
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToughening of epoxy thermosets by self-assembled nanostructures of amphiphilic comb-like random copolymers
Li-Cheng Jheng *a,
Ting-Yu Changb,
Chin-Ting Fana,
Tsung-Han Hsiehb,
Feng-Ming Hsiehc and
Wan-Ju Huanga
*a,
Ting-Yu Changb,
Chin-Ting Fana,
Tsung-Han Hsiehb,
Feng-Ming Hsiehc and
Wan-Ju Huanga
aDepartment of Chemical and Materials Engineering, National Kaohsiung University of Science and Technology, Kaohsiung, Taiwan, ROC. E-mail: lcjheng@nkust.edu.tw; Fax: +886 7 3830674; Tel: +886 7 3814526 ext.15148
bDepartment of Mold and Die Engineering, National Kaohsiung University of Science and Technology, Kaohsiung, Taiwan, ROC
cMaterial and Chemical Research Laboratories, Industrial Technology Research Institute, Hsinchu, Taiwan, ROC
First published on 15th November 2023
Abstract
Amphiphilic comb-like random copolymers synthesized from poly(ethylene glycol) methyl ether methacrylate (PEGMMA) and stearyl methacrylate (SMA) with PEGMMA contents ranging between 30 wt% and 25 wt% were demonstrated to self-assemble into various well-defined nanostructures, including spherical micelles, wormlike micelles, and vesicle-like nanodomains, in anhydride-cured epoxy thermosets. In addition, the polymer blends of the comb-like random copolymer and poly(stearyl methacrylate) were prepared and incorporated into epoxy thermosets to form irregularly shaped nanodomains. Our research findings indicate that both the comb-like random copolymers and polymer blends are suitable as toughening modifiers for epoxy. When added at a concentration of 5 wt%, both types of modifiers lead to substantial improvements in the tensile toughness (>289%) and fracture toughness of epoxy thermosets, with minor reductions in their elastic modulus (<16%) and glass transition temperature (<6.1 °C). The fracture toughness evaluated in terms of the critical stress intensity factor (KIC) and the strain energy release rate (GIC) increased by more than 67% and 131% for the modified epoxy thermosets containing comb-like random copolymers.
1 Introduction
Epoxy thermosets have numerous advantages, including superior thermal and chemical stability, good corrosion resistance, and ease of processing, which make them widely used in various applications. However, their densely crosslinked networks often result in intrinsic brittleness. To address this issue and improve the poor fracture toughness of epoxy thermosets, various modifiers that can generate microdomains or nanodomains in the epoxy matrix for restricting the crack growth have been developed. They include liquid rubbers,1–3 core–shell rubbers,4,5 inorganic nanoparticles,6,7 amphiphilic block copolymers,8–12 and so on. Among these modifiers, amphiphilic block copolymers represent a relatively recent class of modifiers known for “toughening by nanostructures”. They can significantly enhance the fracture toughness of epoxy without sacrificing properties like the glass transition temperature and elastic modulus at relatively low modifier concentrations (≤5 wt%).13–15 In comparison to other modifiers, amphiphilic block copolymers are considered highly competitive for optimizing epoxy thermoset properties to meet the demands of various applications.The development of nanostructured morphologies in epoxy thermosets modified with block copolymers can occur through either a self-assembly approach before the curing reaction8,9 or a reaction-induced microphase separation (RIMPS) process during the curing reaction16,17 or a combination of self-assembly and RIMPS mechanisms.18 In most cases, well-defined micellar nanostructures (e.g., spherical micelles, wormlike micelles, vesicles, or mixtures of them) in epoxies are obtained from the self-assembly of an amphiphilic block copolymer that is composed of at least one block miscible with epoxy (epoxy-philic) and at least one block immiscible with epoxy (epoxy-phobic).13 The miscibility between a polymer and an epoxy precursor depends on the difference between their solubility parameters. Certain polymers, characterized by solubility parameters in proximity to those of epoxy precursors (e.g., DEGBA) can be regarded as epoxy-philic polymers. Examples of such polymers include poly(ethylene oxide) (PEO), poly(methylmethacrylate) (PMMA), and polycaprolactone (PCL).19 Ever since Bates and Hillmyer introduced the pioneering concept of self-assembled nanophases derived from PEO-b-PEP within epoxy thermosets in 1997, a diverse range of PEO-based block copolymers (e.g., PEO-b-PPO, PEO-b-PDMS, PEO-b-PHO, PEO-b-PBO, PEO-b-PPO-b-PEO) with the ability to generate nanostructured morphologies in epoxy thermosets through self-assembly has been subject to extensive investigation.8,20–25 Among them, poly(ethylene oxide)-b-poly(butylene oxide) (PEO-b-PBO) has been successfully commercialized and is offered by Dow Chemical Company under the trade name of Fortegra™ 100.23,26 Another commercially available modifier based on amphiphilic block copolymers for toughening epoxy, is a PMMA-based triblock copolymer, poly(methyl methacrylate)-b-poly(butyl acrylate)-b-poly(methyl methacrylate) (PMMA-b-PBA-b-PMMA) manufactured by Kuraray and Arkema.27,28 Aside from PEO-based and PMMA-based block copolymers, Zheng and colleagues have synthesized several PCL-based triblock copolymers capable of forming nanophases within epoxy thermosets through self-assembly. These include PCL-b-PDMS-b-PCL,29 PCL-b-PEEE-b-PCL,30 and PCL-b-PE-b-PCL.31 Furthermore, a distinct class of amphiphilic modifiers based on glycidyl methacrylate (PGMA)-derived block copolymers has been introduced for the enhancement of epoxy thermosets.32,33 For example, Zhou recently incorporated amphiphilic triblock copolymers poly(glycidyl methacrylate)-b-poly(dimethylsiloxane)-b-poly(glycidyl methacrylate) (PGMA-b-PDMS-b-PGMA) consisting of reactive epoxy-miscible PGMA blocks into epoxy to obtain nanostructured epoxy thermosets with controlled nanodomain morphology.34 Beyond the aforementioned amphiphilic modifiers, we believe there exist undiscovered amphiphilic copolymers with the potential to enhance the toughness of epoxy resins, worthy of further exploration.
In contrast to amphiphilic block copolymers, amphiphilic random copolymers are considered less likely to rearrange into highly ordered nanostructures in bulk or dilute solutions because of their ill-defined properties and broader dispersity.35 Despite that, a series of methacrylate-based amphiphilic random copolymers bearing hydrophilic poly(ethylene glycol) (PEG) and hydrophobic alkyl pendants (e.g., PEGMA/RMA and PEGMA/DMA random copolymers) can precisely self-assemble into uniform micelles in water, demonstrated by Terashima and coworkers.36–40 Recently, they also reported that PEGA/ODA random copolymers can form well-defined vesicles, thermoresponsive micelles, and reverse micelles in water or hexane.38 On the other hand, Iborra et al. suggested an amphiphilic random copolymer synthesized from poly(ethylene glycol) methyl ether methacrylate (PEGMMA) and lauryl methacrylate (LMA) to be used as the macrosurfactant for dispersing carbon nanotubes in water.41 Additionally, Fang et al. polymerized comb-like random copolymers from poly(propylene glycol) acrylate (PPGA) and 2-(dimethylamino)ethyl methacrylate (DMA) as polymeric dispersants for dispersing SiO2 particles in organic media.42 These examples indicate that amphiphilic random copolymers are possible to be used as surfactants, emulsion agents, or polymeric dispersants for various applications, especially for methacrylate-based or acrylate-based random copolymers with a comb-like architecture. The dispersion effectiveness or assembly behavior of amphiphilic random copolymers may not be as good as that of amphiphilic block copolymers. Nonetheless, the synthesis of random copolymers can be accomplished via a one-step copolymerization process, which is comparably simpler and less time-consuming than the synthesis of block copolymers that involves sequential controlled polymerizations or coupling reactions.
To date, there are few efforts made to study the modifiers based on amphiphilic random copolymers for toughening epoxy thermosets by ordered nanostructures. In the present work, we synthesized a series of amphiphilic comb-like random copolymers (CRCPs) from PEGMMA and stearyl methacrylate (SMA) with varying PEGMMA contents using thermally-initiated free radical polymerization. By varying the copolymer composition, these CRCPs consisting of epoxy-miscible PEGMMA and epoxy-immiscible SMA pendants achieved the self-assembly into various micellar nanostructures in epoxy thermosets. Previous research conducted by Thio has suggested that it's feasible to adjust the structure and dimensions of nanodomains within epoxy thermosets by manipulating the epoxy-hydrophilic content of the modifier through either the binary blending of amphiphilic block copolymers with distinct compositions or the polymer blending of an amphiphilic block copolymer with a homopolymer of its epoxy-immiscible block in different blend ratios.21,43 Accordingly, we further prepared a polymer blend of P(PEGMMA-co-SMA) and a homopolymer synthesized from SMA as an alternative modifier for toughening epoxy. The morphology of secondary nanophases rearranged by CRCP or polymer blend modifiers in anhydride-cured epoxy thermosets was identified using a transmission electron microscope (TEM). The tensile toughness, impact strengths, and plane-strain fracture toughness of the modified epoxy thermosets were evaluated through tensile, impact, and single-edge notched bending (SENB) tests, respectively. To demonstrate the effectiveness of CRCP-based modifiers in enhancing the toughness of epoxy thermosets, a comparative analysis was performed with a commercially available block copolymer (BCP) modifier, Fortegra™100.
2 Experimental
2.1 Materials
Poly(ethylene glycol) methyl ether methacrylate (PEGMMA) (average Mn 2000 g mol−1, 50 wt% in H2O) and stearyl methacrylate (SMA) were purchased from Sigma-Aldrich. Before use, the PEGMMA monomer needs to be isolated by removing the water with a rotary evaporator. The MEHQ inhibitor within the SMA monomer was filtered out by passing the liquid monomer through a basic alumina column. The solvents and reagents for polymer synthesis include toluene (Sigma-Aldrich), methanol (Macron Fine Chemicals), 2,2′-azobis(2-methylpropionitrile) (AIBN) (Otsuka chemical), and 1-dodecanethiol (Aldrich) were used as received without further purifications.The materials used for preparing epoxy thermosets include an epoxy monomer diglycidyl ether of bisphenol A (DGEBA) (Araldite LY556), a curing agent methyl tetrahydrophthalic anhydride (MTHPA) (Aradur 917), and a curing accelerator 1-methyl imidazole (DY070). All of them were industrial-grade and produced by Huntsman. The BCP modifier is the commercially available product Fortegra 100 manufactured by Dow Chemical. Fortegra 100 is a diblock copolymer poly(ethylene oxide-b-butylene oxide) (PEO-b-PBO) with appropriately designed molecule weight and composition.23
2.2 Preparation of comb-like random copolymer (CRCP) and polymer blend modifiers
We synthesized a series of CRCP from PEGMMA and SMA with three different PEGMMA contents of 30 wt%, 27 wt%, and 25 wt% as the following procedures. In a three-necked reactor with magnetically stirring under a nitrogen atmosphere, 30 g of SMA and a predetermined amount of PEGMMA were dissolved in 40 g of toluene at room temperature. To initiate the free radical polymerization and control the polymer molecular weight, a thermal initiator AIBN and a chain transfer agent 1-dodecanethiol were added into the solution in a molar ratio of SMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AIBN:1-dodecanethiol = 40:1:3. Then, the reactor was heated to 70 °C in an oil bath to undergo the polymerization reaction for 18 h. After the polymerization, the solvent of the solution was removed by a rotary evaporator to collect the resulting polymer. For purification, we washed the resulting polymer with methanol several times to remove the unreacted monomers. Subsequently, it was dried in a vacuum oven at 90 °C overnight. Finally, the polymer product P(PEGMMA-co-SMA) was obtained and stored in a vial in an ambient condition. The CRCP modifiers were designated as CRCP-30, CRCP-27, and CRCP-25, reflecting their respective PEGMMA content in the composition.
:AIBN:1-dodecanethiol = 40:1:3. Then, the reactor was heated to 70 °C in an oil bath to undergo the polymerization reaction for 18 h. After the polymerization, the solvent of the solution was removed by a rotary evaporator to collect the resulting polymer. For purification, we washed the resulting polymer with methanol several times to remove the unreacted monomers. Subsequently, it was dried in a vacuum oven at 90 °C overnight. Finally, the polymer product P(PEGMMA-co-SMA) was obtained and stored in a vial in an ambient condition. The CRCP modifiers were designated as CRCP-30, CRCP-27, and CRCP-25, reflecting their respective PEGMMA content in the composition.
A polymer blend modifier was prepared by mixing CRCP-30 with a homopolymer of poly(stearyl methacrylate) (PSMA) in a specific ratio using mechanical stirring at 55 °C for 30 min. The PSMA homopolymer was synthesized from SMA without adding 1-dodecanethiol, following similar polymerization and purification procedures for the synthesis of P(PEGMMA-co-SMA). In this work, two polymer blend modifiers with the PEGMMA contents of 27 wt% and 25 wt% were prepared, denoted Blend-27 and Blend-25, respectively.
2.3 Preparation of modified epoxy thermosets
The chemical structures of the ingredients of modified epoxy thermosets are presented in Scheme 1. The compositions of the CRCP, polymer blend, and BCP modifiers adopted in this work are listed in Table 1.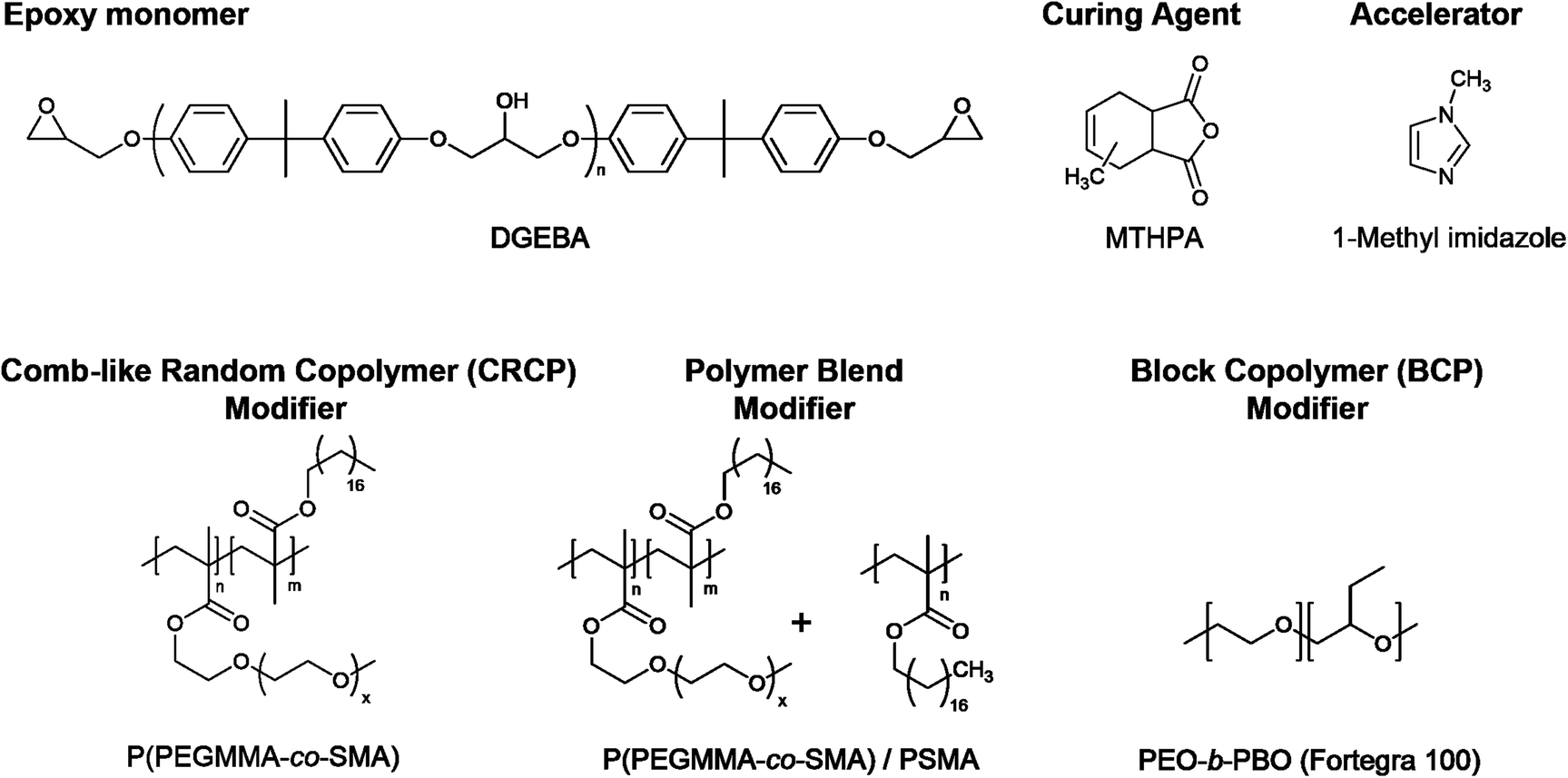 | ||
| Scheme 1 Chemical structures of ingredients of the modified epoxy thermosets. | ||
| Modifier | Polymer | Type | PEGMMA content |
|---|---|---|---|
| CRCP-30 | P(PEGMMA-co-SMA) | Comb-like random copolymer | 30 wt% |
| CRCP-27 | P(PEGMMA-co-SMA) | Comb-like random copolymer | 27 wt% |
| CRCP-25 | P(PEGMMA-co-SMA) | Comb-like random copolymer | 25 wt% |
| Blend-27 | P(PEGMMA-co-SMA)/PSMA | Polymer blend | 27 wt% |
| Blend-25 | P(PEGMMA-co-SMA)/PSMA | Polymer blend | 25 wt% |
| BCP (Fortegra 100) | PEO-b-PBO | Block copolymer | N/A |
Before a thermal curing process, a desired amount of the modifier (5 wt% content with respect to the neat epoxy thermoset) was mixed with the epoxy monomer DEGBA (Araldite LY556) by mechanical stirring for at least 10 min until the mixture appeared homogeneous. Then, the curing agent MTHPA (Aradur 917) and the accelerator 1-methyl imidazole (DY070) were added to the mixture and stirred for 10 min. To meet the stoichiometric ratio of epoxy to anhydride, the weight ratio of Araldite LY556:Aradur 917:DY070 was set to be 100:90:2. The bubbles within the mixture were removed by centrifugation at 3000 rpm for 5 min, followed by degassing under reduced pressure at 50 °C for 30 min. After pouring into a preheated stainless mold, the mixture was pre-cured at 80 °C for 4 h and subsequently post-cured at 140 °C for 8 h in an oven. A plate of the modified epoxy thermoset in 420 mm × 300 mm × 6.2 mm was obtained after release from the mold.
2.4 Characterizations
The polymers synthesized in this work were characterized by nuclear magnetic resonance (NMR) spectroscopy, Fourier transform infrared (FTIR) spectroscopy, and gel permeation chromatography (GPC) analyses to confirm their chemical structures and molecular weights. The NMR analysis was performed on a Brucker AVANCE 600 MHz spectrometer for a polymer solution sample dissolved in chloroform-d1. The FTIR analysis was conducted on a PerkinElmer Spectrome ONE spectrometer in a transmittance mode for a polymer sample coated on a KBr pellet to record a spectrum ranging from 400 cm−1 to 4000 cm−1 by scanning 16 times with a resolution of 2 cm−1. The GPC analysis for a polymer sample was carried out on a GPC system consisting of a Hitachi Elite LaChrom L-2490 RI Detector and a Hitachi Elite LaChrom L-2130 Pump, which used tetrahydrofuran as the eluent at an elution rate of 0.5 mL min−1 and a series of polystyrene polymers with various molecular weights as the calibration standards.The nanostructures of secondary phases formed in the modified epoxy thermosets were analyzed by TEM. The modified epoxy thermosets were microtomed by a Leica Ultra-microtome machine equipped with a diamond knife to obtain the sliced samples with a thickness of less than 70 nm. The sliced samples were placed on a copper grid and stained with OsO4. The TEM images were taken on a JEM-2100F at an accelerating voltage of 120 kV. To further understand the toughening mechanisms of various modifiers, a scanning electron microscope (SEM) was used to observe the fracture surfaces of the modified epoxy thermosets, which were obtained after the single-edge notched bending (SENB) test. All the fracture surfaces were coated with an ultra-thin layer of gold by sputtering. The SEM observations were carried out on a JEOL JSM 6800F at an accelerating voltage of 20 kV. The observation area on the fracture surface was located at the crack propagation region.
2.5 Measurements of thermal and mechanical properties of epoxy thermosets
The glass transition temperature (Tg) and thermal degradation temperature (Td) of a thermoset sample were measured by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), respectively. The DSC analysis was operated on a TA Instruments DSC 2920 in a temperature range between 20 °C and 250 °C. The DSC trace of the second heating run was recorded at a heating rate of 10 °C min−1. To detect a distinguishable glass transition of the second heating run, a cooling run prior to it at a cooling rate higher than 30 °C min−1 is necessary. The TGA thermogram of a thermoset sample was recorded on a TA SDT-Q600 at a heating rate of 10 °C min−1 from 50 °C to 750 °C with a nitrogen flow of 20 mL min−1.The toughness of the modified epoxy thermosets was evaluated in terms of their tensile toughness, impact strengths, and plane-strain fracture toughness through tensile, impact, and SENB tests, respectively. The tensile test was performed on a universal material testing machine QC-H51A2 equipped with a 20 kN load cell with a crosshead displacement speed of 1.0 mm min−1 at room temperature. The tensile test specimens were machined into a dogbone shape in accordance with ASTM D638 by a GX-6060FC CNC mill. The elastic modulus of an epoxy thermoset specimen was determined by the initial slope of the stress–strain curve in the strain range between 0% and 1%. The area underneath the stress–strain curve before the strain at break can calculate the tensile toughness of an epoxy thermoset specimen. Additionally, the measured data of at least six specimens were averaged to yield valid results of elastic modulus, tensile stress, and tensile toughness.
The Charpy impact test was carried out following ASTM D6110 on an impact tester. The dimensions of an impact test specimen were 62 mm × 12 mm × 6 mm. The SENB test was conducted on a universal material testing machine QC-H51A2 with a crosshead displacement speed of 2.0 mm min−1. A pre-crack in a SENB specimen can be generated by hitting its notch tip with a razor blade. We selected at least six specimens that fulfill the selection criterion of ASTM D5045 to complete the SENB test and obtain an average result of the critical stress intensity factor (KIC) for each of the modified epoxy thermosets. The critical stress intensity factors (KIC) are determined from the maximum loading at failure (PQ), the specimen thickness (B), the specimen width (W), and the shape factor (f(a/W)) using eqn (1) and (2). Meanwhile, the energy release rate (GIC) can be calculated from KIC, the Poisson's ratio (ν), and the elastic modulus (E) according to eqn (3).
 | (1) |
 | (2) |
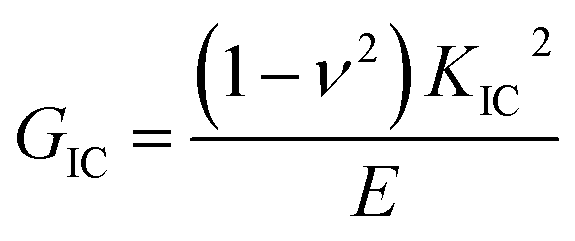 | (3) |
3 Results and discussion
Three comb-like random copolymers of P(PEGMMA-co-SMA) with different PEGMMA contents (30 wt%, 27 wt%, and 25 wt%) and a homopolymer of PSMA were synthesized using thermally initiated free radical polymerization. The polymer syntheses were accomplished through one-step copolymerization, as shown in Scheme 2. The GPC profiles of the synthesized polymers are presented in Fig. 1(a). The weight averaged molecular weights (Mw) of CRCP-30, CRCP-27, and CRCP-25 were measured by GPC analysis to be 13300 g mol−1, 13500 g mol−1, and 127000 g mol−1, respectively. The polydispersity index (PDI) values of these P(PEGMMA-co-SMA) copolymers were all close to 1.8. The Mw of PSMA was 32600 g mol−1, and its PDI was 2.5.
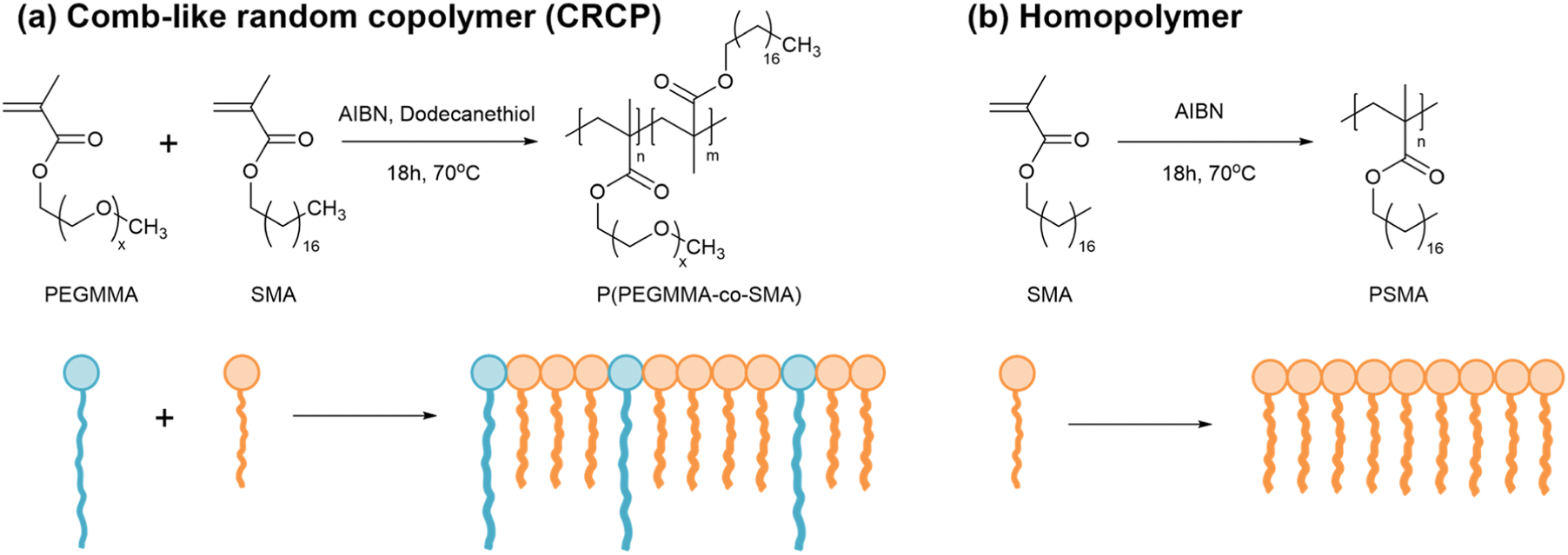 | ||
| Scheme 2 Syntheses of (a) comb-like random copolymers of P(PEGMMA-co-SMA) and (b) a homopolymer of PSMA. | ||
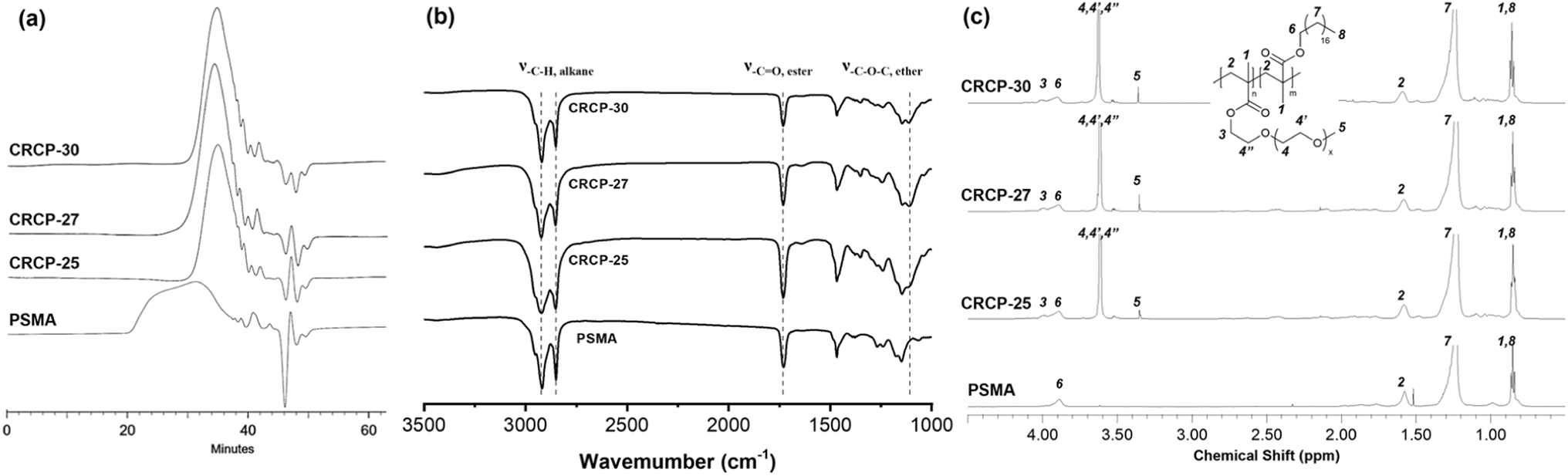 | ||
| Fig. 1 (a) GPC profiles, (b) FTIR spectra, and (c) 1H NMR spectra of the comb-like random copolymers of P(PEGMMA-co-SMA) with different PEGMMA contents (CRCP-25, CRCP-27, CRCP-30) and the homopolymer PSMA synthesized in this work. | ||
Fig. 1(b) shows the FTIR spectra of CRCP-30, CRCP-27, CRCP-25, and PSMA. The absence of the absorption peak at 1630 cm−1 (ν–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) corresponding to the vibrations of carbon–carbon double bonds for the methacrylate monomers SMA and PEGMMA indicated that the polymerization was successful, and all the unreacted monomers were removed from the polymer products after purifications. Meanwhile, three absorption peaks at 2925 cm−1 (ν–C–H, alkane), 2850 cm−1 (ν–C–H, alkane), and 1730 cm−1 (ν–CO, ester) were detected for all the polymers, as expected. The characteristic peak for P(PEGMMA-co-SMA) existed at 1108 cm−1 (ν–C–O, ether), which are attributed to the stretching of the ether moieties of PEGMMA.
C) corresponding to the vibrations of carbon–carbon double bonds for the methacrylate monomers SMA and PEGMMA indicated that the polymerization was successful, and all the unreacted monomers were removed from the polymer products after purifications. Meanwhile, three absorption peaks at 2925 cm−1 (ν–C–H, alkane), 2850 cm−1 (ν–C–H, alkane), and 1730 cm−1 (ν–CO, ester) were detected for all the polymers, as expected. The characteristic peak for P(PEGMMA-co-SMA) existed at 1108 cm−1 (ν–C–O, ether), which are attributed to the stretching of the ether moieties of PEGMMA.
The 1H NMR spectra of the synthesized polymers are presented in Fig. 1(c). The signals at chemical shift δ = 0.86 ppm (H1 and H8) and at δ = 3.35 ppm (H5) are attributed to the methyl protons as indicated. The two signals belonging to the aliphatic methylene protons in the SMA side chain (H7) and the polymer backbone (H2) are located at δ = 1.23 ppm and δ = 1.59 ppm, respectively. Meanwhile, the signal corresponding to the methylene protons adjacent to the alkoxy moiety of ester in the SMA side chain (H6) and PEGMMA side chain (H3) can be observed in the range between 3.85 ppm and 4.15 ppm. The signal at δ = 3.62 ppm was assigned to the methylene protons of ether groups (H4, H4′, and H4′′). Apart from the signal assignments, the molar ratio of PEGMMA in P(PEGMMA-co-SMA) can be calculated by the integral ratio of signal H6 + H3 and signal H5. The PEGMMA contents in CRCP-30, CRCP-27, and CRCP-25 were determined to be 5.88 mol% (30.4 wt%), 6.28 mol% (26.0 wt%), and 7.47 mol% (24.3 wt%), respectively. If compared by weight percentage, the PEGMMA measured ratios are consistent with the PEGMMA feed ratio. In addition, we confirmed through NMR integration analysis that the number average molecular weight of PEGMMA monomer is 1884 g mol−1, which is close to the molecular weight grade claimed by the supplier (2000 g mol−1).
The TEM micrographs in Fig. 2(a) obviously revealed that the comb-like random copolymers of P(PEGMMA-co-SMA) can be rearranged into well-defined nanostructures in anhydride-cured epoxy thermosets. The nanostructure of the modified epoxy thermosets varied with the modifier's epoxy-philic PEGMMA content ranging from 30 wt% to 25 wt%. For example, a spherical micellar structure was observed in the epoxy thermoset containing 5 wt% of CRCP-30. Meanwhile, the CRCP-27 modified epoxy thermosets displayed a morphology characterized by the coexistence of spherical and worm-like micelles. The CRCP-25 modifiers produced vesicle-like nanodomains along with some coexisting micelles in the epoxy matrix. On the other hand, it was found that the polymer blend modifiers (Blend-27 and Blend-25) rearranged into irregularly shaped nanodomains in nonuniform size within the thermosets. These nanodomains of the polymer blend inclusions were primarily formed by swelling the SMA core of CRCP-30 spherical micelles with the PSMA homopolymer. It is noted that the nanodomain size of Blend-25 was slightly larger than that of Blend-27, attributable to the higher PSMA content present in Blend-25.
 | ||
| Fig. 2 (a) TEM micrographs showing the nanostructured morphologies of the anhydride-cured epoxy thermosets modified with 5 wt% of CRCP-30, CRCP-27, CRCP-30, Blend-27, and Blend-25. (b) Schematic illustration of various nanostructures formed by the rearrangements of the CRCP and polymer blend modifiers. (c) Photos of the mixtures of the DEGBA epoxy precursor and the modifiers (without curing agent) before thermal curing. (d) Photos of the modified epoxy thermosets after thermal curing. | ||
On the other hand, Fig. 2(c) shows that the uncured epoxy mixture containing CRCP-30 was transparent and hazy, the mixture having CRCP-27 looked translucent and milky, as well as the other mixtures containing either CRCP-25, Blend-27, or Blend-25 exhibited opaque. After the thermal curing process, we can still see through both the CRCP-30 and CRCP-27 modified epoxy thermosets, as shown in Fig. 2(d). The other modified epoxy thermosets remained opaque in appearance. This result suggests that the rearrangements of the comb-like random copolymers or the polymer blends into nanostructures in epoxy take place before thermal curing and probably followed a self-assembly mechanism8 rather than a reaction-induced microphase separation (RIMPS) mechanism.44
As mentioned earlier, Terashima and his coworkers demonstrated that well-defined and self-assembled nanostructures (e.g., spherical micelles and vesicles) in water can be developed with amphiphilic random copolymers bearing hydrophilic PEG and hydrophobic alkyl pendants.37–39 In fact, the chemical structures of their amphiphilic random copolymers are similar to P(PEGMMA-co-SMA) used in this work. The solubility parameter of the DGEBA epoxy precursor (20.7 MPa0.5)45 is close to the calculated value of PEGMMA (18.74 MPa0.5)46 but far from that of PSMA (15.6 MPa0.5).47 In this case, PEGMMA pendants can be considered as epoxy-philic segments of comb-like random copolymers. We suppose that the big difference in miscibility between PEGMMA and SMA pendants with respect to the reactive solvent DGEBA facilitates the self-assembly of P(PEGMMA-co-SMA) into micellar nanostructures in epoxies before curing.
The glass transition temperature (Tg) of an epoxy thermoset was determined by the DSC analysis. As shown in Fig. 3(a), the neat epoxy thermoset exhibited a Tg at 137.4 °C. The Tgs of the epoxy thermosets modified with the comb-like random copolymers of P(PEGMMA-co-SMA) or the polymer blends of P(PEGMMA-co-SMA)/PSMA ranged between 136.2 °C and 131.3 °C, higher than that of the epoxy thermoset containing Fortegra 100 (122.7 °C). The decrease in Tg was modest (less than 6.1 °C), revealing that our synthesized modifiers cause minor plasticization effects on the softening of epoxy thermosets. On the other hand, the thermal degradation temperature (Td) of an epoxy thermoset, which refers to the temperature at 5 wt% weight loss, was analyzed by TGA. As presented in Fig. 3(b), the Tds of the neat epoxy and modified epoxy thermosets ranged from 275.3 °C to 345.6 °C. The decreased amount of Td mainly depends on the PEGMMA content.
 | ||
| Fig. 3 (a) DSC curves and (b) TGA thermograms of the neat epoxy and modified epoxy thermosets with a modifier content of 5 wt%. | ||
The representatives stress–strain curves of the neat epoxy and modified epoxy thermosets were presented in Fig. 4(a). We found that all the elastic moduli of the modified epoxy thermosets decreased compared to that of the neat epoxy, as summarized in Fig. 4(b). The presence of the 5 wt% modifier resulted in a 6% to 16% decrease in elastic modulus, indicating that the stiffness of epoxy thermosets was compromised to some extent. Compared to the smaller CRCP-27 and CRCP-30 nanodomains, the bigger CRCP-25 vesicle-like nanodomains may occupy more volume in the epoxy thermoset when the modifier content is identical. The higher effective volume fraction of the secondary phase may lead to the more significant plasticization effect of the epoxy thermosets. This could explain the relatively considerable decrease in elastic modulus and Tg for the CRCP-25 modified epoxy thermoset. Our finding is similar to the result reported by Thio et al. that the modified epoxy thermosets containing the vesicle morphology exhibited a more considerable decrease in elastic modulus compared to the ones containing wormlike or spherical micelles.21
 | ||
| Fig. 4 (a) Representative stress–strain curves and (b) elastic modulus results for the neat epoxy thermoset and the modified epoxy thermosets containing 5 wt% of CRCP-30, CRCP-27, CRCP-25, Blend-27, Blend-25, and BCP Fortegra 100. | ||
As shown in Fig. 5(a), the tensile toughness of epoxy thermosets was considerably improved after incorporating with the modifiers. The increase in tensile toughness is more than 289% (from 0.54 kJ m−3 to 2.10 kJ m−3) for all the modified epoxy thermosets. However, the spherical micelles (CRCP-30) seem to improve the tensile toughness more effectively compared to other micellar nanostructures (CRCP-27 and CRCP-25). The reason may lie in the fact that the spherical micelles of CRCP-30 with more epoxy-philic PEGMMA content in the compositions possess a higher surface area (smaller in size) and better interfacial interaction with the epoxy matrix. On the other hand, it is found that the epoxy thermosets modified with the polymer blends (Blend-27 and Blend-25) achieved higher tensile toughness (higher strain) compared to the epoxy thermosets modified with the comb-like random copolymers (CRCP-30, CRCP-27, and CRCP-25). We supposed that the van der Waals interaction between SMA pendants of P(PEGMMA-co-SMA) and PSMA in the core of the polymer blend nanodomain might increase its cohesive strength, allowing the polymer blend's nanodomains to resist the tensile deformation easier compared to the comb-like random copolymer modifier's nanodomains.
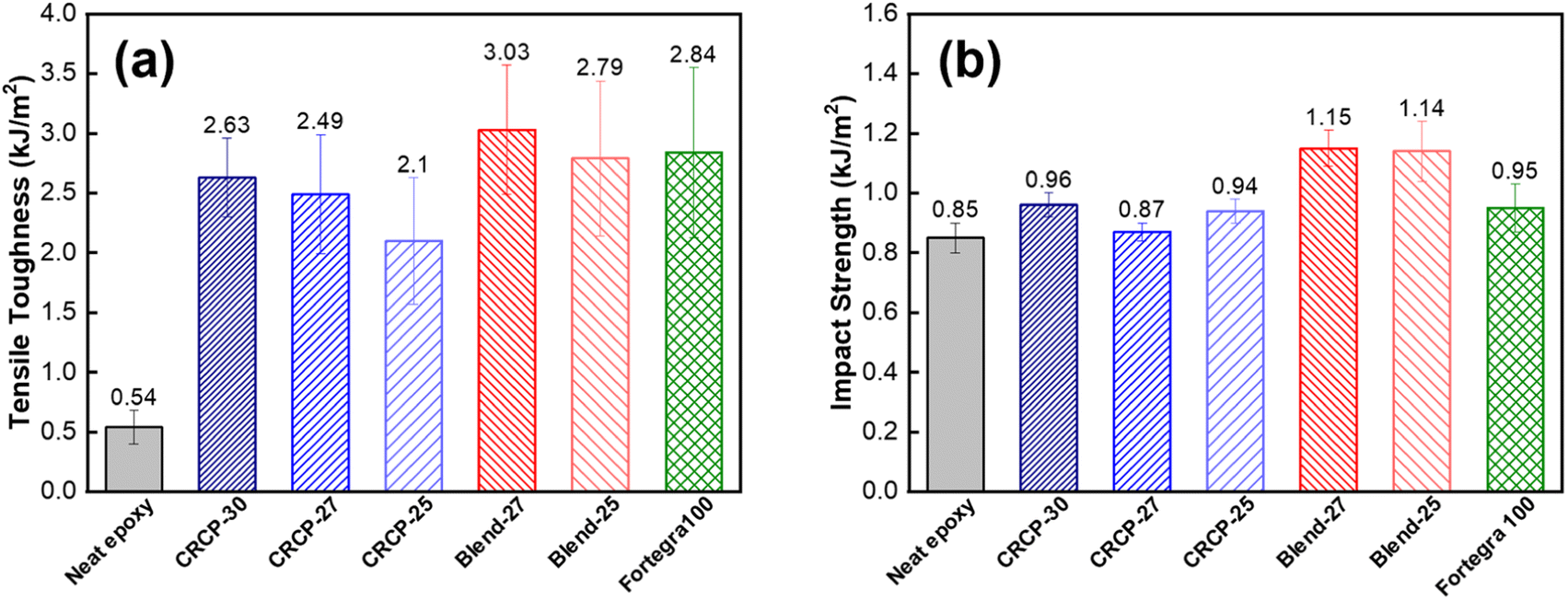 | ||
| Fig. 5 Measured results of (a) tensile toughness and (b) impact strength for the neat epoxy thermoset and the modified epoxy thermosets containing 5 wt% of CRCP-30, CRCP-27, CRCP-25, Blend-27, Blend-25, and BCP Fortegra 100. | ||
The impact strength is the measure of energy absorbed by a material before fracturing under a high rate of deformation. As presented in Fig. 5(b), the addition of 5 wt% modifiers only improved the impact strength of the epoxy thermoset to a small extent (by less than 34%). Owing to the amphiphilic modifiers used in this work acting as unreactive additives for epoxy, the lack of covalent interaction at the interface between the epoxy matrix and secondary phase domains may result in insignificant plastic deformations during impact in such an extremely short time. This could be one of the reasons for the limited improvement in the impact strength of the modified epoxy thermosets.
The fracture toughness in terms of the critical stress intensity factor (KIC) and the strain energy release rate (GIC) refers to the ability of a material containing a crack to resist fracture. The measured results of KIC and GIC for the neat epoxy and modified epoxy thermosets are compared in Fig. 6(a) and (b). For the neat epoxy thermosets in the DGEBA/MTHPA system, the measured values of KIC and GIC are both consistent with our previously reported values.48 Many researchers suggested that toughening of epoxy with amphiphilic block copolymers mainly depends on the morphologies of self-assembled nanodomains.11,13,43,45,49,50 Spherical micelles are generally less efficient in enhancing the fracture toughness of epoxy resins than vesicles and worm-like micelles because spherical micelles have smaller domain sizes and cannot induce significant plastic deformations of the matrix.15,21,51 Our result for comb-like random copolymers is in line with the previous findings for amphiphilic block copolymers. We found that the CRCP-25 modified epoxy thermoset containing vesicle-like nanodomains achieved a greater GIC value than the other CRCP modified epoxy thermosets containing spherical or worm-like micelles. Although the difference in measured KIC values between these CRCP modified epoxy thermosets is not significant, the lower elastic modulus of the CRCP-25 modified epoxy thermoset allow it to dissipate more fracture energy during crack propagation compared to the others, according to eqn (3).
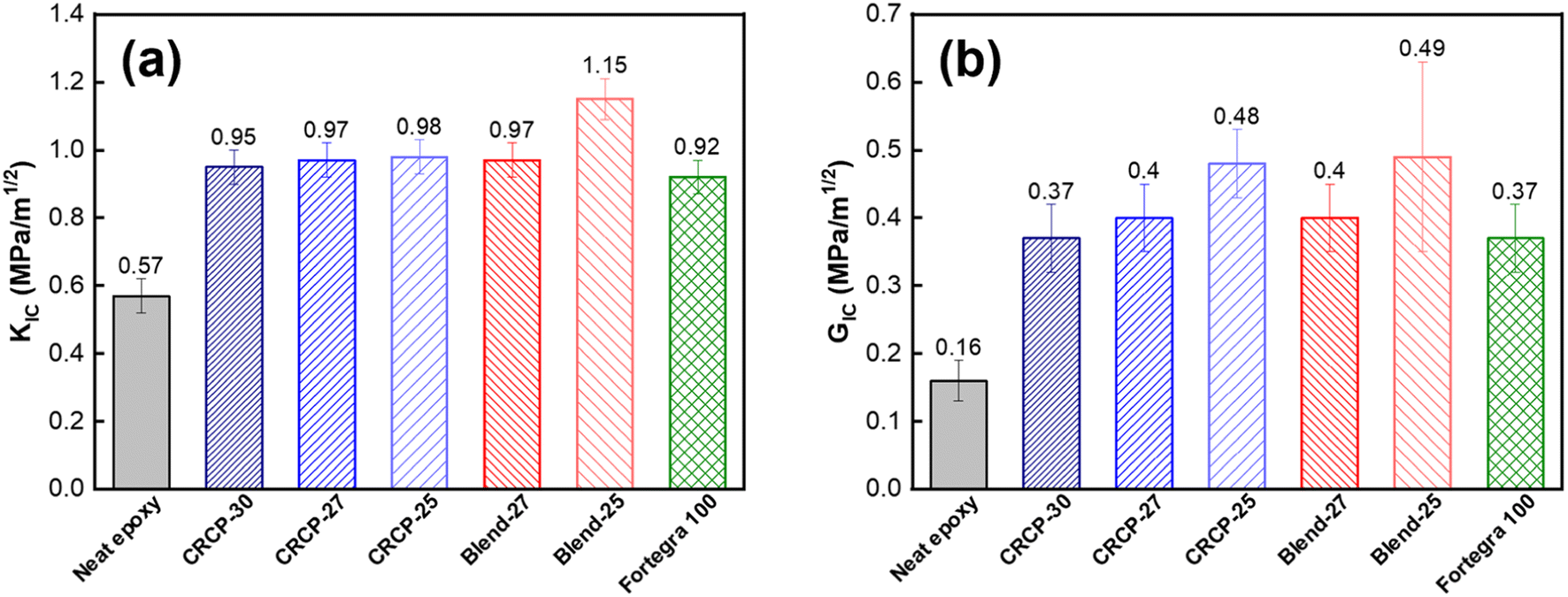 | ||
| Fig. 6 Measured results of (a) the critical stress intensity factor (KIC), and (b) the strain energy release rate (GIC) for the neat epoxy thermoset and the modified epoxy thermosets containing 5 wt% of CRCP-30, CRCP-27, CRCP-25, Blend-27, Blend-25, and BCP Fortegra 100. | ||
Of all the modifiers prepared in this work, the Blend-25 modifier, which forms irregularly shaped nanodomains within the epoxy resin matrix, was found to be the most effective in improving the fracture toughness of the epoxy resin. Compared to neat epoxy resin, the KIC and GIC increased by 102% and 225% respectively. The KIC measured value (1.15 MPa m−1/2) of the Blend-25 modified epoxy thermoset is as high as the KIC reported value of a PMMA resin.52 Surprisingly, it can be seen in Fig. 6(b) that the GIC values of the modified epoxy thermosets were almost the same when the PEGMMA content of the modifier was identical, regardless of using CRCP or polymer blend modifiers. This finding implied that the toughening effectiveness of our amphiphilic random copolymer based modifiers for anhydride-cured epoxy thermosets could be adjusted by their PEGMMA content through either copolymerization or blending approaches.
It is worth noting that both the comb-like random copolymer modifier and the polymer blend modifier are comparable to the commercially available block copolymer modifier (Fortegra™ 100) in enhancing the fracture toughness of epoxy thermosets. The KIC of the anhydride-cured bisphenol-A based epoxy thermoset modified with 5 wt% Fortegra 100 was measured to be 0.92 MPa m−1/2. This KIC value is quite close to the KIC of anhydride-cured bisphenol-F based epoxy thermoset incorporated with 4 wt% Fortegra 100 (0.94 MPa m−1/2) reported recently by Bajpai.26 Accordingly, we believe that the amphiphilic comb-like random copolymers and their polymer blends have the potential to be the alternative to amphiphilic block copolymers as toughening modifiers for epoxy.
The fracture surfaces of the SENB specimens consisting of various modifiers were examined by SEM to further understand the energy dissipation mechanisms responsible for the restriction of crack propagation. As observed in Fig. 7(a)–(c), all CRCP-modified epoxy thermosets showed fracture surfaces with step and leaf-like structures in the crack propagation region, indicating modest plastic deformations of the epoxy matrix. These matrix deformations were mainly caused by cavitations of nanodomains in response to the stress near the crack tip followed by a shear yielding.11,13,50 Meanwhile, the leaf-like patterns reflect massive local disruptions of the epoxy matrix due to crack deflection.23 Furthermore, the larger-sized vesicle-like nanodomains (CRCP-25) resulted in relatively pronounced step heights on the fracture surface compared to the smaller-sized spherical and worm-like micelles (CRCP-30 and CRCP-27). This finding may account for the slightly higher KIC of the CRCP-25 modified epoxy thermoset than the others.
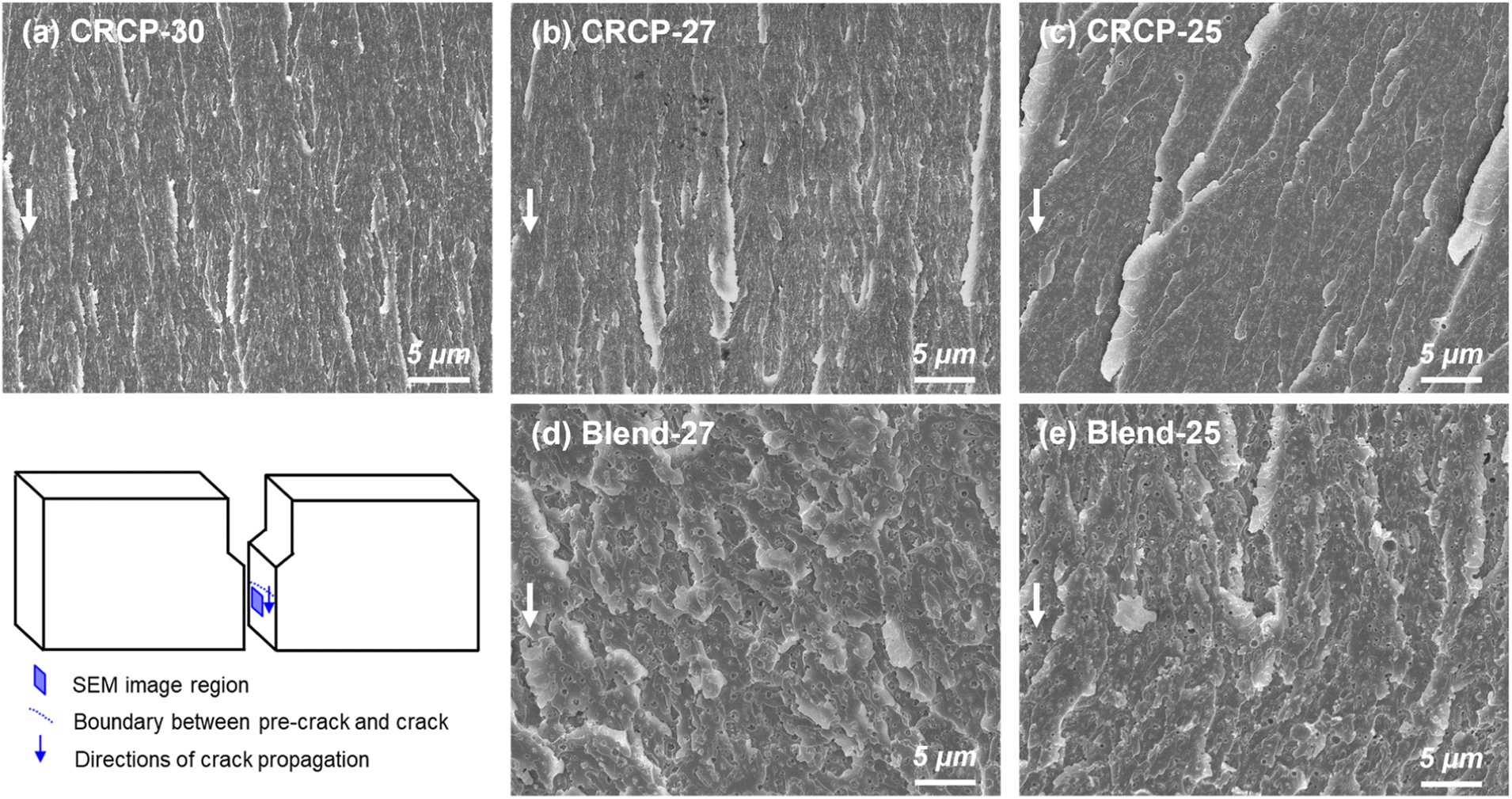 | ||
| Fig. 7 Cross-sectional SEM micrographs showing the fracture surface of SENB specimens at the crack propagation region for the (a) CRCP-30, (b) CRCP-27, (c) CRCP-25, (d) Blend-27, and (e) Blend-25 modified epoxy thermosets with a modifier content of 5 wt%. Arrows indicate the crack propagation directions. | ||
In contrast to the leaf-like morphology of the fracture surface of the CRCP modified epoxy thermosets, the polymer blend (Blend-27 and Blend-25) modified epoxy thermosets displayed relatively uneven and rough fracture surfaces with highly terraced morphologies, as shown in Fig. 7(d) and (e). The uneven and rough fracture surfaces indicate significant plastic deformations of the epoxy matrix, which may be caused by interfacial debonding and cavitations of the nanodomains with irregular shapes and sizes. We speculate that mechanisms such as crack deflection and frictional interlocking of crack wakes might be responsible for dissipating energy, resulting in the terraced morphology observed on the fracture surface, referring to Dean's explanation.53
4 Conclusion
We have developed a new type of toughening modifier for epoxy thermosets, which features amphiphilic comb-like random copolymers bearing epoxy-philic PEGMMA and epoxy-phobic SMA pendants. By varying the PEGMMA content from 30 wt% to 25 wt%, the comb-like random copolymer of P(PEGMMA-b-SMA) can be rearranged into various well-defined micellar structures (e.g., spherical micelles, worm-like micelles, and vesicle-like nanodomains) through self-assembly in anhydride-cured epoxy thermosets. Other than the copolymerization approach, we also employed the polymer blending approach to adjust the modifier's PEGMMA content to alter the nanostructured morphology of modified epoxy thermosets. Even though the polymer blend modifier did not generate highly ordered nanostructures in epoxy thermosets as the CRCP modifier did, both modifiers at a 5 wt% concentration were found to produce remarkable improvements in tensile toughness and fracture toughness of epoxy thermosets without significantly affecting their stiffness and glass transition temperature (Tg). Furthermore, our modifiers have exhibited comparable toughening effects to the commercially available block copolymer modifier (Fortegra™ 100) in enhancing the toughness of anhydride-cured epoxy thermosets, demonstrating their application potential.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The financial support from the National Science and Technology Council (Taiwan, ROC) through the project MOST 109-2622-E-992-029 is appreciated. The authors are grateful for the support from Mr Tsung-Jen Wang and Taiwan Surfactant Corp. Also, the authors would like to thank Mrs Bi-Yun Lin from the Instrument Center of National Cheng Kung University for operating the Bruker Avance 600 NMR spectrometer.References
- H. Kargarzadeh, I. Ahmad and I. Abdullah, in Micro- and Nanostructured Epoxy/Rubber Blends, ed. C. S. Sabu Thomas, and R. Thomas, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2014, ch. 2, pp. 31–52, DOI:10.1002/9783527666874.ch2.
- R. Thomas, J. Abraham, S. Thomas P and S. Thomas, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 2531–2544 CrossRef CAS.
- N. Chikhi, S. Fellahi and M. Bakar, Eur. Polym. J., 2002, 38, 251–264 CrossRef CAS.
- E. R. Mafi and M. Ebrahimi, Polym. Eng. Sci., 2008, 48, 1376–1380 CrossRef CAS.
- D. Quan and A. Ivankovic, Polymer, 2015, 66, 16–28 CrossRef CAS.
- B. B. Johnsen, A. J. Kinloch, R. D. Mohammed, A. C. Taylor and S. Sprenger, Polymer, 2007, 48, 530–541 CrossRef CAS.
- T. H. Hsieh, A. J. Kinloch, K. Masania, A. C. Taylor and S. Sprenger, Polymer, 2010, 51, 6284–6294 CrossRef CAS.
- M. A. Hillmyer, P. M. Lipic, D. A. Hajduk, K. Almdal and F. S. Bates, J. Am. Chem. Soc., 1997, 119, 2749–2750 CrossRef CAS.
- P. M. Lipic, F. S. Bates and M. A. Hillmyer, J. Am. Chem. Soc., 1998, 120, 8963–8970 CrossRef CAS.
- F. Meng, S. Zheng, W. Zhang, H. Li and Q. Liang, Macromolecules, 2006, 39, 711–719 CrossRef CAS.
- J. Liu, H.-J. Sue, Z. J. Thompson, F. S. Bates, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2008, 41, 7616–7624 CrossRef CAS.
- J. Yang, Q. Zhou, K. Shen, N. Song and L. Ni, RSC Adv., 2018, 8, 3705–3715 RSC.
- L. Ruiz-Pérez, G. J. Royston, J. P. A. Fairclough and A. J. Ryan, Polymer, 2008, 49, 4475–4488 CrossRef.
- J. M. Dean, N. E. Verghese, H. Q. Pham and F. S. Bates, Macromolecules, 2003, 36, 9267–9270 CrossRef CAS.
- J. M. Dean, R. B. Grubbs, W. Saad, R. F. Cook and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2444–2456 CrossRef CAS.
- Y. Xiang, S. Xu and S. Zheng, Eur. Polym. J., 2018, 98, 321–329 CrossRef CAS.
- F. Meng, S. Zheng, H. Li, Q. Liang and T. Liu, Macromolecules, 2006, 39, 5072–5080 CrossRef CAS.
- Z. Heng, X. Zhang, Y. Chen, H. Zou and M. Liang, Chem. Eng. J., 2019, 360, 542–552 CrossRef CAS.
- H. Garate, N. J. Morales, S. Goyanes and N. B. D'Accorso, in Handbook of Epoxy Blends, ed. J. Parameswaranpillai, N. Hameed, J. Pionteck and E. M. Woo, Springer International Publishing, Cham, 2017, pp. 841–881, DOI:10.1007/978-3-319-40043-3_31.
- J. Wu, Y. S. Thio and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 1950–1965 CrossRef CAS.
- Y. S. Thio, J. Wu and F. S. Bates, Macromolecules, 2006, 39, 7187–7189 CrossRef CAS.
- Q. Guo, F. Chen, K. Wang and L. Chen, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 3042–3052 CrossRef CAS.
- T. Li, M. J. Heinzer, L. F. Francis and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 189–204 CrossRef CAS.
- B. J. Rohde, T. E. Culp, E. D. Gomez, J. Ilavsky, R. Krishnamoorti and M. L. Robertson, Macromolecules, 2019, 52, 3104–3114 CrossRef CAS.
- L. Cano, D. H. Builes and A. Tercjak, Polymer, 2014, 55, 738–745 CrossRef CAS.
- A. Bajpai and B. Wetzel, J. Compos. Sci., 2019, 3, 68 CrossRef CAS.
- M. Asada, S. Oshita, Y. Morishita, Y. Nakashima, Y. Kunimitsu and H. Kishi, Polymer, 2016, 105, 172–179 CrossRef CAS.
- R. Matadi Boumbimba, C. Froustey, P. Viot and P. Gerard, Composites, Part B, 2015, 76, 332–342 CrossRef CAS.
- Z. Xu and S. Zheng, Polymer, 2007, 48, 6134–6144 CrossRef CAS.
- D. Hu, C. Zhang, R. Yu, L. Wang and S. Zheng, Polymer, 2010, 51, 6047–6057 CrossRef CAS.
- C. Zhang, L. Li and S. Zheng, Macromolecules, 2013, 46, 2740–2753 CrossRef CAS.
- V. Rebizant, V. Abetz, F. Tournilhac, F. Court and L. Leibler, Macromolecules, 2003, 36, 9889–9896 CrossRef CAS.
- N. Hameed, Q. Guo, Z. Xu, T. L. Hanley and Y.-W. Mai, Soft Matter, 2010, 6, 6119–6129 RSC.
- Q. Zhou, Q. Liu, N. Song, J. Yang and L. Ni, Eur. Polym. J., 2019, 120, 109236 CrossRef CAS.
- L. Li, K. Raghupathi, C. Song, P. Prasad and S. Thayumanavan, Chem. Commun., 2014, 50, 13417–13432 RSC.
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Macromolecules, 2014, 47, 589–600 CrossRef CAS.
- Y. Hirai, T. Terashima, M. Takenaka and M. Sawamoto, Macromolecules, 2016, 49, 5084–5091 CrossRef CAS.
- G. Hattori, M. Takenaka, M. Sawamoto and T. Terashima, J. Am. Chem. Soc., 2018, 140, 8376–8379 CrossRef CAS PubMed.
- T. Ikami, Y. Kimura, M. Takenaka, M. Ouchi and T. Terashima, Polym. Chem., 2021, 12, 501–510 RSC.
- M. Hibino, K. Tanaka, M. Ouchi and T. Terashima, Macromolecules, 2022, 55, 178–189 CrossRef CAS.
- A. Iborra, G. Díaz, D. López, J. M. Giussi and O. Azzaroni, Eur. Polym. J., 2017, 87, 308–317 CrossRef CAS.
- T. Fang, M. Huo, Z. Wan, H. Chen, L. Peng, L. Liu and J. Yuan, RSC Adv., 2017, 7, 2513–2519 RSC.
- Y. S. Thio, J. Wu and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 1125–1129 CrossRef CAS.
- D. Hu and S. Zheng, Eur. Polym. J., 2009, 45, 3326–3338 CrossRef CAS.
- S. Maiez-Tribut, J. P. Pascault, E. R. Soulé, J. Borrajo and R. J. J. Williams, Macromolecules, 2007, 40, 1268–1273 CrossRef CAS.
- Q. Wu, A. Tiraferri, H. Wu, W. Xie and B. Liu, ACS Omega, 2019, 4, 19799–19807 CrossRef CAS PubMed.
- D. J. Walsh and G. L. Cheng, Polymer, 1984, 25, 499–502 CrossRef CAS.
- L. C. Jheng, I. H. Wang, T. H. Hsieh, C. T. Fan, C. H. Hsiao, C. P. Wu, M. T. Leu and T. Y. Chang, J. Appl. Polym. Sci., 2021, 138, 50096 CrossRef CAS.
- T. Smart, H. Lomas, M. Massignani, M. V. Flores-Merino, L. R. Perez and G. Battaglia, Nano Today, 2008, 3, 38–46 CrossRef CAS.
- J. Liu, Z. J. Thompson, H.-J. Sue, F. S. Bates, M. A. Hillmyer, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Macromolecules, 2010, 43, 7238–7243 CrossRef CAS.
- X. Yang, F. Yi, Z. Xin and S. Zheng, Polymer, 2009, 50, 4089–4100 CrossRef CAS.
- J. M. de Souza, H. N. Yoshimura, F. M. Peres and C. G. Schön, Polym. Test., 2012, 31, 834–840 CrossRef.
- J. M. Dean, P. M. Lipic, R. B. Grubbs, R. F. Cook and F. S. Bates, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2996–3010 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |