
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of pyrazolo[4,3-d]thiazole derivatives containing α-aminophosphonate as potential Mur A inhibitors against MDR pathogens with radiosterilization and molecular modeling simulation†
Ahmed Ragab *a,
Seham A. Ibrahimb,
Dina S. Aboul-Magd*c and
Mohamed H. Barenb
*a,
Seham A. Ibrahimb,
Dina S. Aboul-Magd*c and
Mohamed H. Barenb
aChemistry Department, Faculty of Science (Boys), Al-Azhar University, Nasr City, Cairo 11884, Egypt. E-mail: ahmed_ragab7@ymail.com; ahmed_ragab@azhar.edu.eg
bChemistry Department, Faculty of Science, Tanta University, Tanta 31527, Egypt. E-mail: seham.ibrahim@science.tanta.edu.eg; sehamabdelatif@yahoo.com
cDrug Radiation Research Department, National Center for Radiation Research and Technology (NCRRT), Egyptian Atomic Energy Authority, Egypt. E-mail: dina.a.a.soliman@gmail.com
First published on 29th November 2023
Abstract
The present study involves the synthesis of a new series of α-aminophosphonate derivatives in good yields with a simple workup via the Kabachnik–Fields reaction using lithium perchlorate (LiClO4) as a catalyst to facilitate the reaction. All the newly synthesized compounds were confirmed using various physical, spectroscopic, and analytical data, and the obtained results correlated with the proposed molecular structure. The in vitro antimicrobial activities of each compound were evaluated against different clinical isolates. The results indicated that among these derivatives, two compounds (5a and 5b) were the most active and displayed potent activity with MICs in the range from 0.06 to 0.25 μg mL−1 compared with fosfomycin and fluconazole as standard antibiotics. Moreover, the synthesized phosphonates displayed a broad spectrum of bactericidal and fungicidal activities depending on MICs, MBCs/MFCs, and the time-kill kinetics. In addition, the checkerboard assay showed synergistic and partial synergistic activities between the active compounds combined with fosfomycin and fluconazole. Furthermore, the SEM images showed distinct ruptures of the OM integrity of the FOS-R E. coli at their MICs, which was further indicated by the increased EtBr accumulation within the bacterial cells. Moreover, active derivatives revealed MurA inhibitory activity with IC50 values of 3.8 ± 0.39 and 4.5 ± 0.23 μM compared with fosfomycin (IC50 = 12.7 ± 0.27 μM). To our surprise, exposing 5a and 5b compounds to different gamma radiation doses revealed that 7.0 kGy eradicated the microbial load completely. Finally, the results of quantum chemical study supported the binding mode obtained from the docking study performed inside the active site of MurA (PDB: 1UAE), suggesting that these phosphonates may be promising safe candidates for MDR infection therapy clinical trials with no toxic effects on the normal human cells.
1. Introduction
Antibiotic resistance is one of the biggest threats to the global healthcare system, according to the World Health Organization.1 The α-aminophosphonates (AAPs) are an important class of organophosphorus compounds with structural amino acid analogs where a carboxylic group is substituted by phosphonic acid or related groups.2 As a result of their remarkable biological and physical properties and their value as synthesis intermediates, AAPs have been used in various fields, including manufacturing, agriculture, and medicine. They are discovered to be antibacterial,3 anti-Alzheimer,4 antiviral,5 anti-inflammatory,6 anti-HIV,7 antitumor,8 anti-proliferative and apoptosis-inducing,9 herbicidal,10 and insecticidal11 agents. Additionally, the phosphonates and α-aminophosphonates make up an intriguing class of substances that have been used to synthesize fire retardants,12–14 dispersants, corrosion inhibitors,15,16 dental additives,17,18 and deposition prevention agents.19Numerous methods have been developed for synthesizing phosphonates and α-aminophosphonates due to the many applications.20,21 Many drugs containing phosphorus are released as drugs or prodrugs in order to achieve higher selectivity and bioavailability.22 For example, clindamycin phosphate I (broad-spectrum antibiotic),23 tedizolid phosphate antibiotic II (inhibits bacterial protein synthesis),24 fosfluconazole III (antifungal agent),25 dexamethasone phosphate IV and prednisolone phosphate V (anti-inflammatory agents),26 etoposide phosphate VI (has antitumor activity by inhibiting DNA synthesis),27 and sofosbuvir VII (antiviral drug and a hepatitis C protease inhibitor)28 (Fig. 1). Moreover, the presence of heterocyclic moieties in the structure of α-aminophosphonates appears to have substantially impacted biological activities. Additionally, introducing these heterocyclic rings, such as quinazoline, pyrazole, thiazole, pyridine, and chromene, in the structure is particularly helpful in enhancing their activity.29 For that reason, the synthesis of α-aminophosphonates has attracted considerable attention, and significant progress has been made in developing more efficient methods for the synthesis of these compounds.30 Two main pathways have been developed for the synthesis of α-aminophosphonates: the Pudovik and Kabachnik–Fields reactions. The Kabachnik–Fields reaction involved the nucleophilic addition of an amine to a carbonyl compound followed by the addition of a dialkyl or diaryl phosphite to the resulting imine in either one-step or multi-step operation using Lewis acids as catalysts.31,32 The Kabachnik–Fields reaction still gains interest due to the challenging synthetic procedures and the potential biological activity of the resulting α-aminophosphonic derivatives.33
 | ||
| Fig. 1 Drugs containing a phosphonate group and their biological target. | ||
As a result, novel synthesis has been established for the management of thiazolyl-pyrazole derivatives, allowing for their chemical diversity, preferred biological efficacy, and low toxicity for medical purposes. Various thiazolyl-pyrazole analogues have been synthesized and investigated for biological activity as cyclooxygenase inhibitors, antioxidants, analgesics, and anti-inflammatory drugs, as well as their potential as antibacterial, antifungal, antioxidant, and anticancer agents.34–37 Targeting the biosynthetic steps in the bacterial cell wall assembly is a promising way to produce new antibacterial agents, as many clinically important antibiotics such as penicillins and vancomycin demonstrate.38 MurA is important in the first cytoplasmic step of peptidoglycan biosynthesis. It is a UDP-N-acetylglucosamine enolpyruvyl transferase that catalyzes the transfer of enolpyruvate from phosphoenolpyruvate (PEP) to the 30-hydroxy group of uridine diphospho-N-acetylglucosamine (UNAG).39 This transfer originated by binding UNAG to the enzyme's active site, followed by the binding of PEP. Cys115 and Asp305 are two amino acids that are essential in forming this product, particularly for the deprotonation of the C3 atom of the tetrahedral intermediate and for the final product release.40 MurA is a validated antibacterial drug discovery target. It represents selective targeting because it is only present in bacteria and is neither required nor present in human cells.40 Sterilization of pharmaceutics is required to be safely and effectively used. It is a unique critical procedure. The anonymous “sterilization” is defined as the use of a suitably designed, validated and controlled process to inactivate or reduce the viable microorganisms, determined by the sterility assurance level (SAL) < 10−6, within the target product.41 One of the most widely used methods is gamma radiation via a source of radiation such as Cobalt-60 (60Co). The use of gamma radiation has many advantages such as easy penetration into the product, ensuring sterility and effectiveness independent of temperature and pressure.42,43
Based on the previous data and in continuation of our works aiming to synthesize heterocyclic molecules with remarkable biological activities,44–47 we aimed to investigate the use of LiClO4 as a catalyst to synthesize novel thiazolyl-pyrazole heterocycle-containing α-aminophosphonate derivatives via a “one-pot” three-component reaction as excellent non-cytotoxic alternatives for synthesizing α-aminophosphonates. Moreover, the newly designed derivatives were evaluated for their antimicrobial activity (i.e., MIC, MBC/MFC, time-killing kinetics, and drug combination studies) against MDR microbial isolates. The effects of phosphonates on the outer membrane of E. coli were studied using SEM analysis and ethidium bromide (EtBr) dye accumulation assay. The evaluation of MurA inhibition activity as a mode of action was confirmed by molecular docking and quantum chemical studies.
2. Result and discussion
2.1. Chemistry
In the present work, a new series of α-aminophosphonate derivatives 3, 4, 5a, 5b, 6a and 6b were synthesized in good yields by a one-pot three-component reaction of 5-aminopyrazolethiazole, aldehyde derivatives, and diethyl and/or diphenyl phosphite at room temperature in the presence of lithium perchlorate (LiClO4) as a catalyst via the Kabachnik–Fields reaction (Schemes 2 and 3).First, 5-amino-1H-pyrazolo[4,3-d]thiazole derivative 2 was prepared by the reaction of 5-amino pyrazole derivative 1 with bromine and potassium thiocyanate as described previously48 (Scheme 1). Then, a molar ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1) of 5-amino-1H-pyrazolo[4,3-d]thiazole derivative 2, trialkyl phosphite, and appropriate aldehyde in dichloromethane (CH2Cl2) was stirred at room temperature for 48 h in the presence of anhydrase lithium perchlorate (LiClO4) as the one-pot three-component reaction, and the progress of the reaction was monitored by TLC until the reaction was complete, as described in Schemes 2 and 3. The mechanism for synthesizing α-aminophosphonates using LiClO4 as a catalyst is illustrated in Scheme S1.† First, the reaction started by activating the carbonyl group of aldehydes with the Lewis acid catalyst (LiClO4), followed by the condensation of the carbonyl group attached with primary amines to afford the Schiff base. Then the nitrogen of the Schiff base that is formed in the first step donated a pair of electrons to make a coordinate bond with LiClO4. This coordination makes nitrogen positively charged and therefore induces a partial positive charge on sp2 carbon. In addition, the free pair of electrons of phosphorus attack the partially positively charged carbon, followed by the elimination of phenol or ethanol, and afford cyclic α-aminophosphonates.
:1:1) of 5-amino-1H-pyrazolo[4,3-d]thiazole derivative 2, trialkyl phosphite, and appropriate aldehyde in dichloromethane (CH2Cl2) was stirred at room temperature for 48 h in the presence of anhydrase lithium perchlorate (LiClO4) as the one-pot three-component reaction, and the progress of the reaction was monitored by TLC until the reaction was complete, as described in Schemes 2 and 3. The mechanism for synthesizing α-aminophosphonates using LiClO4 as a catalyst is illustrated in Scheme S1.† First, the reaction started by activating the carbonyl group of aldehydes with the Lewis acid catalyst (LiClO4), followed by the condensation of the carbonyl group attached with primary amines to afford the Schiff base. Then the nitrogen of the Schiff base that is formed in the first step donated a pair of electrons to make a coordinate bond with LiClO4. This coordination makes nitrogen positively charged and therefore induces a partial positive charge on sp2 carbon. In addition, the free pair of electrons of phosphorus attack the partially positively charged carbon, followed by the elimination of phenol or ethanol, and afford cyclic α-aminophosphonates.
 | ||
| Scheme 1 Synthesis of new 1H-pyrazolo[4,3-d]thiazol-5-amine derivative 2. | ||
 | ||
| Scheme 2 Synthetic route for novel ethyl α-aminophosphonates 3 and 4. | ||
 | ||
| Scheme 3 Synthetic route for novel α-aminophosphonates 5a, 5b, 6a and 6b. | ||
The chemical structures of α-aminophosphonate derivatives 3, 4, 5a, 5b, 6a and 6b were characterized by IR, 1H NMR, 13C NMR, mass spectroscopy and elemental analyses, and their data are presented in the experimental section. The IR spectra of compounds 3, 4, 5a, 5b, 6a and 6b showed the NH band in the range of 3320–3378 cm−1. The sharp band observed in the range of 1237–1277 cm−1 is attributed to P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and a band for P–C stretching occurred in the range of 769–779 cm−1. The 1H NMR spectra of the synthesized derivatives exhibited a characteristic signal at δ 9.29–9.72 ppm assigned to the –NH group and a signal in the range δ 5.97–7.64 ppm as a singlet signal assigned to P–CH of all the title compounds. Moreover, for diethyl phosphonate derivatives 3 and 4, the methylene and methyl protons of P–O–CH2CH3 displayed as a quartet at δ 3.85 ppm and triplet at δ 1.05–1.19 ppm. The 13C NMR spectra of diethyl phosphonate derivatives 3 and 4 revealed chemical shifts for methylene and methyl carbons observed at δ 66.80–66.81 ppm and δ 12.23–22.05 ppm, respectively. Moreover, the chemical shift for P–CH carbons was observed at δ 59.58–66.84 ppm. The compounds were analyzed by mass spectroscopy, and M+ ions were observed in the expected m/z values.
O, and a band for P–C stretching occurred in the range of 769–779 cm−1. The 1H NMR spectra of the synthesized derivatives exhibited a characteristic signal at δ 9.29–9.72 ppm assigned to the –NH group and a signal in the range δ 5.97–7.64 ppm as a singlet signal assigned to P–CH of all the title compounds. Moreover, for diethyl phosphonate derivatives 3 and 4, the methylene and methyl protons of P–O–CH2CH3 displayed as a quartet at δ 3.85 ppm and triplet at δ 1.05–1.19 ppm. The 13C NMR spectra of diethyl phosphonate derivatives 3 and 4 revealed chemical shifts for methylene and methyl carbons observed at δ 66.80–66.81 ppm and δ 12.23–22.05 ppm, respectively. Moreover, the chemical shift for P–CH carbons was observed at δ 59.58–66.84 ppm. The compounds were analyzed by mass spectroscopy, and M+ ions were observed in the expected m/z values.
2.2. Biological evaluation
| Cpd. no. | Average diameter of inhibition zone (ZI) in mm | |||||
|---|---|---|---|---|---|---|
| Bacteria | Fungi | |||||
| Gram +ve | Gram −ve | |||||
| E. faecalisKA247 | S. aureusKA112 | E. coliKA780 | P. mirabilisKA320 | C. albicansKA205 | C. tropicalisKA205 | |
| a ±: standard error mean; each value is the mean of three values; FO: fosfomycin; FCA: fluconazole; NA: non-applicable; −ve: no growth detected; CLSI breakpoints: zone diameter for fosfomycin “M100, 30th edition, 2020” (≥16 [susceptible], 13 to 15 [intermediate], and ≤12 [resistant] mm); zone diameter for fluconazole “M27-A4, 4th edition, 2017” (≥19 [susceptible], 15 to 18 [intermediate], and ≤14 [resistant] mm). | ||||||
| 2 | 17 ± 0.00 | 17 ± 0.39 | 17 ± 0.62 | 16 ± 0.79 | 17 ± 0.16 | 16 ± 0.63 |
| 3 | 18 ± 0.05 | 18 ± 0.31 | 19 ± 0.14 | 17 ± 0.71 | 17 ± 0.88 | 17 ± 0.48 |
| 4 | 16 ± 0.80 | 17 ± 0.00 | 17 ± 0.54 | 16 ± 0.34 | 16 ± 0.67 | 16 ± 0.54 |
| 5a* | 24 ± 0.20 | 24 ± 0.51 | 25 ± 0.28 | 23 ± 0.44 | 22 ± 0.76 | 22 ± 0.43 |
| 5b** | 23 ± 0.88 | 24 ± 0.03 | 25 ± 0.00 | 23 ± 0.78 | 22 ± 0.22 | 21 ± 0.32 |
| 6a | 18 ± 0.74 | 19 ± 0.00 | 19 ± 0.66 | 18 ± 0.50 | 20 ± 0.89 | 20 ± 0.20 |
| 6b | 22 ± 0.55 | 23 ± 0.77 | 24 ± 0.86 | 22 ± 0.12 | 21 ± 0.91 | 21 ± 0.70 |
| FO | 17 ± 0.34 | 18 ± 0.32 | 11 ± 0.23 | 19 ± 0.13 | NA | NA |
| FCA | NA | NA | NA | NA | 17 ± 0.99 | 17 ± 0.15 |
| DMSO | −ve | −ve | −ve | −ve | −ve | −ve |
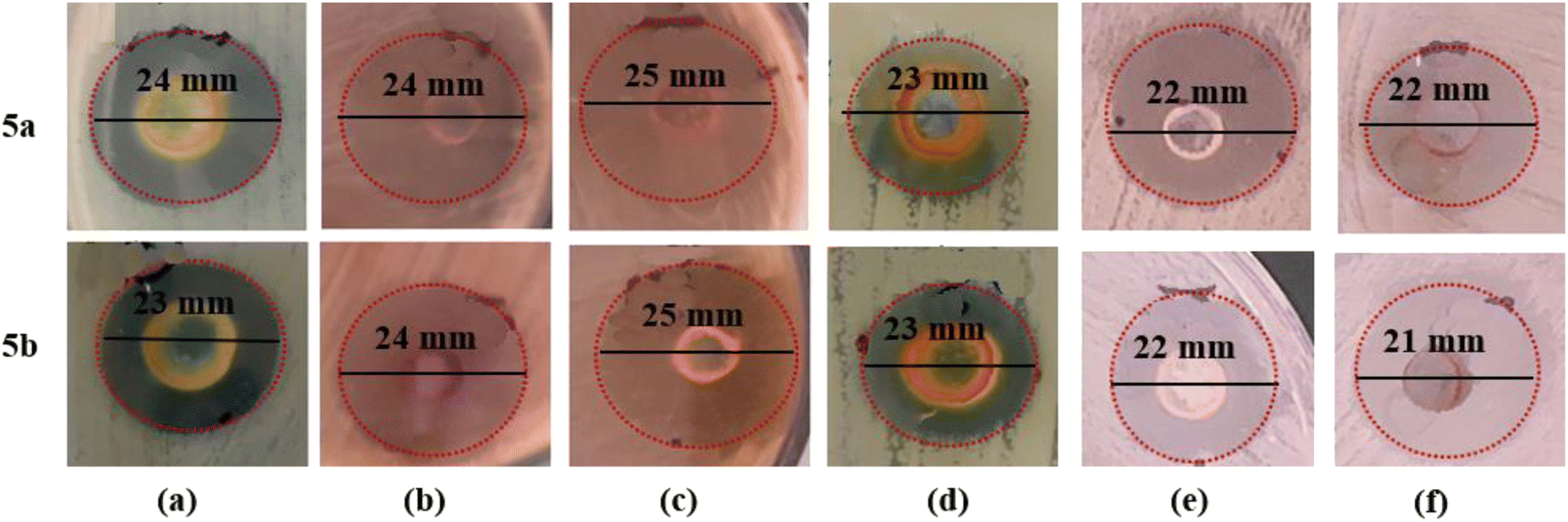 | ||
| Fig. 2 Diameter of inhibition zones of the most active compounds 5a and 5b against (a) E. faecalisKA247, (b) S. aureusKA112, (c) E. coliKA780, (d) P. mirabilisKA320, (e) C. albicansKA205, and (f) C. tropicalisKA205. | ||
| Cpd. no. | MIC (mean ± SEM) (μg mL−1) (MBC/MIC or MFC/MIC) | |||||
|---|---|---|---|---|---|---|
| Bacteria | Fungi | |||||
| Gram +ve | Gram −ve | |||||
| E. faecalisKA247 | S. aureusKA112 | E. coliKA780 | P. mirabilisKA320 | C. albicansKA205 | C. tropicalisKA205 | |
| a ±: standard error mean; each value is the mean of three values; FO: fosfomycin; FCA: fluconazole; NA: non-applicable; −ve: no growth detected; CLSI breakpoints: MIC breakpoints (M100, 30th edition, 2020) for fosfomycin (≤64 [susceptible], 128 [intermediate], and ≥256 [resistant] μg mL−1); MIC breakpoints for fluconazole (M27-A4, 4th edition, 2017): (≤2 [susceptible], 4 [intermediate], and ≥8 [resistant] μg mL−1). | ||||||
| 2 | 8 (1) | 8 (0.5) | 4 (0.5) | 8 (1) | 1 (0.5) | 1 (2) |
| 3 | 2 (2) | 1 (2) | 1 (1) | 2 (1) | 1 (1) | 1 (0.5) |
| 4 | 16 (1) | 8 (2) | 8 (1) | 16 (1) | 2 (0.5) | 2 (1) |
| 5a* | 0.125 (0.5) | 0.06 (0.5) | 0.06 (0.5) | 0.125 (0.5) | 0.125 (0.5) | 0.125 (1) |
| 5b** | 0.125 (1) | 0.06 (1) | 0.06 (0.5) | 0.25 (0.5) | 0.125 (1) | 0.5 (0.5) |
| 6a | 2 (0.5) | 1 (1) | 1 (0.5) | 2 (1) | 0.5 (0.5) | 0.5 (1) |
| 6b*** | 0.25 (0.5) | 0.125 (0.5) | 0.125 (0.5) | 0.25 (1) | 0.25 (1) | 0.5 (1) |
| FO | 8 (1) | 8 (0.4) | 256 (1) | 16 (1) | NA | NA |
| FCA | NA | NA | NA | NA | 2 (1) | 4 (0.5) |
| DMSO | −ve | −ve | −ve | −ve | −ve | −ve |
The structure–activity relationship (SAR) exhibited that modification of 5-amino-1H-pyrazolo[4,3-d]thiazole derivative 2 with a phosphonate group enhancement antimicrobial activity in general. In addition, diphenyl α-aminophosphonate derivatives 5 and 6 demonstrated a higher activity than that of diethyl α-aminophosphonates 3 and 4. In terms of comparison, the presence of an electron-withdrawing group as a chlorine atom in benzyl phosphonate derivative 3 showed MIC values ranging from 1 to 2 μg mL−1 and MBC (1–4 μg mL−1) and MFC (0.5–1 μg mL−1), while replacing a chlorine atom with N,N-dimethyl amine (electron donating group) at the same position as compound 4 causes a dramatic decrease in the activity on both bacterial and fungal isolates, but still promising than fosfomycin (except in case of E. faecalisKA247) and fluconazole with MIC values of 8–16 μg mL−1, MBC of 8–16 μg mL−1 and MFC of 1–2 μg mL−1, and these results indicate that the presence of an electron-withdrawing group in the para position to benzyl phosphonate is desirable. Interestingly, replacing diethyl groups in compound 4 with diphenyl groups that appeared in compound 5b improved the antimicrobial activity with 64–133 folds against bacterial isolates and 4–16 folds against fungal isolates based on MIC values.
Furthermore, grafting the hydroxyl group in the ortho position to benzyl phosphonate in compound 5a showed enhancement in the inhibitory activity against P. mirabilisKA320 (MIC = 0.125 μg mL−1) compared to N,N-dimethyl amine in the para position as compound 5b (MIC = 0.25 μg mL−1) and increased the minimal bactericidal (MBC) and fungicidal (MFC) activity to a two-fold value against E. faecalisKA247 and S. aureusKA112 and that may be related to increasing the electron density that occurs by a hydroxyl group in the ortho position rather than a para position to N,N-dimethyl amine. In addition, replacing aromatic phenyl in methyl phosphonate with heterocyclic rings as thiophene core 6a and pyrrole core 6b exhibited good antimicrobial activity, but less than the benzyl phosphonate group. In addition, the presence of the NH group in pyrrole revealed a higher activity with MIC values of 0.125–0.25 μg mL−1 against bacteria (∼8 folds) and 0.25–0.50 μg mL−1 (∼1–2 folds) against fungal isolates, and this activity may be related to sulfur that has higher electronegativity and, therefore, a lesser tendency to donate its lone pair.
Moreover, it has been exploited that MBC/MFC is the lowest concentration of antibiotic to kill a certain bacterium/fungus. It involves adding extra steps performed after the determination of MIC. The antimicrobial agents are considered bactericidal/fungicidal if the MBC or MFC ratio is not greater than four times the MIC. The tested phosphonate compounds showed bactericidal and fungicidal effects. In addition, both fosfomycin and fluconazole showed bactericidal and fungicidal activities against different isolates.
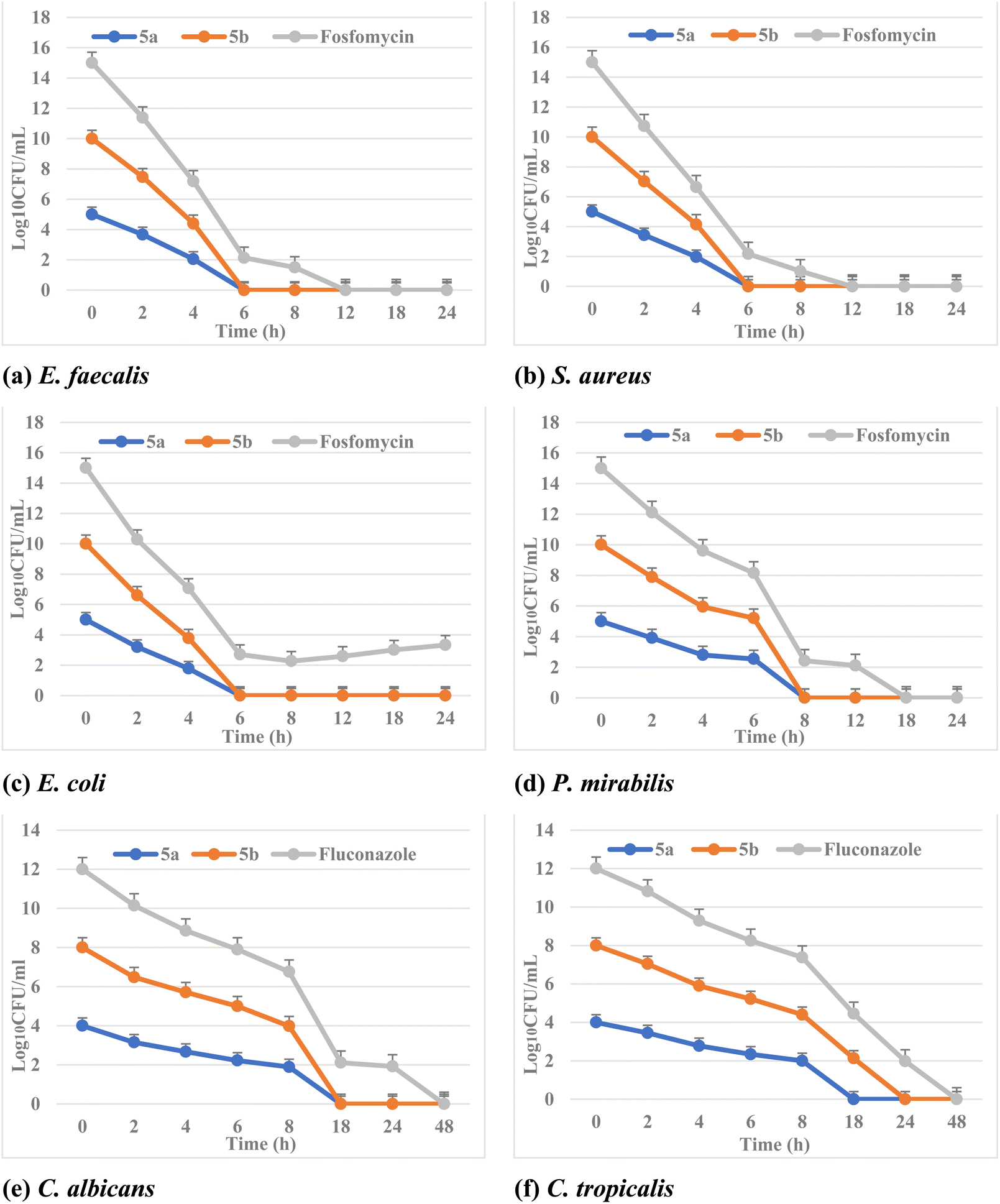 | ||
| Fig. 3 Time-kill curve of different MDR bacterial and fungal isolates (a) E. faecalisKA247, (b) S. aureusKA112, (c) E. coliKA780, (d) P. mirabilisKA320, (e) C. albicansKA205 and (f) C. tropicalisKA205 with 5a, 5b, fosfomycin and fluconazole at the concentration of MIC. | ||
The synergistic activity was determined according to the fractional inhibitory concentration index (ΣFICI), in which the value of ΣFICI ≤ 0.5 indicates synergism between the compound and the antibiotic. Indeed, as shown in Table 3, both phosphonate compounds 5a and 5b exhibited ΣFICI ≤ 0.5 and from 0.5 < FICI > 1 in the drug combinations reported (i.e., able to re-sensitize the isolates to antibiotics), suggesting synergistic and partial synergistic antimicrobial activities. Finally, these phosphonate derivatives 5a and 5b considerably reduced the MIC of fosfomycin and fluconazole against all the tested clinical isolates.
| Cpd. no. | Fractional inhibitory concentration index (ΣFICI)/effect | |||||
|---|---|---|---|---|---|---|
| Bacteriaa | Fungib | |||||
| Gram +ve | Gram −ve | |||||
| E. faecalisKA247 | S. aureusKA112 | E. coliKA780 | P. mirabilisKA320 | C. albicansKA205 | C. tropicalisKA205 | |
| a Combination of 5a or 5b with fosfomycin.b Combination of 5a or 5b with fluconazole; FICI ≤ 0.5 (synergism); 0.5 < FICI > 1 (partial synergism); 1 ≤ FICI < 2 (indifference); FICI ≥ 2 (antagonism). | ||||||
| 5a* | 0.5/S | 0.25/S | 0.25/S | 0.5/S | 0.75 | 0.5/S |
| 5b** | 0.5/S | 0.5/S | 0.25/S | 0.25/S | 0.75/P | 0.75/P |
 | ||
| Fig. 4 Microscopic observation of the morphological alterations of fosfomycin-resistant E. coliKA780 treated with DMSO (1%) and with phosphonate compounds (a) 5a and (b) 5b for 12 h using a scanning electron microscope (SEM). | ||
As shown in Fig. 5, adding PAβN increases the accumulation of EtBr in the E. coli strain. Similarly, the addition of phosphonates showed an increase in the accumulation of EtBr at their different MIC levels. Finally, it can be concluded that the phosphonate treatment caused a dose-dependent increase in the EtBr uptake, indicating evidence of the dramatic effect of phosphonates on the integrity of the outer membrane (OM) integrity.
 | ||
| Fig. 5 EtBr accumulation in E. coli Kam3-AcrB. E. coli was treated with phosphonate compounds 5a and 5b at a concentration ranging from 0.06 μg mL−1 to 2 μg mL−1 or DMSO (0.1%) for 12 h, followed by the addition of EtBr to give a final conc. of 4 μg mL−1 and RFU was determined. | ||
| Tested compound | BNL | Vero | ||
|---|---|---|---|---|
| IC50 | Viability % | IC50 | Viability % | |
| a S.D.: standard deviation, each value is the mean of three values; BNL: mouse normal liver cells; and Vero: green monkey kidney. | ||||
| 5a | >100 | 101.9 ± 0.58 to 94.35 ± 0.86 | >100 | 100.2 ± 0.82 to 81.13 ± 2.6 |
| 5b | >100 | 99.84 ± 0.6 to 73.02 ± 1.54 | >100 | 99.72 ± 2.16 to 94.5 ± 2.23 |
| DOX | 14.7 | 99.26 ± 0.9 to 7.09 ± 0.38 | 0.68 | 101 ± 2.28 to 1.34 ± 0.18 |
| Compound no. | RAa,b % (IC50 in μM) | RAa,c % (IC50 in μM) |
|---|---|---|
| a RA: residual activity determined at 100 μM.b Time of preincubation at 0 min.c Time of incubation at 10 min. | ||
| 5a | 32 ± 0.44 (41 ± 0.25) | 0 (3.8 ± 0.39) |
| 5b | 39 ± 0.61 (45 ± 0.78) | 4 ± 0.55 (4.5 ± 0.23) |
| FO | 52 ± 0.55 (52 ± 0.33) | 10 ± 0.92 (12.7 ± 0.27) |
As given in Table 5, compound 5a displayed significant inhibition of MurA enzyme from E. coli, with RAs of 0% and IC50 = 3.8 μM. Compound 5b displayed a RA value of 7% with IC50 corresponding to 4.5 μM. These findings suggested the significant inhibition of MurA derived from E. coli displaying IC50 < 5 μM. Fosfomycin (FO) showed an IC50 value of 12.7 μM. However, these results are in accordance with the ability of these molecules to inhibit the development of FOS-R E. coli cells tested in this study, as was evidenced by the MIC values and time-kill kinetics. The inhibitory activity of phosphonate compounds might be attributed to the ability of active phosphonates to affect the bacterial OM integrity and, therefore, allow the penetration of phosphonates into the bacterial cell, as shown in Fig. 4.
| Dose (kGy) | Total count (CFU g−1) | |||
|---|---|---|---|---|
| 5a | 5b | |||
| Bacteria | Fungi | Bacteria | Fungi | |
| Control (non-irradiated) | 3 × 103 | 2 × 101 | 2 × 103 | 3 × 101 |
| 3.0 | 3 × 101 | 1 × 103 | 2 × 101 | 1 × 102 |
| 5.0 | 0 | 0 | 1 × 101 | 0 |
| 7.0 | 0 | 0 | 0 | 0 |
| 10.0 | 0 | 0 | 0 | 0 |
| 15.0 | 0 | 0 | 0 | 0 |
| 20.0 | 0 | 0 | 0 | 0 |
2.3. Computational studies
The docking results indicated that the most active phosphonate derivatives 5a and 5b revealed binding energy S = −23.46 and −20.11 kcal mol−1 with good binding with specified receptor targets compared with fosfomycin, S = −10.89 kcal mol−1. The most active phosphonate derivatives 5a showed one hydrogen bond backbone acceptor between the residue Val163 and oxygen of the phosphonate group with a bond length of 1.94 Å and a strength of 17%. In addition, the hydrogen atom of the hydroxyl group attached to the ortho position in benzylidene phosphonate could form a hydrogen bond sidechain acceptor with Ser162 amino acid in the receptor target. Moreover, the diphenyl α-aminophosphonate derivatives 5a formed two arene–cation interactions between Arg91 and Arg120 with phenyl of benzylidene and phenyl phosphonate, respectively Fig. 6a. In addition, a hydrophobic interaction was observed between the phenyl group at N1 of pyrazole and phenyl of 4-nitrophenyl at C3 of pyrazole, as well as phenyl of phenyl phosphonate and appeared as blue background in the 2D structure.
 | ||
| Fig. 6 Two-dimensional and three-dimensional structures of most active diphenyl α-aminophosphonate derivatives 5a and 5b inside the active site of Mur A (PDB: 1AUE) (a) 5a/1AUE and (b) 5b/1AUE. | ||
Furthermore, the α-aminophosphonate derivatives 5b exhibited binding energy S = −20.11 kcal mol−1 through a one arene–cation interaction between Arg120 and the phenyl group of 4-nitrophenyl at C3 of pyrazole, in addition to a great number of hydrophobic interactions Fig. 6b. The fosfomycin as a positive control demonstrated the lowest binding energy S = −10.89 kcal mol−1 through two hydrogen bonds from the sidechain (Arg91 and Ser162) and one hydrogen bond from the backbone (Val163). The oxygen of phosphonate in fosfomycin formed two hydrogen bond sidechains with residues Arg91 and Ser162 with a bond length of 1.97 Å (39%) and 3.24 Å (21%). In addition, the residue Val163 was bound to the oxygen of ethylene oxide through hydrogen bond backbone acceptors with a bond length of 2.06 Å and strength of 28% (all docking figures are displayed in full size in ESI† file).
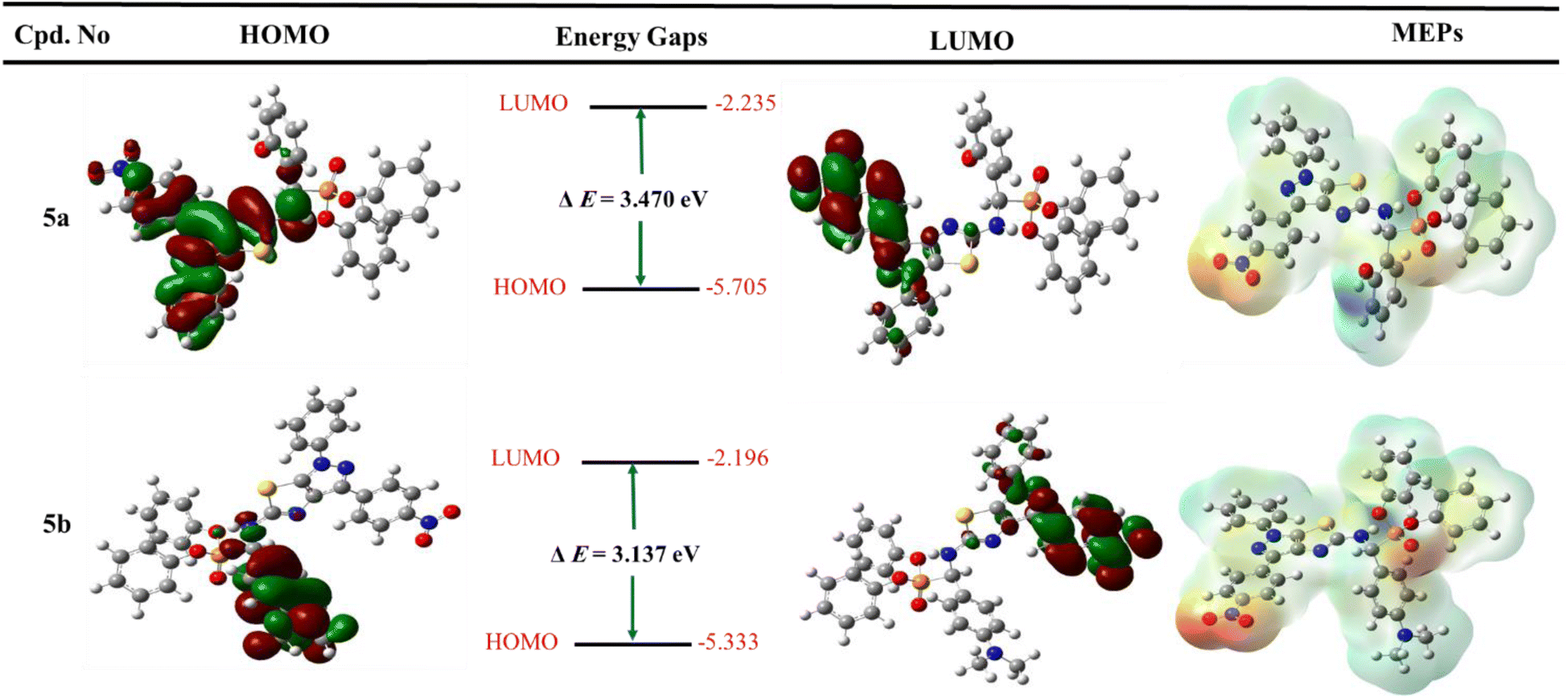 | ||
| Fig. 7 Graph represented frontier molecular orbitals (FMOs) and their energies with molecular electrostatic potential (MEP maps) for the most active derivatives 5a and 5b. | ||
Furthermore, the two phenyl groups of diphenyl phosphonates do not include both HOMOs and LUMOs. It suggested that these hydrophobic groups regulate the properties of physicochemical compounds. In addition, the HOMO and LUMO orbitals for fosfomycin (positive control) have dispersed over all structures, except the methyl group at the ethylene oxide ring that does not cove in the LUMO orbital (all figures in complete size are represented in the ESI† file).
To determine the charge distributed over the most active two diphenyl α-aminophosphonates 5a and 5b, the molecular electrostatic potential maps were generated and represented in Fig. 7. In addition, MEP maps explained the electrostatic interaction between the designed derivative and biological targets and the orientation of drug candidates in terms of their activity.60,61 The negative positions on the molecules showed the oxygen of nitro group, oxygen of phosphonate, and nitrogen of pyrazole and thiazole fragments and represented as red color. Furthermore, the positive charge is represented by blue color and displayed on the NH group at C-2 of pyrazolo[4,3-d]thiazole derivatives and hydrogen of the hydroxyl group at 5a. In addition, the neutral charge is distributed at all aryl groups and appears as green color. Finally, the quantum chemical study supports the binding mode obtained from the previous docking study.
3. Conclusion
In summary, a new series of α-aminophosphonates derivatives containing a pyrazolo[4,3-d]thiazole pharmacophore have been synthesized and fully characterized by 1H/13C NMR, IR, and mass spectroscopy, as well as elemental analysis. The newly synthesized derivatives were assessed in vitro for antimicrobial evaluation as evidence of the concept of designing new antimicrobial agents containing new pharmacophore moieties. A panel of MDR clinical isolates, including Gram +ve, Gram −ve bacteria, and fungi, were selected representing the most common UTI microorganisms. All the synthesized derivatives showed promising in vitro antimicrobial activity, with compounds 5a and 5b as the most potent microbicidal compounds, as represented by the ZOIs, MICs, MBCs and MFCs compared with fosfomycin and fluconazole in addition to the time-kill assay. Moreover, the most active derivatives 5a and 5b showed synergistic activity upon their combination with fosfomycin and fluconazole, except in case of 5b, which showed partial synergistic activity when combined with fluconazole against the Candida isolates. In addition, it has been shown that the phosphonate compounds 5a and 5b act by disrupting the OM integrity of the bacterial cell walls as evidenced by both SEM analysis and EtBr accumulation assay. In addition, both 5a and 5b showed non-cytotoxicity against two normal cell lines, indicating their safety margin to the normal cells. Furthermore, these derivatives exhibited a potent inhibitory activity against the E. coli MurA enzyme compared with fosfomycin. The gamma sterilization of both compounds displayed a potent eradication effect on the active compounds' microbial load at 7.0 kGy without affecting the physicochemical properties of the antimicrobial agents. Finally, the molecular docking simulation of the promising derivatives 5a and 5b revealed binding energies S = −23.46 and −20.11 kcal mol−1 with good binding with specified receptor target compared with the fosfomycin S = −10.89 kcal mol−1, with different types of interactions inside the active site of MUR A (PDB: 1UAE). In addition, the FMOs, including HOMO, LUMO, and energy band gap, as well as the graph of MEP maps displayed the electronic distribution over the molecules identifying positive and negative regions that interact with the biological target. In conclusion, the designed phosphonate moiety could be a potential non-cytotoxic antimicrobial agent in treating UTIs as MurA inhibitors.4. Experimental section
4.1. Materials and instrumentation
All reagents of analytical grade were purchased from Sigma-Aldrich and used without further purification. All melting points were measured using a Gallenkamp melting point apparatus without corrections. The infrared spectra were recorded using a PerkinElmer FTIR 1430 spectrophotometer by the KBr disk technique. The 1H NMR spectra were recorded using a Bruker AC spectrometer (500 MHz) at 25 °C in DMSO-d6 with TMS as an internal standard, and chemical shifts are reported in ppm as δ values and 13C NMR at 126 MHz. Mass spectra were recorded using a Finnigan MAT 8222 EX mass spectrometer at 70 eV. The elemental analyzer was performed at Microanalytical center at Cairo University and the values found to be within ±0.4% of the theoretical ones unless otherwise indicated. The reaction progress was monitored by thin-layer chromatography (TLC) using benzene/acetone (3/1 by volume) as eluent.4.2. Chemistry
:1 to give compound 2.Yield: 76%; M.p.: over 360 °C; IR ν/max (cm−1): 3357 (NH2), 3095 (CH–Ar), 2966 (CH–aliph), 1565 (CN), 682 (C–S); 1H NMR (500 MHz, DMSO-d6) δ/ppm 8.56 (dd, J = 8.2, 1.4 Hz, 1H, Ar–H), 8.25 (t, J = 9.0 Hz, 1H, Ar–H), 7.88 (t, J = 10.5 Hz, 1H, Ar–H), 7.68 (d, J = 10.5 Hz, 1H, Ar–H), 7.63–7.61 (m, 1H, Ar–H), 7.59 (d, J = 2.5 Hz, 1H, Ar–H), 7.58 (t, J = 2.5 Hz, 1H, Ar–H), 7.55 (d, J = 3.0 Hz, 1H, Ar–H), 7.53 (d, J = 3.5 Hz, 1H, Ar–H), 7.41 (s, 2H, NH2 exchangeable with D2O); 13C NMR (126 MHz, DMSO-d6) δ/ppm 158.28 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –NH2), 148.96 (CN), 143.01, 140.73, 135.82, 130.97, 130.33, 130.24, 129.67, 129.48, 129.17, 127.59, 127.42, 127.26, 117.44, 116.38 (Ar–Cs); MS (EI) m/z: calcd for C16H11N5O2S [M]+, 337.06; found, 337.75; Anal. Calcd. for C16H11N5O2S (337.063): C, 56.96; H, 3.29; N, 20.76; found: C, 56.75; H, 3.42; N, 20.58.
–NH2), 148.96 (CN), 143.01, 140.73, 135.82, 130.97, 130.33, 130.24, 129.67, 129.48, 129.17, 127.59, 127.42, 127.26, 117.44, 116.38 (Ar–Cs); MS (EI) m/z: calcd for C16H11N5O2S [M]+, 337.06; found, 337.75; Anal. Calcd. for C16H11N5O2S (337.063): C, 56.96; H, 3.29; N, 20.76; found: C, 56.75; H, 3.42; N, 20.58.
N), 1252 (PO); 1061 (P–O–C), 769 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.72 (s, 1H, NH exchangeable with D2O), 8.13 (d, 4H, Ar–H), 7.83 (d, 2H, Ar–H), 7.64 (s, 1H, CH-methane), 7.44 (d, 4H, Ar–H), 6.96 (t, 3H, Ar–H), 3.85 (q, 4H, 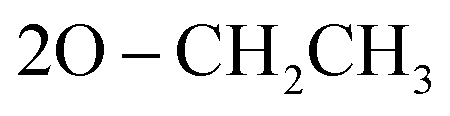 ), 1.05 (t, 6H,
), 1.05 (t, 6H,  ); 13C NMR (126 MHz, DMSO-d6) δ/ppm 145.52, 144.19 (2C = N), 138.65, 137.16, 135.85, 132.21, 130.28, 130.07, 128.92, 128.46, 125.39, 122.55, 120.27, 119.74, 119.37, 117.43, 112.53, 108.90 (Ar–Cs), 66.80 (
); 13C NMR (126 MHz, DMSO-d6) δ/ppm 145.52, 144.19 (2C = N), 138.65, 137.16, 135.85, 132.21, 130.28, 130.07, 128.92, 128.46, 125.39, 122.55, 120.27, 119.74, 119.37, 117.43, 112.53, 108.90 (Ar–Cs), 66.80 (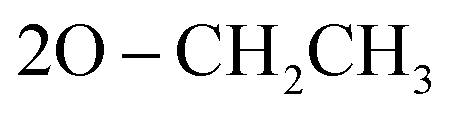 ), 59.58 (CH), 12.23 (
), 59.58 (CH), 12.23 (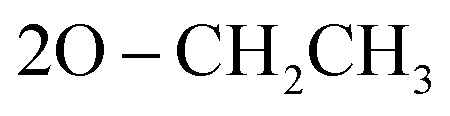 ); MS (EI) m/z: calcd for C27H25ClN5O5PS [M+], 598.01; found, 598.56; Anal. Calcd. for C27H25ClN5O5PS (702.73): C, 54.23; H, 4.21; N, 11.71; found: C, 54.34; H, 4.19; N, 11.83.N), 1277 (PO); 1095 (P–O–C), 776 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.53 (s, 1H, NH exchangeable with D2O), 8.21 (d, J = 8.5 Hz, 2H, Ar–H), 8.07 (d, J = 9.0 Hz, 1H, Ar–H), 7.57 (d, J = 9.0 Hz, 3H, Ar–H), 7.53 (d, J = 6.5 Hz, 1H, Ar–H), 7.45 (t, J = 7.75 Hz, 3H, Ar–H), 6.90 (s, 1H, CH-methane), 6.66 (d, J = 9.0 Hz, 3H, Ar–H), 3.85 (q, J = 6.5 Hz, 4H,
); MS (EI) m/z: calcd for C27H25ClN5O5PS [M+], 598.01; found, 598.56; Anal. Calcd. for C27H25ClN5O5PS (702.73): C, 54.23; H, 4.21; N, 11.71; found: C, 54.34; H, 4.19; N, 11.83.N), 1277 (PO); 1095 (P–O–C), 776 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.53 (s, 1H, NH exchangeable with D2O), 8.21 (d, J = 8.5 Hz, 2H, Ar–H), 8.07 (d, J = 9.0 Hz, 1H, Ar–H), 7.57 (d, J = 9.0 Hz, 3H, Ar–H), 7.53 (d, J = 6.5 Hz, 1H, Ar–H), 7.45 (t, J = 7.75 Hz, 3H, Ar–H), 6.90 (s, 1H, CH-methane), 6.66 (d, J = 9.0 Hz, 3H, Ar–H), 3.85 (q, J = 6.5 Hz, 4H, 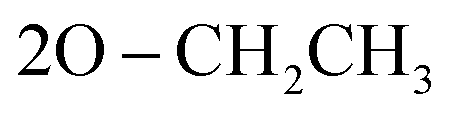 ), 2.91 (s, 6H, 2CH3), 1.19 (t, J = 7.0 Hz, 3H,
), 2.91 (s, 6H, 2CH3), 1.19 (t, J = 7.0 Hz, 3H,  ), 1.13 (t, J = 7.0 Hz, 3H,
), 1.13 (t, J = 7.0 Hz, 3H,  ); 13C NMR (126 MHz, DMSO-d6) δ/ppm 146.41, 143.75 (2C = N), 139.31, 137.87, 133.88, 133.11, 130.64, 129.18, 129.00, 128.57, 124.81, 122.97, 122.73, 121.94, 119.98, 119.71 (Ar–Cs), 66.81 (
); 13C NMR (126 MHz, DMSO-d6) δ/ppm 146.41, 143.75 (2C = N), 139.31, 137.87, 133.88, 133.11, 130.64, 129.18, 129.00, 128.57, 124.81, 122.97, 122.73, 121.94, 119.98, 119.71 (Ar–Cs), 66.81 (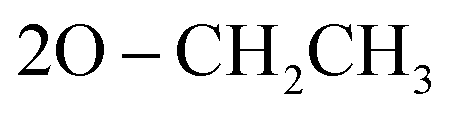 ), 64.00 (CH), 35.33 (2CH3), 22.05 (
), 64.00 (CH), 35.33 (2CH3), 22.05 ( ); MS (EI) m/z: calcd for C29H31N6O5PS [M+], 606.63; found, 606.98; Anal. Calcd. for C29H31N6O5PS (606.638): C, 57.42; H, 5.15; N, 13.85; found: C, 57.55; H, 5.09; N, 13.76.N), 1251 (PO); 1077 (P–O–C), 778 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.98 (s, 1H, OH exchangeable with D2O), 9.67 (s, 1H, NH exchangeable with D2O), 8.15 (d, J = 7.5 Hz, 2H, Ar–H), 8.03 (d, J = 8.5 Hz, 2H, Ar–H), 7.48 (d, J = 7.0 Hz, 3H, Ar–H), 7.23 (d, J = 8.0 Hz, 3H, Ar–H), 7.16 (d, J = 8.0 Hz, 2H, Ar–H), 7.10 (t, J = 8.0 Hz, 6H, Ar–H), 6.99 (t, J = 7.5 Hz, 4H, Ar–H), 6.62 (t, J = 8.25 Hz, 1H, Ar–H), 5.97 (s, 1H, CH-methane); 13C NMR (126 MHz, DMSO-d6) δ/ppm 149.86 (–OH), 148.61, 147.16 (2C = N), 140.60, 139.50, 134.76, 132.87, 130.98, 130.12, 129.88, 129.54, 129.38, 129.07, 129.00, 128.86, 125.77, 120.59, 118.34, 115.05 (Ar–Cs), 66.81 (CH); MS (EI) m/z: calcd for C35H26N5O6PS [M+], 675.65; found, 675.80; Anal. Calcd. for C35H26N5O6PS (675.65): C, 62.22; H, 3.88; N, 10.37; found: C, 62.45; H, 3.64; N, 10.12.N), 1237 (PO); 1067 (P–O–C), 779 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.52 (s, 1H, NH exchangeable with D2O), 8.16 (d, J = 8.5 Hz, 2H, Ar–H), 8.06 (d, J = 8.5 Hz, 2H, Ar–H), 7.58 (d, J = 7.5 Hz, 3H, Ar–H), 7.52 (d, J = 7.5 Hz, 1H, Ar–H), 7.41 (t, J = 8.5 Hz, 3H, Ar–H), 7.27 (d, J = 6.5 Hz, 3H, Ar–H), 7.13 (t, J = 6.5 Hz, 3H, Ar–H), 6.99 (t, J = 6.75 Hz, 2H, Ar–H), 6.86 (s, 1H, CH-methane), 6.72 (d, J = 7.0 Hz, 3H, Ar–H), 6.65 (t, J = 7.0 Hz, 1H, Ar–H), 2.91 (s, 6H, 2CH3); 13C NMR (126 MHz, DMSO-d6) δ/ppm 146.41, 143.75 (2C = N), 139.31, 137.87, 133.88, 133.11, 130.64, 129.18, 129.00, 128.57, 124.81, 122.97, 122.73, 121.94, 119.98, 119.71 (Ar–Cs), 64.00 (CH), 35.33 (2CH3); MS (EI) m/z: calcd for C37H31N6O5PS [M] +, 702.73; found, 702.99; Anal. Calcd. for C37H31N6O5PS (702.73): C, 63.24; H, 4.45; N, 11.96; found: C, 63.09; H, 4.74; N, 11.71.N), 1255 (PO); 1076 (P–O–C), 776 (P–CH);1H NMR (500 MHz, DMSO-d6) δ/ppm 9.72 (s, 1H, NH exchangeable with D2O), 8.17 (d, J = 7.5 Hz, 2H, Ar–H), 8.05 (d, J = 6.0 Hz, 2H, Ar–H), 7.86 (t, J = 7.75 Hz, 1H, Ar–H), 7.70 (d, J = 8.0 Hz, 2H, Ar–H), 7.50 (d, J = 7.5 Hz, 2H, Ar–H), 7.40 (t, J = 7.75 Hz, 3H, Ar–H), 7.27 (t, d, J = 7.5 Hz, 3H, Ar–H), 7.12 (d, J = 7.5 Hz, 2H, Ar–H), 7.00 (t, d, J = 7.75 Hz, 3H, Ar–H), 6.64 (d, J = 7.5 Hz, 2H, Ar–H), 5.99 (s, 1H, CH-methane); 13C NMR (126 MHz, DMSO-d6) δ/ppm 149.35, 147.92 (2C = N), 146.50, 144.83, 140.62, 138.62, 136.99, 134.20, 129.49, 128.88, 128.03, 121.95, 121.18, 120.65, 114.41, 66.84 (CH); MS (EI) m/z: calcd for C33H24N5O5PS2 [M+], 665.67; found, 665.74; Anal. Calcd. for C33H24N5O5PS2 (665.67): C, 59.54; H, 3.63; N, 10.52; found: C, 59.49; H, 3.71; N, 10.52.N), 1264 (PO); 1069 (P–O–C), 776 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.29 (s, 1H, NH exchangeable with D2O), 7.42 (t, J = 7.75 Hz, 1H, Ar–H), 7.33 (s, 1H, NH exchangeable with D2O), 7.28 (d, J = 8.5 Hz, 2H, Ar–H), 7.25 (d, J = 8.0 Hz, 2H, Ar–H), 7.20 (t, J = 7.0 Hz, 3H, Ar–H), 7.13 (t, J = 8.25 Hz, 6H, Ar–H), 7.08 (d, J = 7.5 Hz, 2H, Ar–H), 7.02 (d, J = 8.0 Hz, 2H, Ar–H), 6.99 (d, J = 9.5 Hz, 2H, Ar–H), 6.64 (d, J = 7.5 Hz, 2H, Ar–H), 6.00 (s, 1H, CH-methane); 13C NMR (126 MHz, DMSO-d6) δ/ppm 149.86, 148.61 (2C = N), 147.16, 140.60, 139.50, 134.76, 132.87, 131.92, 131.51, 130.98, 130.12, 129.88, 129.54, 129.44, 129.38, 129.07, 129.00, 128.86, 125.77, 120.59, 118.34, 115.05, 66.81 (CH); MS (EI) m/z: calcd for C33H25N6O5PS [M+], 648.63; found, 648.73; Anal. Calcd. for C33H25N6O5PS (648.63): C, 61.11; H, 3.89; N, 12.96; found: C, 61.25; H, 3.91; N, 12.86.
); MS (EI) m/z: calcd for C29H31N6O5PS [M+], 606.63; found, 606.98; Anal. Calcd. for C29H31N6O5PS (606.638): C, 57.42; H, 5.15; N, 13.85; found: C, 57.55; H, 5.09; N, 13.76.N), 1251 (PO); 1077 (P–O–C), 778 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.98 (s, 1H, OH exchangeable with D2O), 9.67 (s, 1H, NH exchangeable with D2O), 8.15 (d, J = 7.5 Hz, 2H, Ar–H), 8.03 (d, J = 8.5 Hz, 2H, Ar–H), 7.48 (d, J = 7.0 Hz, 3H, Ar–H), 7.23 (d, J = 8.0 Hz, 3H, Ar–H), 7.16 (d, J = 8.0 Hz, 2H, Ar–H), 7.10 (t, J = 8.0 Hz, 6H, Ar–H), 6.99 (t, J = 7.5 Hz, 4H, Ar–H), 6.62 (t, J = 8.25 Hz, 1H, Ar–H), 5.97 (s, 1H, CH-methane); 13C NMR (126 MHz, DMSO-d6) δ/ppm 149.86 (–OH), 148.61, 147.16 (2C = N), 140.60, 139.50, 134.76, 132.87, 130.98, 130.12, 129.88, 129.54, 129.38, 129.07, 129.00, 128.86, 125.77, 120.59, 118.34, 115.05 (Ar–Cs), 66.81 (CH); MS (EI) m/z: calcd for C35H26N5O6PS [M+], 675.65; found, 675.80; Anal. Calcd. for C35H26N5O6PS (675.65): C, 62.22; H, 3.88; N, 10.37; found: C, 62.45; H, 3.64; N, 10.12.N), 1237 (PO); 1067 (P–O–C), 779 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.52 (s, 1H, NH exchangeable with D2O), 8.16 (d, J = 8.5 Hz, 2H, Ar–H), 8.06 (d, J = 8.5 Hz, 2H, Ar–H), 7.58 (d, J = 7.5 Hz, 3H, Ar–H), 7.52 (d, J = 7.5 Hz, 1H, Ar–H), 7.41 (t, J = 8.5 Hz, 3H, Ar–H), 7.27 (d, J = 6.5 Hz, 3H, Ar–H), 7.13 (t, J = 6.5 Hz, 3H, Ar–H), 6.99 (t, J = 6.75 Hz, 2H, Ar–H), 6.86 (s, 1H, CH-methane), 6.72 (d, J = 7.0 Hz, 3H, Ar–H), 6.65 (t, J = 7.0 Hz, 1H, Ar–H), 2.91 (s, 6H, 2CH3); 13C NMR (126 MHz, DMSO-d6) δ/ppm 146.41, 143.75 (2C = N), 139.31, 137.87, 133.88, 133.11, 130.64, 129.18, 129.00, 128.57, 124.81, 122.97, 122.73, 121.94, 119.98, 119.71 (Ar–Cs), 64.00 (CH), 35.33 (2CH3); MS (EI) m/z: calcd for C37H31N6O5PS [M] +, 702.73; found, 702.99; Anal. Calcd. for C37H31N6O5PS (702.73): C, 63.24; H, 4.45; N, 11.96; found: C, 63.09; H, 4.74; N, 11.71.N), 1255 (PO); 1076 (P–O–C), 776 (P–CH);1H NMR (500 MHz, DMSO-d6) δ/ppm 9.72 (s, 1H, NH exchangeable with D2O), 8.17 (d, J = 7.5 Hz, 2H, Ar–H), 8.05 (d, J = 6.0 Hz, 2H, Ar–H), 7.86 (t, J = 7.75 Hz, 1H, Ar–H), 7.70 (d, J = 8.0 Hz, 2H, Ar–H), 7.50 (d, J = 7.5 Hz, 2H, Ar–H), 7.40 (t, J = 7.75 Hz, 3H, Ar–H), 7.27 (t, d, J = 7.5 Hz, 3H, Ar–H), 7.12 (d, J = 7.5 Hz, 2H, Ar–H), 7.00 (t, d, J = 7.75 Hz, 3H, Ar–H), 6.64 (d, J = 7.5 Hz, 2H, Ar–H), 5.99 (s, 1H, CH-methane); 13C NMR (126 MHz, DMSO-d6) δ/ppm 149.35, 147.92 (2C = N), 146.50, 144.83, 140.62, 138.62, 136.99, 134.20, 129.49, 128.88, 128.03, 121.95, 121.18, 120.65, 114.41, 66.84 (CH); MS (EI) m/z: calcd for C33H24N5O5PS2 [M+], 665.67; found, 665.74; Anal. Calcd. for C33H24N5O5PS2 (665.67): C, 59.54; H, 3.63; N, 10.52; found: C, 59.49; H, 3.71; N, 10.52.N), 1264 (PO); 1069 (P–O–C), 776 (P–CH); 1H NMR (500 MHz, DMSO-d6) δ/ppm 9.29 (s, 1H, NH exchangeable with D2O), 7.42 (t, J = 7.75 Hz, 1H, Ar–H), 7.33 (s, 1H, NH exchangeable with D2O), 7.28 (d, J = 8.5 Hz, 2H, Ar–H), 7.25 (d, J = 8.0 Hz, 2H, Ar–H), 7.20 (t, J = 7.0 Hz, 3H, Ar–H), 7.13 (t, J = 8.25 Hz, 6H, Ar–H), 7.08 (d, J = 7.5 Hz, 2H, Ar–H), 7.02 (d, J = 8.0 Hz, 2H, Ar–H), 6.99 (d, J = 9.5 Hz, 2H, Ar–H), 6.64 (d, J = 7.5 Hz, 2H, Ar–H), 6.00 (s, 1H, CH-methane); 13C NMR (126 MHz, DMSO-d6) δ/ppm 149.86, 148.61 (2C = N), 147.16, 140.60, 139.50, 134.76, 132.87, 131.92, 131.51, 130.98, 130.12, 129.88, 129.54, 129.44, 129.38, 129.07, 129.00, 128.86, 125.77, 120.59, 118.34, 115.05, 66.81 (CH); MS (EI) m/z: calcd for C33H25N6O5PS [M+], 648.63; found, 648.73; Anal. Calcd. for C33H25N6O5PS (648.63): C, 61.11; H, 3.89; N, 12.96; found: C, 61.25; H, 3.91; N, 12.86.4.3. Biological evaluation
| ΣFICI = (MIC of drug A in combinstion/MIC of drug A) + (MIC of drug B in combination/MIC of drug B) |
In which, drug A is the antibiotic and drug B is the target compound; FICI ≤ 0.5 (synergism); 0.5 < FICI > 1 (partial synergism); 1 ≤ FICI < 2 (indifference); FICI ≥ 2 (antagonism).
4.4. Molecular docking simulation
The molecular docking study for the most active diphenyl α-aminophosphonate derivatives 5a, 5b and fosfomycin (as positive control) was carried out inside the active site of UDP-N-acetylglucosamine enolpyruvyl transferase (MUR A, PDB: 1UAE). The 3D crystal structure has been retrieved from the protein data bank (https://www.rcsb.org/structure/1uae. Accessed on 21/3/2023),57 where this protein is isolated from E. coli with resolution =1.80 Å and present in the complex with a co-crystallized ligand uridine-diphosphate-n-acetylglucosamine (UD1). The docking simulation was performed using the Molecular Operating Environmental (MOE) software75–77 version 10.2009. The validation process was performed using the triangle matcher placement, and London dG as rescoring 1 with refinement known as forcefield and retain thirty pose, as well as removing the duplication according to the reported method.78 The validation process exhibited that the co-crystallized ligand showed binding energy S =−17.69 kcal mol−1 with RMSD = 1.53 Å, where the superimposition of the co-crystallized ligand (turquoise) and the re-docked co-crystallized (green) are described in the ESI† file. The co-crystallized ligand (UD1) exhibited five hydrogen bond sidechain acceptors with bond lengths as Arg91 (1.75 Å, 84%), ser162 (1.92 Å, 45%), Lys160 (1.74 Å, 62%), and Arg120 (1.97 Å with 51% and 2.07 Å with 23%). Moreover, the UD1 displayed two sidechain donors with residues Asp305 (1.90 Å, 35%) and Asp123 (1.73 Å, 24%) and three hydrogen bond backbone acceptor with residues Gly164 (1.99 Å, 43%), Val163 (1.84 Å, 63%), and Val327 (1.81 Å, 18%). The structure of the diphenyl α-aminophosphonate derivatives 5a, 5b and fosfomycin was built using ChemBioDraw 2014, and the structures were exported to MOE; then, the structures were subjected to protonated 3D, and energy was minimized using gradient 0.0001, as described previously.79,804.5. Quantum chemical studies
The quantum chemical studies, including frontier molecular orbital model (FMOs) and electrostatic potential, were calculated using density functional theory (DFT) in Gaussian 09W and visualized using Gaussian View 6.0. In addition, the molecules were optimized using hybrid functional B3LYP and basis set 6-31G(d), as described previously.81–83Conflicts of interest
The authors declare no conflicts of interest.References
- A. Y. Alzahrani, Y. A. Ammar, M. Abu-Elghait, M. A. Salem, M. A. Assiri, T. E. Ali and A. Ragab, Bioorg. Chem., 2022, 119, 105571 CrossRef CAS PubMed.
- E. D. Naydenova, P. T. Todorov, P. I. Mateeva, R. N. Zamfirova, N. D. Pavlov and S. B. Todorov, Amino Acids, 2010, 39, 1537–1543 CrossRef CAS PubMed.
- T. N. M. An, N. Van Cuong, N. M. Quang, T. V. Thanh and M. Alam, ChemistrySelect, 2020, 5, 6339–6349 CrossRef CAS.
- S. Shaikh, P. Dhavan, G. Pavale, M. M. V Ramana and B. L. Jadhav, Bioorg. Chem., 2020, 96, 103589 CrossRef CAS PubMed.
- H. Patnala, H. S. Abbo, K. M. Potla, S. J. J. Titinchi and S. Chinnam, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 1035–1039 CrossRef CAS.
- B. Sujatha, S. Mohan, C. Subramanyam and K. P. Rao, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 1110–1113 CrossRef CAS.
- A. K. Bhattacharya, K. C. Rana, C. Pannecouque and E. De Clercq, ChemMedChem, 2012, 7, 1601–1611 CrossRef CAS PubMed.
- M. A. Azzam, H. A. L. El-Boraey and I. E. T. El-Sayed, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 195, 339–347 CrossRef CAS.
- R.-Z. Huang, C.-Y. Wang, J.-F. Li, G.-Y. Yao, Y.-M. Pan, M.-Y. Ye, H.-S. Wang and Y. Zhang, RSC Adv., 2016, 6, 62890–62906 RSC.
- J. Che, X. Xu, Z. Tang, Y. Gu and D. Shi, Bioorg. Med. Chem. Lett., 2016, 26, 1310–1313 CrossRef CAS PubMed.
- Z. Jiang, J. Zhao, B. Gao, S. Chen, W. Qu, X. Mei, C. Rui, J. Ning and D. She, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1026–1037 CrossRef CAS.
- H. Singh and A. K. Jain, J. Appl. Polym. Sci., 2009, 111, 1115–1143 CrossRef CAS.
- H. Ai, K. Xu, H. Liu, M. Chen and X. Zhang, J. Appl. Polym. Sci., 2009, 113, 541–546 CrossRef CAS.
- J. Canadell, B. J. Hunt, A. G. Cook, A. Mantecón and V. Cádiz, Polym. Degrad. Stab., 2007, 92, 1482–1490 CrossRef CAS.
- M. Salasi, T. Shahrabi, E. Roayaei and M. Aliofkhazraei, Mater. Chem. Phys., 2007, 104, 183–190 CrossRef CAS.
- K. Kavipriya, S. Rajendran, J. Sathiyabama and A. S. Prabha, Eur. Chem. Bull., 2012, 1, 366–374 CAS.
- N. Moszner, F. Zeuner, U. K. Fischer and V. Rheinberger, Macromol. Chem. Phys., 1999, 200, 1062–1067 CrossRef CAS.
- I. Cabasso, J. Smid and S. K. Sahni, J. Appl. Polym. Sci., 1990, 41, 3025–3042 CrossRef CAS.
- L. H. Taboada, M. Guzmann, K. Neubecker and A. Göthlich, US Pat., 12/520642, 2010 Search PubMed.
- M. Ordóñez, H. Rojas-Cabrera and C. Cativiela, Tetrahedron, 2009, 65, 17–49 CrossRef PubMed.
- C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed.
- H. Yu, H. Yang, E. Shi and W. Tang, Med. Drug Discovery, 2020, 8, 100063 CrossRef CAS PubMed.
- F. Cambazard, J. Eur. Acad. Dermatol. Venereol., 1998, 11, S20–S27 CrossRef PubMed.
- J. M. Rybak and K. Roberts, Infect. Dis. Ther., 2015, 4, 1–14 CrossRef PubMed.
- K. Richardson, K. Cooper, M. S. Marriott, M. H. Tarbit, F. Troke and P. J. Whittle, Rev. Infect. Dis., 1990, 12, S267–S271 CrossRef CAS PubMed.
- K. Papangkorn, K. R. Truett, A. T. Vitale, C. Jhaveri, D. K. Scales, C. S. Foster, A. Montieth, J. W. Higuchi, B. Brar and W. I. Higuchi, Curr. Eye Res., 2019, 44, 185–193 CrossRef CAS PubMed.
- D. R. Budman, L. N. Igwemezie, S. Kaul, J. Behr, S. Lichtman, P. Schulman, V. Vinciguerra, S. L. Allen, J. Kolitz and K. Hock, J. Clin. Oncol., 1994, 12, 1902–1909 CrossRef CAS PubMed.
- S. Pol, M. Corouge and A. Vallet-Pichard, Hepatic Med. Evid. Res., 2016, 8, 21–26 CrossRef PubMed.
- B. Litim, A. Djahoudi, S. Meliani and A. Boukhari, Med. Chem. Res., 2022, 31, 60–74 CrossRef CAS PubMed.
- G. Sravya, A. Balakrishna, G. V. Zyryanov, G. Mohan, C. S. Reddy and N. Bakthavatchala Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196, 353–381 CrossRef CAS.
- C. S. Transactions, Chem. Sci. Trans., 2019, 8, 359–367 Search PubMed.
- G. Gao, M.-N. Chen, L.-P. Mo and Z.-H. Zhang, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 528–532 CrossRef CAS.
- P. R. Varga and G. Keglevich, Molecules, 2021, 26, 2511 CrossRef CAS PubMed.
- S. A. Ibrahim, E. A. Fayed, H. F. Rizk, S. E. Desouky and A. Ragab, Bioorg. Chem., 2021, 116, 105339 CrossRef CAS PubMed.
- G. S. Masaret, ChemistrySelect, 2021, 6, 974–982 CrossRef CAS.
- A. R. Sayed, S. M. Gomha, F. M. Abdelrazek, M. S. Farghaly, S. A. Hassan and P. Metz, BMC Chem., 2019, 13, 116 CrossRef PubMed.
- S. J. Takate, A. D. Shinde, B. K. Karale, H. Akolkar, L. Nawale, D. Sarkar and P. C. Mhaske, Bioorg. Med. Chem. Lett., 2019, 29, 1199–1202 CrossRef CAS PubMed.
- H. Barreteau, A. Kovač, A. Boniface, M. Sova, S. Gobec and D. Blanot, FEMS Microbiol. Rev., 2008, 32, 168–207 CrossRef CAS PubMed.
- M. Hrast, I. Sosič, R. Šink and S. Gobec, Bioorg. Chem., 2014, 55, 2–15 CrossRef CAS PubMed.
- A. Gautam, P. Rishi and R. Tewari, Appl. Microbiol. Biotechnol., 2011, 92, 211–225 CrossRef CAS PubMed.
- T. Van Cauwenbergh, E. Theys, D. Stroeykens, B. Croonenborghs, A. Gillet, A. DeMent, A. Van Schepdael and E. Haghedooren, J. Pharm. Sci., 2022, 111, 2011–2017 CrossRef CAS PubMed.
- E. A. Fayed, M. Mohsen, S. M. A. El-Gilil, D. S. Aboul-Magd and A. Ragab, J. Mol. Struct., 2022, 1262, 133028 CrossRef CAS.
- S. A. Ibrahim, H. F. Rizk, D. S. Aboul-Magd and A. Ragab, Dyes Pigm., 2021, 193, 109504 CrossRef CAS.
- A. Ragab, M. S. Abusaif, N. A. Gohar, D. S. Aboul-Magd, E. A. Fayed and Y. A. Ammar, Bioorg. Chem., 2023, 131, 106307 CrossRef CAS PubMed.
- S. A. Ibrahim, A. Ragab and H. A. El-Ghamry, Appl. Organomet. Chem., 2022, 36, 1–17 Search PubMed.
- H. F. Rizk, M. A. El-Borai, A. Ragab, S. A. Ibrahim and M. E. Sadek, Polycyclic Aromat. Compd., 2023, 43, 500–522 CrossRef CAS.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, Drug Dev. Res., 2023, 84, 3–24 CrossRef CAS PubMed.
- H. F. Rizk, M. A. El-Borai, A. Ragab and S. A. Ibrahim, J. Iran. Chem. Soc., 2020, 17, 2493–2505 CrossRef CAS.
- Y. A. Ammar, J. A. Micky, D. S. Aboul-Magd, S. M. A. Abd El-Hafez, S. A. Hessein, A. M. Ali and A. Ragab, Chem. Biol. Drug Des., 2023, 101(2), 245–270 CrossRef CAS PubMed.
- M. M. Abdelgalil, Y. A. Ammar, G. A. M. Elhag Ali, A. K. Ali and A. Ragab, J. Mol. Struct., 2023, 1274, 134443 CrossRef CAS.
- H. Mohammad, A. S. Mayhoub, M. Cushman and M. N. Seleem, J. Antibiot., 2015, 68, 259–266 CrossRef CAS PubMed.
- A. Ragab, M. S. Abusaif, D. S. Aboul-Magd, M. M. S. Wassel, G. A. M. Elhagali and Y. A. Ammar, Drug Dev. Res., 2022, 1305–1330 CrossRef CAS PubMed.
- A. Ragab, S. A. Fouad, Y. A. Ammar, D. S. Aboul-Magd and M. S. Abusaif, Antibiotics, 2023, 12(1), 128 CrossRef CAS PubMed.
- Y. A. Ammar, A. A. Farag, A. M. Ali, S. A. Hessein, A. A. Askar, E. A. Fayed, D. M. Elsisi and A. Ragab, Bioorg. Chem., 2020, 99, 103841 CrossRef CAS PubMed.
- W.-J. Lu, H.-J. Lin, T. K. Janganan, C.-Y. Li, W.-C. Chin, V. N. Bavro and H.-T. V. Lin, Int. J. Mol. Sci., 2018, 19(4), 1000 CrossRef PubMed.
- T. J. Opperman, S. M. Kwasny, K. Hong-Suk, S. T. Nguyen, H. Chad, D. Sanjay, G. C. Walker, N. P. Peet, N. Hiroshi and T. L. Bowlin, Antimicrob. Agents Chemother., 2014, 58, 722–733 CrossRef PubMed.
- https://www.rcsb.org/structure/1t2w, accessed on 20/7/2022.
- A. A. Ali, H. Abd El-Wahab, M. S. Abusaif, A. Ragab, O. A. Abdel-Jaid, E. A. Eldeeb and Y. A. Ammar, Pigm. Resin Technol., 2023 DOI:10.1108/PRT-12-2022-0141.
- S. A. El-Kalyoubi, A. Ragab, O. A. Abu Ali, Y. A. Ammar, M. G. Seadawy, A. Ahmed and E. A. Fayed, Pharmaceuticals, 2022, 15(3), 367 CrossRef PubMed.
- R. Ayman, M. S. Abusaif, A. M. Radwan, A. M. Elmetwally and A. Ragab, Eur. J. Med. Chem., 2023, 249, 115138 CrossRef CAS PubMed.
- D. M. Elsisi, A. Ragab, A. A. Elhenawy, A. A. Farag, A. M. Ali and Y. A. Ammar, J. Mol. Struct., 2022, 1247, 131314 CrossRef CAS.
- R. Humphries, A. M. Bobenchik, J. A. Hindler and A. N. Schuetz, J. Clin. Microbiol., 2021, 59(12), e0021321 CrossRef PubMed.
- B. D. Alexander, G. W. Procop, P. Dufresne, J. Fuller and M. A. Ghannoum, Clin. Lab. Stand. Inst., 2017, 3, M27-A3 Search PubMed.
- B. T. Tsuji, J. C. Yang, A. Forrest, P. A. Kelchlin and P. F. Smith, J. Antimicrob. Chemother., 2008, 62, 156–160 CrossRef CAS PubMed.
- H. Ali Mohamed, Y. A. Ammar, G. A. M. Elhagali, H. A. Eyada, D. S. Aboul-Magd and A. Ragab, ACS Omega, 2022, 7, 4970–4990 CrossRef CAS PubMed.
- S. Kumar, V. Saini, I. K. Maurya, J. Sindhu, M. Kumari, R. Kataria and V. Kumar, PLoS One, 2018, 13, e0196016 CrossRef PubMed.
- A. G. Dale, J. Hinds, J. Mann, P. W. Taylor and S. Neidle, Biochemistry, 2012, 51, 5860–5871 CrossRef CAS PubMed.
- A. R. Brown, K. A. Ettefagh, D. Todd, P. S. Cole, J. M. Egan, D. H. Foil, T. N. Graf, B. D. Schindler, G. W. Kaatz and N. B. Cech, PLoS One, 2015, 10, e0124814 CrossRef PubMed.
- A. Perdih, M. Hrast, H. Barreteau, S. Gobec, G. Wolber and T. Solmajer, Bioorg. Med. Chem., 2014, 22, 4124–4134 CrossRef CAS PubMed.
- B. Plackett, Nature, 2020, 586, S50+ CrossRef CAS.
- S. Abdou and S. Ebraheem, Egypt J. Radiat. Res. Appl., 2018, 31, 137–142 Search PubMed.
- Ş. Çolak, Evolution of Ionizing Radiation Research, 2015, vol. 12, pp. 281–306 Search PubMed.
- S. H. M. Husseiny and F. A. Helimish, World Appl. Sci. J., 2012, 19, 847–855 Search PubMed.
- A. Özer, S. Turker, S. Çolak, M. Korkmaz, E. Kiliç and M. Özalp, Interv. Med. Appl. Sci., 2013, 5, 122–130 Search PubMed.
- H. Ali Mohamed, Y. A. Ammar, G. A. M. Elhagali, H. A. Eyada, D. S. Aboul-Magd and A. Ragab, J. Mol. Struct., 2023, 1287(5), 135671 CrossRef.
- E. S. A. E. H. Khattab, A. Ragab, M. A. Abol-Ftouh and A. A. Elhenawy, J. Biomol. Struct. Dyn., 2022, 40, 1–19 CrossRef PubMed.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, RSC Adv., 2023, 13, 9281–9303 RSC.
- K. Rožman, S. Lešnik, B. Brus, M. Hrast, M. Sova, D. Patin, H. Barreteau, J. Konc, D. Janežič and S. Gobec, Bioorg. Med. Chem. Lett., 2017, 27, 944–949 CrossRef PubMed.
- M. Eldeeb, E. F. Sanad, A. Ragab, Y. A. Ammar, K. Mahmoud, M. M. Ali and N. M. Hamdy, Biomedicines, 2022, 10, 722 CrossRef CAS PubMed.
- A. Ragab, Y. A. Ammar, A. Ezzat, A. M. Mahmoud, M. Basseem, I. Mohamed, A. S. El-tabl and R. S. Farag, Comput. Biol. Med., 2022, 145, 105473 CrossRef CAS PubMed.
- K. E. Saadon, N. M. H. Taha, N. A. Mahmoud, G. A. M. Elhagali and A. Ragab, J. Iran. Chem. Soc., 2022, 19, 3899–3917 CrossRef CAS.
- R. Ayman, A. M. Radwan, A. M. Elmetwally, Y. A. Ammar and A. Ragab, Arch. Pharm., 2023, 365, 2200395 CrossRef PubMed.
- M. S. Abusaif, A. M. Hyba, Y. A. Ammar, M. A. Salem, D. M. Elsisi and A. Ragab, J. Taiwan Inst. Chem. Eng., 2023, 153, 105207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07040a |
| This journal is © The Royal Society of Chemistry 2023 |