
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic routes to trifluoromethylphenyl diazirine photolabeling reagents containing an alkyne substituent (TPDYNE) for chemical biology applications†
Mingxing Qiana,
Yuanjian Xua and
Douglas F. Covey *abcd
*abcd
aDepartment of Developmental Biology, Washington University in St. Louis, 660 S. Euclid Ave., St. Louis, MO 63110, USA. E-mail: dcovey@wustl.edu
bDepartment of Anesthesiology, Washington University in St. Louis, 660 S. Euclid Ave., St. Louis, MO 63110, USA
cDepartment of Psychiatry, Washington University in St. Louis, 660 S. Euclid Ave., St. Louis, MO 63110, USA
dTaylor Family Institute for Innovative Psychiatric Research, Washington University in St. Louis, 660 S. Euclid Ave., St. Louis, MO 63110, USA
First published on 14th December 2023
Abstract
The trifluoromethylphenyl diazirine (TPD) group is widely used in photoaffinity labeling studies. The TPDYNE group (TPD with an additional alkyne substituent on the phenyl ring) enables the use of click chemistry in conjunction with photoaffinity labeling and expands the utility of the TPD group. New methods for preparing previously known as well as new TPDYNE reagents are reported. Additional methods for preparation of a TPDYNE precursor from which the TPDYNE group can be generated once the precursor is attached to the molecule of interest are also described. Procedures for attaching the TPDYNE or TPDYNE precursor to carboxyl, amino, hydroxyl and alkyne groups are demonstrated using steroids as examples.
The trifluoromethylphenyl diazirine (TPD) group is a widely used photoaffinity labeling (PAL) group for chemical biology applications.1,2 We previously incorporated the TPD group into analogues of both neurosteroid and cholesterol analogues for photolabeling studies.3,4 In cases where we wanted to have the option to use click chemistry in conjunction with photolabeling we incorporated an alkyne group into the steroid portion of the analogues.4 This strategy entails finding a location for the alkyne in the steroid that does not negate biological activity and can require lengthy steroid synthesis. Realizing that placing the alkyne group on the TPD moiety would obviate the development of chemical methods for modifying the steroid, we searched the literature for previous reports of alkyne modified TPD groups.
Two alkyne modified TPD precedents were found in the literature. The first modification involved the initial preparation of the difluoro alkyne 1 (Fig. 1) and its subsequent attachment to an aromatic ring to ultimately produce compound 2.5 Although we were able to efficiently attach difluoro alkyne 1 to t-butyl(4-iodobenzyl)dimethylsilane to obtain 1-(4-((tert-butyldimethylsilyl)methyl)phenyl)-2,2-difluorobut-3-yn-1-one, the conversion of the ketone group to the diazirine group was not successful. Consequently, we turned our attention to the second reported modification of a TPD group which involves placing the alkyne group on the aromatic ring of the TPD group (3a, 3b).6 As reported, compound 3b was incorporated into a dantrolene analogue and shown to photolabel proteins involved in physiological Ca2+ release from sarcoplasmic reticulum of skeletal muscle and compound 3b was incorporated into a nucleoside analogue to photolabel microRNA targets.6,7 Both of these prior studies demonstrate that alkyne modified TPD groups (hereafter referred to as TPDYNE groups) are effective PAL groups.
 | ||
| Fig. 1 Literature precedents for trifluoromethylphenyl diazirines modified with an alkyne group (1, 2, 3a and 3b)5–7 and those synthesized herein (3a–3e) by new synthetic procedures. | ||
Use of the TPDYNE group in PAL studies has been limited to the studies cited and general methods for attachment of the TPDYNE group to different functional groups have not been reported. Using a newly developed synthetic route we prepared the previously reported compounds 3a and 3b as well as the new alkyne TPDYNEs 3c–3e. We also developed a synthetic route to TPDYNE precursors from which the TPDYNE can be generated once the precursor is attached to the probe of interest. The precursors are useful when the TPDYNE group cannot be efficiently attached to the probe of interest. General methods for the attachment of these PAL groups to carboxylic acid, amine, hydroxyl and alkyne groups are described using steroids as examples.
TPDYNEs 3a–3e were prepared from 3,5-dibromobenzoic acid methyl ester 4 as detailed in Fig. 2. Reduction of ester 4 with DIBAL-H gave alcohol 5 (93% crude) and subsequent protection of the hydroxyl group with the TBS group gave silyl ether 6 (∼100%). Treatment of compound 6 with 1.05 equivalents of n-BuLi and paraformaldehyde gave alcohol 7 (46%) and the newly formed hydroxyl group was protected as the MOM ether 8 (90%). The remaining bromo substituent in compound 8 was replaced by a trifluoromethyl ketone group using n-BuLi and 1-trifluoroacetyl piperidine yielding compound 9 (82%). Trifluormethyl ketone 9 was converted into the oxime 10 (64%), tosylated to give compound 11 (85%) and then transformed into the trifluoromethyl diazirine 12 in the standard manner (liquid NH3 followed by Et3N, I2; 88%). After removal of the TBS group using TBAF (94%), trifluoromethyl diazirine 13 was converted by MnO2 oxidation followed by a Bestmann–Ohira reaction to TPDYNE 14 (80% for the two steps). Removal of the MOM protecting group using HCl generated in situ from acetyl chloride in MeOH yielded TPDYNE 3a (88%). Treatment of compound 3a with CBr4/PPh3 converted it to TPDYNE 3b (∼100%). Treatment of compound 3a with I2, PPh3 and imidazole gave TDPYNE 3c (74%). Reaction of compound 3c with NaN3 produced TPDYNE 3d (78%) and reduction of TPDYNE 3d with PPh3 yielded TPDYNE 3e (50%).
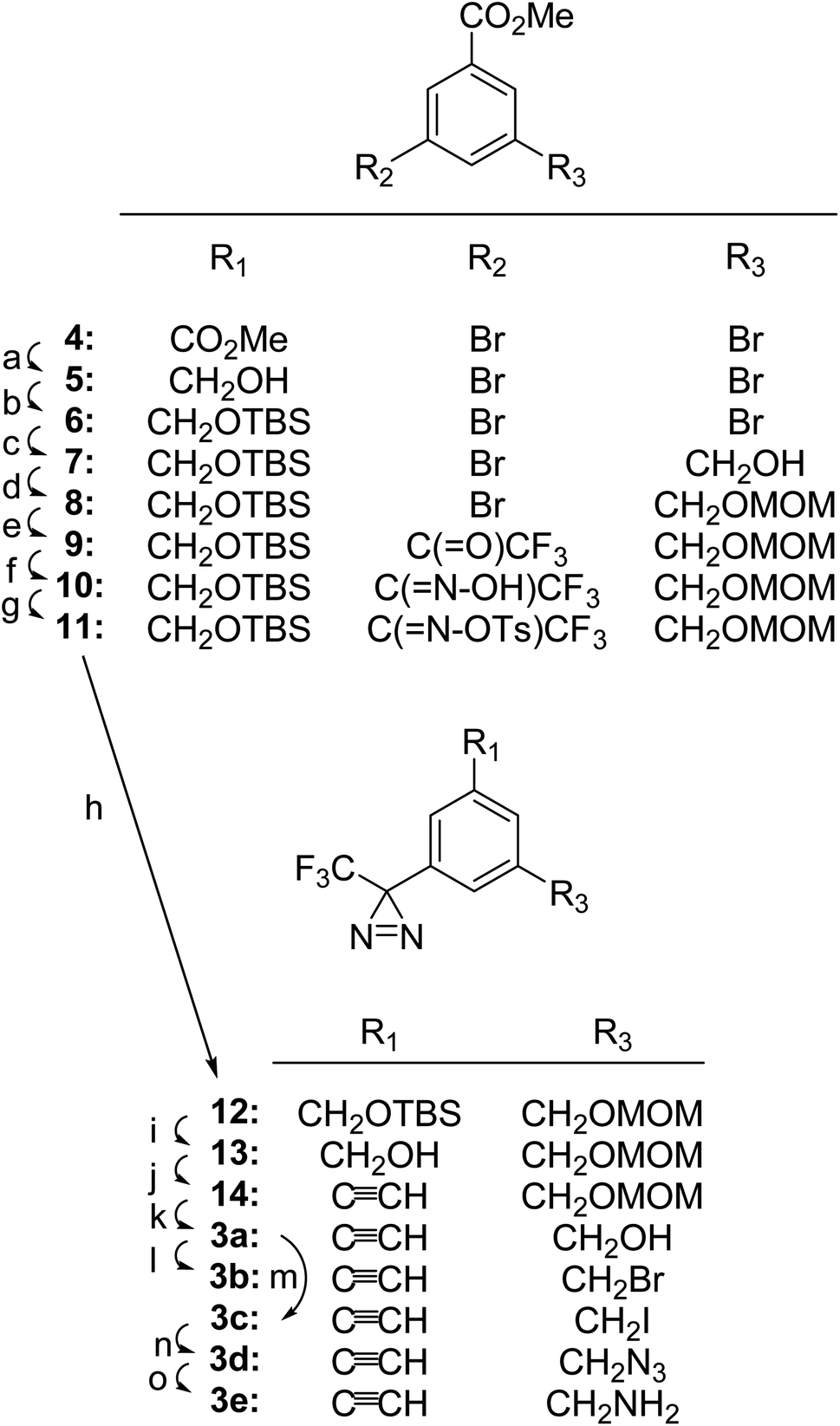 | ||
| Fig. 2 Synthetic route for the conversion of 3,5-dibromobenzoate (4) to TPDYNES 3a–3e. Reagents and yields: (a) CH2Cl2, DIBAL-H in hexanes, −45 °C then −20 °C, 2 h (93% crude); (b) TBSCl, imidazole, DMF, 23 °C, 16 h (∼100%); (c) (i): THF, 1.05 eq. n-BuLi in THF, −78 °C, 1 h; (ii): paraformaldehyde, THF, −78 °C then 23 °C 20 h (46%); (d) MOMCl, i-(Pr2)NEt, CH2Cl2, 23 °C, 16 h (90%); (e) (i): n-BuLi in THF, THF, −78 °C, 1 h; (ii) 1-trifluoroacetyl piperidine in THF, −78 °C, 1.5 h (82%); (f) HONH2·HCl, EtOH/Pyr, reflux, 16 h (64%); (g) Et3N, TsCl, CH2Cl2, 0 °C, 2 h (85%); (h) (i): liq. NH3, CH2Cl2, −78 °C to 23 °C, 16 h; (ii): MeOH, Et3N, I2, 10 min (88%); (i) TBAF, THF, 23 °C, 16 h (94%); (j) (i): MnO2, THF, 23 °C, 4 h; (ii): MeOH/THF, K2CO3, dimethyl (1-diazo-2-oxo-propyl)phosphonate, 23 °C, 16 h under N2 (80%); (k) MeOH, 5–7% HCl, 23 °C, 2 h (88%); (l) CBr4, PPh3, CH2Cl2, 23 °C, 1 h (∼100%); (m) PPh3, imidazole, I2, CH2Cl2, 23 °C, 1 h (74%); (n) NaN3, DMF, 70 °C, 3 h (78%); (o) PPh3, THF/H2O, 23 °C, 16 h (50%). | ||
As described below, we found that TPDYNE 3b or 3c, unlike the analogous TPD compounds, could not be attached in an ether linkage to a hydroxyl group in an acceptable yield. To overcome this difficulty a synthetic route to a TPDYNE precursor from which the TPDYNE group could be formed after the precursor is attached to a hydroxyl group was developed. Fig. 3 shows the synthetic route for the TPDYNE 17. Compound 7 is an intermediate in the synthesis of TPDYNE precursor 17 and it was obtained using a different method from the one shown for its preparation in Fig. 2. This alternate method for obtaining compound 7 does not require the use of n-BuLi when the method is carried out on a large scale. Accordingly, dimethyl 5-bromoisophthalate 15 was reduced to diol 16 using DIBAL-H (93%). Using 1 equivalent of TBSCl, the mono silylated alcohol 7 was obtained in 36% yield. Both unreacted diol 16 and the disilyated diol also formed in the reaction can be recycled if desired to obtain additional alcohol 7. Compound 7 was converted to compound 8 (90%) and then converted to compound 13 as described in Fig. 2. Compound 13 was reacted with PPh3 and I2 to give iodinated TPDYNE precursor 17 (73%). Iodide 17 was converted into azide 18 using NaN3 (90%) for use as described later.
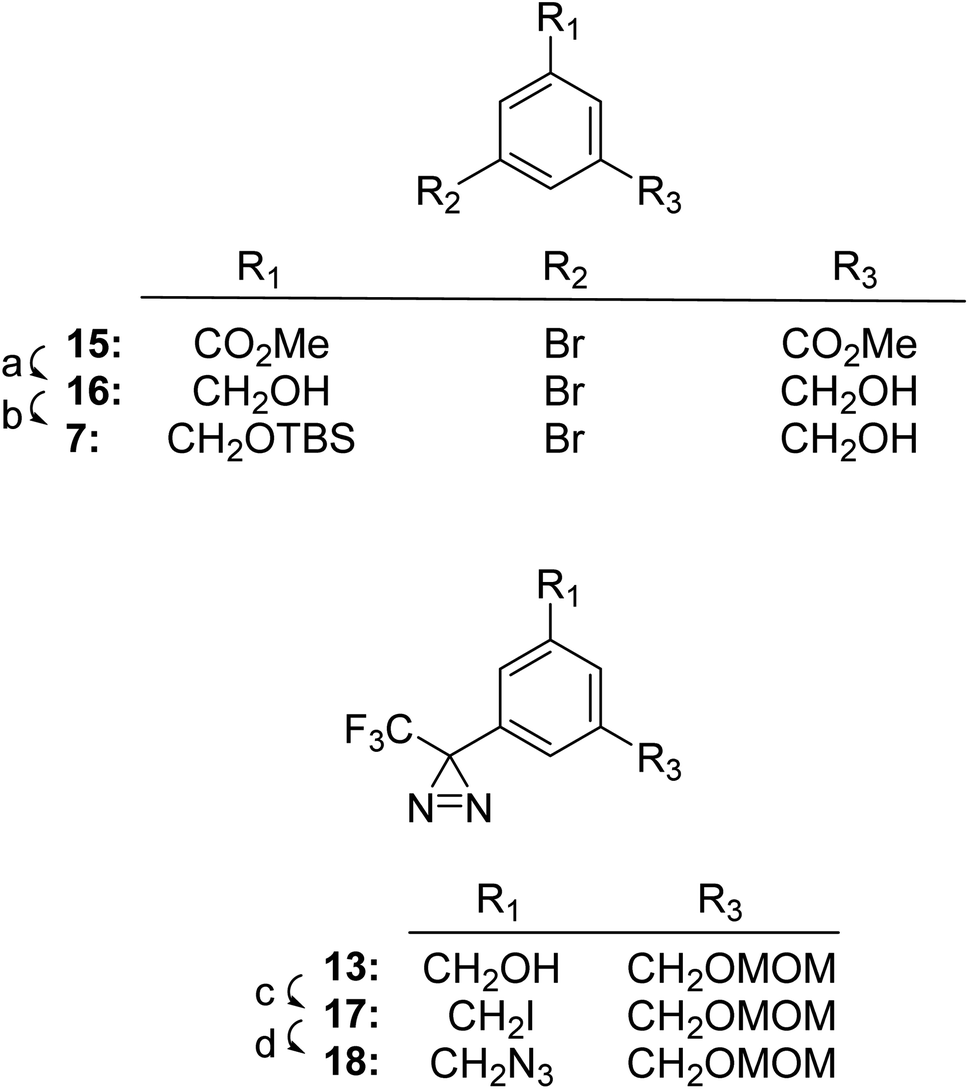 | ||
| Fig. 3 Synthetic route for the preparation of TPDYNE precursors 13–17. Reagents and yields: (a) CH2Cl2, DIBAL-H in hexanes, −45 °C then −20 °C, 4 h (93%); (b) 1 eq. TBSCl, imidazole, CH2Cl2, 23 °C, 16 h (36%); (c) PPh3, imidazole, I2, CH2Cl2, 1 h (73%); (d) NaN3, DMF, 70 °C, 3 h (90%). | ||
Reaction conditions for attachment of the TPDYNE group to compounds with an amino or carboxyl were established using steroids containing these substituents at the 17β-position (Fig. 4). Using LiOH in DMF, aminosteroid 19 was reacted with TPDYNE 3c to give steroid 20 (56%) and the MOM ether was removed using HCl in MeOH to obtain steroid 21 (47%). Steroid carboxylic acid 22 was coupled to TPDYNE 3a in an ester linkage using N,N′-dicyclohexylcarbodiimide to give steroid 23 (92%). Steroid 22 was coupled to TPDYNE 3e in an amide linkage by first reacting the steroid with N-hydroxysuccinimide and N,N′-dicyclohexylcarbodiimide to give the intermediate N-succinimidyl ester (88%) and then reacting this intermediate with TPDYNE 3e in DMF to give steroid 24 (52%).
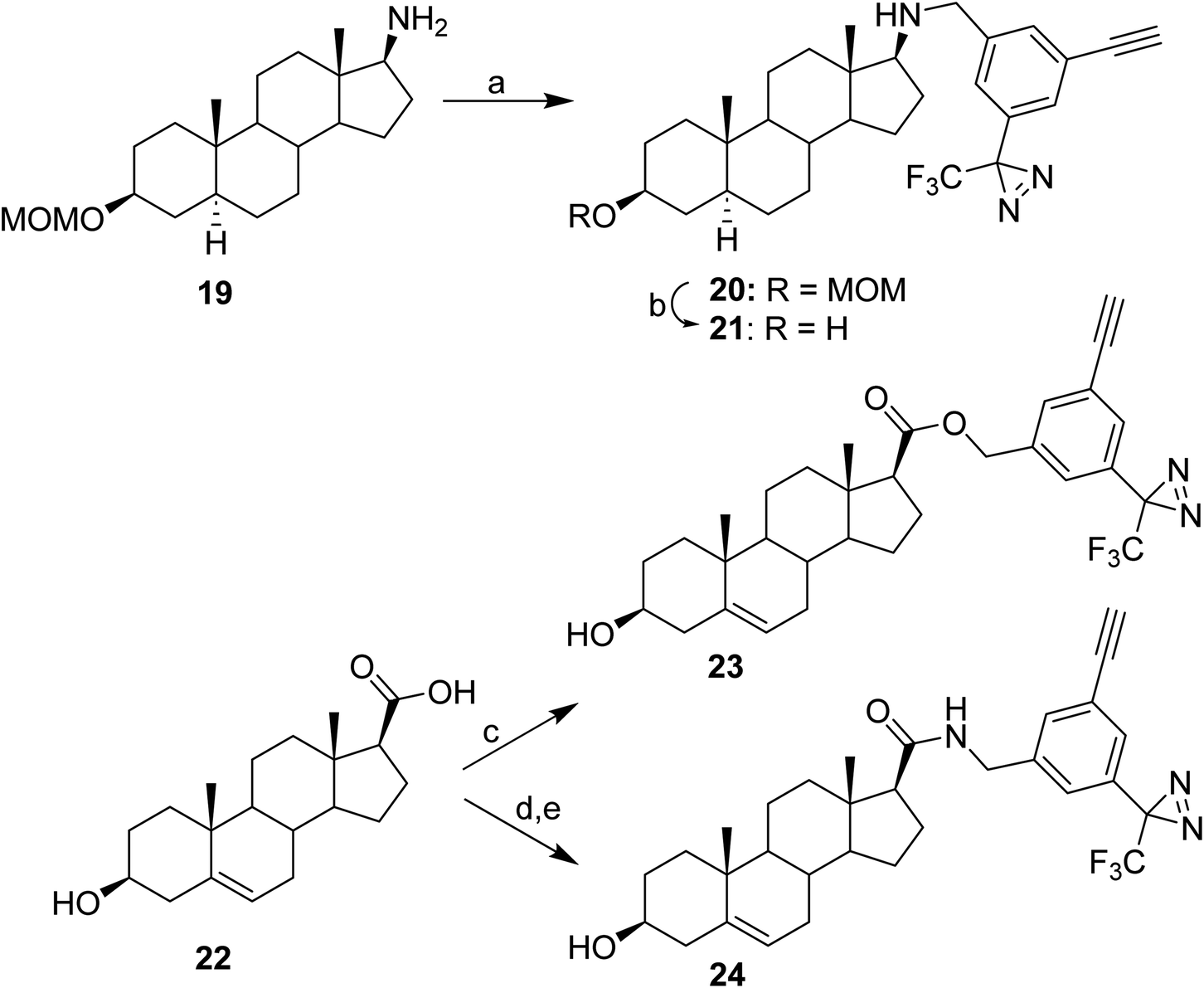 | ||
| Fig. 4 Synthetic procedures for the attachment of the TPDYNE group to carboxylic acid and amine groups using steroids as examples. Reagents and yields: (a) LiOH, DMF, 3c, 23 °C, 16 h (56%); (b) (i): MeOH, AcCl, 23 °C, 3 h; (ii): 3 N NaOH, pH ∼10 (47%); (c) DMAP, N,N′-dicyclohexylcarbodiimide, 3a, 23 °C, 16 h (88%); (d) N-hydroxysuccinimide, N,N′-dicyclohexylcarbodiimide, 23 °C, 16 h (92%); (e) 3e, DMF, 23 °C, 16 h (52%). | ||
As noted above, TPDYNE 3c could not be attached to a steroid 17β-hydroxyl group in an ether linkage in yields of more than ∼5%. Attempted reaction conditions included treating steroid 25 with either NaH or KH in THF at room temperature for 1 h followed by the addition of either 3b or 3c and then reflux for 16 h or continuing the reaction at room temperature for 16 h. Consequently, we used the TPDYNE precursor 17 to obtain a hydroxyl group modified with a TPDYNE group in an ether linkage (Fig. 5). Using KH in THF, TPDYNE precursor 17 was reacted with steroid 25 to give steroid 26 (90%). Removal of both MOM protecting groups from steroid 26 using HCl in MeOH gave steroid 27 (∼100%). Oxidation of steroid 27 with MnO2 gave steroid 28 which was immediately reacted with Bestmann–Ohira reagent to give steroid 29 (48% from steroid 27).
 | ||
| Fig. 5 Synthetic procedures for attachment of a TPDYNE precursor to a hydroxyl group and its elaboration to attached TPDYNE using a steroid example. Reagents and yields: (a) KH, THF, 15 min; (ii): reflux, overnight (90%); (b) MeOH, AcCl, 23 °C, 3 h (∼100%); (c) (i): MnO2, THF, rt, 5 h; (ii): THF/MeOH, K2CO3, dimethyl (1-diazo-2-oxo-propyl)phosphonate, 23 °C, 16 h under N2 (48% from steroid 27). | ||
In some circumstances where the nucleophilic groups in the molecule of interest cannot be used to attach a photolabeling group without losing activity a strategy is needed whereby the TPDYNE group can be attached by means other than a nucleophilic addition reaction. One such strategy if the molecule of interest contains an alkyne group would be to attach a TPDYNE azide containing reagent to it in a click reaction. TPDYNE 3d, which was prepared only as an intermediate to prepare TPDYNE 3e, could possibly be used for this purpose. However, reaction of compound 3e with itself in the click reaction would likely lead to a poor yield of the desired click reaction with the molecule of interest. To avoid this problem the TPDYNE precursor 18 (Fig. 3) can be used in a click reaction and the TPDYNE group can be formed subsequently using the same sequence (27 to 29) described in Fig. 5. Thus, TPDYNE precursor 18 was attached to steroid 30 in a click reaction (Fig. 6) to form steroid 31 (50%). The MOM group was removed using HCl in MeOH to give steroid 32 (76%). Oxidation of steroid 32 with MnO2 gave aldehyde 33 (52%) and the TPDYNE modified steroid 34 was formed from the aldehyde using the Bestmann–Ohira reagent (50%).
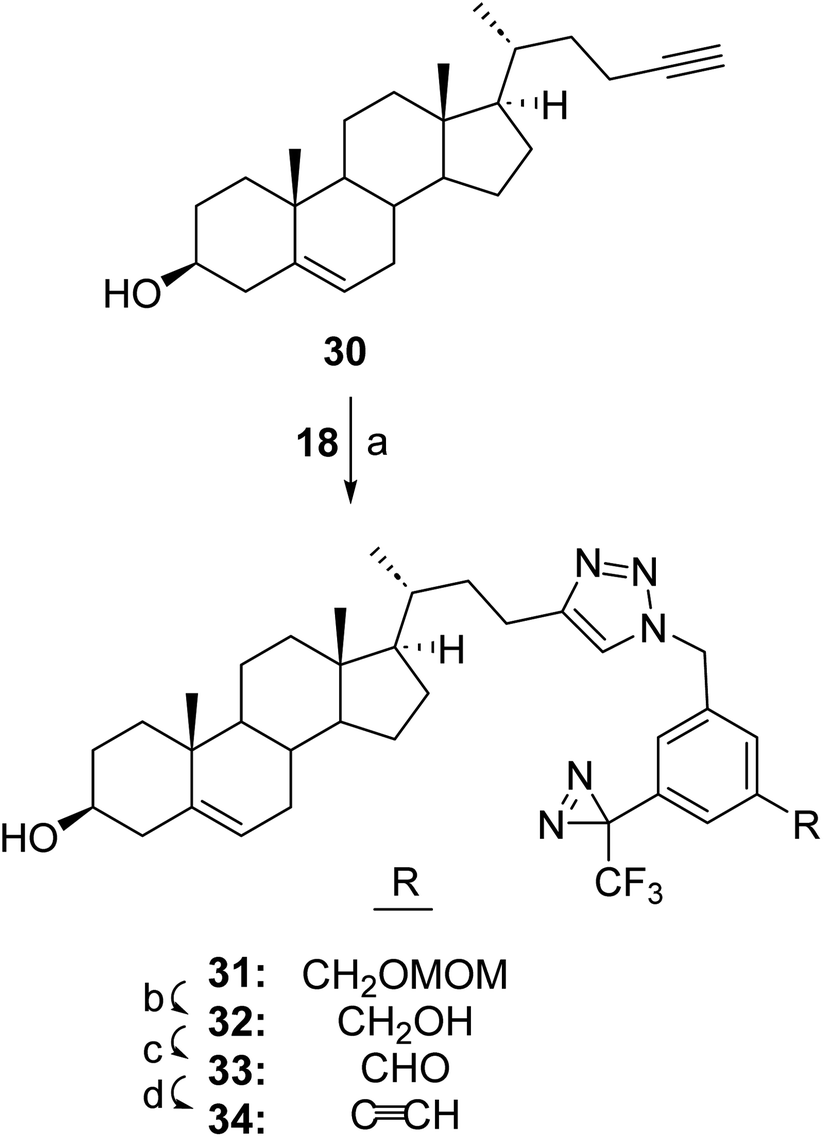 | ||
| Fig. 6 Synthetic procedures for the attachment of a TPDYNE precursor to an alkyne group and its elaboration to attached TPDYNE group using a steroid example. Reagents and yields: (a) CuSO4, sodium ascorbate, 23 °C, 72 h, (50%); (b) MeOH, AcCl, reflux, 2 h (76%); (c) MnO2, THF, 23 °C, overnight, (52%); (d) THF/MeOH, K2CO3, dimethyl (1-diazo-2-oxo-propyl)phosphonate, 23 °C, 16 h under N2 (50%). | ||
In conclusion, we report new synthetic methods for the preparation of a group of TPDYNE reagents. Additionally, we report reaction conditions for the attachment these reagents to hydroxyl, amino and carboxylic acid and alkyne groups using steroids as examples. We believe the methods reported will lead to an increased use of the TPDYNE group in chemical biology applications.
Experimental procedures
(3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)methanol (3a)
To a stirred solution of compound 14 (188 mg, 0.66 mmol) in MeOH (20 mL) was added 5–7% of HCl (generated in situ from acetyl chloride) in MeOH (10 mL) and the reaction was stirred at 23 °C for 1.5 h. Water was added and the product was extracted into CH2Cl2 (100 mL × 3). The combined extracts were washed with water (50 mL), then aqueous NaHCO3 (100 mL), dried over anhydrous Na2SO4 and the solvent removed. The residue was purified by flash column chromatography (silica gel, eluted with 10–25% of EtOAc in hexanes) to afford compound 3a (ref. 6) (188 mg, 80%): 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.24 (s, 1H), 7.14 (s, 1H), 4.66 (s, 2H), 3.12 (s, 1H), 2.09 (s, br, 1H); 13C NMR (100 MHz, CDCl3) δ 142.1, 131.3, 129.8, 129.2, 124.8, 123.3, 123.2 (q, J = 274.7 Hz), 82.1, 78.7, 64.0, 28.8 (q, J = 41.2 Hz).3-(3-(Bromomethyl)-5-ethynylphenyl)-3-(trifluoromethyl)-3H-diazirine (3b)
To a stirred solution of compound 3a (95 mg, 0.4 mmol) in CH2Cl2 (10 mL) was added CBr4 (332 mg, 1 mmol) and PPh3 (275 mg, 1.05 mmol) at 23 °C. After 1 h, the reaction solution was directly purified by flash column chromatography (silica gel, eluted with 5% of EtOAc in hexanes) to afford product 3b (ref. 6) (121 mg, ∼100%): 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 1H), 7.26 (s, 1H), 7.15 (s, 1H), 4.40 (s, 2H), 3.15 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 139.1, 133.7, 130.2, 129.9, 127.0, 123.8, 120.4 (q, J = 275 Hz), 81.6, 79.2, 31.2, 28.3 (q, J = 40.4 Hz).3-(3-Ethynyl-5-(iodomethyl)phenyl)-3-(trifluoromethyl)-3H-diazirine (3c)
To a stirred solution of compound 3a (75 mg, 0.33 mmol) in CH2Cl2 (10 mL) was added PPh3 (113 mg, 0.43 mmol), imidazole (68 mg, 1 mmol) and I2 (118 mg, 0.47 mmol). After 1 h, the reaction solution was directly purified by flash column chromatography (silica gel, eluted with 10% of EtOAc in hexanes) to afford product 3c (81 mg, 74%): 1H NMR (400 MHz, CDCl3) δ 7.55 (s, 1H), 7.21 (s, 1H), 7.13 (s, 1H), 4.37 (s, 2H), 3.16 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 140.8, 133.4, 130.2, 129.4, 126.7, 123.7, 123.1 (q, J = 275 Hz), 81.7, 79.1, 28.2 (q, J = 40.5 Hz).3-(3-(Azidomethyl)-5-ethynylphenyl)-3-(trifluoromethyl)-3H-diazirine (3d)
To a stirred solution of compound 3c (77 mg, 0.22 mmol) in DMF (4 mL) was added NaN3 (65 mg, 1 mmol). The reaction was heated to 70 °C for 3 h. Water (10 mL) was added and the product was extracted into EtOAc (2 × 100 mL). The combined extracts were washed with brine (2 × 30 mL), dried over anhydrous Na2SO4 and the solvent removed. The residue was purified by flash column chromatography (silica gel, 5% EtOAc in hexanes) to afford product 3d (45 mg, 78%): 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.30 (s, 1H), 7.10 (s, 1H), 4.37 (s, 2H), 3.17 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 137.0, 132.5, 132.2, 129.8, 126.0, 123.8, 123.2 (q, J = 275 Hz), 81.7, 79.2, 53.7, 28.3 (q, J = 41 Hz).(3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)methanamine (3e)
To a stirred solution of compound 3d (45 mg, 0.17 mmol) in THF/water (10 mL/1 mL) was added PPh3 (445 mg, 1.7 mmol) at 23 °C. The reaction was stirred at 23 °C for 16 h. Water (10 mL) was added and the product was extracted into EtOAc (2 × 100 mL). The combined extracts were dried over anhydrous Na2SO4 and the solvent removed. The residue was purified by flash column chromatography (silica gel, eluted with 50% EtOAc in CH2Cl2) to afford product 3e (20 mg, 50%): 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.22 (s, 1H), 7.11 (s, 1H), 3.86 (s, 2H), 3.12 (s, 1H), 1.80 (s, br, 2H); 13C NMR (100 MHz, CDCl3) δ 144.2, 132.0, 129.7, 128.6, 125.4, 123.3 (q, J = 275 Hz), 123.2, 82.3, 78.5, 45.6, 28.4 (q, J = 41 Hz).(3,5-Dibromophenyl)methanol (5)
To a stirred solution of methyl 3,5-dibromobenzoate (4, 12.5 g, 42.5 mmol) in CH2Cl2 (150 mL) was added DIBAL-H (1.0 M in heptane, 85 mL, 85 mmol) at −45 °C and then raised to −20 °C for 2 h. 1 N HCl was slowly added at 23 °C and stirring continued for 1 h. The product was extracted into CH2Cl2 (2 × 150 mL). The combined extracts were washed with aqueous NaHCO3, water, dried over anhydrous Na2SO4 and the solvent was removed. The residue containing product 5 (11.1 g, 93% crude) was converted to compound 6 without purification or characterization.tert-Butyl((3,5-dibromobenzyl)oxy)dimethylsilane (6)
To a stirred solution of crude compound 5 (11.1 g, 41.4 mmol) in DMF (100 mL) was added tert-butyldimethylsilyl chloride (8.1 g, 53.8 mmol) and imidazole (107 mmol, 7.28 g) at 23 °C and the reaction was stirred for 16 h. Aqueous NaHCO3 was added and the product was extracted into EtOAc (350 mL). The EtOAc was washed with brine (3 × 100 mL), dried over anhydrous Na2SO4 and the solvent removed. The residue was purified by flash column chromatography (silica gel, eluted with 2% of EtOAc in hexanes) to afford product 6 (15.8 g, ∼100%): 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1H), 7.40 (s, 2H), 4.68 (s, 2H), 0.96 (s, 9H), 0.12 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 146.5, 132.4 (2 × C), 127.6 (2 × C), 122.8, 63.5, 25.9 (3 × C), 18.4, −5.3 (2 × C).(3-Bromo-5-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)methanol (7)
To a stirred solution of compound 6 (16.1 g, 42.4 mmol) in THF (200 mL) was slowly added n-BuLi (2.5 M in THF, 17.8 mL, 44.5 mmol) at −78 °C. After 1 h, paraformaldehyde (12 g, 400 mmol) was added, and the mixture was warmed to 23 °C for 20 h. Aqueous NaHCO3 (150 mL) was added and the product was extracted into EtOAc (2 × 150 mL). The combined extracts were dried over anhydrous Na2SO4, removed and the residue was purified by flash column chromatography (silica gel, eluted with 25% EtOAc in hexanes) to afford the product 7 (6.49 g, 46%): 1H NMR (400 MHz, CDCl3) δ 7.40 (s, 2H), 7.21 (s, 1H), 4.71 (s, 2H), 4.67 (s, 2H), 0.96 (s, 9H), 0.13 (d, J = 1.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 143.9, 142.9, 128.3, 128.1, 122.8, 122.5, 64.4, 64.1, 25.9 (3 × C), 18.4, −5.3 (2 × C).Second procedure for the preparation of compound 7 from compound 16
To a stirred solution of compound 16 (14.2 g, 65 mmol) in DMF (200 mL) was added tert-butyldimethylsilyl chloride (9.75 g, 65 mmol) and imidazole (8.84 g, 130 mmol) at 23 °C. After 16 h, the solvent was removed and the residue was purified by flash column chromatography (silica gel, eluted with 10% EtOAc in hexanes) to give product 7 (7.8 g, 36%): 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1H), 7.32 (s, 1H), 7.14 (s, 1H) 4.67 (s, 2H), 4.54 (s, 2H), 3.10–3.04 (m, 1H), 0.95 (s, 9H), 0.11 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 143.7, 142.9, 128.1, 128.0, 122.7, 122.4, 64.1, 64.0, 25.9 (3 × C), 18.3, −5.4(2 × C).((3-Bromo-5-((methoxymethoxy)methyl)benzyl)oxy)(tert-butyl)dimethylsilane (8)
To a stirred solution of compound 7 (6.49 g, 19.7 mmol) in CH2Cl2 (40 mL) was added (i-Pr)2NEt (8.4 mL, 60 mmol) and methoxymethyl chloride (3.04 mL, 40 mmol) at 23 °C. After 16 h, aqueous NaHCO3 (100 mL) was added and the product was extracted into EtOAc (2 × 150 mL). The combined extracts were dried over anhydrous Na2SO4, the solvent removed and the residue was purified by flash column chromatography (silica gel, eluted with 10% of EtOAc in hexanes) to afford product 8 (6.65 g, 90%): 1H NMR (400 MHz, CDCl3) δ 7.39 (s, 2H), 7.21 (s, 1H), 4.70 (s, 4H), 4.55 (s, 2H), 3.41 (s, 3H), 0.94 (s, 9H), 0.10 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 143.8, 140.0, 129.0, 128.2, 123.7, 122.4, 95.7, 68.2, 64.1, 55.4, 25.9 (3 × C), 18.3, −5.3 (2 × C).1-(3-(((tert-Butyldimethylsilyl)oxy)methyl)-5-((methoxymethoxy)methyl)phenyl)-2,2,2-trifluoroethan-1-one (9)
To a stirred solution of compound 8 (1.0 g, 2.67 mmol) in THF (200 mL) was added n-BuLi (1.6 M in THF, 2.2 mL, 3.5 mmol) at −78 °C. After 1 h, 1-trifluoroacetyl piperidine (1.62 g, 9 mmol) in THF (8 mL) was added through an addition funnel over 45 min. After addition, the reaction was stirred at −78 °C for 90 min. Aqueous NaHCO3 was added at −78 °C and the reaction was warmed to 23 °C for 30 min. The product was extracted into EtOAc (150 mL × 2). The combined extracts were dried over anhydrous Na2SO4, the solvent removed and the residue was purified by flash column chromatography (silica gel, eluted with 5–10% of EtOAc in hexanes) to afford product 9 (855 mg, 82%): 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1H), 7.94 (s, 1H), 7.65 (s, 1H), 4.81 (s, 2H), 4.72 (s, 2H), 4.65 (s, 2H), 3.41 (s, 3H), 0.95 (s, 9H), 0.11 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 180.6 (q, J = 35.1 Hz), 143.1, 139.4, 132.0, 130.1, 127.6, 126.7, 118.1 (q, J = 291 Hz), 96.0, 68.3, 63.9, 55.4, 25.8 (3 × C), 18.3, −5.4 (2 × C).1-(3-(((tert-Butyldimethylsilyl)oxy)methyl)-5-((methoxymethoxy)methyl)phenyl)-2,2,2-trifluoroethan-1-one oxime (10)
To a stirred solution of compound 9 (871 mg, 2.56 mmol) in EtOH (15 mL) and pyridine (15 mL) was added hydroxylamine hydrochloride at 23 °C. The reaction was refluxed for 16 h. The solvent was removed and the residue was purified by flash column chromatography (silica gel, eluted with 10–20% of EtOAc in hexanes) to afford oxime 10 (583 mg, 64%). The major isomer had: 1H NMR (400 MHz, CDCl3) δ 9.50 (s, 1H), 7.43 (s, 2H), 7.39 (s, 1H), 4.80 (s, 2H), 4.75 (s, 2H), 4.66 (s, 2H), 3.44 (s, 3H), 0.95 (s, 9H), 0.12 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 147.4 (q, J = 32.1 Hz), 142.1, 138.2, 127.3, 126.6, 126.4, 125.5, 122.0 (q, J = 274 Hz), 95.7, 68.8, 64.4, 55.4, 25.8 (3 × C), 18.3, −5.3 (2 × C).1-(3-(((tert-Butyldimethylsilyl)oxy)methyl)-5-((methoxymethoxy)methyl)phenyl)-2,2,2-trifluoroethan-1-one O-tosyl oxime (11)
Tosyl chloride (573 mg, 3 mmol) was added to a stirred, ice cold solution of oxime 10 (583 mg, 1.43 mmol), Et3N (0.84 mL, 6 mmol), and DMAP (30 mg) in CH2Cl2 (20 mL) and the reaction was stirred at 0 °C for 2 h. Aqueous NaHCO3 (100 mL) was added and the product was extracted into CH2Cl2 (2 × 100 mL). The combined extracts were dried over anhydrous Na2SO4, the solvent removed and the residue was purified by flash column chromatography (silica gel, eluted with 10–15% of EtOAc in hexanes) to afford product 11 (683 mg, 85%). The major isomer had: 1H NMR (400 MHz, CDCl3) δ 7.89–7.88 (m, 2H), 7.46–7.25 (m, 5H), 4.76 (s, 2H), 4.70 (s, 2H), 4.61 (s, 2H), 3.39 (s, 3H), 2.45 (s, 3H), 0.93 (s, 9H), 0.09 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 154.2 (q, J = 32.8 Hz), 146.0, 142.7, 138.9, 131.2, 129.8, 129.7, 129.2, 129.0, 128.1, 125.8, 124.8, 124.6, 120.9 (q, J = 277 Hz), 95.8, 73.2, 64.0, 55.4, 25.8 (3 × C), 21.7, 18.2, −5.4 (2 × C).3-(3-(((tert-Butyldimethylsilyl)oxy)methyl)-5-((methoxymethoxy)methyl)phenyl)-3-(trifluoromethyl)-3H-diazirine (12)
Condensed anhydrous ammonia (10 mL) was added to a stirred, −78 °C solution of compound 11 (683 mg, 1.22 mmol) in CH2Cl2 (20 mL) and the reaction was slowly warmed to 23 °C and stirring continued for 16 h. Water was added and the product was extracted into CH2Cl2 (2 × 100 mL). The CH2Cl2 was dried over anhydrous Na2SO4 and removed to give the crude diaziridine intermediate which was used without purification. The crude diaziridine was dissolved in stirred MeOH (20 mL) and Et3N (1 mL) was added. MeOH saturated with I2 was added in portions until a brown color persisted. Excess I2 was quenched with aqueous Na2S2O3 (10 mL) and water (20 mL) was added. The product was extracted into CH2Cl2 (100 mL × 2). The combined extracts were dried over anhydrous Na2SO4, the solvent removed and the residue was purified by flash column chromatography (silica gel, eluted with 15% of EtOAc in hexanes) to afford diazirine 12 (435 mg, 88%): 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.20 (s, 1H), 7.02 (s, 1H), 4.76 (s, 2H), 4.71 (s, 2H), 4.59 (s, 2H), 3.42 (s, 3H), 0.97 (s, 9H), 0.12 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 142.8, 138.9, 129.4, 126.2, 123.9, 123.5 (q, J = 275 Hz), 123.2, 95.9, 68.6, 64.2, 55.4, 29.7 (q, J = 40.4 Hz) 25.8 (3 × C), 18.3, −5.4 (2 × C).(3-((Methoxymethoxy)methyl)-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)methanol (13)
To a stirred solution of compound 12 (435 mg, 1.08 mmol) in THF (15 mL) was added tetra n-butyl ammonium fluoride (1.0 M in THF, 2 mL, 2 mmol) at 23 °C. After 16 h, THF was removed and the residue was purified by flash column chromatography (silica gel, eluted with 25–35% of EtOAc in hexanes) to afford product 13 (293 mg, 94%): 1H NMR (400 MHz, CDCl3) δ 7.39 (s, 1H), 7.13 (s, 1H), 7.11 (s, 1H), 4.69 (s, 2H), 4.66 (s, 2H), 4.57 (s, 2H), 3.40 (s, 3H), 2.67 (s, br, 1H); 13C NMR (100 MHz, CDCl3) δ 142.2, 139.2, 129.6, 127.1, 124.6, 123.9, 123.4 (q, J = 274.6 Hz), 95.9, 68.5, 64.2, 55.4, 28.5 (q, J = 40.4 Hz).3-(3-Ethynyl-5-((methoxymethoxy)methyl)phenyl)-3-(trifluoromethyl)-3H-diazirine (14)
Compound 13 (293 mg, 1.01 mmol) was added to a stirred suspension of activated MnO2 (870 mg, 10 mmol) in THF (15 mL) at 23 °C for 4 h. Anhydrous MeOH (10 mL) was added, followed by K2CO3 (1.38 g, 10 mmol) and dimethyl (1-diazo-2-oxo-propyl)phosphonate (0.3 mL, 2 mmol) and stirring was continued for 16 h at 23 °C under N2. The crude mixture was filtered through a Celite pad and the pad was washed with CH2Cl2 (100 mL). The CH2Cl2 was removed and the residue was re-dissolved in CH2Cl2 (50 mL) which was washed with aqueous NaHCO3 (20 mL), brine (20 mL), dried over anhydrous Na2SO4 and the solvent was removed. The residue was purified by flash column chromatography (silica gel, eluted with 5–10% of EtOAc in hexanes) to afford diazirine 14 (188 mg, 80%): 1H NMR (400 MHz, CDCl3) δ 7.51 (s, 1H), 7.26 (s, 1H), 7.14 (s, 1H), 4.70 (s, 2H), 4.56 (s, 2H), 3.39 (s, 3H), 3.12 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 139.5, 132.2, 129.7, 129.2, 125.7, 123.3, 120.5 (q, J = 274.6 Hz), 96.0, 82.1, 78.7, 68.0, 55.5, 28.3 (q, J = 46.4 Hz).(5-Bromo-1,3-phenylene)dimethanol (16)
To a stirred solution of dimethyl 5-bromoisophthalate (15, 25 g, 83 mmol) in CH2Cl2 (100 mL) was added DIABL-H (1 M in heptane, 400 mL, 400 mmol) at −45 °C. After addition, the reaction was stirred at −20 °C for 4 h. EtOEt was added and the reaction was brought to 0 °C. Water (16 mL) was slowly added followed by 15% NaOH solution (16 mL) and water (40 mL). The mixture was warmed to 23 °C and stirred for 15 min. MeOH (100 mL) was added and stirring continued for 15 min. After filtration to remove solids, the solvent was removed and the residue was purified by flash column chromatography (silica gel, eluted with 5% methanol in CH2Cl2) to give product 16 (16.8 g, 93%): 1H NMR (400 MHz, CD3OD): δ 7.37 (s, 2H), 7.22 (s, 1H), 4.53 (s, 4H); 13C NMR (100 MHz, CD3OD) δ 145.4, 129.5 (2 × C), 124.9 (2 × C), 123.4, 64.4 (2 × C).3-(3-(Iodomethyl)-5-((methoxymethoxy)methyl)phenyl)-3-(trifluoromethyl)-3H-diazirine (17)
To a stirred solution of compound 13 (930 mg, 3.21 mmol) in CH2Cl2 (15 mL) was added PPh3 (1.26 g, 4.8 mmol), imidazole (435 mg, 6.4 mmol) and then I2 (1.22 g, 4.8 mmol added in eight portions). After 1 h, the reaction solution was directly purified by flash column chromatography (silica gel, eluted with 5% EtOAc in hexanes) to give product 17 (940 mg, 73%): 1H NMR (400 MHz, CDCl3) δ 7.43 (s, 1H), 7.11 (s, 1H), 7.07 (s, 1H), 4.71 (s, 2H), 4.57 (s, 2H), 4.41 (s, 2H), 3.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 140.5, 139.9, 130.0, 129.0, 125.7, 124.8, 123.3 (q, J = 275 Hz), 95.9, 68.1, 55.4, 28.4 (q, J = 40.4 Hz), 3.5.3-(3-(Azidomethyl)-5-((methoxymethoxy)methyl)phenyl)-3-(trifluoromethyl)-3H-diazirine (18)
To a stirred solution of compound 17 (250 mg, 0.63 mmol) in DMF (10 mL) was added NaN3 (130 mg, 2 mmol). The reaction was heated to 70 °C and stirred for 3 h. Water (20 mL) was added and the product was extracted into EtOAc (2 × 100 mL). The combined extracts were washed with brine (30 mL × 2),dried over anhydrous Na2SO4, the solvent removed and the residue was purified by flash column chromatography (silica gel, eluted with 5% EtOAc in hexanes) to give product 18 (180 mg, 90%): 1H NMR (400 MHz, CDCl3) δ 7.37 (s, 1H), 7.15 (s, 1H), 7.06 (s, 1H), 4.71 (s, 2H), 4.60 (s, 2H), 4.37 (s, 2H), 3.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 139.9, 136.7, 130.0, 128.3, 125.3, 125.1, 123.3 (q, J = 275 Hz), 95.9, 68.2, 55.4, 54.1, 28.5 (q, J = 40.4 Hz).(17β)-N-(3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)-3-(methoxymethoxy)-5α-androstan-17β-amine (20)
To a stirred solution of the 3-methoxymethoxy ether of 17β-amino-5α-androstan-3β-ol (ref. 8) (19, 65 mg, 0.193 mmol) in DMF (4 mL) was added LiOH·H2O (101 mg, 0.6 mmol) and 4 Å molecular sieves (150 mg) at 23 °C. The suspension was stirred for 30 min, compound 3c (81 mg, 0.231 mmol) was added and the reaction was stirred at 23 °C for 16 h. The mixture was directly purified by flash column chromatography (silica gel, 10–20% of EtOAc in hexanes) to afford steroid 20 (60 mg, 56%): 1H NMR (400 MHz, CDCl3) δ 7.52 (s, 1H), 7.21 (s, 1H), 7.15 (s, 1H), 4.69 (s, 2H), 3.83 (dd, J = 14.0, 6.6 Hz, 2H), 3.53–3.39 (m, 1H), 3.37 (s, 3H), 3.12 (s, 1H), 2.50 (t, J = 8.2 Hz, 1H), 2.03–0.60 (m, 23H), 0.83 (s, 3H), 0.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 142.7, 132.6, 129.4, 128.5, 126.1, 122.9, 120.6 (q, J = 275 Hz), 94.5, 82.5, 78.3, 76.2, 68.2, 55.1, 54.5, 53.1, 51.7, 44.9, 42.9, 38.0, 37.0, 35.7, 35.4, 35.2, 31.8, 29.5, 28.6, 28.4 (q, J = 40 Hz), 23.7, 20.9, 12.2, 11.9.17β-((3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)amino)-5α-androstan-3β-ol (21)
To a stirred solution of steroid 20 (60 mg, 0.108 mmol) in MeOH (10 mL) was added acetyl chloride (1 mL) at 23 °C. After 3 h, aqueous NaOH was slowly added until pH 11. The product was extracted into CH2Cl2 (100 mL × 2). The combined extracts were dried over anhydrous Na2SO4, the solvent was removed and the residue was purified by flash column chromatography (silica gel, eluted with 25% of EtOAc in hexanes) to afford product 21 (314 mg, 47%): 1H NMR (400 MHz, CDCl3) δ 7.51 (s, 1H), 7.21 (s, 1H), 7.14 (s, 1H), 3.80 (dd, J = 14.0, 7.0 Hz, 2H), 3.60–3.56 (m, 1H), 3.12 (s, 1H), 2.50 (t, J = 8.5 Hz, 1H), 2.00–0.61 (m, 24H), 0.82 (s, 3H), 0.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 142.6, 132.7, 129.4, 128.5, 126.1, 122.9, 120.6 (q, J = 275 Hz), 82.5, 78.3, 71.2, 68.2, 54.5, 53.2, 51.7, 44.8, 42.9, 38.1, 38.0, 37.0, 35.5, 35.4, 31.7, 31.5, 29.5, 28.6, 28.0, 23.7, 20.9, 12.3, 11.9.3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl-3β-hydroxyandrost-5-ene-17β-carboxylate (23)
To a stirred solution of steroid 22 (45 mg, 0.142 mmol), compound 3a (41 mg, 0.17 mmol) and 4-dimethylaminopyridine (35 mg, 0.285 mmol) in CH2Cl2 (10 mL) was added N,N′-dicyclohexylcarbodiimide (35 mg, 0.17 mmol) at 23 °C. After 16 h, the CH2Cl2 was removed and the residue was purified by flash column chromatography (silica gel, 30% of EtOAc in hexanes) to afford product 23 (67 mg, 88%): 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.26 (s, 1H), 7.18 (s, 1H), 5.34–5.29 (m, 1H), 5.13 (q, J = 13.3 Hz, 2H), 3.55–3.47 (m, 1H), 3.14 (s, 1H), 2.43 (t, J = 9.3 Hz, 1H), 2.32–0.68 (m, 20H), 1.00 (s, 3H), 0.64 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 173.7, 140.8, 137.8, 132.5, 129.8, 128.5, 126.0, 123.4, 123.2 (q, J = 275 Hz), 121.3, 81.9, 78.9, 71.6, 64.5, 56.2, 55.1, 50.0, 44.2, 42.2, 38.2, 37.2, 36.5, 31.9, 31.7, 31.5, 29.7 (q, J = 41 Hz), 24.5, 23.6, 20.8, 19.3, 13.3.N-(3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)-3β-hydroxyandrost-5-ene-17β-carboxamide (24)
To a solution of 3β-hydroxyandrost-5-ene-17β carboxylic acid (ref. 9) (22, 130 mg, 0.41 mmol) in THF (10 mL) was added N-hydroxysuccinimide (115 mg, 1 mmol) and N,N′-dicyclohexylcarbodiimide (206 mg, 1 mmol at 23 °C). After 16 h, the mixture was filtered through a fritted funnel and washed with cold EtOAc (20 mL). The EtOAc was removed and the residue was purified by flash column chromatography (silica gel, 40% EtOAc in hexanes) to afford the previously reported N-succinimidyl ester9 (156 mg, 92%): 1H NMR (400 MHz, CDCl3) δ 5.37–5.35 (m, 1H), 3.54–3.53 (m, 1H), 2.84 (d, J = 1.7 Hz, 4H), 2.65 (t, J = 9.4 Hz, 1H), 2.35–0.96 (m, 22H), 1.03 (s, 3H), 0.85 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.3 (2 × C), 140.9, 121.2, 71.6, 56.3, 52.4, 49.9, 45.1, 42.2, 37.6, 37.2, 36.5, 32.0, 31.7, 31.6, 25.6 (2 × C), 24.6, 24.0, 21.0, 19.4, 12.8.To a stirred solution of previously reported N-succinimidyl ester of steroid 22 (22 mg, 0.053 mmol) in DMF (4 mL) was added compound 3e (14 mg, 0.0586 mmol) at 23 °C. After 16 h, DMF was removed and the residue was purified by flash column chromatography (silica gel, 45% of EtAOc in hexanes) to afford product 24 (15 mg, 52%): 1H NMR (400 MHz, CDCl3) δ 7.45 (s, 1H), 7.22 (s, 1H), 7.07 (s, 1H), 5.72 (t, J = 5.2 Hz, 1H), 5.36–5.34 (m, 1H), 4.56–4.32 (m, 2H), 3.56–3.47 (m, 1H), 3.13 (s, 1H), 2.33–0.86 (m, 20H), 1.02 (s, 3H), 0.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 172.9, 140.8, 140.4, 132.4, 129.9, 129.0, 125.7, 123.5, 121.3, 78.8, 71.7, 57.1, 56.5, 50.1, 43.9, 42.7, 42.2, 38.6, 37.2, 36.5, 33.9, 31.9, 31.8, 31.6, 29.7, 25.6, 24.9, 24.5, 23.7, 20.9, 19.4, 13.1.
3-(3-(((-3β-(Methoxymethoxy)-5α-androstan-17β-yl)oxy)methyl)-5-((methoxymethoxy)methyl)phenyl)-3-(trifluoromethyl)-3H-diazirine (26)
To a stirred solution of the 3-methyoxymethoxy ether of 5α-androstane-3β,17β-diol (25, 212 mg, 0.63 mmol) in THF (20 mL) was added KH (∼1 mmol in mineral oil) at 23 °C. The mixture was stirred for 15 min and then compound 17 was added (470 mg, 1.2 mmol) in THF (5 mL). The mixture was refluxed at 60 °C overnight. Water (20 mL) was added and the product was extracted into EtOAc (2 × 100 mL). The combined extracts were dried over anhydrous Na2SO4, the solvent removed and the residue was purified by flash column chromatography (silica gel, eluted with 10% EtOAc in hexanes) to give product 26 (350 mg, 90%): 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.13 (s, 1H), 7.08 (s, 1H), 4.72 (s, 2H), 4.69 (s, 2H), 4.59 (s, 2H), 4.54 (s, 2H), 3.58–3.43 (m, 1H), 3.42 (s, 3H), 3.38 (s, 3H), 0.83 (s, 3H), 0.83 (s, 3H), 1.95–0.76 (m, 23H); 13C NMR (100 MHz, CDCl3) δ 140.7, 139.0, 129.3, 127.5, 124.4, 124.3, 123.5 (q, J = 275 Hz), 95.9, 94.5, 88.7, 76.2, 70.7, 68.5, 55.4, 55.1, 54.5, 51.1, 44.9, 43.1, 37.9, 37.0, 35.7, 35.2, 35.2, 31.6, 28.6, 28.6, 28.2 (q, J = 41.5 Hz), 27.8, 23.4, 20.8, 12.2, 11.8.17β-((3-(Hydroxymethyl)-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)oxy)-5α-androstan-3β-ol (27)
To a stirred solution of steroid 26 (350 mg, 0.57 mmol) in MeOH (20 mL) was added acetyl chloride (3 mL) at 23 °C. After 3 h, water was added and the product was extracted into CH2Cl2 (2 × 50 mL). The combined extracts were dried over anhydrous Na2SO4, the solvent removed and the residue was purified by flash column chromatography (silica gel, eluted with 25–50% EtOAc in hexanes) to give product 27 (300 mg, ∼100%): 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 1H), 7.09 (s, 1H), 7.06 (s, 1H), 4.67 (s, 2H), 4.52 (s, 2H), 3.57–3.55 (m, 1H), 3.39–3.35 (m, 1H), 0.81 (s, 3H), 0.81 (s, 3H), 2.01–0.61 (m, 24H); 13C NMR (100 MHz, CDCl3) δ 141.9, 140.7, 129.4, 126.6, 124.1, 123.5, 123.4 (q, J = 275 Hz), 88.8, 71.3, 70.8, 64.5, 54.4, 51.1, 44.8, 43.1, 38.0, 37.9, 37.0, 35.5, 35.2, 31.6, 31.4, 28.5, 28.2 (q, J = 41.5 Hz), 27.8, 23.3, 20.9, 12.3, 11.8.3-(((-3β-Hydroxy-5α-androstan-17β-yl)oxy)methyl)-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzaldehyde (28)
To a solution of steroid 27 (300 mg, 0.57 mmol) in THF (20 mL) was added activated MnO2 (496 mg, 5.7 mmol) at 23 °C. The mixture was stirred for 5 h. The mixture was filtered through Celite, the THF was removed and the residue (280 mg) was directly converted to steroid 29 without purification or characterization.17β-((3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)oxy)-5α-androstan-3β-ol (29)
To a stirred solution of steroid 28 (280 mg) in THF (5 mL) and MeOH (5 mL) was added dimethyl-1-diazo-2-oxopropyl phosphonate (0.4 mL) and K2CO3 (690 mg, fine powder) at 23 °C and stirring was continued for 16 h. The MeOH was removed and water was added. The product was extracted into EtOAc (2 × 50 mL). The combined extracts were dried over anhydrous Na2SO4, the solvent was removed and the residue was purified by flash column chromatography (silica gel, eluted with 25% EtOAc in hexanes) to give steroid 29 (140 mg, 48% from steroid 27): 1H NMR (400 MHz, CDCl3) δ 7.47 (s, 1H), 7.20 (s, 1H), 7.14 (s, 1H), 4.49 (s, 2H), 3.60–3.55 (m, 1H), 3.37–3.33 (m, 1H), 3.12 (s, 1H), 0.81 (s, 6H), 1.99–0.59 (m, 23H); 13C NMR (100 MHz, CDCl3) δ 140.8, 131.6, 129.5, 128.8, 125.1, 123.3 (q, J = 275 Hz), 123.0, 88.9, 82.3, 78.4, 71.2, 70.3, 54.4, 51.1, 44.8, 43.1, 38.1, 37.9, 37.0, 35.5, 35.2, 31.6, 31.4, 28.5, 28.3 (q, J = 41.5 Hz), 27.8, 23.3, 20.8, 12.3, 11.8.17β-((R)-4-(1-(3-((Methoxymethoxy)methyl)-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)-1H-1,2,3-triazol-4-yl)butan-2-yl)androst-5-en-3β-ol (31)
To a solution of 26,27-dinorcholest-5-en-24-yne-3β-ol (ref. 10) (30, 120 mg, 0.34 mmol) and compound 18 (180 mg, 0.57 mmol) in THF (10 mL) was added sodium ascorbate (198 mg, 1 mmol) and CuSO4.5H2O (125 mg, 0.5 mmol) dissolved in water (5 mL) at 23 °C. After 72 h, the product was extracted into CH2Cl2 (2 × 20 mL). The combined organic extracts were dried over anhydrous Na2SO4, the sovent was removed and the residue was purified by flash column chromatography (silica gel, eluted with 50% EtOAc in hexanes) to give product 31 (115 mg, 50%): 1H NMR (400 MHz, CDCl3) δ 7.25 (s, 1H), 7.18 (s, 1H), 7.15 (s, 1H), 6.96 (s, 1H), 5.46 (s, 2H), 5.31 (d, J = 4.3 Hz, 1H), 4.67 (s, 2H), 4.53 (s, 2H), 3.52–3.46 (m, 1H), 3.36 (s, 3H), 0.98 (s, 3H), 0.96 (d, J = 6.6 Hz, 3H), 0.64 (s, 3H), 2.78–0.88 (m, 26H); 13C NMR (100 MHz, CDCl3) δ 149.6, 140.8, 140.2, 136.2, 130.2, 128.1, 125.6, 124.9, 123.2 (q, J = 275 Hz), 121.4, 120.4, 96.0, 71.5, 68.1, 56.6, 55.7, 55.4, 53.3, 50.0, 42.3, 42.2, 39.7, 37.2, 36.4, 35.6, 35.5, 31.8 (2 × C), 31.5, 28.4 (q, J = 41.5 Hz), 28.1, 24.1, 22.3, 21.0, 19.3, 18.4, 11.7.17β-((R)-4-(1-(3-(Hydroxymethyl)-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)-1H-1,2,3-triazol-4-yl)butan-2-yl)androst-5-en-3β-ol (32)
To a solution of compound 31 (115 mg, 0.17 mmol) in MeOH (10 mL) was added 6 N HCl (1 mL). The reaction was refluxed 2 h and cooled to 23 °C. A 10% NaOH solution was added dropwise to reach pH 8–9. The product was extracted into CH2Cl2 (3 × 20). The combined organic extracts were dried over anhydrous Na2SO4, the CH2Cl2 was removed and the residue was purified by flash column chromatography (silica gel, eluted with EtOAc) to give product 32 (80 mg, 76%): 1H NMR (400 MHz, CDCl3) δ 7.29 (s, 1H), 7.19 (s, 1H), 7.16 (s, 1H), 6.96 (s, 1H), 5.45 (s, 2H), 5.33 (d, J = 4.3 Hz, 1H), 4.67 (s, 2H), 3.53–3.47 (m, 1H), 0.98 (s, 3H), 0.96 (d, J = 6.6 Hz, 3H), 0.65 (s, 3H), 2.74–0.87 (m, 27H); 13C NMR (100 MHz, CDCl3) δ 149.6, 143.4, 140.7, 136.1, 130.2, 127.4, 124.8, 124.7, 123.2 (q, J = 275 Hz), 121.6, 120.5, 71.7, 63.8, 56.7, 55.7, 53.4, 50.0, 42.3, 42.2, 39.7, 37.2, 36.4, 35.6, 35.5, 31.8 (2 × C), 31.6, 28.4 (q, J = 41.5 Hz), 28.2, 24.2, 22.4, 21.0, 19.3, 18.5, 11.8.3-((4-((3R)-3-(3β-Hydroxy-androst-5-en-17-yl)butyl)-1H-1,2,3-triazol-1-yl)methyl)-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzaldehyde (33)
To a solution of compound 32 (80 mg, 0.13 mmol) in THF (5 mL) was added activated MnO2 (326 mg, 3.75 mmol) at 23 °C. The reaction mixture was efficiently stirred overnight and then filtered through Celite. The THF was removed and the residue was purified by flash column chromatography (silica gel, eluted with 50% EtOAc in hexanes) to give product 33 (43 mg, 52%): 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 7.77 (s, 1H), 7.68 (s, 1H), 7.30 (s, 1H), 7.25 (s, 1H), 5.57 (s, 2H), 5.33 (d, J = 4.3 Hz, 1H), 4.67 (s, 2H), 3.52–3.48 (m, 1H), 0.99 (s, 3H), 0.98 (d, J = 6.6 Hz, 3H), 0.65 (s, 3H), 2.78–0.82 (m, 24H); 13C NMR (100 MHz, CDCl3) δ 190.2, 140.7, 137.6, 131.4, 130.9, 129.7, 127.6, 123.2 (q, J = 275 Hz), 121.6, 120.5, 71.7, 56.7, 55.8, 52.8, 50.0, 42.3, 42.2, 39.7, 37.2, 36.4, 35.6, 35.5, 31.8 (2 × C), 31.6, 31.5, 28.4 (q, J = 41.5 Hz), 28.2, 24.2, 22.6, 22.4, 21.0, 19.4, 18.5, 11.8.17β-((R)-4-(1-(3-Ethynyl-5-(3-(trifluoromethyl)-3H-diazirin-3-yl)benzyl)-1H-1,2,3-triazol-4-yl)butan-2-yl)androst-5-en-3β-ol (34)
To a solution of steroid 33 (43 mg, 0.07 mmol) in THF (2 mL) and MeOH (2 mL) was added dimethyl-1-diazo-2-oxopropyl phosphonate (0.1 mL) and K2CO3 (70 mg, fine powder) at 23 °C. After 16 h, the solvent was removed and water was added. The product was extracted into EtOAc (2 × 20 mL). The combined organic extracts were dried over anhydrous Na2SO4, the solvent was removed and the residue was purified by flash chromatography (silica gel, eluted with 50% EtOAc in hexanes) to give product 34 (21 mg, 50%): 1H NMR (400 MHz, CD3OD) δ 7.37 (s, 1H), 7.29 (s, 1H), 7.19 (s, 1H), 7.00 (s, 1H), 5.46 (s, 2H), 5.34 (d, J = 4.3 Hz, 1H), 3.54–3.48 (m, 1H), 3.15 (s, 1H), 1.00 (s, 3H), 0.98 (d, J = 6.6 Hz, 3H), 0.65 (s, 3H), 2.82–0.83 (m, 26H); 13C NMR (100 MHz, CD3OD) δ 149.8, 140.7, 136.5, 132.4, 130.5, 130.2, 125.8, 124.2, 123.2 (q, J = 275 Hz), 121.6, 120.4, 81.4, 79.7, 71.7, 56.7, 55.8, 53.0, 50.0, 42.3, 42.2, 39.7, 37.2, 36.4, 35.6, 35.5, 31.8 (2 × C), 31.6, 28.4 (q, J = 41.5 Hz), 28.2, 24.2, 22.4, 21.0, 19.4, 18.5, 11.8.Author contributions
DFC and MQ took part in the conceptualization of the study. MQ and YX carried out the chemical synthesis and characterization of the compounds. DFC wrote the original manuscript. All authors contributed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by Grant 1 P50 MH122379 from the National Institutes of Health and the Taylor Family Institute for Innovative Psychiatric Research.References
- J. R. Hill and A. A. B. Robertson, J. Med. Chem., 2018, 61, 6945–6963 CrossRef CAS PubMed.
- A. V. West, G. Muncipinto, H.-Y. Wu, A. C. Huang, M. T. Labenski, L. H. Jones and C. M. Woo, J. Am. Chem. Soc., 2021, 143, 6691–6700 CrossRef CAS PubMed.
- Z.-W. Chen, J. R. Bracamontes, M. M. Budelier, A. L. Germann, D. J. Shin, K. Kathiresan, M. Qian, B. Manion, W. W. L. Cheng, D. E. Reichert, G. Akk, D. F. Covey and A. S. Evers, PLoS Biol., 2019, 17, 3000157 CrossRef PubMed.
- K. Krishnan, M. Qian, M. Feltes, Z.-W. Chen, S. Gale, L. Wang, Y. Sugasawa, D. E. Reichert, J. E. Schaffer, D. S. Ory, A. S. Evers and D. F. Covey, ACS Chem. Biol., 2021, 16, 1493–1507 CrossRef CAS PubMed.
- N. S. Kumar and R. N. Young, Bioorg. Med. Chem., 2009, 17, 5388–5395 CrossRef CAS PubMed.
- T. Hosoya, T. Hiramatsu, T. Ikemoto, H. Aoyama, T. Ohmae, M. Endo and M. Suzuki, Bioorg. Med. Chem. Lett., 2005, 15, 1289–1294 CrossRef CAS PubMed.
- K. Nakamoto, Y. Akao and Y. Ueno, Bioorg. Med. Chem. Lett., 2018, 28, 2906–2909 CrossRef CAS PubMed.
- S. D. Taylor and J. Harris, Steroids, 2011, 76, 1098–1102 CrossRef CAS PubMed.
- I. Cerny, M. Budesinsky, V. Pouzar and P. Drasar, Collect. Czech. Chem. Commun., 2001, 66, 933–946 CrossRef CAS.
- H. J. Lee, W. Zhang, D. Zhang, Y. Yang, B. Liu, E. L. Barker, K. K. Buhman, L. V. Slipchenko, M. Dai and J.-X. Cheng, Sci. Rep., 2015, 5, 7930 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07260f |
| This journal is © The Royal Society of Chemistry 2023 |