DOI:
10.1039/D3RA06262G
(Paper)
RSC Adv., 2023,
13, 32363-32370
Facile synthesis of telechelic poly(phenylene sulfide)s by means of electron-deficient aromatic sulfonium electrophiles†
Received
14th September 2023
, Accepted 18th October 2023
First published on 3rd November 2023
Abstract
We report the facile synthesis of telechelic poly(phenylene sulfide) (PPS) derivatives bearing functional groups at both termini. α,ω-Dihalogenated dimethyl-substituted PPS were obtained in high yield with a high degree of end-functionalization by using soluble poly(2,6-dimethyl-1,4-phenylenesulfide) (PMPS) and 4,4′-dihalogenated diphenyl disulfide (X-DPS, X = Cl, Br) as a precursor and an end-capping agent, respectively. Further end-functionalization is achieved through cross-coupling reactions; particularly, the Kumada–Tamao cross-coupling reaction of bromo-terminated telechelic PMPS and a vinylated Grignard reagent afforded end-vinylated PMPS with thermosetting properties. This synthetic approach can be applied to the preparation of various aromatic telechelic polymers with the desired structures and functionalities.
Introduction
The precise control of polymer structures from the first- to the higher-order levels is a key issue in polymer chemistry. In order to synthesize polymers with desired functions and structures, various methods including controlled or sterically confined polymerization1–4 and post-polymerization modification5,6 should be made full use. Among these methods, end-functionalization of polymers is an important transformation technique because it enables the connection of multiple segments for bioconjugation,7,8 surface modification,9,10 and others. Telechelic polymers, which are end-functionalized polymers with α,ω-terminal reactive groups, are also useful reactive precursors of other polymers, such as triblock copolymers11–14 and cyclic polymers.15–17 They are generally synthesized through living polymerization with reactive end groups (ex. atom transfer radical polymerization, reversible addition–fragmentation polymerization, and chain-growth polycondensation) or post-polymerization modification.18,19 Cyclic–linear topology transformation via dynamic covalent chemistry20,21 is another strategy for telechelic polymer synthesis. However, these methods are limited to the polymers that always have α,ω-reactive termini or bear reactive bonds in every repeating unit. To the best of our knowledge, although there are some modification techniques for polymers with unreactive groups,22,23 such transformation reactions have not been adopted in end-functionalization applications, including telechelic polymer synthesis.
Among unreactive polymers, poly(phenylene sulfide) (PPS) is an insoluble super-engineering plastics that is difficult to end-functionalize. To make PPS functionalizable, we investigated the properties and the usage of PPS derivatives synthesized by oxidative polymerization of aromatic disulfides.24–26 As they are soluble and have “single” interchain disulfide bonds, α-end-functionalized derivatives27,28 and diblock PPS-based copolymers24 can be synthesized efficiently. However, this method cannot be applied to multi-end functionalization because of the lack of reactive end groups. To overcome this issue, we used three-centered sulfonium cations with electron-withdrawing substituents,29 which can react as Friedel–Crafts electrophiles for aromatic rings, as an end-capping reagent for the oxidative polymerization of diphenyl disulfide to prepare α,ω-dihalogenated oligo(phenylene sulfide)s.30 However, because of the poor solubility, low molecular weight (pentamer to hexamer), low degree of functionalization (60–90%), and low yield (32–52%) of the products, this method is far from a versatile and efficient method for end-functionalization.
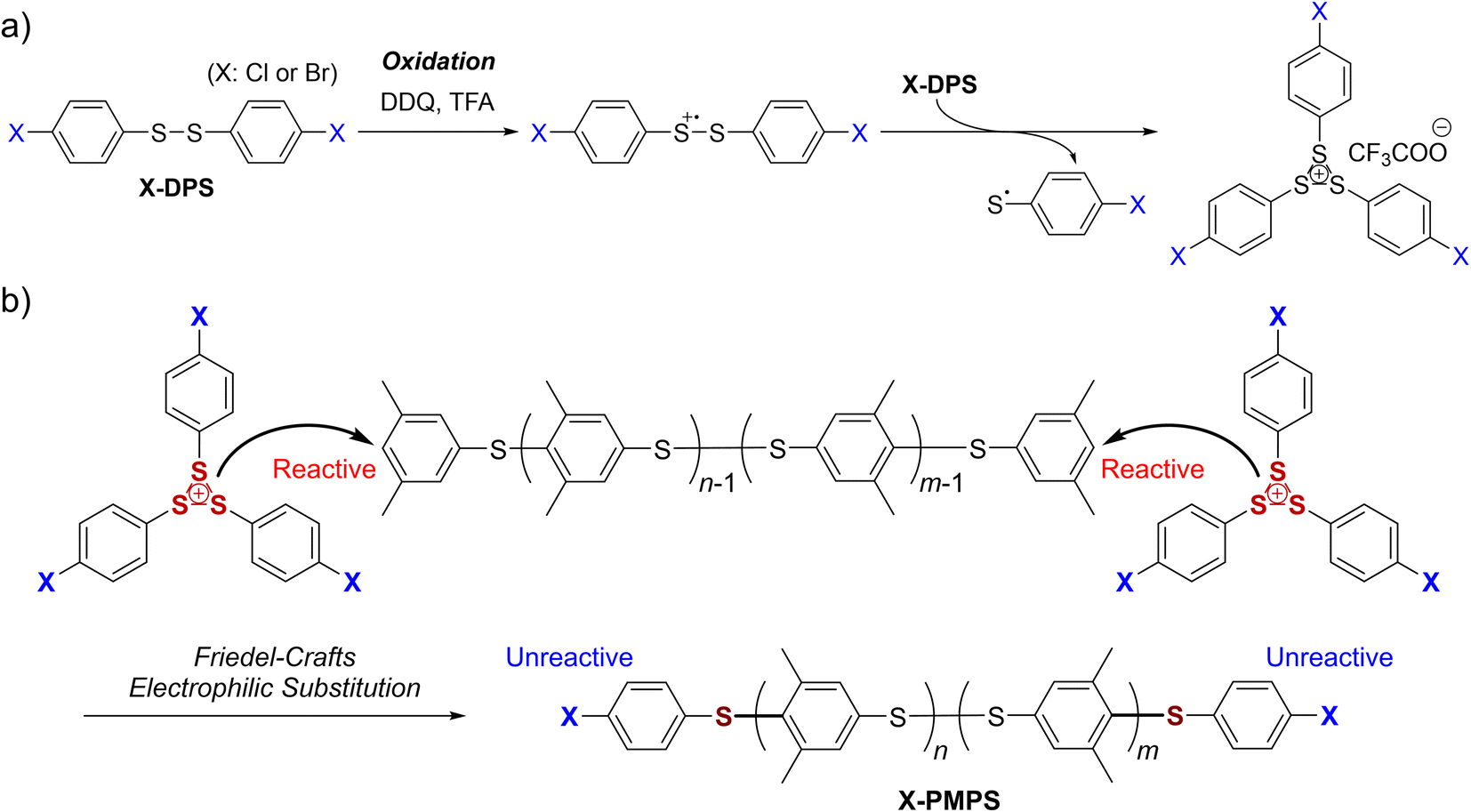
Herein, we report the facile synthesis of telechelic PPS derivatives resulting in high conversion and high yield by using a highly soluble and structure-confined methylated PPS derivative, poly(2,6-dimethyl-1,4-phenylenesulfide) (PMPS). As the in situ generated sulfonium cation X-DPS+ (Fig. 1a) did not self-polymerize but instead selectively reacted with another electron-rich aromatic compound, both termini of the PPS derivatives could be efficiently functionalized (Fig. 1b). We further demonstrated that these α,ω-dihalogenated PPS derivatives could be further end-vinylated via cross-coupling reactions.
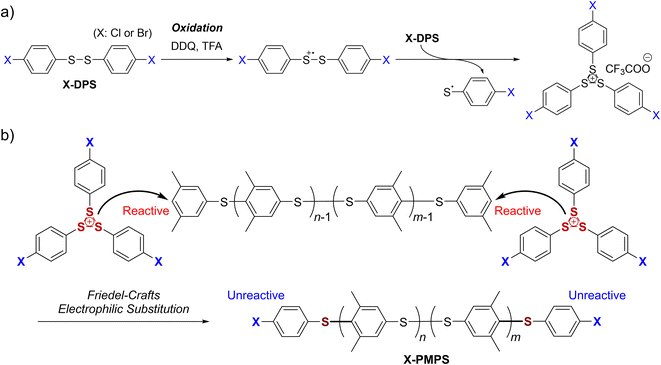 |
| | Fig. 1 End-functionalization of poly(2,6-dimethyl-1,4-phenylenesulfide) (PMPS) with 4,4′-halogenated diphenyl disulfide (X-DPS): (a) oxidation of X-DPS to form an unreactive sulfonium cation (X-DPS+). (b) End-capping of reactive PMPS to afford halogen-substituted PMPS (X-PMPS′). | |
Experimental section
Reagents
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ), dichloromethane, 4,4′-dichlorodiphenyl disulfide (Cl-DPS), bis(4-bromophenyl) disulfide (Br-DPS), tetrabutylammonium tetrafluoroborate (TBABF4), tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4), cesium carbonate, 4-vinylphenylboronic acid, [1,3-bis(diphenylphosphino)propane]nickel(II) dichloride (Ni(dppp)Cl2), and vinylmagnesium bromide (ca. 1 M solution in THF) were purchased from Tokyo Chemical Industry Co. (TCI). Methanol was purchased from Kokusan Chemical Co. or TCI. Hydrochloric acid, potassium hydroxide, tetrahydrofuran (THF), hexane, sodium borohydride, and toluene were purchased from Kanto Chemical Co. Trifluoroacetic acid (TFA) was purchased FUJIFILM Wako Pure Chemical Co. Bis(3,5-dimethylphenyl)disulfide (3,5-DPS) was prepared according to the previous report.24
Synthesis of Cl-terminated PMPS by one-pot polymerization/functionalization
The procedure was arranged according to our previous report30 with several modifications. To a 20 mL flask, DDQ (1.18 g, 5.3 mmol) was dispersed in dichloromethane (3.5 mL) and TFA (133 μL, 1.75 mmol) was added. Then, 3,5-DPS (0.72 g, 2.63 mmol) was added and stirred at room temperature to proceed with the polymerization. After 1 hour, Cl-DPS (0.75 g, 2.63 mmol) was added and was further stirred for 15 hours at room temperature for the termination. The solution was diluted with dichloromethane (15 mL) and was precipitated in methanol containing 5 vol% hydrochloric acid (500 mL). The solid was collected by filtration after subsequent washing with methanol, potassium hydroxide aqueous, and water. The solid was redissolved in THF (20 mL) and was purified by reprecipitating in hexane (500 mL). The precipitate was collected by filtration, was washed with hexane, and was dried in vacuo to obtain Cl-PMPS as a whitish powder (0.24 g, yield: 33%, Mn = 4.5 × 103 (NMR), Mn = 4.5 × 103 (SEC), Mw/Mn = 1.3 (SEC), End-functionalization degree: 100%).
Synthesis of Cl-terminated PMPS by post-polymerization modification
A typical procedure has been described as follows (Run 1, Table 1). To a 30 mL flask, Cl-DPS (0.57 g, 2 mmol) was dissolved in dichloromethane (20 mL) and TFA (765 μL, 10 mmol) and DDQ (0.45 g, 2 mmol) were added and stirred at room temperature to prepare Cl-DPS+ solution. Then, PMPS (0.26 g, 2 mmol of unit) was added and further stirred for 20 hours at room temperature. The solution was precipitated in methanol containing 5 vol% hydrochloric acid (500 mL) and the solid was collected by filtration and was washed with methanol, potassium hydroxide aqueous, and water. The solid was then reprecipitated with THF/hexane (15 mL/300 mL), and the filtrate was collected, was washed with hexane, and was dried in vacuo to obtain Cl-PMPS′ as a whitish powder (0.20 g, yield: 75%).
Table 1 Properties of Cl-PMPS′ synthesized by post-polymerization modification
| Run |
[Cl-DPS]/M |
[PMPS]/M |
[TFA]/M |
Yield/% |
Mna/×103 |
Mnb/×103 |
Mwb/×103 |
Mw/Mnb/— |
Mn of precursor PMPSb/×103 |
Degree of end-functionalization/% |
| Determined by 1H NMR spectroscopy. Determined by SEC in chloroform. Not determined because of the main-chain degradation (side reaction). |
| 1 |
0.1 |
0.1 |
0.5 |
75 |
8.7 |
8.6 |
17.0 |
2.0 |
6.8 |
99 |
| 2 |
0.05 |
0.2 |
0.25 |
44 |
11.1 |
4.9 |
6.6 |
1.3 |
4.6 |
44 |
| 3 |
0.1 |
0.2 |
1.0 |
51 |
N.D.c |
2.3 |
4.4 |
1.9 |
4.6 |
N.D.c |
Synthesis of Br-Terminated PMPS
A typical procedure has been described as follows (Run 4, Table 2). To a 50 mL flask, Br-DPS (1.88 g, 5 mmol) was dissolved in dichloromethane (25 mL), TFA (0.83 mL, 12.5 mmol), and DDQ (1.14 g, 5 mmol) were added and stirred at room temperature. PMPS (0.68 g, 5 mmol of unit) was further added and stirred at room temperature for 40 hours at room temperature. The solution was precipitated in methanol containing 5 vol% hydrochloric acid (1000 mL) and the solid was collected by filtration, was washed with methanol, potassium hydroxide aqueous, and water, and was dried in vacuo. The solid was reprecipitated with THF/hexane (20 mL/800 mL) and the precipitate was collected, was washed with hexane, and was dried in vacuo to obtain Br-PMPS (0.63 g, yield: 91%).
Table 2 Properties of Br-PMPS synthesized by post-polymerization modificationa
| Run |
[Br-DPS]/M |
[PMPS]/M |
Reaction time/h |
Yield/% |
Mnb/×103 |
Mnc/×103 |
Mwc/×103 |
Mw/Mnc/— |
Mn of precursor PMPSc/×103 |
Degree of end-functionalization/% |
| Temp. = r.t., [TFA] = 0.5 M. Determined by 1H NMR spectroscopy. Determined by SEC in chloroform. |
| 1 |
0.1 |
0.1 |
20 |
45 |
7.1 |
4.9 |
6.4 |
1.3 |
4.6 |
70 |
| 2 |
0.1 |
0.2 |
20 |
58 |
9.4 |
4.0 |
6.0 |
1.5 |
4.6 |
53 |
| 3 |
0.1 |
0.1 |
40 |
71 |
8.1 |
7.3 |
12.9 |
1.8 |
7.1 |
91 |
| 4 |
0.2 |
0.2 |
40 |
91 |
6.3 |
6.3 |
13.1 |
2.1 |
7.1 |
99 |
Post-functionalization of Br-PMPS by Kumada–Tamao cross-coupling reactions
To a 30 mL flask, Br-PMPS (Run 4, Table 2) (96 mg, 10 μmol of Br) and Ni(dppp)Cl2 (11 mg, 20 μmol) was added and dissolved in THF (7.6 mL) under argon atmosphere. 1 M vinylmagnesium bromide solution in THF (0.4 mL, 0.4 mmol) was further added and stirred for 24 hours at room temperature. The solution was precipitated in methanol containing 5 vol% hydrochloric acid (300 mL). The precipitate was collected by filtration, was washed with methanol and water, and was dried in vacuo to obtain V-PMPS′ as a whitish powder (28 mg, yield: 29%, Mn = 5.1 × 103, Mw/Mn = 2.3).
Results and discussion
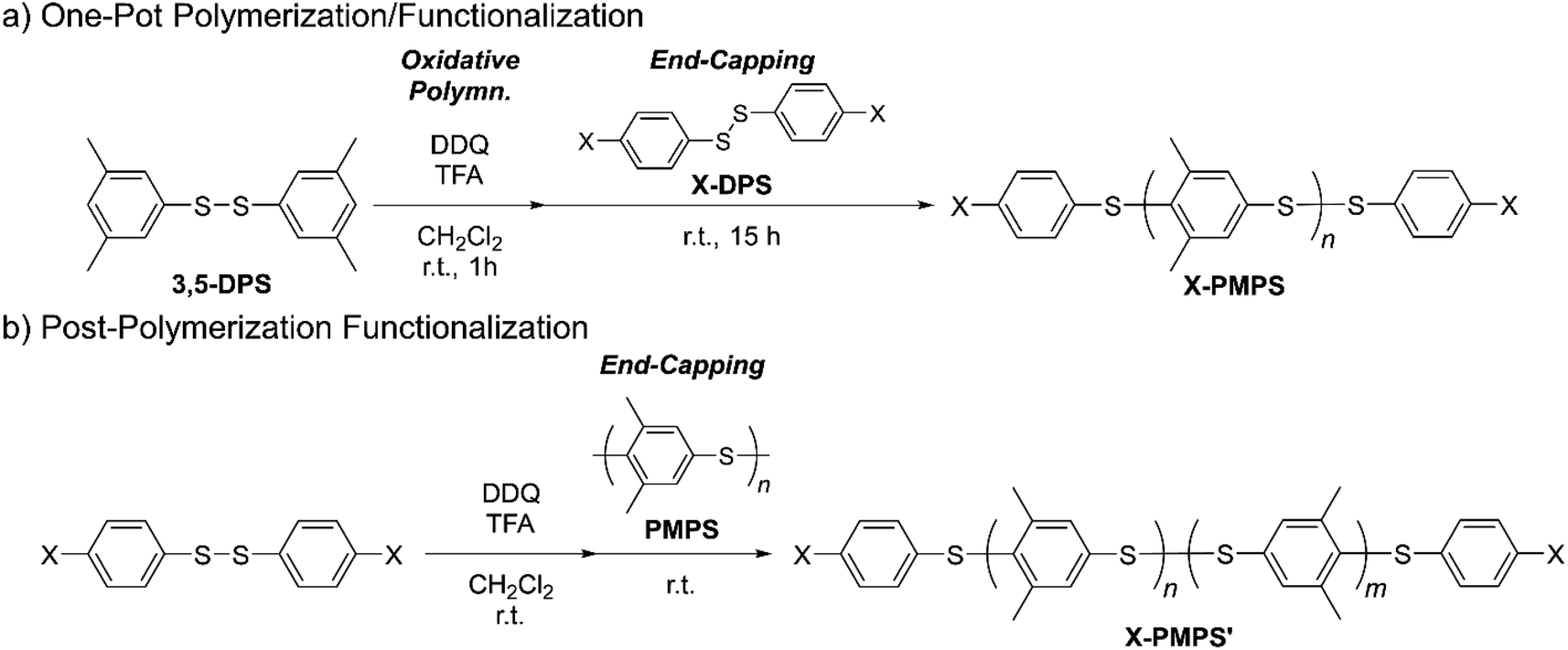
One-pot synthesis of Cl-terminated telechelic PMPS
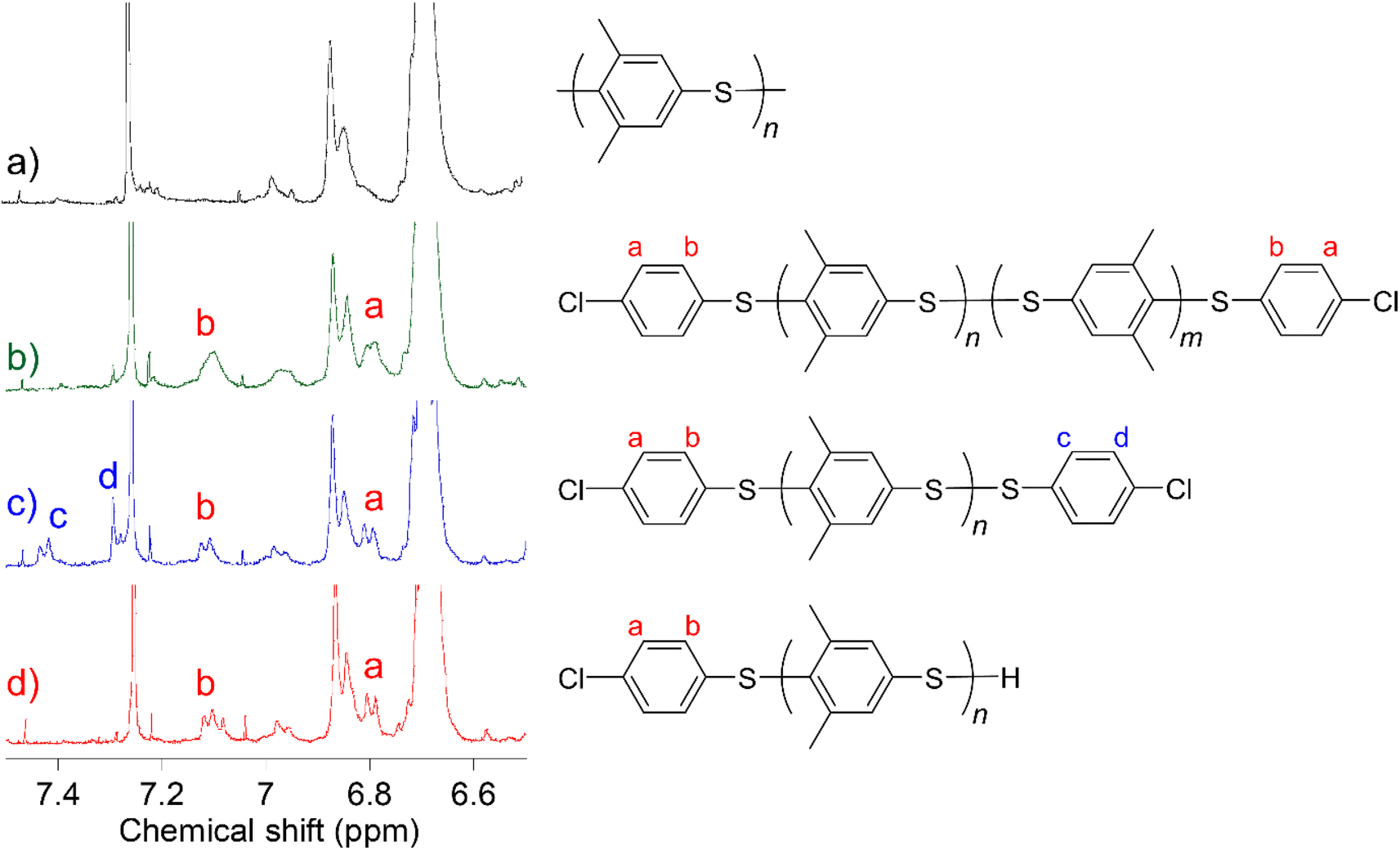
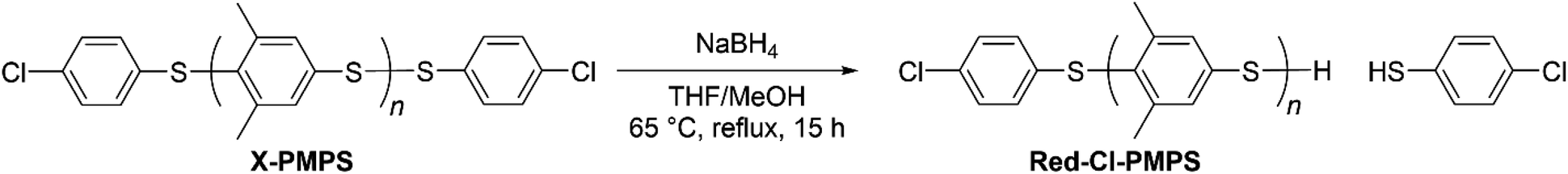
First, we synthesized α,ω-dichlorinated dimethyl-substituted PPS (Cl-PMPS) by a one-pot preparation method involving oxidative polymerization of 3,5-DPS and subsequent addition of 4,4′-dichlorophenyl disulfide (Cl-DPS) for in situ end-capping (Scheme 1a, X = Cl). After the reaction, the 1H NMR spectrum revealed several new doublet signals of aromatic groups with high degree of end-functionalization (100%) that were different from those of PMPS and Cl-DPS, indicating the efficient progress of the end-capping reaction (Fig. 2, detailed characterization was described in Fig. 3). In the IR spectrum of Cl-PMPS, the absorption peaks ascribed to the 1,4-phenylene C–H (δC–H: 820 cm−1) and C–Cl (νC–Cl: 740 cm−1) bonds were observed (Fig. S1†). Additionally, the DOSY NMR spectrum of Cl-PMPS showed the same diffusion coefficient for all aromatic peaks, indicating the presence of 4-chlorophenyl units at the polymer chain ends (Fig. S2†). As the oxidative homopolymerization of Cl-DPS did not proceed owing to the higher oxidation potential (see Section 1.2 of the ESI† for details), Cl-DPS+ selectively reacted with its dimethyl-substituted aromatic counterpart, leading to successful end-capping. However, this one-pot functionalization resulted in the low yield (33%) and low molecular weight (Mn = 4.5 × 103) of the polymer, which were similar results to our previous report, presumably owing to the presence of unreacted 3,5-DPS and the low-molecular-weight oligomers due to the early termination of the polymerization.
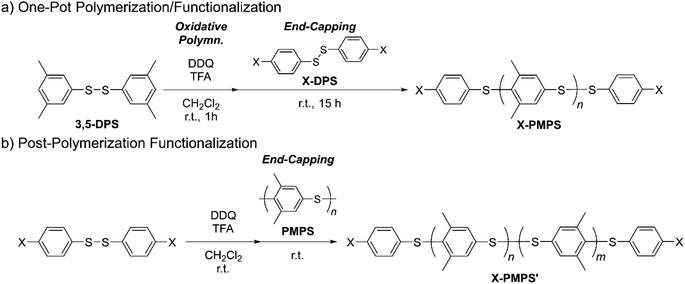 |
| | Scheme 1 Synthetic route for dihalogenated telechelic PMPS (X: Cl or Br). | |
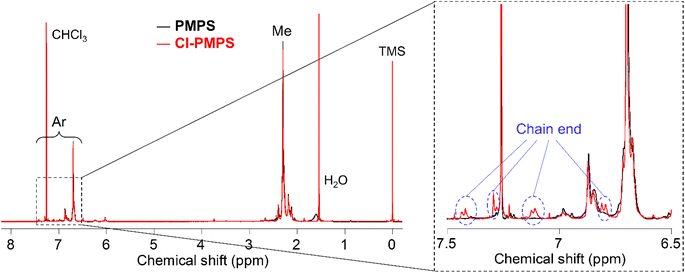 |
| | Fig. 2 Overall (left) and expanded (right) 1H NMR spectra of PMPS (same as the sample in Fig. S3†) and Cl-terminated PMPS (Cl-PMPS) in chloroform-d. | |
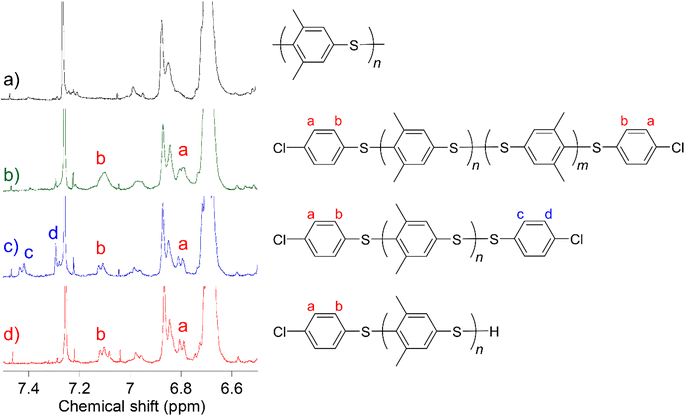 |
| | Fig. 3 Expanded 1H NMR spectra of (a) PMPS, (b) Cl-PMPS′, (c) Cl-PMPS, and (d) Red-Cl-PMPS in chloroform-d. | |
Post-polymerization synthesis of Cl-terminated telechelic PMPS
To increase the efficiency of the end-capping reactions, we applied this electrophilic substitution reaction to the post-polymerization modification of PMPS. A concentrated solution of Cl-DPS+ was first prepared by mixing Cl-DPS, DDQ, and TFA in dichloromethane. In contrast to the highly reactive and unstable phenylbis(thiophenyl)sulfonium cation,31 Cl-DPS+ was unreactive to Cl-DPS and did not polymerize even in the presence of excessive counterions owing to the electron-withdrawing chlorine substituents at the para positions. The subsequent addition of PMPS instead of 3,5-DPS to Cl-DPS+ solution ensured the selective addition of the halogenated thiophenylene fragments to the end of PMPS chains, leading to a higher yield and higher molecular weight of the end-functionalized products (Cl-PMPS′).
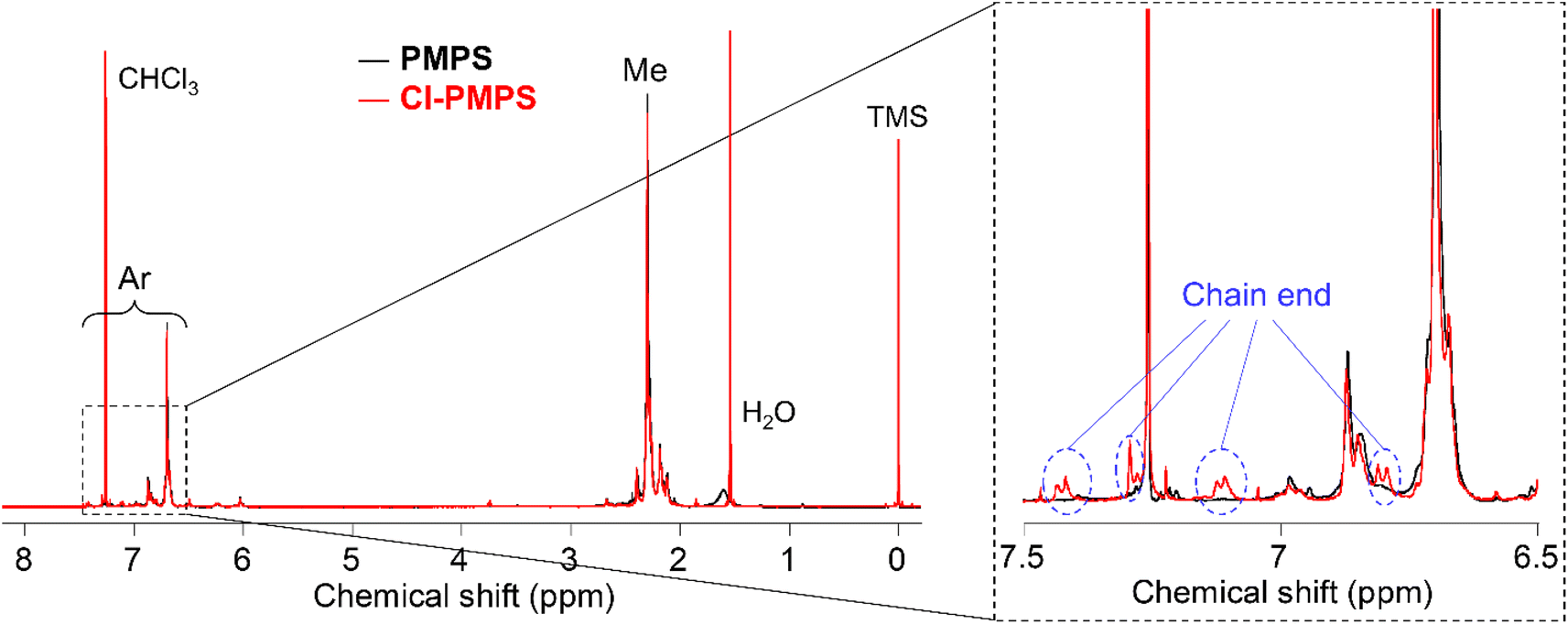
The presence of end chlorinated phenylene units was confirmed from the 1H NMR spectrum (Fig. S3†), in which the signals of the end aromatics were almost at similar chemical shifts, as in the case of Fig. 2 (the details were discussed in Fig. 3). The IR spectra also indicated the existence of chlorinated terminal groups, which is the same as in the case discussed in the Former section (Fig. S4†). The yield and degree of end-functionalization were the highest (75% and 99%, respectively) at low PMPS and high TFA concentrations (Run 1, Table 1). These results suggested that the end-capping reaction proceeds more effectively in the presence of excessive electrophile relative to the reactive chain-end of PMPS. Therefore, using macro-reagents as a reactant is more effective to achieve higher yields and higher molecular weight compared with the case for the one-pot reaction system.
End structure characterization of Cl-terminated telechelic PMPS
To investigate the mechanism of the end-capping reaction, we characterized the 1H NMR signals of the terminal groups of each telechelic polymer. To understand the difference between the end aromatic signals, the disulfide bond of Cl-PMPS was reduced to obtain thiol-terminated Red-Cl-PMPS (Scheme 2) and its 1H NMR spectrum was measured to investigate the contribution of the disulfide end-group (Fig. 3). Two downfield signals (7.43 and 7.29 ppm; assigned as c and d, respectively) in the spectrum of Cl-PMPS disappeared after the reduction; otherwise, the spectrum of Red-Cl-PMPS was mostly identical to that of Cl-PMPS′. Therefore, Cl-PMPS′ has an interchain disulfide bond in the middle whereas Cl-PMPS has a disulfide bond at the chain end bearing a chlorinated thiophenylene. This is presumably because the exchange reaction between the end disulfide bond of the oligomer and Cl-DPS+ proceeds predominantly in the one-pot reaction system.30
 |
| | Scheme 2 Reduction of disulfide bond of Cl-PMPS for structural examination. | |
Synthesis of Br-terminated telechelic PMPS
Subsequently, we applied this end-capping system to the synthesis of bromo-terminated PMPS (Br-PMPS) synthesis because Br is a more reactive leaving group than Cl and is suitable for further post-polymerization functionalization (e.g., coupling reactions).
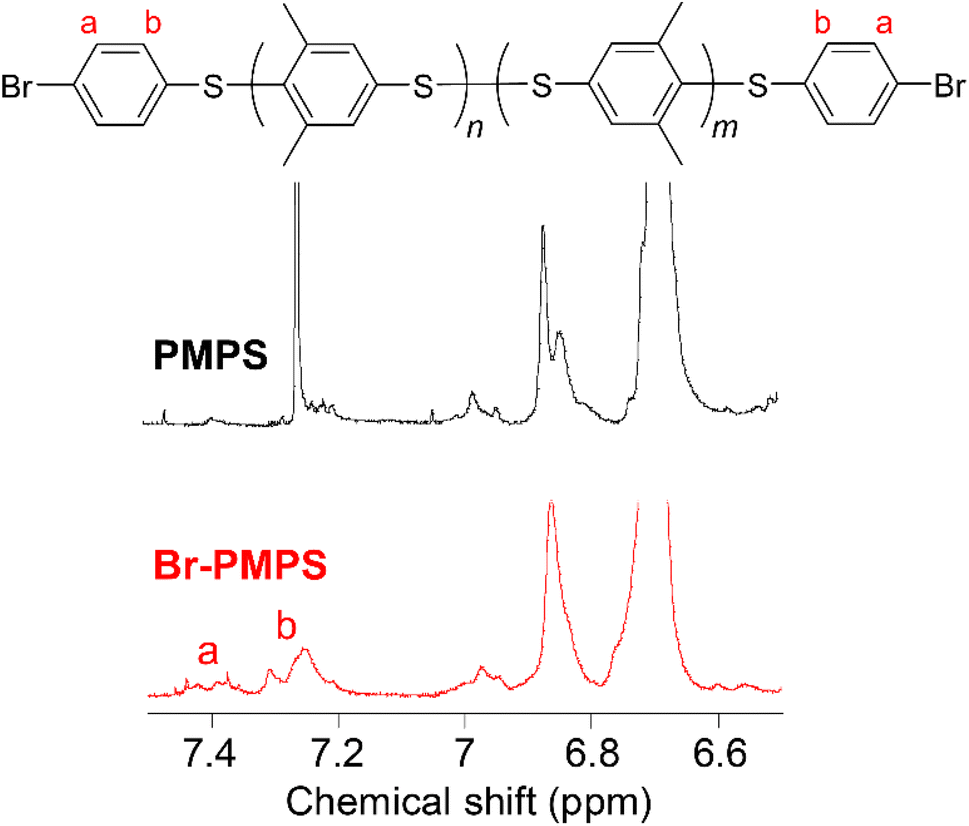
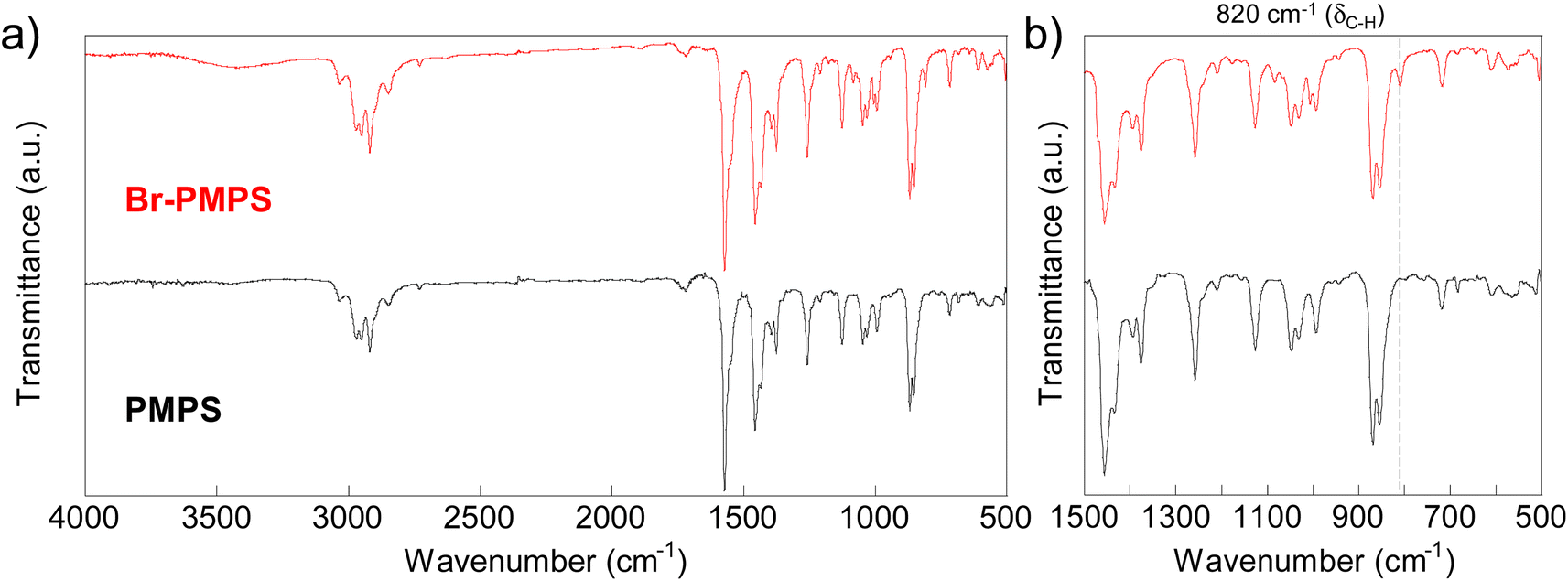
Br-PMPS was synthesized via post-polymerization modification by adding PMPS to the Br-DPS-derived sulfonium cation (Br-DPS+) solution in dichloromethane (Table 2). Two new signals in the 1H NMR spectrum of Br-PMPS corresponding to the end aromatic groups appeared in a more downfield region (7.42–7.26 ppm) than those in the 1H NMR spectrum of Cl-PMPS′ (Fig. 4). This trend held the same end-capping mechanism as in the case of Cl-PMPS′ synthesis, yielding halogenated terminal groups without adjacent disulfide bonds (vide ante). While the end-capping reaction under the optimized conditions for the Cl-PMPS′ synthesis resulted in a low degree of end-functionalization (70%, Run 1 for Table 2), the reaction with high reactant concentrations and longer reaction time (Run 4 for Table 2) yielded Br-PMPS with the highest degree of functionalization (99%) and highest yield (91%). Cl-DPS only has a slightly higher oxidation potential than Br-DPS based on the cyclic voltammograms (Fig. S10†), which indicates a similar reactivity for producing X-DPS+. Thus, the need for harsh reaction conditions for Br-PMPS may be ascribed to the steric effect regarding X-DPS+ during the electrophilic substitution. We also confirmed the nature of the terminal groups of Br-PMPS based on the presence of 1,4-phenylene C–H (δC–H: 820 cm−1) absorption peak in the IR spectrum (Fig. 5) and the same diffusion coefficient for all the polymer signals in the DOSY-NMR spectrum (Fig. S11†). Such bromine-containing PPS derivatives are useful for reactive macro-reagents which can contribute to versatile structure transformation.
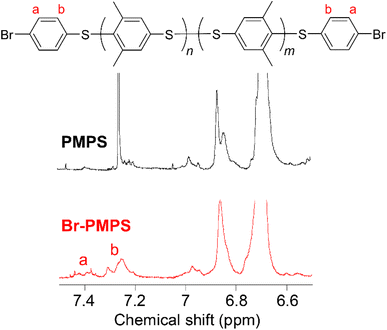 |
| | Fig. 4 Expanded 1H NMR spectra of PMPS in chloroform-d and bromo-terminated PMPS (Br-PMPS) (Run 4 in Table 2) in dichloromethane-d2. The overall spectrum for Br-PMPS is described in Fig. S8.† | |
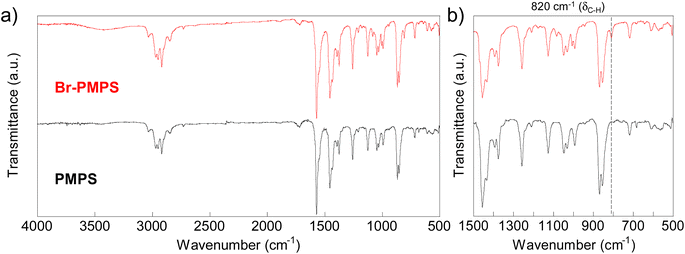 |
| | Fig. 5 IR spectra of PMPS and Br-PMPS: (a) Overall (4000–500 cm−1) and (b) expanded (1500–500 cm−1) spectra. | |
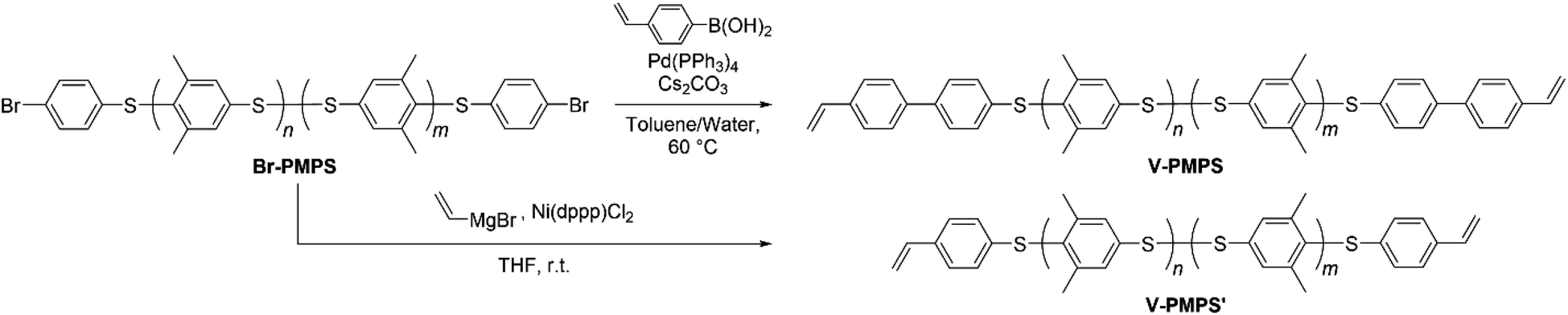
Synthesis of end-vinylated PMPS by cross-coupling reactions
We further performed the post-polymerization modification of Br-PMPS via several cross-coupling reactions to obtain vinyl-end-functionalized PMPS (Scheme 3). First, we conducted the Suzuki–Miyaura cross-coupling reaction of Br-PMPS and 4-vinylphenylboronic acid with a Pd catalyst (route for V-PMPS). While the 1H NMR spectrum showed the signals with larger integrals for the aromatics and broadened signals for the end groups, the peaks of the vinyl groups were not clearly observed (Fig. S12 and S13†). The IR spectrum of Br-PMPS also showed no absorptions characteristic of vinyl groups (Fig. S14†). These results were ascribed to the strong interactions between the Pd catalyst and sulfide/disulfide bonds32,33 of the PMPS chain during the coupling reaction, which produced a brownish product with a small number of vinyl end groups.
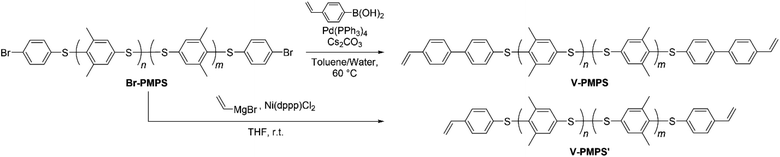 |
| | Scheme 3 End-functionalization of Br-PMPS with vinyl groups via Suzuki–Miyaura cross-coupling (yielding V-PMPS) or Kumada–Tamao cross-coupling (yielding V-PMPS′). | |
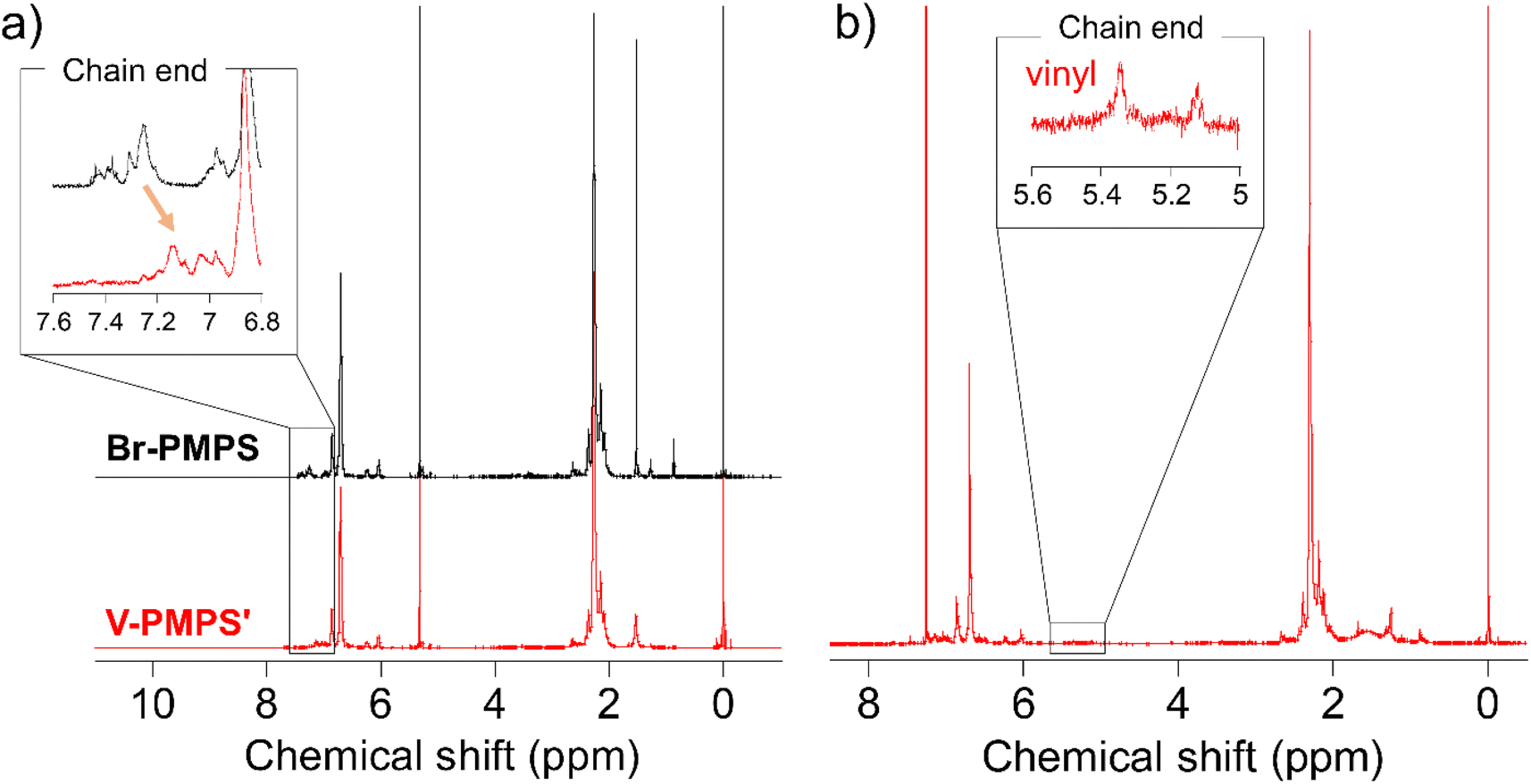
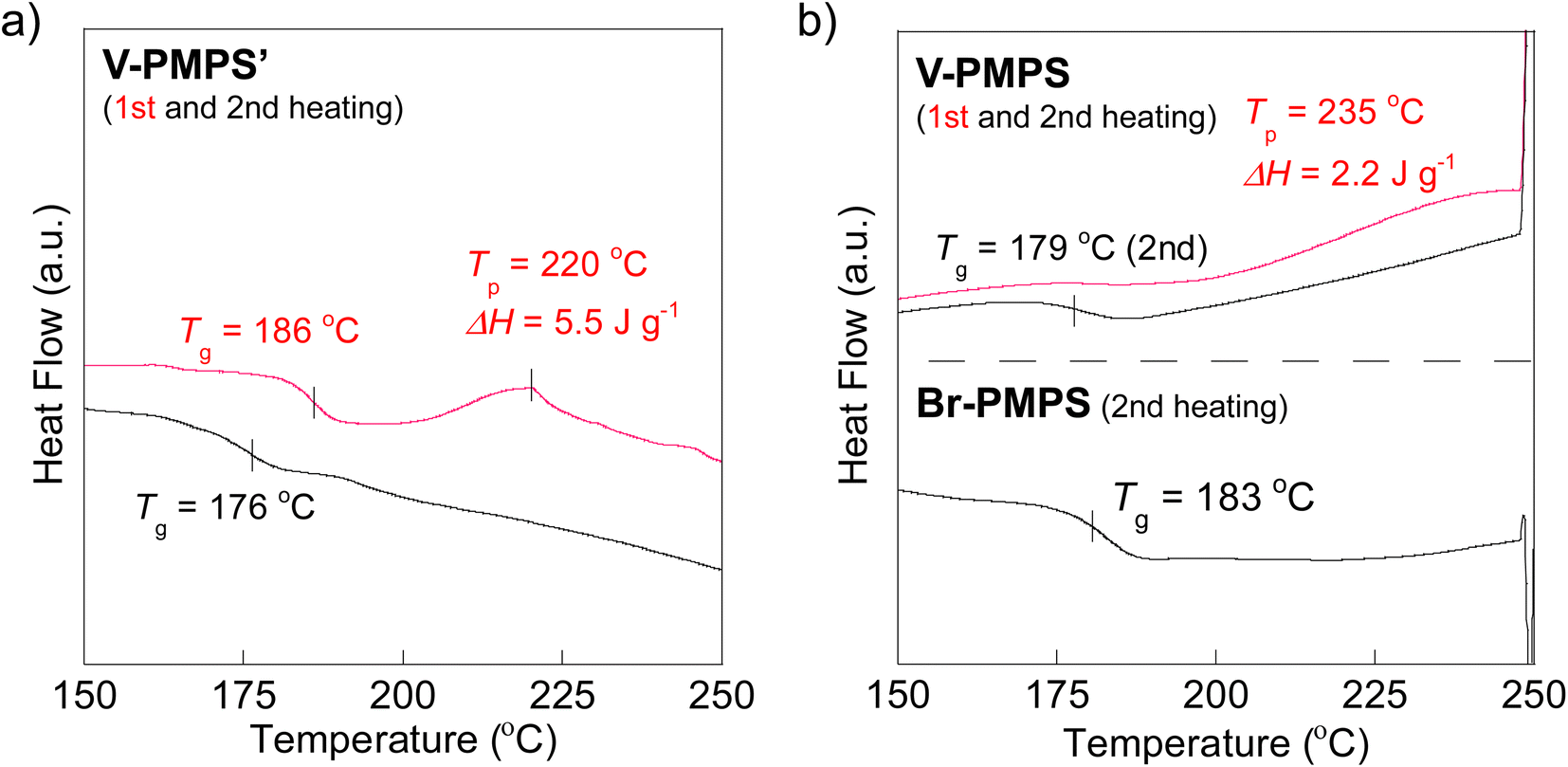
To overcome this issue, we chose the Kumada–Tamao cross-coupling reaction with a Ni catalyst as an alternative because of its feasibility even in the presence of thioethers.34 The coupling reaction of Br-PMPS and vinyl magnesium bromide with Ni(dppp)Cl2 gave the product as a whitish powder. After the reaction, the 1H NMR signals of the end 4-vinylphenyl aromatic protons (7.14 and 7.03 ppm) were observed (Fig. 6a and S15a†). Simultaneously, two signals ascribed to the end vinyl protons (5.34 and 5.12 ppm) appeared with a degree of end-functionalization of 38% (Fig. 6b and S15b†). Notably, the conversion rate of the end Br groups to vinyl groups was low despite the absence of the signals of the end 4-bromophenyl groups, presumably because of the exchange reaction between the Grignard-inserted PMPS end and excess vinyl magnesium bromide, which quenched the Grignard reagents and eliminated the Br termini to give phenyl end groups.35 The reaction progress was monitored from the absorption peak of the vinylated C–H groups in the IR spectrum (Fig. S16†). The DSC thermogram of V-PMPS′ represented both glass transition and the exothermic peak (enthalpy change ΔH = 5.5 J g−1) ascribed to the thermosetting of the vinyl groups in the first heating, whereas only glass transition (Tg = 176 °C) was occurred in the second heating (Fig. 7a). Similar exothermic peak was also observed in the DSC for V-PMPS with the smaller ΔH values (2.2 J g−1) (Fig. 7b), suggesting its smaller vinyl end-functionalization efficiency than V-PMPS′. Such thermosetting PPS is applicable for preparing crosslinking networks, especially in optical fields such as transparent and high-refractive-index thermosetting resins.36
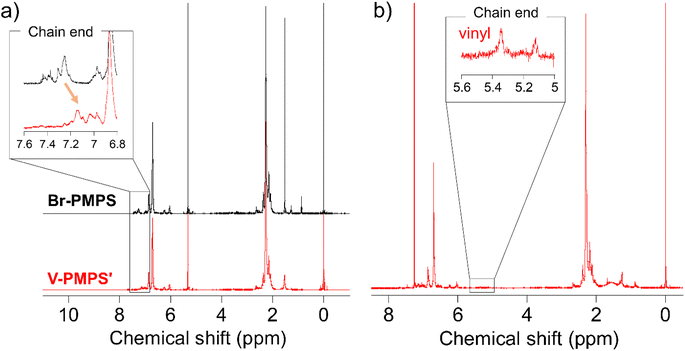 |
| | Fig. 6 1H NMR spectra of (a) Br-PMPS (Run 4 in Table 2) and V-PMPS′ in dichloromethane-d2 and (b) V-PMPS′ in chloroform-d. End-vinylation was confirmed from the upfield shift of the signals of terminal aromatic protons and presence of the signals of terminal vinyl groups. | |
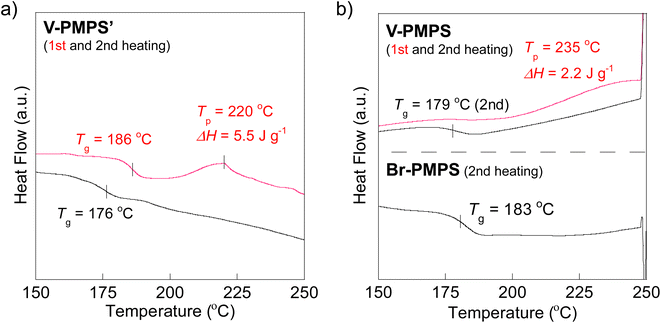 |
| | Fig. 7 DSC thermograms before and after the cross-coupling end-functionalization: (a) V-PMPS′ (1st and 2nd heating). (b) V-PMPS (1st and 2nd heating) and Br-PMPS (Run 3 in Table 2) (2nd heating). | |
Conclusions
In conclusion, we demonstrated the facile synthesis of α,ω-dihalogenated telechelic PPS derivatives using the halogenated DPS-derived sulfonium cation X-DPS+ (X: Cl or Br) as the end-capping agent. End-capping of PMPS was successful for both one-pot polymerization/functionalization and post-polymerization modification, especially for the latter, which afforded telechelic polymers with a higher yield, functionality, and molecular weight. Br-terminated telechelic PMPS could be further end-functionalized to afford thermosetting end-vinylated PMPS via the Kumada–Tamao cross-coupling reaction. To the best of our knowledge, this paper provides a simple, the most quantitative, and the highest-yield telechelic polymer synthesis without any use of reactive groups compared with the previous methods. This method can be applied to various (common) aromatic polymers to obtain novel functionalities through side/end-functionalization in a simple manner. On aiming at the industrial production of such functionalized aromatic polymers, investigation toward a larger scale should be further necessary. Also, we intend to expand this strategy to the synthesis of PPS derivatives with hetero-telechelic ends or unique structures (e.g., bottlebrush, cyclic, and figure-eight-shaped polymers), which will be our next targets.
Conflicts of interest
There are no financial conflicts to declare.
Acknowledgements
This work was partially supported by Grants-in-Aid for Scientific Research (No. 21H04695 (K. O.), 22K18335 (K. O.), and 22KJ2927 (S. W.)) from MEXT, Japan, and a NEDO project “Research and development of advanced materials and measurements for millimeter-wave and terahertz bands”. S. W. acknowledges the Grant-in-Aid from Mizuho Foundation for the Promotion of Sciences.
Notes and references
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120–5156 CrossRef CAS PubMed.
- A. J. Teator, T. P. Varner, P. C. Knutson, C. C. Sorensen and F. A. Leibfarth, ACS Macro Lett., 2020, 9, 1638–1654 CrossRef CAS PubMed.
- Y. Kametani, F. Tournilhac, M. Sawamoto and M. Ouchi, Angew. Chem., Int. Ed., 2020, 59, 5193–5201 CrossRef CAS PubMed.
- N. Hosono, S. Mochizuki, Y. Hayashi and T. Uemura, Nat. Commun., 2020, 11, 3573 CrossRef CAS PubMed.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed.
- L. Tao, J. Liu, J. Xu and T. P. Davis, Chem. Commun., 2009, 6560–6562 RSC.
- S. M. Henry, A. J. Convertine, D. S. W. Benoit, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2009, 20, 1122–1128 CrossRef CAS PubMed.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Müller, Macromolecules, 2009, 42, 3707–3714 CrossRef CAS.
- P. Tao, Y. Li, A. Rungta, A. Viswanath, J. Gao, B. C. Benicewicz, R. W. Siegel and L. S. Schadler, J. Mater. Chem., 2011, 21, 18623 RSC.
- M. R. Kember, J. Copley, A. Buchard and C. K. Williams, Polym. Chem., 2012, 3, 1196–1201 RSC.
- K. Dan and S. Ghosh, Polym. Chem., 2014, 5, 3901–3909 RSC.
- T. Yan and D. Guironnet, Angew. Chem., Int. Ed., 2020, 59, 22983–22988 CrossRef CAS PubMed.
- S. Inagaki and T. Higashihara, Polym. Chem., 2022, 13, 3613–3618 RSC.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS PubMed.
- Q. Tang, Y. Wu, P. Sun, Y. Chen and K. Zhang, Macromolecules, 2014, 47, 3775–3781 CrossRef CAS.
- L. Zhang, Y. Wu, S. Li, Y. Zhang and K. Zhang, Macromolecules, 2020, 53, 8621–8630 CrossRef CAS.
- M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Prog. Polym. Sci., 2011, 36, 455–567 CrossRef CAS.
- T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950–1968 CrossRef CAS PubMed.
- H. Yokochi, R. Takashima, D. Aoki and H. Otsuka, Polym. Chem., 2020, 11, 3557–3563 RSC.
- T. Katoh, T. Suzuki, Y. Ohta and T. Yokozawa, Polym. Chem., 2022, 13, 794–800 RSC.
- J. B. Williamson, S. E. Lewis, R. R. Johnson, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef CAS PubMed.
- E. R. King, S. B. Hunt, L. J. Hamernik, L. E. Gonce, J. S. Wiggins and J. D. Azoulay, JACS Au, 2021, 1, 1342–1347 CrossRef CAS PubMed.
- M. Jikei, J. Katoh, N. Sato, K. Yamamoto, H. Nishide and E. Tsuchida, Bull. Chem. Soc. Jpn., 1992, 65, 2029–2036 CrossRef CAS.
- S. Watanabe, T. Takayama, H. Nishio, K. Matsushima, Y. Tanaka, S. Saito, Y. Sun and K. Oyaizu, Polym. Chem., 2022, 13, 1705–1711 RSC.
- S. Watanabe and K. Oyaizu, Bull. Chem. Soc. Jpn., 2023, 96, 1108–1128 CrossRef CAS.
- S. Watanabe and K. Oyaizu, ACS Appl. Polym. Mater., 2021, 3, 4495–4503 CrossRef CAS.
- S. Watanabe and K. Oyaizu, Macromolecules, 2022, 55, 2252–2259 CrossRef CAS.
- F. Aida and K. Oyaizu, Chem. Lett., 2016, 45, 102–109 CrossRef CAS.
- E. Tsuchida, K. Yamamoto, K. Oyaizu, F. Suzuki, A. S. Hay and Z. Y. Wang, Macromolecules, 1995, 28, 409–416 CrossRef CAS.
- E. Tsuchida, K. Yamamoto, H. Nishide, S. Yoshida and M. Jikei, Macromolecules, 1990, 23, 2101–2106 CrossRef CAS.
- S. Yun, S. Lee, S. Yook, H. A. Patel, C. T. Yavuz and M. Choi, ACS Catal., 2016, 6, 2435–2442 CrossRef CAS.
- Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed.
- P.-S. Lin, Y. Shoji, S. N. Afraj, M. Ueda, C.-H. Lin, S. Inagaki, T. Endo, S.-H. Tung, M.-C. Chen, C.-L. Liu and T. Higashihara, ACS Appl. Mater. Interfaces, 2021, 13, 31898–31909 CrossRef CAS PubMed.
- A. Kiriy, V. Senkovskyy and M. Sommer, Macromol. Rapid Commun., 2011, 32, 1503–1517 CrossRef CAS PubMed.
- L. Fang, J. Sun, X. Chen, Y. Tao, J. Zhou, C. Wang and Q. Fang, Macromolecules, 2020, 53, 125–131 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Hiromichi Nishio and
Kenichi Oyaizu
,
Hiromichi Nishio and
Kenichi Oyaizu