
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe quantitative pyrrole protection of L-phenylalanine/L-phenylalaninol in aqueous media and rationally updating the mechanisms of the Clauson-Kaas reaction through DFT study†
Yuan Qin‡
a,
Pei Cao‡ a,
Virinder S. Parmarbc,
Yonghong Liua,
Chenghai Gao*a and
Kai Liu*a
a,
Virinder S. Parmarbc,
Yonghong Liua,
Chenghai Gao*a and
Kai Liu*a
aGuangxi Key Laboratory of Marine Drugs/Institute of Marine Drugs, Guangxi University of Chinese Medicine, Nanning 530200, P.R. China. E-mail: gaoch@gxtcmu.edu.cn; kailiu@outlook.com
bNanoscience Program, CUNY Graduate Center and Department of Chemistry and Environmental Science, Medgar Evers College, The City University of New York, 1638 Bedford Avenue, New York, NY 11225, USA
cAmity Institute of Click Chemistry and Research Studies, Amity University, Sector 125, Noida 201313, Uttar Pradesh, India
First published on 11th December 2023
Abstract
The classical Paal–Knorr reaction is a prominent tool that can be adopted under biocompatible conditions covering various γ-dicarbonyls for either chemical biology or drug discovery. Meanwhile, the relatively mild conditions for larger molecules within biological systems have not been employed to obtain N-substituted pyrrole derivatives from simpler chiral amino acids/alcohols. The Clauson-Kaas methodology of a standard two-phase acidic mixture buffered with acetate salts was generally required for the time-consuming catalytic condensation of 2,5-dimethoxytetrahydrofuran and fast removal of pyrrolyl products after formation to inhibit their racemization. To achieve a large amount of tethered pyrrole pendants based on L-phenylalanine to construct bioactive ureas as ASK1 and PI3K inhibitors, one quick and highly efficient protocol was realized in an almost neutral and benign aqueous condition. This protocol proceeds within only 15 minutes at 90 °C, achieving nearly quantitative conversion to the final pyrrolyl product via convenient and facile column-free purification. The detailed mechanistic studies by DFT method proposed a new series involving the pathway by the initiation of a zwitterionic species/intermediate for a subsequently much more efficient self-driven pyrrole-formation. This was inconsistent with the traditional kinetic modelling of ring opening to furnish a carbocation, or the utilization of succinaldehyde/dihydroxytetrahydrofuran as a dialdehyde synthetic equivalent. In addition to the relationally neighbouring intramolecular catalytic effect from the amino acid, the crucial “H-bridge” interplay of water, along with the suggestion of a biomimetic route features similar to the N-glycosylation of carbohydrates, probably indicates the totally different reaction courses. The auto-catalysis ability of L-phenylalanine was also extensively investigated by comparisons on the details relating to L-phenylalaninol.
Introduction
Biological systems starting from natural amino acids within tissues and cells exhibit variously complicated mechanisms. The related elaborate modifications generally play significant roles throughout nearly all facets of life as the crucial material basis at different structural levels.1 The artistic transmutation from simple amino acid monomers to the variant folding patterns leads to variously minute functions, encouraging scientists to simulate mother nature's ingenuity.2 As a case in point, the diversified peptides probably form under plausible prebiotic aqueous phosphorylation, while avoiding the assistance of additional condensing agents.3Synthetic chemistry has demonstrated the potential to be adopted in biocompatible conditions. The classical Paal–Knorr reaction has attracted much attention since its first report, which involves the condensation of γ-dicarbonyls to afford pyrroles.1 N-Pyrrolyl alanine derivatives were obtained by the Clauson-Kaas condensing reagent, 2,5-dimethoxytetrahydrofuran (DMTHF). The readily available 1 exhibited remarkable efficiency as an attractive choice for bioconjugations.4a The 1,4-diketone probe 2 displayed prospects for future development as further generation tools, in chemical and biological applications for protein labeling lysine-rich objects, including BSA, KLH, or LC-8.4b
A lysine-specific probe ONAyne 3 represents a singular addition mode for studying LDEs, particularly regarding its pyrrole adduct with endogenous FA.4c,d It can supply opportunities extended to other bioactive γ-dicarbonyls, including natural products like ophiobolin A 4;4c polygodial or other drimane sesquiterpenoids, such as BH3 mimetic 5;4e the rearranged spongian diterpenes;4c the reactive metabolites of furan-containing xenobiotics;4c together with dopamine-originated toxic dicatecholaldehyde derivative 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4f (Fig. 1).
4f (Fig. 1).
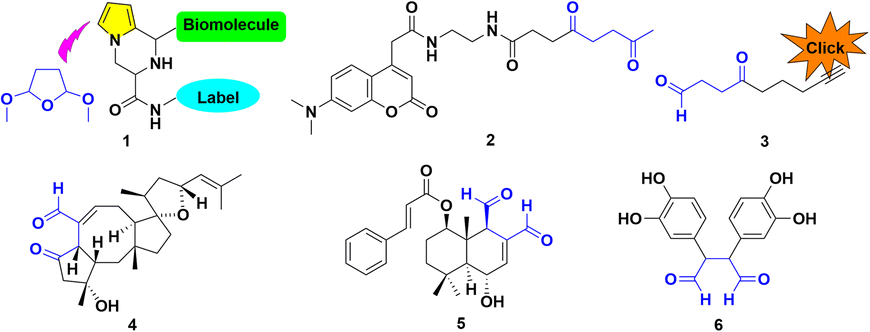 | ||
| Fig. 1 Representative γ-dicarbonyls of the surrogate DMTHF for pyrrolyl functionalization in a biological system. | ||
The typical pyrrole ring generally exists in natural or non-natural drug candidates, synthetic intermediates, and optoelectronic polymers covering agriculture, food, medicinal and pharmaceutical chemistry.5 The pyrrolyl side chains have been extensively adopted in peptidomimetics. The enantiomerically pure moiety 7 was introduced into suitable peptides from γ-ketoaldehyde.6a,b 1,3-Acetonylcyclohexandione was cyclized to deliver 8 through the Paal–Knorr ring closure.6c A series of pyrrole derivatives can facilitate garlic greening.7a The chemical structures of pink-red pigments in macerated onion were tentatively elucidated as a basic polymethine framework in 9.7b The buildings of structurally unique polyheterocyclic cores like 10 are facilitated on the basis of diversity-oriented essence.8a,b A dual nickel- and photoredox-catalyzed modular approach affords the enantioenriched N-benzylic heterocycles 11.9a The reduction of acids generates corresponding alcohols, while a ruthenium complex necessitates the higher H2 pressure and punctilious control of temperature.9b Therefore, one alternative strategy to 12 was also implemented flexibly from commercial amino alcohols directly9c (Fig. 2).
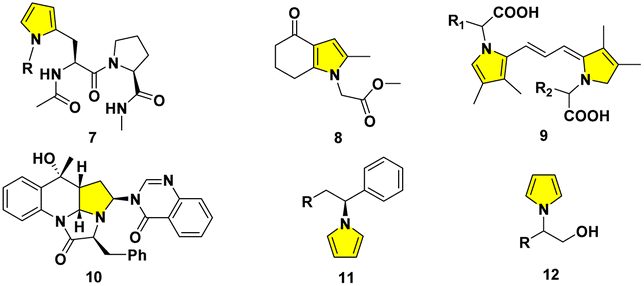 | ||
| Fig. 2 Pyrrolyl derivatives in peptidomimetics, natural products and synthetic heterocycles. | ||
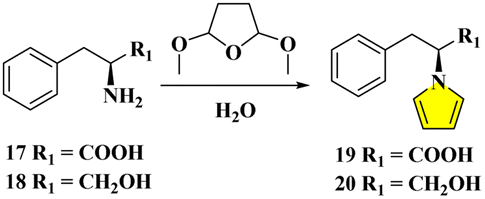
Due to the epimerizable centers, enantiomerically pure primary amines like amino acids/alcohols generally convert into pyrrolyl derivatives in a mixture of aqueous acid and 1,2-dichloroethane. The racemizing effect of the acid is buffered by acetate. While the acid is required for condensation, its continuous existence is deleterious. Thus, the fast product removal from the acidic medium would inhibit the racemization as effective practice.9d,e However, the procedures suffer from certain drawbacks, such as the prolonged heating time with large amounts of acids in volatile/hazardous solvents for the dehydrative cyclization, and the utilization of non-recyclability of catalysts, albeit in lower to moderate yields accompanying tedious purifications9 (Fig. 3). Although the developments of more efficient, simple, economical, environmentally benign protocols containing improvements and optimizations have been continuously reported,10 astonishingly, the standard Clauson-Kaas two-phase system was still adopted to date. Herein, we report our design for the direct pyrrolyl protection of L-phenylalanine 17 by DMTHF only in aqueous solution with nearly quantitative conversion to 19. When succinaldehyde was employed, the N-substituted pyrrole derivative 20 from L-phenylalaninol 18 could be delivered with a similarly perfect transformation. The mechanisms were studied by DFT techniques in detail (Fig. 3).
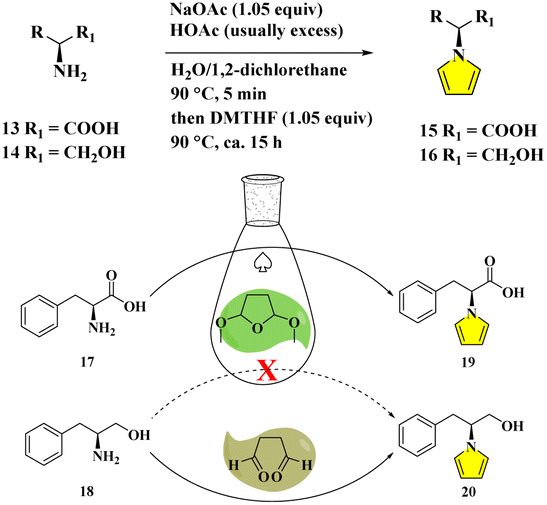 | ||
| Fig. 3 Classical Clauson-Kaas procedures (upper sketch), and the present greener modification with the template of L-phenylalanine/L-phenylalaninol (below diagram). | ||
Results and discussion
Optimization for the facile pyrrole protection of L-phenylalanine
Belonging to the family of hexacyclic quinazolinone alkaloids, bioactive chaetominines have received much attention among chemists and biochemists.11 On the basis of 3D-QSAR, molecular docking, pharmacophore model and molecular dynamics simulations,12 we designed chaetominine mimics in continuous pursuit of ASK1 and PI3K inhibitors.13 Based on the structure virtual screening, the L-phenylalanine derivative 19 was chosen as a promising intermediate through rational dockings, in addition to the comparison with the PI3K pan-inhibitor copanlisib.14 Thus, we initiated the intramolecular cyclization of photogenerated azaxylylenes,8 for which a large amount of tethered pyrrole pendants was needed for post modifications.At the onset of optimization, the conduction of the common buffering system hampers the quick access to 19 with low yields and separation difficulties even after 16 h reflux at 90 °C. The yield of 40% approached those from preceding data reported in the recent references.8b,9a,b By chance, in the absence of acetate buffer, one attempted condensation in water alone with 10% critic acid (CA) under the same refluxing temperature over 2 h resulted in the dramatic increase of yield to 72%, after removing polymeric by-products via column chromatography (Fig. 4).
 | ||
| Fig. 4 Optimization of Clauson-Kaas reactions in water for the template of L-phenylalanine/L-phenylalaninol with DMTHF. | ||
Because of this obvious amelioration in which the final oily product could deposit directly, we considered that similar results might be obtained by simply employing water as the reaction medium. Furthermore, we considered reducing the loading of the catalyst CA from 10% to a point as low as possible to minimize the deleterious influence on the epimerizable centers of the reactants/products, and also to inhibit the side reactions. Thus, an evaluation of the catalytic ability of CA was performed by reducing the catalyst down to 3.75%, where the standard conditions (90 °C/2 h) were firstly implemented with 17 at 0.1 mmol scale using 300 μL water as the sole solvent. The facile purification of the oily product was realized by the aid of centrifuge separation, followed by the conventional extraction between EtOAc/H2O. To our delight, the crude 13C NMR by rapid real-time sample analysis was very pure with a perfect crude yield that was near quantitative transformation, with only a trace amount of EtOAc appearing as residual impurities (see ESI†).
Being encouraged by this optimistic result, we continued to test the CA loading at 0.75% with the variation of heating time for 1 h, 30 min, 15 min (Fig. 5, entries 1, 2, 3), respectively, without the distinct change for crude yields (around 93%) and purities. We then chose 30 min as one fixed parameter, and set the CA loading at 0.3%, 0.1%, 0.05%, 0.01% (Fig. 5, entries 4, 5, 6, 7), respectively. Again, there were no apparent losses for crude yields (around 92%) and purities. Ultimately, the reaction was run at 90 °C over 30 min, without any acidic catalysts (Fig. 5, entry 8). The conversion was still fluent without any evident decrease for crude yields (94%) and purities. As the direct control experiment, one slightly visible inferior yield (77%) was found when the reaction was run at 90 °C over 15 min in pure water only (Fig. 5, entry 9). However, the crude 13C NMR spectra still showed fine signals without the disturbance of impurities after the convenient centrifugal purification. More pleasingly, the reaction can be successfully scaled up to 24.2 mmol scale, affording 19 in 91% yield, with no loss of reactivity or selectivity. Albeit, in order to drive this reaction to full completion, the heating time was extended to 1 h. The auto-catalysis of 17 was extensively investigated by comparisons on the minute particulars of 18 relating to DMTHF and succinaldehyde (See ESI, Fig. S1†).
 | ||
| Fig. 5 Crude 13C NMR spectra for the N-protected products of L-phenylalanine after simplified centrifugal isolation. Reaction temperature was 90 °C for all entries. 0.75% CA, 1 h (entry 1); 0.75% CA, 30 min (entry 2); 0.75% CA, 15 min (entry 3); 0.3% CA, 30 min (entry 4); 0.1% CA, 30 min (entry 5); 0.05% CA, 30 min (entry 6); 0.01% CA, 30 min (entry 7); 0% CA, 30 min (entry 8); 0% CA, 15 min (entry 9). For the crude parallel 1H NMR, see ESI.† | ||
Mechanistic studies by comprehensive computational modelling
The fast and highly efficient procedure, together with the nearly quantitative conversion to the final pyrrolyl product, probably implies totally different reaction pathways compared with the canonical ones involving either the initial ring opening to furnish carbocation, or succinaldehyde/dihydroxytetrahydrofuran as dialdehyde synthetic equivalents. These latter ones were afforded generally by the assistance of variously “stronger” catalysts (Brønsted acids, such as HOAc, Amberlite resin, metals, nano-organo reagents etc.) at higher temperature (around 100 °C) for longer heating times (2 h to 16 h or even more).15a For instance, one equivalent racemic tartaric acid was even adopted in 1,2-dichloroethane/H2O over 1 h at 80 °C with the yield of 61% for DL-Phenylalanine.15bAs to the difficulties being encountered for the usual protonation of DMTHF, we deduced that the primary amine perhaps could nucleophilically attack the DMTHF ring. However, the calculated apparent activation energy via this proposed direct SN2 step is about 48.9 kcal mol−1. It sounds absolutely too high as one reasonable proposition. The new inferred chemical intermediate was a zwitterion like the polar specie of the pyridinium-type salt previously reported, which needs comparably harsh and strict conditions.16 Thus, we focused more attention to the reaction media that was infinitely close to the prebiotic system to some extent. The structural similarity for the acetal/ketal units among DMTHF and natural carbohydrates gives the informative clues for us to choose the N-glycosylation as referring models. The first protecting group-free synthesis of N-glycosyl carbamates has been realized by D-glucose with n-butyl carbamate being catalyzed in 2.4 M HCl/AcOEt.17 The one-pot conversion of sustainable D-ribose with L-amino acid methyl esters produced pyrrole-2-carbaldehydes in reasonable yields under pressurized conditions with the assistance of oxalic acid in DMSO.18 Although malonic acid was still adopted as the necessary catalyst, the amadori compound N-(1-deoxy-D-fructos-1-yl)-L-proline exhibits exemplarily much higher isomerization rates than D-fructose through the C-1 substituent-mediated intramolecular catalytic effect.19 The intrinsic origin of C/N-ribonucleoside through the reaction of 2,4,6-triaminopyrimidine (TAP) with D-ribose in aqueous solution was understood by employing detailed density functional theory (DFT) studies. The water molecule plays a crucial “H-bridge” role by the hydrogen atom relay within the model structure of the nucleobase being beneficial for the glycosidic bonds formation. In general, the pathways proceed over three steps, including nucleophilic addition, dehydration and cyclization.20 At this point, we took full consideration of the components reflecting the neighbouring intramolecular catalytic function from the amino acid, along with the “H-bridge” interplay of water20,21 to the reaction path simulation model.
The nucleophilic assault to the DMTHF ring, accompanied by the release of methanol (partial acetal hydrolysis to Int1), was expedited by the assistance of water combined with the weak H+ liberated from the amino acid, where the energy barrier decreased dramatically to 9.4 kcal mol−1 (Fig. S3 of ESI†). The following H-shift afforded the N,O-acetal Int2, which was activated by the neighbouring intramolecular catalytic –COOH. Thus, this step led to the hydrolytic cleavage for the furan ring in aqueous media to give the imine salt Int3′. The subsequent 1,3 H-shift furnished the more stable enamine Int3 to experience the hydrolysis for the removal of methanol. The intramolecular cyclization of enamine Int4 gave the semi-acetal Int5, and the dehydrative aromatization allowed the final N-pyrrole protection (Fig. 6).
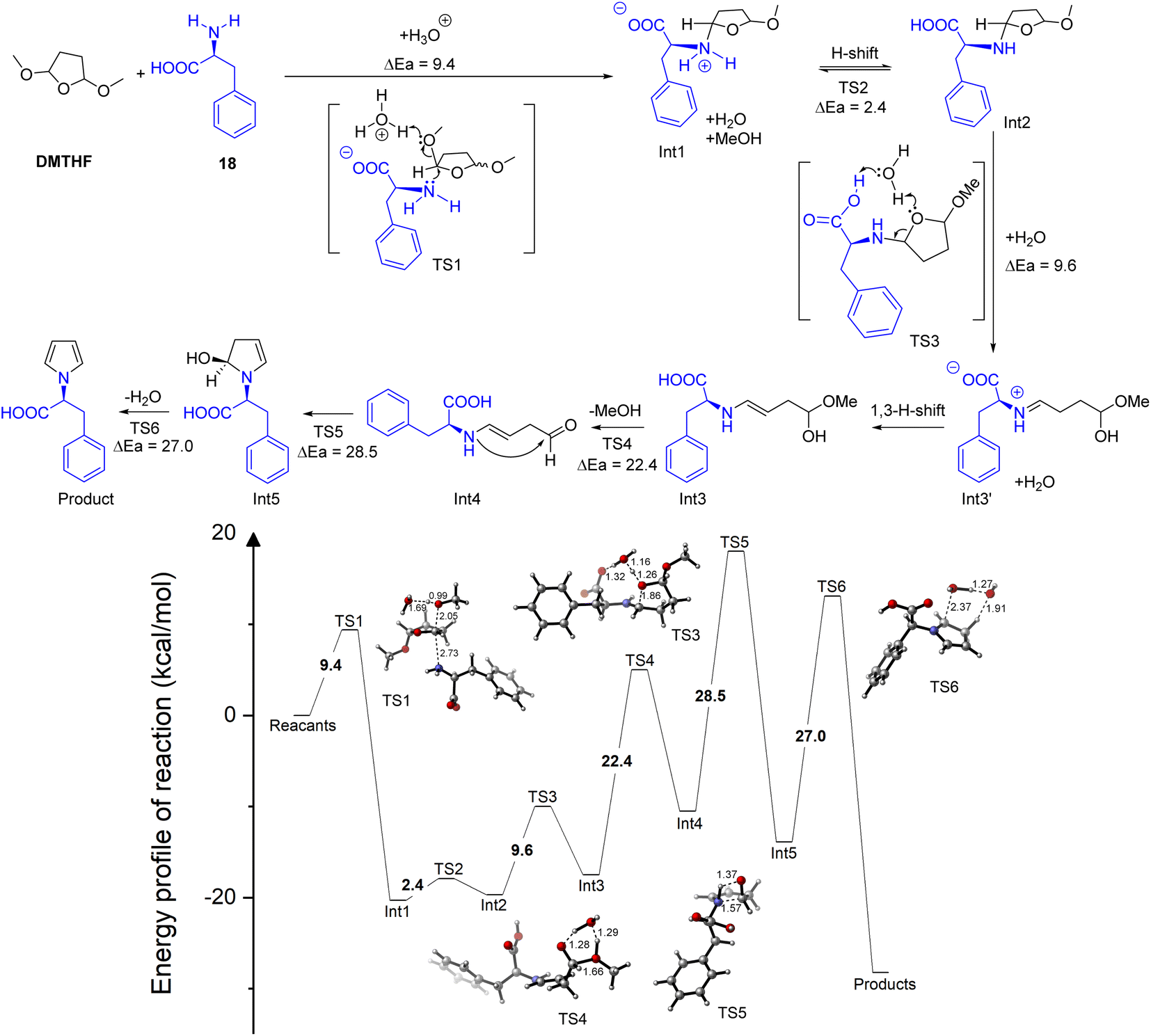 | ||
| Fig. 6 Proposed paths and reaction energy barriers (kcal mol−1) for the generation of the N-pyrrolyl derivative from L-phenylalanine. | ||
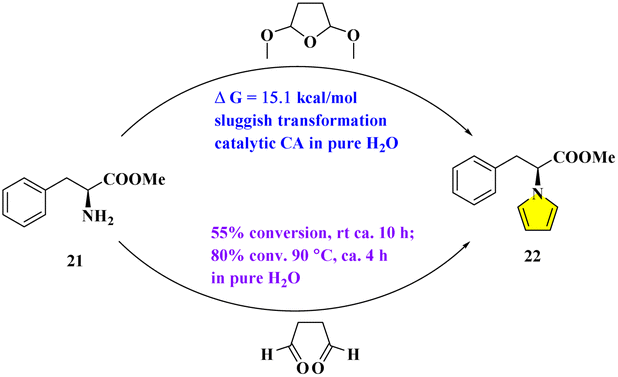
To further explore the reaction mechanisms above, the L-phenylalanine methyl ester (21) (whose acid moiety –COOH is masked by –Me to form ester –COOMe) was chosen. The following reaction was then carried out with DMTHF in pure H2O at 90 °C for 30 min. After the usual workup by extraction of chloroform, the NMR of the crude reactants in CDCl3 displayed only the signals of the starting materials, including the methyl ester 21 and DMTHF. The corresponding energy barrier of this SN2 initiation step was 45.4 kcal mol−1, as predicted by theoretical calculation (shown in Fig. S4 of ESI†). Compared with the one reported in Fig. 5, entry 8, the intramolecular catalytic effect from the amino acid was thus confirmed.
Next, the reaction of 21 with DMTHF and 0.75% CA was run in H2O at 90 °C for 30 min. This reaction is used to test the influence of an external acid by direct comparison to see whether the “H-bridge” role of water (TS1 in Fig. 6) is crucial or not. After the similar workup, the NMR of the crude reactants in CDCl3 demonstrated only the signals of the starting precursors again. It seems that we can exclude the possibility to generate acetal (or dialdehyde) firstly, followed by the pyrrole formation with amine assisted by the outside “H+” (from the acid moiety or water combined with the weak H+ liberated from the amino acid).
Finally, the reaction of 21 with DMTHF and 10% CA was put in D2O directly at 90 °C for 2 h. Although the reaction did not proceed as usual, as monitored by TLC, the total reactants in D2O were sent for non-invasive detection via real-time NMR analysis. To our astonishment, besides the three starting components including the methyl ester, DMTHF, and catalytic CA, the remaining 13C NMR signals match those of the intermediate for protonated 21, which is probably unstable with the corresponding NMR shifts being mimicked and predicted by Mnova software (version 14.0). Moreover, the direct comparisons with spectral data relating to the hydrochloride salt of 21 were also made.22 As suggested by the similar calculation under acid condition, the energy barrier of this step was reduced to 15.1 kcal mol−1 (Fig. S4 of ESI†), indicating that this SN2 reaction could be triggered by acids in theory. This conclusion is well supported by the reactions containing 21 and its structural analogues, which were reported in the traditional two-phase media by the aid of acidic catalysts.23 However, the markedly different microenvironment was hypothesized to be very crucial to facilitate the sequences being likely involved inside.
The reaction between 21 and succinaldehyde was also testified in H2O at room temperature for 10 h. After the general workup, the NMR of the crude reactants in CDCl3 exhibited the signals of the starting methyl ester 21 and pyrrolyl product 22 in the ratio of about 5/6. It is worth mentioning that the transformation to the ultimate targeting pyrrolyl product was not improved dramatically even at 90 °C for 4 h. While pyrrolyl product 22 was enhanced in crude NMR with the proportion of 80%, some uncharacteristic side reactions were also detected by TLC (Fig. 7). By contrast, the similar reaction between L-phenylalaninol and succinaldehyde progressed with much higher efficiency, either at rt (83% yield) or with heating (near 100% yield) (See ESI, Table S1†). At this point, we may deduce with some certainty that the inner hydrogen bonds within the examples (–COOH or –OH) of both L-phenylglycine and L-phenylglycinol could be essentially beneficial, during the procedures of either Clauson-Kaas or Paal–Knorr reactions. On the contrary, the lack of assistance by neighbouring functional groups in the case of L-phenylalanine methyl ester 21 led to the much more sluggish conversion to the pyrrolyl ring.
 | ||
| Fig. 7 Mechanistic studies through the template of the L-phenylalanine methyl ester with DMTHF and succinaldehyde. | ||
The magic of the “H-bridge” interplay was demonstrated in the following histogram for detailed exhibition of the much lower energy barrier found at the initiating stage (Fig. 8). The proposed mechanisms of the Paal–Knorr procedure being similarly inferred for 18 were discussed in ESI.†24
 | ||
| Fig. 8 Comparison of the energy barriers regulated by the interplay of water combined with the weak H+ (distance in Å). | ||
Experimental
Experimental procedures and analytical data of all of the compounds, the copies of their 1H, 13C NMR spectra, along with the calculation method, calculated energies and Cartesian coordinates, are available in the ESI.†Conclusions
In summary, the direct catalytic pyrrole protection of L-phenylalanine/L-phenylalaninol in aqueous media has been developed. This methodology highlights the simplicity of combining DMTHF or aqueous succinaldehyde, amines to deliver pyrrole-tethered chiral motifs under mild conditions. The DFT calculations deduced the self-driven procedures, which matched the experimental outcome. The neighbouring intramolecular catalytic function from the amino acid, the crucial “H-bridge” role of water, reflected the N-glycosylation mimicking characters relational to the biogenic pathway involving structurally similar carbohydrates. The synthetic applicability was testified for the gram-scale synthesis for post-modifications. This protocol provides a valuable alteration to the classical two-phase buffer system, thus securing pyrrole derivatives against the Achilles heels containing asymmetrical centers that are especially prone to epimerization. Efforts to widen the application scopes in this orientation are currently underway in our laboratory.Author contributions
P. Cao, Y. Liu, C. Gao and K. Liu conceived the project; Y. Qin and P. Cao performed the experiments and analyzed the data; K. Liu and V. S. Parmar conducted the DFT calculations and held discussions. Y. Qin, P. Cao, V. S. Parmar and K. Liu prepared and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank the following financial supports provided by the Natural Science Foundation of Guangxi (Grant No. including 2020GXNSFAA297112, 2020GXNSFGA297002, 2020GXNSFFA297005), the Specific Research Project of Guangxi for Research Bases and Talents (AD21075003), the Special Fund for Hundred Talents Program for Universities in Guangxi (Gui2019-71), the Special Fund for Bagui Scholars of Guangxi (05019055) and Doctoral Research Foundation of GXUCM (2019BS020), the “GuiPai Traditional Chinese Medicine Inheritance and Innovation Team” Project (2022A007), and the Development Program of High-level Talent Team under Qihuang Project of GXUCM (No. 2021004).Notes and references
- A. Kornienko and J. J. La Clair, Nat. Prod. Rep., 2017, 34, 1051–1060 RSC.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- (a) C. Gibard, S. Bhowmik, M. Karki, E.-K. Kim and R. Krishnamurthy, Nat. Chem., 2018, 10, 212–217 CrossRef CAS; (b) A. Osumah and R. Krishnamurthy, ChemBioChem, 2021, 22, 3001–3009 CrossRef CAS.
- (a) S. Pomplun, M. Y. H. Mohamed, T. Oelschlaegel, C. Wellner and F. Bergmann, Angew. Chem., Int. Ed., 2019, 58, 3542–3547 CrossRef CAS; (b) R. Dasari, J. J. La Clair and A. Kornienko, ChemBioChem, 2017, 18, 1792–1796 CrossRef CAS; (c) M.-R. Wang, J.-Y. He, J.-X. He, K.-K. Liu and J. Yang, Chem. Sci., 2021, 12, 14557–14563 RSC; (d) J.-X. He, Z.-C. Fei, L. Fu, C.-P. Tian, F.-C. He, H. Chi and J. Yang, Nat. Chem. Biol., 2022, 18, 904–912 CrossRef CAS PubMed; (e) F. Daressy, F. Malard, L. Seguy, V. Guérineau, C. Apel, V. Dumontet, A. Robert, A.-C. Groo, M. Litaudon, J. Bignon, S. Desrat, A. Malzert-Fréon, J. Wiels, E. Lescop and F. Roussi, ChemMedChem, 2021, 16, 1789–1798 CrossRef; (f) J. W. Werner-Allen, J. F. DuMond, R. L. Levine and A. Bax, Angew. Chem., Int. Ed., 2016, 55, 7374–7378 CrossRef CAS.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, RSC Adv., 2021, 11, 13585–13601 RSC.
- (a) A. A. Dörr and W. D. Lubell, J. Org. Chem., 2012, 77, 6414–6422 CrossRef; (b) A. Douchez, A. Geranurimi and W. D. Lubell, Acc. Chem. Res., 2018, 51, 2574–2588 CrossRef CAS PubMed; (c) R.-K. Li, Q.-L. Yang, Y. Liu, D.-W. Li, N.-Y. Huang and M.-G. Liu, Chin. Chem. Lett., 2016, 27, 345–348 CrossRef CAS.
- (a) D. Wang, H. Nanding, N. Han, F. Chen and G. Zhao, J. Agric. Food Chem., 2008, 56, 1495–1500 CrossRef CAS; (b) E. J. Lee, Y. H. Rezenom, D. H. Russell, B. S. Patil and K. S. Yoo, Food Chem., 2012, 131, 852–861 CrossRef CAS.
- (a) N. N. B. Kumar, O. A. Mukhina and A. G. Kutateladze, J. Am. Chem. Soc., 2013, 135, 9608–9611 CrossRef CAS; (b) O. A. Mukhina, D. M. Kuznetsov, T. M. Cowger and A. G. Kutateladze, Angew. Chem., Int. Ed., 2015, 54, 11516–11520 CrossRef CAS.
- (a) C. Pezzetta, D. Bonifazi and R. W. M. Davidson, Org. Lett., 2019, 21, 8957–8961 CrossRef CAS; (b) A. Saito, S. Yoshioka, M. Naruto and S. Saito, Adv. Synth. Catal., 2020, 362, 424–429 CrossRef CAS; (c) M. Leroux, W.-Y. Huang, Y. Lemke, T. J. Koller, K. Karaghiosoff and P. Knochel, Chem.–Eur. J., 2020, 26, 8951–8957 CrossRef CAS PubMed; (d) C. W. Jefford, F. de Villedon de Naide and K. Sienkiewicz, Tetrahedron: Asymmetry, 1996, 7, 1069–1076 CrossRef CAS; (e) B. S. Gourlay, P. P. Molesworth, J. H. Ryan and J. A. Smith, Tetrahedron Lett., 2006, 47, 799–801 CrossRef CAS.
- (a) H. Cho, R. Madden, B. Nisanci and B. Török, Green Chem., 2015, 17, 1088–1099 RSC; (b) A. Balakrishna, A. Aguiar, P. J. M. Sobral, M. Y. Wani, J. Almeida e Silva and A. J. F. N. Sobral, Catal. Rev. - Sci. Eng., 2019, 61, 84–110 CrossRef CAS; (c) S. Alvi and R. Ali, Org. Biomol. Chem., 2021, 19, 9732–9745 RSC; (d) G. Xie, A. Lazarev and B. Török, Green Chem., 2023, 25, 1582–1587 RSC.
- (a) Q.-L. Peng, S.-P. Luo, X.-E. Xia, L.-X. Liu and P.-Q. Huang, Chem. Commun., 2014, 50, 1986–1988 RSC; (b) Z.-Y. Mao, H. Geng, T.-T. Zhang, Y.-P. Ruan, J.-L. Ye and P.-Q. Huang, Org. Chem. Front., 2016, 3, 24–37 RSC; (c) H. Geng and P.-Q. Huang, Chem. Rec., 2019, 19, 523–533 CrossRef CAS.
- (a) X. X. Peng, K. R. Feng and Y. J. Ren, RSC Adv., 2017, 7, 56344–56358 RSC; (b) D. Halder, S. Das, R. Aiswarya and R. S. Jeyaprakash, RSC Adv., 2022, 12, 21452–21467 RSC; (c) M. Castelli, S. A. Serapian, F. Marchetti, A. Triveri, V. Pirota, L. Torielli, S. Collina, F. Doria, M. Freccero and G. Colombo, RSC Med. Chem., 2021, 12, 1491–1502 RSC.
- (a) Y. Xie, P. Cao, Y. Qin, X. Wu, B. Huang, K. Liu and Y. Liu, Comput. Biol. Chem., 2022, 99, 107712 CrossRef CAS; (b) K. Liu, Y. Xie, Y. Qin, V. S. Parmar, Y. Liu and P. Cao, Org. Chem. Front., 2023, 10, 3182–3192 RSC.
- B. Vanhaesebroeck, M. W. D. Perry, J. R. Brown, F. André and K. Okkenhaug, Nat. Rev. Drug Discovery, 2021, 20, 741–769 CrossRef CAS.
- (a) D. K. Singh and R. Kumar, Beilstein J. Org. Chem., 2023, 19, 928–955 CrossRef CAS; (b) E. Tokumaru, A. Tengeiji, T. Nakahara and I. Shiina, Chem. Lett., 2015, 44, 1768–1770 CrossRef CAS.
- (a) H. Fujioka, T. Okitsu, Y. Sawama, N. Murata, R. Li and Y. Kita, J. Am. Chem. Soc., 2006, 128, 5930–5938 CrossRef CAS PubMed; (b) H. Fujioka, T. Okitsu, T. Ohnaka, R. Li, O. Kubo, K. Okamoto, Y. Sawama and Y. Kita, J. Org. Chem., 2007, 72, 7898–7902 CrossRef CAS.
- Y. Ichikawa, D. Kaneno, N. Saeki, T. Minami, T. Masuda, K. Yoshida, T. Kondo and R. Ochi, Carbohydr. Res., 2021, 505, 108280 CrossRef CAS PubMed.
- S. Cho, L. Gu, I. J. IN, B. Wu, T. Lee, H. Kim and S. Koo, RSC Adv., 2021, 11, 31511–31525 RSC.
- M. Kaufmann, P. M. Meissner, D. Pelke, C. Mügge and L. W. Kroh, Carbohydr. Res., 2016, 428, 87–99 CrossRef CAS.
- W. Wang, F. Huang, C. Sun, J. Liu, X. Sheng and D. Chen, Phys. Chem. Chem. Phys., 2017, 19, 10413–10426 RSC.
- L. Legnani, A. Darù, A. X. Jones and D. G. Blackmond, J. Am. Chem. Soc., 2021, 143, 7852–7858 CrossRef CAS PubMed.
- (a) J. Li and Y. Sha, Molecules, 2008, 13, 1111–1119 CrossRef CAS PubMed; (b) A. Hartono, U. E. Aronu and H. F. Svendsen, Energy Procedia, 2011, 4, 209–215 CrossRef CAS.
- (a) J. P. Lellouche, G. Senthil, A. Joseph, L. Buzhansky, I. Bruce, E. R. Bauminger and J. Schlesinger, J. Am. Chem. Soc., 2005, 127, 11998–12006 CrossRef CAS PubMed; (b) A. S. Demir, N. T. Subasi and E. Sahin, Tetrahedron: Asymmetry, 2006, 17, 2625–2631 CrossRef CAS.
- S. Abbat, D. Dhaked, M. Arfeen and P. V. Bharatam, RSC Adv., 2015, 5, 88353–88366 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra06595b |
| ‡ Y. Qin and P. Cao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |