
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of WOx and dopants (ZrO2 and SiO2) on CeO2-based nanostructure catalysts in the selective oxidation of benzyl alcohol to benzaldehyde under ambient conditions
Ganeshbabu Bathulaab,
Surjyakanta Ranacd,
Siddaramagoud Bandallaa,
Vijaykumar Dosarapua,
Satyanarayana Mavurapu a,
Aswathi Rajeevan V. V.e,
Bhaskar Sharmaf,
Sreekantha B. Jonnalagadda*d,
Mallesham Baithy*e and
Chandra Shekar Vasam*af
a,
Aswathi Rajeevan V. V.e,
Bhaskar Sharmaf,
Sreekantha B. Jonnalagadda*d,
Mallesham Baithy*e and
Chandra Shekar Vasam*af
aDepartment of Pharmaceutical Chemistry, Telangana University, Nizamabad-503322, TS, India. E-mail: csvasamsa@gmail.com
bDepartment of Chemistry, Satavahana University, Karimnagar-505002, TS, India
cFunGlass – Centre for Functional and Surface Functionalized Glass & University of Antwerp, Belgium
dSchool of Chemistry & Physics, University of KwaZulu-Natal, Durban, South Africa. E-mail: Jonnalagaddas@ukzn.ac.za
eDepartment of Chemistry, School of Science, Gandhi Institute of Technology and Management (GITAM) University, Hyderabad – 502329, Telangana, India. E-mail: mbaithy@gitam.edu
fDepartment of Chemistry, Guru Ghasidas Vishwavidyalaya, Bilaspur-495009, CG, India
First published on 12th December 2023
Abstract
Herein, the efficacy of WOx-promoted CeO2–SiO2 and CeO2–ZrO2 mixed oxide catalysts in the solvent-free selective oxidation of benzyl alcohol to benzaldehyde using molecular oxygen as an oxidant is reported. We evaluated the effects of the oxidant and catalyst concentration, reaction duration, and temperature on the reaction with an aim to optimize the reaction conditions. The as-prepared CeO2, CeO2–ZrO2, CeO2–SiO2, WOx/CeO2, WOx/CeO2–ZrO2, and WOx/CeO2–SiO2 catalysts were characterized by X-ray diffraction (XRD), N2 adsorption–desorption, Raman spectroscopy, temperature-programmed desorption of ammonia (TPD-NH3), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM). These characterisation results indicated that the WOx/CeO2–SiO2 catalyst possessed improved physicochemical (i.e., structural, textural, and acidic) properties owing to the strong interactivity between WOx and CeO2–SiO2. A higher number of Ce3+ ions (Iu′′′/ITotal) were created with the WOx/CeO2–SiO2 catalyst than those with the other catalysts in this work, indicating the generation of a high number of oxygen vacancies. The WOx/CeO2–SiO2 catalyst exhibited a high conversion of benzyl alcohol (>99%) and a high selectivity (100%) toward benzaldehyde compared to the other promoted catalysts (i.e., WOx/CeO2 and WOx/CeO2–ZrO2), which is attributed to the smaller particle size of the WOx and CeO2 and their high specific surface area, more significant number of acidic sites, and superior number of oxygen vacancies. The WOx/CeO2–SiO2 catalyst could be quickly recovered and utilized at least five times without suffering any appreciable activity loss.
1 Introduction
The selective oxidation of aromatic alcohols to aldehydes is an important reaction in basic research and industrial processes.1–4 Among aldehydes, benzaldehyde (BAL) is a versatile intermediate and highly-valuable chemical that is widely used in the pharmaceutical, dyestuff, food, cosmetic, and agrochemical industries.5–9 However, the wide range of applications of benzyl alcohol (BOL) oxidation to BAL depends on improving its cost effectiveness and sustainability, which is intricately linked to the use of green oxidants, solvents, and catalysts.Among the oxidants, hydrogen peroxide (H2O2), tert-butyl hydroperoxide (t-BOOH), and molecular oxygen (O2) have been selected and broadly studied. However, H2O2- and t-BOOH-based oxidation reactions show several serious disadvantages.1 Significantly, H2O2 decomposes quickly under mild conditions, often requiring stabilizers, such as acetic acid and phosphoric acid. The storage and transportation of H2O2 is also a problem that needs to be addressed owing to its poor stability.10 Even though persulfates (peroxymonosulfate and peroxydisulfate) are highly favourable inexpensive solid oxidants, their practical use to oxidize BOL is limited because of environmental concerns. Concerning the issues with t-BOOH, it is temperature sensitive and can spontaneously decompose under certain conditions, leading to safety issues during its handling and storage. There may also be selectivity issues in certain reactions owing to the generation of multiple radical species, which can lead to the formation of unsolicited by-products. These limitations highlight the need for alternative oxidants that can offer better efficiency, selectivity, and safety profiles. Campbell et al.11 reported that molecular oxygen (O2) is a perfect oxidant for catalytic oxidation reactions. The catalytic oxidation of BOL has also been performed in various organic solvents, including dimethyl sulfoxide (DMSO), toluene, diethoxyethane, benzene, dimethyl formamide (DMF), and chloroform.12–15 These procedures generally produce unwanted organic waste, and the solvents and oxidants raise severe environmental issues, which motivated the search for alternative catalytic oxidation processes.16–18 Most researchers have focused on inexpensive and non-toxic reagents adhering to “green chemistry” principles.19 Due to the solid economic and environmental motivations for using molecular oxygen, many chemical industries often use aerobic oxidation processes. Therefore, employing clean and inexpensive oxidants, such as molecular oxygen, in combination with supported or promoted metal oxide nanoparticulate catalysts seems to be appropriate for the solvent-free selective oxidation of various alcohols.14
For this purpose, various researchers have explored competent and reusable solid catalysts, such as Rh, Au, Pt, Pd, Co, Cu, Mo, and W.1,20 Based on these studies, elements with different oxidation states can produce a reversible redox cycle, enabling reactive oxygen species to be generated from the support matrix and preservation of the intrinsic active sites.40 Tungsten oxide (WOx) is an abundant and inexpensive transition metal with different oxidation states, which is often used as an alternative to noble metals in various oxidation reactions and has demonstrated promising catalytic efficiency.1 Especially, promoted WOx materials are one of the essential classes of solid acid catalysts used in various chemical industries due to their non-toxic nature, strong acidity, notable stability in water, hydrogen, and oxygen atmospheres, and excellent recyclability performance.21–23 The hydroxyl groups on the support material can also display a Brønsted acidic nature, but their interaction with the WOx sites will improve the acidic strength.20 Besides, it is noticeable that the CeO2-based mixed oxide support materials can play an essential role in various oxidation reactions (e.g. three-way catalysis, soot oxidation, water–gas-shift reaction, and oxidation of amines and alcohols),22–26 because of their unique redox properties, ample oxygen vacancies, sufficient acid–base properties, smaller crystalline size, and higher specific surface area than pristine CeO2.27–29 Then, the selection of suitable isovalent/aliovalent Lewis acidic cations to incorporate into the crystal lattice of CeO2 is critical since the oxide source (metal/non-metal) of an isovalent/aliovalent cation will influence the physicochemical properties of the CeO2. Hence, various iso- and aliovalent metal/non-metal cations have been investigated as dopant agents for CeO2 to improve the thermal stability and oxygen storage capacity. Specifically, Si4+ and Zr4+ are both known for their ability to modify the electronic and structural properties of host materials. Incorporating Si4+ and Zr4+ ions into ceria-based catalysts can induce lattice distortions, create oxygen vacancies, and alter the redox behaviour, which can collectively enhance the catalytic performance.
Therefore, our work was designed to demonstrate the efficacy of WOx-promoted isovalent Si4+ and Zr4+ (50% wt)-doped CeO2-based catalysts in the solvent-free selective oxidation of benzyl alcohol in the presence of green and inexpensive molecular oxygen (O2) as an oxidant. For this task, we prepared pure CeO2, CeO2–ZrO2, CeO2–SiO2, WOx/CeO2, WOx/CeO2–ZrO2, and WOx/CeO2–SiO2 catalysts (abbreviated as Ce, CeZr, CeSi, W/Ce, W/CeZr, and W/CeSi oxides) by precipitation and wet-impregnation procedures. The prepared catalysts were characterized by X-ray powder diffraction (XRD), N2 adsorption–desorption, Raman spectroscopy, temperature-programmed desorption of ammonia (NH3-TPD), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM), to determine their structural, textural, and acid properties. We investigated the catalyst's efficiency in the selective oxidation of benzyl alcohol in a simple batch reactor under solvent-free conditions. We also tried correlating the structure–activity relationship between the catalytic efficiency and characterisation properties.
2 Experimental section
2.1. Preparation of the catalyst
CeZr and CeSi oxide solid solutions with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 molar ratio were synthesised from ultra-dilute aqueous solution by a simple co-precipitation method. In brief, to obtain CeZr and CeSi samples, the required quantities of Ce(NO3)3·6H2O [99.99%, Aldrich], ZrO(NO3)2·xH2O [99%, Aldrich], tetraethyl orthosilicate (TEOS) [Sigma-Aldrich] were dissolved in de-ionised water and stirred for 30 min. Meanwhile, the NH3 aqueous solution was prepared and added drop-wise to the above-mentioned mixed solutions to reach a suitable pH of pH 9. The precipitate was decanted, centrifugated, washed several times with de-ionised water to remove weakly adherent impurities on the surface, and dried in a hot air oven at 120 °C for 12 h. The dried products CeZr and CeSi oxides were pulverised using an agate mortar. Later, they were calcined in a muffle furnace at 500 °C for 5 h at a heating rate of 5 °C min−1 in static air. The same procedure was also used to obtain the pristine CeO2 material.
:50 molar ratio were synthesised from ultra-dilute aqueous solution by a simple co-precipitation method. In brief, to obtain CeZr and CeSi samples, the required quantities of Ce(NO3)3·6H2O [99.99%, Aldrich], ZrO(NO3)2·xH2O [99%, Aldrich], tetraethyl orthosilicate (TEOS) [Sigma-Aldrich] were dissolved in de-ionised water and stirred for 30 min. Meanwhile, the NH3 aqueous solution was prepared and added drop-wise to the above-mentioned mixed solutions to reach a suitable pH of pH 9. The precipitate was decanted, centrifugated, washed several times with de-ionised water to remove weakly adherent impurities on the surface, and dried in a hot air oven at 120 °C for 12 h. The dried products CeZr and CeSi oxides were pulverised using an agate mortar. Later, they were calcined in a muffle furnace at 500 °C for 5 h at a heating rate of 5 °C min−1 in static air. The same procedure was also used to obtain the pristine CeO2 material.
The WOx-promoted CeO2-based mixed oxide catalysts W/CeZr and W/CeSi oxides were prepared using the previously reported procedure. For example, the dried materials of the doped CeO2, i.e. CeSi or CeZr samples, were first dispersed in 30 mL of de-ionised water by constant stirring. Then, the ammonium meta-tungstate hydrate ((NH4)6H2W12O40·H2O) solution was added drop-wise to the dispersed solution. Further, the solution mixture was stirred for 3 h at room temperature and then the temperature was slowly increased to 110 °C to slowly evaporate off the excess water. The fine powder obtained was oven dried overnight at 110 °C and then calcined at 650 °C for 5 h in static air.
2.2. Catalyst characterisation
A Rigaku multiplex diffractometer equipped with a nickel-filtered Cu Kα (λ = 0.15418 nm) light and a scintillation counter detector was used to collect the powder X-ray diffraction data of the catalyst materials/samples. The catalyst materials' scanned 2 theta values from 2°–80° were obtained at a scan rate of 2° per minute. The prepared catalyst samples' average crystallite size was calculated using the Scherrer equation. A Micromeritics ASAP 2010 instrument was used to measure the BET specific surface area at a temperature of −196 °C. Before the investigation, each catalyst sample was degassed at 150 °C for 4 h.The TEM data of the catalyst materials were collected using a JEOL JEM-2100f instrument employing a slow-scanning CCD camera and an electron beam accelerating voltage of 80 kV. The testing sample preparation was done by sonicating in acetone for 30 min. Later, a drop of the sonicated sample was placed on a Cu grid supporting the porous carbon film and allowed to dry.
The XPS data of all the catalysts were gathered using a Thermo K-5 Alpha XPS instrument at a pressure of 1 × 10−7 Torr with an Al Kα (1486.6 eV) source. The binding energy data were charge-corrected compared to the adventitious carbon (C 1s) peak at 284.6 eV. The Raman spectral data of the catalysts were obtained using a Horiba Jobin-Yvon HR800 Raman spectrometer that was equipped with a confocal microscope, liquid N2-cooled charge-coupled device (CCD) detector, and a 632 nm of Ar+ ion laser.
The acidic sites present on the catalyst surface were measured in NH3-TPD experiments conducted on a Micromeritics Autochem 2910 device. Before the measurements, the catalyst was degassed at 300 °C in a He stream for physisorption of 5% NH3 balanced He gas for 45 min. Later the physisorbed NH3 gas was removed by helium flush. Continuous gas desorption was monitored with a TCD detector, and the area under the peak was assessed. The chemisorption process of NH3 was measured under helium flow at a flow rate of 20 mL min−1 and a heating rate of 10 °C min−1 in the range of 50 °C to 800 °C.
2.3. Activity measurements of the catalysts
The catalytic oxidation of benzyl alcohol was conducted in a 25 mL three-necked round-bottom flask (RBF) connected with a reflux condenser and a thermometer under oxygen bubbling. First, 5 mmol of benzyl alcohol and 12 wt% of the catalyst were added to the flask. Then the mixture's temperature was raised to the desired level under continuous O2 bubbling and constant stirring (800 rpm). When the reaction was completed, the catalyst and the reaction mixture were separated by centrifugation. The liquid products were identified by a GC-MS instrument that was fixed with a DB-5 capillary column and flame ionisation detector (FID). These operations were carried out under kinetically controlled conditions. The below equations measured the conversions of each reactant and selectivity for each product.
where nit → moles of product “i” formed, nRt=0 → initial moles of reactant “R”, nRt → final moles of reactant “R”.

3 Results and discussion
3.1. Characterisation of the catalysts
The powder X-ray diffraction patterns of the tungsten-promoted and unpromoted catalyst samples (Ce, CeZr, CeSi, W/Ce, W/CeZr, and W/CeSi oxides) calcined at 500 °C are depicted in Fig. 1. | ||
| Fig. 1 X-ray diffraction patterns of the promoted and unpromoted CeO2 (Ce), CeO2–ZrO2 (CeZr), CeO2–SiO2 (CeSi), WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) samples. | ||
The pristine CeO2 sample exhibited a diffraction line corresponding to the cubic structure of CeO2 (PDF-ICDD 34-0394).30 The Zr4+ and Si4+ ions in CeZr and CeSi samples also showed similar diffraction patterns as the pure CeO2 characteristic phase. More interestingly, the CeZr and CeSi catalyst samples exhibited broad diffraction lines. These significant observations validated that the crystallite sizes of the CeZr and CeSi oxides were appreciably smaller, possibly due to the crystal lattice's expansion or contraction. The 2θ value of the mixed oxides CeO2 was shifted towards the higher angle side when compared with pure CeO2. The change in the diffraction lines was determined by comparing the ionic radii of the incorporated ions (Zr4+ ∼0.084 nm; Si4+ ∼0.054 nm) and host ionic radii (Ce4+ ∼0.097 nm). Owing to the above observation, the CeZr and CeSi oxide catalysts displayed a lattice contraction when substituting the Ce4+cation with the lower-sized Zr4+ and Si4+ cations.31–33 The doped samples' estimated lattice parameters decreased when compared to pure CeO2 (Table 1). The crucial observations, including peak shift, change in the lattice parameter, and the nonappearance of XRD lines for ZrO2 and SiO2, verified the formation of the corresponding CeO2 solid solutions. The WOx-promoted CeO2-based samples also showed similar XRD patterns of the materials. This could be due to the finely dispersed or amorphous nature of WOx on the surface of the support. Table 1 summarises the estimated crystallite sizes of all the prepared catalysts. In addition, the average crystallite sizes and specific surface areas of the doped CeO2-based samples indicated the incorporated metal oxide role in the textural properties of CeO2 (Table 1). Notably, incorporating Zr4+ and Si4+ metal ions into CeO2 caused a reduction in crystallite growth.
| Catalyst | SBETa (m2 g−1) | Pa (nm) | Va (cc g−1) | Crystallite sizeb (nm) | Acidic sitesc (μmol g−1) |
|---|---|---|---|---|---|
| a From N2 adsorption–desorption analysis.b From XRD analysis.c NH3-TPD analysis. | |||||
| CeO2 | 41 | 9.81 | 0.113 | 9.1 | 33 |
| CeSi | 144 | 3.40 | 0.083 | 4.7 | 47 |
| CeZr | 86 | 4.11 | 0.079 | 5.6 | 48 |
| W/Ce | 27 | 6.37 | 0.064 | 7.2 | 79 |
| W/CeZr | 79 | 4.65 | 0.083 | 4.5 | 97 |
| W/CeSi | 120 | 3.75 | 0.090 | 3.8 | 118 |
Fig. 2 presents the Raman spectral profiles of the synthesised W/CeSi and W/CeZr catalyst samples subjected to calcination at 500 °C. These profiles represent the amorphous state of the deposited WOx on the CeZr and CeSi mixed oxide support and exhibit three broad Raman bands with lower intensity in the range of ∼250–350, ∼810, and ∼980 cm−1. According to the earlier reports, the first one (∼250–350 cm−1) corresponded to the W5+–O bond and the bands at 810 cm−1 were due to a superposition of the W6+–O band, respectively.30 The band at ∼980 cm−1 was assigned to the W6+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of the terminal oxygen atoms.30 Concerning amorphous WOx, the primary crystal structure could be explained by forming the W6+O octahedral structure. These involved short W6+O bonds and long W6+–O bonds, leading to distorted octahedral clusters, where the W+6O bonds were projected onto the outer surface of the clusters.32 These results well correlated with the XRD analysis results (Fig. 1). A strong Raman band at ∼463 cm−1 was also observed in the above catalyst materials' spectra. This band was due to the F2g Raman vibrational mode of the fluorite structure of CeO2, consistent with the XRD results (Fig. 2).31–33 Another Raman peak was also observed at ∼605 cm−1, specifying an Ov band (oxygen vacancy defect sites) in the doped-ceria lattice.30,31 Also, no Ov band was noticed in the pure CeO2 sample. This notable observation revealed the beneficial role of the incorporated ions in enhancing the oxygen vacancy defect sites in the mixed oxide samples. As evidenced by Fig. 2, the wavenumber difference between the F2g and Ov bands was ∼136 cm−1 due to the oxygen vacancy from the intrinsic defects when converting Ce4+ to Ce3+.34 The broadened Raman spectra showed that incorporating the metal/non-metal cations ions into the CeO2 crystal lattice resulted in a shift of 4 F2g band to lower wavenumbers. These observations show that the incorporation of isovalent cations with different ionic radii (e.g. Si4+ and Zr4+) leads to disorder in the CeO2 lattice. Hence, the Ce–O band frequency was changed for the CeZr and CeSi mixed oxide catalyst samples.35,36 This shift corresponded to the change in the forces between the atoms in the lattice due to the presence of vacancy defects induced by the dopant. Therefore, these observations could indicate the leading cause for the enhanced vacancy defects in the CeSi and CeZr samples.
O stretching mode of the terminal oxygen atoms.30 Concerning amorphous WOx, the primary crystal structure could be explained by forming the W6+O octahedral structure. These involved short W6+O bonds and long W6+–O bonds, leading to distorted octahedral clusters, where the W+6O bonds were projected onto the outer surface of the clusters.32 These results well correlated with the XRD analysis results (Fig. 1). A strong Raman band at ∼463 cm−1 was also observed in the above catalyst materials' spectra. This band was due to the F2g Raman vibrational mode of the fluorite structure of CeO2, consistent with the XRD results (Fig. 2).31–33 Another Raman peak was also observed at ∼605 cm−1, specifying an Ov band (oxygen vacancy defect sites) in the doped-ceria lattice.30,31 Also, no Ov band was noticed in the pure CeO2 sample. This notable observation revealed the beneficial role of the incorporated ions in enhancing the oxygen vacancy defect sites in the mixed oxide samples. As evidenced by Fig. 2, the wavenumber difference between the F2g and Ov bands was ∼136 cm−1 due to the oxygen vacancy from the intrinsic defects when converting Ce4+ to Ce3+.34 The broadened Raman spectra showed that incorporating the metal/non-metal cations ions into the CeO2 crystal lattice resulted in a shift of 4 F2g band to lower wavenumbers. These observations show that the incorporation of isovalent cations with different ionic radii (e.g. Si4+ and Zr4+) leads to disorder in the CeO2 lattice. Hence, the Ce–O band frequency was changed for the CeZr and CeSi mixed oxide catalyst samples.35,36 This shift corresponded to the change in the forces between the atoms in the lattice due to the presence of vacancy defects induced by the dopant. Therefore, these observations could indicate the leading cause for the enhanced vacancy defects in the CeSi and CeZr samples.
 | ||
| Fig. 2 Raman spectra of the oxides of CeO2 (Ce), CeO2–ZrO2 (CeZr), CeO2–SiO2 (CeSi), WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) samples. | ||
The textural characteristics of the prepared catalysts were further studied by measuring the N2-sorption and BJH curves at −196 °C, as shown in Fig. 3 and 4. The BET specific surface areas, BJH pore volumes, and pore diameters are given in Table 1. As noted in Fig. 3, all the prepared catalyst samples showed a type-IV isotherm with an H1-hysteresis loop, indicating the mesoporous nature of the W/CeZr and W/CeSi oxide catalysts. The hysteresis loops indicated the improved pore size and connectivity of the W/CeZr and W/CeSi oxide catalysts with a homogeneous distribution.21,36 The BJH pore size distribution curves suggested that the oxides of CeO2, CeZr, and CeSi, and the W/CeZr oxide catalysts displayed a unimodal pore-size distribution. In contrast, the W/Ce and W/CeSi mixed oxide catalysts exhibited a bimodal pore-size distribution. The specific surface area of the pure CeO2 was 41 m2 g−1. On the other hand, the doped-CeO2 samples had a significantly enhanced specific surface area by incorporating the metal oxides (i.e. CeSi = 144 m2 g−1 and CeZr = 86 m2 g−1). The BET specific surface area of the WOx-promoted catalysts was found to be higher than for the unpromoted ones. Specifically, the CeSi and W/CeSi oxide catalysts showed a higher BET surface area and smaller average BJH pore size than the other samples. Therefore, a high surface area is advantageous for surface adsorption during catalytic reactions.
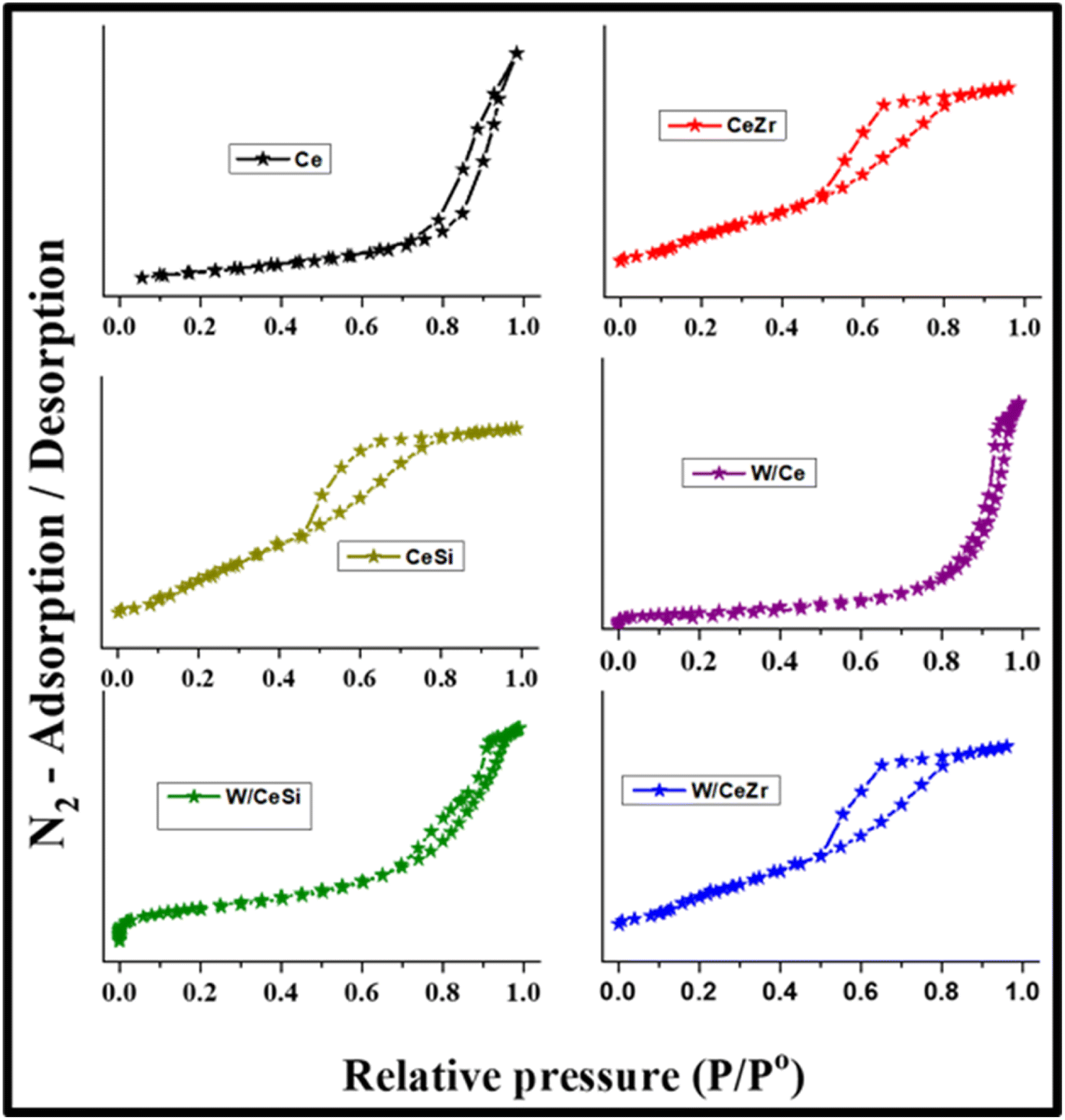 | ||
| Fig. 3 N2 adsorption–desorption isotherms of CeO2 (Ce), CeO2–ZrO2 (CeZr), CeO2–SiO2 (CeSi), WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) samples. | ||
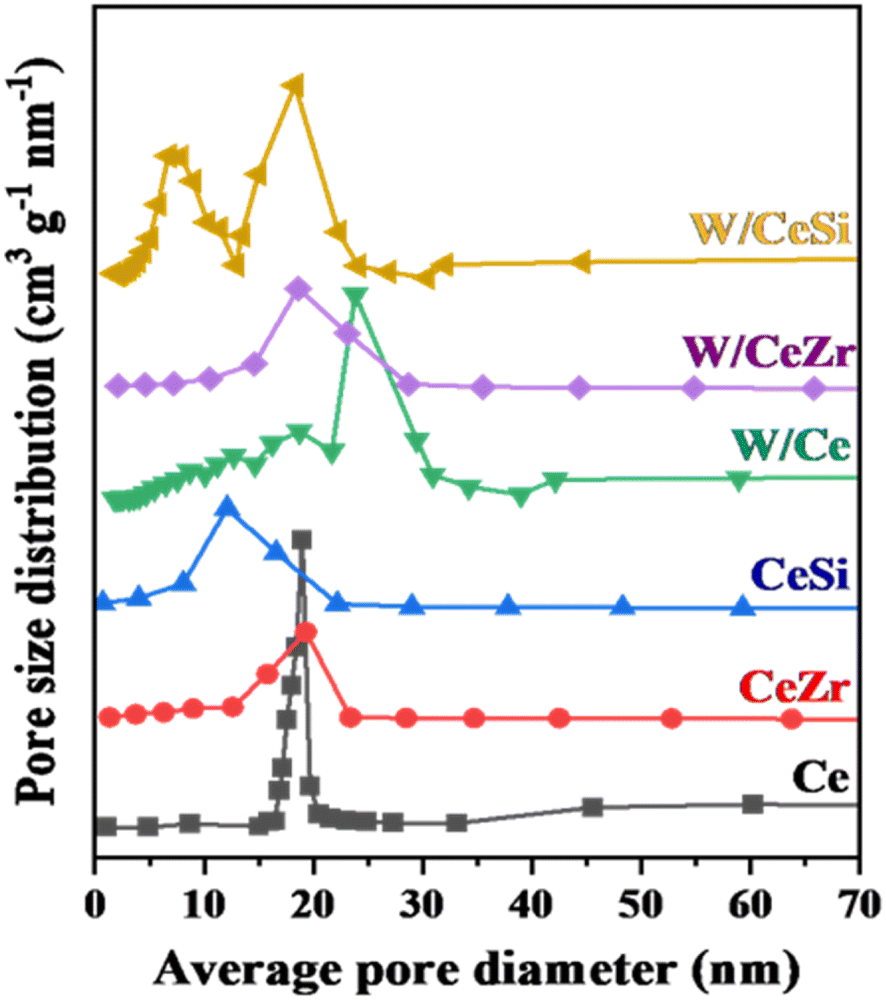 | ||
| Fig. 4 BJH pore-size distribution profiles of CeO2 (Ce), CeO2–ZrO2 (CeZr), CeO2–SiO2 (CeSi), WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) samples. | ||
Fig. 5 displays the NH3 temperature-programmed desorption (NH3-TPD) profiles of the synthesised CeO2, CeSi, CeZr, W/Ce, W/CeZr, and W/CeSi catalysts that were calcined at 500 °C. As shown in Fig. 5, the catalysts exhibited variable NH3-desorption peaks due to the change in their activation energies for desorbing ammonia from various types of acidic sites on the catalyst surface. Generally, the various acid sites can be classified as weak (<370 °C), medium (370–600 °C), and strong acid sites (>600 °C).37 The WOx-promoted CeO2-based mixed oxide samples showed all three types of acidic sites. The estimated amount of acidic sites is presented in Table 1. Among them, the W/CeSi mixed oxide catalyst revealed a higher concentration and strength of acidic sites than the W/Ce and W/CeZr samples due to its higher specific surface area. The higher surface area can result in a larger number of active centres assuming a homogeneous and equivalent surface distribution, but the surface area itself may not directly impact the strength of the acidic centres. However, the strength of the acidic sites is typically associated with their chemical nature and interactions with the surrounding environment (WOx species), rather than the surface area. The enhanced strength of the promoted catalysts was more prominent due to the WOx species on the CeSi and CeZr mixed oxide surface. Generally, a higher specific surface area is a crucial property to produce more active sites on a support, which agrees with the BET and XRD results (Table 1). These results were evidenced here by the WOx promoter's significant role in enhancing the strength and concentration of the acidic sites, which are vital for achieving a better catalytic performance.
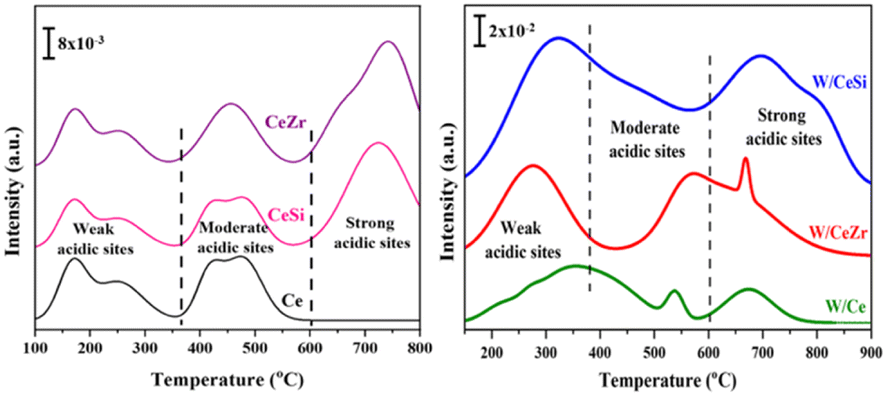 | ||
| Fig. 5 NH3-TPD profiles of CeO2 (Ce), CeO2–ZrO2 (CeZr), CeO2–SiO2 (CeSi), WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) samples. | ||
Next, X-ray photoelectron spectroscopy analysis was used to gain information on the synthesised catalysts' surfaces (i.e. the elemental values, oxidation states, and atomic concentrations). Fig. 6A illustrates the Gaussian fitting of the Ce 3d XPS results of the WOx-promoted doped-CeO2 catalysts. In the figures, the spin-splitting peaks attributed to Ce 3d3/2 and Ce 3d5/2 spin-splitting components are represented by “v” and “u”. The binding energy peaks marked with v (88.19 eV), v′′ (888.22 eV), v′′′ (897.76 eV), u (900.25 eV), u′′ (906.89 eV), and u′′′ (916.23 eV) correspond to the surface Ce4+ species of the 3d10 4f0 electronic configuration, whereas the peaks marked with v′ and u′ are due to the surface Ce3+ species of the 3d10 4f1 initial electronic configuration. Hence, these results suggest that all the catalyst samples exhibited both the Ce4+ and Ce3+ oxidation states. An investigation of the u′′′ (Ce4+) peak was thus essential to determine the reduction of Ce4+ since it did not overlap with the remaining peaks. The Ce3+ amount on the catalyst surface was estimated by considering the intensity ratio of the u′′′ peak to the total intensity of the Ce 3d peak (Iu′′′/ITotal), whereby a smaller value obtained would indicate a higher amount of Ce3+ on the catalyst surface. The calculated Iu′′′/ITotal values were 0.0916, 0.0846, and 0.0813 for W/Ce, W/CeZr, and W/CeSi samples. As can be seen in Fig. 6A, the intensity of the u′′′ peak was lower for the W/CeSi sample in comparison to that of the W/Ce and W/CeZr oxide samples. The decreased surface Ce4+/Ce3+ ratio was the result of doping Zr4+ and Si4+ ions into the CeO2 lattice to create oxygen vacancy sites on the catalyst surface. Therefore, the W/CeSi oxide sample exhibited a higher amount of Ce3+ on the catalyst surface, which was in good agreement with the XRD and Raman spectra results (Fig. 1 and 2).36 Besides, adding WOx to the CeZr and CeSi mixed oxide catalyst samples could considerably improve the amount of Ce3+ ions on the catalyst surface. Thus, it is probable that WOx also played a role in the transformation from Ce+4 to Ce3+. The Ce3+ species could create a significant charge variance, as well as oxygen vacancies, and chemisorbed oxygen on the catalyst surface, contributing to the excellent catalytic performance and suggesting a powerful synergistic effect between the W, Ce, and Si species.38
 | ||
| Fig. 6 XPS patterns of (A) Ce 3d spectra, and (B) O 1s spectra for WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) samples. | ||
Fig. 6 illustrates the O 1s core-level spectra of the W/Ce, W/CeZr, and W/CeSi oxide catalyst samples. The spectra demonstrated different types of oxygen species in all the materials. The two peaks observed at ∼529.6 and ∼531.9 eV were related to the lattice oxygen and various adsorbed oxygen species on the catalyst surface (i.e. hydroxyl and/or carbonyl groups), respectively. The peak at a value greater than 532 eV was correlated with the O2− species in the Si–O bond. In this study, the lattice oxygen environment of the CeZr and CeSi oxide samples was significantly changed compared to that of pure CeO2 catalyst sample, as evidenced by the shift in the lattice oxygen binding energy peak towards the lower side.39 The W/CeSi oxide sample exhibited a higher swing of the binding energy towards the downside than the W/CeZr mixed oxide sample. Hence, the lattice oxygen of the W/CeSi mixed oxide catalyst became more labile with the lower binding energy, which was conducive to creating oxygen vacancy sites.40–42
Fig. 7A presents the W 4f core-level spectra of the W/Ce, W/CeZr, and W/CeSi oxide catalyst samples. The W 4f peaks could be mainly observed as two doublets at 35.68 and 34.15 eV for the different oxidation states of the tungsten species present in the above-said catalysts. The first doublet was found at 35.68 and 37.78 eV for the W 4f7/2 and W 4f5/2 spin–orbit splitting, respectively. The binding energy difference for the spin–orbit splitting was at ∼2.1 eV, which shows a characteristic property of the W6+ species present in the synthesised WO3-promoted doped CeO2 catalysts. The second binding energies were found at 34.15 and 36.94 eV corresponding to the W5+ species present in the catalyst samples, which were in agreement with the Raman results (Fig. 2) and previous reports.43
 | ||
| Fig. 7 XPS patterns of the (A) W 4f spectra, and (B) Zr 3d and Si 2p spectra of WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) oxide samples. | ||
We observed a slight shift in the binding energy from the XPS spectra because of the interaction between the WOx and the support matrix. Fig. 7B depicts the Zr 3d core-level spectra of the W/CeZr oxide catalyst samples. The deconvoluted binding energy peaks observed at ∼184.8 (Zr 3d3/2) and ∼182.2 eV (Zr 3d5/2) indicated the presence of Zr4+ ions.44 The binding energy of Zr 3d was shifted towards a higher binding energy side than that in pure ZrO2, which further confirmed that Zr4+ ions were incorporated into the CeO2 lattice and formed CeZr solid solutions, which well correlated with the XRD and Raman results.45 The Si 2p XPS profile of the W/CeSi sample could be observed at ∼103.2–103.6 eV in Fig. 7B, which indicated the presence of Si4+ species in the lattice of the CeSi oxide.39
The morphology and sizes of the particles of the W/CeSi catalyst were obtained by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), as shown in Fig. 8(A–D). The morphology of the W/CeSi sample revealed a homogeneous sphere-like structure nanocluster. A fine dispersion of WO3 on the CeSi oxide surface was evident from the SEM images and ascribed to the higher specific surface area from the incorporation of CeSi oxide. The W/CeSi sample had an average particle size range of 28–40 nm (Fig. 8A and B). The TEM images of W/CeSi are depicted in Fig. 8(C and D). A spherical-type structure (Fig. 8C) with an average particle size of ∼5–10 nm was observed. Fig. 8D shows the HR-TEM images of the W/CeSi catalyst, which indicated that the CeO2 possessed a hexagonal shape and the WOx a spherical shape of nanoparticles built by the random packing of the crystallites. Both the WOx and CeO2 nanoparticles exhibited clear lattice fringes. Moreover, the lattice interplanar distances of 0.375 and 0.308 nm for the W/CeSi oxide catalysts were assigned to the (020) and (111) lattice planes of WOx and CeO2, respectively. These results abundantly evidenced the formation of WOx and CeO2 nanoparticles.46
 | ||
| Fig. 8 (A and B) SEM images of WOx/CeO2–SiO2 (W/CeSi) and (C and D) TEM images of the W/CeSi oxide sample. | ||
3.2. Catalytic activity study
A solvent-free selective catalytic oxidation of benzyl alcohol over the WOx-promoted doped CeO2 catalysts was performed at 140 °C with O2 bubbling through in a three-necked round-bottom flask. The results are presented in Fig. 9. Lower and moderate benzyl alcohol conversions were observed over the pristine CeO2 and doped CeO2 catalysts, respectively. The catalytic activity of the pristine CeO2 was low, with ∼33% benzyl alcohol conversion. After incorporating the isovalent cations (Zr4+ and Si4+ in the form of ZrO2 and SiO2), the catalytic efficiency of the doped-CeO2 samples was enhanced considerably, demonstrating the positive role of ZrO2 and SiO2 during the selective oxidation of alcohols. The conversion of benzyl alcohol was found to be ∼43% and 47% for the CeZr and CeSi catalysts, respectively.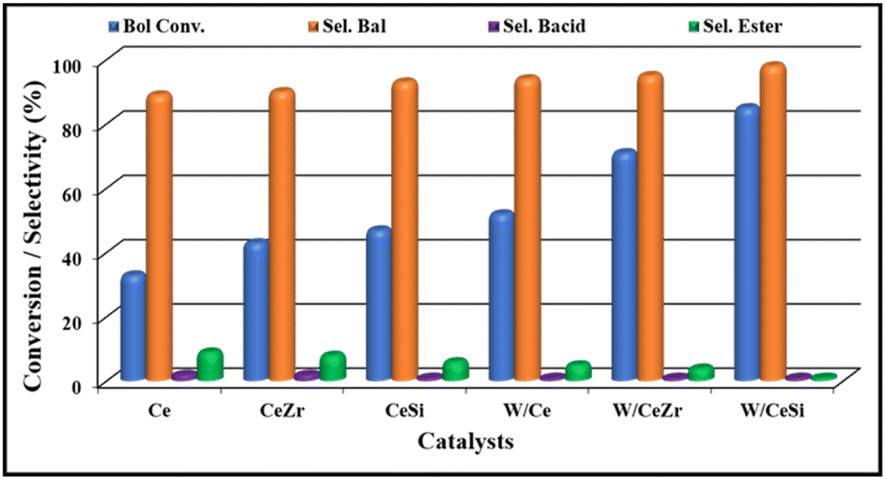 | ||
| Fig. 9 The catalytic oxidation of benzyl alcohol over CeO2 (Ce), CeO2–ZrO2 (CeZr), CeO2–SiO2 (CeSi), WOx/CeO2 (W/Ce), WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) oxide catalysts. Reaction conditions: 10 wt% of catalyst, 5 mmol of benzyl alcohol, O2 bubbling (20 mL min−1), 140 °C of temperature, 4 h of reaction time, and 800 rpm of stirring speed. | ||
These results indicate that the CeZr and CeSi oxide catalysts possessed a higher number of acidic sites and enhanced oxygen vacancy sites compared to the pure CeO2 catalyst. Furthermore, the selectivity for the formation of benzaldehyde by the CeZr and CeSi catalysts was slightly higher than for the undoped, i.e. pure CeO2, catalysts, which was attributable to the controlled oxidation of the benzyl alcohol by the doped CeO2 catalysts. Next, to examine the influence of the WOx promoter on the conversion and selectivity in the above catalytic process, tungsten-promoted W/Ce, W/CeZr, and W/CeSi mixed oxides were employed as catalysts. According to our observation, the benzyl alcohol conversions were 52%, 71%, and 85%, and the benzaldehyde selectivities were 94%, 96%, and >98%, respectively. Overall, the W/CeSi catalyst showed a higher conversion of benzyl alcohol and higher selectivity for benzaldehyde than the other promoted catalysts (i.e. W/Ce and W/CeZr). This phenomenon could be attributed to the smaller-sized particles of WOx and CeO2, higher specific surface areas, more acidic sites, and superior oxygen vacancies. Raman analysis showed that the W/CeSi oxide catalyst had more surface oxygen defect sites than the other samples (Fig. 9). Related to the influence of the size of the WOx and CeO2 nanoparticles on the oxidation of alcohol, the number of low coordinated atoms and increased active sites enhanced the adsorption of benzyl alcohol on the surface of the catalyst, and decreased the activation energy barrier for selective transformation.28 The NH3-TPD results indicated the higher strength and concentration of the W/CeSi oxide catalyst than all the other oxide catalysts investigated, which were critical factors for achieving a higher conversion of alcohol in the oxidation reaction. Therefore, the attractive physicochemical properties and promotional effects of the W/CeSi oxide catalyst were crucial for their superior catalytic efficiency during the solvent-free oxidation of benzyl alcohol.
The effects of the reaction time on benzyl alcohol conversion and benzaldehyde selectivity over the W/CeZr and W/CeSi catalysts were examined (Fig. 10). It was found that the W/CeSi catalyst exhibited a greater conversion rate than the W/CeZr catalyst at each specified reaction time point. The conversion rate increased with the reaction duration, approaching a constant level after 6 h. At 5 h reaction time, both catalysts showed an ideal conversion and selectivity. At this point, W/CeZr displayed a selectivity and conversion of ∼87%, whereas W/CeSi had a selectivity and conversion of ∼96%. Therefore, a reaction time of 5 h was considered the optimum for the high conversion of benzyl alcohol and the high selectivity to benzaldehyde.
 | ||
| Fig. 10 The effect of the reaction time on the catalytic oxidation of benzyl alcohol over WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) catalysts. Reaction conditions: 10 wt% of catalyst, 5 mmol of benzyl alcohol, O2 bubbling (20 mL min−1), 140 °C of temperature, and 800 rpm of stirring speed. | ||
Fig. 11 reveals how the reaction temperature effected the benzyl alcohol conversion and benzaldehyde selectivity over the W/CeZr, and W/CeSi catalysts. The BOL achieved 32% and 23% conversions at 80 °C reaction temperature over the W/CeSi and W/CeZr catalysts, respectively, producing benzaldehyde as the major product. At 100 °C, the BOL conversions for the W/CeSi and W/CeZr catalysts were 50% and 38%, respectively. When the temperature was increased to 140 °C, the conversions of benzyl alcohol increased to ∼85% and ∼71% with the W/CeSi and W/CeZr catalysts, respectively. However, the benzaldehyde selectivity was significantly decreased over the W/CeZr catalyst and not altered over the W/CeSi catalyst. These results indicated that the W/CeSi catalyst was more selective towards benzaldehyde than the W/CeZr catalyst. When the temperature was further increased to 180 °C, the conversion rate of benzyl alcohol gradually rose until it almost equalised, and the selectivity of benzaldehyde decreased significantly for both cases. Therefore, the temperature of 140 °C was considered the optimum reaction temperature for the catalytic oxidation of benzyl alcohol.
 | ||
| Fig. 11 The effect of temperature on the catalytic oxidation of benzyl alcohol over WOx/CeO2–ZrO2 (W/CeZr) and WOx/CeO2–SiO2 (W/CeSi) catalysts. Reaction conditions: 10 wt% of catalyst, 5 mmol of benzyl alcohol, O2 bubbling (20 mL min−1), 4 h of reaction time, and 800 rpm of stirring speed. | ||
Fig. 12 presents the effect of the catalyst loading during the selective oxidation of benzyl alcohol over the W/CeZr and W/CeSi catalysts. The conversion rate for both catalysts was increased gradually with the increasing catalyst wt%, and the selectivity for benzaldehyde was stable up to 10 wt%. Further, when the loading was increased to 12 wt%, the selectivity towards BAL by W/CeSi remained unchanged, while the selectivity with the W/CeZr catalyst decreased. Therefore, the optimum catalyst loading of 12 wt% of W/CeSi oxide was considered best to selectively oxidise benzyl alcohol to benzaldehyde.
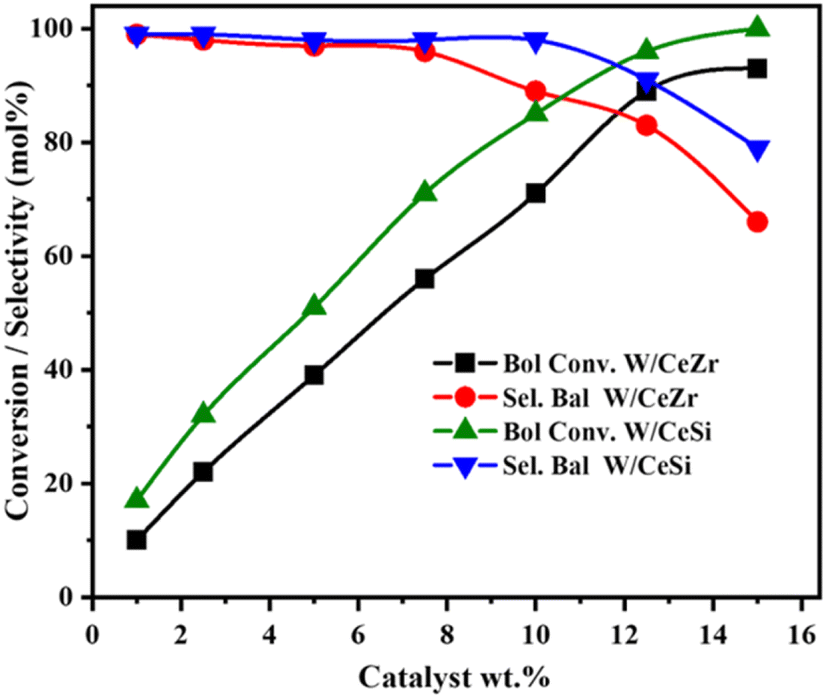 | ||
| Fig. 12 The effect of catalyst loading on the catalytic oxidation of benzyl alcohol over WOx/CeO2–ZrO2 (W/CeZr), and WOx/CeO2–SiO2 (W/CeSi) catalysts. Reaction conditions: 5 mmol of benzyl alcohol, O2 bubbling (20 mL min−1), 140 °C of temperature, 4 h of reaction time, and 800 rpm of stirring speed. | ||
Before the recycling experiment, the catalyst was filtered after the initial run, washed with acetone, and dried at 80 °C. The recycling experiment (Fig. 13) was done with the W/CeSi catalyst since it showed higher selectivity towards benzaldehyde. This effort produced a promising result of 85% benzyl alcohol conversion and 98% selectivity for benzaldehyde. The second cycle also displayed nearly the same results. A conversion of 82% BOL was achieved in the 5th cycle, with 96% BAL product selectivity. The catalyst material was thus stable for up to 5th run without significant change in the activity. After the 5th cycle, the conversion rate (69%) and selectivity (86%) both decreased. This outcome showed that the heterogeneous W/CeSi oxide is a highly efficient catalyst in oxidising benzyl alcohol selectively to benzaldehyde under solvent-free aerobic conditions.
 | ||
| Fig. 13 Catalyst recycling study for the oxidation of benzyl alcohol using the WOx/CeO2–SiO2 (W/CeSi) catalyst. Reaction conditions: 10 wt% of catalyst, 5 mmol of benzyl alcohol, O2 bubbling (20 mL min−1), 140 °C of temperature, 4 h of reaction time, and 800 rpm of stirring speed. | ||
 | ||
| Scheme 1 Possible reaction mechanism for the selective catalytic oxidation of benzyl alcohol to benzaldehyde over the WOx/CeO2–SiO2 (W/CeSi) oxide catalyst. | ||
| Catalyst | Conditions | Activity performance | Ref. | |
|---|---|---|---|---|
| Conv. (%) | Sel. (%) | |||
| CeO2 NPs | T = 50 °C, t = 6 h | 68 | 95 | 51 |
| Au–Pd/CeO2 | T = 120 °C, t = 3 h | 88 | 89 | 51 |
| Mn0.5/(BaxCe1−x)0.5 | T = 70 °C, t = 6 h | 55.0 | 51.9 | 52 |
| Ce 0.8Zr 0.2O2 | T = 120 °C, t = 4 h | 92.5 | 100 | 53 |
| Pd/CeO2 | T = 90 °C, t = 4 h | 72.6 | 92.3 | 54 |
| Pd/CeO2-meso | T = 90 °C, t = 1 h | 62 | 62 | 54 |
| CZRu | T = 90 °C, t = 24 h | 61–90 | 100 | 55 |
| CeO2–Sm2O3/SiO2 | T = 25 °C, t = 24 h | 53.4 | 91.4 | 56 |
| WOx/CeO2–SiO2 | T = 140 °C, t = 5 h | 99 | 100 | This work |
4 Conclusions
An efficient solvent-free and base-free heterogeneous selective catalytic oxidation of benzyl alcohol to benzaldehyde was achieved and the conditions optimised using WOx-promoted CeO2–SiO2 catalysts (W/CeSi) in the presence of inexpensive and green molecular O2 as an oxidant. This observation was supported by comparing the results obtained with Ce, CeZr, CeSi, W/Ce, and W/CeZr oxide catalysts. In conclusion, the tungsten-promoted mixed oxide catalyst, i.e. W/CeSi oxide catalyst, showed higher benzyl alcohol conversion and selectivity for benzaldehyde than the other promoted catalysts. The smaller particle size of the WOx and CeO2, higher specific surface area, more significant number of acidic sites, and superior oxygen vacancies could help explain the excellent catalytic activity of the tungsten-promoted W/CeSi oxide catalyst. Both the WOx promoter and SiO2 exhibited vital roles in the selective oxidation of benzyl alcohol. Furthermore, the W/CeSi oxide catalyst was recyclable during the oxidation process of benzyl alcohol without any activity loss for up to five cycles.Author contributions
Conceptualization: SB Jonnalagadda, M. Baithy, and C. S. Vasam; methodology: G. Bathula, S. Rana, S. Bandalla, software: V. Dosarapu, Aswathi R. V. V., B. Sharma; validation: G. Bathula, B. Sharma; formal analysis: G. Bathula, S. Mavurapu, Aswathi R. V. V.; investigation: G. Bathula, V. Dosarapu, S. Bandalla; B. Sharma, re-sources: S. B. Jonnalagadda, M. Baithy, and C. S. Vasam; data curation: M. Baithy and C. S. Vasam; writing – original draft preparation: SB Jonnalagadda, M. Baithy, G. Bathula, C. S. Vasam; writing – review & editing: SB Jonnalagadda, M. Baithy, and C. S. Vasam; visualisation: S. Rana, S. Bandalla, S. Mavurapu; supervision: S. B. Jonnalagadda, M. Baithy, and C. S. Vasam.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support received from Indo-South Africa joint project Ref. No. DST/INT/SA/P-15/2011.References
- Y. Wu, L.-H. Kong, W.-Ti. Ge, W.-J. Zhang, Z.-Y. Dong, X.-J. Guo, X. Yan, Y. Chen and W.-Z. Lang, J. Catal., 2022, 413, 668–680 CrossRef CAS.
- P. S. F. Musere, A. Rahman, V. Uahengo, J. Naimhwaka, L. Daniel, S. V. H. S. Bhaskurani and S. B. Jonnalagadda, J. Cleaner Prod., 2021, 312, 127581 CrossRef CAS.
- R. Pagadala, S. Maddila, S. Rana and S. B. Jonnalagadda, RSC Adv., 2014, 4, 6602–6607 RSC.
- M. S. S. Adam, O. S. Abdel-Rahman and M. M. Makhlouf, J. Mol. Struct., 2023, 1272, 134164 CrossRef CAS.
- M. Kermani, F. Mohammadi, B. Kakavandi, A. Esrafili and Z. Rostamifasih, J. Phys. Chem. Solids, 2018, 117, 49–59 CrossRef CAS.
- Y. Liu, R. Luo, Y. Li, J. Qi, C. Wang, J. Li, X. Sun and L. Wang, Chem. Eng. J., 2018, 347, 731–740 CrossRef CAS.
- A. Pennetier, W. Y. Hernandez, B. T. Kusema and S. Streiff, Appl. Catal., A, 2021, 624, 118301 CrossRef CAS.
- W. Ma, N. Wang, Y. Du, T. Tong, L. Zhang, K.-Y. A. Lin and X. Han, Chem. Eng. J., 2019, 356, 1022–1031 CrossRef CAS.
- J. Miao, J. Sunarso, X. Duan, W. Zhou, S. Wang and Z. Shao, J. Hazard. Mater., 2018, 349, 177–185 CrossRef CAS PubMed.
- X. Huang, X. Wang, X. Wang, X. Wang, M. Tan, W. Ding and X. Lu, J. Catal., 2013, 301, 217–226 CrossRef CAS.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- S. Marx and A. Baiker, J. Phys. Chem. C, 2009, 113, 6191–6201 CrossRef CAS.
- D. Riemer, B. Mandaviya, W. Schilling, A. C. Götz, T. Kühl, M. Finger and S. Das, ACS Catal., 2018, 8, 3030–3034 CrossRef CAS.
- C. E. Chan-Thaw, A. Savara and A. Villa, Catalysts, 2018, 8, 431 CrossRef.
- H. A. Duarte, P. J. Luggren, J. Zelin, V. K. Diez and J. I. Di Cosimo, Catal. Today, 2022, 394–396, 178–189 CrossRef CAS.
- M. Sutradhar, L. M. D. R. S. Martins, M. F. C. G. da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2015, 301–302, 200–239 CrossRef CAS.
- X. Bao, H. Li, Z. Wang, F. Tong, M. Liu, Z. Zheng, P. Wang, H. Cheng, Y. Liu, Y. Dai, Y. Fan, Z. Li and B. Huang, Appl. Catal., B, 2021, 286, 119885 CrossRef CAS.
- R. Bulanek, H. Sheng-Yang, P. Knotek and L. Capek, Stud. Surf. Sci. Catal., 2008, 174, 1295–1298 CrossRef.
- M. Pagliaro, S. Campestrini and R. Ciriminna, Chem. Soc. Rev., 2005, 34, 837–845 RSC.
- P. Sudarsanam, N. K. Gupta, B. Mallesham, N. Singh, P. N. Kalbande and B. M. Reddy, ACS Catal., 2021, 11, 13603–13648 CrossRef CAS.
- B. Mallesham, P. Sudarsanam, G. Raju and B. M. Reddy, Green Chem., 2013, 15, 478–489 RSC.
- B. G. Rao, P. Sudarsanam, B. Mallesham and B. M. Reddy, RSC Adv., 2016, 6, 95252–95262 RSC.
- P. Sudarsanam, B. Hillary, M. H. Amin, S. B. A. Hamid and S. K. Bhargava, Appl. Catal., B, 2016, 185, 213–224 CrossRef CAS.
- E. T. Saw, U. Oemar, M. L. Ang, H. Kus and S. Kawi, Catal. Sci. Technol., 2016, 6, 5336–5349 RSC.
- L. Lisi, G. Landi and V. Di Sarli, Catalysts, 2020, 10, 1–16 RSC.
- V. Di Sarli, G. Landi, A. Di Benedetto and L. Lisi, Top. Catal., 2021, 64, 256–269 CrossRef CAS.
- B. M. Reddy, L. Katta and G. Thrimurthulu, Chem. Mater., 2010, 22, 467–475 CrossRef CAS.
- P. Sudarsanam, B. Mallesham, P. S. Reddy, D. Großmann, W. Grünert and B. M. Reddy, Appl. Catal., B, 2014, 144, 900–908 CrossRef CAS.
- P. Sudarsanam, A. Rangaswamy and B. M. Reddy, RSC Adv., 2014, 4, 46378–46382 RSC.
- P. Sudarsanam, B. Mallesham, D. N. Durgasri and B. M. Reddy, RSC Adv., 2014, 4, 11322–11330 RSC.
- B. Mallesham, P. Sudarsanam, B. V. S. Reddy and B. M. Reddy, Appl. Catal., B, 2016, 181, 47–57 CrossRef CAS.
- M. Gies, F. Michel, C. Lupó, D. Schlettwein, M. Becker and A. Polity, J. Mater. Sci., 2021, 56, 615–628 CrossRef CAS.
- G. N. Vayssilov, M. Mihaylov, P. S. Petkov, K. I. Hadjiivanov and K. M. Neyman, J. Phys. Chem. C, 2011, 115, 23435–23454 CrossRef CAS.
- J. Wang, S. Yin, M. Komatsu and T. Sato, J. Eur. Ceram. Soc., 2005, 25, 3207–3212 CrossRef CAS.
- J. D. Lessard, I. Valsamakis and M. Flytzani-Stephanopoulos, Chem. Commun., 2012, 48, 4857–4859 RSC.
- B. Mallesham, P. Sudarsanam and B. M. Reddy, Catal. Sci. Technol., 2014, 4, 803–813 RSC.
- A. Boréave, A. Auroux and C. Guimon, Microporous Mater., 1997, 11, 275–291 CrossRef.
- R. Fan, Z. Li, Y. Wang, C. Zhang, Y. Wang, Z. Ding, X. Guo and R. Wang, RSC Adv., 2020, 10, 5845–5852 RSC.
- B. Mallesham, P. Sudarsanam, B. V. S. Reddy, B. G. Rao and B. M. Reddy, ACS Omega, 2018, 3, 16839–16849 CrossRef CAS PubMed.
- J. Ftouni, A. Muñoz-Murillo, A. Goryachev, J. P. Hofmann, E. J. M. Hensen, L. Lu, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Catal., 2016, 6, 5462–5472 CrossRef CAS.
- X. C. Sun, K. Yuan, W.-D. Hua, Z.-R. Gao, Q. Zhang, C.-Y. Yuan, H.-C. Liu and Y.-W. Zhang, ACS Catal., 2022, 12, 11942–11954 CrossRef CAS.
- K. K. Patra, Z. Liu, H. Lee, S. Hong, H. Song, H. G. Abbas, Y. Kwon, S. Ringe and J. Oh, ACS Catal., 2022, 12, 10973–10983 CrossRef CAS.
- Y. Tao, O. De Luca, B. Singh, A. J. Kamphuis, J. Chen, P. Rudolf and P. P. Pescarmona, Mater. Today Chem., 2020, 18, 100373 CrossRef CAS.
- X. Hao, X. Zhang, Y. Xu, Y. Zhou, T. Wei, Z. Hu, L. Wu, X. Feng, J. Zhang, Y. Liu, D. Yin, S. Ma and B. Xu, J. Colloid Interface Sci., 2023, 643, 282–291 CrossRef CAS PubMed.
- B. Liu, C. Li, G. Zhang, Y. Xuesi, S. S. C. Chuang and Z. Li, ACS Catal., 2018, 8, 10446–10456 CrossRef CAS.
- C. Sui, L. Xing, X. Cai, Y. Wang, Q. Zhou and M. Li, Catalysts, 2020, 10, 243 CrossRef CAS.
- J. Kodchase, C. Chanloi, P. Khemthong, B. Uapipatankul, M. Ehara and K. Bobuatong, Catalysts, 2021, 11, 720 CrossRef.
- J. Liu, X. P. Wu, S. Zou, Y. Dai, L. Xiao, X. Q. Gong and J. Fan, J. Phys. Chem. C, 2014, 118, 24950–24958 CrossRef CAS.
- S. Bandalla, V. Dosarapu, G. B. Bathula, M. Ravula, J. Yadagiri, P. Gogoi, M. Baithy, S. B. Jonnalagadda and C. S. Vasam, Mol. Catal., 2022, 533, 112759 CrossRef CAS.
- S. Najafishirtari, K. Friedel Ortega, M. Douthwaite, S. Pattisson, G. J. Hutchings, C. J. Bondue, K. Tschulik, D. Waffel, B. Peng, M. Deitermann, G. W. Busser, M. Muhler and M. Behrens, Chem.–Eur. J., 2021, 27, 16833 CrossRef PubMed.
- X. Li, J. Feng, M. Perdjon, R. Oh, W. Zhao, X. Huang and S. Liu, Appl. Surf. Sci., 2020, 505, 144473 CrossRef CAS.
- R. K. More, N. R. Lavande and P. M. More, Catal. Commun., 2018, 116, 52–56 CrossRef CAS.
- C. A. Akinnawo, N. Bingwa and R. Meijboom, Catal. Commun., 2020, 145, 106115 CrossRef CAS.
- F. Meng, M.-Y. Wang, F. Wang, B. Xue and J. Xu, Appl. Catal., A, 2023, 665, 119384 CrossRef.
- E. Aneggi, F. Campagnolo, J. Segato, D. Zuccaccia, W. Baratta, J. Llorca and A. Trovarelli, Mol. Catal., 2023, 540, 113049 CrossRef CAS.
- A. S. Priya, K. R. S. Devi and N. J. Venkatesha, J. Australas. Ceram. Soc., 2020, 56, 217–225 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |