
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFive and six membered heterocyclic rings endowed with azobenzene as dual EGFRT790M and VEGFR-2 inhibitors: design, synthesis, in silico ADMET profile, molecular docking, dynamic simulation and anticancer evaluations†
Kurls E. Anwera,
Sanadelaslam S. A. El-Hddad b,
Nour E. A. Abd El-Sattarac,
Ahmed El-morsyd,
Fathalla Khedre,
Samy Mohamadyf,
Doaa E. Keshekgh,
Samir A. Salamai,
Khaled El-Adl*ej and
Noura S. Hanafyj
b,
Nour E. A. Abd El-Sattarac,
Ahmed El-morsyd,
Fathalla Khedre,
Samy Mohamadyf,
Doaa E. Keshekgh,
Samir A. Salamai,
Khaled El-Adl*ej and
Noura S. Hanafyj
aDepartment of Chemistry, Faculty of Science, Ain Shams University, Abbassia, Cairo, Egypt
bPharmaceutical Chemistry Department, Faculty of Pharmacy, Omar Almukhtar University, Libya
cBasic & Medical Sciences Department, Faculty of Dentistry, Alryada University for Science & Technology, Egypt
dPharmaceutical Chemistry Department, College of Pharmacy, The Islamic University, Najaf, Iraq
ePharmaceutical Medicinal Chemistry and Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Nasr City 11884, Cairo, Egypt. E-mail: khaled.eladl@hu.edu.eg; eladlkhaled74@azhar.edu.eg; eladlkhaled74@yahoo.com
fPharmaceutical Chemistry Department, Faculty of Pharmacy, The British University in Egypt, Cairo, Egypt
gDepartment of Biology, Jumum College University, Umm Al-Qura University, P.O. Box 7388, Makkah 21955, Sudia Arabia
hAgriculture Genetic Engineering Research Institute (AGERI), Agriculture Research Centre, Giza, Egypt
iDivision of Biochemistry, Department of Pharmacology, College of Pharmacy, Taif University, P.O. Box 11099, Taif 21944, Kingdom of Saudi Arabia
jPharmaceutical Chemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt
First published on 4th December 2023
Abstract
Novel azobenzene scaffold-joined heterocyclic isoxazole, pyrazole, triazole, and/or triazine moieties have been developed and synthesized utilizing microwave and traditional methods. Our compounds were tested for growth inhibition of A549, MCF-7, HCT-116, and HepG2 tumors by dual targeting the VEGFR-2 and EGFRT790M enzymes. The suggested compound's manner of binding with EGFRT790M and VEGFR-2 active sites was explored through molecular design and MD modeling. The information from the results of the biological screening and the docking studies was highly correlated. The A549 cell line was the one that responded to the novel compound's effects most effectively. Having IC50 values of 5.15, 6.37, 8.44 and 6.23 μM, respectively, 14 was the most effective derivative on the four A549, MCF-7, HCT116 and HepG2 cancer cells. It had greater activity than erlotinib and slightly inferior activities on the tested cell lines than sorafenib, respectively. The cytotoxicity of the most effective derivatives, 5, 6, 10 and 14, was evaluated against typical VERO cell lines. Having IC50 values ranging from 42.32 to 55.20 μM, the results showed that the investigated drugs have modest toxicity against VERO normal cells. Additionally all derivatives were assessed for their dual VEGFR-2 and EGFRT790M inhibitory effects. Among them, derivatives 14, 5 and 10 were established as the greatest inhibitors of VEGFR-2 at IC50 values of 0.95, 1.25 and 1.50 μM correspondingly. As well, derivatives 14, 6, 5 and 10 could inhibit EGFRT790M activity demonstrating strongest effects with IC50 = 0.25, 0.35, 0.40 and 0.50 μM respectively. Furthermore, the ADMET profile was evaluated for compounds 5, 6, 10 and 14 in contrast to reference drugs sorafenib and erlotinib.
1 Introduction
A rational drug design tool for drug candidates could be achieved by molecular hybridization, which includes combination of two or more pharmacophoric groups or chemical units by either linking or fusing with each other to form new hybrid molecules, and their choice is based on their recognized bio-profiles in the hope that this hybridization could produce compounds with more powerful pharmacological activities. This strategy is often used since some of these hybrid derivatives showed enhanced efficacy and had a safer toxicity profile than their parent drug candidates.1,2Nitrogenous-heterocyclic rings are one of the utmost momentous groups in medicinal chemistry, and have lately been shown to introduce remarkably complicated biological characteristics. Due to their ability to mimic and interact with many biological components and produce amazing pharmacological effects, they serve as a fundamental scaffold in many drugs.3,4
As anticancer drugs that target EGFR-TK, several isoxazole derivatives have been identified.5 Additionally, a variety of pyrazole compounds have been implicated in the prevention of cancer as dual EGFR and VEGFR-2 TK inhibitors.6 Triazole derivatives are being utilized to treat various kinds of illnesses, including cancer.4,7 There are many anticancer medications with the 1,2,4-triazole ring on the market, including vorozole, letrozole and anastrozole (Fig. 1).4 Additionally, the triazine scaffold serves as a fundamental framework for the syntheses of several bioactive molecules with wide-ranging medical applications.1,2 Numerous studies examined the progress that had been made in the triazine candidates that acted via various protein kinases inhibition, including tubulin polymerization, EGFR (EGFRWT and EGFRT790M), hDHFR, human topoisomerase IIα, carbonic anhydrase (CA), PI3Kα/mTOR, and CDK2, to have highly promising antitumor activity.8,9 FDA-approved triazine-containing anticancer medications include tretamine, gedatolisib, KY-04031, altretamine, enasidenib, and HL 010183 (Fig. 1).4
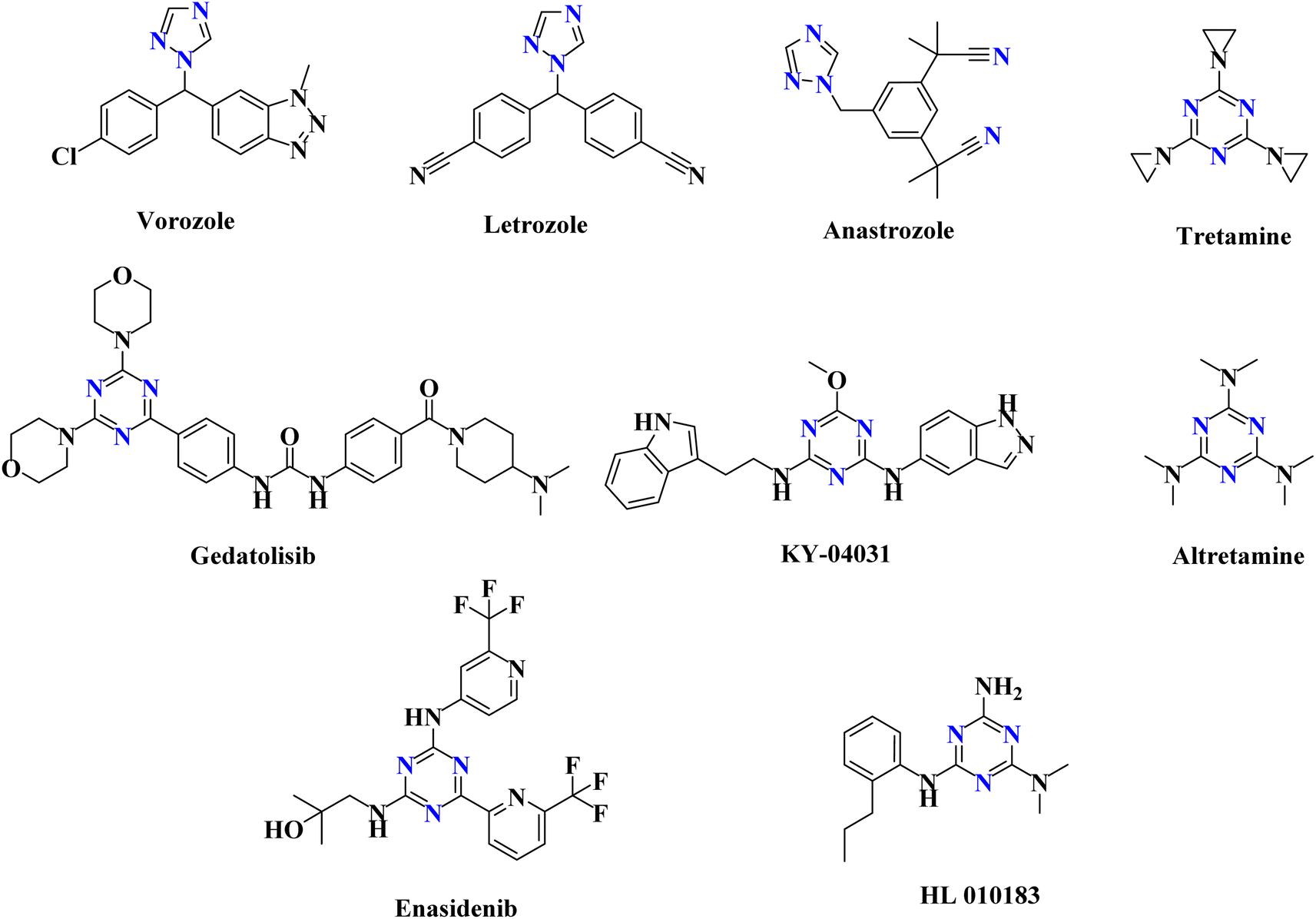 | ||
| Fig. 1 FDA-approved anticancer medications with 1,2,4-triazole and triazine rings. | ||
Azobenzenes, contain N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N linkages, are endowed with diversity of uses as antituberculotic, antitumor, antiviral, antidiabetic, anti-inflammatory, and antimicrobial agents.10 The potential of azobenzene to act as a trigger in several biomedical fields exhibited a thorough review.11,12 Azobenzene photo-switch can be utilized in protein structure and function photo-control, as well as kinases, proteases,13 ion channels,14 and G-coupled-protein receptor.15 Particularly, azobenzene has been integrated with PROTACs for target proteins photo-controlled degradation.16 Azobenzene is also frequently employed to accurately manipulate lipid membranes biophysics (e.g. permeability and fluidity).17 Azobenzenes have also been used to regulate nucleic acids for enzymatic reactions and gene expression photo-regulation.18 Moreover, different derivatives containing azobenzene moieties were reported as VEGFR-2 inhibitors.19
N linkages, are endowed with diversity of uses as antituberculotic, antitumor, antiviral, antidiabetic, anti-inflammatory, and antimicrobial agents.10 The potential of azobenzene to act as a trigger in several biomedical fields exhibited a thorough review.11,12 Azobenzene photo-switch can be utilized in protein structure and function photo-control, as well as kinases, proteases,13 ion channels,14 and G-coupled-protein receptor.15 Particularly, azobenzene has been integrated with PROTACs for target proteins photo-controlled degradation.16 Azobenzene is also frequently employed to accurately manipulate lipid membranes biophysics (e.g. permeability and fluidity).17 Azobenzenes have also been used to regulate nucleic acids for enzymatic reactions and gene expression photo-regulation.18 Moreover, different derivatives containing azobenzene moieties were reported as VEGFR-2 inhibitors.19
EGFR and VEGFR-2 are bounded in different disorders pathogenesis and different sets of carcinoma growth. Both have strong correlations and split common lower down signaling routes. The inter-correlations amidst VEGFR-2 and EGFR has been well-recognized: inhibition of signaling pathway of VEGFR-2 enhances the anticancer activity of EGFR inhibitors, whilst stimulating VEGFR-2 is independent on EGFR signaling which obstructs the EGFR inhibitors.20 Therefore, blocking both signaling pathways of VEGFR and EGFR synchronously appears to be an excellent strategy in treatment of cancer.21–23
1.1 Structure-based and rationale design
The rational design of the target compounds kept inhibitors of both EGFR and VEGFR-2 in the essential pharmacophoric form.A novel series of azobenzene endowed with heterocyclic moieties were designed and synthesized according to the essential VEGFR-2 inhibitors pharmacophoric structures as a continuation of our efforts to discover new agents for anticancer treatment.24–40 Four key characteristics were found to be shared by sorafenib and different VEGFR-2 inhibitors (Fig. 2);41–43 the terminal hydrophobic part which occupy the allosteric hydrophobic fissure forming hydrophobic bonds, pharmacophore linker with amino or urea functional groups that bind to the essential amino acids Glu883 and Asp1044, central hydrophobic spacer and a flat hetero aromatic ring system.
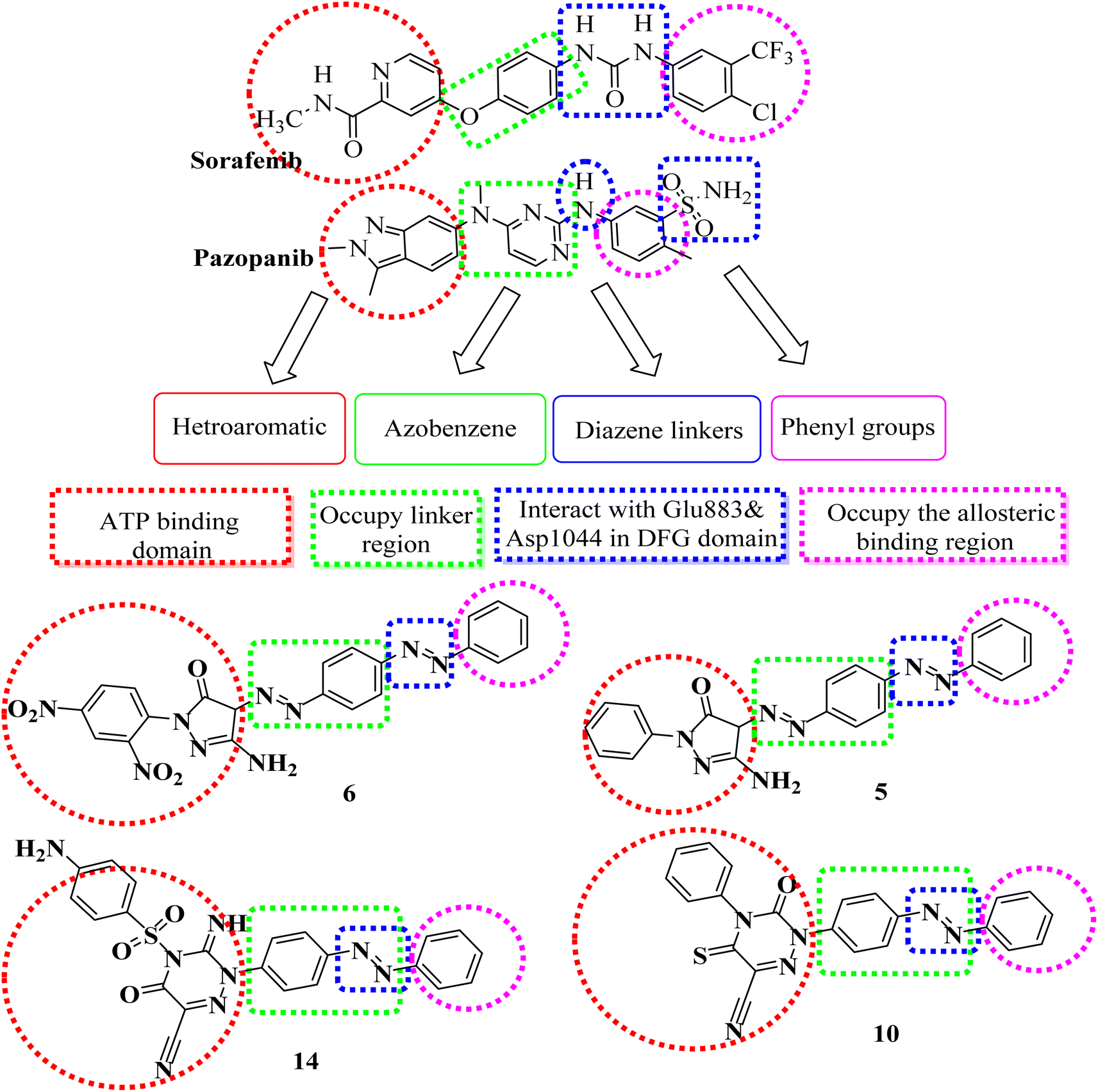 | ||
| Fig. 2 Pharmacophoric and structural similarities of some designed compounds and VEGFR-2 inhibitors. | ||
Our molecular design synthetic strategy focused on bioisosteric modification of sorafenib & pazopanib as inhibitors of VEGFR-2 at the different four parts (Fig. 2).
However, our novel compounds also displayed the EGFR-TKIs pharmacophoric structures, such as: (i) a lipophilic head that resides in the hydrophobic area. (ii) The adenine adherence pocket forms an interaction with a hetero aromatic ring. (iii) H-bond donor/and or acceptor42 e.g. NH group, which might interact at the linker domain with an amino acid to create strong hydrogen bonds. (iv) A hydrophobic tail that connects to the hydrophobic region II,44,45 (Fig. 3). Our investigation into SAR of synthetic anti-cancer compounds with combined EGFRT790M and VEGFR-2 preventive events was inspired by all of these alterations.
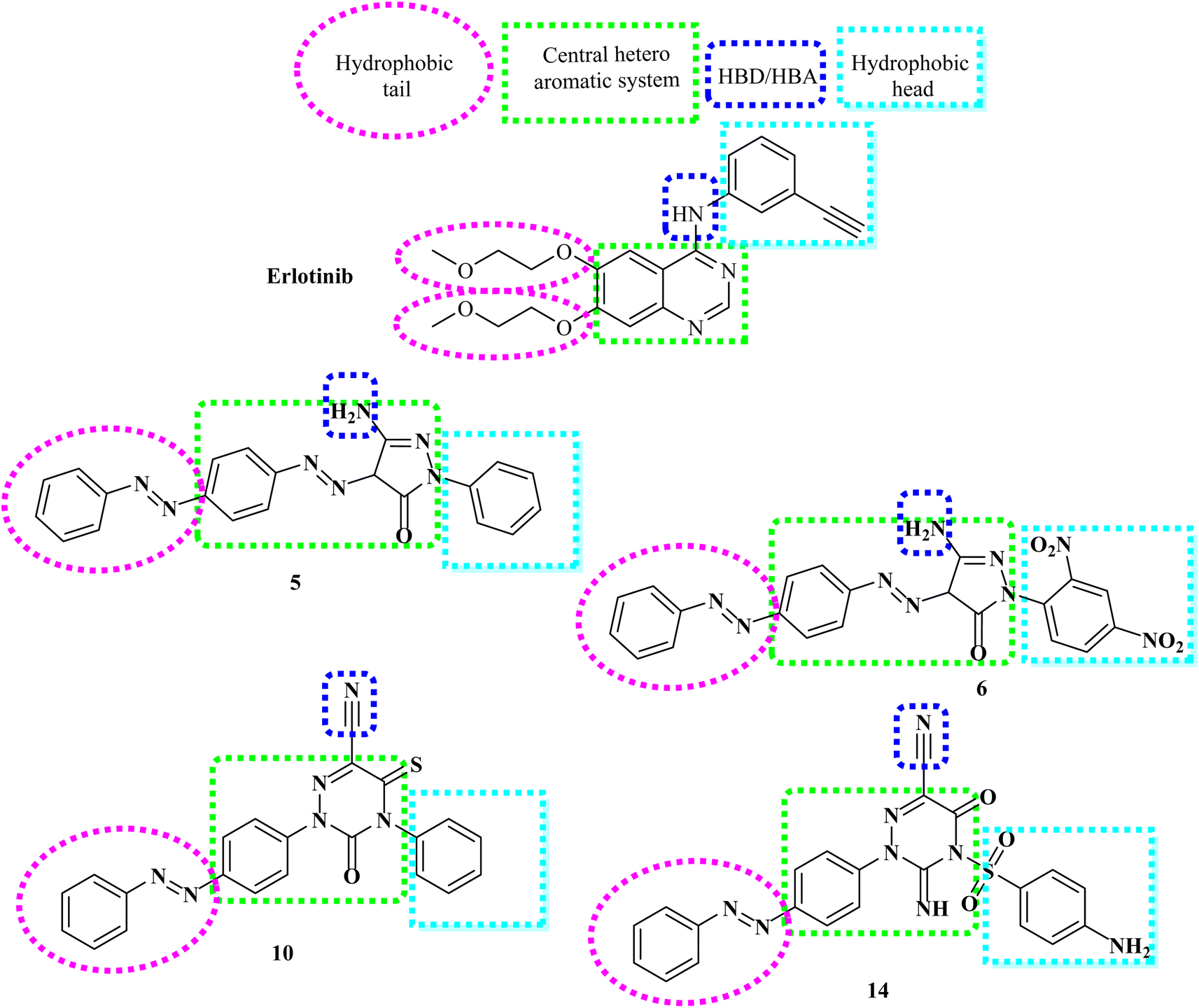 | ||
| Fig. 3 Design rationale based on structural characteristics of EGFR inhibitors. | ||
2 Results and discussion
2.1 Chemistry
Syntheses of new isoxazolone, pyrazalone, 1,2,4-triazole and 1,2,4-triazine derivatives following the reactions descripted in (Schemes 1–3). The starting material ethyl 2-cyano-2-{[4-(phenyldiazenyl)phenyl]-diazenyl}acetate (2) was synthesized via treatment of 4-aminoazobenzene with NaNO2 and HCl at 0–5 °C giving the corresponding diazonium chloride 1 which undergo coupling with ethyl cyanoacetate to produce the starting material 2 (Scheme 1). Compound 2 IR spectrum illustrated characteristic bands for C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N at 2225, CO at 1741, and 2 NN at 1489 and 1470. Also, the 1H-NMR and 13C-NMR spectra of compound 2 were consistent with the formed structure.
N at 2225, CO at 1741, and 2 NN at 1489 and 1470. Also, the 1H-NMR and 13C-NMR spectra of compound 2 were consistent with the formed structure.
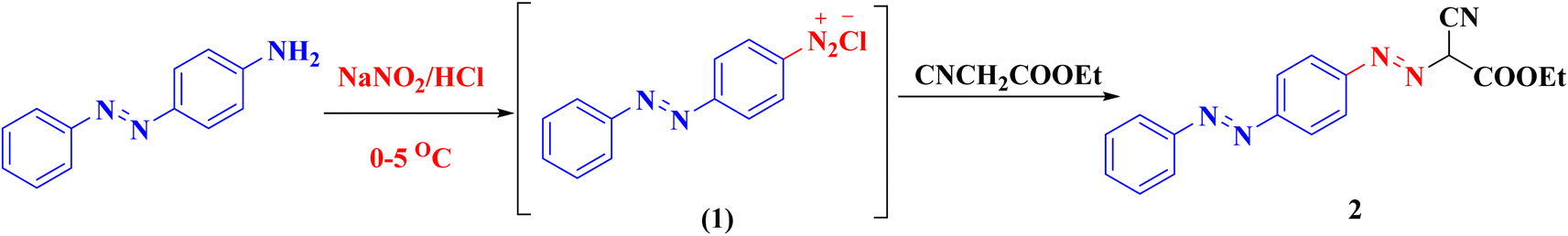 | ||
| Scheme 1 Synthesis of compound 2. | ||
Compound 2 was cyclized with hydroxylamine hydrochloride in the existence of anhydrous K2CO3 to give the targeted isoxazolone 3. This reaction proceeds via the nucleophilic attack of NH2 on cyano group then nucleophilic addition of OH group on CO group followed by elimination of one molecule of ethanol. The structure of 3 showed IR absorption bands at 3461, 3346 (NH2), 3190 (NH), 1686 cm−1 (CO). Compound 3 proton NMR showed 7.51–7.93 (m, 9H, Ar-H), 8.60 (s, 2H, NH2, D2O exchangeable), and 15.95 (s, 1H, NH, D2O exchangeable).
Also, compound 2 underwent cyclization with hydrazine hydrate, phenyl hydrazine, 2,4-dinitrophenyl hydrazine (DNP) and semicarbazide hydrochloride to obtain the corresponding pyrazolone derivatives 4–7, respectively. The proposed structures of 4–7 were confirmed by the disappearance of cyano bands and ester carbonyl bands but presence of NH2, NH and pyrazolone carbonyl bands, in their IR spectra.
Alternatively, cyclization of compound 2 with thiosemicarbazide was interestingly proceeds in another manner to produce 1,2,4-triazole derivative 9 instead of the expected pyrazolone derivative 8 (ESI†). Analytical and spectroscopic examinations of 9 led to the characterization of its chemical composition and structure. For instance, the compound's IR spectra showed CN band at 2226 cm−1, but no NH2 or CO bands. The proton NMR of 9 disclosed a signal at 4.49, 7.19 and 8.62 ppm specific for the 3NH groups as well as its 13C-NMR revealed a singlet signal at 181.6 ppm specific for CS group (Scheme 2).
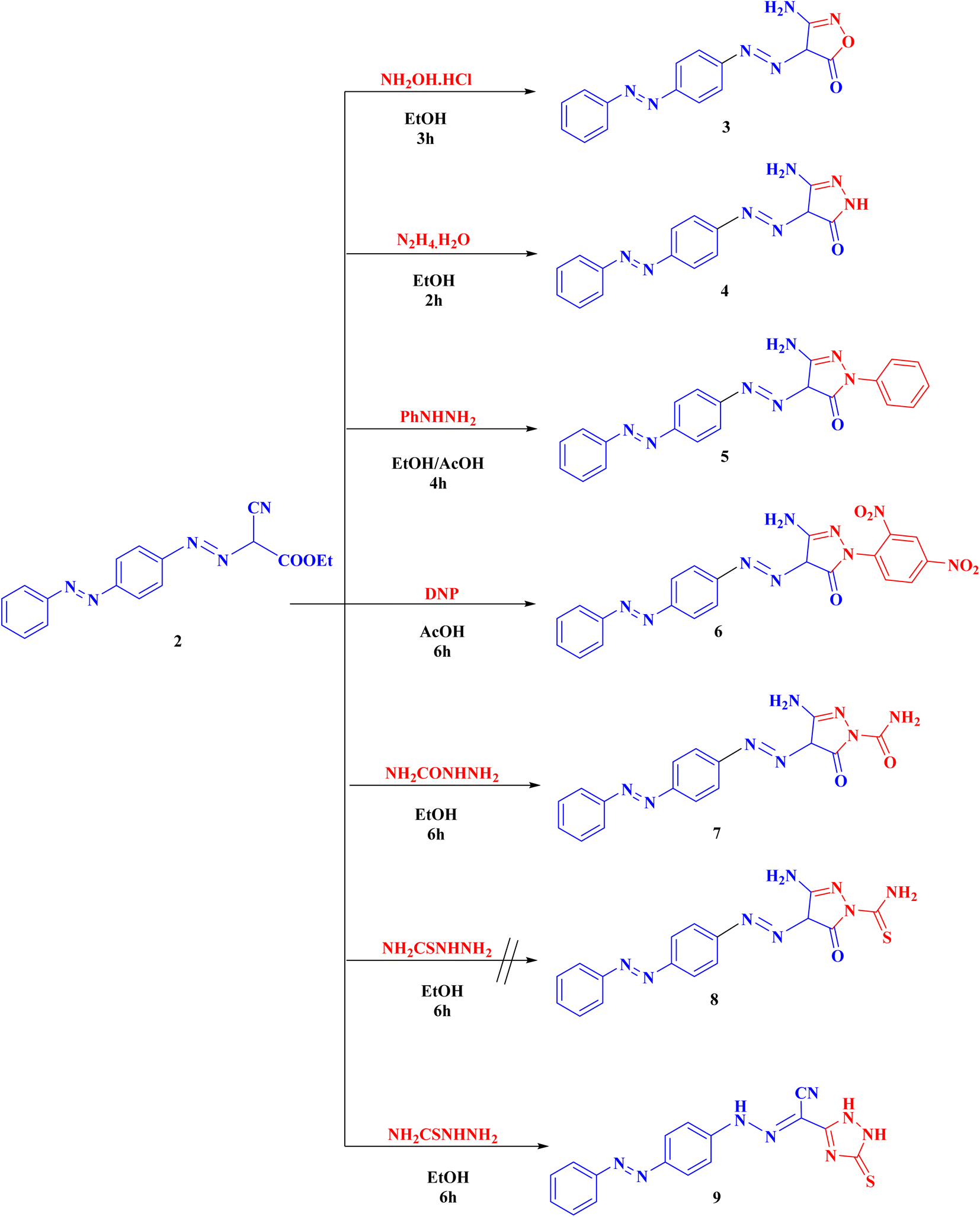 | ||
| Scheme 2 Syntheses of the targeted compounds (3–9). | ||
Compound 2 can be used as a fundamental intermediate for the syntheses of new 1,2,4-triazine derivatives via its cyclization reactions with phenyl isothiocyanate, thiourea, urea, guanidine hydrochloride, and/or sulphaguanidine to form compounds 10–14, respectively. Compound 10 was cyclized by nucleophilic attack of the NH group on the CS of the isothiocyanate group followed by intramolecular cyclization with removal of one ethanol molecule. Compounds 11–14 were obtained (Scheme 3) through the nucleophilic attack on the ester carbonyl by the NH group followed by loss of one ethanol molecule, then intramolecular rearrangement with elimination of one molecule of ammonia leading to ring cyclization via nucleophilic addition (ESI†).
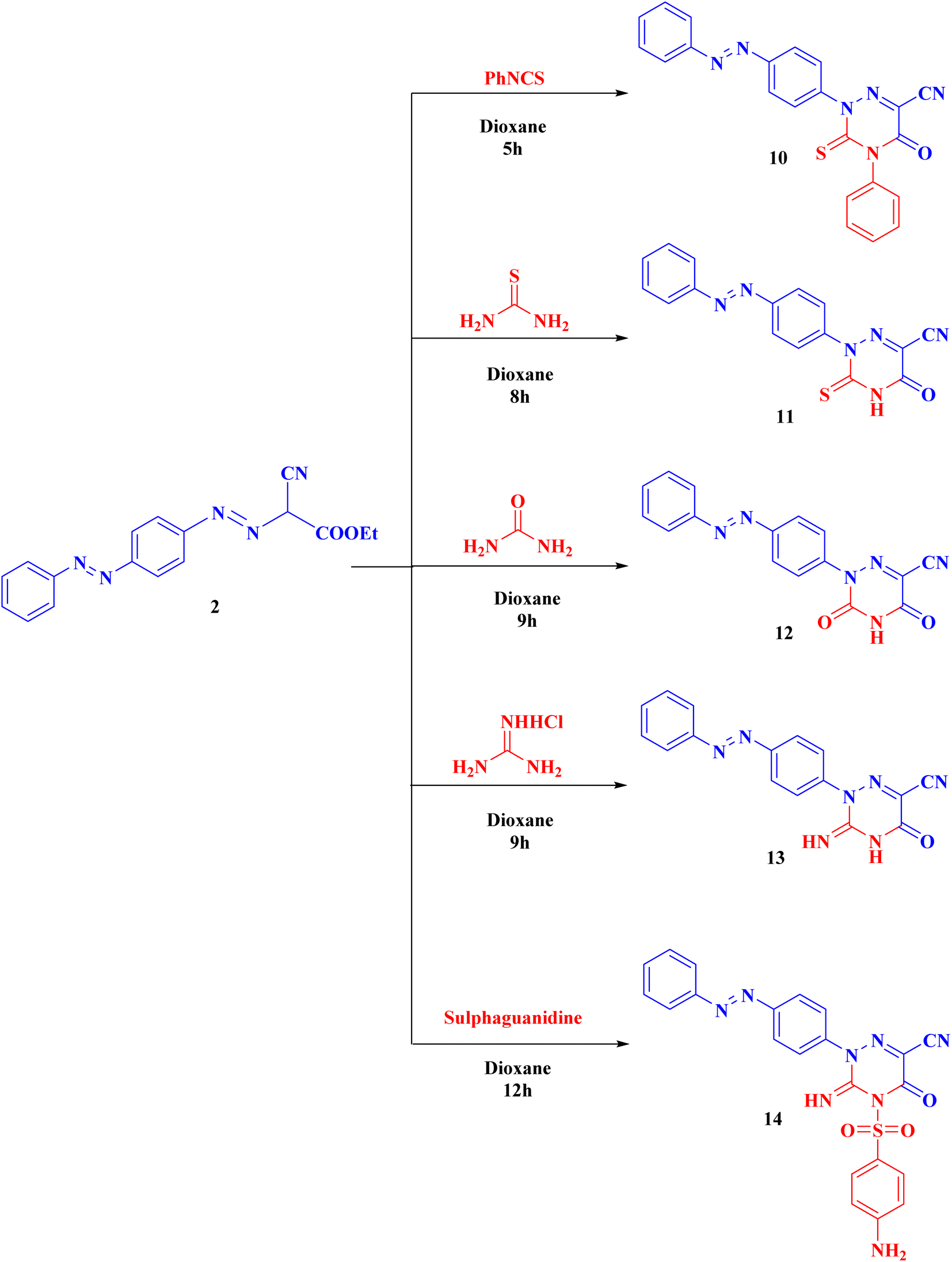 | ||
| Scheme 3 Syntheses of the targeted derivatives (10–14). | ||
Our compounds were produced utilising microwave technology and conventional methods46–50 (ESI†).
2.2 Docking studies
Molsoft program was used in this modeling research. Both EGFRT790M (PDB ID 3W2O)45 and VEGFR-2 (PDB ID 4ASD)51 were subjected to manipulation in each experiment.| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 3 | −85.25 | 10 | −91.59 |
| 4 | −93.97 | 11 | −77.72 |
| 5 | −99.64 | 12 | −78.46 |
| 6 | −98.92 | 13 | −82.37 |
| 7 | −85.71 | 14 | −102.62 |
| 9 | −85.89 | Sorafenib | −99.50 |
Five H-bonds and −99.50 kcal mol−1 were found for sorafenib. It made one H-bond with Asp1046 (1.50 Å), 2 hydrogen bonds with Glu885 (2.75 Å and 1.77 Å) and 2 hydrogen bonding interfaces with Cys919 (2.51 Å and 2.10 Å). The hydrophobic groove created by Leu1035, Cys1045, Val848 and Lys868 is wrapped by the central phenyl linker. Additionally, the hydrophobic channel formed by Ile892, Ile888, Hie1026, Glu885, Cys1045, and Asp1046 was dominated by the distal 3-trifluromethyl-4-chlorophenyl array. In excess, N-methylpicolinamide set assimilated inside the cleft constructed by Cys919, Phe918, Glu917, Lys920, Val848, Leu1035 and Leu840 (Fig. 4).
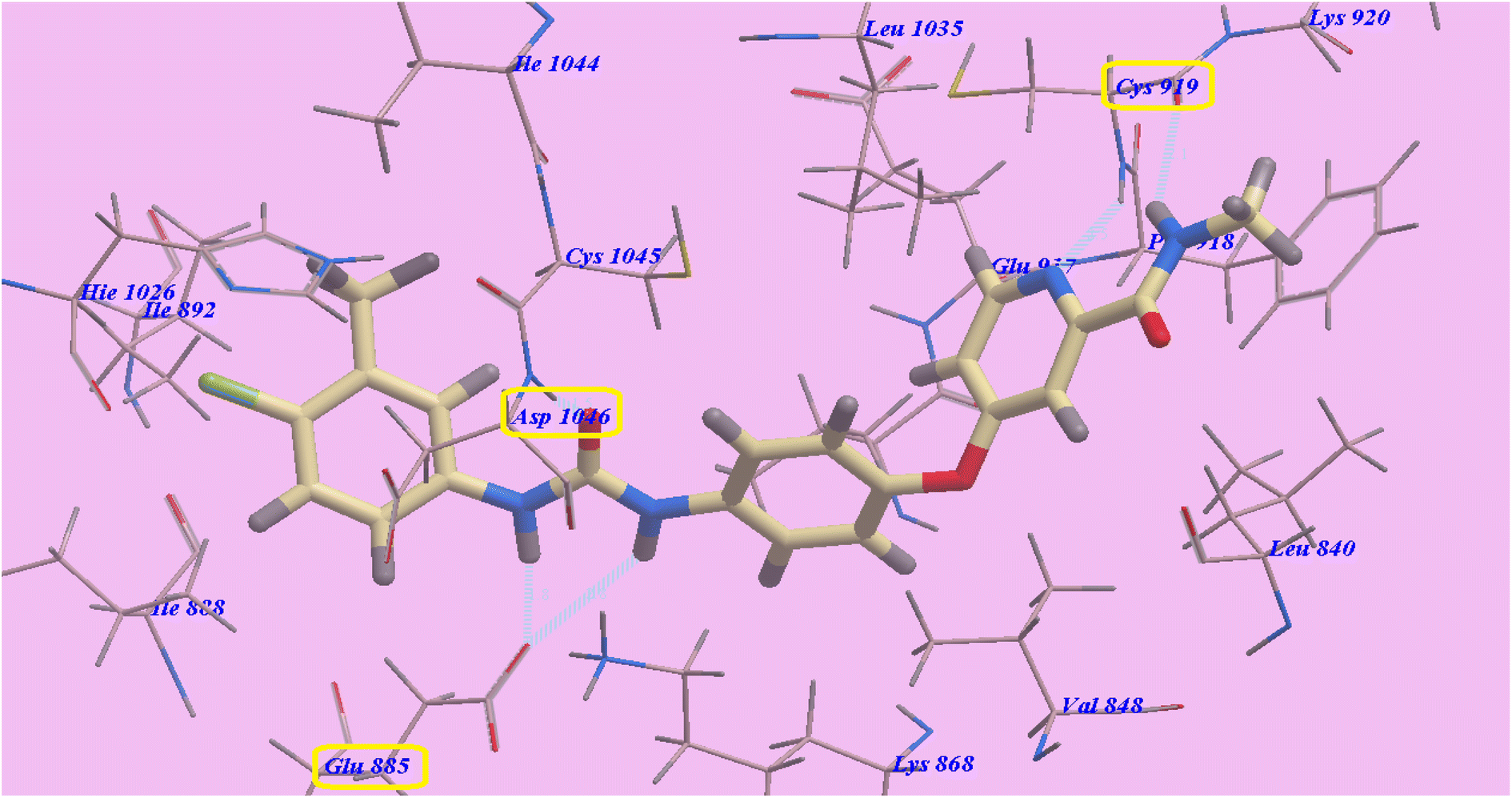 | ||
| Fig. 4 Predicted 4ASD binding mechanism of sorafenib. | ||
Compound 14 exhibited −102.62 kcal mol−1 and 5 hydrogen bonds. The SO2 group exhibited two hydrogen bonds with Cys919 (2.45 Å and 2.79 Å). The NH2 group showed one H-bond with Leu840 (2.73 Å). Moreover, the cyano group displayed one H-bond with Ile849 (2.19 Å). Also the diazene linker displayed one hydrogen bond with Asp1046 (1.87 Å). The socket created by Lys868, Leu1035, Val848, Ile849, Leu840, Ly920, Phe918, Cys919, and Glu917 was filled by the heterocyclic p-aminophenylsulfonyltriazine molecule. As well, the hydrophobic groove shaped by Leu1035, Lys868, Glu917, Cys1045 and Asp1046 has an azobenzene spacer built into it. The hydrophobic vessel generated by Cys1045, Ile892, Asp1046, Hie1026, Ile888, Glu885, Ile1044 and Glu885 was likewise filled by the terminal benzene ring (Fig. 5).
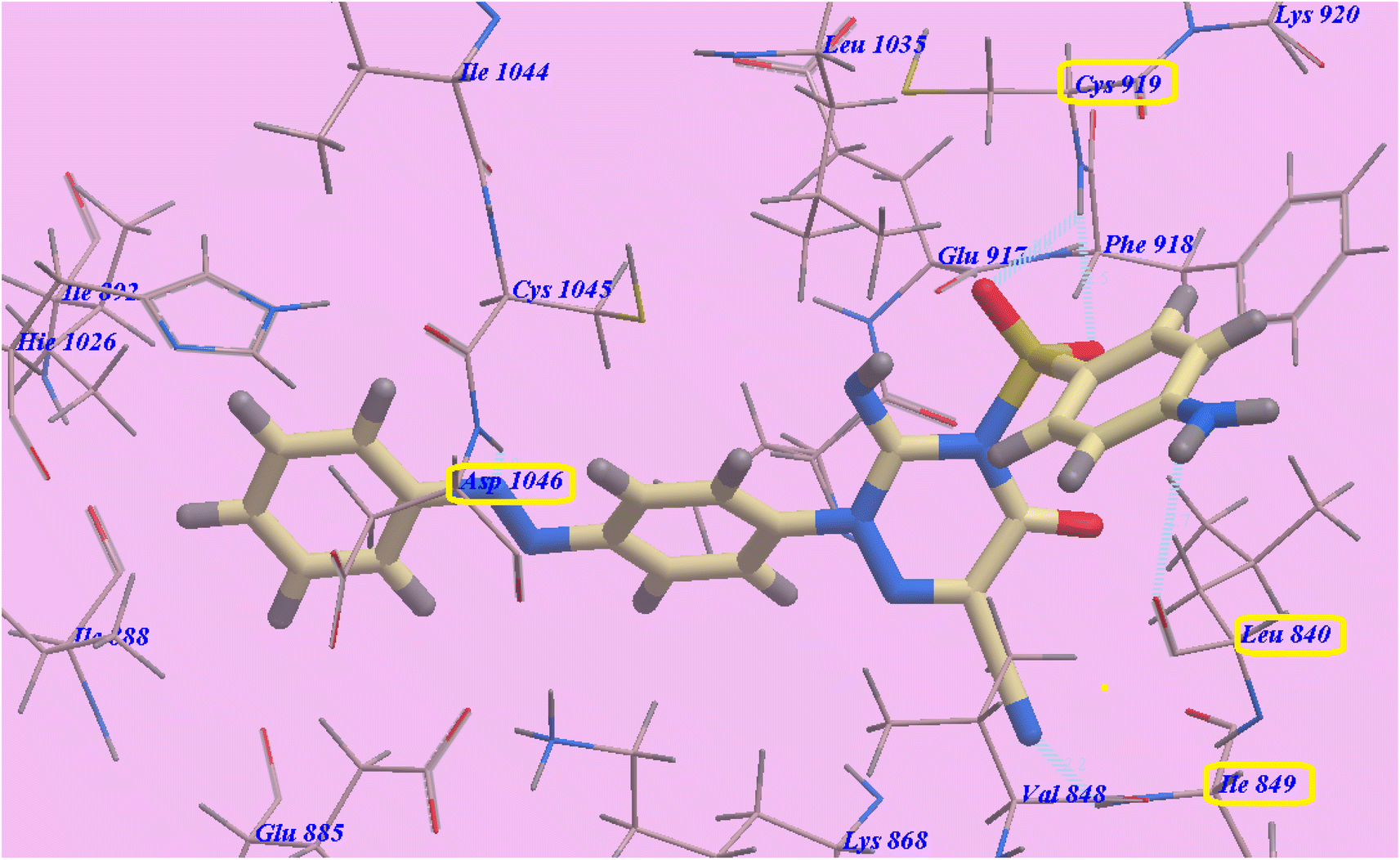 | ||
| Fig. 5 Compound 14's predicted binding mode with 4ASD. | ||
Compounds 5 showed nearly the same binding mode of 14 with −78.04 kcal mol−1 and 4 H-bonds. It formed one H-bond with Cys919 (2.47 Å), two hydrogen bonds with Glu917 (1.68 Å and 1.69 Å) and one H-bond with Asp1046 (1.68 Å). 5-Amino-2-phenylpyrazol-3-one occupies the hollow formed via Lys868, Val848, Leu1035, Ile849, Cys919, Phe918, Lys920, Leu840 and Glu917. While compound 10 exhibited −76.05 kcal mol−1 and 4 H-bonds. It linked to Cys919 through two H-bonds (1.68 Å and 2.99 Å), one hydrogen bond with Glu917 (2.79 Å) and one H-bond with Asp1046 (2.07 Å). The heterocyclic 5-oxo-4-phenyl-3-thioxo-1,2,4-triazine-6-carbonitrile occupies the channel formed via Lys868, Val848, Leu1035, Ile849, Cys919, Phe918, Lys920, Leu840 and Glu917 (ESI†).
| Compound | ΔG [kcal mol−1] | Compound | ΔG [kcal mol−1] |
|---|---|---|---|
| 3 | −63.49 | 10 | −76.05 |
| 4 | −66.21 | 11 | −60.33 |
| 5 | −78.04 | 12 | −67.58 |
| 6 | −82.90 | 13 | −65.36 |
| 7 | −71.84 | 14 | −87.16 |
| 9 | −66.15 | Erlotinib | −82.77 |
Four H-bondings and −82.77 kcal mol−1 were seen with Erlotinib. The quinazoline scaffold is linked to Valine726 and Methionine793 through one H binding each with 2.97 Å and 1.82 Å respectively. One 2-methoxyethoxy moiety is linked to Cys797 through one H binding (2.05 Å). Also NH spacer is linked to Thr854 through H-bond (2.99 Å). The hydrophobic domain I created with Ile759, Phe723, Gly724, Val726, Glu762, Leu777, Thr854, Glu791, Met790, and Asp855 was linked to the 3-ethynylphenyl head. In addition, the 2-methoxyethoxy end made hydrophobic interactions with the groove II that was created by Val 845, Met 793, Leu 718, Pro 794, and Leu 844 (Fig. 6).
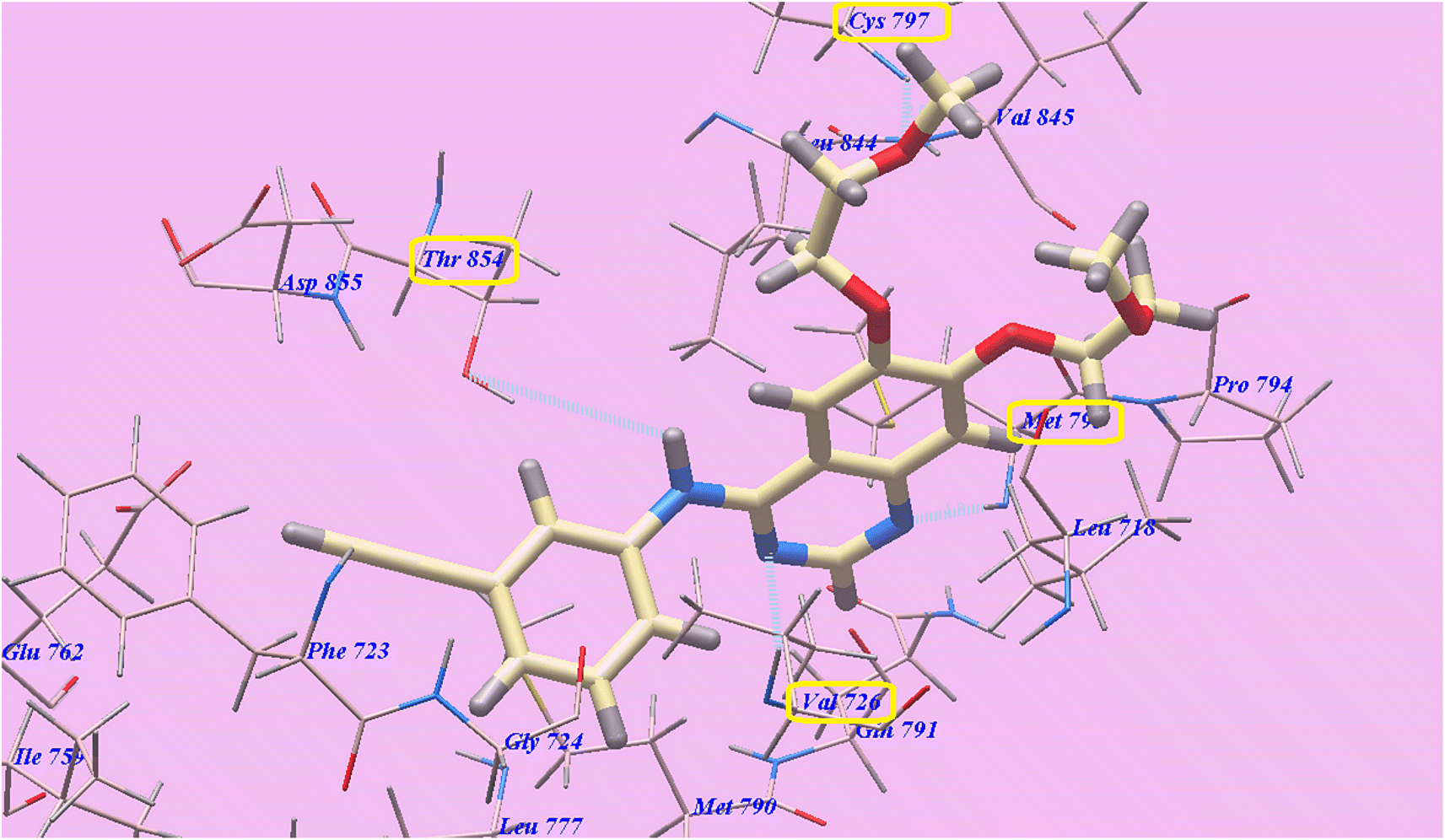 | ||
| Fig. 6 Erlotinib's anticipated binding style with 3W2O. | ||
Compound 14 exhibited 6 H-bonds and −87.16 kcal mol−1. The NH2 group linked with Glu762 through two H-bonds (1.71 Å and 2.60 Å). Also SO2 exhibited two hydrogen bonds with Gly724 (2.56 Å and 2.67 Å). Additionally, the cyano group linked with Lys754 through H-bond (2.77 Å). Furthermore the triazine ring linked with Thr854 through hydrogen bond (2.99 Å). The azobenzene end occupies the hydrophobic region II built via Pro794, Met793, Leu718, Val726, Val845, Cys797 and Leu844. Moreover, the 5-amino-2-phenylpyrazol-3-one head occupies the hydrophobic zone I created by Leu788, Glu762, Ile759, Gly724, Phe723 and Asp855 (Fig. 7).
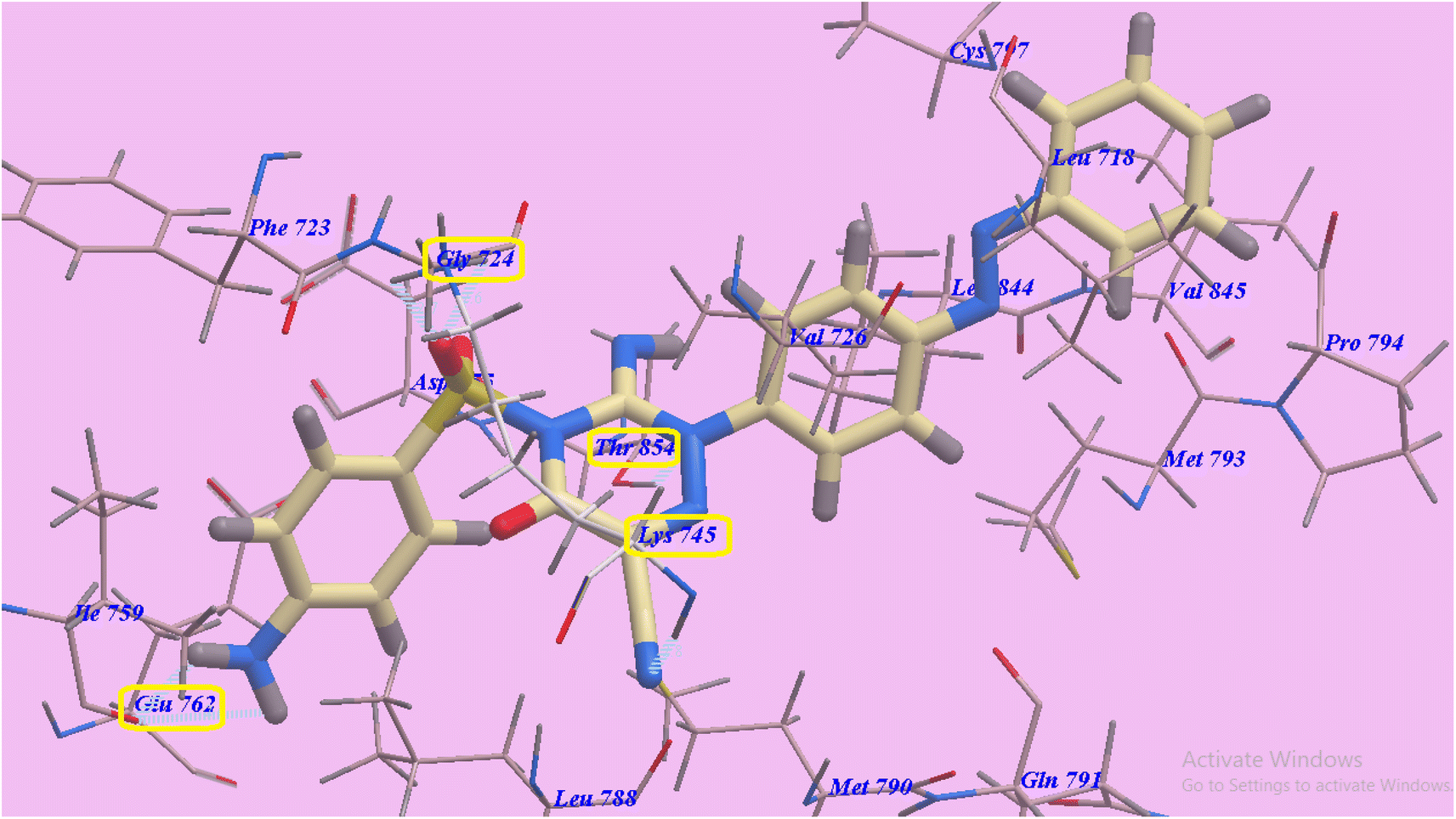 | ||
| Fig. 7 Anticipated binding style for 14 with 3W2O. | ||
Structure 6 is approximately the same as 14 with 4H-bonds and −82.90 kcal mol−1. It made 4 hydrogen bonds with Asp855 (2.36 Å), Thr854 (1.75 Å and 2.98 Å), and Lys745 (2.14 Å). The azobenzene end occupies the hydrophobic region II built via Pro794, Met793, Leu718, Val726, Cys797 and Leu844. Moreover, 5-amino-2-(2,4-dinitrophenyl)pyrazol-3-one head filled the hydrophobic zone I created by Leu788, Glu762, Ile759, Gly724, Phe723 and Asp855. Also derivative 5 showed three H-bonding interactions and −78.04 kcal mol−1. It made 3 hydrogen bonds with Asp855 (2.47 Å) and Thr854 (2.06 Å and 2.97 Å) (ESI†).
2.3 Molecular dynamics simulation
The highly active derivatives 5, 6, 10 and 14 in the proteins VEGFR-2 and EGFRT790M were simulated using molecular dynamics (MD). By utilizing Amber's MM/GBSA.py script and the trajectory, the receptor-ligand binding energy was calculated.52 Additionally, the employed as positive controls were sorafenib and erlotinib. With the help of GAFF2 (ref. 53 and 54) and the force field AMBERff14SB for the protein, ligand force fields were produced.55 The monitored root mean square deviation (RMSD) validated the studied inhibitor compound's considerable global stability inside the target's recognized active site throughout the 30 ns all-atom MD runs (Fig. 8). A given ligand's molecular divergence from a defined original/reference structure is estimated using RMSD. The selected MD simulation procedure was valid, and it gave a respectable suggestion of the stability of the ligand-target interaction.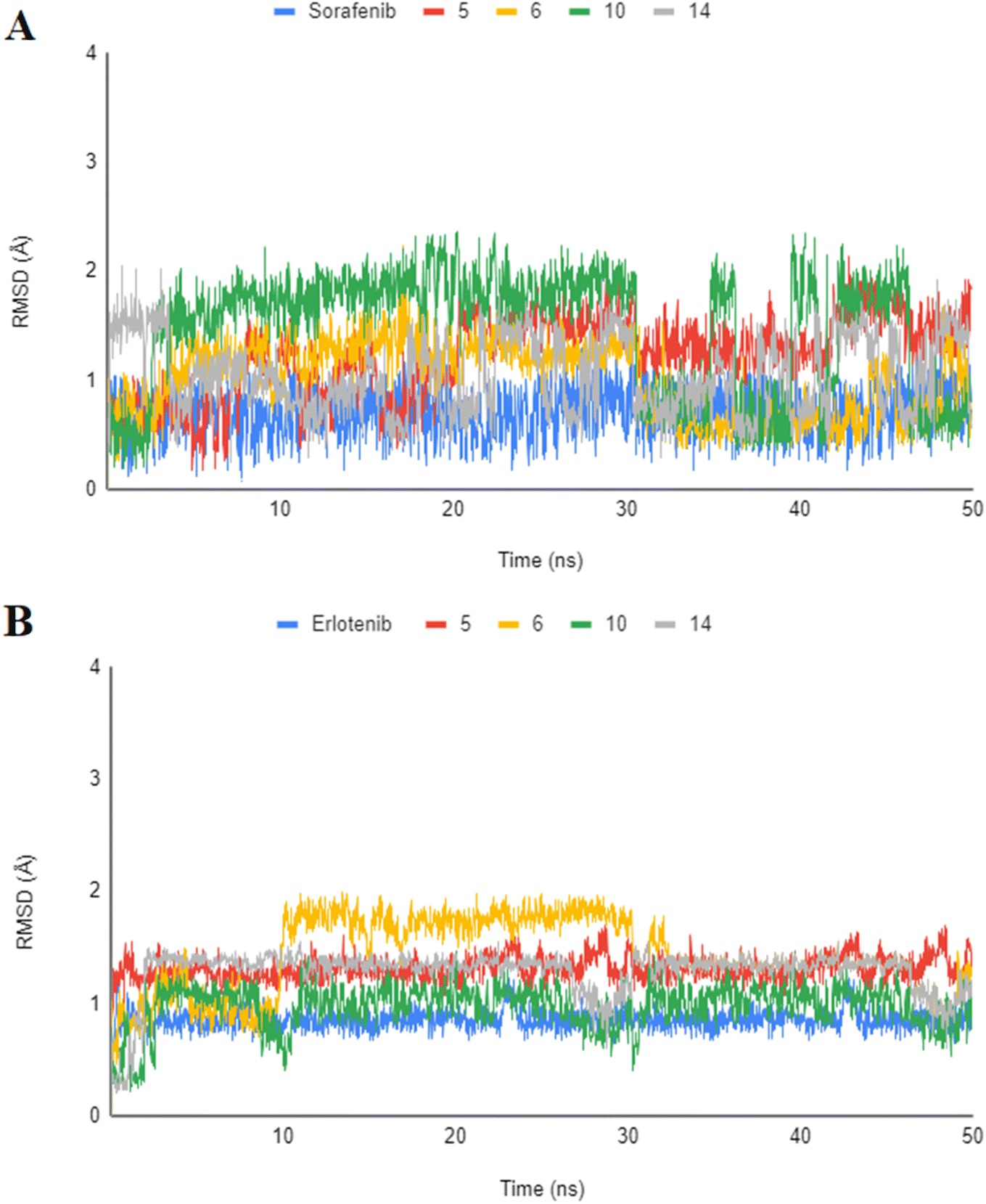 | ||
| Fig. 8 Analysis of RMSD throughout 50 ns for the ligand–protein complexes all-atom MD simulation. (A) VEGFR-2 protein RMSD; (B) EGFRT790M protein RMSD. | ||
2.4 Biological testing
| Comp. | IC50a (μM) | ||||||
|---|---|---|---|---|---|---|---|
| HepG2 | HCT116 | MCF-7 | A549 | VERO | VEGFR-2 | EGFRT790M | |
| a IC50 values are the mean ± S.D. of three separate experiments.b NT = Not tested. | |||||||
| 3 | 19.00 ± 4.4 | 16.75 ± 3.7 | 17.55 ± 4.5 | 25.50 ± 3.5 | NTb | 3.25 ± 0.50 | 2.10 ± 0.25 |
| 4 | 10.55 ± 3.0 | 11.27 ± 3.1 | 11.33 ± 3.3 | 10.85 ± 1.5 | NTb | 1.85 ± 0.10 | 1.20 ± 0.35 |
| 5 | 7.80 ± 0.7 | 8.12 ± 0.9 | 6.98 ± 1.1 | 6.50 ± 1.5 | 44.60 ± 0.31 | 1.25 ± 0.50 | 0.40 ± 0.35 |
| 6 | 8.17 ± 2.6 | 10.10 ± 2.9 | 9.11 ± 2.7 | 5.88 ± 0.5 | 45.15 ± 0.43 | 1.60 ± 0.50 | 0.35 ± 0.15 |
| 7 | 15.60 ± 3.5 | 17.50 ± 4.0 | 14.25 ± 3.5 | 8.50 ± 0.5 | NTb | 2.35 ± 0.20 | 0.90 ± 0.25 |
| 9 | 14.50 ± 2.8 | 16.75 ± 3.3 | 13.80 ± 3.0 | 12.25 ± 2.5 | NTb | 2.00 ± 0.10 | 1.35 ± 0.15 |
| 10 | 6.81 ± 1.9 | 9.23 ± 2.2 | 7.28 ± 2.4 | 7.35 ± 1.5 | 42.32 ± 0.31 | 1.50 ± 0.10 | 0.50 ± 0.25 |
| 11 | 35.64 ± 4.8 | 37.93 ± 4.6 | 39.22 ± 4.8 | 30.25 ± 3.5 | NTb | 3.85 ± 0.50 | 2.50 ± 0.35 |
| 12 | 21.10 ± 4.9 | 22.50 ± 4.8 | 30.25 ± 4.8 | 9.25 ± 1.5 | NTb | 3.50 ± 0.50 | 0.95 ± 0.35 |
| 13 | 20.50 ± 4.2 | 20.05 ± 3.5 | 23.05 ± 3.8 | 15.50 ± 2.5 | NTb | 3.35 ± 0.50 | 1.50 ± 0.15 |
| 14 | 6.23 ± 2.3 | 8.44 ± 2.6 | 6.37 ± 2.5 | 5.15 ± 1.5 | 55.20 ± 0.32 | 0.95 ± 0.10 | 0.25 ± 0.15 |
| Sorafenib | 4.00 ± 0.33 | 5.05 ± 0.50 | 5.58 ± 0.55 | 4.04 ± 0.33 | NTb | 0.84 ± 0.04 | NTb |
| Erlotinib | 7.73 ± 0.67 | 13.91 ± 1.3 | 8.20 ± 0.34 | 5.49 ± 0.45 | NTb | NTb | 0.24 ± 0.22 |
With reverence to the HepG2 cell lines, derivatives 5, 6 and 10 demonstrated very high anticancer effects with IC50 = 7.80, 8.17 and 6.81 μM respectively. Compounds 3, 4, 7 and 9, with IC50 ranging from 10.55 to 19.00 μM, presented good cytotoxicity. Furthermore, derivatives 12 and 13, with IC50 = 21.10 and 20.50 μM respectively, showed moderate cytotoxicity. Additionally, derivative 11 with IC50 = 35.64 μM, demonstrated the lowest cytotoxicity.
Assessment of cytotoxicity on HCT-116 cell lines revealed that derivatives 5 and 10 demonstrated very high anticancer effects with IC50 = 8.12 and 9.23 μM respectively. Compounds 3, 4, 6, 7 and 9, with IC50 ranging from 10.10 to 17.50 μM, presented good cytotoxicity. Furthermore, compounds 12 and 13, with IC50 = 22.50 and 20.05 μM respectively, showed moderate cytotoxicity. Additionally, derivative 11 with IC50 = 37.93 μM, demonstrated the lowest cytotoxicity.
Assessment of cytotoxicity on MCF-7 cell lines showed that derivatives 5, 6 and 10 demonstrated very high anticancer effects with IC50 = 6.98, 9.11 and 7.28 μM respectively. Compounds 3, 4, 7 and 9, with IC50 ranging from 11.33 to 17.55 μM, displayed good cytotoxicity. In addition, 13, with IC50 = 23.05 μM, showed moderate cytotoxicity. Finally, compounds 11 and 12 with IC50 = 39.22 and 30.25 μM respectively, demonstrated the lowest cytotoxicity.
Assessment of cytotoxicity against A549 cell lines discovered that derivatives 5, 6, 7, 10 and 12 with IC50 ranging from 5.88 to 9.25 μM displayed very high anticancer effects. Compounds 4, 9 and 13, with IC50 = 10.85, 12.25 and 15.50 μM respectively, presented good cytotoxicity. Besides, compound 3 with IC50 = 25.50 μM, demonstrated moderate cytotoxicity. Finally, compound 11 with IC50 = 30.25 μM correspondingly, presented the lowest cytotoxicity.
Furthermore, the greatest active compounds 5, 6, 10 and 14 were examined against normal VERO cell lines for their cytotoxicity. The results discovered that the tested derivatives with IC50 values ranging from 42.32 to 55.20 μM showed low toxicity against VERO normal cells. Compounds 5, 6, 10 and 14 are respectively, 5.72, 5.49, 6.39 and 6.86 fold times more toxic in toxic in HepG-2 than in VERO normal cells. Uniformly, derivatives 5, 6, 10 and 14 are consequently 5.53, 4.47, 4.96 and 7.68 folds toxic in HCT-116 than in normal VERO cells. Furthermore, structures 5, 6, 10 and 14 are respectively 6.21, 4.59, 5.81 and 5.76 folds toxic in MCF-7 than in ordinary VERO cells. Furthermore, products 5, 6, 10 and 14 are respectively 8.86, 6.54, 8.67 and 10.72 folds toxic in A549 than in ordinary VERO cells.
Derivatives 14, 6, 5 and 10 could inhibit EGFRT790M activity demonstrating strongest effects with IC50 = 0.25, 0.35, 0.40 and 0.50 μM respectively. Derivatives 4, 7, 9, 12 and 13 considerably inhibited EGFRT790M at IC50 ranging from 0.90 to 1.50 μM. Conversely compounds 3 and 11 moderately inhibited EGFRT790M at IC50 values of 2.10 and 2.50 μM respectively.
We are able to categorize the tested derivatives into two categories based on the structure of the synthesized derivatives and the data shown in Table 3. The first category consists of compounds 3–9, which include heterocyclic rings with five members that are isoxazole, pyrazole, or triazole. Generally, the pyrazole derivatives exhibited higher activities than triazole and isoxazole derivatives respectively. The isoxazole derivative 3 exhibited the lowest activities against HepG2, A549 and MCF-7 but against HCT116 it showed anticancer activity equipotent to that of triazole derivative 9. The tiazole 9 revealed higher activities than pyrazole 7 against the tested cell lines except A549. Against HepG2, MCF-7, and HCT116 cell lines, pyrazole derivative 5 with a more lipophilic phenyl group shown stronger activities than 6 with lipophilic electron withdrawing 2,4-dinitro substituents, less lipophilic pyrazole derivative 4 without a phenyl ring, and 7 with carboxamide, whereas against A549 the order of activity is 6 > 5 > 7 > 4.
The second group is compounds 10–14 which containing triazine heterocyclic ring; where the more lipophilic derivative 14 with electron withdrawing 4-aminophenylsulfone and 3-imino showed higher activities than 10 with lipophilic 4-phenyl and 3-thioxo, less lipophilic 13 with 3-imino and no phenyl ring, 12 with 3-oxo and no phenyl ring and 11 with 3-thioxo and no phenyl ring against HepG2, HCT116 and MCF-7 but against A549 the order is 14 > 10 > 12 > 13 > 11 (Fig. 9).
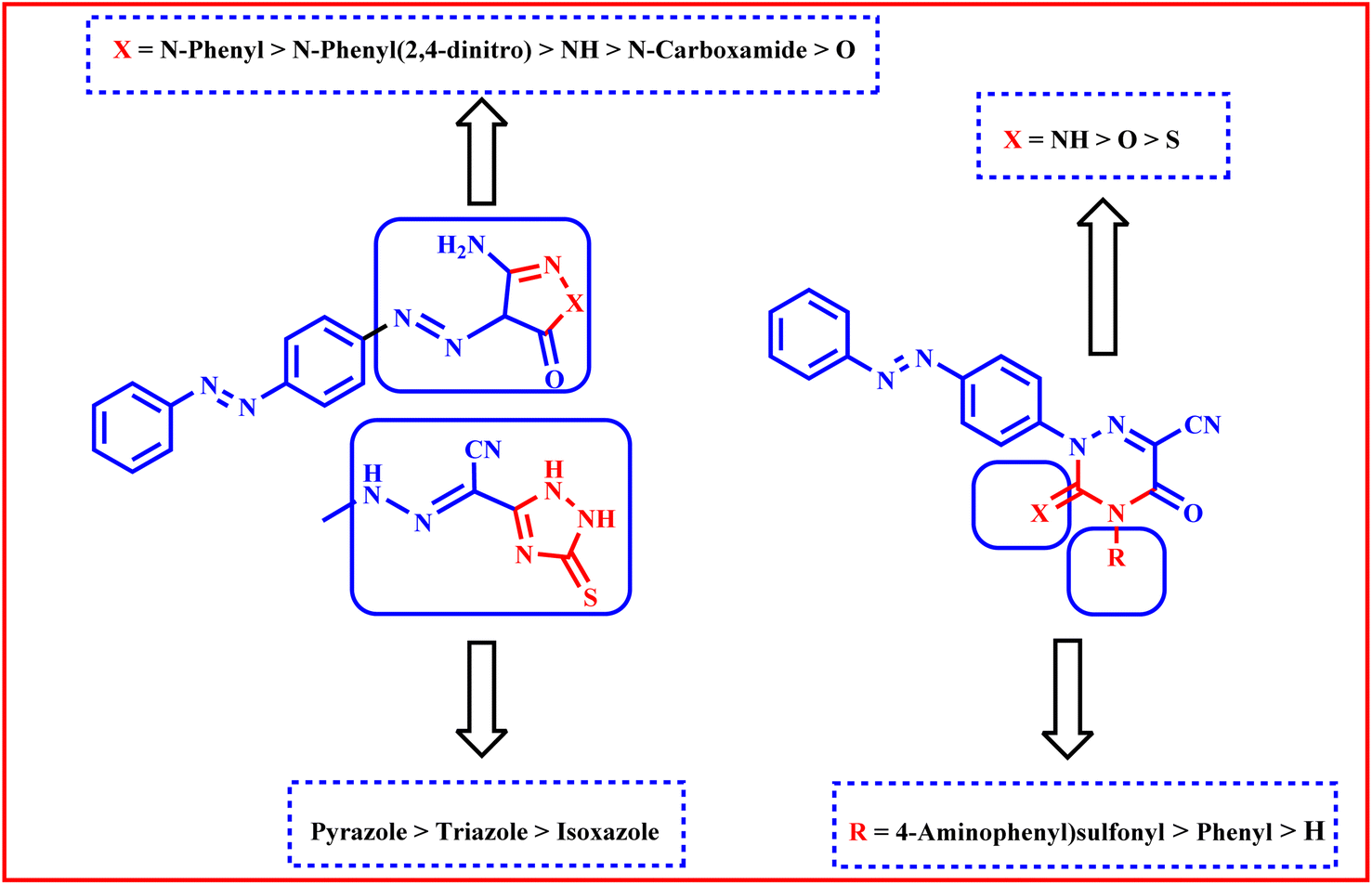 | ||
| Fig. 9 Schematic presentation for SAR study. | ||
2.5 In silico ADMET profile
The highly active compounds 5, 6, 7, 9, 10 and 14 were subjected to in silico study of their proposed ADMET profile and the physicochemical characters evaluation. It was expected using pkCSM descriptor algorithm procedures62 and corresponding to Lipinski's rule of five.63 For the compounds that comply with at least three rules, the favorable absorption characteristics were predictable: (i) hydrogen bond donors should not exceed five, (ii) partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) should not exceed five, (iii) molecular weight should not exceed 500, (iv) hydrogen bond acceptors should not exceed ten. The traditional anticancer drug sorafenib and our derivatives 6, 10, and 14 all violate one rule in the published work, however our novel molecules 5, 7, 9 and erlotinib don't.
P) should not exceed five, (iii) molecular weight should not exceed 500, (iv) hydrogen bond acceptors should not exceed ten. The traditional anticancer drug sorafenib and our derivatives 6, 10, and 14 all violate one rule in the published work, however our novel molecules 5, 7, 9 and erlotinib don't.
Our compounds have good human gastrointestinal tract (GIT) absorption (84.583 to 100), which implies that they are more simply able to cross different biological membranes,64 according to the results collected (Table 4). As a result, they may exhibit an astonishingly high bioavailability through GIT. Our synthetic compounds can penetrate the central nervous system (CNS) (CNS permeability range: −1.644 to −2.654).
| Parameter | 5 | 6 | 7 | 9 | 10 | 14 | Sorafenib | Erlotinib |
|---|---|---|---|---|---|---|---|---|
| a + = yes while − = No. | ||||||||
| Physicochemical properties | ||||||||
| Molecular weight | 383.531 | 473.409 | 350.342 | 348.395 | 410.462 | 472.49 | 464.831 | 393.443 |
| LogP |
4.8733 | 4.6897 | 2.7474 | 4.22247 | 5.03977 | 2.61975 | 5.5497 | 3.4051 |
| Acceptors | 7 | 11 | 8 | 7 | 8 | 11 | 4 | 7 |
| Rotatable bonds | 5 | 7 | 4 | 5 | 4 | 5 | 5 | 10 |
| Surface area | 166.655 | 195.961 | 147.464 | 147.506 | 176.375 | 192.802 | 185.111 | 169.532 |
| Donors | 1 | 1 | 2 | 3 | 0 | 2 | 3 | 1 |
|
||||||||
| Absorption | ||||||||
| Water solubility | −5.124 | −4.759 | −3.805 | −3.934 | −6.312 | −3.651 | −4.822 | −4.736 |
| Caco2 permeability | 0.894 | −1.077 | −0.412 | −0.607 | 0.385 | −0.113 | 0.689 | 1.431 |
| GIT absorption | 100 | 96.005 | 86.122 | 84.583 | 95.101 | 86.405 | 89.043 | 94.58 |
| Inhibitor of P-glycoprotein II | + | + | − | + | + | + | + | + |
| Inhibitor of P-glycoprotein I | + | + | + | − | + | + | + | + |
| Substrate for P-glycoprotein | + | + | + | + | + | + | + | − |
| Skin permeability | −2.685 | −2.735 | −2.802 | −2.775 | −2.723 | −2.735 | −2.767 | −2.741 |
|
||||||||
| Distribution | ||||||||
| CNS permeability | −1.764 | −2.263 | −2.424 | −2.257 | −1.644 | −2.654 | −1.655 | −3.216 |
| BBB permeability | −0.816 | −1.858 | −1.137 | −1.59 | −0.963 | −1.522 | −1.726 | −0.745 |
| VDss (human) | 0.002 | −0.171 | −0.427 | −0.912 | −0.136 | −0.814 | −0.625 | 0.199 |
|
||||||||
| Metabolism | ||||||||
| CYP3A4 substrate | + | + | + | + | + | + | + | + |
| CYP2D6 substrate | − | − | − | − | − | − | − | − |
| Inhibition of CYP2C9 | + | + | − | + | + | + | + | + |
| Inhibition of CYP1A2 | + | − | + | + | + | − | + | + |
| Inhibition of CYP3A4 | + | + | − | + | + | + | + | + |
| Inhibition of CYP2C19 | + | + | − | + | + | + | − | − |
| Inhibition of CYP2D6 | − | − | − | − | − | − | + | + |
|
||||||||
| Excretion | ||||||||
| Renal OCT2 subst | − | − | + | − | − | − | − | − |
| Clearance | −0.407 | −0.426 | −0.309 | −0.011 | −0.152 | 0.103 | −0.219 | 0.702 |
|
||||||||
| Toxicity | ||||||||
| Skin sensitization | − | − | − | − | − | − | − | − |
| Hepatotoxic effect | − | − | − | + | + | + | + | + |
| Chronic toxic activity (LOAEL) | 0.91 | 1.345 | 1.025 | 1.507 | 0.732 | 0.132 | 1.198 | 1.37 |
| Acute toxic activity (LD50) | 2.103 | 2.489 | 2.281 | 2.583 | 2.191 | 2.627 | 2.538 | 2.393 |
| hERG II inhibitor | + | + | − | + | + | + | + | + |
| hERG I inhibitor | − | − | − | − | − | − | − | − |
| Max. Tolerated dose | 0.116 | −0.235 | −0.433 | 0.234 | 0.557 | 0.128 | 0.549 | 0.839 |
| AMES toxicity | + | + | − | − | + | + | − | − |
It is generally known that erlotinib, sorafenib, and our derivatives 5, 6, 9, 10 and 14 may inhibit CYP3A4, the main enzyme responsible for drug metabolism. The overall clearance, which is a crucial consideration in choosing dosage intervals, predicted elimination. Compared to sorafenib and our drugs, which had extremely low clearance values, the findings exhibited that erlotinib had superior clearance rates. Erlotinib should thus have shorter dose intervals since it might be eliminated more quickly. In contrast to erlotinib, the synthesized compounds showed a slow clearance rate, which denotes a prolonged half-life and wider dosage windows. The final analyzed factor for the ADMET profile is toxicity. As shown in Table 4, the disadvantage of unintended hepatotoxic effects was shared by sorafenib, erlotinib, and our drugs 9, 10 and 14, but not by 5, 6 and 7. The drug with the greatest maximum tolerated dose was erlotinib. The maximum tolerable dosages for sorafenib and our derivatives, however, were lower. Lastly, the acute toxic doses of our novel compounds are nearly the same as that of sorafenib and erlotinib. Also, the oral chronic toxic dose of compound 9 is higher than that of both sorafenib and erlotinib while that of compound 6 is higher than that of sorafenib and nearly equipotent to that of erlotinib. These data revealed that our derivatives have good therapeutic index.
3 Conclusion
In conclusion, novel isoxazole, pyrazole, triazole, and/or triazine heterocyclic moieties attached to the azobenzene scaffold have been designed, synthesized, and tested on HCT-116, MCF-7, HepG2, and A549 for their anticancer activities targeting both EGFRT790M and VEGFR-2 enzymes. The suggested compound's mechanism of interaction with the EGFRT790M and VEGFR-2 receptors was examined through molecular design. The information from the biological screening and the results of the docking studies were strongly linked. Compound 14 was the most potent derivative against the four A549, HepG2, MCF-7 and HCT116 cell lines with IC50 = 5.15, 6.236.37 and 8.44 μM respectively. It presented slightly lower activities than sorafenib, (IC50 = 4.00, 5.05, 5.58 and 4.04 μM respectively), and revealed higher activities than erlotinib (IC50 = 7.73, 13.91, 8.20 and 5.49 μM respectively) against the four cell lines. With reverence to the HepG2 cell lines, derivatives 5, 6 and 10 demonstrated very good anticancer effects with IC50 = 7.80, 8.17 and 6.81 μM respectively. Assessment of cytotoxicity against HCT-116 cell lines revealed that derivatives 5 and 10 demonstrated very good anticancer effects with IC50 = 8.12 and 9.23 μM respectively. Evaluation of cytotoxicity against MCF-7 cell lines showed that derivatives 5, 6 and 10 demonstrated very good anticancer effects with IC50 = 6.98, 9.11 and 7.28 μM respectively. When derivatives 5, 6, 7, 10 and 12 were tested for cytotoxicity against A549 cell lines, it was found that they had excellent anticancer effects, with IC50 values ranging from 5.88 to 9.25 μM. The cytotoxicity of the most powerful derivatives 5, 6, 10 and 14, was evaluated against normal VERO cell lines. With IC50 values ranging from 42.32 to 55.20 μM, the results showed that the investigated drugs have modest toxicity against VERO normal cells. In addition all compounds were selected to assess their dual VEGFR-2 and EGFRT790M inhibitory effects. Among them, compounds 14, 5 and 10 were found to be the most potent that inhibited VEGFR-2 at IC50 value of 0.95, 1.25 and 1.50 μM correspondingly. As well, derivatives 14, 6, 5 and 10 could inhibit EGFRT790M activity demonstrating strongest effects with IC50 = 0.25, 0.35, 0.40 and 0.50 μM respectively. Additionally, the highly active derivatives 5, 6, 10 and 14 in both VEGFR-2 and EGFRT790M proteins were subjected to a molecular dynamics (MD) simulation in order to evaluate our molecular modeling findings. Furthermore, the ADMET profiles of the most active compounds 5, 6, 7, 9, 10 and 14 were assessed in comparing to sorafenib and erlotinib as the reference drugs. The data obtained indicated that our derivatives could be beneficial as a future design and optimization template to create more powerful dual VEGFR-2 and EGFRT790M inhibitors with stronger anticancer analogues.4 Experimental
4.1 Chemistry
1H NMR and 13C NMR spectra were recorded on a Bruker 400 and 100 MHZ-NMR respectively, with a spectrophotometer at Microanalytical Center, Ain Shams University.Orange crystals from ethanol, m.p. = 250–252 °C; IR (KBr, ν, cm−1) 2225 (CN), 1741 (CO), 1489, 1470 (NN); 1H-NMR (DMSO-d6) δ: 1.24 (t, 3H, CH3CH2), 1.85 (s, 1H, –NN–CH), 4.15 (q, 2H, CH3CH2), 7.43–7.84 (m, 9H, Ar-H); 13C-NMR (DMSO-d6) δ: 15.3, 58.9, 98.7, 118.4, 120.2 (2), 122.6 (2), 124.3 (2), 129.8 (2), 130.8, 148.2, 152.8, 166.2, 174.7; anal. calcd for C17H15N5O2: C, 63.55; H, 4.67; N, 21.81; found: C, 63.66; H, 4.59; N, 21.84.
4.1.2.1 3-Amino-4-{2-[4-(phenyldiazenyl)phenyl]hydrazono}isoxazol-5(4H)-one (3). Orange crystals from methanol, m.p. >300 °C; IR (KBr, ν, cm−1) 3461, 3346 (NH2), 3190 (NH), 1686 (C
O), 1597, 1545 (CN), 1417 (NN); 1H-NMR (DMSO-d6) δ: 7.51–7.93 (m, 9H, Ar-H), 8.60 (s, 2H, NH2, D2O exchangeable), 15.95 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 111.9, 117.5 (2), 122.9 (2), 124.6 (2), 129.9 (2), 131.9, 145.4, 149.2, 152.6, 160.6 and 170.7; MS (m/z): 308 (M+, 29.23%), 271 (37.04%), 196 (100%, base peak), 161 (63.98%), 96 (80.35%), 76 (51.94%); anal. calcd for C15H12N6O2: C, 58.44; H, 3.90; N, 27.27; found: C, 58.29; H, 3.78; N, 27.33.
4.1.2.2 5-Amino-4-{2-[4-(phenyldiazenyl)phenyl]hydrazono}-2,4-dihydro-3H-pyrazol-3-one (4). Black crystals from methanol, m.p. >300 °C; IR (KBr, ν, cm−1) 3378, 3324 (NH2), 3183 (NH), 1682 (C
O), 1635, 1604 (CN), 1566 (NN); 1H-NMR (DMSO-d6) δ: 5.95 (s, 2H, NH2, D2O exchangeable), 7.54–7.97 (m, 9H, Ar-H), 10.62 (s, 1H, NH, D2O exchangeable), 13.05 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 111.9, 117.2 (2), 122.9 (2), 124.7, 129.9, 131.6 (2), 143.9, 145.4, 149.2, 152.5, 160.6 and 161.3; anal. calcd for C15H13N7O: C, 58.63; H, 4.23; N, 31.92; found: C, 58.49; H, 4.33; N, 32.04.
4.1.2.3 5-Amino-2-phenyl-4-{2-[4-(phenyldiazenyl)phenyl]hydrazono}-2,4-dihydro-3H-pyrazol-3-one (5). Reddish brown crystals from acetic acid, m.p. >300 °C; IR (KBr, ν, cm−1) 3418 (broad, NH2), 3179 (NH), 1683 (C
O), 1469, 1444 (NN); 1H-NMR (DMSO-d6) δ: 7.56–7.99 (m, 16H, 14 Ar-H & NH2 (D2O exchangeable)), 12.48 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 105.9, 106.5, 111.8, 117.1, 117.3 (2), 122.9 (2), 124.7 (2), 129.9 (2), 131.7 (2), 131.8 (2), 144.0, 145.0, 149.1, 152.5, 161.2; MS (m/z): 383 (M+, 2.22%), 367 (100%, base peak), 366 (60.07%), 175 (35.79%), 115 (84.99%), 91 (97.54%); anal. calcd for C21H17N7O: C, 65.80; H, 4.44; N, 25.59; found: C, 65.74; H, 4.35; N, 26.06.
4.1.2.4 5-Amino-2-(2,4-dinitrophenyl)-4-{2-[4-(phenyldiazenyl)phenyl]hydrazono}-2,4-dihydro-3H-pyrazol-3-one (6). Brown crystals from acetic acid, m.p. = 188–190 °C; IR (KBr, ν, cm−1) 3445 (broad, NH2), 3164 (NH), 1685 (C
O), 1469, 1444 (NN); 1H-NMR (DMSO-d6) δ: 7.55–7.99 (m, 14H, 12 Ar-H & NH2, (D2O exchangeable)), 12.49 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 105.5, 108.5, 112.1, 117.3 (2), 122.8 (2), 122.9 (2), 124.7 (2), 129.9 (2), 131.6, 138.5, 141.9, 145.8, 149.8, 152.5, 155.3, 161.4; MS (m/z): 473 (M+, 1.88%), 386 (23.95%), 162 (35.91%), 103 (100%, base peak), 77 (92.77%); anal. calcd for C21H15N9O5: C, 53.28; H, 3.17; N, 26.64; found: C, 53.33; H, 3.06; N, 26.70.
4.1.2.5 3-Amino-5-oxo-4-{[4-(phenyldiazenyl)phenyl]diazenyl}-4,5-dihydro-1H-pyrazole-1-carboxamide (7). Brown crystals from methanol, m.p. = 214–216 °C; IR (KBr, ν, cm−1) 3381, 3325 (NH2), 3186 (NH), 1683 (C
O), 1636, 1604 (CN), 1495 (NN); 1H-NMR (DMSO-d6) δ: 5.86 (s, 2H, NH2, D2O exchangeable), 7.53–7.94 (m, 11H, 9 Ar-H & NH2 (D2O exchangeable)), 11.59 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 116.2 (2), 122.9 (2), 124.7 (2), 125.9, 129.9 (2), 131.6, 145.1, 148.7, 150.3, 152.6, 158.8, 160.0; MS (m/z): 350 (M+, 15.31%), 301 (100%, base peak), 300 (44.03%), 265 (83.38%), 126 (74.51%); anal. calcd for C16H14N8O2: C, 54.86; H, 4.00; N, 32.00; found: C, 54.97; H, 3.89; N, 32.12.
4.1.2.6 N-[4-(Phenyldiazenyl)phenyl]-5-thioxo-2,5-dihydro-1H-1,2,4-triazole-3-carbohydrazonoyl-cyanide (9). Orange crystals from acetone, m.p. = 266–268 °C; IR (KBr, ν, cm−1) 3500–2500 (broad, NH), 2226 (C
N), 1625 (CN), 1522 (NN), 1252 (CS); 1H-NMR (DMSO-d6) δ: 4.49 (s, 1H, NH, D2O exchangeable), 7.19–7.99 (m, 10H, 9 Ar-H & NH (D2O exchangeable)), 8.62 (s, 2H, 2NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 105.4, 112.3, 117.4 (2), 122.9 (2), 124.7 (2), 129.9 (2), 131.6, 146.1, 149.0, 152.5, 161.6, 181.6; MS (m/z): 348 (M+, 11.00%), 326 (15.09%), 184 (37.37%), 177 (100%, base peak), 169 (48.79%), 65 (65.76%); anal. calcd for C16H12N8S: C, 55.17; H, 3.45; N, 32.18; S, 9.20; found: C, 55.02; H, 3.54; N, 32.13; S, 9.31.
4.1.3.1 5-Oxo-4-phenyl-2-[4-(phenyldiazenyl)phenyl]-3-thioxo-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile (10). Orange crystals from ethanol, m.p. 284–286 °C; IR (KBr, ν, cm−1) 2224 (C
N), 1683 (CO), 1596 (CN); 1H-NMR (DMSO-d6) δ: 7.13–7.99 (m, 14H, Ar-H); 13C-NMR (DMSO-d6) δ: 117.2, 117.3, 122.8, 124.1 (4), 124.7 (2), 124.9, 128.9 (4), 129.2 (2), 129.9, 139.9, 149.1, 152.5, 180.1; anal. calcd for C22H14N6OS: C, 64.39; H, 3.41; N, 20.49; S, 7.80; found: C, 64.21; H, 3.49; N, 20.61; S, 7.77.
4.1.3.2 5-Oxo-2-[4-(phenyldiazenyl)phenyl]-3-thioxo-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile (11). Reddish brown crystals from acetone, m.p. >300 °C; IR (KBr, ν, cm−1) 2224 (C
N), 1680 (CO), 1622, 1598 (CN), 1520 (NN), 1237 (CS); 1H-NMR (DMSO-d6) δ: 7.56–7.99 (m, 9H, Ar-H), 11.27 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 105.8, 111.8, 113.9, 116.0, 117.2 (2), 122.9 (2), 124.7 (2), 129.9 (2), 131.8, 149.1, 152.5, 201.5; anal. calcd for C16H10N6OS: C, 57.49; H, 2.99; N, 25.15; S, 9.58; found: C, 57.55; H, 3.08; N, 25.03; S, 9.44.
4.1.3.3 3,5-Dioxo-2-[4-(phenyldiazenyl)phenyl]-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile (12). Brown crystals from methanol, m.p. 280–284 °C; IR (KBr, ν, cm−1) 3177 (NH), 2225 (C
N), 1680 (CO), 1598 (CN), 1522 (NN); 1H-NMR (DMSO-d6) δ: 7.49–8.00 (m, 9H, Ar-H), 12.44 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 113.9, 116.0, 117.2 (2), 122.9 (2), 124.7 (2), 129.9 (2), 131.7, 143.9, 145.2, 149.1, 152.5, 161.2; anal. calcd for C16H10N6O2: C, 60.38; H, 3.14; N, 26.42; Found: C, 60.49; H, 3.07; N, 26.55.
4.1.3.4 3-Imino-5-oxo-2-[4-(phenyldiazenyl)phenyl]-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile (13). Brown crystals from acetic acid, m.p. 276–278 °C; IR (KBr, ν, cm−1) 3174 (NH), 2222 (C
N), 1679 (CO), 1597 (CN), 1520 (NN); 1H-NMR (DMSO-d6) δ: 7.36–8.06 (m, 9H, Ar-H), 12.64 (s, 2H, 2NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 105.7, 111.9, 117.3 (2), 122.9 (2), 124.7 (2), 129.9 (2), 131.7, 144.0, 145.4, 149.1, 152.5, 161.3; MS (m/z): 317 (M+, 19.14%), 269 (100%, base peak), 250 (60.16%), 188 (84.71%), 174 (51.62%); anal. calcd for C16H11N7O: C, 60.57; H, 3.47; N, 30.91; found: C, 60.51; H, 3.33; N, 31.06.
4.1.3.5 4-[(4-Aminophenyl)sulfonyl]-3-imino-5-oxo-2-[4-(phenyldiazenyl)phenyl]-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile (14). Brown crystals from methanol, m.p. >300 °C; IR (KBr, ν, cm−1) 3366, 3329 (broad, NH2), 3215 (NH), 2225 (C
N), 1681 (CO), 1621, 1599 (CN), 1501 (NN); 1H-NMR (DMSO-d6) δ: 5.67 (s, 2H, NH2, D2O exchangeable), 6.53–7.99 (m, 13H, Ar-H), 9.68 (s, 1H, NH, D2O exchangeable); 13C-NMR (DMSO-d6) δ: 105.9, 106.5, 111.8, 117.1, 117.3 (2), 122.8, 122.9 (2), 124.6, 124.7 (2), 129.9 (2), 131.7, 131.8, 144.0, 145.0, 149.1, 152.5, 160.7, 161.2; MS (m/z): 472 (M+, 7.13%), 451 (20.61%), 340 (24.14%), 312 (32.65%), 266 (100%, base peak); anal. calcd for C22H16N8O3S: C, 55.93; H, 3.40; N, 23.73; S, 6.78; found: C, 56.06; H, 3.37; N, 23.70; S, 6.89.
4.2 Conventional and microwave methods comparison
In the microwave reactions, the same reactants amounts in the thermal technique were used. The reaction completion was monitored by using TLC. The reaction mixtures were washed with ethanol and crystallized from the suitable solvent. Anton Paar (monowave 300) was used for microwave irradiation reactions using borosilicate glass vials of 10 mL.4.3 Docking studies
Both VEGFR-2 (PDB ID 4ASD)51 and EGFRT790M (PDB ID 3W2O)45 were used by Molsoft program to carry out docking studies (ESI†).4.4 Molecular dynamics simulation
The highly active derivatives 5, 6, 10 and 14 were simulated using molecular dynamics (MD) in VEGFR-2 and EGFRT790M With the help of GAFF2 (ref. 52 and 53) (ESI†).4.5 Biological testing
Conflicts of interest
There is no interest to declare.Acknowledgements
We would like to acknowledge the Deanship of Scientific Research, Taif University for funding this work.References
- R. Reddyrajula, U. Dalimba and S. M. Kumar, Molecular hybridization approach for phenothiazine incorporated 1,2,3-triazole hybrids as promising antimicrobial agents: design, synthesis, molecular docking and in silico ADME studies, Eur. J. Med. Chem., 2019, 168, 263–282, DOI:10.1016/j.ejmech.2019.02.010.
- M. R. C. de Castro, R. F. Naves, A. Bernardes, C. C. da Silva, C. N. Perez, A. F. Moura, M. O. de Moraes and F. T. Martins, Tandem chalcone-sulfonamide hybridization, cyclization and further Claisen–Schmidt condensation: tuning molecular diversity through reaction time and order and catalyst, Arabian J. Chem., 2020, 13, 1345–1355, DOI:10.1016/j.arabjc.2017.11.005.
- L. M. Moreno, J. Quiroga, R. Abonia, A. Lauria, A. Martorana, H. Insuasty and B. Insuasty, Synthesis, biological evaluation, and in silico studies of novel chalcone- and pyrazoline-based 1,3,5-triazines as potential anticancer agents, RSC Adv., 2020, 10, 34114, 10.1039/D0RA06799G.
- H. E. Hashem, A. E.-G. E. Amr, E. S. Nossier, M. M. Anwar and E. M. Azmy, New Benzimidazole-, 1,2,4-Triazole-, and 1,3,5-Triazine-Based Derivatives as Potential EGFRWT and EGFRT790M Inhibitors: Microwave-Assisted Synthesis, Anticancer Evaluation, and Molecular Docking Study, ACS Omega, 2022, 7(8), 7155–7171, DOI:10.1021/acsomega.1c06836.
- E. T. Warda, I. A. Shehata, M. B. El-Ashmawy and N. S. El-Gohary, New series of isoxazole derivatives targeting EGFR-TK: synthesis, molecular modeling and antitumor evaluation, Bioorg. Med. Chem., 2020, 28(21), 115674, DOI:10.1016/j.bmc.2020.115674.
- N. M. Saleh, M. G. El-Gazzar, H. M. Aly and R. A. Othman, Novel Anticancer Fused Pyrazole Derivatives as EGFR and VEGFR-2 Dual TK Inhibitors, Front. Chem., 2020, 7, 917, DOI:10.3389/fchem.2019.00917.
- R. Dahmani, M. Manachou, S. Belaidi, S. Chtita and S. Boughdiri, Structural characterization and QSAR modeling of 1,2,4-triazole derivatives as α-glucosidase inhibitors, New J. Chem., 2021, 45, 1253–1261, 10.1039/D0NJ05298A.
- A. El-Faham, M. Farooq, Z. Almarhoon, R. Abd Alhameed, M. A. M. Wadaan, B. G. de la Torre and F. Albericio, Di-and trisubstituted s-triazine derivatives: synthesis, characterization, anticancer activity in human breast-cancer cell lines, and developmental toxicity in zebrafish embryos, Bioorg. Chem., 2020, 94, 103397, DOI:10.1016/j.bioorg.2019.103397.
- B. Zhang, Q. Zhang, Z. Xiao, X. Sun, Z. Yang, Q. Gu, Z. Liu, T. Xie, Q. Jin, P. Zheng, S. Xu and W. Zhu, Design, synthesis and biological evaluation of substituted 2-(thiophen-2-yl)-1,3,5-triazine derivatives as potential dual PI3Kα/mTOR inhibitors, Bioorg. Chem., 2020, 95, 103525, DOI:10.1016/j.bioorg.2019.103525.
- I. Karakaya, Synthesis and Characterization of Azobenzene Derived from 8-aminoquinoline in Aqueous Media, J. Turk. Chem. Soc., Sect., 2022, 9(1), 85–114 CrossRef CAS.
- J. Zhu, T. Guo, Z. Wang and Y. Zhao, Triggered azobenzene-based prodrugs and drug delivery systems, J. Controlled Release, 2022, 345, 475–493, DOI:10.1016/j.jconrel.2022.03.041.
- M. Di Martino, L. Sessa, M. Di Matteo, B. Panunzi, S. Piotto and S. Concilio, Azobenzene as Antimicrobial Molecules, Molecules, 2022, 27, 5643, DOI:10.3390/molecules27175643.
- K. Hüll, J. Morstein and D. Trauner, In vivo photopharmacology, Chem. Rev., 2018, 118, 10710–10747 CrossRef PubMed.
- O. Leippe and J. A. Frank, Designing azobenzene-based tools for controlling neurotransmission, Curr. Opin. Struct. Biol., 2019, 57, 23–30 CrossRef PubMed.
- A. E. Berizzi and C. Goudet, Strategies and considerations of G-protein-coupled receptor photopharmacology, Adv. Pharmacol., 2020, 88, 143–172 CAS.
- P. Wu and D. Manna, Optochemical control of protein degradation, ChemBioChem, 2020, 21, 2250–2252 CrossRef CAS PubMed.
- J. Morstein, A. C. Impastato and D. Trauner, Photoswitchable lipids, ChemBioChem, 2021, 22, 73–83 CrossRef CAS PubMed.
- J. Li, X. Wang and X. Liang, Modification of nucleic acids by azobenzene derivatives and their applications in biotechnology and nanotechnology, Chem.–Asian J., 2014, 9, 3344–3358 CrossRef CAS PubMed.
- N. M. Saleh, M. S. A. El-Gaby, K. El-Adl and N. E. A. Abd El-Sattar, Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors, Bioorg. Chem., 2020, 104, 104350, DOI:10.1016/j.bioorg.2020.104350.
- M. L. D. C. Barbosa, L. M. Lima, R. Tesch, C. M. R. SantAnna, F. Totzke, M. H. G. Kubbutat, C. Schachtele, S. A. Laufer and E. J. Barreiro, Novel 2-chloro-4-anilino-quinazoline derivatives as EGFR and VEGFR-2 dual inhibitors, Eur. J. Med. Chem., 2014, 71, 1–14 CrossRef PubMed.
- A. Garofalo, L. Goossens, A. Lemoine, S. Ravez, P. Six, M. Howsam, A. Farce and P. Depreux, Med. Chem. Commun., 2011, 2, 65–72, 10.1039/C0MD00183J.
- S. Wilhelm, et al., Discovery and development of sorafenib: a multikinase inhibitor for treating cancer, Nat. Rev. Drug Discovery, 2006, 5(10), 835–844 CrossRef CAS PubMed.
- A. Pircher, et al., Biomarkers in tumor angiogenesis and anti-angiogenic therapy, Int. J. Mol. Sci., 2011, 12(10), 7077–7099 CrossRef CAS PubMed.
- M. H. El-Shershaby, K. M. El-Gamal, A. H. Bayoumi, K. El-Adl, H. E. A. Ahmed and H. S. Abulkhair, Synthesis, antimicrobial evaluation, DNA gyrase inhibition, and in silico pharmacokinetic studies of novel quinoline derivatives, Arch. Pharm., 2020, e2000277, DOI:10.1002/ardp.202000277.
- A. Turky, A. H. Bayoumi, F. F. Sherbiny, K. El-Adl and H. S. Abulkhair, Unravelling the anticancer potency of 1,2,4-triazole-N-arylamide hybrids through inhibition of STAT3: synthesis and in silico mechanistic studies, Mol. Diversity, 2020, 10131, DOI:10.1007/s11030-020-10131-0.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem, S. I. Eissa, H. Elkady and I. H. Eissa, Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents, Bioorg. Chem., 2020, 104, 104218, DOI:10.1016/j.bioorg.2020.104218.
- K. El-Adl, et al., Design, synthesis, molecular docking and anticancer evaluations of 5-benzylidenethiazolidine-2,4-dione derivatives targeting VEGFR-2 enzyme, Bioorg. Chem., 2020, 102, 104059, DOI:10.1016/j.bioorg.2020.104059.
- A. G. A. El-Helby, H. Sakr, I. H. Eissa, A. A. Al-Karmalawy and K. El-Adl, Benzoxazole/benzothiazole-derived VEGFR-2 inhibitors: design, synthesis, molecular docking, and anticancer evaluations, Arch. Pharm., 2019, 352(12), 1900178 CrossRef CAS PubMed.
- K. El-Adl, et al., Design, synthesis, docking, anticancer and in silico pharmacokinetic studies of novel 5-([4-chloro/2,4-dichloro]benzylidene)thiazolidine-2,4-dione derived VEGFR-2 inhibitors, Arch. Pharm., 2020, e202000279, DOI:10.1002/ardp.202000279.
- A.-G. A. El-Helby, H. Sakr, R. R. A. Ayyad, K. El-Adl, M. M. Ali and F. Khedr, Design, Synthesis, In Vitro Anti-cancer Activity, ADMET Profile and Molecular Docking of Novel Triazolo [3, 4-a] phthalazine Derivatives Targeting VEGFR-2 Enzyme, Anti-Cancer Agents Med. Chem., 2018, 18(8), 1184–1196 CrossRef CAS PubMed.
- A. G. A. El-Helby, R. R. A. Ayyad, H. Sakr, K. El-Adl, M. M. Ali and F. Khedr, Design, Synthesis, Molecular Docking, and Anticancer Activity of Phthalazine Derivatives as VEGFR-2 Inhibitors, Arch. Pharm., 2017, 350(12), 1700240 CrossRef PubMed.
- K. El-Adl, M.-K. Ibrahim, F. Khedr, H. S. Abulkhair and I. H. Eissa, N-substituted-4-phenylphthalazin-1-amine derived VEGFR-2 inhibitors: design, synthesis, molecular docking, and anticancer evaluation studies, Arch. Pharm., 2020, e2000219, DOI:10.1002/ardp.2020002190.
- K. El-Adl, A. A. El-Helby, R. R. Ayyad, H. A. Mahdy, M. M. Khalifa, H. A. Elnagar, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly and I. H. Eissa, Design,synthesis, and anti-proliferative evaluation of new quinazolin-4(3H)-ones as potential VEGFR-2 inhibitors, Bioorg. Med. Chem., 2021, 29, 115872, DOI:10.1016/j.bmc.2020.115872.
- N. S. Hanafy, N. A. A. M. Aziz, S. S. A. El-Hddad, M. A. Abdelgawad, M. M. Ghoneim, A. F. Dawood, S. Mohamady, K. El-Adl and S. Ahmed, Design, synthesis, and docking of novel thiazolidine-2,4-dione multitarget scaffold as new approach for cancer treatment, Arch. Pharm., 2023, e2300137, DOI:10.1002/ardp.202300137.
- N. A. A. M. Aziz, R. F. George, K. El-Adl and W. R. Mahmoud, Design, synthesis, and docking of novel thiazolidine-2,4-dione multitarget scaffold as new approach for cancer treatment, Arch. Pharm., 2022, e2200465, DOI:10.1002/ardp.202200465.
- N. E. A. A. El-Sattar, S. E. S. A. El-Hddad, M. M. Ghobashy, A. A. Zaher and K. El-Adl, Nanogel-mediated drug delivery system for anticancer agent: pH stimuli responsive poly(ethylene glycol/acrylic acid) nanogel prepared by gamma irradiation, Bioorg. Chem., 2022, 127, 105972, DOI:10.1016/j.bioorg.2022.105972.
- H. Elkady, K. El-Adl, H. Sakr, A. S. Abdelraheem, S. I. Eissa and M. A. El-Zahabi, Novel promising benzoxazole/benzothiazole-derived immunomodulatory agents: design, synthesis, anticancer evaluation, and in silico ADMET analysis, Arch. Pharm., 2023, 356(9), e2300097, DOI:10.1002/ardp.202300097.
- D. Adel, K. El-Adl, T. Nasr, T. M. Sakr and W. Zaghary, Pyrazolo[3,4-d]pyrimidine derivatives as EGFRT790M and VEGFR-2 dual TK inhibitors: design, synthesis, molecular docking, ADMET profile and anticancer evaluations, J. Mol. Struct., 2023, 1291, 136047, DOI:10.1016/j.molstruc.2023.136047.
- A. M. Sayed, F. A. Taher, M. R. K. Abdel-Samad, M. S. A. El-Gaby, K. El-Adl and N. M. Saleh, Design, synthesis, molecular docking, in silico ADMET profile and anticancer evaluations of sulfonamide endowed with hydrazone-coupled derivatives as VEGFR-2 inhibitors, Bioorg. Chem., 2021, 108, 104669, DOI:10.1016/j.bioorg.2021.104669.
- N. M. Saleh, A. A. Abdel-Rahman, A. M. Omar, M. M. Khalifa and K. El-Adl, Pyridine-derived VEGFR-2 inhibitors: Rational design, synthesis, anticancer evaluations, in silico ADMET profile, and molecular docking, Arch. Pharm., 2021, 354(8), e2100085, DOI:10.1002/ardp.202100085.
- Q.-Q. Xie, et al., Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2, J. Mol. Graphics Modell., 2009, 27(6), 751–758 CrossRef CAS PubMed.
- K. Lee, et al., Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors, Eur. J. Med. Chem., 2010, 45(11), 5420–5427 CrossRef CAS PubMed.
- V. A. Machado, et al., Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno [3, 2-b] pyridin-7-ylthio) phenyl] ureas: discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors, Bioorg. Med. Chem., 2015, 23(19), 6497–6509 CrossRef CAS PubMed.
- F. Sangande, E. Julianti and D. H. Tjahjono, Ligand-Based Pharmacophore Modeling, Molecular Docking, and Molecular Dynamic Studies of Dual Tyrosine Kinase Inhibitor of EGFR and VEGFR2, Int. J. Mol. Sci., 2020, 21(20), 7779, DOI:10.3390/ijms21207779.
- S. A. Elmetwally, K. F. Saied, I. H. Eissa and E. B. Elkaeed, Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers, Bioorg. Chem., 2019, 88, 102944, DOI:10.1016/j.bioorg.2019.102944.
- B. M. Essa, A. A. Selim, G. H. Sayed and K. E. Anwer, Conventional and microwave-assisted synthesis, anticancer evaluation, 99mTc-coupling and in vivo study of some novel pyrazolone derivatives, Bioorg. Chem., 2022, 125, 105846, DOI:10.1016/j.bioorg.2022.105846.
- N. E. A. Abd El-Sattar, E. H. K. Badawy, E. Z. Elrazaz and N. S. M. Ismail, Discovery of pyrano[2,3-d] pyrimidine-2,4-dione derivatives as novel PARP-1 inhibitors: design, synthesis and antitumor activity, RSC Adv., 2021, 11(8), 4454–4464, 10.1039/D0RA10321G.
- M. K. A. Regal, E. Rafat and N. E. A. Abd El-Sattar, Synthesis, characterization, and dyeing performance of some azo thienopyridine and thienopyrimidine dyes based on wool and nylon, J. Heterocycl. Chem., 2020, 57(3), 1173–1182, DOI:10.1002/jhet.3854.
- K. E. Anwer and G. H. Sayed, Conventional and microwave reactions of 1,3-diaryl-5,4-enaminonitrile-pyrazole derivative with expected antimicrobial and anticancer activities, J. Heterocycl. Chem., 2020, 57(6), 2339–2353, DOI:10.1002/jhet.3946.
- K. E. Anwer, G. H. Sayed and R. M. Ramadan, Synthesis, spectroscopic, DFT calculations, biological activities and molecular docking studies of new isoxazolone, pyrazolone, triazine, triazole and amide derivatives, J. Mol. Struct., 2022, 1256, 132513, DOI:10.1016/j.molstruc.2022.132513.
- F. Khedr, M.-K. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Phthalazine-based VEGFR-2 inhibitors: rationale, design, synthesis, in silico, ADMET profile, docking, and anticancer evaluations, Arch. Pharm., 2021, e2100201, DOI:10.1002/ardp.202100201.
- B. R. Miller III, T. D. J. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, MMPBSA.py: An efficient program for end-state free energy calculations, J. Chem. Theory Comput., 2012, 8(9), 3314–3321 CrossRef PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- S. El-Hddad, M. Sobhy, A. Ayoub and K. El-Adl, In silico molecular docking, dynamics simulation and repurposing of some VEGFR-2 inhibitors based on the SARS-CoV-2-main-protease inhibitor N3, J. Biomol. Struct. Dyn., 2022, 1–15, DOI:10.1080/07391102.2022.2148000.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63, DOI:10.1016/0022-1759(83)90303-4.
- F. M. Freimoser, et al., The MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay is a fast and reliable method for colorimetric determination of fungal cell densities, Appl. Environ. Microbiol., 1999, 65(8), 3727–3729 CrossRef CAS PubMed.
- K. El-Adl, et al., Design, synthesis, molecular docking, and anticancer evaluations of 1-benzylquinazoline-2,4(1H,3H)-dione bearing different moieties as VEGFR-2 inhibitors, Arch. Pharm., 2020, e2000068, DOI:10.1002/ardp.202000068.
- S. M. Abou-Seri, et al., 1-Piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: synthesis and in vitro biological evaluation, Eur. J. Med. Chem., 2016, 107, 165–179 CrossRef CAS PubMed.
- C. Deng, J. Xiong, X. Gu, X. Chen, S. Wu, Z. Wang, D. Wang, J. Tu and J. Xie, Novel recombinant immunotoxin of EGFR specific nanobody fused with cucurmosin, construction and antitumor efficiency in vitro, Oncotarget, 2017, 8(24), 38568–38580 CrossRef PubMed.
- N. A. A. M. Aziz, R. F. George, K. El-Adl and W. R. Mahmoud, Exploration of thiazolidine-2,4-diones as tyrosine kinase inhibitors: design, synthesis, ADMET, docking, and antiproliferative evaluations, Arch. Pharm., 2022, e2200465, DOI:10.1002/ardp.202200465.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures, J. Med. Chem., 2015, 58(9), 4066–4072, DOI:10.1021/acs.jmedchem.5b00104.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25, DOI:10.1016/S0169-409X(96)00423-1.
- A. Beig, R. Agbaria and A. Dahan, Oral Delivery of Lipophilic Drugs: The Tradeoff between Solubility Increase and Permeability Decrease When Using Cyclodextrin-Based Formulation, PLoS One, 2013, 8(10), e68237, DOI:10.1371/journal.pone.0068237.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06614b |
| This journal is © The Royal Society of Chemistry 2023 |