
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding interactions of hydrophobically-modified flavonols with β-glucosidase: fluorescence spectroscopy and molecular modelling study
Liudmyla V. Chepelevaa,
Oleksii O. Demidova,
Arsenii D. Snizhkoa,
Dmytro O. Tarasenko a,
Andrii Y. Chumaka,
Oleksii O. Kolomoitseva,
Volodymyr M. Kotliara,
Eugene S. Gladkovab,
Alexander Kyrychenko*ab and
Alexander D. Roshala
a,
Andrii Y. Chumaka,
Oleksii O. Kolomoitseva,
Volodymyr M. Kotliara,
Eugene S. Gladkovab,
Alexander Kyrychenko*ab and
Alexander D. Roshala
aInstitute of Chemistry, V.N. Karazin Kharkiv National University, 4 Svobody Sq., Kharkiv 61022, Ukraine. E-mail: a.v.kyrychenko@karazin.ua
bState Scientific Institution “Institute for Single Crystals”, National Academy of Sciences of Ukraine, 60 Nauky Ave., Kharkiv 61072, Ukraine
First published on 21st November 2023
Abstract
Natural flavonoids are capable of inhibiting glucosidase activity, so they can be used for treating diabetes mellitus and hypertension. However, molecular-level details of their interactions with glucosidase enzymes remain poorly understood. This paper describes the synthesis and spectral characterization of a series of fluorescent flavonols and their interaction with the β-glucosidase enzyme. To tune flavonol–enzyme interaction modes and affinity, we introduced different polar halogen-containing groups or bulky aromatic/alkyl substituents in the peripheral 2-aryl ring of a flavonol moiety. Using fluorescence spectroscopy methods in combination with molecular docking and molecular dynamics simulations, we examined the binding affinity and identified probe binding patterns, which are critical for steric blockage of the key catalytic residues of the enzyme. Using a fluorescent assay, we demonstrated that the binding of flavonol 2e to β-glucosidase decreased its enzymatic activity up to 3.5 times. In addition, our molecular docking and all-atom molecular dynamics simulations suggest that the probe binding is driven by hydrophobic interactions with aromatic Trp and Tyr residues within the catalytic glycone binding pockets of β-glucosidase. Our study provides a new insight into structure–property relations for flavonol–protein interactions, which govern their enzyme binding, and outlines a framework for a rational design of new flavonol-based potent inhibitors for β-glucosidases.
Introduction
Flavonol-based fluorescent probes represent a class of environmentally sensitive dyes whose emission properties are highly sensitive to their immediate environment.1–7 The notable feature of these probes is that due to the presence of a hydroxyl group at the 3-position of a flavonoid moiety, they are capable of excited-state intramolecular proton transfer (ESIPT) (Scheme 1). A proton transfer reaction occurs through an intramolecular hydrogen bond bridge, resulting in an extremely fast (kESIPT > 1012 s−1) phototautomerization from the normal state (N*) to the tautomer state (T*).7–9 ESIPT leads to the appearance of dual-wavelength emission in flavonols. An essential advantage of ESIPT probes is that an intensity ratio of their normal-to-tautomer emission bands can be tuned by electron donor/acceptor substituents in a flavonol moiety. | ||
| Scheme 1 Scheme of an excited-state intramolecular proton transfer (ESIPT) in flavonol. | ||
The emission life time and quantum yield of flavonols depends strongly on the environment properties.1,8,10 Flavonol-based probes have been utilized as environment-sensitive fluorophores for monitoring solvent polarity1,11,12 and pH,13,14 as well as the determination of water traces in organic solvents.15 The unique optical properties and biocompatibility make flavonols a promising fluorescence probe for studying the structure of amphiphilic polymers16 and biomacromolecules.17 A large fluorescence “turn-on” effect has been observed upon selective binding of flavonols to some proteins, such as lysozymes18 and albumins.17,19 The selectivity of flavonols for certain proteins has been utilized for developing new fluorescence detection assays.17,20,21 Moreover, flavonol-based probes have also demonstrated a low perturbation impact on the lipid membrane physical state,22 so some fluorescent flavonols were utilized for fluorescent cell imaging.23–25
It has recently been demonstrated that flavonol-based probes, such as 4′-fluoroflavonol β-D-glucopyranoside, could be used as a fluorescent indicator of β-glucosidase activity.26,27 β-Glucosidases are a family of hydrolase enzymes that catalyze the hydrolysis of aryl and alkyl β-D-glucosides and cellobioses. β-Glucosidases are present in plants, fungi, animals, and bacteria. In addition, these enzymes are promising reagents in various biotechnological processes because they can catalyze the hydrolysis of many artificial substrates upon biofuel production and oligosaccharide cleavage.28 Regulation of glucosidase activity plays a key role in treating Gaucher's disease29 and the effectiveness of chemotherapy for breast cancer.30 Therefore, many glycosidase inhibitors have been isolated from natural sources or synthesized to optimize their inhibitory potency.31–37 Inhibition of the catalytic activity of glycosidase enzymes has great potential in developing highly potent and specific drugs against such diseases as diabetes, cancer, and viral infections.37,38
In this work, we studied the binding interaction of a series of synthetic flavonols with a β-glucosidase enzyme using fluorescence titration measurements and computational chemistry tools, such as molecular docking and molecular dynamics simulations. A series of flavonol derivatives was designed by varying different substituents in the peripheral 2-aryl moiety. The fluorescence titration of the flavonols with β-glucosidase in aqueous solution revealed pronounced protein-induced fluorescence “turn-on” effects, which are characteristic for the probe binding to the hydrophobic water-free cavity of the protein. In addition, the favorable binding mode of the flavonols into β-glucosidase enzymes taken from different sources was identified by molecular docking calculations. Finally, the stability of the docked flavonol–enzyme complexes was further re-examined by all-atom molecular dynamics simulations in an explicit water solution.
Experimental section
Chemistry
 | ||
| Scheme 2 Chemical structure and preparation of flavonols 2a–f. | ||
2-Aryl-3-hydroxy-4H-chromen-4-ones 2a–f were synthesized by the following method. For the synthesis of the target 2-aryl-3-hydroxy-4H-chromen-4-ones 2a–f, we used the one-pot optimized method27 without obtaining and additional purification of the corresponding intermediate unsaturated ketones. The synthesis of unsaturated ketones 1a–f described elsewhere.39 The general procedures of the synthesis of most typical substances of the series used in our work and typical examples of substance characteristics are given in the experimental section.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CHCl3:n-hexane). 3.4 mL (30 mmol) 30% H2O2 was added dropwise to the mixture. After 30 min mixture was acidified with 10% HCl to pH 3. The obtained precipitate was filtered off and washed by water and n-hexane. The crud product was re-crystalized from the corresponding solvents.
:1 CHCl3:n-hexane). 3.4 mL (30 mmol) 30% H2O2 was added dropwise to the mixture. After 30 min mixture was acidified with 10% HCl to pH 3. The obtained precipitate was filtered off and washed by water and n-hexane. The crud product was re-crystalized from the corresponding solvents.
2-Phenyl-3-hydroxy-4H-chromen-4-one (2a). Yield 1.92 g (81%), mp 170.0–172.0 °C (ethanol). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 9.65 (s), 8.23 (d, J = 7.40 Hz, 2H), 8.13 (dd, J = 8.04, 1.12 Hz, 1H), 7.82–7.76 (m, 2H), 7.60–7.56 (m, 2H), 7.53–7.46 (m, 2H). 13C NMR (126 MHz, DMSO-d6), δ, ppm: 173.0, 154.6, 145.2, 139.1, 133.7, 131.3, 129.9, 128.5, 127.6, 124.8, 124.5, 121.3, 118.4. Mass spectrum, m/z (Irel, %): 239 [M + H]+ (100). Found, %: C 75.67; H 4.18. C15H10O3. Calculated, %: C 75.62; H 4.23. These data are in accordance with the literature.27
2-(4-Chlorophenyl)-3-hydroxy-4H-chromen-4-one (2b). Yield 1.45 g (53%), beige crystals, mp 201.0–201.5 °C (ethanol). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 9.8 (bs, 1H, OH), 8.25 (d, J = 8.4 Hz, 2H), 8.12 (d, J = 8.1 Hz, 1H), 7.82 (dd, 1H), 7.78 (t, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.48 (t, 1H). 13C NMR (126 MHz, DMSO-d6), δ, ppm: 172.9, 154.4, 143.9, 139.3, 134.4, 133.8, 130.1, 129.2, 128.6, 124.7, 124.5, 121.2, 118.3. Mass spectrum, m/z (Irel, %): 157.0 (31), 273.0 [M + H]+ (100), 275.0 (25). Found, %: C 66.11; H 3.29. C15H9ClO3. Calculated, %: C 66.07; H 3.33. These data are in accordance with the literature.40
2-(4-(Trifluoromethyl)phenyl)-3-hydroxy-4H-chromen-4-one (2c). Yield 1.89 g (62%), beige crystals, mp 186.5–187.0 °C (ethanol). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 10.0 (bs, 1H, OH), 8.35 (d, J = 8.4 Hz, 2H), 8.06 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.74 (dd, 1H), 7.67 (d, 1H), 7.42 (t, 1H). 13C NMR (126 MHz, DMSO-d6), δ, ppm: 173.5, 154.9, 143.5, 140.6, 135.6, 134.4, 128.5, 125.7, 125.6, 125.3, 125.0, 121.7, 118.8. Mass spectrum, m/z (Irel, %): 58.5 (27), 307.2 [M + H]+ (100). Found, %: C 62.69; H 2.91. C16H9F3O3. Calculated, %: C 62.75; H 2.96. These data are in accordance with the literature.27
2-(4-Benzyloxyphenyl)-3-hydroxy-4H-chromen-4-one (2d). Yield 2.92 g (85%), yellowish crystals, mp 175.0–176.0 °C (ethanol). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 9.45 (s, 1H), 8.24–8.17 (m, 3H), 8.11 (dd, J = 8.0, 1.6 Hz, 1H), 7.83–7.71 (m, 3H), 7.53–7.30 (m, 8H), 7.21 (dd, J = 8.6, 1.6 Hz, 3H), 5.21 (s, 2H). 13C NMR (126 MHz, DMSO-d6), δ, ppm: 173.1, 159.9, 154.9, 145.9, 138.7, 137.1, 133.9, 129.8, 128.9, 128.4, 128.3, 125.2, 124.9, 124.2, 121.8, 118.8, 115.3, 69.8. Mass spectrum, m/z (Irel, %): 345 [M + H]+ (100). Found, %: C 76.69; H 4.70. C22H16O4. Calculated, %: C 76.73; H 4.68. These data are in accordance with the literature.41
2-(3,4-Dibenzyloxyphenyl)-3-hydroxy-4H-chromen-4-one (2e). Yield 3.92 g (87%), pale yellow crystals, mp 145.0–146.0 °C (ethanol). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 9.48 (s, 1H), 8.10 (d, J = 7.9 Hz, 1H), 7.96–7.91 (m, 1H), 7.91–7.85 (m, 1H), 7.83–7.71 (m, 2H), 7.61–7.16 (m, 12H), 5.23 (d, J = 9.4 Hz, 4H). 13C NMR (126 MHz, DMSO-d6), δ, ppm: 173.1, 154.8, 150.2, 148.2, 145.7, 138.8, 137.5, 137.3, 134.0, 128.9, 128.4, 128.3, 128.1, 125.2, 124.9, 124.4, 122.4, 121.7, 118.8, 114.2, 70.9, 70.3, 60.2. Mass spectrum, m/z (Irel, %): 451 [M + H]+ (100). Found, %: C 77.36; H 4.88. C29H22O5. Calculated, %: C 77.32; H 4.92. These data are in accordance with the literature.42
6-(4-(3-Hydroxy-4-oxo-4H-chromen-2-yl)phenoxy)hexanoic acid (2f). Yield 1.66 g (45%), yellow solid, mp 185.5–186.0 °C (ethanol
:DMF 3:1). 1H NMR (400 MHz, DMSO-d6), δ, ppm: 12.0 (bs, 1H, COOH), 10.5 (bs, 1H, OH), 8.23–8.16 (m, 2H, Ar), 8.09 (d, 1H, Ar), 7.75 (d, 2H, Ar), 7.44 (t, 1H, Ar), 7.10 (d, 2H, Ar), 4.06–4.16 (m, 2H, CH2), 2.26–2.19 (m, 2H, CH2), 1.76–1.71 (m, 2H, CH2), 1.60–1.52 (m, 2H, CH2), 1.46–1.40 (m, 2H, CH2). 13C NMR (126 MHz, DMSO-d6), δ, ppm: 179.8, 159.6, 158.8, 143.3, 138.7, 134.6, 130.0, 129.4, 128.6, 123.6, 119.7, 119.2, 72.8, 38.9, 33.6, 30.3, 29.5. Mass spectrum, m/z (Irel, %): 157.0 (12), 369.2 [M + H]+ (100). Found, %: C 68.42; H 5.50. C21H20O6. Calculated, %: C 68.47; H 5.47.
Materials and methods
1H and 13C NMR spectra (400 and 100 MHz) were recorded on Bruker Avance 400 and Varian MR-400 spectrometers in DMSO-d6. 1H and 13C chemical shifts were reported relative to residual protons and the carbon atoms of the solvent (2.49 and 39.5 ppm, respectively) as the internal standard. The LCMS spectra were recorded using a chromatography/mass spectrometric system that consists of a high-performance liquid chromatography Agilent 1100 Series equipped with a diode matrix and a mass selective detector Agilent LC/MSD SL, column SUPELCO Ascentis Express C18 2.7 μm 4.6 mm × 15 cm. Elemental analysis was realized on a EuroVector EA-3000 instrument. TLC was performed using Polychrom SI F254 plates. Melting points of all synthesized compounds were determined with a Hanon Instruments automatic melting point apparatus MP450 in open capillary tubes.According to the HPLC MS data, all synthesized compounds are >95% pure. All solvents and reagents were commercial grade and, if required, purified in accordance with the standard procedures. Starting unsaturated ketones were synthesized as described in ref. 43.
β-Glucosidase enzyme (β-D-glucoside glucohydrolase) from almonds were purchased as lyophilized powder with >98% purity from Sigma-Aldrich.
Spectroscopic measurements
UV-vis absorption spectra were measured using Agilent Cary 3500 UV-Vis Multicell Spectrophotometer. Fluorescence spectra were recorded using a Hitachi 850 steady-state fluorescence spectrometer equipped with double-grating excitation and emission monochromators. The fluorescence measurements were made in the neutral aqueous solution in a 10 × 10 mm cuvette maintained at 20 °C.Fluorescence assay for β-glucosidase activity
β-Glucosidase enzymatic activity was tested using a fluorescent indicator based on flavonol β-D-glucoside. The fluorescence assay principle relies on the enzymatic cleavage (hydrolysis) of a glycosidic bond of a weakly fluorescent indicator, which results in the release of a brightly fluorescent flavonol aglycone fluorophore that is readily extracted by an organic phase. The details of this assay and its working principles have recently been described in detail elsewhere.26,27In brief, a stock solution of a flavonol β-D-glucoside was prepared by dissolving its small aliquot in DMSO. A stock solution of β-glucosidase from almond was prepared by dissolving an enzyme in an aqueous phosphate buffer with pH = 6.86. Next, both aliquots were transferred to 1 mL of a buffer solution (pH = 6.86) and were used for kinetic studies of enzymatic cleavage. Concentrations of a flavonol β-D-glucoside indicator and β-glucosidase enzyme ranged from 5.0 to 7.0 × 10−5 mol L−1.
The enzymatic hydrolysis reaction was carried out while stirring the mixture at a constant temperature. During the hydrolysis process, after reaction initialization, aliquots of 0.1 mL were taken at 1, 2, 6, 10, 15, and 30 min. Immediately after sampling, each aliquot was transferred to 2.5 mL of dichloromethane and intensively shaken. Using extraction, a mixture of the original flavonol β-D-glucoside and its hydrolyzed derivative – a flavonol aglycone, were separated from the enzyme. Next, the fluorescence spectra of the mixture of the flavonol β-D-glucoside and the corresponding aglycone were recorded in the organic phase.
The fluorescence was excited at 360 nm and the fluorescence spectra were recorded in the range of 380–600 nm using a Hitachi 850 spectrofluorimeter. The emission intensity (IA) at the maximum of the long-wavelength fluorescence band at 525 nm was used for quantitative kinetics studies. The value of IA, which is proportional to the aglycone concentration and inversely proportional to the glucoside concentration, was used to determine the reaction order and calculate the rate constant of the enzymatic hydrolysis.
Molecular docking setup
The molecular docking setting up, addition of hydrogen, the calculation of the Gasteiger charges of the receptor and ligands were performed using the AutoDock Tools (ADT) software, version 1.5.7.44 Molecular docking calculations were performed with the AutoDock Vina 1.1.2 software.45 The 3D X-ray structure of Paenibacillus polymyxa β-glucosidase B (BglB, PDB ID: 2O9R),46 Raucaffricine β-glucosidase (rBG, PDB ID: 4A3Y),47 Thermogota maritima β-glucosidase (TmGH1, PDB ID: 1OD0),48 and human cytosolic β-glucosidase (hCBG, PDB ID: 2JFE)49 were downloaded from the RCSB Protein Data Bank.Ligand–protein interactions were studied by the semi-flexible molecular docking, so that the receptor was kept rigid and the ligand molecules were conformational flexible. The docking details of glucosidases are summarized in Table 1. Docking cell size was 45 × 45 × 45 and the grid point spacing set to 0.375 Å, respectively. For all runs, the number of binding modes was set to 9 and the exhaustiveness to 256. For each ligand, three independent runs were performed using different random seeds. The best docking mode corresponds to the largest ligand-binding affinity.
Molecular dynamic simulations
The X-ray structure of β-glucosidase was obtained from the Protein Data Bank. Co-crystalized ligands and water molecules were removed from the X-ray data. Protonation states for all titratable residues were set at their default state at pH = 7. The molecular topology of a protein was built by using pdb2gmx GROMACS tool. LigParGen web-server was used to generate the molecular topology for flavonols.50,51 The OPLS-AA force field was used for the proteins and the flavonols.52 TIP3P model53 was used to treat explicit water molecules.An initial structure of a probe–protein complex was taken from the corresponding most favourable structure obtained by the molecular docking calculations. The complex was solvated in cubic cell with a size of 9 nm. The system was first pre-equilibrated at NVT ensemble for 2 ns, during which probe and protein structure was positionally restrained. Second, the free unconstrained equilibration of a probe–protein complex was carried out for 0.1 ns at an NPT ensemble, during which a MD box cell size was allowed to relax. The final equilibrated configuration of the system was used to sample productive MD simulations for 200 ns.
All productive MD simulations were carried out for an NPT ensemble. The reference temperature of 298.15 K was kept constant using the v-rescale weak coupling scheme54 with the coupling constant τT = 0.1 ps. The constant pressure of 1 atm was maintained using the Parrinello–Rahman barostat with the coupling constant τP = 1 ps. The initial atomic velocities were generated with a Maxwellian distribution at the given absolute temperature. Periodic boundary conditions were applied to all three directions of the simulated box. Electrostatic interactions were simulated with the particle mesh Ewald (PME) approach55 using the long-range cut-off of 1.1 nm. The cut-off distance of Lennard–Jones interactions was also equal to 1.1 nm. The MD simulation time step was 2 fs with the neighbour list updates every 10 fs. All bond lengths were kept constant using the LINCS routine.56,57 The MD simulations were carried out using the GROMACS set of programs, version 2019.4.58
Molecular graphics and visualization were performed using VMD 1.9.3.59
Results and discussion
Spectral properties of flavonols 2a–f
The optical properties of flavonol derivatives have been examined in various solvents.4,11,60 A numerous studies demonstrated that their fluorescence properties are essentially dependent on the environment, so that upon going from non-polar solvents, such as hexane, to polar protic water solution, their fluorescence quantum yield decreases significantly from 0.4 up to 0.02, respectively.10,11,26,27 Table 2 summarizes the spectral properties of flavonols 2a–f in water. A key feature of emission of 2a–f is a high Stokes shift of their long-wavelength band varying from 8100 up to 9925 cm−1, which originates from the ESIPT tautomer.| Flavonol | Absorption maximum, nm | Fluorescence maximum (normal/tautomer), nm | Fluorescence Stokes shift, cm−1 |
|---|---|---|---|
| 2a | 343 | 520 | 9925 |
| 2b | 366 | 540 | 8800 |
| 2c | 355 | 540 | 9650 |
| 2d | 362 | 445/540 | 5150/9100 |
| 2e | 379 | 443/547 | 3810/8100 |
| 2f | 356 | 445/542 | 5620/9640 |
Fluorescence studying protein–flavonol binding interaction
Fluorescence properties of flavonols 2a–f were studied in aqueous neutral solution in the presence and the absence of β-glucosidase (Fig. 1). Despite the wide use of flavonols in biomedical applications, their solubility in water is known to be very low, being about (0.12, 0.5, and <0.01) g L−1 at 20 °C for rutin, naringin, and quercetin, respectively.61,62 Therefore, the flavonols were added into the solution as a small aliquot of their DMSO stocks. Next, to study flavonol–enzyme binding interaction, we utilized fluorescence titration protocol, in which the concentration of flavonols in water solution was fixed at ∼1–2 × 10−5 M and the concentration of glucosidase was varied in a range from 0 up to 7 × 10−4 M, respectively.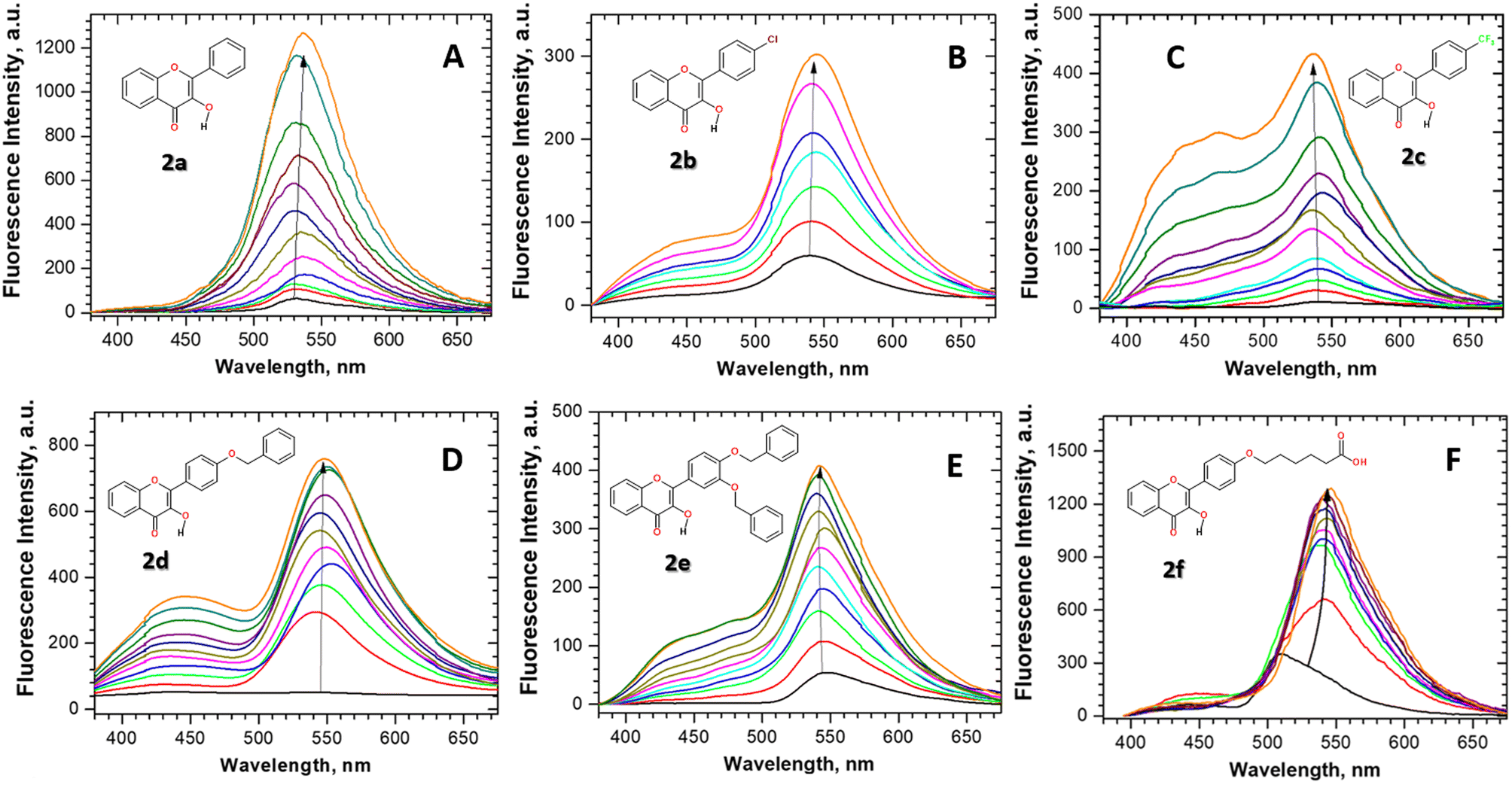 | ||
| Fig. 1 (A–F) Fluorescence titration of flavonols 2a–f with β-glucosidase in the neutral aqueous solution at 298 K. The fluorescence was excited at 360 nm. The arrow corresponds to increase in the fluorescence intensity of the flavonols upon the increase in β-glucosidase concentration. In the typical titration experiment, the flavonol concentration was kept fixed at ∼1–2 × 10−5 M, whereas the β-glucosidase concentration was varied in a range from 0.5 × 10−5 up to 7 × 10−4 M, respectively. | ||
Fluorescence titration experiments and emission spectra of flavonol 2a–f in the aqueous solution are summarized in Fig. 1. As can be seen, significant fluorescence changes, such as fluorescence enhancement and the appearance of new emission bands, were observed for flavonol 2a–f upon titration with β-glucosidase stocks. The increase in the fluorescence intensity of an environment-sensitive probe is known as the protein-induced “turn-on” effect. The observed spectral changes of flavonols 2a–f are indicative of changes in the local environment around the probe. Upon binding to the protein, a flavonol probe leaves a polar aqueous solution and penetrates a nonpolar hydrophobic environment inside a protein pocket. The fluorescence increase is associated with the loss of water-induced fluorescence quenching of 2a–f, which might be due to deep penetration of the flavonols into the hydrophobic, water-free region of the β-glucosidase enzyme pocket.
The detailed analysis of spectral changes in the studied flavonols demonstrates that the titration of 2a was mainly accompanied by the increase in the emission of the tautomer band at 545 nm (Fig. 1A). In contrast, more complex spectral changes were observed for the cases of substituted flavonols 2b–f. These changes included the redistribution of normal-to-tautomer intensity at 440/545 nm and the appearance of the new band at 514 nm (Fig. 1B–F). Measuring the emission spectrum at pH > 10 allowed us to assign that the new emission band at 514 nm belongs to an anionic form (A) of a flavonol (Fig. 2).
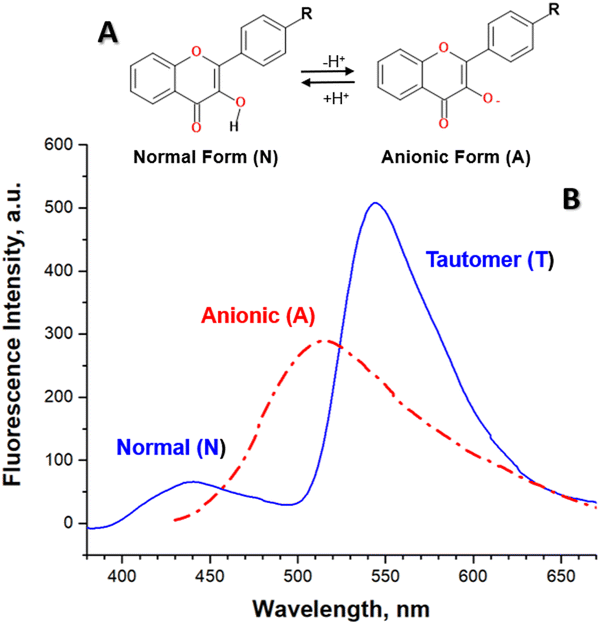 | ||
| Fig. 2 (A) Scheme of acid–base equilibrium in flavonols in an aqueous solution. (B) The fluorescence spectra of flavonol 2d in the neutral (excitation at 360 nm, solid blue) and the base aqueous solution (excitation at 420 nm, dash-dotted red). | ||
The appearance of acid–base equilibrium in flavonols (Fig. 2A) has been observed for its other derivatives in aqueous and non-aqueous polar protic solvents.9–11
The quantitative interpretation of protein–ligand interactions can be analysed using site-specific or nonspecific binding models, such as Michaelis–Menten, Hill or Scatchard formalisms.63–66 Taking into account that the binding behaviour of numerous ligands to enzymes of a glucosidase family has been well-characterized and, in most cases, demonstrated the single-site specific binding to the primary catalytic pocket,67–71 we first considered “one site-specific binding” model.63
Relative fluorescence intensities of flavonol 2a–f were plotted as a function of the concentration of β-glucosidase, as shown in Fig. 3.
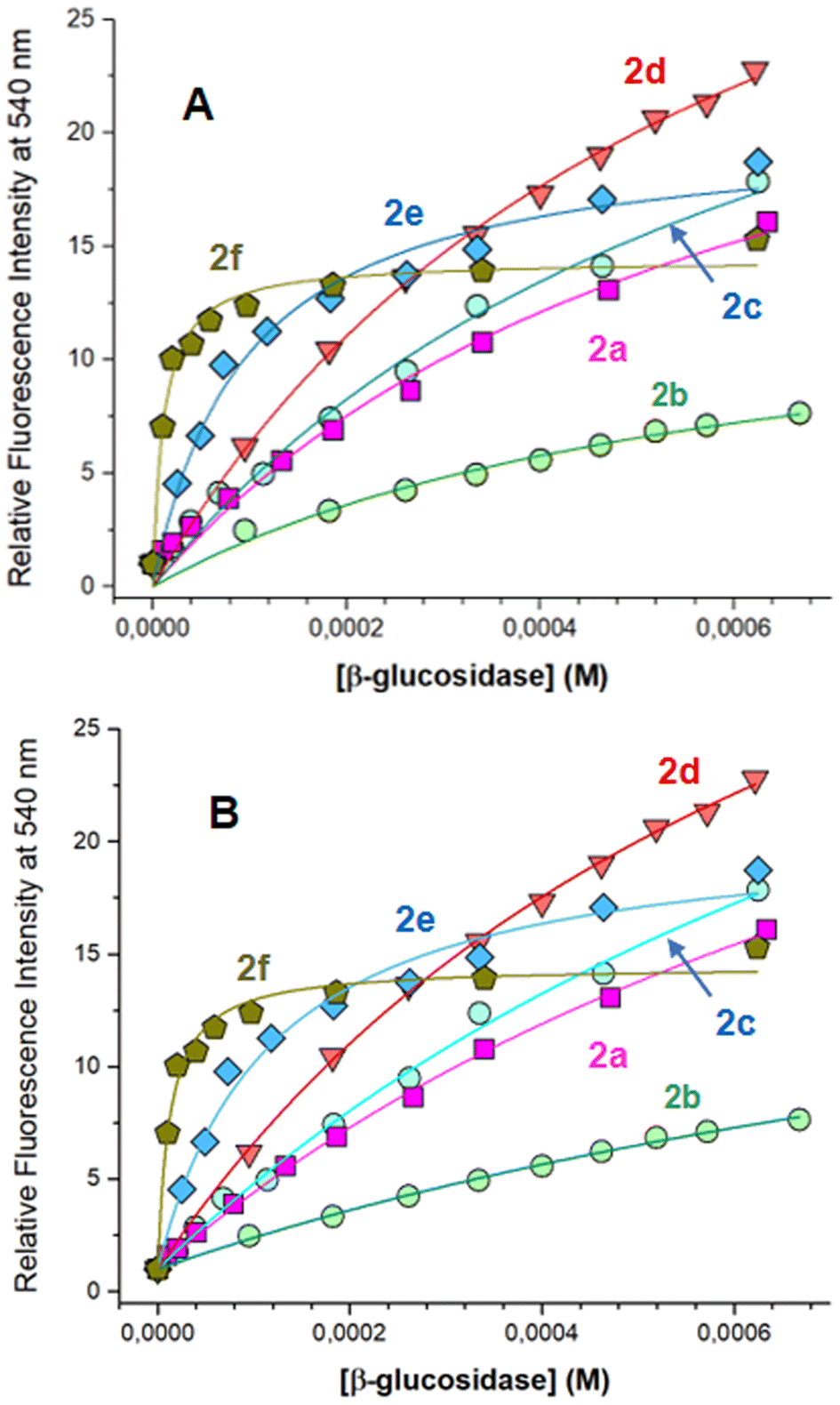 | ||
| Fig. 3 Relative fluorescence intensities of flavonol 2a–f in aqueous solution are plotted as a function of the concentration of β-glucosidase. Relative intensities (Ii/I0, Ii is the apparent fluorescence intensity in the presence of β-glucosidase, and I0 is the intensity in the solution with no β-glucosidase) were determined at fixed wavelengths of 540 nm. Titration profiles were fitted to eqn (1) (A) and eqn (3) (B) to determine the binding parameters, as summarized in Tables 3 and 4. | ||
First, the fluorescence titration data were analyzed using nonlinear regression with “one site-specific binding” model by eqn (1):
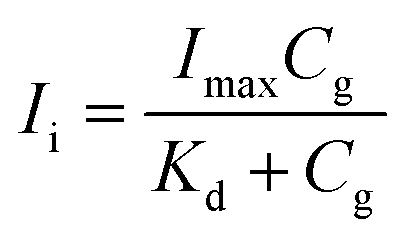 | (1) |
The binding constant of ligand–protein interaction can be expressed by eqn (2).
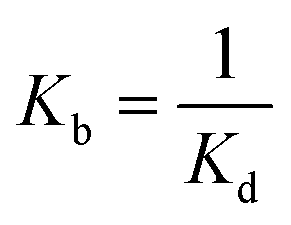 | (2) |
Fig. 3A shows the titration data were well fitted by “one site-specific binding” model using eqn (1). From these data, the binding constant Kb for flavonol–enzyme interaction was summarized in Table 3.
| Flavonol | Imax | Binding constant Kb, mM |
|---|---|---|
| 2a | 30.9 | 1.61 × 103 |
| 2b | 14.4 | 1.69 × 103 |
| 2c | 36.3 | 1.47 × 103 |
| 2d | 43.9 | 1.67 × 103 |
| 2e | 20.2 | 1.04 × 104 |
| 2f | 14.4 | 1.01 × 105 |
As seen from Table 3, introducing substituents 4′-Cl and 4′-CF3 into a flavonol moiety has little effect on the binding constant of flavonols 2b and 2c, respectively. Surprisingly, small changes in the binding constant were also observed after introducing the bulk 4′-O-benzyl group in flavonol 2d. However, a significant increase in the binding rate was found for flavonols 2e and 2f, bearing large and bulky 3′,4,′-dibenzyloxy and 4′-hexylcarboxylic fragments (Table 3). These findings suggest that in the studied series, the binding interaction with β-glucosidase was mainly driven by van-der-Waals interactions and hydrophobic forces.
Considering the critical role of hydrophobic forces in the flavonol–enzyme binding, the interactions between a flavonol probe and the enzyme can alternatively be regarded as probe partitioning from the aqueous solution into the hydrophobic region of the protein. Therefore, we re-analyzed our titration experiments by using partitioning formalism.
The apparent partition coefficient Kp was determined by fitting fluorescence intensities, Ii, measured at the fixed wavelength 540 nm, to eqn (3):
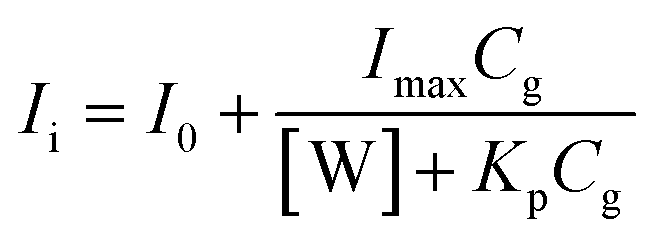 | (3) |
|
ΔG = −RTlnKp
| (4) |
Fig. 3B shows the analysis of the fluorescence titration data by a “partitioning” model using eqn (3). As can be seen, the titration data were well fitted by eqn (3). One can note that interactions of flavonols 2a–c with β-glucosidase were characterized by the similar partitioning coefficient Kp (Table 4). Whereas flavonols 2e–f favor the partitioning from water to the protein environment. The same trends are seen in the Gibbs free energy ΔG of partitioning, so that flavonols 2a–d are characterized with ΔG from −6.3 to −6.6 kcal mol−1, whereas the essential increase ΔG up to −7.7 and −9.1 kcal mol−1 observed for 2e and 2f (Table 4), respectively.
Protein homology in β-glucosidase family
Because the high-resolution 3D structure of commercial almonds β-glucosidase used in our experimental studies is currently unavailable, we first considered the available X-ray glucosidase structures from other sources. β-Glucosidase family includes enzymes with different activities widely distributed among all sorts of living organisms, which are capable of the common ability to hydrolyze β-glucosidic linkages of disaccharides, oligosaccharides or conjugated saccharides.72 Among the members of this family are bacterial and fungal cellobiases that play an essential role in cellulolytic hydrolysis.73Therefore, to carry out molecular docking analysis of flavonol–glucosidase interactions, we used β-glucosidase enzymes taken from various sources, such as Paenibacillus polymyxa β-glucosidase B (BglB), Raucaffricine β-glucosidase (rBG), Thermogota maritima β-glucosidase (TmGH1) and human cytosolic β-glucosidase (hCBG), respectively. Despite the difference in amino acid residues around their glycone binding pockets, the sequence alignment of these enzymes revealed a high identity of above 90%, which is characteristic of the many members of this family.46,49,73,74
Fig. 4 shows the structure overlap of three different β-glucosidases. Despite some structure variations seen at peripheral protein regions, all enzymes are characterized by main common features: (i) they have a deep hydrophobic pocket capable of accommodating substrate molecules during cellulolytic hydrolysis (Fig. 4). (ii) The catalytic active site is composed of two glutamate (Glu) residues placed in close proximity to one another (Fig. 4). The 3D structure of BlgB, TmGH1, rBG, and hCBG glucosidases is well-resolved, so that these structures have already been used as a receptor model for molecular docking calculations.75–79
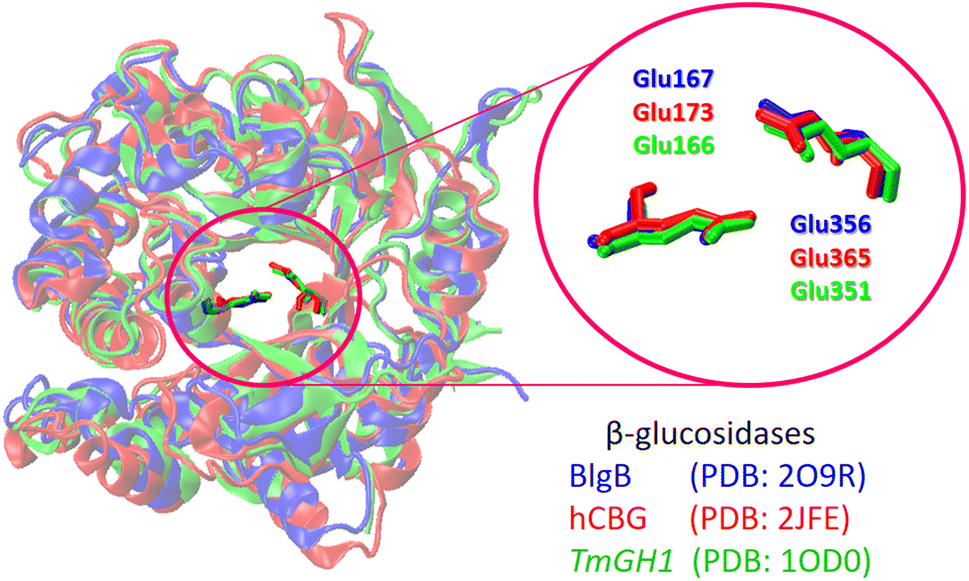 | ||
| Fig. 4 Overlap of the protein structure and the conservative catalytic residues for β-glucosidases from different sources. The enzyme and the corresponding catalytic Glu residues are shown as color-coded sticks. | ||
Molecular docking of flavonols against β-glucosidase
The structural and energetic parameters of flavonol interactions with β-glucosidases were examined using four different protein structures (Table 1). Molecular docking calculations demonstrate that all studied flavonols bind strongly and are able to insert deeply into a central cavity of β-glucosidase. Examples of binding modes of flavonols 2a–f are shown for Thermogota maritima β-glucosidase (TmGH1) in Fig. 5 and 6. Molecular docking suggests that the binding affinity of 2a–f towards β-glucosidase depends on the nature of peripheral substituents R and R1 located in the 4′-aryl ring of the flavonols (Scheme 2). On the other hand, the choice of the β-glucosidase structure has little effect. Molecular docking results of flavonols 2a–f with β-glucosidase from four different sources are summarized in Table 5.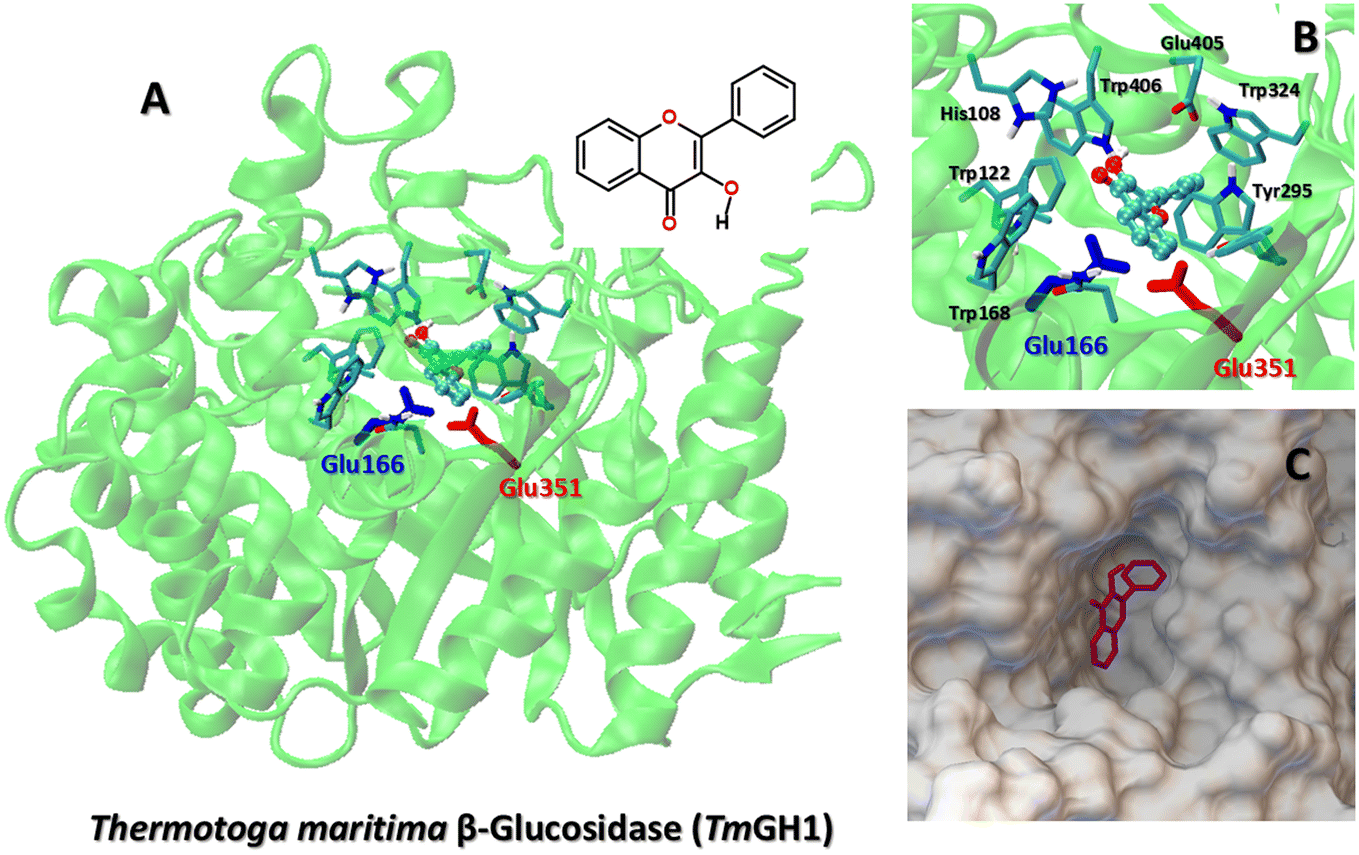 | ||
| Fig. 5 (A) The best binding mode of flavonol 2a to Thermogota maritima β-glucosidase (TmGH1, PDB 1OD0). The catalytic residues Glu166 and Glu351 are shown as color-coded sticks. (B) The docking pose of flavonol 2a in the enzyme pocket of TmGH1. (C) The insert shows the binding mode of 2a and key enzyme residues. | ||
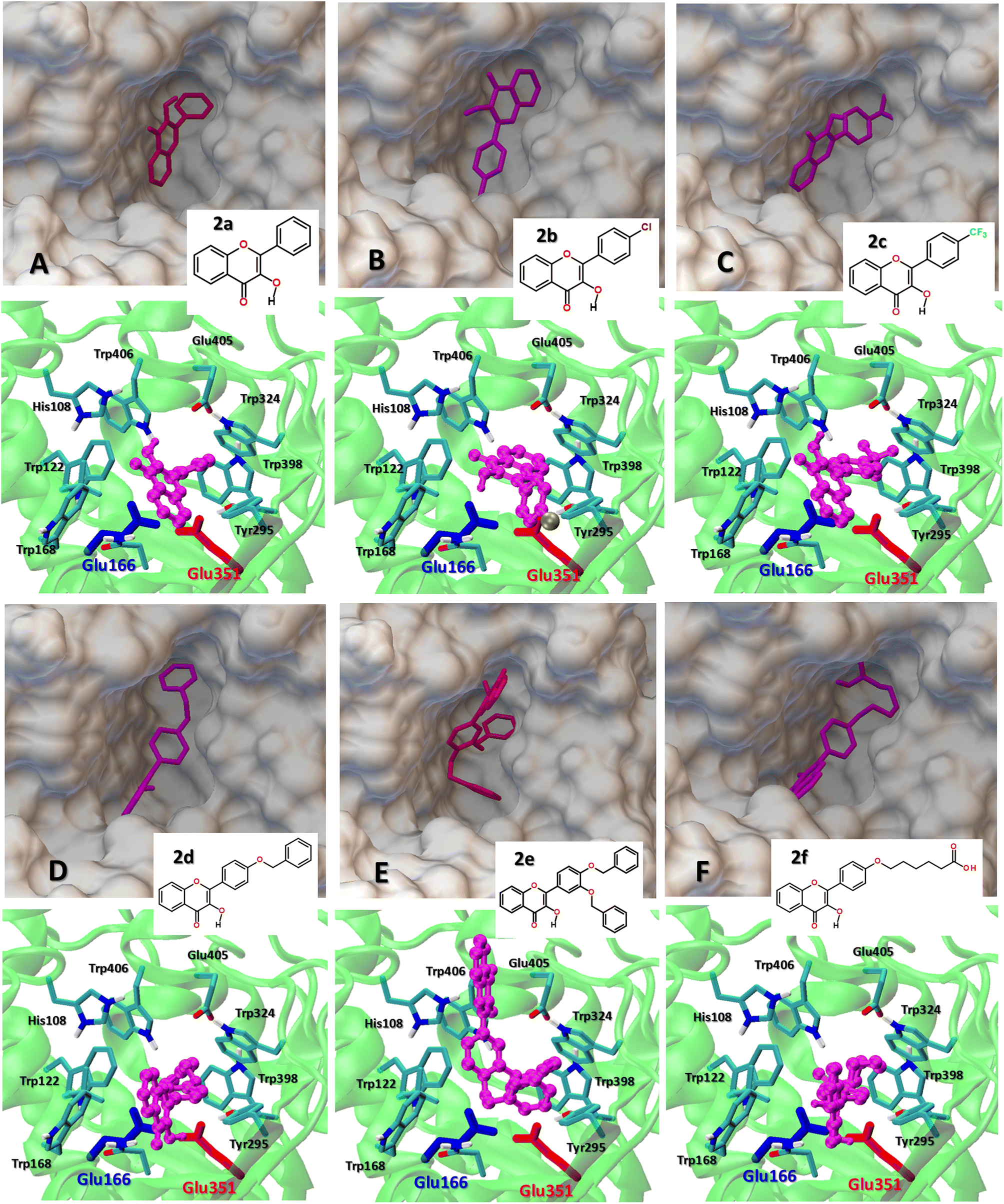 | ||
| Fig. 6 (A–F) The comparison of the best binding mode of the studied flavonol 2a–f at Thermogota maritima β-glucosidase (TmGH1, PDB 1OD0). The catalytic residues Glu166 and Glu351 are shown as color-coded sticks. (Top) The insert shows the top view of the location of flavonols 2a–f within the protein binding pocket. (Bottom) The side view of the flavonol–enzyme binding interaction within the active center region. | ||
| Flavonol | Binding affinity, kcal mol−1 | |||
|---|---|---|---|---|
| Human cytosolic | Paenibacillus polymyxa | Thermotoga maritima | Raucaffricine | |
| β-Glucosidase | β-Glucosidase | β-Glucosidase | β-Glucosidase | |
| (PDB: 2JFE) | (PDB: 2O9R) | (PDB: 1OD0) | (PDB: 4A3Y) | |
| Studied flavonols | ||||
| 2a | −8.8 | −8.2 | −8.5 | −8.8 |
| 2b | −9.0 | −8.4 | −8.3 | −8.6 |
| 2c | −9.8 | −9.1 | −9.2 | −9.6 |
| 2d | −10.4 | −9.0 | −9.3 | −9.3 |
| 2e | −11.6 | −11.1 | −11.0 | −9.6 |
| 2f | −8.7 | −9.1 | −9.5 | −8.7 |
|
||||
| Known inhibitors | ||||
| Noeuromycin | −6.1 | −6.8 | −6.3 | −6.8 |
| Isofagomine | −5.7 | −6.0 | −5.5 | −5.8 |
| Cas1 | −7.4 | −7.7 | −7.8 | −7.5 |
| Cas2 | −7.0 | −7.7 | −7.6 | −7.4 |
| Imd1 | −5.8 | −6.8 | −6.2 | −6.0 |
| Imd2 | −8.2 | −9.6 | −9.2 | −8.0 |
One can note that, depending on the nature of the substitutions R and R1, the binding affinity of flavonols 2a–f varies in a range from −8.2 up to −9.6 kcal mol−1. The etherification of the peripheral 3′ and 4′-hydroxyl groups in the 2-aryl moiety with the hydrophobic benzyl substituent significantly increases the affinity of the flavonols 2d–e towards the β-glucosidase enzymes, so that the binding affinity of 2e increases up to −11.7 kcal mol−1 (Table 5).
TmGH1 enzyme has a deep hydrophobic pocket with an active site (about 16 Å deep) (Fig. 5 and 6). The two catalytic glutamate residues, the acid/base Glu166 and the nucleophile Glu352 are located deep within the active site. It can be seen that all studied flavonols 2a–f bind close to the catalytic dyad Glu166–Glu351. The ligand binding is driven by hydrophobic interactions with aromatic Trp122, Trp168, Trp295, Trp324, Trp398, Trp406 as well as Tyr295, respectively. Moreover, the receptor-bound flavonols 2a–f shield the active site residues Glu166 and Glu351 and restrict the accessibility of other substrate molecules toward the catalytic center. Such a binding mode of 2a–f holds promise of their high inhibitory potency against glucosidase activity.
It has been shown that the binding of bulk-size ligands into the glucosidase active site was able to inhibit the hydrolyze-type activity of these enzymes. The inhibition of α-glucosidases by natural flavonoids, such as combretol, morin,80,81 myricetin,68,81,82 ombuin, quercetin,31,68,80,82 and retusin,31 has been reported by using experimental and computational techniques. In addition, there are some reports that ligand binding within the enzyme cavity had complex multi-mode behavior, so that substrate binding occurred into different sub-sites of the β-glucosidase pocket.83 Some studies reported that catalytic Glu residues did not interact with the scissile glycosidic bond of a ligand, indicating a shallow binding mode, where the substrate initially binds before moving into the active site. Such a mode has been reported for β-glucosidases in several GH families.84 Moreover, it has been shown that the binding thermodynamics parameters of 1-deoxynojirimycin and isofagomine inhibitors towards almond β-glucosidase and Thermogota maritima β-glucosidase (TmGH1) have similar values,48 so despite the absence of the high-resolution X-ray structure of almond β-glucosidase, it can be well modeled by the corresponding TmGH1 structure.
Therefore, we compared the molecular docking binding affinity of the studied flavonols 2a–f with those of well-known inhibitors. For this purpose, we performed comparative re-docking of a series of structurally different inhibitors, summarized in Scheme 3. Compounds Imd1 and Imd2 are known to be among the most potent glycosidase inhibitors reported to date. It has been demonstrated that adding the hydrophobic phenethyl moiety in Imd2 improves its binding by 20–80-fold, possibly due to significantly better entropic contributions to binding.85
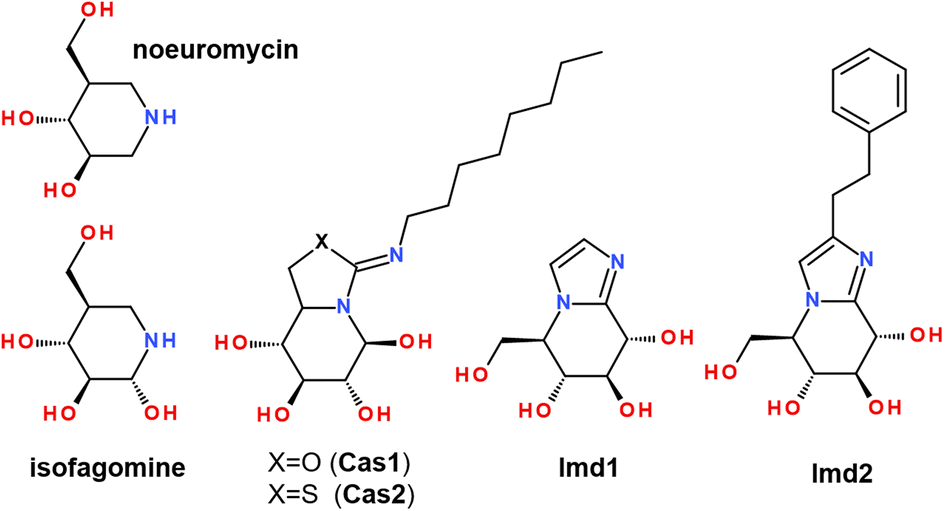 | ||
| Scheme 3 Structure of the known inhibitors of enzymes from the β-glucosidase family. | ||
It has been noticed that the peripheral phenyl rings of an inhibitor molecule could be faced towards the solvent-exposed region of an enzyme, which played an essential effect in anti-β-glucosidase activity.34 Our molecular docking demonstrates that, in terms of the binding affinity, the studied flavonols are among strong β-glucosidase binders with the comparable and higher affinity than available inhibitors Imd2 and Cas2 (Table 5).
Determining β-glucosidase activity
To test if the strong binding affinity of the hydrophobically-modified flavonols, such as 2e and 2f, led to steric blockage of the enzymatic activity of β-glucosidase, we used flavonol-based fluorescent indicator – flavonol β-D-glucopyranoside 3 (Fig. 7).26,27 The indicator has a β-D-glucopyranoside moiety attached to the 3-OH group of flavonol and it is characterized by weak emission. However, upon the enzymatic cleavage of the glycosidic bond, the indicator emission is essentially enhanced due to the appearance of bright aglycone fluorescence.26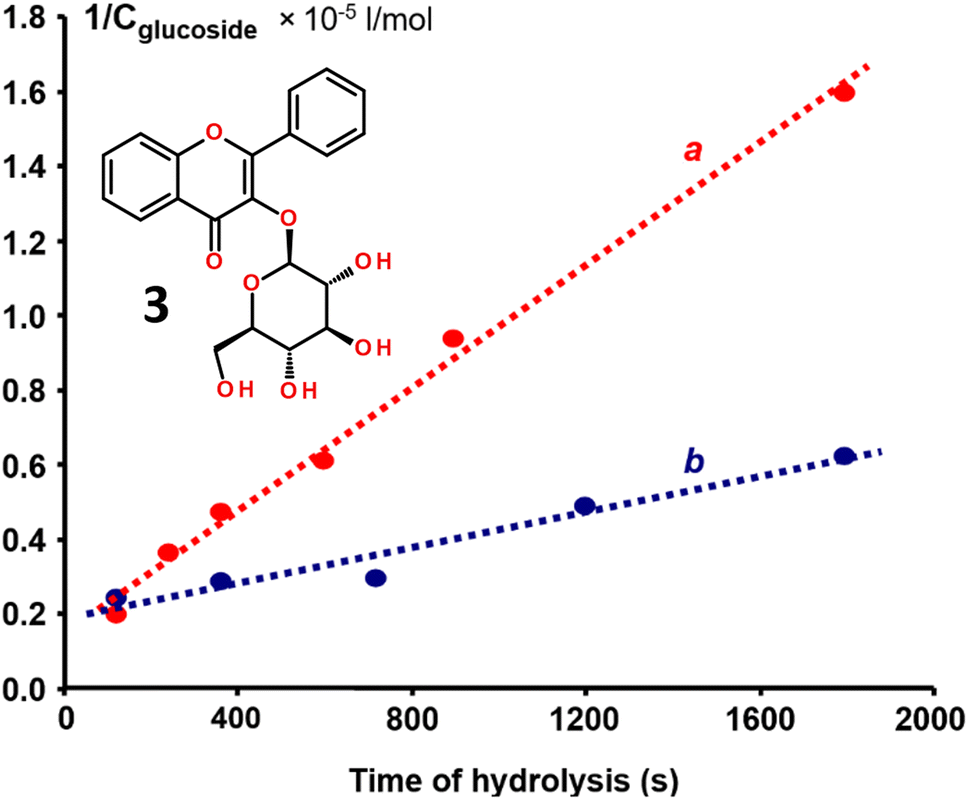 | ||
| Fig. 7 The 2nd order kinetics plots for the enzymatic cleavage reaction of a fluorescent indicator, 3-hydroxyflavone glucoside 3 by β-glucosidase: (a) the free enzyme without any inhibitor and (b) the enzyme in the presence of flavonol 2e. | ||
The enzymatic activity of β-glucosidase was measured using a flavonol β-D-glucopyranoside indicator 3 in the absence and the presence of flavonol 2e. The latter experiment was carried out by incubating β-glucosidase solution with flavonol 2e for up to 1 h, followed by the enzymatic activity test.
The kinetic analysis of the enzymatic reaction demonstrated that, for the free β-glucosidase, the reaction rate constant k of the indicator hydrolysis was found to be 81.52 L mol−1 s−1 (Fig. 7). After the binding flavonol 2e to β-glucosidase, re-measuring of the enzyme activity revealed that the rate constant k was decreased to 23.46 L mol−1 s−1, indicating to a 3.5-time decrease in the enzymatic activity caused by flavonol 2e.
Molecular mechanism of flavonol–enzyme interactions
While molecular docking provides valuable information about the favorable binding mode and the ligand affinity towards an enzyme, its major drawback is the lack of explicit solvation effects. Therefore, the stability of the docking-derived flavonol–enzyme complexes in bulk water solution was further re-examined by all-atom molecular dynamics (MD) simulations (Fig. 8A). MD simulations can provide detailed information about the conformational changes and fluctuations in a protein and a ligand.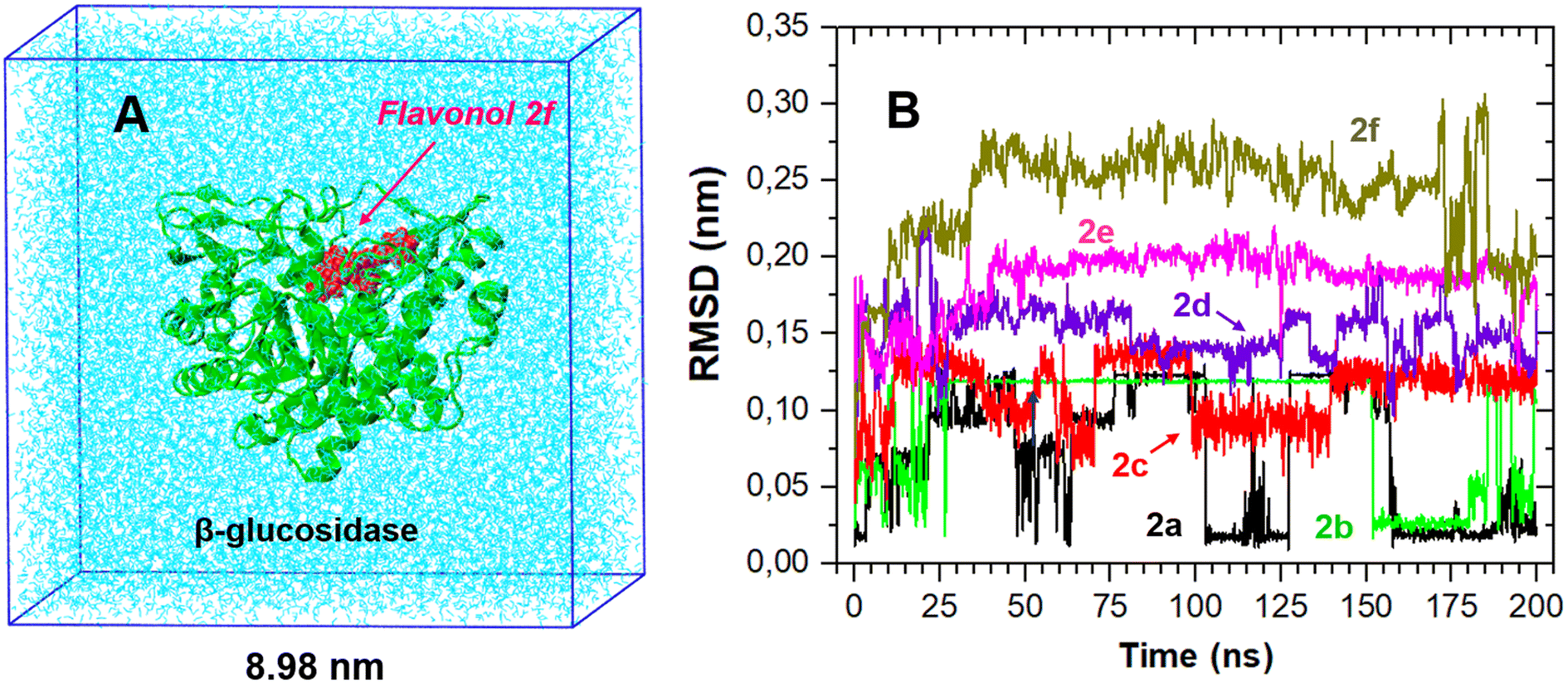 | ||
| Fig. 8 (A) An example of MD simulations of the flavonol–enzyme complex for 2f in explicit water solution. The initial structure of the complex was taken as the most favorable structure from the molecular docking calculations. (B) RMSD time traces of the coordinate positions of flavonols 2a–f with the respect to their initial docked structure. | ||
The best-docked conformation of flavonols 2a–f based on molecular docking was used as the initial conformation for MD sampling. The structural stability of the bound conformation of flavonol in its complex with the enzyme was evaluated by calculating the root mean square displacement (RMSD) over 200 ns. RMSD measures the deviation of a set of current coordinates of a molecule to a reference set of coordinates. RMSD time traces for the given flavonol were calculated by least-square fitting the ligand structure (τ2) to its initially-docked structure (τ1 = 0) by eqn (5):
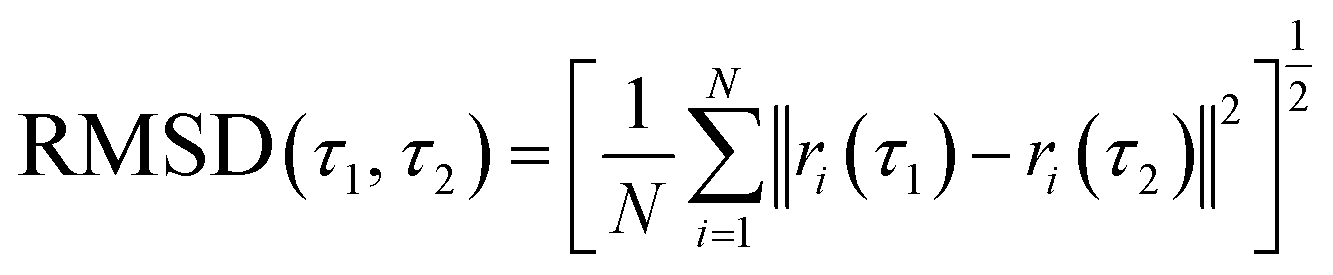 | (5) |
The RMSD time traces for flavonols 2a–f in its complex with β-glucosidase are shown in Fig. 8B. The analysis of these time traces demonstrates different mobility of the studied compounds within the ligand–protein complex; however, all studied complexes remained stable during the MD sampling. The RMSD plots suggest that flavonols 2a–c were located near their initial docked positions during all time. Flavonols 2d–f revealed some high-amplitude mobility, which indicates some changes in their conformations and binding locations concerning the initially docked poses.
The above findings suggest that an energy balance between hydrophobic van-der-Waals and electrostatic interactions drove the observed strong binding of 2d–f to β-glucosidases. Therefore, to examine the polar fragments and hydrophobic spots on the surface of the studied flavonols 2a–f, we performed DFT calculations of their ground-state structure and electronic properties, as summarized in Fig. 9. The ESP maps demonstrate that flavonols 2a–f have some polar regions near the carbonyl oxygen atoms of a chromone moiety. In contrast, most surface areas of probes have a relatively neutral hydrophobic character. These findings further support their strong hydrophobically-driven binding behavior towards the protein environment.
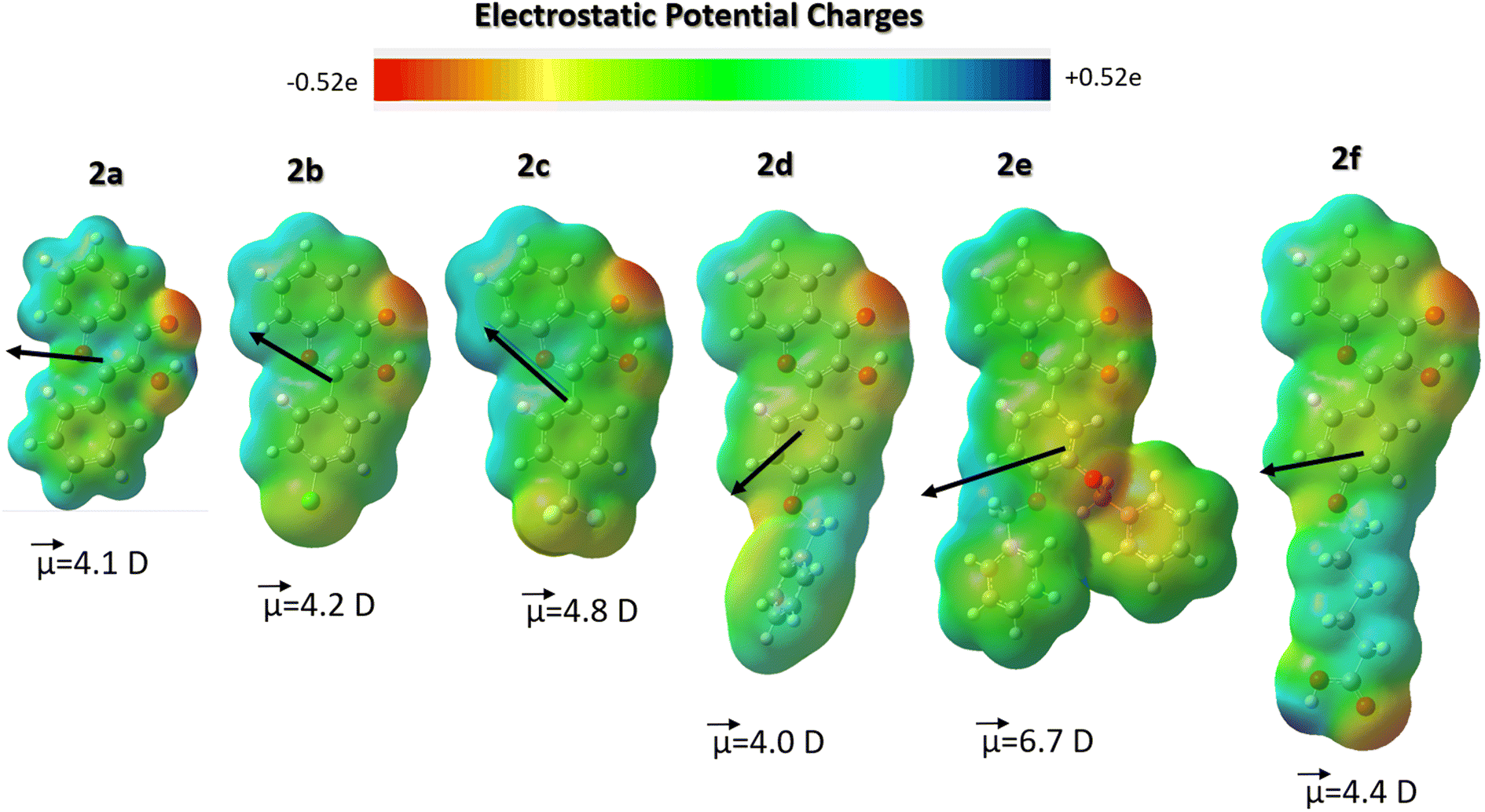 | ||
| Fig. 9 Comparison of electrostatic potential (ESP) maps with an isosurface value of 0.010 e Å−3, showing charge density distributions across flavonols 2a–f. A ground-state dipole moment is shown as a vector and its absolute value is given in Debye (D). The DFT calculations were carried out at the B3LYP/cc-pVDZ level. | ||
Summary and perspectives
In summary, we have synthesized a series of fluorescent flavonol derivatives 2a–f containing different polar halogen-containing groups or bulky aromatic/alkyl substituents in the 2-aryl ring with the goal of tuning their hydrophobic/hydrophilic interactions with β-glucosidase enzyme. Using fluorescence titration experiments, we found that, upon adding almonds β-glucosidase, all studied flavonols revealed protein-induced fluorescence “turn-on” effects, which is indicative of their binding to the enzyme. The quantitative analysis of the binding isotherms demonstrated that flavonols 2e and 2f had the highest binding constants compared to unsubstituted or halogen-substituted derivatives 2a and 2b, which suggests that hydrophobic forces mostly drove the strong binding of 2e and 2f to β-glucosidase. In terms of the binding affinity, the studied flavonols belong to strong β-glucosidase binders, so they may also inhibit the enzyme activity. Indeed, using a fluorescent assay, we demonstrated that the binding of flavonol 2e to β-glucosidase decreased its enzymatic activity up to 3.5 times.Molecular aspects of flavonol–enzyme interactions were in-depth analyzed by molecular docking calculations, which demonstrate that the studied flavonols bind to the catalytic active site of β-glucosidase, so that they block and shield the key catalytic Glu residues of the enzyme from contacts with other substrate molecules. In addition, these calculations further support our suggestion that the substrate binding is governed by hydrophobic interactions with several aromatic Trp and Tyr residues within the catalytic glycone binding pockets of β-glucosidase. In addition, all-atom MD simulations of the docked conformations of flavonols 2a–f in its complex with β-glucosidase in explicit water solution confirmed their high structural stability. Finally, we believe that our study provides a novel insight into structure–property relations for flavonol–protein interactions, which drives and governs their enzyme binding and outlines a framework for a rational design of new flavonol-based potent inhibitors for β-glucosidase enzymes.
Author contributions
Liudmyla V. Chepeleva: methodology, data, writing – original draft. Oleksii O. Demidov: data, resources, writing – original draft. Arsenii D. Snizhko: data, resources. Dmytro O. Tarasenko: data, resources. Andrii Y. Chumak: data, resources. Oleksii O. Kolomoitsev: methodology, resources, writing – original draft. Volodymyr M. Kotliar: conceptualization, methodology. Eugene S. Gladkov: methodology, writing – original draft. Alexander Kyrychenko: conceptualization, methodology, writing – review & editing. Alexander D. Roshal: conceptualization, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. O. T., O. O. K., A. Y. C., E. S. G., A. V. K., A. D. R. acknowledge Grant 2020.02/0016 (0121U112517) “Indicators based on chromone derivatives for fluorescent determination of β-glucosidase activity” from the National Research Foundation of Ukraine.References
- I. E. Serdiuk and A. D. Roshal, Exploring double proton transfer: a review on photochemical features of compounds with two proton-transfer sites, Dyes Pigm., 2017, 138, 223–244 CrossRef CAS.
- A. S. Klymchenko, Solvatochromic and Fluorogenic Dyes as Environment-Sensitive Probes: Design and Biological Applications, Acc. Chem. Res., 2017, 50, 366–375 CrossRef CAS.
- S. Höfener, P. C. Kooijman, J. Groen, F. Ariese and L. Visscher, Fluorescence behavior of (selected) flavonols: a combined experimental and computational study, Phys. Chem. Chem. Phys., 2013, 15, 12572–12581 RSC.
- A. Y. Chumak, V. O. Mudrak, V. M. Kotlyar and A. O. Doroshenko, 4′-Nitroflavonol fluorescence: excited state intramolecular proton transfer reaction from the non-emissive excited state, J. Photochem. Photobiol., A, 2021, 406, 112978 CrossRef CAS.
- Z. Xu, X. Zhao, M. Zhou, Z. Zhang, T. Qin, D. Wang, L. Wang, X. Peng and B. Liu, Donor engineering on flavonoid-based probes to enhance the fluorescence brightness in water: design, characterization, photophysical properties, and application for cysteine detection, Sens. Actuators, B, 2021, 345, 130367 CrossRef CAS.
- T. Qin, B. Liu, Y. Huang, K. Yang, K. Zhu, Z. Luo, C. Pan and L. Wang, Ratiometric fluorescent monitoring of methanol in biodiesel by using an ESIPT-based flavonoid probe, Sens. Actuators, B, 2018, 277, 484–491 CrossRef CAS.
- T. Qin, B. Liu, Z. Xu, G. Yao, H. Xu and C. Zhao, Flavonol-based small-molecule fluorescent probes, Sens. Actuators, B, 2021, 336, 129718 CrossRef CAS.
- A. D. Roshal, J. A. Organero and A. Douhal, Tuning the mechanism of proton-transfer in a hydroxyflavone derivative, Chem. Phys. Lett., 2003, 379, 53–59 CrossRef CAS.
- I. E. Serdiuk and A. D. Roshal, Single and double intramolecular proton transfers in the electronically excited state of flavone derivatives, RSC Adv., 2015, 5, 102191–102203 RSC.
- V. G. Pivovarenko, Multi-parametric sensing by multi-channel molecular fluorescent probes based on excited state intramolecular proton transfer and charge transfer processes, BBA Adv., 2023, 3, 100094 CrossRef CAS.
- A. O. Doroshenko, A. V. Kyrychenko, O. M. Valyashko, V. N. Kotlyar and D. A. Svechkarev, 4′-Methoxy-3-hydroxyflavone excited state intramolecular proton transfer reaction in alcoholic solutions: intermolecular versus intramolecular hydrogen bonding effect, J. Photochem. Photobiol., A, 2019, 383, 111964 CrossRef CAS.
- A. P. Demchenko, S. Ercelen, A. D. Roshal and A. S. Klymchenko, Excited-State Proton Transfer Reaction in a New Benzofuryl 3-Hydroxychromone Derivative: The Influence of Low-Polar Solvents, Pol. J. Chem., 2002, 76, 1287–1299 CAS.
- V. F. Valuk, G. Duportail and V. G. Pivovarenko, A wide-range fluorescent pH-indicator based on 3-hydroxyflavone structure, J. Photochem. Photobiol., A, 2005, 175, 226–231 CrossRef CAS.
- X. Poteau, G. Saroja, C. Spies and R. G. Brown, The photophysics of some 3-hydroxyflavone derivatives in the presence of protons, alkali metal and alkaline earth cations, J. Photochem. Photobiol., A, 2004, 162, 431–439 CrossRef CAS.
- W. Liu, Y. Wang, W. Jin, G. Shen and R. Yu, Solvatochromogenic flavone dyes for the detection of water in acetone, Anal. Chim. Acta, 1999, 383, 299–307 CrossRef CAS.
- D. Svechkarev, A. Kyrychenko, W. M. Payne and A. M. Mohs, Probing the self-assembly dynamics and internal structure of amphiphilic hyaluronic acid conjugates by fluorescence spectroscopy and molecular dynamics simulations, Soft Matter, 2018, 14, 4762–4771 RSC.
- B. Liu, Y. Pang, R. Bouhenni, E. Duah, S. Paruchuri and L. McDonald, A step toward simplified detection of serum albumin on SDS-PAGE using an environment-sensitive flavone sensor, Chem. Commun., 2015, 51, 11060–11063 RSC.
- K. A. Bertman, C. S. Abeywickrama, H. J. Baumann, N. Alexander, L. McDonald, L. P. Shriver, M. Konopka and Y. Pang, A fluorescent flavonoid for lysosome detection in live cells under “wash free” conditions, J. Mater. Chem. B, 2018, 6, 5050–5058 RSC.
- X. Jin, X. Sun, X. Di, X. Zhang, H. Huang, J. Liu, P. Ji and H. Zhu, Novel fluorescent ESIPT probe based on flavone for nitroxyl in aqueous solution and serum, Sens. Actuators, B, 2016, 224, 209–216 CrossRef CAS.
- K. A. Bertman, C. S. Abeywickrama, A. Ingle, L. P. Shriver, M. Konopka and Y. Pang, A Fluorescent Flavonoid for Lysosome Imaging: the Effect of Substituents on Selectivity and Optical Properties, J. Fluoresc., 2019, 29, 599–607 CrossRef CAS.
- I. E. Serdiuk, M. Reszka, A. Synak, B. Liberek and P. Bojarski, Determination of low-activity hydrolases using ESIPT fluorescent indicators on silver surfaces, Dyes Pigm., 2018, 149, 224–228 CrossRef CAS.
- S. Concilio, M. Di Martino, A. M. Nardiello, B. Panunzi, L. Sessa, Y. Miele, F. Rossi and S. Piotto, A Flavone-Based Solvatochromic Probe with a Low Expected Perturbation Impact on the Membrane Physical State, Molecules, 2020, 25, 3458 CrossRef CAS.
- L. McDonald, B. Liu, A. Taraboletti, K. Whiddon, L. P. Shriver, M. Konopka, Q. Liu and Y. Pang, Fluorescent flavonoids for endoplasmic reticulum cell imaging, J. Mater. Chem. B, 2016, 4, 7902–7908 RSC.
- B. Liu, J. Wang, G. Zhang, R. Bai and Y. Pang, Flavone-Based ESIPT Ratiometric Chemodosimeter for Detection of Cysteine in Living Cells, ACS Appl. Mater. Interfaces, 2014, 6, 4402–4407 CrossRef CAS.
- B. Liu, L. McDonald, Q. Liu, X. Bi, J. Zheng, L. Wang and Y. Pang, A flavonoid-based light-up bioprobe with intramolecular charge transfer characteristics for wash-free fluorescence imaging in vivo, Sens. Actuators, B, 2016, 235, 309–315 CrossRef CAS.
- I. E. Serdiuk, M. Reszka, H. Myszka, K. Krzymiński, B. Liberek and A. D. Roshal, Flavonol-based fluorescent indicator for determination of β-glucosidase activity, RSC Adv., 2016, 6, 42532–42536 RSC.
- M. Reszka, I. E. Serdiuk, K. Kozakiewicz, A. Nowacki, H. Myszka, P. Bojarski and B. Liberek, Influence of a 4′-substituent on the efficiency of flavonol-based fluorescent indicators of β-glycosidase activity, Org. Biomol. Chem., 2020, 18, 7635–7648 RSC.
- A. T. Adetunji, F. B. Lewu, R. Mulidzi and B. Ncube, The biological activities of β-glucosidase, phosphatase and urease as soil quality indicators: a review, J. Soil Plant Nutr., 2017, 17, 794–807 CrossRef CAS.
- T. D. Butters, Gaucher disease, Curr. Opin. Struct. Biol., 2007, 11, 412–418 CrossRef CAS.
- X. Zhou, Z. Huang, H. Yang, Y. Jiang, W. Wei, Q. Li, Q. Mo and J. Liu, β-Glucosidase inhibition sensitizes breast cancer to chemotherapy, Biomed. Pharmacother., 2017, 91, 504–509 CrossRef CAS.
- U. Ghani, M. Nur-e-Alam, M. Yousaf, Z. Ul-Haq, O. M. Noman and A. J. Al-Rehaily, Natural flavonoid α-glucosidase inhibitors from Retama raetam: enzyme inhibition and molecular docking reveal important interactions with the enzyme active site, Bioorg. Chem., 2019, 87, 736–742 CrossRef CAS.
- S. P. Schröder, L. Wu, M. Artola, T. Hansen, W. A. Offen, M. J. Ferraz, K.-Y. Li, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée, G. J. Davies and H. S. Overkleeft, Gluco-1H-imidazole: A New Class of Azole-Type β-Glucosidase Inhibitor, J. Am. Chem. Soc., 2018, 140, 5045–5048 CrossRef.
- P.-S. Tseng, C. Ande, K. W. Moremen and D. Crich, Influence of Side Chain Conformation on the Activity of Glycosidase Inhibitors, Angew. Chem., Int. Ed., 2023, 62, e202217809 CrossRef CAS.
- X. Liu, F. Li, L. Su, M. Wang, T. Jia, X. Xu, X. Li, C. Wei, C. Luo, S. Chen and H. Chen, Design and synthesis of novel benzimidazole-iminosugars linked a substituted phenyl group and their inhibitory activities against β-glucosidase, Bioorg. Chem., 2022, 127, 106016 CrossRef CAS.
- G. Legler, in Advances in Carbohydrate Chemistry and Biochemistry, ed. R. S. Tipson and D. Horton, Academic Press, 1990, vol. 48, pp. 319–384 Search PubMed.
- V. H. Lillelund, H. H. Jensen, X. Liang and M. Bols, Recent Developments of Transition-State Analogue Glycosidase Inhibitors of Non-Natural Product Origin, Chem. Rev., 2002, 102, 515–554 CrossRef CAS.
- A. K. Maurya, V. Mulpuru and N. Mishra, Discovery of Novel Coumarin Analogs against the α-Glucosidase Protein Target of Diabetes Mellitus: Pharmacophore-Based QSAR, Docking, and Molecular Dynamics Simulation Studies, ACS Omega, 2020, 5, 32234–32249 CrossRef CAS.
- T. D. Heightman and A. T. Vasella, Recent Insights into Inhibition, Structure, and Mechanism of Configuration-Retaining Glycosidases, Angew. Chem., Int. Ed., 1999, 38, 750–770 CrossRef CAS.
- O. O. Demidov, E. S. Gladkov, A. V. Kyrychenko and A. D. Roshal, Synthetic and Natural Flavonols as Promising Fluorescence Probes for β-Glucosidase Activity Screening, Funct. Mater., 2022, 29, 252–262 CAS.
- S. H. White, W. C. Wimley, A. S. Ladokhin and K. Hristova, Protein folding in membranes: determining the energetics of peptide-bilayer interactions, Methods Enzymol., 1998, 295, 62–87 CAS.
- T. Roy, S. T. Boateng, S. Banang-Mbeumi, P. K. Singh, P. Basnet, R.-C. N. Chamcheu, F. Ladu, I. Chauvin, V. S. Spiegelman, R. A. Hill, K. G. Kousoulas, B. M. Nagalo, A. L. Walker, J. Fotie, S. Murru, M. Sechi and J. C. Chamcheu, Synthesis, inverse docking-assisted identification and in vitro biological characterization of flavonol-based analogs of fisetin as c-Kit, CDK2 and mTOR inhibitors against melanoma and non-melanoma skin cancers, Bioorg. Chem., 2021, 107, 104595 CrossRef CAS.
- F. A. A. van Acker, J. A. Hageman, G. R. M. M. Haenen, W. J. F. van der Vijgh, A. Bast and W. M. P. B. Menge, Synthesis of Novel 3,7-Substituted-2-(3′,4′-dihydroxyphenyl)flavones with Improved Antioxidant Activity, J. Med. Chem., 2000, 43, 3752–3760 CrossRef CAS.
- Z. Qiang, W. Chun, L. Yunping and P. Wenchen, Synthesis and antibacterial activity of flavonols, Chin. J. Appl. Environ. Biol., 2017, 23, 232–237 Search PubMed.
- D. S. Goodsell, G. M. Morris and A. J. Olson, Automated docking of flexible ligands: applications of Autodock, J. Mol. Recognit., 1996, 9, 1–5 CrossRef CAS.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS.
- P. Isorna, J. Polaina, L. Latorre-García, F. J. Cañada, B. González and J. Sanz-Aparicio, Crystal Structures of Paenibacillus polymyxa β-Glucosidase B Complexes Reveal the Molecular Basis of Substrate Specificity and Give New Insights into the Catalytic Machinery of Family I Glycosidases, J. Mol. Biol., 2007, 371, 1204–1218 CrossRef CAS.
- L. Xia, M. Ruppert, M. Wang, S. Panjikar, H. Lin, C. Rajendran, L. Barleben and J. Stöckigt, Structures of Alkaloid Biosynthetic Glucosidases Decode Substrate Specificity, ACS Chem. Biol., 2012, 7, 226–234 CrossRef CAS.
- D. L. Zechel, A. B. Boraston, T. Gloster, C. M. Boraston, J. M. Macdonald, D. M. G. Tilbrook, R. V. Stick and G. J. Davies, Iminosugar Glycosidase Inhibitors: Structural and Thermodynamic Dissection of the Binding of Isofagomine and 1-Deoxynojirimycin to β-Glucosidases, J. Am. Chem. Soc., 2003, 125, 14313–14323 CrossRef CAS.
- S. Tribolo, J.-G. Berrin, P. A. Kroon, M. Czjzek and N. Juge, The Crystal Structure of Human Cytosolic β-Glucosidase Unravels the Substrate Aglycone Specificity of a Family 1 Glycoside Hydrolase, J. Mol. Biol., 2007, 370, 964–975 CrossRef CAS.
- L. S. Dodda, I. Cabeza de Vaca, J. Tirado-Rives and W. L. Jorgensen, LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands, Nucleic Acids Res., 2017, 45, W331–W336 CrossRef CAS.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations, J. Phys. Chem. B, 2017, 121, 3864–3870 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, Canonical Sampling through Velocity Rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: an N × log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A Linear Constraint Solver for Molecular Simulations, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- B. Hess, P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, Flexible, and Free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- M. I. Lvovskaya, A. D. Roshal, A. O. Doroshenko, A. V. Kyrychenko and V. P. Khilya, Fluorescence behavior of chromones containing several protolytic centers. 3-Thiazolylchromones: emission band assignment and pH dependent effects, Spectrochim. Acta, Part A, 2006, 65, 397–405 CrossRef CAS.
- L. Chebil, C. Humeau, J. Anthoni, F. Dehez, J.-M. Engasser and M. Ghoul, Solubility of Flavonoids in Organic Solvents, J. Chem. Eng. Data, 2007, 52, 1552–1556 CrossRef CAS.
- W. Koenigs and E. Knorr, Ueber einige Derivate des Traubenzuckers und der Galactose, Ber. Dtsch. Chem. Ges., 1901, 34, 957–981 CrossRef.
- D. W. Miller and K. A. Dill, Ligand binding to proteins: the binding landscape model, Protein Sci., 1997, 6, 2166–2179 CrossRef CAS.
- J. D. McGhee and P. H. von Hippel, Theoretical aspects of DNA-protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS.
- T. L. Hill, Cooperativity Theory in Biochemistry: Steady-State and Equilibrium Systems, Springer-Verlag, New York, Berlin, Heidelberg, Tokyo, 1985 Search PubMed.
- R. Gesztelyi, J. Zsuga, A. Kemeny-Beke, B. Varga, B. Juhasz and A. Tosaki, The Hill equation and the origin of quantitative pharmacology, Archive for History of Exact Sciences, 2012, 66, 427–438 CrossRef.
- S. Kuusk and P. Väljamäe, When substrate inhibits and inhibitor activates: implications of β-glucosidases, Biotechnol. Biofuels, 2017, 10, 7 CrossRef.
- C. He, X. Liu, Z. Jiang, S. Geng, H. Ma and B. Liu, Interaction Mechanism of Flavonoids and α-Glucosidase: Experimental and Molecular Modelling Studies, Foods, 2019, 8, 355 CrossRef CAS.
- M. Fu, W. Shen, W. Gao, L. Namujia, X. Yang, J. Cao and L. Sun, Essential moieties of myricetins, quercetins and catechins for binding and inhibitory activity against α-Glucosidase, Bioorg. Chem., 2021, 115, 105235 CrossRef CAS.
- X. Ding, Y. Yu and Z. Ding, Interaction between active compounds from Rosa roxburghii Tratt and β-glucosidase: characterization of complexes and binding mechanism, LWT, 2022, 165, 113707 CrossRef CAS.
- D. Liu, X. Cao, Y. Kong, T. Mu and J. Liu, Inhibitory mechanism of sinensetin on α-glucosidase and non-enzymatic glycation: insights from spectroscopy and molecular docking analyses, Int. J. Biol. Macromol., 2021, 166, 259–267 CrossRef CAS.
- Y. Bhatia, S. Mishra and V. S. Bisaria, Microbial β-Glucosidases: Cloning, Properties, and Applications, Crit. Rev. Biotechnol., 2002, 22, 375–407 CrossRef CAS.
- J. R. Ketudat Cairns, B. Mahong, S. Baiya and J.-S. Jeon, β-Glucosidases: multitasking, moonlighting or simply misunderstood?, Plant Sci., 2015, 241, 246–259 CrossRef CAS.
- S. He and S. G. Withers, Assignment of Sweet Almond β-Glucosidase as a Family 1 Glycosidase and Identification of Its Active Site Nucleophile, J. Biol. Chem., 1997, 272, 24864–24867 CrossRef CAS.
- A. D. Snizhko, A. V. Kyrychenko and E. S. Gladkov, Synthesis of Novel Derivatives of 5,6,7,8-Tetrahydro-quinazolines Using of α-Aminoamidines and In Silico Screening of Their Biological Activity, Int. J. Mol. Sci., 2022, 23, 3781 CrossRef CAS.
- G.-Y. Chen, H. Zhang and F.-Q. Yang, A simple and portable method for β-glucosidase activity assay and its inhibitor screening based on a personal glucose meter, Anal. Chim. Acta, 2021, 1142, 19–27 CrossRef CAS.
- M. Kazmi, S. Zaib, A. Ibrar, S. T. Amjad, Z. Shafique, S. Mehsud, A. Saeed, J. Iqbal and I. Khan, A new entry into the portfolio of α-glucosidase inhibitors as potent therapeutics for type 2 diabetes: design, bioevaluation and one-pot multi-component synthesis of diamine-bridged coumarinyl oxadiazole conjugates, Bioorg. Chem., 2018, 77, 190–202 CrossRef CAS.
- S. Riaz, I. U. Khan, M. Yar, M. Ashraf, T. U. Rehman, A. Shaukat, S. B. Jamal, V. C. M. Duarte and M. J. Alves, Novel pyridine-2,4,6-tricarbohydrazide derivatives: design, synthesis, characterization and in vitro biological evaluation as α- and β-glucosidase inhibitors, Bioorg. Chem., 2014, 57, 148–154 CrossRef CAS.
- S. Khan, T. Pozzo, M. Megyeri, S. Lindahl, A. Sundin, C. Turner and E. N. Karlsson, Aglycone specificity of Thermotoga neapolitana β-glucosidase 1A modified by mutagenesis, leading to increased catalytic efficiency in quercetin-3-glucoside hydrolysis, BMC Biochem., 2011, 12, 11 CrossRef CAS.
- C. Proença, M. Freitas, D. Ribeiro, E. F. T. Oliveira, J. L. C. Sousa, S. M. Tomé, M. J. Ramos, A. M. S. Silva, P. A. Fernandes and E. Fernandes, α-Glucosidase inhibition by flavonoids: an in vitro and in silico structure–activity relationship study, J. Enzyme Inhib. Med. Chem., 2017, 32, 1216–1228 CrossRef.
- L. Zeng, G. Zhang, S. Lin and D. Gong, Inhibitory Mechanism of Apigenin on α-Glucosidase and Synergy Analysis of Flavonoids, J. Agric. Food Chem., 2016, 64, 6939–6949 CrossRef CAS.
- K. Tadera, Y. Minami, K. Takamatsu and T. Matsuoka, Inhibition of α-Glucosidase and α-Amylase by Flavonoids, J. Nutr. Sci. Vitaminol., 2006, 52, 149–153 CrossRef CAS.
- S. Horikoshi, W. Saburi, J. Yu, H. Matsuura, J. R. K. Cairns, M. Yao and H. Mori, Substrate specificity of glycoside hydrolase family 1 β-glucosidase AtBGlu42 from Arabidopsis thaliana and its molecular mechanism, Biosci., Biotechnol., Biochem., 2021, 86, 231–245 CrossRef.
- R. Uehara, R. Iwamoto, S. Aoki, T. Yoshizawa, K. Takano, H. Matsumura and S.-i. Tanaka, Crystal structure of a GH1 β-glucosidase from Hamamotoa singularis, Protein Sci., 2020, 29, 2000–2008 CrossRef CAS.
- T. M. Gloster, S. Roberts, G. Perugino, M. Rossi, M. Moracci, N. Panday, M. Terinek, A. Vasella and G. J. Davies, Structural, Kinetic, and Thermodynamic Analysis of Glucoimidazole-Derived Glycosidase Inhibitors, Biochemistry, 2006, 45, 11879–11884 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, A. R. van Buuren, E. Apol, P. J. Meulenhoff, D. P. Tieleman, A. L. T. M. Sijbers, K. A. Feenstra, R. van Drunen and H. J. C. Berendsen, Gromacs User Manual Version 4.5.4, 2010, https://www.gromacs.org/ Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |