
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosorption of uranyl ions from aqueous solutions by soluble renewable polysaccharides
Oshrat Levy-Ontman *a,
Ofra Paz-Tal*b,
Yaron Alfia and
Adi Wolfsona
*a,
Ofra Paz-Tal*b,
Yaron Alfia and
Adi Wolfsona
aDepartment of Chemical Engineering, Shamoon College of Engineering, Basel/Bialik Sts., Beer-Sheva 8410001, Israel. E-mail: oshrale@sce.ac.il; Tel: +972-8-6475636
bNuclear Research Center, Negev, Beer-Sheva 84190, Israel. E-mail: ofrapt@gmail.com; Tel: +972-8-6567514
First published on 8th December 2023
Abstract
Polysaccharides derived from natural sources have been offered as environment friendly sorbents for the adsorption of heavy metals. We present a simple technique to remove uranyl ions from aqueous solutions by using representative polysaccharides. The adsorption efficiency of UO22+ decreased in the following order: xanthan gum > kappa > iota/guar gum, for instance, the efficiencies after sorption of 30 min with 500 mg per L uranyl acetate and 0.03 g of the corresponding polysaccharide were: 89.7%, 85.2%, 79.1% and 77.1%. Lowering the acidity in the system decreased the sorption efficiency with all the polysaccharides, and reducing the ratio between the amount of uranyl ions and the amount of polysaccharide increased the sorption efficiencies, e.g., using 500 mg per L uranyl acetate and 0.05 g of the corresponding polysaccharide (xanthan gum, kappa, iota, guar gum) yielded after 30 min sorption efficiencies of 94.3%, 91.5%, 89.0% and 87.7%, respectively. FTIR, SEM-EDS and TGA analyses verified the presence of uranium in the polysaccharides and showed that the uranyl ions were interacting with the different functional groups. Moreover, the addition of uranyl ions to the polysaccharides caused a sharp decrease in viscometry measurements. In addition, the measurements showed that the addition of uranyl lowered both modules, G′ and G′′, and made the solution more liquid.
1. Introduction
Uranium, the heaviest naturally occurring element on Earth, is the primary fuel for nuclear reactors.1,2 It is also used for other applications, from the space industry to medicine. Uranium ore extraction from the ground and subsequent use as a nuclear fuel leads to the production of large amounts of wastewater that contains uranium ions, mainly as a hexavalent cation (U(VI)).3,4As a radioactive substance, a large amount of uranium, above natural levels, can cause different health effects,5 such as liver and kidney disease.6 Moreover, long exposure to uranium radionuclides may also cause cancer.7 The World Health Organization has therefore listed U(VI) as a carcinogen and determined that its concentration in water should not exceed 50 mg L−1. Nonetheless, the US Environmental Protection Agency has recommended that 20 mg per L 238U be set as the standard for drinking water. In addition, wastewater that contains uranium is also hazardous for ecosystems.8,9 A variety of methods exist for the removal from wastewater of uranium or uranyl ions (UO22+), the latter formed by the hydrolysis of uranium.3,4,10,11 The most studied and applicable methods are extraction,12–14 flotation,15,16 precipitation,17,18 membrane filtration,19,20 electrodialysis21 and adsorption.22–27 Among these techniques, adsorption based on different organic and inorganic adsorbents was found to be the simplest, most effective and most versatile method.
Biosorbents, living or dead biomass (e.g., algae, yeast and bacteria) and their derived products, such as polysaccharide-based materials, have also been shown to adsorb different contaminates from wastewater, among them radioactive ions,28 including uranium.26,29 Insofar as they are renewable, biodegradable and usually hydrophilic polymers, polysaccharides produced from biological sources are considered safe for use. As such, they have numerous applications,30 including in the food,31 pharmaceuticals32 and cosmetics33 industries. This wide applicability is due to the variability in the characteristics of each polysaccharide, which depends on its functional groups and its structure. Among its other qualities, these properties also render them good adsorbents for heavy metal ions.34–36 Moreover, they are readily available and usually more cost-effective than synthetic adsorbents.
Different polysaccharide-based systems have been used to remove U(VI) from aqueous solutions that resemble wastewater. For example, Marqués et al. reported that exopolysaccharide from Pseudomonas sp. was able to remove uranium from an aqueous solution with a maximum uptake of 96 μg U per mg polymer.37 In addition, Kazy et al. used extracellular polysaccharide (EPC) produced by a Pseudomonas aeruginosa strain to remove uranium from an aqueous solution.38 The maximum sorption observed was 985 mg U per g polymer at pH 5.0 due to the strong binding of uranium with the carboxylic groups of the uronic acids in the polymer, while at lower pH values, amino and hydroxyl groups were responsible for the bulk of metal binding. Sakaguchi et al. studied the adsorption of uranium from seawater by titanium(IV)–cellulose–xanthate or titanium(IV)–starch–xanthate, which yielded an uptake of about 77% when 1 g of the adsorbent was suspended in 5 L of natural seawater with constant stirring at 30 °C for three days.39 In later work, alginate beads that were prepared in a CaCl2 solution were successfully used for uranium recovery from aqueous solutions. The adsorption efficiency and distribution constant (Kd) for uranium ions under optimized experimental conditions were 91 ± 1% and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 043 ± 834 mL g−1, respectively.40 In other studies, a chitosan–glucan complex41 and different cross-linked chitosan systems,41–43 which were insoluble in wastewater, and a magnetic biosorbent composed of nanoparticles of magnetite covered with chitosan,44 were also successfully tested. Furthermore, water insoluble polysaccharides like chitin and chitosan were also shown to be efficient biosorbents.45,46
043 ± 834 mL g−1, respectively.40 In other studies, a chitosan–glucan complex41 and different cross-linked chitosan systems,41–43 which were insoluble in wastewater, and a magnetic biosorbent composed of nanoparticles of magnetite covered with chitosan,44 were also successfully tested. Furthermore, water insoluble polysaccharides like chitin and chitosan were also shown to be efficient biosorbents.45,46
In this study, we report on the sorption of UO22+ from aqueous solutions using natural and water-soluble polysaccharides as adsorbents. The separation process is very fast and straightforward, and the adsorption capacities, which were detected after precipitation and filtration of the different polysaccharides with the uranyl ions, were effective and dependent on polysaccharide type.
2. Experimental
2.1 Materials
All of the polysaccharides used in this study (guar gum (G), xanthan gum (X), iota (I), kappa (Κ)), uranyl acetate dihydrate (UO2(CH3COO)2·2H2O), and other chemicals (analytical grades) were purchased from Sigma-Aldrich.2.2 Preparation of polysaccharide solutions
One g of polysaccharide (G/X/I/Κ) was dissolved in 100 mL double-distilled water (DDW) and mixed for 1 h, yielding 1% w/v aqueous solution of polysaccharide. When I and Κ were used, the mixture was heated to 50 °C and stirred for another 1 h.2.3 Sorption experiments
In a typical procedure, 1–5 mL of 1% w/v aqueous solution of polysaccharides were added to 5–9 mL of an aqueous solution of U(VI) (500–900 mg L−1), which were prepared from a uranyl acetate stock solution of 1000 mg L−1 via proper dilution, and then mixed for 30 min at room temperature. (In all of the experiments, the final volume was 10 mL). Then, 4 × 10 mL of cold ethanol was added to the aqueous solution, after which the solution was mixed and the solid was removed by centrifugation and filtration. It worth mentioned that the amount of ethanol that used to allow full and ease separation of the polysaccharide and the UO22+ ion within it, should be optimized to make the system more sustainable. Finally, the ethanol was evaporated from the solution, and the concentration of U(VI) that was left in the solution was detected using ICP-OES (SPECTRO, model ARCOS, error limit ± 10%) and UV-Vis analysis (Agilent, model 8453, wavelength accuracy < ±0.5 nm) using xylenol orange as indicator.47Each sorption experiment was performed five times, and each displayed yield is the mean of five experiments. In addition, two control sets, one containing only the uranyl solution at an appropriate concentration and the other with the polysaccharides and without the uranyl solution, were also run for comparison.
The sorption efficiency was calculated as follows:
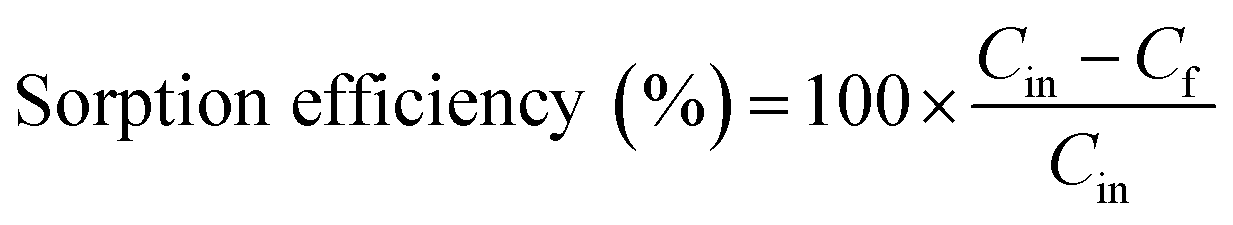
2.4 Characterization methods
3. Results and discussion
The nature of the polysaccharide, i.e., its structure and the functional groups on its backbone, and the characteristics of the ion to be removed, i.e., its size and charge, are used to detect the ion sorption efficiency. Recently, we proposed a novel method for the recovery of europium ions (Eu3+) from aqueous solutions by adding an aqueous solution of readily available, commercial polysaccharides to a europium solution.48 Eu3+, which could associate with the sulfate, carboxylic and hydroxyl groups on the backbone of the polysaccharide, was preferably adsorbed to the more acidic groups. Thus, it was suggested that the same method can also be employed with renewable polysaccharides to recover UO22+ ions from aqueous solutions.The sorption of UO22+ was investigated by using four representative polysaccharides with different functional groups: G with only hydroxyl groups, X with hydroxyl and carboxylic groups, and carrageenan types I and Κ with hydroxyl and ester sulfate groups. As UO22+ sorption efficiency reached equilibrium after 30 min, the investigation began with a study of the effects of both the UO22+ concentration and the amount of polysaccharide on the sorption efficiency after 30 min at 298 K and pH = 4.5. The results in Table 1 show that at polysaccharide amounts of 0.03 g and above, X exhibited the highest sorption efficiency, followed by Κ with slightly lower yields, while the use of I and G resulted in additional small decreases in the sorption efficiency under all the conditions. As previously noted, the difference in sorption efficiency between the four polysaccharides might be attributed to the different functional groups on the polymers. The carboxylic groups on X seem to be slightly more effective sorption agents of UO22+ compared to the ester sulfate groups and the hydroxyl on the two carrageenans and to the hydroxyl groups in G. In addition, surprisingly, Κ, which has one sulfate ester group, exhibited higher sorption efficiencies than I, which has two sulfate groups. This finding may be explained by the different molecular structure of the two carrageenans – though both polysaccharides form three-fold, right-handed double helice conformations composed of parallel chains, I has a longer helix pitch than Κ, and the chain positions differ significantly. In addition, I forms weaker and more elastic gels than Κ,49,50 which may affect the functional group availability.51 Notably, the higher sorption efficiencies attributed to Κ compared to that of I differ from our former findings, which demonstrated that I is a better Eu3+ sorbent than Κ.48,51 The observed differences in sorption behaviour are the manifestation of the dependence of that behaviour on both the nature of the polysaccharide and the characteristics of the metal ions.
| Polysaccharide | Sorption efficiencies (%) ± 3% | |||
|---|---|---|---|---|
| U(VI) (mg L−1):PS (g) |
||||
| 900:0.01 |
500:0.05 |
700:0.03 |
500:0.03 |
|
| Guar gum (G) | 57.9 | 87.7 | 77.1 | 82.2 |
| Iota (I) | 58.9 | 89.0 | 79.1 | 80.7 |
| Kappa (Κ) | 66.7 | 91.5 | 85.2 | 88.9 |
| Xanthan gum (X) | 65.4 | 94.3 | 89.7 | 95.0 |
As expected, all four polysaccharides exhibited higher sorption efficiencies when the metal concentration in the solution was lower or when the polysaccharide amount was higher (Table 1). This observation implies that reducing the UO22+/polysaccharide ratio increased the sorption efficiency, as the number of adsorbing functional groups on the polysaccharide relative to the number of adsorbed ions increased. Above a concentration of 700 mg L−1, however, it seems that there are not enough available binding sites in the polysaccharides, thus causing the yields to decline markedly. As such, in solutions that contained a high uranyl concentration (900 mg L−1) and a low amount of polysaccharides (0.01 g), the sorption efficiencies that were detected for G, I, Κ, and X were 57.9%, 58.9%, 66.7%, and 65.4%, respectively (Table 1).
The adsorption kinetics of the two polysaccharides that exhibited the highest sorption efficiencies, X and K, were determined by using a pseudo-first order and a pseudo-second order kinetic models. The expressions of the two models after linearization are given in eqn (1) and (2), respectively, where qt and qe are the uranyl adsorption capacity (mg g−1) at any time t (min) and at equilibrium, and, k1 (min−1) is pseudo-first order rate constant and k2 (g mg−1 min−1) is pseudo-second order rate constant.
| ln(qe − qt) = ln(qe) − k1t | (1) |
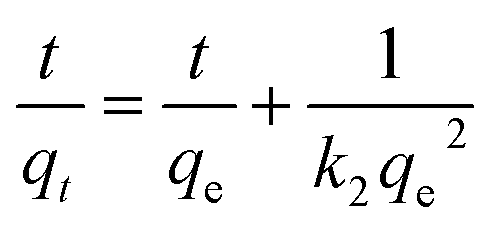 | (2) |
The results of modelling are presented in Fig. 1. It can be seen that the sorption with both polysaccharides fits to pseudo-second order model (with high R2 values) indicating that chemisorption is the rate-limiting mechanism of the process, and that uranyl was chemically adsorbed to the polysaccharides.
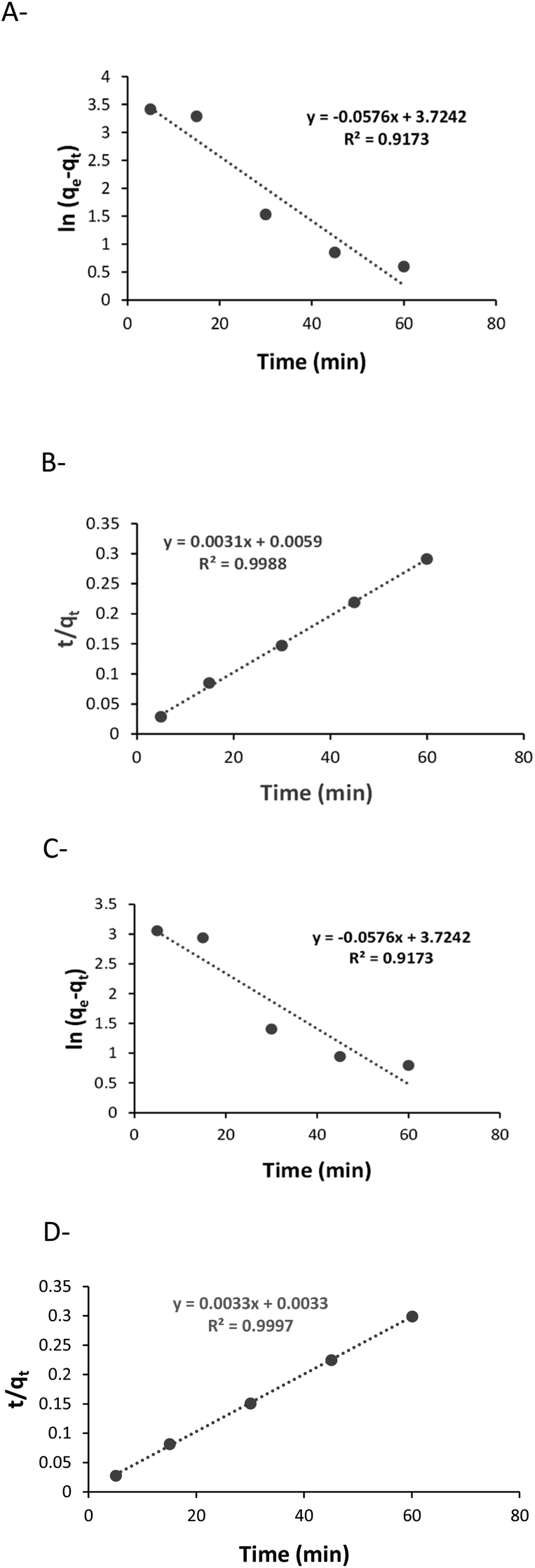 | ||
| Fig. 1 Kinetics of uranyl sorption: (A) pseudo-first order with X, (B) pseudo-second order with X, (C) pseudo-first order with K, (D) pseudo-second order with K. | ||
3.1 FTIR analysis
ATR-FTIR spectroscopy was used to gain knowledge about the interaction between the UO22+ and the different functional groups on the polysaccharides, before and after the sorption, the latter of which is designated as U–PS (polysaccharide), e.g., the I after adsorption of UO22+ ions was labelled U–I.ATR-FTIR spectra of pristine G vs. U–G are shown in Fig. 2. As expected, the spectrum of pristine G contains the typical bands in accordance with the G structure: a broad band assigned to the –OH stretching at 3333 cm−1; a peak at 2920 cm−1 that confirmed –CH2– asymmetric stretching vibrations; and a peak at 1403 cm−1 that confirmed the C–H stretching vibrations.52 In addition, the ring stretching of mannose and bending vibration of the OH group and associated water molecules were observed at 1640 cm−1 and the –CH2– deformation at 1384 cm−1. Finally, the peaks in the spectrum between 863 and 1151 cm−1 indicated the highly coupled stretching vibrations of C–O–C, C–C–O, C–OH, and (1–4) and (1–6) linkages of galactose and mannose units.53,54 Yet when UO22+ ions were adsorbed to the G (U–G, in Fig. 2), the broad peak at 1640 cm−1 shifted to 1650 cm−1 and became sharper, and the peak at 1151 cm−1 shifted to 1145 cm−1. These changes indicate that the hydroxyl groups of C–OH and UO22+ interacted with each other. Moreover, a new U–G band was observed at 907 cm−1, which is attributed to O–U–O stretching, also indicating that the metal ions were adsorbed via the hydroxyl groups on the polysaccharide.
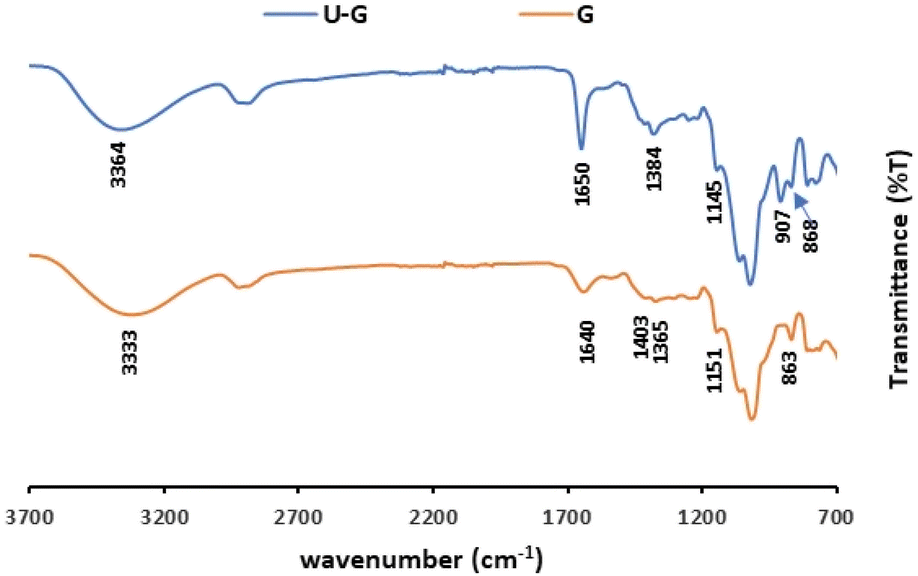 | ||
| Fig. 2 ATR-FTIR spectra of pristine G and U–G (sorption conditions: 700 mg per L UO22+, 3 mL of 1% w/v G). | ||
The FTIR spectra of pristine I and Κ and their corresponding analogues, U–I and U–K, are shown in Fig. 3. As expected, in their pristine states, the two polysaccharides present the same characteristic bands that are indicative of the basic chemical bonding in the carrageenans, specifically: a peak at 3200–3400 cm−1 due to the assigned hydrogen O–H stretching vibration bonds; C–O–C of 3,6-anhydro-D-galactose at 1068 cm−1 and 929/923 cm−1, respectively; O–SO3 stretching vibration at D-galactose-4-sulfate for I and Κ at 1373/1424 cm−1 and 844/847 cm−1, respectively; and at 804 cm−1 for D-galactose-2-sulfate.55,56 However, following UO22+ sorption, the H–O–H deformation bands of I and Κ at 1634 cm−1 and 1627 cm−1, respectively, shifted to 1652 cm−1 and 1653 cm−1, respectively.57 Also note that the bands at 3200–3500 cm−1 that were observed following the addition of UO22+ and that correspond to the O–H stretching vibrations were less intense than those observed in both of the pristine polysaccharides. The reduction in the band intensities could be attributed to reduced hydrogen bonds. Indeed, the interaction of the uranyl ions with the oxygen of the hydroxyl groups can lead to the lower availability of the OH groups that could otherwise self-assemble the polymer chains. In the 1100–1300 cm−1 range, the characteristic S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band of the ester sulfate (OSO symmetric vibration), which appears in pristine I at 1204 cm−1 and in pristine Κ at 1220 cm−1, shifted to a higher wavenumber of 1210 cm−1 in U–I and to double peaks at 1226 cm−1 and 1236 cm−1 in U–K.58 For O–SO3 stretching vibrations at D-galactose-4-sulfate, the broad peak of pristine I at 918 cm−1 split into two sharp peaks at 929 and 898 cm−1 in U–I. These findings indicate that the UO22+ is also interacting with the ester sulfate groups. Moreover, the band at 987 cm−1, which corresponds to pristine K, shifted to a lower frequency (968 cm−1) after sorption, and the peak at 867 cm−1, which corresponds to galactose-4-sulfate, was not observed. Therefore, it can be suggested that the ester sulfate groups were involved in the complexation of the uranyl ions to the Κ matrix.
O band of the ester sulfate (OSO symmetric vibration), which appears in pristine I at 1204 cm−1 and in pristine Κ at 1220 cm−1, shifted to a higher wavenumber of 1210 cm−1 in U–I and to double peaks at 1226 cm−1 and 1236 cm−1 in U–K.58 For O–SO3 stretching vibrations at D-galactose-4-sulfate, the broad peak of pristine I at 918 cm−1 split into two sharp peaks at 929 and 898 cm−1 in U–I. These findings indicate that the UO22+ is also interacting with the ester sulfate groups. Moreover, the band at 987 cm−1, which corresponds to pristine K, shifted to a lower frequency (968 cm−1) after sorption, and the peak at 867 cm−1, which corresponds to galactose-4-sulfate, was not observed. Therefore, it can be suggested that the ester sulfate groups were involved in the complexation of the uranyl ions to the Κ matrix.
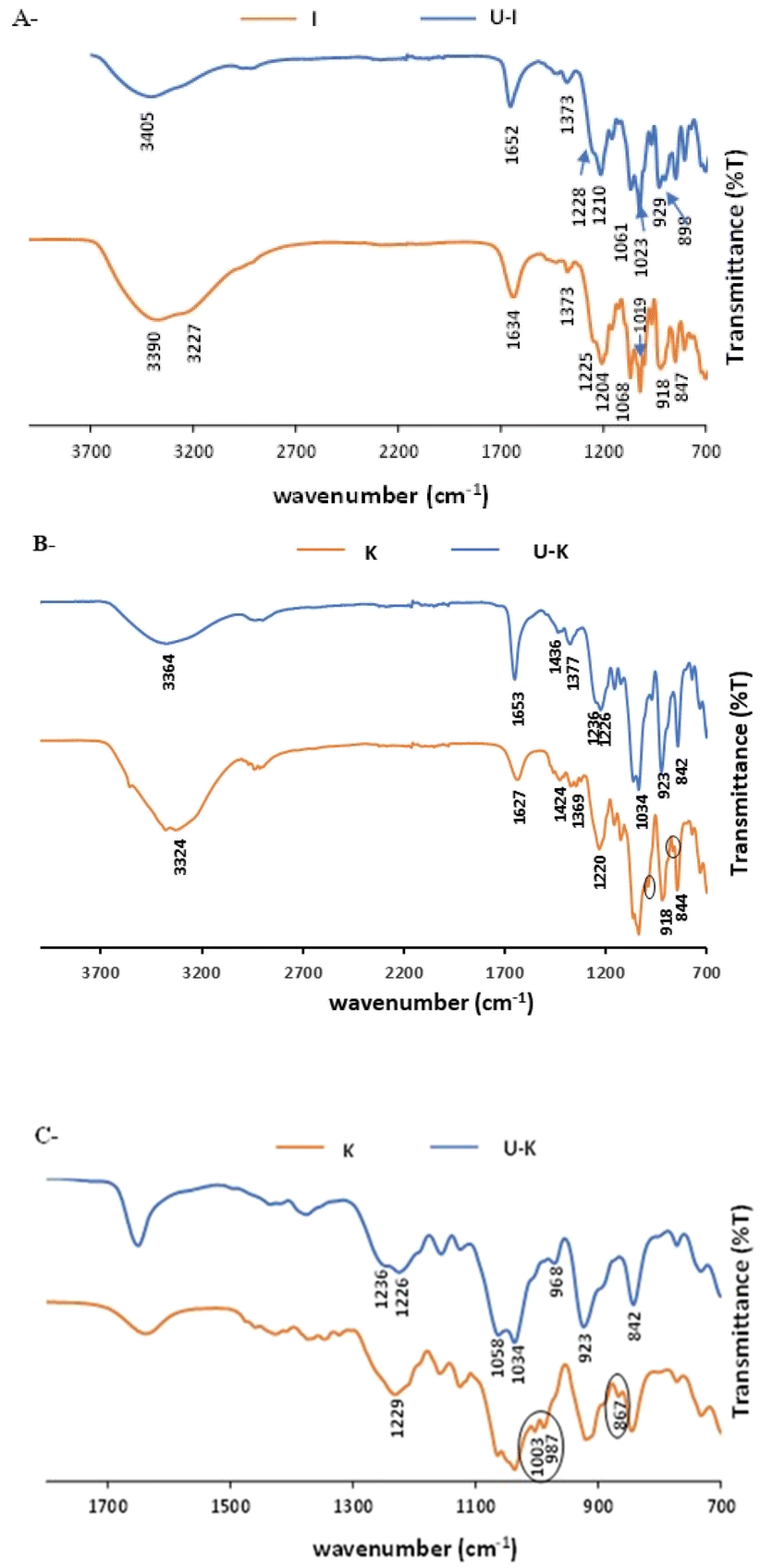 | ||
| Fig. 3 ATR-FTIR spectra of pristine polysaccharides and their analogues: (A) I and U–I, (B) Κ and U–K in the range of 700–3800 cm−1, and (C) Κ and U–K in the range of 700–1800 cm−1. (Sorption conditions: 700 mg per L UO22+, 3 mL of 1% w/v polysaccharide). | ||
The ATR-FTIR spectra of pristine X and U–X are shown in Fig. 4. As expected, the spectrum of pristine X contains bands in the region of 850–950 cm−1 corresponding to the skeleton mode of the anomeric configuration and glycosidic linkages. It also contains several other typical bands: at 3310 cm−1, which is due to the O–H axial deformation; at 2850–2910 cm−1 that is attributed to the symmetric and asymmetric stretching vibrations of the C–H group in the methyl and methylene groups; at 1718 cm−1 due to the CO stretching ester vibrations; and at 1614 cm−1 and 1393 cm−1 corresponding to asymmetrical and symmetrical stretching vibrations of carboxylate ion, respectively. As illustrated in Fig. 3, the 1016 cm−1 band, which is related to C–O stretching of primary alcohols,59 shifted to 1025 cm−1 after UO22+ sorption. In addition, a new band appears in U–X, at 1645 cm−1, assigned to CO stretching vibrations from pyruvate. These bands indicate that the adsorption caused changes in the axial deformation of the C–O part of the enols, which may indicate interaction of the UO22+ via the carboxylic groups. Moreover, the band at 1393 cm−1, which corresponds to the C–O symmetric stretching of the carboxylate anion,60 broadened after binding to UO22+, an observation that may be due to the intermolecular hydrogen bonds between the polysaccharide chains. Furthermore, after sorption of the metal ions, a new peak appears at 915 cm−1 that can be assigned to the bending vibration of δU–OH, the asymmetric stretching vibration of ν3,61 which might implies on interactions between uranyl ions and OH of carboxyl group, or as in G, the same part of UO22+ was adsorbed via the hydroxyl groups on the polysaccharide.
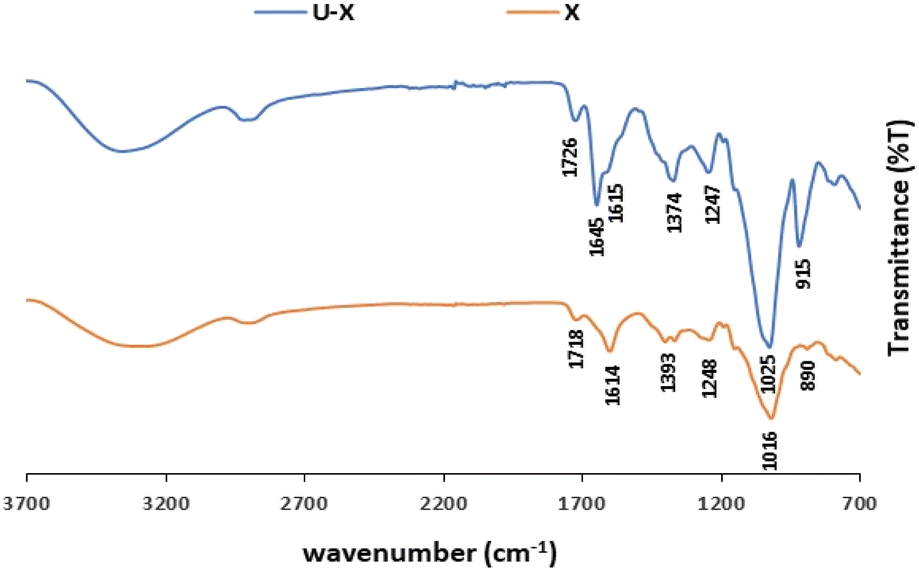 | ||
| Fig. 4 ATR-FTIR spectra of pristine X and U–X (sorption conditions: 700 mg per L UO22+, 3 mL of 1 w/v% X). | ||
As noted previously in the literature, the nature of metal ion coordination with the different functional groups on the polysaccharides varies among the different ions, and it is also altered by the metal ion concentration in the solution and by the solution pH.62,63 We also found that, compared to the carboxylic groups on X and the hydroxyl groups on G, Eu3+ had stronger electrostatic interactions with the ester sulfate groups of I, and at low pH values, increased functional group protonation led to decreased sorption efficiencies.48 However, as the ionic radius of UO22+ is larger and its charge differs from that of Eu3+, it probably interacts differently with the different functional groups. In addition, polysaccharide structure can also affect its sorption ability. As such, higher sorption efficiencies were obtained for X, which not only has different functional groups compared to those of the other tested polysaccharides, also possesses branched glycan residues with carboxylic groups on its backbone that are more available to adsorb uranyl ions.
3.2 SEM-EDS analysis
SEM-EDS analysis also confirmed that the metal ions are adsorbed to the different polysaccharides (Fig. 5). As expected, all of the U–polysaccharide composites contain mainly carbon (C) and oxygen (O) from the native polymer and potassium (K) and/or calcium (Ca) as counter ions to the acidic groups on the polysaccharides. In addition, in all of the samples, relatively high amounts of uranium U(VI) were also detected in relative contents that are in agreement with the results of the sorption experiments: X > Κ > I/G.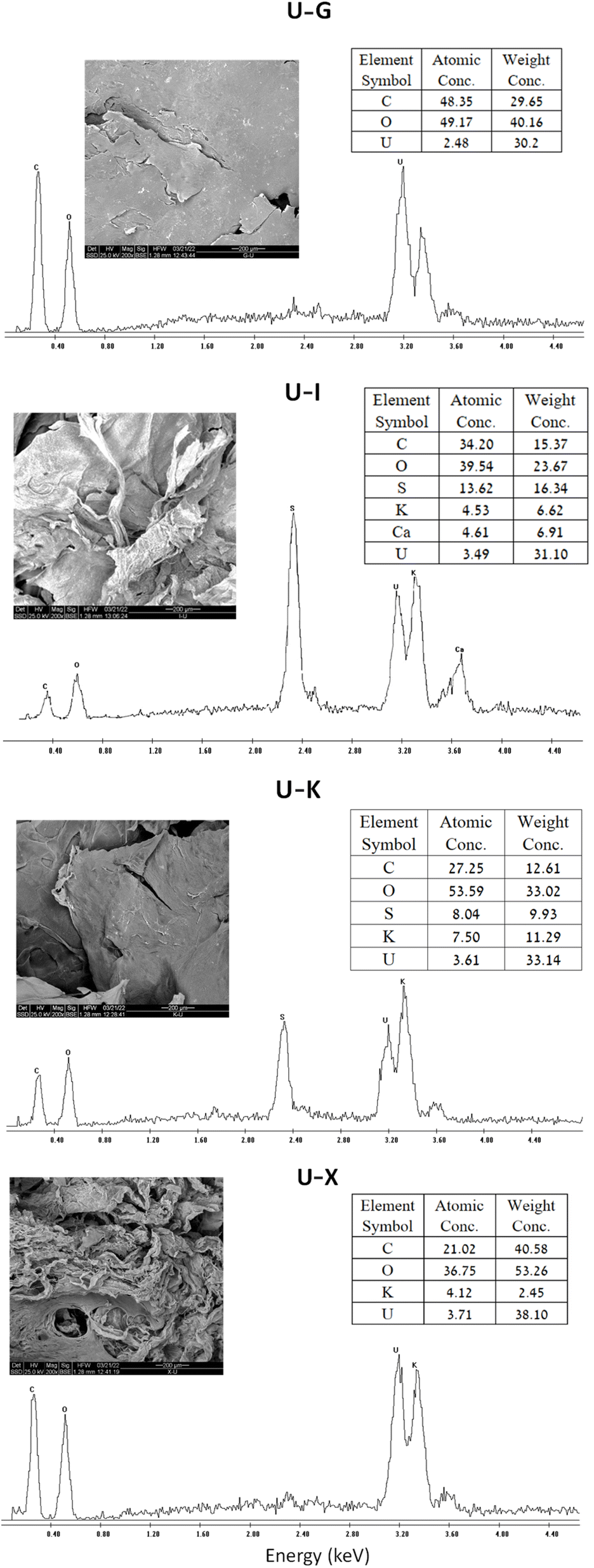 | ||
| Fig. 5 SEM image at ×200 magnification, EDS spectrum and tabulated results of polysaccharides before and after UO22+ adsorption (sorption conditions: 700 mg per L UO22+, 3 mL of 1% w/v polysaccharide). | ||
3.3 TGA analysis
TGA and TG-DTA (differential thermal analysis) analyses of the four polysaccharides (G/X/I/Κ) before and after UO22+ sorption is presented in Table 2, which summarizes the percentage of mass loss for each stage (% wt loss), the onset temperature and the differential thermogravimetric DTG.| Step | G | U–G | ||||
|---|---|---|---|---|---|---|
| Onset temp. (°C) | DTG (°C) | Wt loss (%) | Onset temp. (°C) | DTG (°C) | Wt loss (%) | |
| a Sorption conditions: 700 mg per L UO22+, 3 mL of 1% w/v polysaccharide. | ||||||
| Step 1 | — | 73.55 | 6.02 | — | 79.01 | 3.85 |
| Step 2 | 149.61 | 314.90 | 80.66 | 166.75 | 268.91 | 37.93 |
| Step 3 | — | — | — | 318.06 | 334.20 | 20.43 |
| 404.85 | ||||||
| Residue | 13.31 | 37.75 | ||||
| X | U–X | |||||
| Step 1 | — | 114.64 | 4.36 | — | 68.85 | 6.23 |
| Step 2 | 176.84 | 275.17 | 65.68 | 154.65 | 273.19 | 33.55 |
| Step 3 | — | — | — | 350.34 | 348.39 | 11.07 |
| Step 4 | — | — | — | 600.5 | 857.25 | 6.04 |
| Residue | 29.94 | 43.14 | ||||
| I | U–I | |||||
| Step 1 | — | 87.44 | 6.92 | — | 71.49 | 9.67 |
| Step 2 | 137.24 | 164.84 | 25.02 | 156.67 | 222.92 | 18.59 |
| Step 3 | 184.98 | 256.15 | 12.81 | 300.91 | 356.73 | 6.87 |
| Step 4 | 307.36 | 351.05 | 12.11 | 393.72 | 443.93 | 10.29 |
| Step 5 | 599.95 | 747.71 | 16.61 | 509.72 | 601.06 | 13.26 |
| Residue | 26.54 | 37.05 | ||||
| K | U–K | |||||
| Step 1 | — | 69.19 | 9.31 | — | 76.24 | 7.07 |
| Step 2 | 146.58 | 259.39 | 22.39 | 156.67 | 278.44 | 28.40 |
| Step 3 | 277.71 | 321.94 | 23.10 | 395.73 | 440.94 | 22.82 |
| Step 4 | 500.64 | 747.92 | 17.04 | — | — | — |
| Residue | 28.16 | 41.72 | ||||
In general, the TGA thermograms of the different polysaccharides, before and after UO22+ sorption, present the same mass weight trend. In the first stage, the water desorption stage, at temperatures below 200 °C, the total weight declined by about 5–10%.64 This step, mainly due to the interactions between the water molecules and the hydroxyl groups on the surface of the polymer backbone, confirms the hydrophilic character of the various polysaccharides. The second stage, at temperatures above 200 °C, involves several steps wherein the polysaccharides decompose until their residue reached a constant mass (∼50–80% weight loss) up to ∼350 °C. This is attributed to the carbohydrate backbone fragmentation and to the de-polymerization process. For the carrageenans (I and K), these degradation steps are also associated with the degradation of the sulfate ester groups from the pendant chains attached to the polymeric backbone.65,66 Furthermore, for Κ and I, a third stage was observed above 700 °C (step 5) that may be due to the decomposition of inorganic salts.67
The TGA and TG-DTA curves of all of the polysaccharides after sorption of UO22+ showed higher thermal stability, and the residue after sorption for X, Κ, I and G increased from 29.94% to 43.14%, 28.16% to 41.72%, 26.54% to 37.05%, and 13.31% to 37.75%, respectively. The main difference observed in thermal behavior was in stage 2, the so-called degradation stage, during which the main pyrolytic process occurred and different volatile components were gradually released. These results suggest that the polysaccharides become more thermally stable after sorption.68
Furthermore, the maximum decomposition peaks of Κ and I shifted to higher temperatures after UO22+ sorption, as reflected in the maximum peak in the DTG curves: 259.39 °C to 278.44 °C and 164.84 °C to 222.99 °C, respectively. This finding may be ascribed to the loss of –OSO3– and UO22+ groups and may also be due to the carbohydrate backbone fragmentation. The temperature for G, however, decreased after sorption from 314.90 °C to 268.97 °C, and for X, a minor change was observed, from 275.17 °C to 273.19 °C. This finding implies that changes occurred in the molecular structures of X and G, and indeed, the ordered or disordered state of X chains is known to be controlled by temperature and ionic strength.69 It would therefore seem that, after sorption, thermal changes are caused by weak intramolecular interaction forces (e.g., hydrogen bonds). Notably, after the sorption process, all of the polysaccharides exhibited a new, broad peak in the range of 300–400 °C that may be attributed to the decomposition of uranyl in the sample.
The thermal decomposition of uranyl acetate, which was used as the source of uranyl in this study, is known to take place at temperatures between 260 °C and 370 °C.70,71 This decomposition stage could be due to the fission of the UO2–O bonds and to the release of the acetate group from the complex, which can also produce acetate radicals.71 X-ray powder diffraction analysis of the decomposition residues show that in air, uranyl acetate decomposes to U, whereas in nitrogen, the product is mainly non-stoichiometric UO2x+, with small amounts of U. Thus, as the uranyl was adsorbed to the various polysaccharides, without the acetate counter ion, it can be concluded that the peak in the range of 300–400 °C in the U–polysaccharide samples is due to the decomposition of uranyl in the sample that decomposes to U.
3.4 Physicochemical analysis
Viscosity measurements of the aqueous polysaccharide solutions (X/I/Κ sol.) before adsorption and following 30 min of mixing with 700 mg per L uranyl acetate (U–X sol./U–I sol./U–Κ sol.) are shown in Fig. 6. Notably, measuring the viscosity of the U–G sol. was not applicable because the solution was not homogeneous and contained particles. All of the aqueous pristine polysaccharides yielded typical non-Newtonian fluid behavior, indicating that shear stress reduces the viscosity, whereas the viscosity measurements of the solutions of the U–polysaccharide preparations indicated that all of the preparations lost their high viscosity behavior.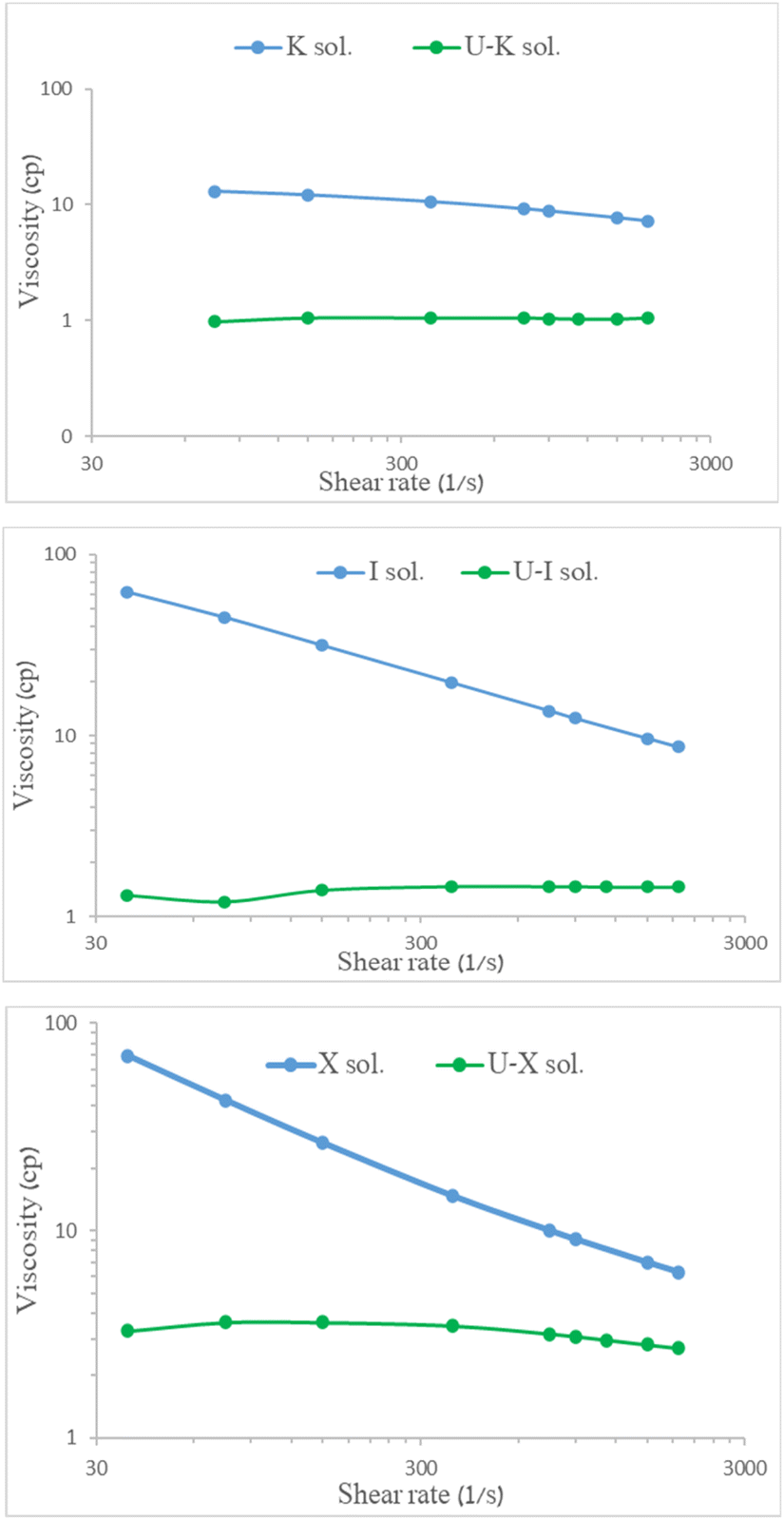 | ||
| Fig. 6 Viscosity of 0.3% w/v polysaccharides before and after UO22+ adsorption at 25 °C as function of shear rate. | ||
To compare the changes that were obtained for each polysaccharide due to UO22+ adsorption, the mechanical properties of each polysaccharide before and after adsorption, i.e., the storage (or elastic) modulus (G′) and viscous (or loss) modulus (G′′), were measured as functions of the angular frequency (Fig. 7).
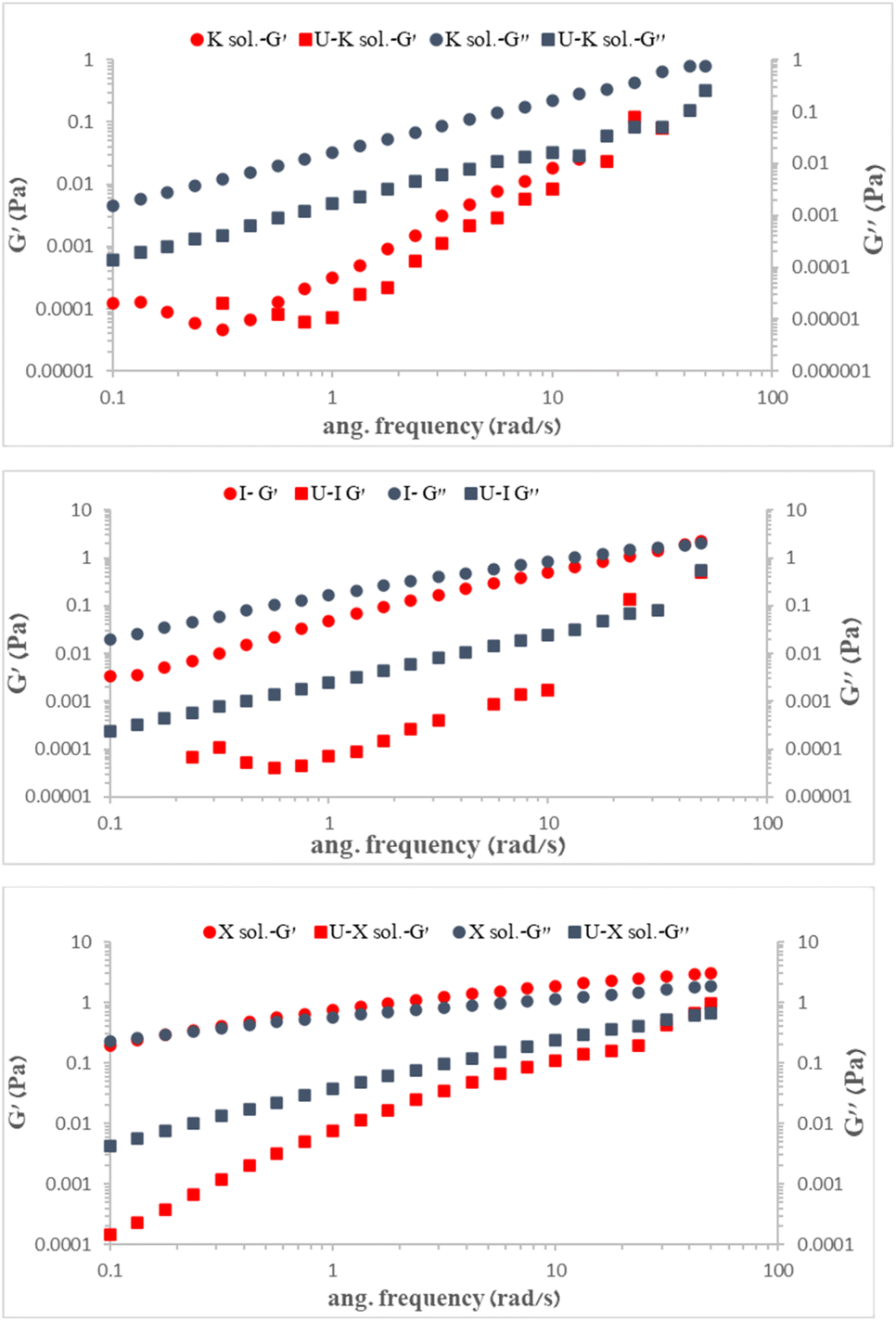 | ||
| Fig. 7 (G′) and (G′′) modulus of 0.3% w/v polysaccharides before and after UO22+ adsorption at 25 °C. | ||
As expected, the presence of uranyl ions reduced both modules, G′ and G′′, in the K, I, and X polysaccharides, with G′′ > G′, indicating electrostatic repulsion caused by UO22+ adsorption and distancing of polysaccharide chains. We suggest that the UO22+ reduced the level of entanglement of the polysaccharides while also increasing the free volume between the chains, thereby lowering both G′ and G′′.
4. Conclusions
The discharge of industrial wastewater containing heavy metals, and in particular uranium, into the environment, affects ecosystems and public health. These metals can be separated from aqueous solutions by various processes, where among these methods, adsorption was found to be highly efficient, with a preference that renewable and biodegradable polysaccharides be exploited as the sorbents.The simple addition of the aqueous polysaccharide solution – of I and K from the carrageenan family, which each contain hydroxyl and ester sulfate groups, X with hydroxyl and carboxylic acid groups, and G with hydroxyl groups only – to UO22+ solutions successfully removed the UO22+. It was found that the structures and the functional groups on the polysaccharide backbone affected the sorption efficiency, which decreased in the order of X > K > I/G. It was also found that lowering the acidity in the system decreased the sorption efficiencies with all of the tested polysaccharides due to protonation of the active sites. Furthermore, reducing the ratio between the amount of UO22+ and the amount of polysaccharide, whether by reducing the amount of UO22+ or by increasing the amount of polysaccharide, increased the sorption efficiencies with all the polysaccharides. Of all the concentrations measured, 0.03 g polysaccharide with 700 mg per L UO22+ was found to be the ratio that provided the maximum adsorption efficiency.
FTIR analysis confirmed that UO22+ was adsorbed via the hydroxyl groups to all of the polysaccharides, and in addition, via the carboxylic groups to X and via the sulfate ester groups to I and Κ. SEM-EDS analysis also verified the presence of UO22+ in the polysaccharide, and the uranium contents were in line with those of the sorption experiments. Moreover, TGA thermograms of the polysaccharides, before and after adsorption, showed that the UO22+ was adsorbed into the polysaccharide. In addition, it indicated the presence of a change in the structure of the material where the residue obtained after adsorption was larger than before the adsorption, suggesting the presence of an inorganic substance in the residue, most likely UO22+.
Rheological measurements that were performed for the aqueous polysaccharide solution with and without UO22+ showed that the viscosity decreased sharply for all of the solutions that contained UO22+, exhibiting Newtonian behaviour. Furthermore, the addition of UO22+ to the polysaccharides lowered both the elasticity model-G′ and the viscosity module-G′′. These findings again indicate the presence of electrostatic repulsion due to the adsorption of the UO22+ and the resultant increased distance that was obtained between the polysaccharide chains.
Author contributions
OL-O, conceptualization, investigation, experiment design, methodology, validation, formal analysis, and writing – original draft, writing – review and editing and supervision; OP-T, conceptualization, investigation, validation, formal analysis, investigation, writing – original draft, writing – review and editing and supervision; YA, conducted the sorption experiments; AW, conceptualization, methodology, investigation, writing – original draft, writing – review and editing, supervision, and project administration.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. C. Miller, Depleted Uranium: Properties, Uses and Health Consequences, CRC Press, Boca Raton, 1st edn, 2006, DOI:10.1201/9781420004564.
- M. Betti, Civil use of depleted uranium, J. Environ. Radioact., 2003, 64(2–3), 113–119 CrossRef CAS PubMed.
- A. Kausar and H. N. Bhatti, J. Chem. Soc. Pak., 2013, 35(3), 1041–1052 Search PubMed.
- J. Li and Y. Zhang, Procedia Environ. Sci., 2012, 13, 1609–1615 CrossRef CAS.
- D. Brugge and V. Buchner, Rev. Environ. Health, 2011, 26(4), 231–249 CAS.
- P. Kurttio, A. Harmoinen, H. Saha, L. Salonen, Z. Karpas, H. Komulainen and A. Auvinen, Am. J. Kidney Dis., 2006, 47(6), 972–982 CrossRef CAS PubMed.
- G. Bjørklund, Y. Semenova, L. Pivina, M. Dadar, M. M. Rahman, J. Aaseth and S. Chirumbolo, Arch. Toxicol., 2020, 94(5), 1551–1560 CrossRef PubMed.
- R. Pereira, S. Barbosa and F. P. Carvalho, Environ. Geochem. Health, 2014, 36(2), 285–301 CrossRef CAS PubMed.
- P. Henner, Bioaccumulation of Radionuclides and Induced Biological Effects in situations of chronic exposure of ecosystems a uranium case study, in Loads and Fate of Fertilizer Derived Uranium, ed. L. J. De Kok and E. Schnug, Backhuys Publishers Leiden, The Netherlands, 2008, pp. 23–33 Search PubMed.
- P. D. Bhalara, D. Punetha and K. Balasubramanian, J. Environ. Chem. Eng., 2014, 2(3), 1621–1634 CrossRef CAS.
- M. Dulama, M. Iordache and M. Deneanu, Proceedings of NUCLEAR 2013 the 6th Annual International Conference on Sustainable Development through Nuclear Research and Education. Part 3/3, 2013, pp. 80–86 Search PubMed.
- X. Yi, Z. Xu, Y. Liu, X. Guo, M. Ou and X. Xu, RSC Adv., 2017, 7(11), 6278–6287 RSC.
- J. Bai, X. Ma, H. Yan, J. Zhu, K. Wang and J. Wang, Microporous Mesoporous Mater., 2020, 306, 110441, DOI:10.1016/j.micromeso.2020.110441.
- L. Kong, Y. Ruan, Q. Zheng, M. Su, Z. Diao, D. Chen, L. Hou, X. Chang and K. Shih, J. Hazard. Mater., 2020, 382, 120784, DOI:10.1016/j.jhazmat.2019.120784.
- K. Shakir, A. F. Elkafrawy, H. F. Ghoneimy, S. G. E. Beheir and M. Refaat, Water Res., 2010, 44(5), 1449, DOI:10.1016/j.watres.2009.10.029.
- C. Aldrich and D. Feng, Miner. Eng., 2010, 13(10–11), 1129–1138 Search PubMed.
- P. Li, P. Chen, G. Wang, L. Wang, X. Wang, Y. Lia, W. Zhanga, H. Jianga and H. Chen, Chem. Eng. J., 2020, 393, 124819, DOI:10.1016/j.cej.2020.124819.
- A. Mellah, S. Chegrouche and M. Barkat, Hydrometallurgy, 2007, 85(2–4), 163–171 CrossRef CAS.
- M. Abubakar, M. N. Tamin, M. A. Saleh, M. B. Uday and N. Ahmad, Ceram. Int., 2016, 42(7), 8212–8220 CrossRef CAS.
- M. G. Torkabad, A. R. Keshtkar and S. J. Safdari, Prog. Nucl. Energy, 2017, 94, 93–100 CrossRef.
- A. Zaheri, A. Moheb, A. Keshtkar and A. Shirani, J. Environ. Health Sci. Eng., 2010, 7(5), 423–430 CAS.
- D. Lakherwal, International Journal of Environmental Research and Development, 2014, 4(1), 41–48 Search PubMed.
- B. Chen, J. Wang, L. Kong, X. Mai, N. Zheng, Q. Zhong, J. Liang and D. Chen, Colloids Surf., A, 2017, 520, 612–621 CrossRef CAS.
- Y. Tan, L. Li, H. Zhang, D. Ding, Z. Dai, J. Xue, J. Liu, N. Hu and Y. Wang, J. Radioanal. Nucl. Chem., 2018, 317(2), 811–824 CrossRef CAS.
- T. S. Anirudhan and P. G. Radhakrishnan, J. Environ. Radioact., 2009, 100(3), 250–257 CrossRef CAS PubMed.
- M. Tsezos and B. Volesky, Biotechnol. Bioeng., 1981, 23(3), 583–604 CrossRef CAS.
- T. Tatarchuk, A. Shyichuk, I. Mironyuk and Mu. Naushad, J. Mol. Liq., 2019, 293, 111563, DOI:10.1016/j.molliq.2019.111563.
- F. Veglio and F. Beolchini, Hydrometallurgy, 1997, 44(3), 301–316 CrossRef CAS.
- N. K. Gupta, A. Sengupta, A. Gupta, J. R. Sonawane and H. Sahoo, J. Environ. Chem. Eng., 2018, 6(2), 2159–2175 CrossRef CAS.
- M. Rinaudo, Polym. Int., 2008, 57(3), 397–430 CrossRef CAS.
- Food Polysaccharides and Their Applications, ed. A. M. Stephen, G. O. Phillips and P. A. Williams, Taylor & Francis Group, CRC Press, Boca Raton, 2nd edn, 2016 Search PubMed.
- G. Franz, Planta Med., 1989, 55(6), 493–497 CrossRef CAS PubMed.
- F. Freitas, V. D. Alves and M. A. Reis, Polysaccharides, 2015, 2017–2043 Search PubMed.
- Y. Na, J. Lee, S. H. Lee, P. Kumar, J. H. Kim and R. Patel, Polym.-Plast. Technol. Mater., 2020, 59(16), 1770–1790 CAS.
- G. Crini, Prog. Polym. Sci., 2005, 30(1), 38–70 CrossRef CAS.
- J. Wang and C. Chen, Biotechnol. Adv., 2009, 27(2), 195–226 CrossRef CAS PubMed.
- A. M. Marqués, R. Bonet, M. D. Simon-Pujol, M. C. Fusté and M. F. Congregado, Appl. Microbiol. Biotechnol., 1990, 34(3), 429–431 CrossRef.
- S. K. Kazy, P. Sar and S. F. D'Souza, Biorem. J., 2008, 12(2), 47–57 CrossRef CAS.
- T. Sakaguchi, A. Nakajima and T. Horikoshi, Nippon Kagaku Kaishi, 1979, 53, 788–792 CrossRef.
- C. Gok and S. Aytas, J. Hazard. Mater., 2009, 168(1), 369–375 CrossRef CAS PubMed.
- R. A. Muzzarelli, Carbohydr. Polym., 2011, 84(1), 54–63 CrossRef CAS.
- Y. Liu, Y. Liu, X. Cao, R. Hua, Y. Wang, C. Pang, M. Hua and X. Li, J. Radioanal. Nucl. Chem., 2011, 290(2), 231–239 CrossRef CAS.
- G. Wang, J. Liu, X. Wang, Z. Xie and N. Deng, J. Hazard. Mater., 2009, 168(2–3), 1053–1058 CrossRef CAS PubMed.
- L. C. B. Stopa and M. Yamaura, Int. J. Nucl. Energy Sci. Technol., 2010, 5(4), 283–289 CrossRef CAS.
- E. Piron and A. Domard, Int. J. Biol. Macromol., 1997, 21(4), 327–335 CrossRef CAS PubMed.
- E. Piron and A. Domard, Int. J. Biol. Macromol., 1998, 22(1), 33–40 CrossRef CAS PubMed.
- B. S. Savvin, Talanta, 1961, 8, 673–685 CrossRef.
- O. Levy-Ontman, C. Yanay, O. Paz Tal and A. Wolfson, Mater. Today Commun., 2022, 32, 104111, DOI:10.1016/j.mtcomm.2022.104111.
- C. M. Iain, Conformational Origins of Polysaccharides Solutions and Gel Properties, Industrial Gums-Polysaccharides and Their Derivatives, ed. R. L. Whistler and J. N. Bemiller, Academic Press, San Diego, 1993, pp. 21–52 Search PubMed.
- R. P. Millane, R. Chandrasekaran, S. Arnott and I. C. Dea, Carbohydr. Res., 1998, 182, 1–17 CrossRef.
- O. Levy-Ontman, C. Yanay, O. Paz Tal and A. Wolfson, J. Polym. Environ., 2023, 31, 2321–2333 CrossRef CAS.
- A. Chetouani, N. Follain, S. Marais, C. Rihouey, M. Elkolli, M. Bounekhel, D. Benachour and D. LeCerf, Int. J. Biol. Macromol., 2017, 97, 348–356 CrossRef CAS PubMed.
- G. Sen, S. Mishra, U. Jha and S. Pal, Int. J. Biol. Macromol., 2010, 47, 164–170 CrossRef CAS PubMed.
- D. Mudgil, S. Barak and B. S. Khatkar, Int. J. Biol. Macromol., 2012, 50, 1035–1039 CrossRef CAS PubMed.
- V. Pascal, R. Besson and C. Schaffer-Lequart, J. Agric. Food Chem., 2004, 52(25), 7457–7463 CrossRef PubMed.
- F. N. Jumaah, N. Mobarak, A. Ahmad, M. A. Ghani and M. Y. A. Rahman, Ionics, 2014, 21(5), 1311–1320 CrossRef.
- S. Karthikeyan, S. Selvasekarapandian, M. Premalatha, S. Monisha, G. Boopathi, G. Aristatil, A. Arun and S. Madeswaran, Ionics, 2016, 23, 2775–2780 CrossRef.
- V. L. Campo, D. F. Kawano, D. B. da Silva and I. Carvalho, Carbohydr. Polym., 2009, 77, 167–180 CrossRef CAS.
- Z. Wang, J. Wu, L. Zhu and X. Zhan, Carbohydr. Polym., 2017, 157, 521–526 CrossRef CAS PubMed.
- P. S. Gils, D. Ray and P. K. Sahoo, Int. J. Biol. Macromol., 2009, 45(4), 364–371 CrossRef CAS PubMed.
- J. Čejka, Infrared spectra and thermal analysis of the uranyl minerals, in Uranium: Mineralogy, Geochemistry and the Environment, ed. P. C. Burns and R. Finch, Rev. Mineral, 1999, vol. 38, pp. 521–622 Search PubMed.
- A. P. Ramos, R. R. Goncalves, O. A. Serra, M. E. D. Zaniquelli and K. Wong, J. Lumin., 2007, 127, 461–468 CrossRef CAS.
- L. Fuks, I. Herdzik-Koniecko, H. Polkowska-Motrenko and A. Oszczak, Int. J. Environ. Sci. Technol., 2018, 15, 2657–2668 CrossRef CAS.
- C. G. Mothé and M. A. Rao, Thermochim. Acta, 2000, 357–358, 9–13 CrossRef.
- D. K. Mishra, J. Tripathy and K. Behari, Carbohydr. Polym., 2008, 71, 524–534 CrossRef CAS.
- M. Vincekovic, A. Pustak, L. Tusek-Bozic, F. Liu, G. Ungar, M. Bujan, I. Smit and N. Filipovic-Vincekovic, J. Colloid Interface Sci., 2010, 341, 117–123 CrossRef CAS PubMed.
- S. Ma, L. Chen, X. Liu, D. Li, N. Ye and L. Wang, Int. J. Green Energy, 2012, 9, 13–21 CrossRef CAS.
- W. A. K. Mahmood, M. M. R. Khan and T. C. Yee, J. Phys. Sci., 2014, 25(1), 123–138 CAS.
- M. Rinaudo and M. Mils, Biopolymers, 1978, 17(11), 2663–2678 CrossRef CAS.
- P. S. Clough, D. Dollimore and P. Grundy, J. Inorg. Nucl. Chem., 1969, 31, 361–370 CrossRef CAS.
- L. Abate, A. Chisari, R. Maggiore and G. Siracusa, J. Therm. Anal., 1983, 27, 139–150 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |