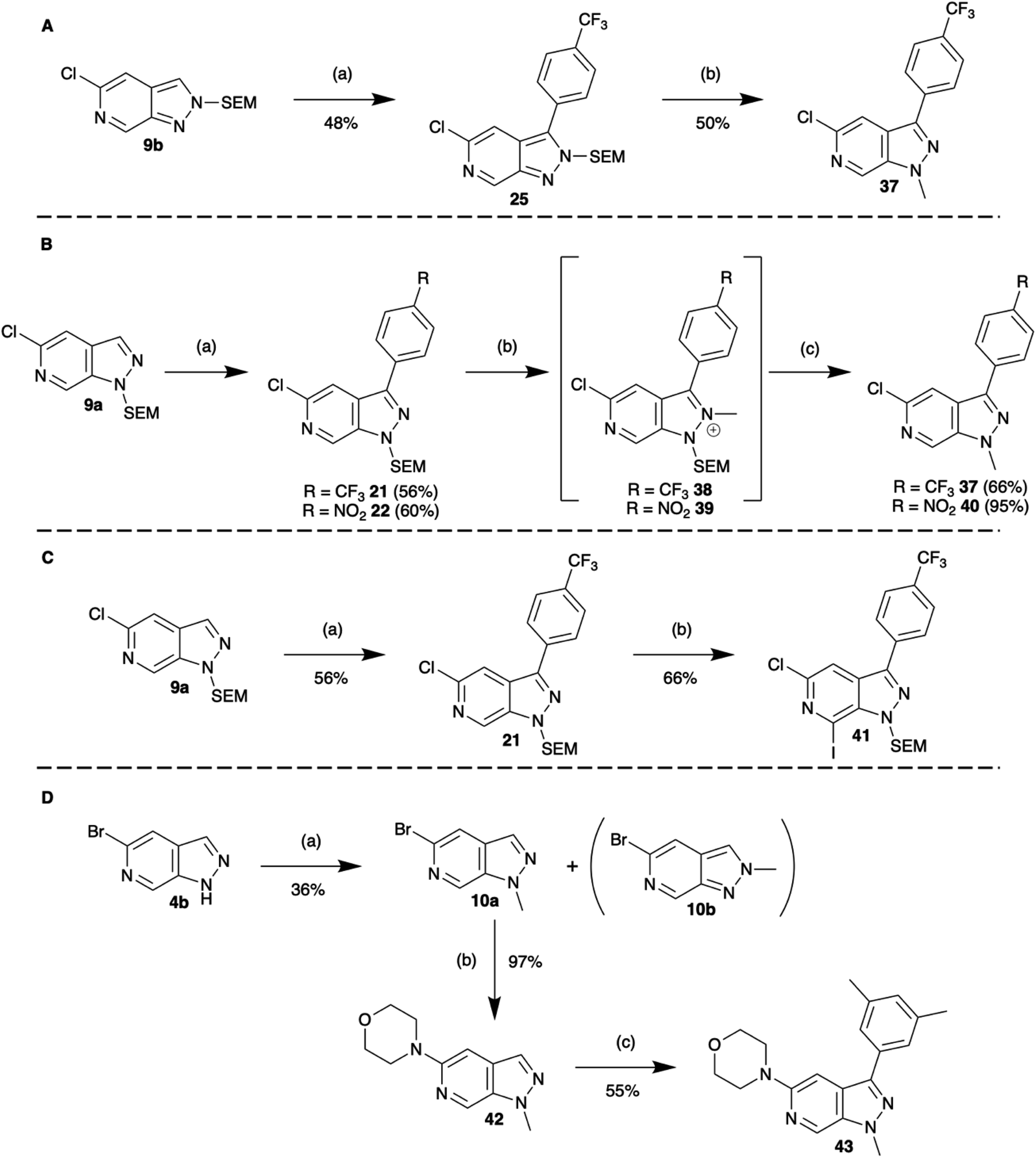 | ||
| Scheme 6 Multiple-vector elaboration sequences. (A) C-3 borylation, Suzuki–Miyaura cross-coupling with a tandem N-methylation/SEM-deprotection, conditions: (a) (1) [Ir(COD)OMe]2, dtbpy, B2pin2, MTBE, 100 °C MW, 2. C7H4F3Br, Pd(dppf)Cl2, Cs2CO3, CuCl, DMAc, 120 °C MW, (b) [Me3O][BF4], EtOAc, rt, 18 h. (B) C-3 borylation, Suzuki–Miyaura cross-coupling with a tandem N-methylation/SEM-deprotection, conditions: (a) (1) [Ir(COD)OMe]2, dtbpy, B2pin2, MTBE, 100 °C MW, 2. ArX, Pd(dppf)Cl2, Cs2CO3, DMAc, 120 °C MW, (b) [Me3O][BF4], EtOAc, rt, 6 h, (c) TFA, DCM, rt, 20 h. (C) C-3 borylation, Suzuki–Miyaura cross-coupling with C-7 TMPMgCl.LiCl metalation and electrophilic trapping with iodine, conditions: (a) (1) [Ir(COD)OMe]2, dtbpy, B2pin2, MTBE, 100 °C MW, (2) C7H4F3Br, Pd(dppf)Cl2, Cs2CO3, DMAc, 120 °C MW, (b) (1) TMPMgCl.LiCl, THF, −40 °C, 30 min (2) I2, THF, rt, 1 h. (D) N-Methylation, Buchwald–Hartwig amination, and C-3 borylation, Suzuki–Miyaura cross-coupling, conditions: (a) MeI, NaH, THF, rt, 45 min, (b) Pd2(dba)3, NHR2, NaOtBu, rac-BINAP, THF, 55 °C, 18 h, (c) (1) [Ir(COD)OMe]2, dtbpy, B2pin2, MTBE, 100 °C MW 30 min (2) C8H9I, Pd(dppf)Cl2, Cs2CO3, DMAc, 120 °C MW 2 h. | ||
However, surprisingly, acid catalysed SEM-deprotection with TFA did not give the expected conversion to N2-methylated compounds, instead, ipso methylation at N-1 was observed and 37 and 40 were isolated (Scheme 6B). It is likely that these observations reflect the greater thermodynamic stability of the 1H-pyrazolo[3,4-c]pyridine core.
A second two stage sequence paired C-3 borylation Suzuki–Miyaura cross-coupling functionalisation with C-7 TMPMgCl.LiCl metalation and electrophilic trapping with iodine to give 41 (Scheme 6C).
Finally, extensions to more elaborate three-vector functionalisation sequences are also possible. Exemplified in Scheme 6D showing a three-step sequence from fragment 4b to compound 43. Importantly, despite the increasing complexity, in each of these processes the yield of each step was comparable to that obtained earlier. Other sequences of elaboration can be endlessly imagined combining the chemistry explored herein to realise an expansive library of heterocyclic compounds.
Conclusion
In summary, we have developed efficient synthetic routes to 5-halo-1H-pyrazolo[3,4-c]pyridine scaffolds and demonstrated how these compounds can be selectively elaborated along multiple reaction vectors. The pyrazolo[3,4-c]pyridine core has begun to attract attention as a therapeutic agent due to its structural similarity to purine.20–23 Purine derivatives have a broad range of applications, as anti-inflammatory, anti-viral, and anti-cancer agents, arising from the prevalence of purine-based biological compounds and, subsequently, the variety of cellular proteins that contain purine-binding pockets.20 Whilst routes have been reported for related products with different substituent patterns around the pyrazolo[3,4-c]pyridine core, most of these require discrete synthetic endeavour.23–25 The chemistry described here allows introduction of functionality at C-3, C-5 and C-7 and as such is complimentary to the recent related report by Silva Júnior et al. which describes alternative C-4 functionalisation of a 5-methoxy analogue.10Importantly, through careful control of reaction sequence it is possible to selectively explore growth in a combination of vectors with no loss in synthetic efficiency.
Overall, the late-stage functionalisation sequences described here provide practical advantages, particularly for the introduction of structural diversity required for fragment-elaboration in an FBDD hit-to-lead evolution pipeline. Extensions of these ideas to other heterocyclic cores are in progress and the results of this will be reported in due course.
Experimental section
All solvents and reagents were purchased from commercial suppliers and used without further purification unless otherwise stated below. Final compound purification by flash column chromatography was performed on a CombiFlash® System from Teledyne Isco equipped with an UV-light detector using prepacked silica RediSep Rf cartridges with the stated solvent gradient. Crude mixtures to be purified were dry loaded onto silica (normal phase) or Celite® 545 (reverse phase) prior to running the column. NMR spectra were recorded on the following instruments: Bruker Neo 700 MHz spectrometer with operating frequencies of 700 MHz for 1H and 175 MHz for 13C; Varian VNMRS-600 with operating frequencies of 600 MHz for 1H and 150 MHz for 13C NMR; and Varian VNMRS-400 with operating frequencies of 400 MHz for 1H and 376 MHz for 19F NMR. Spectra were referenced relative to CDCl3 (δH 7.26 ppm, δC 77.16 ppm), CD3OD (δH 4.87 ppm, δC 49.00 ppm), or DMSO (δH 2.50 ppm, δC 39.52 ppm). Chemical shifts are reported in parts per million (ppm), coupling constants (J) in hertz (Hz) and multiplicity as singlet (s), doublet (d), triplet (t), quartet (q), pentet (p), sextet (s), multiplet (m) or a combination thereof. All J values are JH–H unless otherwise stated. All 1H NMR and 13C NMR spectral assignments were made with the aid of 1H1H COSY, 1H1H NOESY, 1H13C HSQC and 1H13C HMBC NMR experiments. Infra-red spectra were recorded on a PerkinElmer Paragon 1000 FT-IR spectrometer or a PerkinElmer RX FT-IR spectrometer with Golden Gate Diamond ATR apparatus. IR assignments are reported in wavenumbers (cm−1).Melting points were recorded on Thermo Scientific Electrothermal IA9100 Digital Melting Point apparatus. Thin layer chromatography was performed using Merck F254 silica gel 60 aluminium sheets pre-coated with silica gel. High resolution mass spectrometry (HRMS) and liquid chromatography mass spectrometry (LC-MS) were recorded on a Waters TQD mass spectrometer ESI-LC water (0.1% formic acid): MeCN/MeOH, flow rate 0.6 mL min−1 with a UPLC BEH C18 1.7 μm (2.1 mm × 50 mm) column. Gas chromatography mass spectrometry (GCMS) was carried out on a Shimadzu QP2010-Ultra with a temperature gradient 50–300 °C and a hold time of 5 min, using a Rxi-17Sil MS (0.15 μm × 10 m × 0.15 mm) column.
Substrate synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 144.7 (C-5), 138.0 (C-7), 137.8 (C-3), 134.7 (C-7a), 133.8 (C-3a), 114.8 (C-4), 22.6 (C(O)CH3); Vmax (ATR) 1729 (CO), 1390, 1353, 634 cm−1; LC-MS (ES+) [M (35Cl) + H] 196.069, [M (37Cl) + H] 198.007, [M (35Cl) + H − COCH3] 154.035, [M (37Cl) + H − COCH3] 156.011; HRMS (ES+) found [M + H]+ 196.0290, C8H7N3O35Cl requires M 196.0278. The analytical data were consistent with the literature.7
O), 144.7 (C-5), 138.0 (C-7), 137.8 (C-3), 134.7 (C-7a), 133.8 (C-3a), 114.8 (C-4), 22.6 (C(O)CH3); Vmax (ATR) 1729 (CO), 1390, 1353, 634 cm−1; LC-MS (ES+) [M (35Cl) + H] 196.069, [M (37Cl) + H] 198.007, [M (35Cl) + H − COCH3] 154.035, [M (37Cl) + H − COCH3] 156.011; HRMS (ES+) found [M + H]+ 196.0290, C8H7N3O35Cl requires M 196.0278. The analytical data were consistent with the literature.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH 1:100 (8 mL) until acidic pH and concentrated under reduced pressure. The crude product was taken up in H2O (75 mL) then adjusted to pH 10 by addition of aqueous NaOH then extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (75 mL), dried over MgSO4, filtered and concentrated to afford 5-chloro-1H-pyrazolo[3,4-c]pyridine 4a as a white solid (1.5 g, 10 mmol, 95%) with mp 225–226 °C. δH (400 MHz, methanol-d4) 8.80 (1H, s, 7-H), 8.15 (1H, d, J = 1.2 Hz, 3-H), 7.82 (1H, d, J = 1.2 Hz, 4-H); δC (101 MHz, methanol-d4) 141.0 (C-5), 137.6 (C-7a), 135.1 (C-7), 134.2 (C-3), 131.2 (C-3a), 115.6 (C-4); Vmax (ATR) 909, 736, 652 cm−1; LC-MS (ES+) [M (35Cl) + H] 154.113, [M (37Cl) + H] 156.128; HRMS (ES+) found [M + H]+ 154.0167, C6H5N335Cl requires M 154.0172.
:MeOH 1:100 (8 mL) until acidic pH and concentrated under reduced pressure. The crude product was taken up in H2O (75 mL) then adjusted to pH 10 by addition of aqueous NaOH then extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (75 mL), dried over MgSO4, filtered and concentrated to afford 5-chloro-1H-pyrazolo[3,4-c]pyridine 4a as a white solid (1.5 g, 10 mmol, 95%) with mp 225–226 °C. δH (400 MHz, methanol-d4) 8.80 (1H, s, 7-H), 8.15 (1H, d, J = 1.2 Hz, 3-H), 7.82 (1H, d, J = 1.2 Hz, 4-H); δC (101 MHz, methanol-d4) 141.0 (C-5), 137.6 (C-7a), 135.1 (C-7), 134.2 (C-3), 131.2 (C-3a), 115.6 (C-4); Vmax (ATR) 909, 736, 652 cm−1; LC-MS (ES+) [M (35Cl) + H] 154.113, [M (37Cl) + H] 156.128; HRMS (ES+) found [M + H]+ 154.0167, C6H5N335Cl requires M 154.0172.General procedure B – for tandem borylation and Suzuki–Miyaura cross-coupling
[Ir(COD)OMe]2 (0.025 eq.), B2pin2 (1.10 eq.), and dtbpy (0.05 eq.) were sealed in an oven-dried microwave reaction vial and degassed with N2/vacuum cycling. A solution of the SEM-protected pyrazolo[3,4-c]pyridine in anhydrous MTBE (0.4 M) was added under nitrogen. The reaction mixture was heated in a microwave reactor at 100 °C until GCMS analysis showed complete borylation had occurred, then concentrated under reduced pressure to afford the crude boronate ester. To the crude boronate ester was added Cs2CO3(2.00 eq.), Pd(dppf)Cl2(0.025 eq.), the aryl halide (1.10 eq.) and anhydrous DMAc (1 M) under nitrogen. The reaction mixture was heated in a microwave reactor at 120 °C until GCMS analysis showed no boronate ester remained. The reaction mixture was filtered through Celite® and the residue washed with EtOAc. The combined filtrates were concentrated under reduced pressure, and the residue was dissolved in H2O then extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated. The product was purified by silica gel flash column chromatography using the stated solvent system.O asymmetric), 1349 (NO symmetric), 1074, 857, 834, 821 cm−1; LC-MS (ES+) [M (35Cl) + H] 405.282 [M (37Cl) + H] 407.297; HRMS (ES+) found [M + H]+ 405.1153, C18H22N4O3Si35Cl requires M 405.1150.General procedure C – for tandem borylation and Suzuki–Miyaura cross-coupling with CuCl
[Ir(COD)OMe]2 (0.025 eq.), B2pin2 (1.10 eq.), and dtbpy (0.05 eq.) were sealed in an oven-dried microwave reaction vial and degassed with N2/vacuum cycling. A solution of the SEM-protected pyrazolo[3,4-c]pyridine in anhydrous MTBE (0.4 M) was added under nitrogen. The reaction mixture was heated in a microwave reactor at 100 °C until GCMS analysis showed complete borylation had occurred, then concentrated under reduced pressure to afford the crude boronate ester. To the crude boronate ester was added Cs2CO3(1.00 eq.), Pd(OAc)2(0.025 eq.), 1,1′-bis(diphenylphosphino)ferrocene (dppf) (0.050 eq.), CuCl (1.00 eq.), the aryl halide (1.10 eq.) and anhydrous DMAc (1 M) under nitrogen. The reaction mixture was heated in a microwave reactor at 120 °C until GCMS analysis showed no boronate ester remained. The reaction mixture was filtered through Celite® and the residue washed with EtOAc. The combined filtrates were concentrated under reduced pressure, and the residue was dissolved in H2O then extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated. The product was purified by silica gel flash column chromatography using the stated solvent system.General procedure D – for deproto-metalation by TMPMgCl.LiCl and trapping with an electrophile
An oven dried RBF was charged with a solution of the stated substrate (1.00 eq.) in dry THF (0.5 M) and cooled to −40 °C under a nitrogen atmosphere. TMPMgCl.LiCl in THF (2.00 eq.) was added dropwise and the reaction was stirred for 30 min at −40 °C. The corresponding electrophile was added at −40 °C, then the reaction was stirred at room temperature for the time stated. The reaction was quenched with NaHSO3 sat. solution and the crude product extracted with EtOAc, then the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The product was purified by silica gel flash column chromatography using the stated solvent system.General procedure E − for deproto-metalation by TMPMgCl.LiCl and transmetalation to Zn for Negishi cross-coupling
An oven-dried reaction vessel was charged with a solution of the substrate (1.00 eq.) in dry THF (0.5 M), the atmosphere was exchange to nitrogen, and the solution cooled to −40 °C. TMPMgCl.LiCl in THF (2.00 eq.) was added dropwise and the reaction was stirred for 30 minutes at −40 °C. A solution of ZnCl2 (1.00 eq.) in THF (1 M) was added and the reaction stirred for 30 minutes at −40 °C. In a separate oven-dried reaction vessel, a solution of the corresponding (hetero)aryl halide (1.50 eq.) and Pd(PPh3)4 (0.05 eq.) in THF (0.5 M) were stirred at room temperature for 30 minutes. This solution was added to the main reaction mixture at −40 °C, then the reaction was stirred at room temperature overnight. The reaction was quenched with NH4Cl sat. solution and the crude product extracted with EtOAc, then the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The product was purified by silica gel flash column chromatography using the stated solvent system.δH (600 MHz, CDCl3) 8.13 (1H, s, 3′-H), 7.85 (2H, d, J = 7.9 Hz, 2,6-H), 7.81 (2H, d, J = 7.9 Hz, 3,5-H), 7.71 (1H, s, 4′H), 5.38 (2H, s, NCH2O), 3.40 (2H, m, OCH2CH2), 0.77 (2H, m, OCH2CH2Si), −0.07 (9H, s, Si(CH3)3); δC (151 MHz, CDCl3) 143.0 (C-7′), 141.4 (C-4), 140.9 (C-5′), 133.7 (C-3′a), 133.6 (C-7′a), 133.4 (C-3′), 132.1 (C-2,6), 130.2 (C-3,5), 118.3 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 114.3 (C-4′), 113.4 (C-1), 78.4 (NCH2O), 66.8 (OCH2CH2), 17.7 (OCH2CH2Si), −1.5 (Si(CH3)3); Vmax (ATR) 2230 (CN), 1070, 856, 837, 815 cm−1; LC-MS (ES+) [M (35Cl) + H] 385.330 [M (37Cl) + H] 387.307; HRMS found [M + H]+ 385.1250, C19H2235ClN4OSi requires M 385.1251.
N), 114.3 (C-4′), 113.4 (C-1), 78.4 (NCH2O), 66.8 (OCH2CH2), 17.7 (OCH2CH2Si), −1.5 (Si(CH3)3); Vmax (ATR) 2230 (CN), 1070, 856, 837, 815 cm−1; LC-MS (ES+) [M (35Cl) + H] 385.330 [M (37Cl) + H] 387.307; HRMS found [M + H]+ 385.1250, C19H2235ClN4OSi requires M 385.1251.
Author contributions
EVB designed and conducted experiments; EVB and PGS drafted the manuscripts; FE, GCC and PGS conceived the project, designed experiments, and supervised the work; PGS secured the funding. All authors reviewed and edited the manuscript and approved the final submitted version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC (studentship to EVB EP/T518001/1) the GCRF and Newton Fund Consolidation Account (EP/X527713/1) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) - (Grant to GCC 22/05327-7) for financial support of this work; Dr Juan Aguilar(DU), Peter Stokes (DU), and Dr Aileen Congreve (DU) for assistance with compound characterisation data; and Victor Hugo (USP-RP) for guidance with metalation chemistry.References
- P. Kirsch, A. M. Hartman, A. K. H. Hirsch and M. Empting, Mol., 2019, 24, 4309 CrossRef CAS PubMed.
- L. Walsh, D. A. Erlanson, I. J. P. de Esch, W. Jahnke, A. Woodhead and E. Wren, J. Med. Chem., 2023, 66, 1137–1156 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- W. R. Pitt, D. M. Parry, B. G. Perry and C. R. Groom, J. Med. Chem., 2009, 52, 2952–2963 CrossRef CAS PubMed.
- K. Passador, S. Thorimbert and C. Botuha, Synth., 2019, 51, 384–398 CrossRef CAS.
- G. Chessari, R. Grainger, R. S. Holvey, R. F. Ludlow, P. N. Mortenson and D. C. Rees, Chem. Sci., 2021, 12, 11976–11985 RSC.
- D. Chapman and J. Hurst, J. Chem. Soc., Perkin Trans. 1, 1980, 2398–2404 RSC.
- P. E. da Silva Júnior, L. C. D. Rezende, J. P. Gimenes, V. G. Maltarollo, J. Dale, G. H. G. Trossini, F. da Silva Emery and A. Ganesan, RSC Adv., 2016, 6, 22777–22780 RSC.
- D. J. Slade, N. F. Pelz, W. Bodnar, J. W. Lampe and P. S. Watson, J. Org. Chem., 2009, 74, 6331–6334 CrossRef CAS PubMed.
- P. E. da Silva Júnior, S. M. G. de Melo, M. H. de Paula, R. Vessecchi, T. Opatz, J. E. H. Day, A. Ganesan and F. da Silva Emery, Org. Biomol. Chem., 2022, 20, 7483–7490 RSC.
- B. Prasad, R. Adepu, S. Sandra, D. Rambabu, G. R. Krishna, C. M. Reddy, G. S. Deora, P. Misra and M. Pal, Chem. Commun., 2012, 48, 10434–10436 RSC.
- S. Searles and S. Nukina, Chem. Rev., 1959, 59, 1077–1103 CrossRef CAS.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- S. A. Sadler, A. C. Hones, B. Roberts, D. Blakemore, T. B. Marder and P. G. Steel, J. Org. Chem., 2015, 80, 5308–5314 CrossRef CAS PubMed.
- S. A. Sadler, H. Tajuddin, I. A. I. Mkhalid, A. S. Batsanov, D. Albesa-Jove, M. S. Cheung, A. C. Maxwell, L. Shukla, B. Roberts, D. C. Blakemore, Z. Lin, T. B. Marder and P. G. Steel, Org. Biomol. Chem., 2014, 12, 7318–7327 RSC.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- Z. Dong, G. C. Clososki, S. H. Wunderlich, A. Unsinn, J. Li and P. Knochel, Chem.–Eur. J., 2009, 15, 457–468 CrossRef CAS PubMed.
- T. dos Santos, H. P. Orenha, V. E. Murie, R. Vessecchi and G. C. Clososki, Org. Lett., 2021, 23, 7396–7400 CrossRef CAS PubMed.
- M. Balkenhohl, B. Salgues, T. Hirai, K. Karaghiosoff and P. Knochel, Org. Lett., 2018, 20, 3114–3118 CrossRef CAS PubMed.
- V. Giannouli, N. Lougiakis, I. K. Kostakis, N. Pouli, P. Marakos, A.-L. Skaltsounis, S. Nam, R. Jove, D. Horne, R. Tenta, H. Pratsinis and D. Kletsas, Bioorg. Med. Chem. Lett., 2016, 26, 5229–5233 CrossRef CAS PubMed.
- A.-M. Fantel, V. Myrianthopoulos, A. Georgoulis, N. Lougiakis, I. Zantza, G. Lamprinidis, F. Augsburger, P. Marakos, C. E. Vorgias, C. Szabo, N. Pouli, A. Papapetropoulos and E. Mikros, Mol, 2020, 25, 3739 CrossRef CAS PubMed.
- D. Matsuda, Y. Kobashi, A. Mikami, M. Kawamura, F. Shiozawa, K. Kawabe, M. Hamada, K. Oda, S. Nishimoto, K. Kimura, M. Miyoshi, N. Takayama, H. Kakinuma and N. Ohtake, Bioorg. Med. Chem. Lett., 2016, 26, 3441–3446 CrossRef CAS PubMed.
- N. Lougiakis, P. Marakos, N. Poul and J. Balzarini, Chem. Pharm. Bull., 2008, 56, 775–780 CrossRef CAS PubMed.
- A. Papastathopoulos, N. Lougiakis, I. K. Kostakis, P. Marakos, N. Pouli, H. Pratsinis and D. Kletsas, Eur. J. Med. Chem., 2021, 218, 113387 CrossRef CAS PubMed.
- C. Wang, Y. Pei, L. Wang, S. Li, C. Jiang, X. Tan, Y. Dong, Y. Xiang, Y. Ma and G. Liu, J. Med. Chem., 2020, 63, 6066–6089 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimisation of metalation chemistry, experimental procedures, analytical data and 1H and 13C spectra for all compounds synthesised. See DOI: https://doi.org/10.1039/d3ra07458g |
| This journal is © The Royal Society of Chemistry 2023 |