
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of sulfonyl 2-aryl-5-methylenyltetrahydropyrans†
Meng-Yang Chang *abc and
Kuan-Ting Chena
*abc and
Kuan-Ting Chena
aDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 807, Taiwan. E-mail: mychang@kmu.edu.tw
bDepartment of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung 807, Taiwan
cNPUST College of Professional Studies, National Pingtung University of Science and Technology, Pingtung 912, Taiwan
First published on 13th October 2023
Abstract
In this study, the present research describes a high-yield method for the synthesis of sulfonyl 2-aryl-5-methylenetetrahydropyrans by one-pot straightforward DABCO-promoted intramolecular Michael addition of β-sulfonyl styrene with 2-chloromethyl-1-propenol followed by intramolecular alkylation. This Baylis–Hillman-type pathway provides a highly effective stereoselective annulation by forming one carbon–oxygen bond and one carbon–carbon bond.
Introduction
2-Substituted tetrahydropyrans (THPs) are an important class of scaffolds in a wide range of natural products, synthetic intermediates, and bioactive molecules.1,2 Due to its potential applications, developing a new and more efficient one-pot synthetic route to the core framework of 2-functionalized THPs has attracted significant attention and general interest from organic chemists. Among the recent synthetic methods, employing Lewis acid-mediated or transition metal-promoted intramolecular Prins reaction, and Friedel–Crafts cyclization is a major route for synthesizing diversified 2-substituted THPs.3 However, only some examples have been reported on synthesizing 2-substituted THPs by the intermolecular annulation pathway, as shown in Scheme 1. List et al. developed the synthesis of 2-aryl-4-methylene THPs via imino-imidodiphosphate Brønsted acids-catalyzed intermolecular Prins-type (5 + 1) cyclization of aldehydes with the homoallyl alcohol (for a + b bond formations).4a For the involvement of the 3-electron withdrawing group, Makosza and coworkers had documented the synthesis of 2-aryl-3-carboxylate THPs by employing the intermolecular (4 + 2) cyclization of tBuOK-mediated in situ formed δ-halocarbanions with aryl or alkylaldehydes (for b + f bond formations).4b Harrity and co-workers reported the intermolecular (3 + 3) annulation approach to construct 2-alkyl-3-methylenyl THPs via the addition of allylic carbanion (by nBuLi-mediated deprotonation) to monoalkyl epoxide and subsequent Pd(II)/Ti(IV)-promoted intramolecular cyclization (for c + f bond formations).4c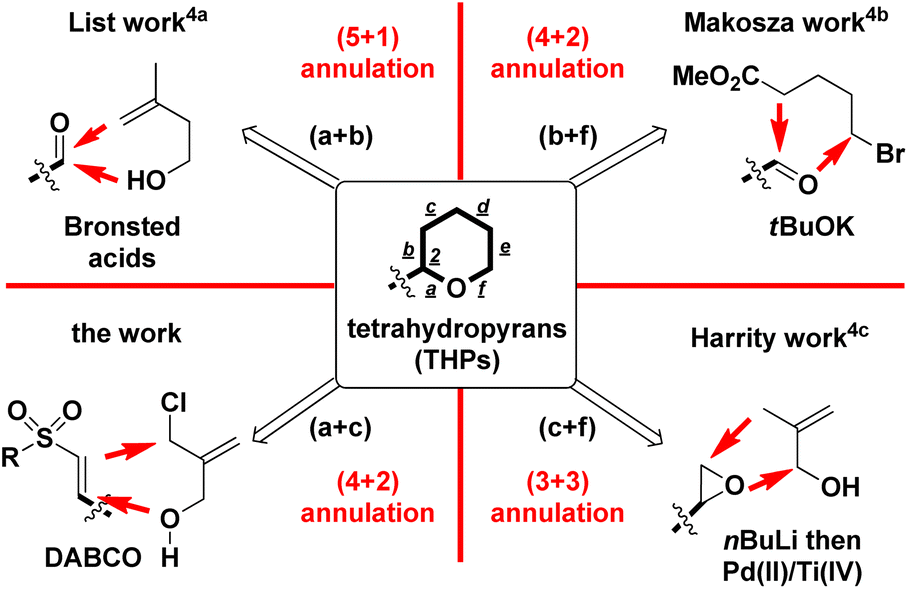 | ||
| Scheme 1 Intermolecular annulation route toward substituted 2-aryltetrahydropyrans. | ||
Therefore, further investigation of novel and efficient methods for generating 2-substituted THPs is still highly desired. Continuing our previous research on the recent synthesis of β-sulfonyl styrenes (prepared from Ac2O-mediated reaction of arylacetaldehydes 1 with RSO2Na),5 we decided to prepare new 2-aryl-5-methylene THPs 5 containing a 3-sulfonyl substituent via the DABCO (i.e. 1,4-diazabicyclo[2.2.2]octane)-mediated intermolecular Michael addition of β-sulfonyl styrenes 2 with 2-chloromethyl-2-propen-1-ol (3). This was followed by sequential intramolecular ring-closure of the resulting 4 during Baylis–Hillman-type process (for a + c bond formations, Scheme 2)6 Sulfone-bearing organic molecules usually exhibit unique and diverse behaviors including chemical, pharmaceutical, and material features. To the best of our knowledge, there have been no reports of DABCO-dependent routes toward the synthesis of sulfonyl 2-aryl-5-methylene THPs and the derivatives.7
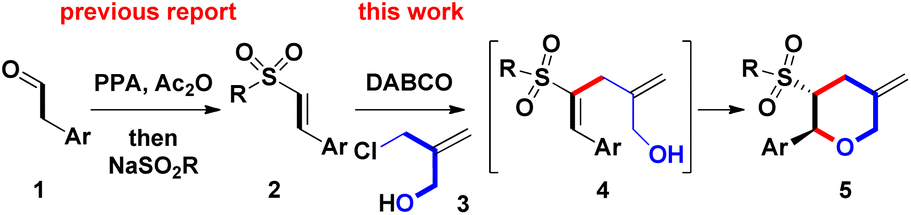 | ||
| Scheme 2 Synthetic route toward 2-aryl-5-methylene THPs 5. | ||
Results and discussion
The initial study for examining the one-step formation of sulfonyl 2-phenyl-5-methylene THP 5a commenced with the treatment of model β-sulfonyl styrene 2a (Ar = Ph, R = Tol, 1 mmol), 2-chloromethyl-2-propen-1-ol (3, 1.0 equiv.), DABCO (20 mol%) in CCl4 (15 mL) for 10 h at 25 °C as shown in Table 1, entry 1. Under the open-atmosphere condition, however, no observation of a reaction, and the starting 2a was recovered as the major component. Prolonging the reaction time from 10 h to 30 and 90 h, only 25 °C yielded trace (8%) amounts of 5a (entries 2 and 3). With the results in mind, the effect of reaction temperature was examined next. In entry 4, by controlling the reaction time as 10 h, the refluxing temperature (77 °C) produced a 78% yield of 5a. The product yield could be increased by a refluxing condition. After adjusting the reaction time from 10 h to 20 h, and 30 h, we observed that 20 h and 30 h produced similar yields (89% and 88%) for forming 5a (entries 5 and 6). Therefore, 77 °C and 20 h were chosen to screen the next reaction condition. Adjusting the catalytic amounts of DABCO from 20 mol% to 10 and 30 mol%, there was no observation of better yields (70% and 85%), as shown in entries 7 and 8. Furthermore, controlling catalytic amounts at 20 mol%, three organo diamines were screened (entries 9–11), including 4-DMAP, DBU, and urea. However, isolation of 5a was still carried out at lower yields (21%, 65%, and 0%). Compared with a tertiary diamine (for DABCO), the results showed that three skeletons of pyridine (for 4-DMAP), amidine (for DBU), and lactam (for urea) were inappropriate to generate 5a.8 We chose DABCO to investigate the sequential reaction conditions from this phenomenon. By maintaining the combination of refluxing CCl4 (77 °C) and 20 h, DME (dimethoxyethane), MeCN, and (CH2Cl)2 having similar boiling points (85 °C, 82 °C, and 84 °C) were selected as the solvents for screening optimal conditions. However, no better yields (48%, 15% and 53%) appeared (entries 12–14). Unexpectedly, entry 15 showed that DMF provided a complex mixture as the major component. Based on the results mentioned above, we realized that DABCO (20 mol%) base, CCl4 solvent and reflux conditions were necessary for an optimal reaction condition (Table 1, entry 5) for the synthesis of 5a. This expeditious synthetic route set up a trans-configuration between the 2-sulfonyl and 3-phenyl groups on 5a, including the bond formations of 1 C–O bond and 1 C–C bond.
|
|||||
|---|---|---|---|---|---|
| Entry | Base (mol%) | Solvent | Temp. | Time | %b |
| a The reactions were run on a 1 mmol scale with 2a (257 mg), 2-chloromethyl-2-propen-1-ol (3, 106 mg, 1.0 equiv.), base (10–40 mol%), solvent (20 mL), temp. (25 °C, reflux), open-vessel conditions.b Isolated yields.c No reaction.d Unidentified and unknown mixture was isolated. | |||||
| 1 | DABCO (20) | CCl4 | 25 | 10 | —c |
| 2 | DABCO (20) | CCl4 | 25 | 30 | —c |
| 3 | DABCO (20) | CCl4 | 25 | 90 | 8 |
| 4 | DABCO (20) | CCl4 | 77 | 10 | 78 |
| 5 | DABCO (20) | CCl4 | 77 | 20 | 89 |
| 6 | DABCO (20) | CCl4 | 77 | 30 | 88 |
| 7 | DABCO (10) | CCl4 | 77 | 20 | 70 |
| 8 | DABCO (30) | CCl4 | 77 | 20 | 85 |
| 9 | 4-DMAP (20) | CCl4 | 77 | 20 | 21 |
| 10 | DBU (20) | CCl4 | 77 | 20 | 65 |
| 11 | Urea (20) | CCl4 | 77 | 20 | —c |
| 12 | DABCO (20) | DME | 85 | 20 | 48 |
| 13 | DABCO (20) | MeCN | 82 | 20 | 15 |
| 14 | DABCO (20) | (CH2Cl)2 | 84 | 20 | 63 |
| 15 | DABCO (20) | DMF | 154 | 20 | —d |
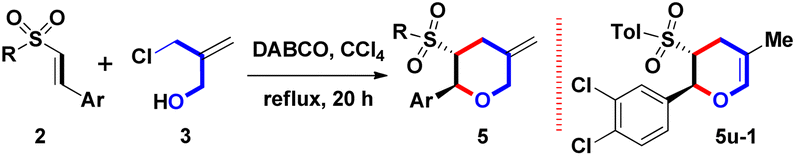
Diversified skeleton 5 was reacted under optimal reaction conditions (Table 1, entry 5) to study the substrate scope and limitations of this DABCO-mediated one-pot route, and give functionalized 5, as shown in Table 2. For the Ar substituent on β-sulfonyl styrenes 2a–2w, next the aromatic ring with diversified electron-neutral, electron-donating, or electron-withdrawing groups was examined, including Ph, 4-FC6H4, 4-MeC6H4, 4-MeOC6H4, 4-CF3C6H4, 4-NO2C6H4, 4-PhC6H4, 2-naphthyl, 3,4-(MeO)2C6H3, 3,4-CH2O2C6H3, 3,4-Cl2C6H3, 3,4,5-(MeO)3C6H2 and 2-thienyl groups (entries 1–23). However, many attempts to afford 5o and 5p failed due to the electron-withdrawing aryl groups (Ar = 4-CF3C6H4 and 4-NO2C6H4) on 2o and 2p could increase the electron-deficient density of the β-position such that DABCO-mediated Michael reaction pathway of A with 3 could be triggered to produce unidentified and unknown mixtures (entries 15 and 16). For the sulfonyl substituent (R) on β-sulfonyl styrenes 2a–2w, the aromatic groups (R = Tol, Ph, 4-FC6H4, 4-MeOC6H4, 3-MeC6H4, 4-EtC6H4, 4-iPrC6H4, 4-nBuC6H4 and 4-tBuC6H4) and the aliphatic groups (R = Me and nBu) were appropriate. The formation of 5a–5n, 5q–5t, and 5v–5w obtained these yields in a range of moderate to good yields (70–90%) by the one-pot condition. Interestingly, only an 80% yield of 5u-1 with an endo-olefin was isolated via the double bond migration (entry 21). The determination of trans-stereochemical structures of 5m and 5n was done by single-crystal X-ray analysis.9
|
||
|---|---|---|
| Entry | 2, R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) , Ar , Ar |
5, %b |
| a The reactions were run on a 1 mmol scale with 2a–2v, 2-chloromethyl-2-propen-1-ol (3, 106 mg, 1.0 equiv.), DABCO (23 mg, 20 mol%), CCl4 (20 mL), 20 h, reflux (77 °C), open-vessel conditions.b Isolated yields.c Unidentified and unknown mixture.d 5u-1 (80%) was isolated. | ||
| 1 | 2a, Tol, Ph | 5a, 89 |
| 2 | 2b, Ph, Ph | 5b, 85 |
| 3 | 2c, 4-FC6H4, Ph | 5c, 80 |
| 4 | 2d, 4-MeOC6H4, Ph | 5d, 84 |
| 5 | 2e, 3-MeC6H4, Ph | 5e, 82 |
| 6 | 2f, 4-EtC6H4, Ph | 5f, 83 |
| 7 | 2g, 4-iPrC6H4, Ph | 5g, 82 |
| 8 | 2h, 4-nBuC6H4, Ph | 5h, 84 |
| 9 | 2i, 4-tBuC6H4, Ph | 5i, 82 |
| 10 | 2j, Me, Ph | 5j, 80 |
| 11 | 2k, nBu, Ph | 5k, 78 |
| 12 | 2l, Tol, 4-FC6H4 | 5l, 70 |
| 13 | 2m, Tol, 4-MeC6H4 | 5m, 86 |
| 14 | 2n, Tol, 4-MeOC6H4 | 5n, 84 |
| 15 | 2o, Tol, 4-CF3C6H4 | 5o, —c |
| 16 | 2p, Tol, 4-NO2C6H4 | 5p, —c |
| 17 | 2q, Tol, 4-PhC6H4 | 5q, 80 |
| 18 | 2r, Tol, 2-naphthyl | 5r, 82 |
| 19 | 2s, Tol, 3,4-(MeO)2C6H3 | 5s, 87 |
| 20 | 2t, Tol, 3,4-CH2O2C6H3 | 5t, 86 |
| 21 | 2u, Tol, 3,4-Cl2C6H3 | 5u, —d |
| 22 | 2v, Tol, 3,4,5-(MeO)3C6H2 | 5v, 90 |
| 23 | 2w, Tol, 2-thienyl | 5w, 84 |
Based on the experimental results, a plausible mechanism for forming 5 was illustrated in Scheme 3. Initially, β-conjugation of 2 with DABCO provided a Baylis–Hillman adduct A. Subsequently, the spontaneous alkylation of the in situ formed α-carbanion of A with 3 led to the formation of B. After removing the α-proton of B, C was generated along with the recovery of DABCO. Under the reaction condition, intramolecular Michael annulation of C yielded D. Finally, proton exchange on D, the stereochemical synthesis of 5 was achieved. From the proposed mechanism, we found that catalytic amounts of DABCO seem to be workable for generating sulfonyl trans-THPs skeleton via a domino Baylis–Hillman-type process under open-vessel conditions.
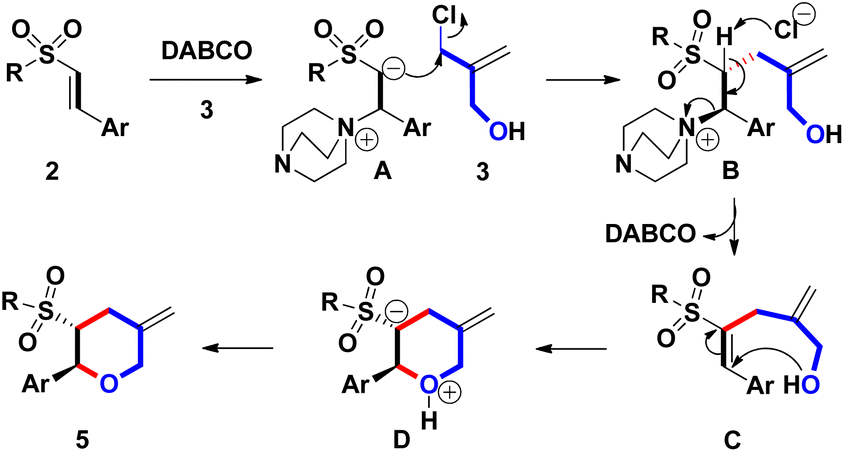 | ||
| Scheme 3 Plausible mechanism. | ||
With the optimal reaction condition (DABCO/refluxing CCl4), β-ketosulfone 6a was selected as another starting materials to displace the previous β-sulfonyl styrene 2a for investigating the synthesis of different sulfonyl skeletons (Scheme 4). Based on our previous work, the β-ketosulfones could be prepared easily from nucleophilic substitution of the commercial available α-bromo acetophenones with sulfonic sodium salts (RSO2Na) in refluxing EtOH.10 DABCO (20 mol%), treatment of 6a with 3 produced sulfonyl dihydropyran (DHP) 7a at a 76% yield via intermolecular α-allylation and intramolecular annulation. Compared with the conversion from β-sulfonyl styrene 2a to sulfonyl THP 5a, β-ketosulfone 6a yielded sulfonyl DHP 7a in the presence of DABCO (20 mol%). According to the optimal condition, two different starting materials possessed complementary methods in synthesizing the pyran family. Hemiacetal I was proposed as the possible intermediate during the one-pot formal (3 + 3) annulation process. After dehydration of I, a compound 7a with a push–pull nature was generated. The DABCO-promoted route formed four DHPs 7b–7e (Ar = 4-FC6H4, 4-CF3C6H4, 4-NO2C6H4, 3,4-Cl2C6H3) in a range of 68–74% yields. In contrast to the unsuccessful result on the formation of 5o and 5p, especially, both electron-withdrawing 4-nirtophenyl and 4-trifluoromethylphenyl groups (for Ar) were well-suitable to provide 7c and 7d in modest yields. Conversely, the 3,4-dichlorophenyl group could yield 7e with a terminal exo-olefin. However, no olefin migration behavior was observed compared with the generation 5u-1 with an endo-olefin.
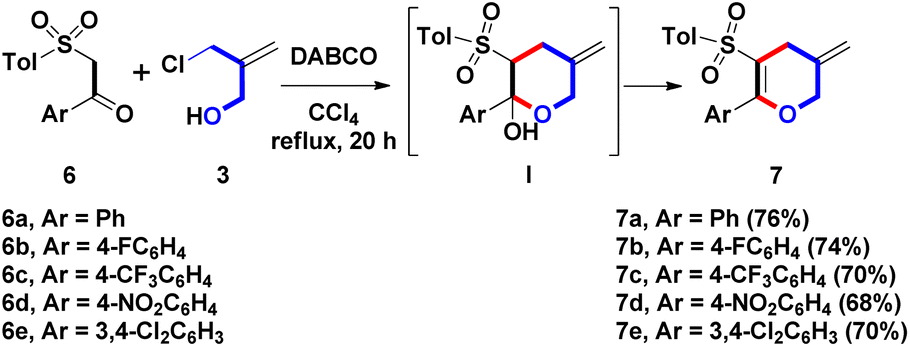 | ||
| Scheme 4 Synthesis of 7a–7e. | ||
Next, the ring-opening of DHP was examined (Scheme 5, eqn (1)). Under the boiling HOAc reaction condition, the hydrolysis of model substrate 7c (Ar = 4-CF3C6H4) was preferred and allowed to proceed; then, acetylation of the resulting primary allylic alcohol afforded 8 in an 80% yield. On the other hand, DABCO-promoted annulation of (Z)-β-sulfonyl styrene and 3 were studied (eqn (2)). By the photo-initiated olefin isomerization of (E)-2q (Ar = 4-PhC6H4), model substrate (Z)-2q-1 was generated.11 Following similar reaction conditions, 9 was isolated in a 71% yield. Compared with the stereochemistry of 5q with the trans-configurated structure (see ESI†), we envisioned that two adjacent stereochemical centers on 9 could be confirmed as cis-configuration. From the above results, we understood that olefinic isomerization of β-sulfonyl styrenes 2 could switch the stereochemistry of 3-sulfonyl-2-aryl THPs 5.
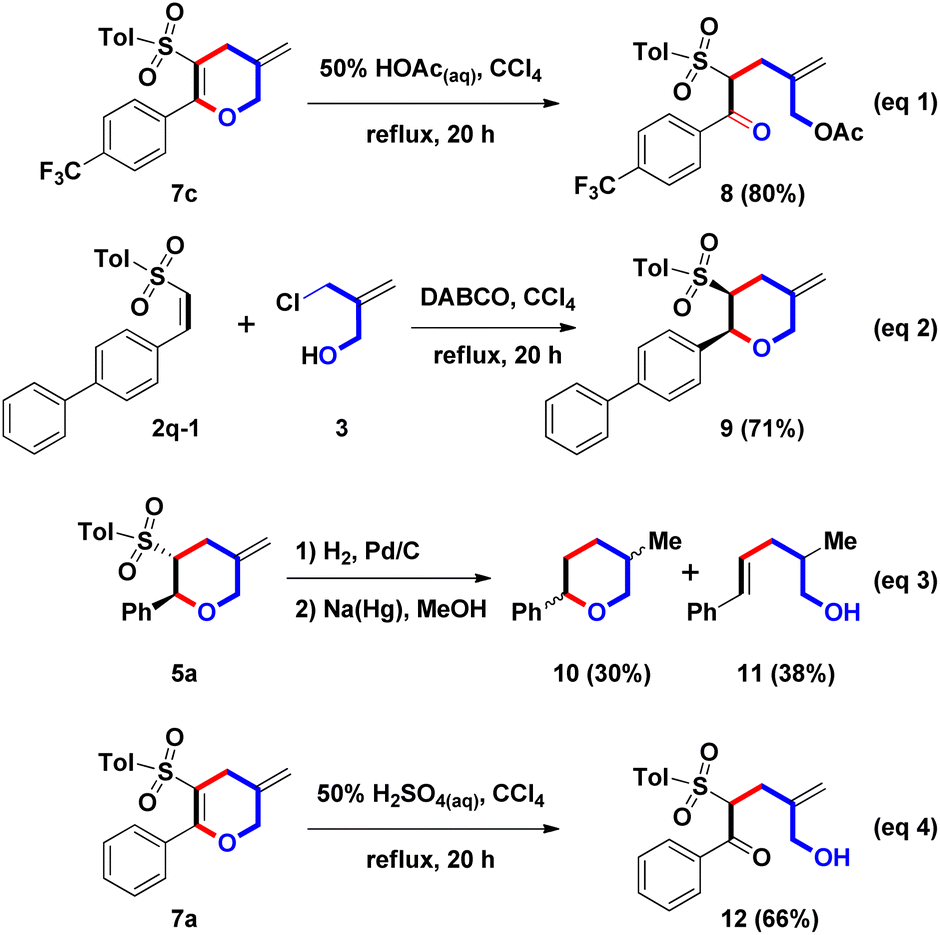 | ||
| Scheme 5 Synthesis of 8–12. | ||
The related Doremox surrogates were prepared (eqn (3)) to extend the synthetic application of this one-pot domino route. Cyclic ether 10 was a member of the rose oxide family, having perfumed properties.12 After the hydrogenation and desulfonation reactions, 5a was converted to 10 with a mixture of cis and trans isomers (ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in a 30% yield via the two-step process. Unexpectedly, a primary alcohol 11 was generated in a 38% yield via the desulfonylative ring-opening process. Although the yield of 10 was low, a new synthetic route could be established. For the diversification of the two double bonds on 7a, the oxidation reaction was chosen as the synthetic application. However, dihydroxylation of 7a with the combination of OsO4 and NMO provided complex and unknown mixture. And, mCPBA-mediated epoxidation of 7a could not produce the desired epoxide product. With these results in hand, we understood that oxidation condition could not differentiate between endo- and exo-olefins. Therefore, we turned the focus to explore the hydrolysis of 7a. By use of 50% H2SO4(aq), ring-opening of 7a (Ar = Ph) was achieved to 12 in a 66% yield. We found that the endo-enol olefin could convert into δ-hydroxy ketone via the ring-opening, and no influence for the exo-olefin. For the formation of two hydrated products 8 and 12, we understood that mono-allylation of β-ketosulfone could be accomplished by the two-steps route, including DABCO-mediated stepwise (4 + 2) annulation followed by the acidic hydrolysis of the resulting sulfonyl DHP.
:1) in a 30% yield via the two-step process. Unexpectedly, a primary alcohol 11 was generated in a 38% yield via the desulfonylative ring-opening process. Although the yield of 10 was low, a new synthetic route could be established. For the diversification of the two double bonds on 7a, the oxidation reaction was chosen as the synthetic application. However, dihydroxylation of 7a with the combination of OsO4 and NMO provided complex and unknown mixture. And, mCPBA-mediated epoxidation of 7a could not produce the desired epoxide product. With these results in hand, we understood that oxidation condition could not differentiate between endo- and exo-olefins. Therefore, we turned the focus to explore the hydrolysis of 7a. By use of 50% H2SO4(aq), ring-opening of 7a (Ar = Ph) was achieved to 12 in a 66% yield. We found that the endo-enol olefin could convert into δ-hydroxy ketone via the ring-opening, and no influence for the exo-olefin. For the formation of two hydrated products 8 and 12, we understood that mono-allylation of β-ketosulfone could be accomplished by the two-steps route, including DABCO-mediated stepwise (4 + 2) annulation followed by the acidic hydrolysis of the resulting sulfonyl DHP.
According to the above experimental results, the effect of π-bond role on 3 was studied next. However, the DABCO-catalyzed (4 + 2) annulation of 2a with 3-chloro-1-propanol (3a) could not produce the expected 13, and only 2a was recovered as major component (Scheme 6, eqn (5)). The possible reason should be the reactivity of aliphatic chloride (for 3a) was lower than allylic chloride (for 3). On the other way, changing the β-aryl (for 2a–2w, Ar) to β-aliphatic (for 2x, Me) group was examined. However, the desired 5x could not be observed, and the major starting material 2a was recovered (eqn (6)). The results indicated that the β-aryl group could provided higher reactivity for the DABCO-catalyzed Michael addition than β-aliphatic group.
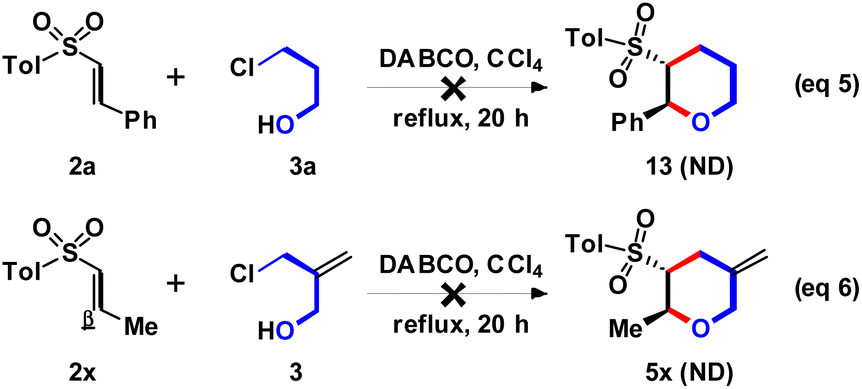 | ||
| Scheme 6 Unsuccessful reactions. | ||
Conclusion
In summary, in this study, a DABCO-promoted synthesis of trans-3-sulfonyl-2-aryl THPs 5 was developed. One-pot formal (4 + 2) annulation of β-sulfonyl styrenes 2 and 3 in CCl4 solvent and reflux conditions furnished moderate to good yields. Moreover, DABCO-promoted synthesis of 3-sulfonyl-2-aryl DHPs 7 via one-pot formal (3 + 3) annulation of β-ketosulfones 6 and 3 was studied. Cyclic ether 10 with rose odorants could be synthesized. The related plausible mechanisms are also proposed. The one-pot open-vessel, atom-economic, convenient process provides a straightforward pathway for efficient formations of one C–O bond and one C–C bond. The structures of the key products were confirmed through X-ray crystallography. Further investigations regarding the synthetic application of β-sulfonyl styrene will be conducted and subsequently published in due course.Experimental
General
All reagents and solvents were obtained from commercial sources and used without further purification. Reactions were routinely carried out under an atmosphere of dry air with magnetic stirring. Products in organic solvents were dried with anhydrous magnesium sulfate before concentration in vacuo. Melting points were determined with a SMP3 melting apparatus. 1H and 13C NMR spectra were recorded on a Varian INOVA-400 spectrometer operating at 400/600 and at 100/125 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) and the coupling constants (J) are given in Hertz. High resolution mass spectra (HRMS) were measured with a mass spectrometer Finnigan/Thermo Quest MAT 95XL. X-ray crystal structures were obtained with an Enraf-Nonius FR-590 diffractometer (CAD4, Kappa CCD).A representative synthetic procedure of skeleton 5 is as follows: DABCO (1,4-diazabicyclo[2.2.2]octane, 23 mg, 20 mol%) was added to a solution of 2 (1.0 mmol) in CCl4 (20 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 10 min. 2-Chloromethyl-2-propen-1-ol (3, 106 mg, 1.0 mmol) was added to the reaction mixture at 25 °C. The reaction mixture was stirred at reflux (77 °C) for 20 h. The reaction mixture was cooled to 25 °C and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 10/1 to 6/1) afforded 5.
5-Methylene-2-phenyl-3-(toluene-4-sulfonyl)tetrahydropyran (5a)
Yield = 89% (292 mg); white solid; mp = 136–138 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H21O3S 329.1211, found 329.1218; 1H NMR (400 MHz, CDCl3): δ 7.26 (d, J = 8.4 Hz, 2H), 7.16–7.08 (m, 5H), 7.01 (d, J = 8.4 Hz, 2H), 5.01 (s, 1H), 5.00 (s, 1H), 4.68 (d, J = 9.2 Hz, 1H), 4.27 (dd, J = 12.4, 1.2 Hz, 1H), 4.11 (d, J = 12.4 Hz, 1H), 3.62–3.56 (m, 1H), 3.05 (dd, J = 14.0, 3.6 Hz, 1H), 2.82 (dd, J = 13.6, 12.4 Hz, 1H), 2.33 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 143.8, 139.8, 137.5, 135.8, 129.2 (2×), 128.5 (2×), 128.3, 128.2 (2×), 128.0 (2×), 112.3, 79.9, 71.9, 65.5, 31.4, 21.5.3-Benzenesulfonyl-5-methylene-2-phenyltetrahydropyran (5b)
Yield = 85% (267 mg); white solid; mp = 149–151 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H19O3S 315.1055, found 315.1059; 1H NMR (600 MHz, CDCl3): δ 7.41–7.36 (m, 3H), 7.23–7.20 (m, 2H), 7.15–7.12 (m, 3H), 7.09–7.07 (m, 2H), 5.03 (d, J = 0.6 Hz, 1H), 5.01 (d, J = 1.2 Hz, 1H), 4.69 (d, J = 9.0 Hz, 1H), 4.28 (dd, J = 12.6, 1.2 Hz, 1H), 4.13 (d, J = 12.6 Hz, 1H), 3.65–3.61 (m, 1H), 3.09 (ddd, J = 13.8, 4.2, 1.2 Hz, 1H), 2.84 (dt, J = 13.8, 1.8 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3): δ 139.7, 138.9, 137.2, 132.8, 128.8, 128.6 (2×), 128.3 (2×), 128.2 (2×), 127.9 (2×), 112.4, 80.0, 71.9, 65.4, 31.2.3-(4-Fluorobenzenesulfonyl)-5-methylene-2-phenyltetrahydropyran (5c)
Yield = 80% (266 mg); white solid; mp = 132–134 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H18FO3S 333.0961, found 333.0952; 1H NMR (400 MHz, CDCl3): δ 7.36–7.31 (m, 2H), 7.20–7.08 (m, 5H), 6.88–6.82 (m, 2H), 5.05 (s, 1H), 5.02 (s, 1H), 4.67 (d, J = 9.2 Hz, 1H), 4.28 (dd, J = 12.4, 0.8 Hz, 1H), 4.12 (d, J = 12.4 Hz, 1H), 3.63–3.56 (m, 1H), 3.13 (ddd, J = 13.6, 4.0, 1.2 Hz, 1H), 2.85 (dd, J = 13.6, 12.8 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 165.1 (d, J = 253.9 Hz), 139.6, 137.1, 135.0, 130.7 (d, J = 9.9 Hz, 2×), 128.9, 128.4 (2×), 128.2 (2×), 115.8 (d, J = 22.7 Hz, 2×), 112.7, 80.0, 72.0, 65.6, 31.1.3-(4-Methoxybenzenesulfonyl)-5-methylene-2-phenyltetrahydropyran (5d)
Yield = 84% (289 mg); white solid; mp = 113–115 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H21O4S 345.1161, found 345.1155; 1H NMR (400 MHz, CDCl3): δ 7.30 (d, J = 8.8 Hz, 2H), 7.14–7.11 (m, 5H), 6.67 (d, J = 9.2 Hz, 2H), 5.01 (s, 1H), 4.99 (d, J = 1.2 Hz, 1H), 4.68 (d, J = 9.2 Hz, 1H), 4.27 (dd, J = 12.4, 0.8 Hz, 1H), 4.12 (d, J = 12.4 Hz, 1H), 3.80 (s, 3H), 3.60–3.54 (m, 1H), 3.06 (dd, J = 14.0, 4.4 Hz, 1H), 2.81 (dd, J = 14.0, 12.8 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 163.1, 139.9, 137.5, 130.8, 130.2 (2×), 128.7, 128.4 (2×), 128.2 (2×), 113.9 (2×), 112.3, 80.0, 71.9, 65.6, 55.6, 31.4.5-Methylene-2-phenyl-3-(toluene-3-sulfonyl)tetrahydropyran (5e)
Yield = 82% (269 mg); white solid; mp = 145–147 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H21O3S 329.1211, found 329.1216; 1H NMR (400 MHz, CDCl3): δ 7.26–7.23 (m, 1H), 7.19–7.05 (m, 8H), 5.04 (s, 1H), 5.01 (s, 1H), 4.67 (d, J = 9.2 Hz, 1H), 4.28 (d, J = 12.8 Hz, 1H), 4.13 (d, J = 12.4 Hz, 1H), 3.66–3.59 (m, 1H), 3.11 (dd, J = 13.6, 4.0 Hz, 1H), 2.84 (dd, J = 13.6, 12.8 Hz, 1H), 2.21 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 139.8, 138.7, 138.6, 137.2, 133.7, 128.7, 128.5, 128.4, 128.2 (2×), 128.1 (2×), 124.8, 112.4, 80.0, 72.0, 65.4, 31.2, 21.0.3-(4-Ethylbenzenesulfonyl)-5-methylene-2-phenyltetrahydropyran (5f)
Yield = 83% (284 mg); white solid; mp = 106–108 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H23O3S 343.1368, found 343.1373; 1H NMR (400 MHz, CDCl3): δ 7.27 (d, J = 8.4 Hz, 2H), 7.15–7.05 (m, 5H), 7.02 (d, J = 8.4 Hz, 2H), 5.03 (s, 1H), 5.00 (d, J = 0.8 Hz, 1H), 4.68 (d, J = 9.6 Hz, 1H), 4.27 (dd, J = 12.8, 1.2 Hz, 1H), 4.13 (d, J = 12.4 Hz, 1H), 3.64–3.57 (m, 1H), 3.09 (ddd, J = 14.0, 4.4, 1.2 Hz, 1H), 2.83 (t, J = 13.6 Hz, 1H), 2.61 (q, J = 7.6 Hz, 2H), 1.21 (t, J = 7.6 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 149.8, 139.9, 137.4, 136.0, 128.7, 128.3 (2×), 128.2 (2×), 128.14 (2×), 128.09 (2×), 112.3, 80.0, 72.0, 65.4, 31.2, 28.8, 15.2.3-(4-Isopropylbenzenesulfonyl)-5-methylene-2-phenyltetrahydropyran (5g)
Yield = 82% (292 mg); white solid; mp = 121–123 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H25O3S 357.1524, found 357.1529; 1H NMR (400 MHz, CDCl3): δ 7.26 (d, J = 8.4 Hz, 2H), 7.13–7.01 (m, 7H), 5.04 (s, 1H), 5.01 (s, 1H), 4.67 (d, J = 9.6 Hz, 1H), 4.27 (d, J = 13.6 Hz, 1H), 4.13 (d, J = 12.8 Hz, 1H), 3.64–3.58 (m, 1H), 3.13 (dd, J = 14.4, 3.6 Hz, 1H), 2.89–2.81 (m, 2H), 1.21 (d, J = 6.8 Hz, 3H), 1.20 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 154.3, 139.9, 137.2, 136.1, 128.7, 128.3 (2×), 128.2 (2×), 128.0 (2×), 126.7 (2×), 112.4, 80.1, 72.0, 65.3, 34.1, 31.1, 23.6, 23.4.3-(4-n-Butylbenzenesulfonyl)-5-methylene-2-phenyltetrahydropyran (5h)
Yield = 84% (311 mg); white solid; mp = 100–102 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H27O3S 371.1681, found 371.1688; 1H NMR (400 MHz, CDCl3): δ 7.26 (d, J = 8.4 Hz, 2H), 7.15–7.05 (m, 5H), 7.04 (d, J = 8.8 Hz, 2H), 5.03 (s, 1H), 5.00 (d, J = 1.2 Hz, 1H), 4.68 (d, J = 9.6 Hz, 1H), 4.27 (dd, J = 12.8, 1.2 Hz, 1H), 4.12 (d, J = 12.8 Hz, 1H), 3.64–3.57 (m, 1H), 3.10 (ddd, J = 14.0, 4.4, 1.2 Hz, 1H), 2.84 (dt, J = 13.6, 0.8 Hz, 1H), 2.56 (t, J = 7.6 Hz, 2H), 1.60–1.51 (m, 2H), 1.38–1.26 (m, 2H), 0.94 (t, J = 7.6 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 148.5, 139.9, 137.3, 136.0, 128.6 (2×), 128.24 (2×), 128.17 (2×), 128.1, 127.9 (2×), 112.3, 80.0, 71.9, 65.4, 35.4, 33.1, 31.2, 22.2, 13.8.3-(4-t-Butyl-benzenesulfonyl)-5-methylene-2-phenyltetrahydropyran (5i)
Yield = 82% (304 mg); white solid; mp = 102–104 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H27O3S 371.1681, found 371.1687; 1H NMR (400 MHz, CDCl3): δ 7.27–7.24 (m, 2H), 7.19–7.16 (m, 2H), 7.12–7.01 (m, 5H), 5.05 (s, 1H), 5.01 (d, J = 1.2 Hz, 1H), 4.67 (d, J = 9.6 Hz, 1H), 4.27 (dd, J = 12.4, 1.2 Hz, 1H), 4.13 (d, J = 12.4 Hz, 1H), 3.64–3.58 (m, 1H), 3.14 (dd, J = 14.0, 4.4 Hz, 1H), 2.85 (t, J = 13.6 Hz, 1H), 1.27 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3): δ 156.5, 139.9, 137.2, 135.7, 128.7, 128.3 (2×), 128.2 (2×), 127.7 (2×), 125.6 (2×), 112.4, 80.1, 72.0, 65.3, 35.0, 31.0, 30.9 (3×).3-Methanesulfonyl-5-methylene-2-phenyltetrahydropyran (5j)
Yield = 80% (202 mg); colorless oil; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H17O3S 253.0899, found 253.0904; 1H NMR (400 MHz, CDCl3): δ 7.50–7.47 (m, 2H), 7.44–7.36 (m, 3H), 5.06 (d, J = 2.0 Hz, 1H), 5.05 (d, J = 1.6 Hz, 1H), 4.69 (d, J = 9.2 Hz, 1H), 4.33 (dd, J = 12.4, 1.2 Hz, 1H), 4.13 (d, J = 12.4 Hz, 1H), 3.33–3.26 (m, 1H), 3.09 (ddd, J = 15.2, 4.4, 1.2 Hz, 1H), 2.76 (dt, J = 12.4, 1.2 Hz, 1H), 2.00 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 139.4, 138.1, 129.5, 129.1 (2×), 128.1 (2×), 112.9, 79.8, 71.9, 66.0, 41.6, 29.7.3-(n-Butane-1-sulfonyl)-5-methylene-2-phenyltetrahydropyran (5k)
Yield = 78% (229 mg); white solid; mp = 101–103 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H23O3S 295.1368, found 295.1374; 1H NMR (400 MHz, CDCl3): δ 7.50–7.47 (m, 2H), 7.43–7.38 (m, 3H), 5.053 (d, J = 1.6 Hz, 1H), 5.047 (s, 1H), 4.67 (d, J = 9.2 Hz, 1H), 4.32 (dd, J = 12.4, 1.6 Hz, 1H), 4.13 (d, J = 12.4 Hz, 1H), 3.31–3.25 (m, 1H), 3.05 (ddd, J = 14.0, 4.4, 1.2 Hz, 1H), 2.76 (t, J = 13.6 Hz, 1H), 1.92–1.75 (m, 2H), 1.58–1.33 (m, 2H), 1.09–0.99 (m, 2H), 0.71 (t, J = 7.2 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 139.7, 138.3, 129.4, 128.9 (2×), 128.1 (2×), 112.7, 80.0, 72.1, 63.5, 53.0, 29.8, 23.5, 21.4, 13.2.2-(4-Fluorophenyl)-5-methylene-3-(toluene-4-sulfonyl)tetrahydropyran (5l)
Yield = 70% (242 mg); white solid; mp = 171–173 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H20FO3S 347.1117, found 347.1124; 1H NMR (400 MHz, CDCl3): δ 7.27 (d, J = 8.0 Hz, 2H), 7.14–7.10 (m, 2H), 7.07 (d, J = 8.0 Hz, 2H), 6.80–6.76 (m, 2H), 5.02 (s, 1H), 5.01 (s, 1H), 4.66 (d, J = 9.6 Hz, 1H), 4.27 (dd, J = 12.8, 1.2 Hz, 1H), 4.11 (d, J = 12.8 Hz, 1H), 3.56–3.50 (m, 1H), 3.03 (ddd, J = 14.0, 4.4, 1.2 Hz, 1H), 2.80 (dd, J = 13.6, 12.8 Hz, 1H), 2.36 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 162.9 (d, J = 245.6 Hz), 144.1, 139.6, 135.8, 133.4, 129.9 (d, J = 8.3 Hz, 2×), 129.3 (2×), 129.0 (2×), 115.1 (d, J = 21.3 Hz, 2×), 112.6, 79.2, 72.1, 65.7, 31.4, 21.5.5-Methylene-3-(toluene-4-sulfonyl)-2-p-tolyltetrahydropyran (5m)
Yield = 86% (294 mg); white solid; mp = 169–171 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H23O3S 343.1368, found 343.1374; 1H NMR (400 MHz, CDCl3): δ 7.25 (d, J = 8.4 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 7.00 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 7.6 Hz, 2H), 5.01 (s, 1H), 4.99 (d, J = 1.2 Hz, 1H), 4.62 (d, J = 9.6 Hz, 1H), 4.26 (dd, J = 12.8, 1.2 Hz, 1H), 4.10 (d, J = 12.8 Hz, 1H), 3.61–3.54 (m, 1H), 3.06 (ddd, J = 13.6, 4.4, 1.2 Hz, 1H), 2.81 (dd, J = 13.6, 12.8 Hz, 1H), 2.35 (s, 3H), 2.25 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 143.6, 139.9, 138.5, 135.9, 134.6, 129.1 (2×), 128.9 (2×), 128.1 (2×), 128.0 (2×), 112.3, 79.7, 71.9, 65.6, 31.3, 21.5, 21.1. Single-crystal X-ray diagram: crystal of compound 5m was grown by slow diffusion of EtOAc into a solution of compound 5m in THF to yield colorless prisms. The compound crystallizes in the orthorhombic crystal system, space group P212121, a = 5.61670(10) Å, b = 15.9231(4) Å, c = 19.6051(4) Å, V = 1753.39(6) Å3, Z = 4, dcalcd = 1.297 g cm−3, F(000) = 1728, 2θ range 2.078–27.006°, R indices (all data) R1 = 0.0354, wR2 = 0.0727.2-(4-Methoxyphenyl)-5-methylene-3-(toluene-4-sulfonyl)tetrahydropyran (5n)
Yield = 84% (301 mg); white solid; mp = 168–170 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H23O4S 359.1317, found 359.1322; 1H NMR (400 MHz, CDCl3): δ 7.24 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 7.01 (d, J = 8.0 Hz, 2H), 6.58 (d, J = 8.4 Hz, 2H), 5.01 (s, 1H), 4.98 (s, 1H), 4.60 (d, J = 9.6 Hz, 1H), 4.25 (d, J = 12.8 Hz, 1H), 4.09 (d, J = 12.8 Hz, 1H), 3.72 (s, 3H), 3.59–3.53 (m, 1H), 3.06 (dd, J = 14.0, 3.6 Hz, 1H), 2.80 (t, J = 13.2 Hz, 1H), 2.32 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 159.8, 143.5, 139.9, 135.9, 129.4, 129.3 (2×), 129.1 (2×), 127.9 (2×), 113.5 (2×), 112.3, 79.4, 71.9, 65.6, 55.1, 31.3, 21.4. Single-crystal X-ray diagram: crystal of compound 5n was grown by slow diffusion of EtOAc into a solution of compound 5n in THF to yield colorless prisms. The compound crystallizes in the monoclinic crystal system, space group P21/n, a = 12.4097(3) Å, b = 9.8815(2) Å, c = 14.6434(3) Å, V = 1774.09(7) Å3, Z = 4, dcalcd = 1.342 g cm−3, F(000) = 760.0, 2θ range 4.008–54.124°, R indices (all data) R1 = 0.0395, wR2 = 0.0890.2-Biphenyl-4-yl-5-methylene-3-(toluene-4-sulfonyl)tetrahydropyran (5q)
Yield = 80% (323 mg); white solid; mp = 114–116 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O3S 405.1524, found 405.1533; 1H NMR (400 MHz, CDCl3): δ 7.49–7.42 (m, 4H), 7.38–7.34 (m, 1H), 7.30–7.25 (m, 4H), 7.19 (d, J = 8.0 Hz, 2H), 6.96 (d, J = 8.0 Hz, 2H), 5.07 (s, 1H), 5.03 (d, J = 0.8 Hz, 1H), 4.71 (d, J = 9.2 Hz, 1H), 4.30 (dd, J = 12.8, 1.2 Hz, 1H), 4.16 (d, J = 12.4 Hz, 1H), 3.69–3.62 (m, 1H), 3.16 (dd, J = 14.0, 4.0 Hz, 1H), 2.87 (t, J = 13.2 Hz, 1H), 2.21 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 143.6, 141.6, 140.5, 139.8, 136.1, 136.0, 129.1 (2×), 128.7 (2×), 128.6 (2×), 127.9 (2×), 127.4, 127.0 (2×), 126.9 (2×), 112.5, 79.8, 72.0, 65.5, 31.1, 21.4.5-Methylene-2-naphthalen-2-yl-3-(toluene-4-sulfonyl)tetrahydropyran (5r)
Yield = 82% (310 mg); white solid; mp = 99–101 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H23O3S 379.1368, found 379.1377; 1H NMR (400 MHz, CDCl3): δ 7.71–7.64 (m, 2H), 7.57 (s, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.46–7.42 (m, 2H), 7.16 (dd, J = 8.4, 1.6 Hz, 1H), 7.10 (d, J = 8.4 Hz, 2H), 6.58 (d, J = 8.4 Hz, 2H), 5.09 (s, 1H), 5.05 (d, J = 1.2 Hz, 1H), 4.81 (d, J = 9.6 Hz, 1H), 4.32 (dd, J = 12.8, 1.2 Hz, 1H), 4.19 (d, J = 12.8 Hz, 1H), 3.74–3.67 (m, 1H), 3.21 (dd, J = 14.0, 4.0 Hz, 1H), 2.90 (t, J = 13.2 Hz, 1H), 2.00 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 143.6, 139.9, 135.6, 134.3, 133.5, 132.9, 128.7 (2×), 128.2, 128.1, 128.0, 127.6 (2×), 127.3, 126.3, 126.0, 125.1, 112.5, 80.2, 72.1, 65.5, 30.9, 21.1.2-(3,4-Dimethoxyphenyl)-5-methylene-3-(toluene-4-sulfonyl)tetrahydropyran (5s)
Yield = 87% (338 mg); white solid; mp = 120–122 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H25O5S 389.1423, found 389.1431; 1H NMR (400 MHz, CDCl3): δ 7.22 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 8.0 Hz, 2H), 6.72 (dd, J = 8.4, 2.0 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.45 (d, J = 2.0 Hz, 1H), 5.04 (s, 1H), 5.00 (s, 1H), 4.58 (d, J = 9.6 Hz, 1H), 4.26 (dd, J = 12.8, 1.2 Hz, 1H), 4.11 (d, J = 12.4 Hz, 1H), 3.80 (s, 3H), 3.66 (s, 3H), 3.59–3.52 (m, 1H), 3.14 (ddd, J = 14.0, 4.4, 1.2 Hz, 1H), 2.82 (t, J = 13.2 Hz, 1H), 2.32 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 149.3, 148.6, 143.7, 139.9, 136.1, 129.5, 128.9 (2×), 127.8 (2×), 121.2, 112.4, 110.5, 110.3, 80.0, 72.0, 65.6, 55.8, 55.4, 31.1, 21.4.5-[5-Methylene-3-(toluene-4-sulfonyl)tetrahydropyran-2-yl]benzo[1,3]dioxole (5t)
Yield = 86% (320 mg); white solid; mp = 191–193 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H21O5S 373.1110, found 373.1115; 1H NMR (400 MHz, CDCl3): δ 7.30 (d, J = 8.4 Hz, 2H), 7.07 (d, J = 8.4 Hz, 2H), 6.72 (dd, J = 8.0, 1.6 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.42 (d, J = 1.6 Hz, 1H), 5.85 (d, J = 1.2 Hz, 1H), 5.80 (d, J = 1.2 Hz, 1H), 5.02 (s, 1H), 5.00 (d, J = 1.2 Hz, 1H), 4.56 (d, J = 9.6 Hz, 1H), 4.25 (dd, J = 12.8, 1.6 Hz, 1H), 4.10 (d, J = 12.4 Hz, 1H), 3.54–3.47 (m, 1H), 3.09 (ddd, J = 14.0, 4.4, 1.2 Hz, 1H), 2.80 (t, J = 13.6 Hz, 1H), 2.36 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 147.8, 147.4, 143.7, 139.8, 136.0, 131.1, 129.1 (2×), 128.0 (2×), 122.6, 112.5, 107.9, 107.8, 101.0, 79.8, 72.0, 65.6, 21.1, 21.5.2-(3,4-Dichlorophenyl)-5-methyl-3-(toluene-4-sulfonyl)-3,4-dihydro-2H-pyran (5u-1)
Yield = 80% (317 mg); colorless oil; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H19Cl2O3S 397.0432, found 397.0441; 1H NMR (400 MHz, CDCl3): δ 7.54 (d, J = 8.0 Hz, 2H), 7.42 (dd, J = 2.0, 0.4 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.28 (dd, J = 8.4, 0.8 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 6.25 (q, J = 1.2 Hz, 1H), 5.40 (d, J = 3.6 Hz, 1H), 3.83–3.78 (m, 1H), 2.63–2.57 (m, 1H), 2.42 (s, 3H), 2.28 (dd, J = 17.6, 6.4 Hz, 1H), 1.60 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 144.9, 138.1, 137.5, 136.0, 132.3, 132.2, 130.0, 129.5 (2×), 128.9, 128.5 (2×), 126.4, 106.6, 72.3, 61.9, 24.9, 21.6, 17.8.5-Methylene-3-(toluene-4-sulfonyl)-2-(3,4,5-trimethoxyphenyl)tetrahydropyran (5v)
Yield = 90% (376 mg); white solid; mp = 141–143 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H27O6S 419.1528, found 419.1533; 1H NMR (400 MHz, CDCl3): δ 7.23 (d, J = 8.4 Hz, 2H), 7.00 (d, J = 8.0 Hz, 2H), 6.25 (s, 2H), 5.05 (s, 1H), 5.00 (s, 1H), 4.54 (d, J = 9.6 Hz, 1H), 4.25 (dd, J = 12.8, 0.4 Hz, 1H), 4.10 (d, J = 12.8 Hz, 1H), 3.72 (s, 3H), 3.67 (s, 6H), 3.60–3.53 (m, 1H), 3.18 (dd, J = 14.0, 3.6 Hz, 1H), 2.82 (t, J = 13.6 Hz, 1H), 2.29 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 152.8 (2×), 143.8, 139.6, 138.0, 136.1, 132.2, 128.8 (2×), 127.7 (2×), 112.5, 105.1 (2×), 80.4, 72.0, 65.2, 60.5, 55.7 (2×), 30.7, 21.3.5-Methylene-2-thiophen-2-yl-3-(toluene-4-sulfonyl)tetrahydropyran (5w)
Yield = 84% (281 mg); white solid; mp = 137–139 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H19O3S2 335.0776, found 335.0782; 1H NMR (400 MHz, CDCl3): δ 7.41 (d, J = 8.0 Hz, 2H), 7.12–7.10 (m, 3H), 6.97 (dd, J = 3.6, 1.2 Hz, 1H), 6.78 (dd, J = 5.2, 3.6 Hz, 1H), 5.07 (d, J = 8.8 Hz, 1H), 4.99 (s, 1H), 4.97 (s, 1H), 4.27 (d, J = 12.8 Hz, 1H), 4.10 (d, J = 12.8 Hz, 1H), 3.58–3.52 (m, 1H), 3.02 (dd, J = 14.0, 4.8 Hz, 1H), 2.81 (dt, J = 13.6, 2.0 Hz, 1H), 2.36 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 144.0, 140.5, 139.1, 135.7, 129.3 (2×), 128.2 (2×), 127.5, 126.6, 126.2, 112.5, 74.3, 71.3, 66.5, 31.1, 21.5.A representative synthetic procedure of skeleton 7 is as follows: DABCO (1,4-diazabicyclo[2.2.2]octane, 23 mg, 20 mol%) was added to a solution of 6 (1.0 mmol) in CCl4 (20 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 10 min. 2-Chloromethyl-2-propen-1-ol (3, 106 mg, 1.0 mmol) was added to the reaction mixture at 25 °C. The reaction mixture was stirred at reflux (77 °C) for 20 h. The reaction mixture was cooled to 25 °C and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 15/1 to 8/1) afforded 7.
3-Methylene-6-phenyl-5-(toluene-4-sulfonyl)-3,4-dihydro-2H-pyran (7a)
Yield = 76% (248 mg); white solid; mp = 158–159 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H19O3S 327.1055, found 327.1063; 1H NMR (400 MHz, CDCl3): δ 7.43 (d, J = 8.4 Hz, 2H), 7.42–7.36 (m, 1H), 7.33–7.26 (m, 4H), 7.15 (d, J = 8.0 Hz, 2H), 5.22 (d, J = 1.2 Hz, 1H), 5.14 (d, J = 0.8 Hz, 1H), 4.49 (s, 2H), 3.30 (s, 2H), 2.37 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 162.2, 143.2, 138.8, 135.9, 133.7, 129.6, 129.4 (2×), 129.1 (2×), 127.4 (2×), 127.3 (2×), 113.7, 113.5, 71.1, 30.2, 21.4.6-(4-Fluorophenyl)-3-methylene-5-(toluene-4-sulfonyl)-3,4-dihydro-2H-pyran (7b)
Yield = 74% (255 mg); white solid; mp = 162–164 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H18FO3S 345.0961, found 345.0970; 1H NMR (400 MHz, CDCl3): δ 7.43 (d, J = 8.4 Hz, 2H), 7.30–7.25 (m, 2H), 7.17 (d, J = 8.0 Hz, 2H), 7.01–6.96 (m, 2H), 5.20 (d, J = 0.8 Hz, 1H), 5.13 (d, J = 0.8 Hz, 1H), 4.47 (s, 2H), 3.26 (br t, J = 1.6 Hz, 2H), 2.36 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 163.4 (d, J = 247.9 Hz), 161.1, 143.4, 138.6, 135.6, 131.5 (d, J = 8.4 Hz, 2×), 129.7 (d, J = 3.0 Hz), 129.2 (2×), 127.2 (2×), 114.4 (d, J = 22.0 Hz, 2×), 113.79, 113.77, 71.1, 30.2, 21.3.3-Methylene-5-(toluene-4-sulfonyl)-6-(4-trifluoromethylphenyl)-3,4-dihydro-2H-pyran (7c)
Yield = 70% (276 mg); white solid; mp = 153–155 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H18F3O3S 395.0929, found 395.0932; 1H NMR (400 MHz, CDCl3): δ 7.57 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 5.24 (d, J = 0.8 Hz, 1H), 5.18 (d, J = 0.8 Hz, 1H), 4.51 (s, 2H), 3.29 (s, 2H), 2.39 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 160.4, 143.8, 138.4, 137.4 (d, J = 1.5 Hz), 135.4, 131.5 (q, J = 32.6 Hz), 129.9 (2×), 129.4 (2×), 127.4 (2×), 124.5 (q, J = 3.8 Hz, 2×), 123.8 (q, J = 271.3 Hz), 114.6, 114.2, 71.4, 30.1, 21.5.3-Methylene-6-(4-nitrophenyl)-5-(toluene-4-sulfonyl)-3,4-dihydro-2H-pyran (7d)
Yield = 68% (252 mg); white solid; mp = 138–140 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H18NO5S 372.0906, found 372.0902; 1H NMR (400 MHz, CDCl3): δ 8.19 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 8.8 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H), 5.24 (s, 1H), 5.18 (s, 1H), 4.51 (s, 2H), 3.23 (s, 2H), 2.41 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 159.4, 148.4, 144.1, 140.3, 138.1, 134.9, 130.5 (2×), 129.6 (2×), 127.4 (2×), 122.7 (2×), 114.7, 114.5, 71.4, 30.1, 21.5.6-(3,4-Dichlorophenyl)-3-methylene-5-(toluene-4-sulfonyl)-3,4-dihydro-2H-pyran (7e)
Yield = 70% (276 mg); white solid; mp = 174–176 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H17Cl2O3S 395.0276, found 395.0281; 1H NMR (400 MHz, CDCl3): δ 7.45 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.0 Hz, 1H), 7.24 (d, J = 2.0 Hz, 1H), 7.1–7.18 (m, 3H), 5.21 (s, 1H), 5.14 (s, 1H), 4.47 (s, 2H), 3.26 (s, 2H), 2.38 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 159.3, 143.8, 138.2, 135.2, 133.8, 133.5, 131.7, 131.0, 129.5, 129.3 (2×), 129.0, 127.2 (2×), 114.7, 114.2, 71.2, 30.0, 21.4.Acetic acid 2-[3-oxo-2-(toluene-4-sulfonyl)-3-(4-trifluoromethylphenyl)propyl]allyl ester (8)
50% HOAc (10 mL) was added to a solution of 7c (394 mg, 1.0 mmol) in CCl4 (20 mL) at 25 °C. The reaction mixture was stirred at reflux (77 °C) for 20 h. The reaction mixture was cooled to 25 °C and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 10/1 to 6/1) afforded 8. Yield = 80% (363 mg); colorless oil; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H22F3O5S 455.1140, found 455.1148; 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 8.0 Hz, 2H), 7.71 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 5.36 (dd, J = 10.4, 3.6 Hz, 1H), 5.00 (s, 1H), 4.85 (s, 1H), 4.46 (d, J = 13.2 Hz, 1H), 4.40 (d, J = 13.2 Hz, 1H), 2.92–2.81 (s, 2H), 2.1 (s, 3H), 1.96 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 190.9, 170.4, 145.8, 139.6, 138.4, 134.9 (q, J = 32.6 Hz), 133.0, 129.68 (2×), 129.64 (2×), 129.3 (2×), 126.0 (q, J = 265.3 Hz), 125.7 (q, J = 3.8 Hz, 2×), 117.2, 68.5, 66.2, 31.3, 21.5, 20.6.2-Biphenyl-4-yl-5-methylene-3-(toluene-4-sulfonyl)tetrahydropyran (9)
DABCO (1,4-diazabicyclo[2.2.2]octane, 23 mg, 20 mol%) was added to a solution of 2q-1 (334 mg, 1.0 mmol) in CCl4 (20 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 10 min. 2-Chloromethyl-2-propen-1-ol (3, 106 mg, 1.0 mmol) was added to the reaction mixture at 25 °C. The reaction mixture was stirred at reflux (77 °C) for 20 h. The reaction mixture was cooled to 25 °C and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 10/1 to 6/1) afforded 9. Yield = 73% (295 mg); White solid; mp = 167–169 °C (recrystallized from hexanes and EtOAc); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25O3S 405.1524, found 405.1530; 1H NMR (400 MHz, CDCl3): δ 7.55–7.52 (m, 2H), 7.46–7.43 (m, 2H), 7.37–7.27 (m, 7H), 6.99 (d, J = 8.0 Hz, 2H), 5.07 (s, 2H), 5.06 (d, J = 2.8 Hz, 1H), 4.56 (d, J = 12.8 Hz, 1H), 4.23 (d, J = 12.8 Hz, 1H), 3.84–3.81 (m, 1H), 3.49 (dd, J = 14.8, 2.0 Hz, 1H), 2.85 (dd, J = 14.8, 2.0 Hz, 1H), 2.25 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 143.4, 140.7, 140.2, 137.7, 137.6, 136.3, 129.1 (2×), 128.7 (2×), 128.2 (2×), 127.3, 126.9 (2×), 126.6 (2×), 126.5 (2×), 113.0, 77.3, 72.3, 64.8, 31.9, 21.4.5-Methyl-2-phenyltetrahydro-pyran (10) and 2-methyl-5-phenylpent-4-en-1-ol (11)
Pd/C (10%, 30 mg) was added to a solution of 5a (164 mg, 0.5 mmol) in EtOAc (20 mL) at 25 °C. Hydrogen gas was installed to the reaction mixture at 25 °C. The reaction mixture was stirred at 25 °C for 20 h, filtered, and the solvent was concentrated. Without further purification, fleshly prepared sodium amalgam (6–8% Na/Hg, 300 mg) was added to a solution of the resulting crude product in MeOH (20 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 10 h, and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 15/1 to 8/1) afforded 10 and 11. For compound 10, ratio, cis/trans = 1:1; yield = 30% (26 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H17O 177.1280, found 177.1286; 1H NMR (400 MHz, CDCl3): δ 7.41–7.13 (m, 5H), 4.32–4.22 (m, 1H), 4.05–3.98 (m, 1H), 3.32–3.20 (m, 1H), 2.05–1.31 (m, 5H), 1.08 (d, J = 6.8 Hz, 3/2H), 0.93 (d, J = 6.8 Hz, 3/2H). Compound 10 was known and their related analytical data (e.g., HRMS and 1H NMR) were identical with those in the reference 11b. For compound 11, yield = 38% (33 mg); colorless liquid; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C12H17O 177.1280, found 177.1288; 1H NMR (400 MHz, CDCl3): δ 7.37–7.28 (m, 4H), 7.23–7.18 (m, 1H), 6.42 (d, J = 15.6 Hz, 1H), 6.23 (dt, J = 15.6, 7.2 Hz, 1H), 3.57 (dd, J = 10.4, 6.0 Hz, 1H), 3.51 (dd, J = 10.4, 6.0 Hz, 1H), 2.38–2.31 (m, 1H), 2.15–2.07 (m, 1H), 1.87–1.79 (m, 1H), 1.47 (br s, 1H), 0.98 (d, J = 6.4 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 137.6, 131.4, 128.7, 128.5 (2×), 127.0, 126.0 (2×), 67.9, 36.9, 36.1, 14.5.
4-(Hydroxymethyl)-1-phenyl-2-tosylpent-4-en-1-one (12)
50% H2SO4 (10 mL) was added to a solution of 7a (326 mg, 1.0 mmol) in CCl4 (20 mL) at 25 °C. The reaction mixture was stirred at reflux (77 °C) for 20 h. The reaction mixture was cooled to 25 °C and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine, dried, filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 8/1 to 2/1) afforded 12. Yield = 66% (227 mg); colorless oil; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H21O4S 345.1161, found 345.1167; 1H NMR (400 MHz, CDCl3): δ 7.97–7.94 (m, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.61–7.57 (m, 1H), 7.48–7.44 (m, 2H), 7.29 (d, J = 8.0 Hz, 2H), 5.46 (dd, J = 10.4, 4.0 Hz, 1H), 5.97 (s, 1H), 4.78 (d, J = 0.8 Hz, 1H), 4.02 (d, J = 13.6 Hz, 1H), 3.98 (d, J = 13.6 Hz, 1H), 2.93–2.83 (s, 2H), 2.43 (s, 3H), 1.66 (br s, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 192.1, 145.5, 143.2, 137.1, 133.9, 133.2, 129.8 (2×), 129.6 (2×), 129.1 (2×), 128.7 (2×), 114.3, 68.6, 66.2, 31.5, 21.7.Author contributions
Meng-Yang Chang: writing – review & editing, supervision. Kuan-Ting Chen: writing – original draft, conceptualization, methodology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Ministry of Science and Technology of the Republic of China (Taiwan) for its financial support (NSTC 112-2113-M-037-016-MY3).Notes and references
- Review articles on synthesis of THPs, see: (a) N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323–3335 RSC; (b) A. B. III Smith, R. J. Fox and T. M. Razler, Acc. Chem. Res., 2008, 41, 675–687 CrossRef; (c) I. M. Pastor and M. Yus, Curr. Org. Chem., 2007, 11, 925–957 CrossRef CAS.
- Examples on synthesis of the biological active THPs, see: (a) S. Umamatheswari, B. Balaji, M. Ramanathan and S. Kabilan, Eur. J. Med. Chem., 2011, 46, 1415–1424 CrossRef CAS PubMed; (b) P. Parthiban, G. Aridoss, P. Rathika, V. Ramkumar and S. Kabilan, Bioorg. Med. Chem. Lett., 2009, 19, 2981–2985 CrossRef CAS PubMed; (c) H. I. El-Subbagh, S. M. Abu-Zaid, M. A. Mahran, F. A. Badria and A. M. Alofaid, J. Med. Chem., 2000, 43, 2915–2921 CrossRef CAS.
- (a) A. P. Jadhav, J.-A. Oh, I.-S. Hwang, H. Yan and C. E. Song, Org. Lett., 2018, 20, 5319–5322 CrossRef CAS; (b) H. Murayama, K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 2039–2041 CrossRef CAS PubMed; (c) S. E. Ammann, G. T. Rice and M. C. White, J. Am. Chem. Soc., 2014, 136, 10834–10837 CrossRef CAS; (d) H. Kim and C. Lee, Org. Lett., 2011, 13, 2050–2053 CrossRef CAS PubMed; (e) A. Aponick and B. Biannic, Org. Lett., 2011, 13, 1330–1333 CrossRef CAS; (f) A. Guérinot, A. Serra-Muns, C. Gnamm, C. Bensoussan, S. Reymond and J. Cossy, Org. Lett., 2010, 12, 1808–1811 CrossRef.
- (a) L. Liu, P. S. J. Kaib, A. Tap and B. List, J. Am. Chem. Soc., 2016, 138, 10822–10825 CrossRef CAS PubMed; (b) M. Barbasiewicz, A. Brud and M. Mąkosza, Synthesis, 2007, 1209–1213 CAS; (c) J. C. R. Brioche, K. M. Goodenough, D. J. Whatrup and J. P. A. Harrity, Org. Lett., 2007, 9, 3941–3943 CrossRef CAS PubMed.
- M.-Y. Chang, Y.-S. Wu and Y.-T. Hsiao, Synthesis, 2018, 50, 4651–4658 CrossRef CAS.
- Examples on synthesis of methylene THPs, see: (a) C.-Y. Ho and L. He, J. Org. Chem., 2014, 79, 11873–11884 CrossRef CAS; (b) K. Okuma, O. Sakai, T. Hayamo, K. Shioji and H. Matsuyama, Bull. Chem. Soc. Jpn., 2005, 78, 2209–2213 CrossRef CAS; (c) J. van der Louw, G. J. J. Out, J. L. van der Baan, F. J. J. de Kanter, F. Bickelhaupt and G. W. Klumpp, Tetrahedron Lett., 1989, 30, 4863–4866 CrossRef CAS; (d) J. van der Louw, J. L. van der Baan, G. J. J. Out, F. J. J. de Kanter, F. Bickelhaupt and G. W. Klumpp, Tetrahedron, 1992, 48, 9901–9916 CrossRef CAS; (e) Y. Huang and X. Lu, Tetrahedron Lett., 1987, 28, 6219–6220 CrossRef CAS.
- Review articles on Baylis–Hillman reactions, see: (a) D. Basavaiah and R. T. Naganaboina, New J. Chem., 2018, 42, 14036–14066 RSC; (b) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659–6690 CrossRef CAS PubMed; (c) D. Basavaiah and G. Veeraraghavaiah, Chem. Soc. Rev., 2012, 41, 68–78 RSC; (d) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811–892 CrossRef CAS.
- (a) J. Ammera, M. Baidyaa, S. Kobayashia and H. Mayr, J. Phys. Org. Chem., 2010, 23, 1029–1035 CrossRef; (b) M. Baidya and H. Mayr, Chem. Commun., 2008, 1792–1794 RSC.
- CCDC 2279054 (5m) and 2279055 (5n) contain the supplementary crystallographic data for this paper.†.
- (a) N.-C. Hsueh, M.-C. Tsai, M.-Y. Chang and H.-Y. Chen, J. Org. Chem., 2019, 84, 15915–15925 CrossRef CAS; (b) M.-Y. Chang and Y.-S. Wu, J. Org. Chem., 2019, 84, 3638–3646 CrossRef CAS PubMed; (c) N.-C. Hsueh, H.-Y. Chen and M.-Y. Chang, J. Org. Chem., 2017, 82, 13324–13332 CrossRef CAS PubMed; (d) M.-Y. Chang, H.-Y. Chen and Y.-L. Tsai, J. Org. Chem., 2019, 84, 326–337 CrossRef CAS.
- Recent example on photoinduced olefin isomerization, see: Q. Zhou, X. Hong, H.-Z. Cui, Y. Sun, B. Zhan, A. Reheman and X.-F. Hou, Tetrahedron Lett., 2020, 152396 CrossRef CAS.
- Synthesis of rose oxide, see: (a) H. Watkins, O. C. Liu and J. A. Krivda, US Pat., US 5219836, 1993 Search PubMed; (b) L. Coulombel, M. Weïwer and E. Duñach, Eur. J. Org Chem., 2009, 5788–5795 CrossRef CAS; (c) V. H. Rawal, S. P. Singh, C. Dufour and C. Michoud, J. Org. Chem., 1991, 56, 5245–5247 CrossRef CAS; (d) V. H. Rawal, S. P. Singh, C. Dufour and C. Michoud, J. Org. Chem., 1993, 58, 7718–7727 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scanned photocopies of NMR spectral data for all compounds and X-ray analysis data of 5m and 5n were supported. CCDC 2279054–2279055. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra06370d |
| This journal is © The Royal Society of Chemistry 2023 |