DOI:
10.1039/D3RA06193K
(Paper)
RSC Adv., 2023,
13, 31674-31686
Internucleotidic bond formation using H-phosphonamidate derivatives and acidic activators†
Received
12th September 2023
, Accepted 21st October 2023
First published on 30th October 2023
Abstract
We previously developed a synthesis method for oligodeoxynucleotides using H-phosphonamidate derivatives as monomers. In this synthesis method, H-phosphonamidate monomers having a heterocyclic amino group as a leaving group reacts with an alcohol to form an internucleotidic H-phosphonate diester in pyridine without any additives upon heating. In this study, H-phosphonamidate reacted to form an internucleotidic linkage in the presence of acidic activators at room temperature. This finding is the first example of the activation of an amino group of an H-phosphonamidate derivative in the presence of acidic activators. In addition, the condensation reaction with acidic activators was accelerated with the use of pyridine as a solvent. Moreover, heterocyclic amino groups that have an electronegative atom, such as S and O, effectively served as a leaving group of the H-phosphonamidate monomer. From the results, we hypothesized that the tautomerization to the phosphite-form of the H-phosphonamidate monomer is an important step of the condensation reaction using acidic activators, and such process is accelerated when using pyridine as the solvent and heterocyclic amino groups as the leaving groups of the H-phosphonamidate monomer. Molecular orbital calculations also support our hypothesis. We optimized the condensation reaction conditions and synthesized dinucleoside phosphorothioate derivatives bearing all nucleobases.
Introduction
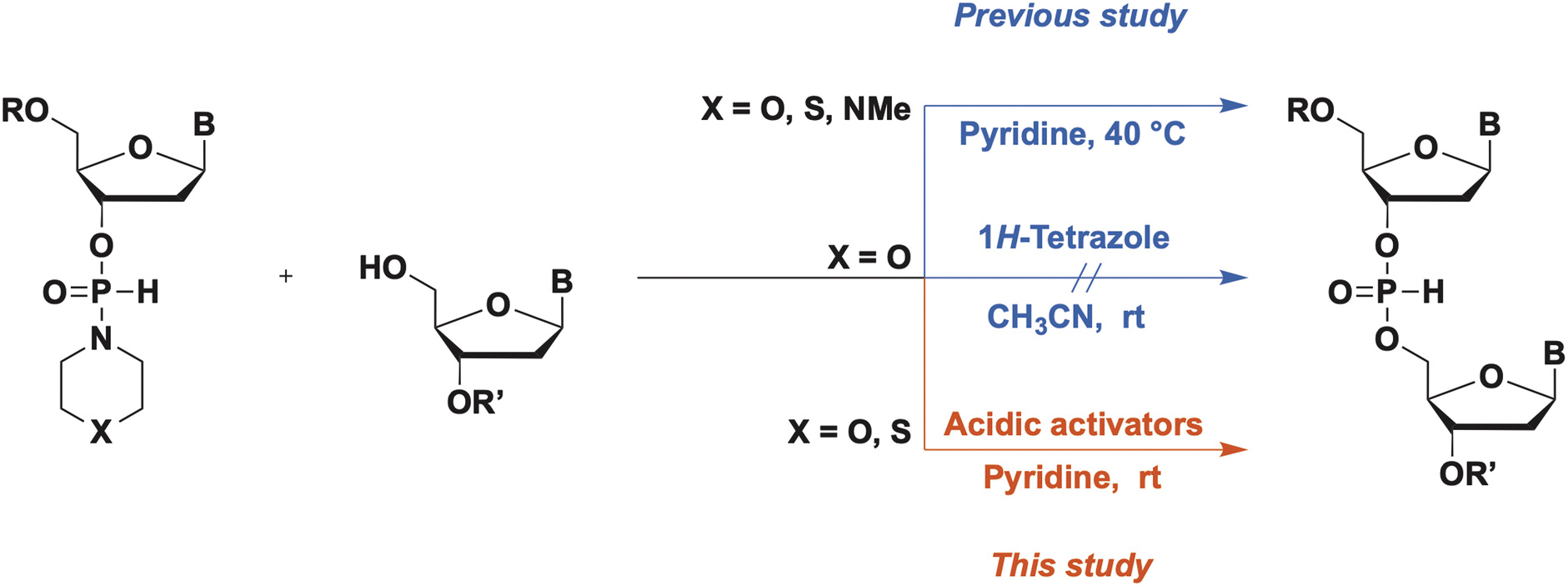
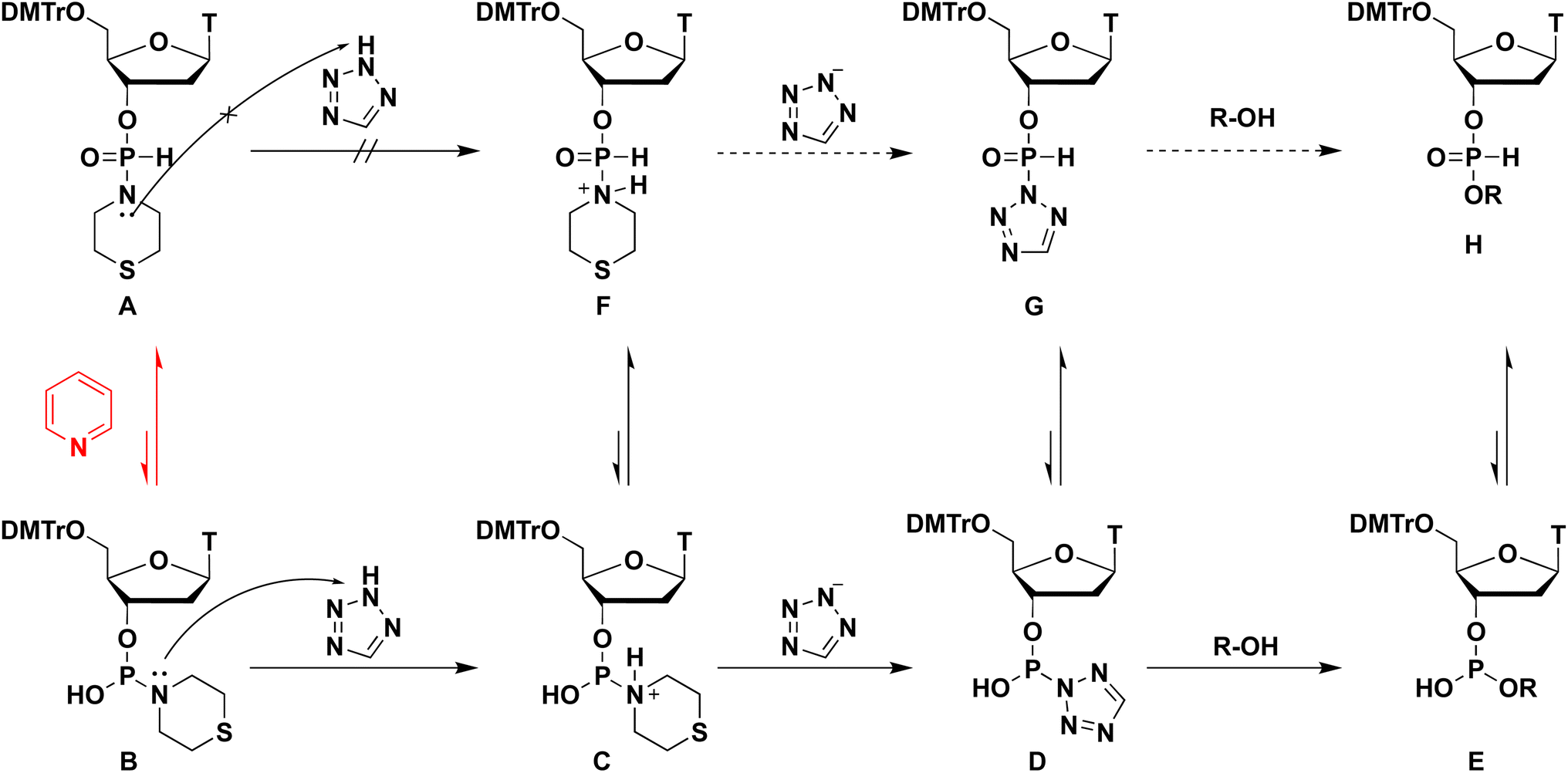
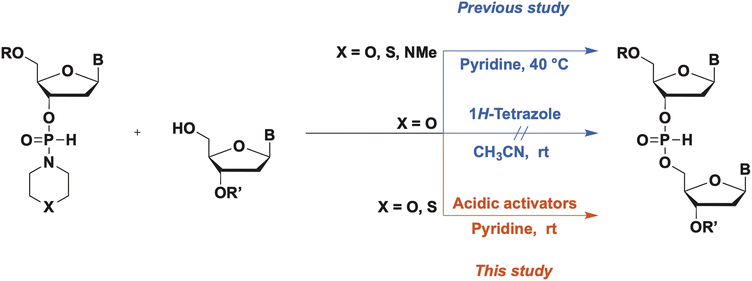
A wide variety of P-modified oligodeoxynucleotides (ODNs) have been developed and used as antisense oligonucleotides,1 and the demand for efficient synthesis methods for diverse kinds of P-modified ODNs is increasing. Thus far, we are focusing on H-phosphonamidate derivatives to develop a novel synthesis method for ODNs. In 1986, H-phosphonamidate derivatives were synthesized by van Boom et al. for the first time.2 Thereafter, a synthesis method for H-phosphonamidate derivatives was established by Stawiński et al.;3–6 however, studies rarely reported the usage of H-phosphonamidate derivatives as monomers for oligonucleotide synthesis. In 1990, we reported a synthesis method that uses a nucleoside 3′-N,N-diisopropylphosphonamidate as a monomer; however, the amino group in the construction of an internucleotidic H-phosphonate diester linkage was not a leaving group.7,8 In that study, the conversion of nucleoside 3′-N,N-diisopropylphosphonamidates to the corresponding aminophosphorochloridites occurred without cleaving the P–N bond. Aminophosphorochloridites were reacted with a 5′-hydroxy group of a nucleoside to obtain phosphoramidite intermediates. Afterward, the P–N bond of the phosphoramidites was hydrolyzed to afford the H-phosphonate diester linkage. Starting from the H-phosphonamidate derivatives, the synthesis of the H-phosphonate diester linkage involved three steps. Recently, we have reported a novel synthesis method for ODNs using H-phosphonamidate derivatives as monomers.9 In this method, H-phosphonamidate monomers having heterocyclic amino groups, such as thiomorpholino, morpholino, and N-methylpiperadino groups, reacted with an alcohol to form an internucleotidic H-phosphonate diester under mild basic conditions without the use of additives (Scheme 1). In the reaction, the heterocyclic amino groups of H-phosphonamidate monomers worked as leaving groups, and one-step formation of the H-phosphonate diester linkage from H-phosphonamidate derivatives was achieved. Moreover, under detritylation conditions, the unreacted H-phosphonamidate monomer was converted into a water-soluble compound that was easily removed by extraction. Thus, the intermediates were purified via simple extractions, and the solution-phase synthesis of trithymidine diphosphorothioate from monomers was achieved with only one silica-gel column chromatography purification. However, this approach is hindered by the low condensation efficacies and long reaction times and heating. To solve this problem, we searched more effective conditions for the activation of H-phosphonamidate monomers. In our previous study, we attempted to activate the morpholino group of H-phosphonamidate derivatives using acidic activators, such as 1H-tetrazole in CH3CN, under conditions similar to those used in the phosphoramidite method. However, the morpholino group of the H-phosphonamidate derivative was not activated in CH3CN.9
 |
| | Scheme 1 Activation of H-phosphonamidate monomers. | |
In this study, an H-phosphonamidate derivative having a thiomorpholino group was activated using acidic activators in CH3CN. This finding was attributed to the higher basicity of the nitrogen atom of the thiomorpholino group (pKaH, pKa of the conjugate acid, of thiomorpholine: 9.1) compared with that of the morpholino group (pKaH of morpholine: 8.3). Based on this result, we optimized the condensation reaction conditions and investigated the activation mechanism of H-phosphonamidate monomers in the presence of acidic activators.
Results and discussion
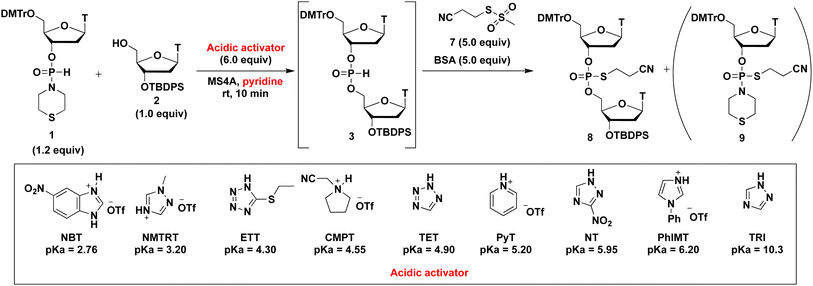
Effect of acidic activators on the condensation reaction
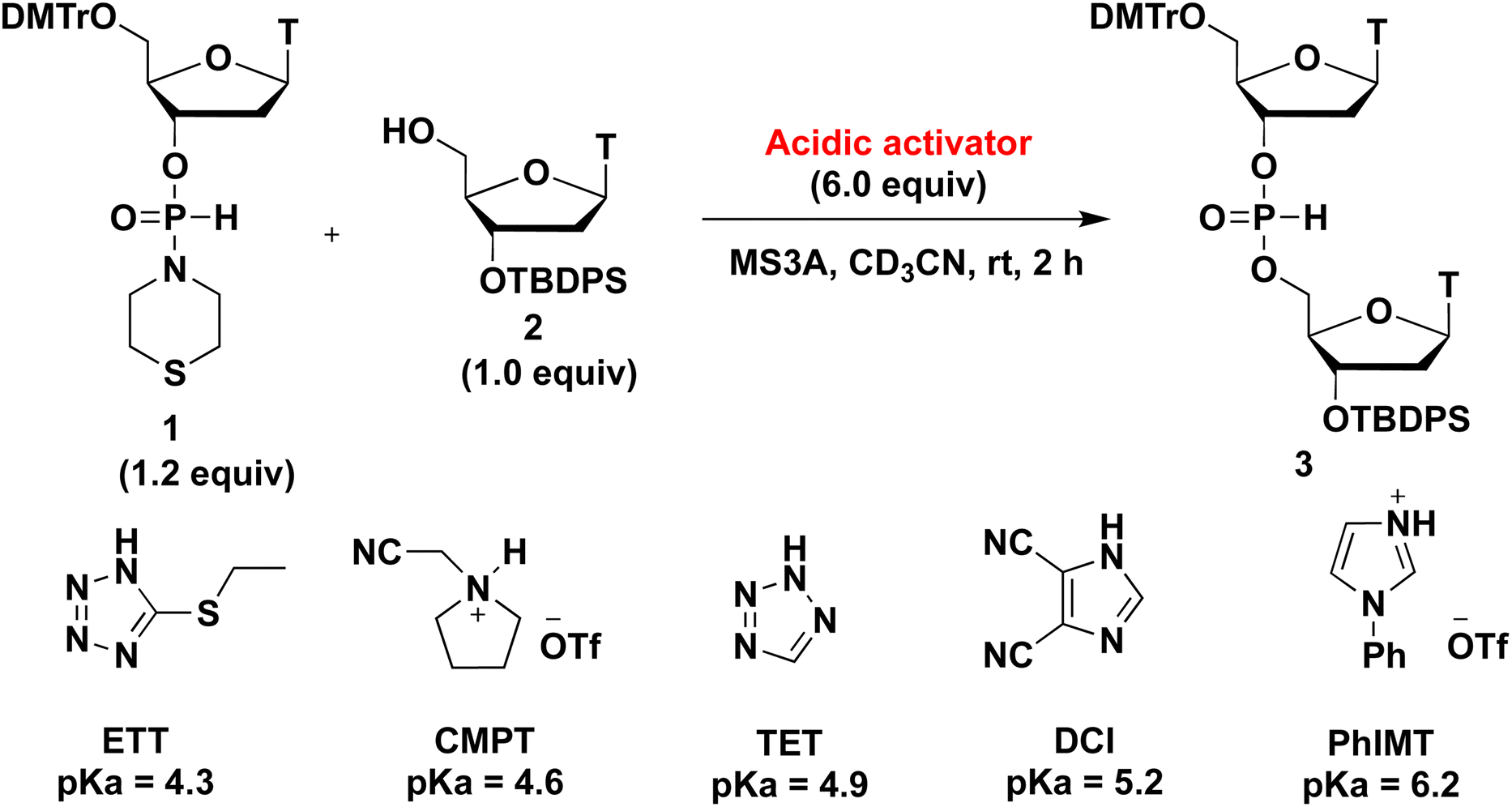
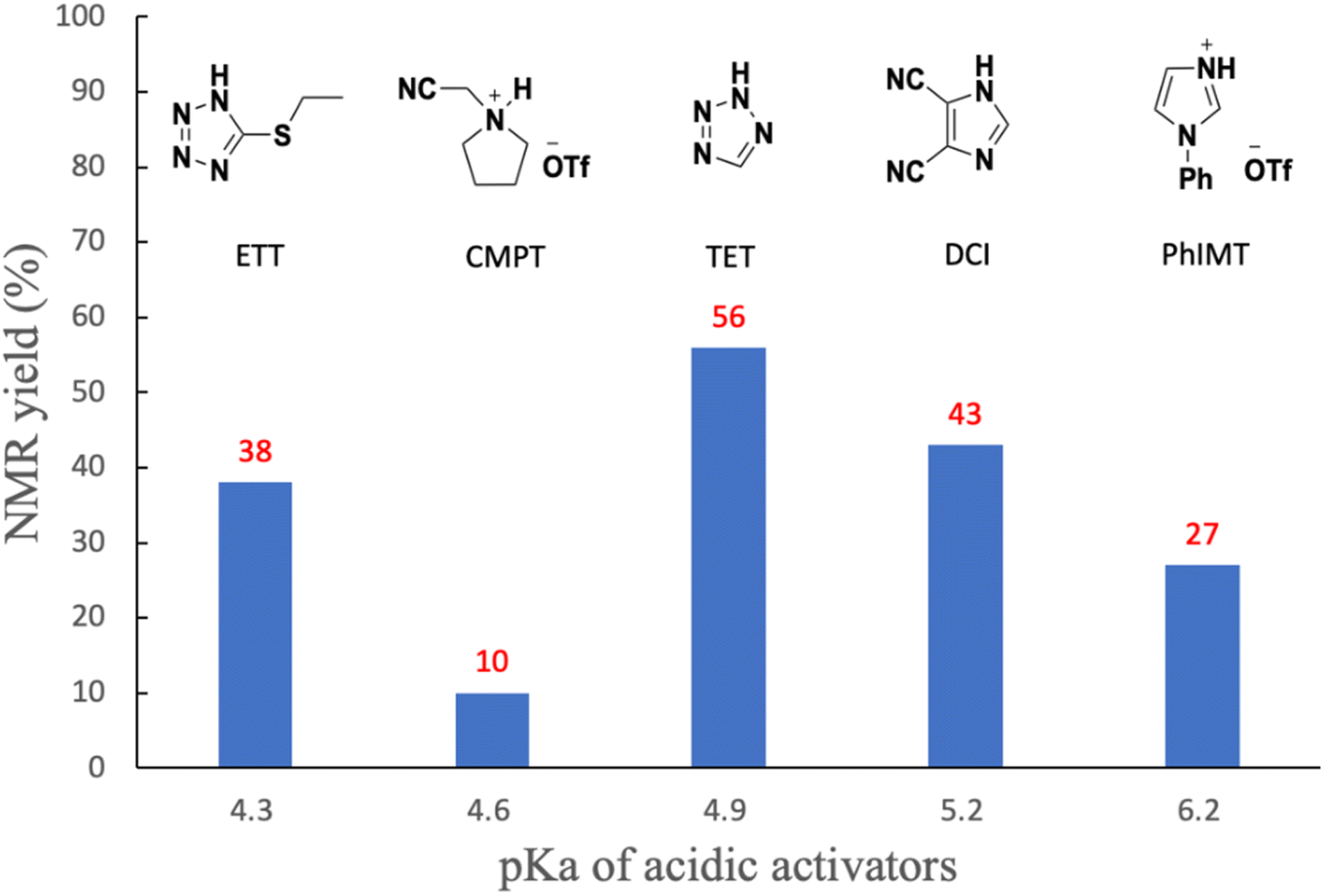
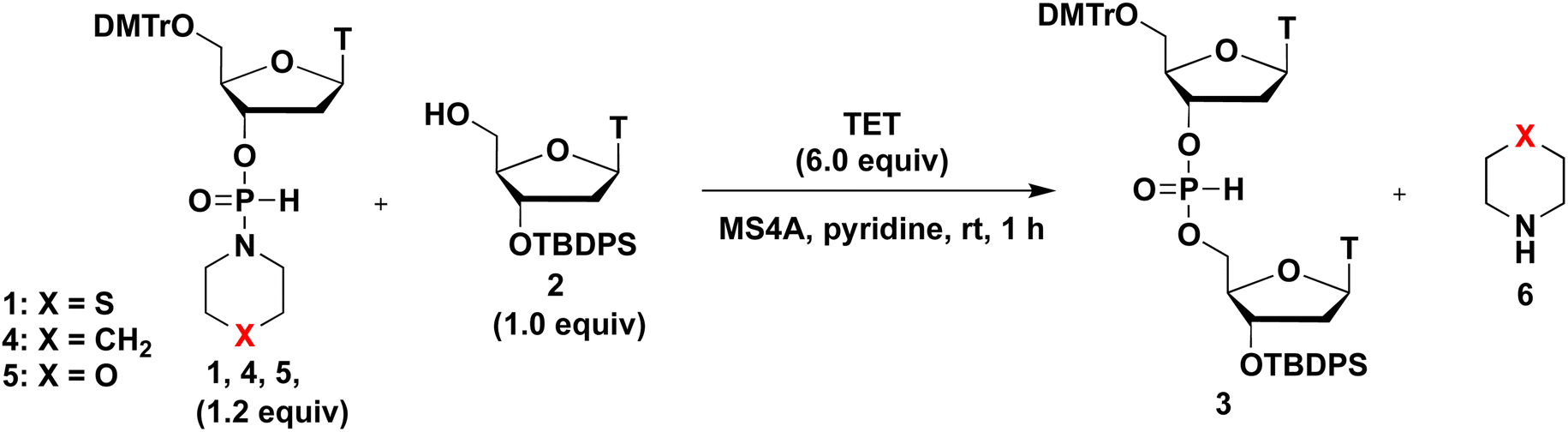
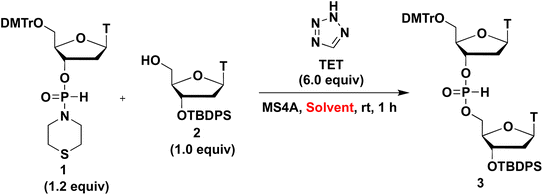
We initially investigated the synthesis of a dithymidine H-phosphonate diester derivative (3) using H-phosphonamidate 1 and a thymidine derivative 2 bearing a 5′-OH group. H-Phosphonamidate 1 was synthesized following our previously reported procedure.9 Next, we activated the thiomorpholino group of H-phosphonamidate monomer using various acidic activators. The 1.2 equivalents of H-phosphonamidate 1 were condensed with 2 bearing the 5′-OH group using acidic activators. CH3CN was used in condensation reaction at room temperature (rt) for 2 h. In all condensation reactions, MS3A or 4A was added to remove H2O in the reaction mixture. The conversion rate was estimated using the 31P nuclear magnetic resonance (NMR) analysis of the reaction mixture, and the results are shown in Table 1. Fig. 1 shows the relationships between the NMR yields and pKa of acidic activators.
Table 1 Investigation of the effect of acidic activators on the condensation reaction
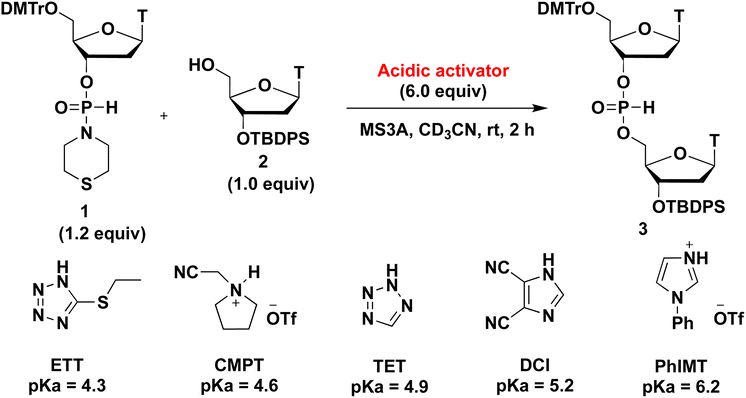
|
| Entry |
Acidic activator |
pKa of acidic activator |
NMR yield (%) |
| 1 |
ETT |
4.3 |
38 |
| 2 |
CMPT |
4.6 |
10 |
| 3 |
TET |
4.9 |
56 |
| 4 |
DCI |
5.2 |
43 |
| 5 |
PhIMT |
6.2 |
27 |
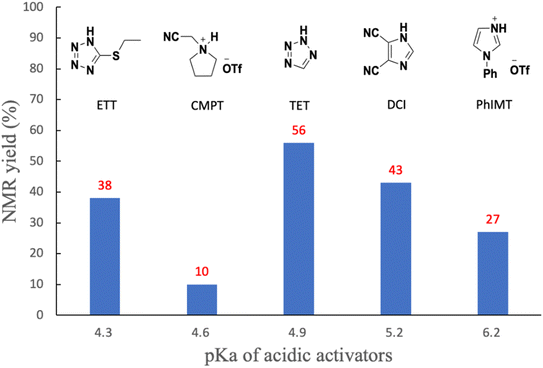 |
| | Fig. 1 Relationships between the pKa of acidic activators and NMR yields. | |
All acidic activators can activate the thiomorpholino group of H-phosphonamidate monomer 1 and gave the desired dithymidine H-phosphonate diester derivative (3). The H-phosphonamidate derivatives having a diisopropylamino group are stable under acidic conditions, such as 0.5% trifluoroacetic acid (TFA) in CHCl3.8 Thus, the results of this study are the first to show the activation of an amino group of an H-phosphonamidate derivative under acidic conditions. We also confirmed the stability of H-phosphonamidate monomer 1 under 0.5% trifluoroacetic acid (TFA) in CHCl3. The H-phosphonamidate monomer 1 was completely hydrolyzed within 10 min. This result indicated that the reactivity of H-phosphonamidate largely depends on the nature of the amino group. As shown in Fig. 1, the best result was observed with TET,10 which showed the middle classes of nucleophilicity and acidity. Conversely, lower results were recorded for ETT11 and PhIMT,12 which presented the highest acidity and nucleophilicity, respectively. Notably, the poorest result was observed with CMPT,13 which is a nonnucleophilic acidic activator. These findings showed that the nucleophilicity and acidity of acidic activators are important factors of condensation reaction.12,14
Effect of solvents on condensation reaction
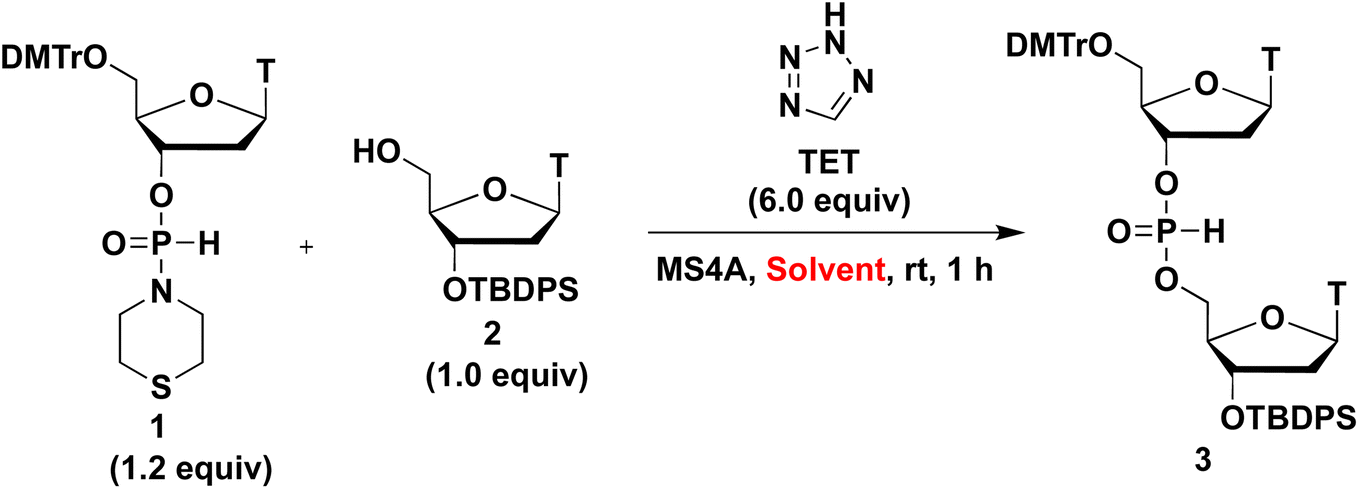
Next, we investigated the effects of solvents on condensation reactions. The 1.2 equivalents of H-phosphonamidate 1 were condensed with 2 bearing the 5′-OH group in the presence of TET as an acidic activator. The condensation reaction was conducted in CH2Cl2, THF, CH3CN, pyridine, and 2,6-lutidine used as solvents at rt for 1 h. 31P NMR analysis of the reaction mixture was performed to estimate the conversion rate, and the results are shown in Table 2.
Table 2 Effects of solvents on the condensation reaction
|
| Entry |
Solvent |
Additives |
NMR yields (%) |
| 1 |
CH2Cl2 |
— |
0 |
| 2 |
THF |
— |
12 |
| 3 |
CH3CN |
— |
22 |
| 4 |
CH3CN |
Pyridine (6 equiv) |
80 |
| 5 |
Pyridine (pKaH = 5.2) |
— |
95 |
| 6 |
2,6-Lutidine (pKaH = 6.6) |
— |
16 |
When the condensation reaction was conducted in CH2Cl2, the reaction did not proceed (entry 1). In the condensation reaction in THF and CH3CN, the reaction proceeded partially (entries 2 and 3). When we added 6 equivalents of pyridine to the mixture using CH3CN solvent, the condensation efficacy was improved (entry 4). Notably, when pyridine was used as a solvent, an almost complete condensation reaction was observed within 1 h (entry 5). The presumed positive effects of pyridine on condensation reactions were as follows. The condensation reaction using H-phosphonamidate derivatives involves their tautomerization to the phosphite-form intermediate B, activation of a thiomorpholino group of intermediate B by TET to afford the tetrazolide intermediate D, and the subsequent nucleophilic attack of the 5′-OH group of 2 to the resultant intermediate D. Excess amounts of pyridine functions as a base and accelerated tautomerization to phosphite-form intermediate B, which promoted the condensation reaction (Scheme 2). From this perspective, we attempted to accelerate the tautomerization to the phosphite-form intermediate by increasing solvent basicity. However, the condensation efficacy decreased (entry 6) when using 2,6-lutidine (pKaH = 6.6), which is more basic than pyridine (pKaH = 5.2), as a solvent. We presumed that the lower basicity of the nitrogen atom of H-phosphonamidate monomer 1 than that of 2,6-lutidine, and deprotonation of TET resulted in the formation of 2,6-lutidinium tetrazolide, which inhibited the activation of the thiomorpholino group of H-phosphonamidate monomer 1. These results indicate the order of pKaH values: pyridine (pKaH = 5.2) < thiomorpholino group of H-phosphonamidate monomer 1 < 2.6-lutidine (pKaH = 6.6). These conditions allowed 2,6-lutidine to work as a base and activated the direct attack of the 5′-OH group of 2 to H-phosphonamidate monomer 1. A similar reaction was revealed in our previous work.
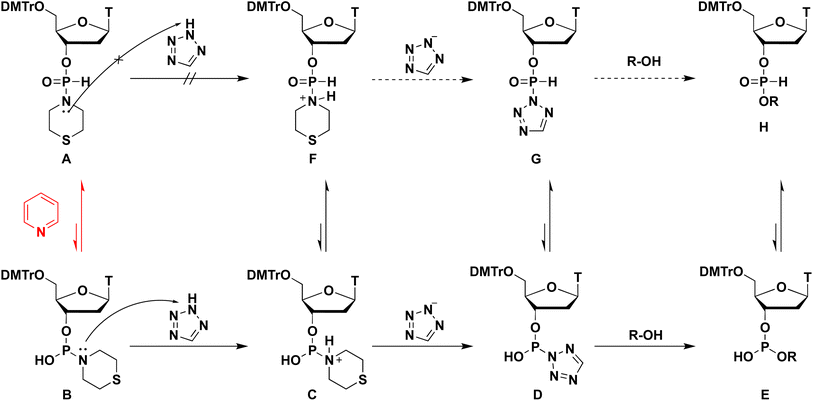 |
| | Scheme 2 Proposed mechanism of the condensation reaction. | |
Effect of leaving groups on the condensation reaction
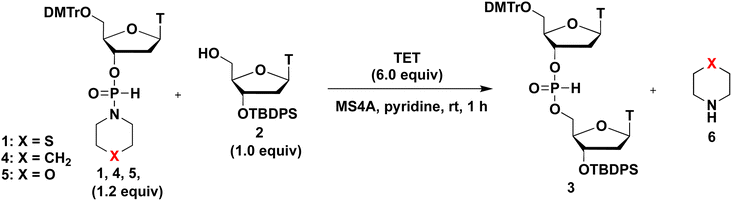
Next, the effects of leaving groups of H-phosphonamidate derivatives on the condensation reaction were investigated. H-Phosphonamidate monomers 4 and 5, which possess piperidino and morpholino group as leaving groups, respectively, were synthesized following literature procedures.3,9 H-Phosphonamidate monomers 4 and 5 were isolated via silica-gel column chromatography. TET was used as an acidic activator to condense 1.2 equivalents of H-phosphonamidate monomers 1, 4, and 5 with 2 bearing the 5′-OH group. The condensation reaction in pyridine was allowed to proceed at rt for 1 h. 31P NMR analysis of the reaction mixture was performed to estimate the conversion rate, and the results are shown in Table 3.
Table 3 Effect of leaving groups on the condensation reaction
|
| Entry |
H-Phosponamidate monomers |
X |
pKaH of 6 |
NMR yield (%) |
| Byproducts were observed. |
| 1 |
4 |
CH2 |
11.1 |
63 |
| 2 |
1 |
S |
9.1 |
95 |
| 3 |
5 |
O |
8.3 |
87a |
When an H-phosphonamidate monomer 4 having a piperidino group was used, the condensation reaction was not completed within 1 h (entry 1). By contrast, when H-phosphonamidate monomers 1 and 5, which have thiomorpholino and morpholino groups, respectively were used, the condensation efficacies were improved (entries 2 and 3). These findings indicate the importance of heterocyclic amino groups having electronegative atom, such as S and O, as leaving groups in the condensation reaction. The basicity of heterocyclic amino groups can be weakened by replacing the carbon atom of the piperidino group with an electronegative atom (Table 3), which seemed to inhibit the activation by acidic activator. However, the opposite results were obtained in this research. This result was attributed to the accelerated tautomerization to the phosphite-form of H-phosphonamidate monomer by the electronegative atom of leaving groups, which eventually enhanced the condensation reaction. Following this explanation, O is more electronegative than S, and H-phosphonamidate monomer 5 having morpholino group seemed to give a better result than H-phosphonamidate monomer 1 having thiomorpholino group. However, the opposite results were obtained in this research. The basicity of the thiomorpholino group is higher than that of the morpholino group, leading to an effective activation by acidic activators. An appropriate balance between the basicity of the heterocyclic amino group and tautomerization to the phosphite-form is important for this reaction. From these perspectives, we concluded that the thiomorpholino group exhibited the most favorable balance. Furthermore, byproducts were also observed in the reactions using H-phosphonamidate monomers 5, as shown by 31P NMR (δ 30.2, 30.1) (Table 3, entries 3: see ESI†). Therefore, the monomer H-phosphonamidate 1 was selected for further investigation.
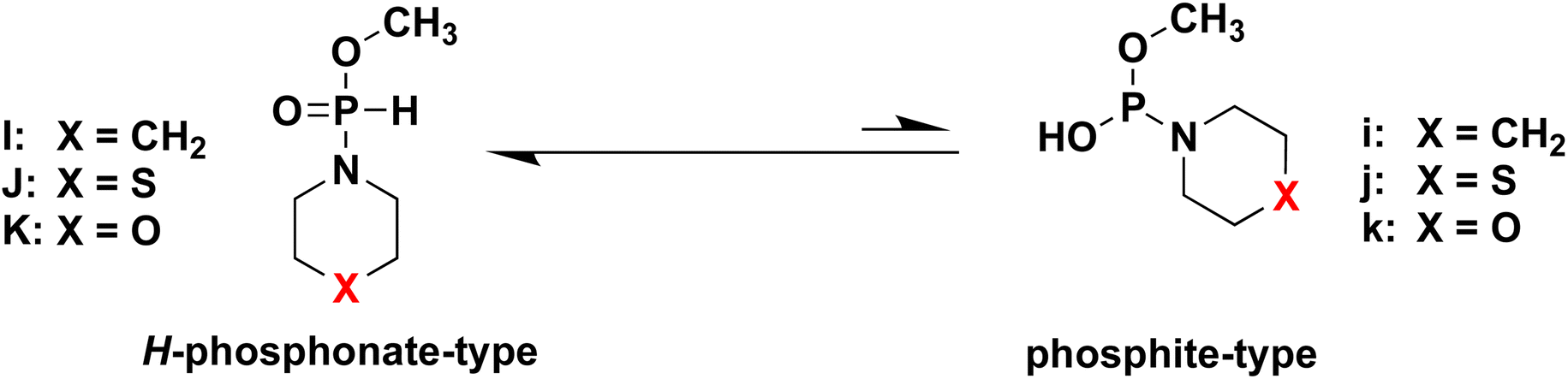
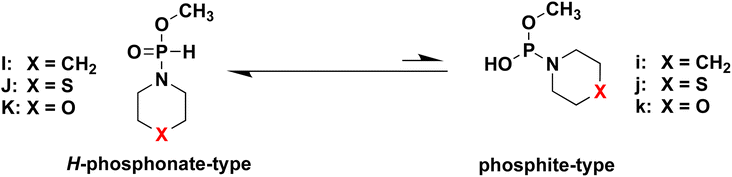
DFT calculations for the tautomers of H-phosphonamidate derivatives
To confirm our hypothesis, we conducted DFT calculations for the two kinds of tautomers of all H-phosphonamidate derivatives. The calculations for the model compounds I, J, K, i, j, and k, which are models of two kinds of tautomers of H-phosphonamidate monomers 4, 1, and 5 (Table 4), were carried out at the B3LYP/6-31G* level.15 In these calculations, the nucleoside residue was substituted for the methyl group. Table 4 shows the total energies and energy differences between two kinds of tautomers. The largest energy differences were observed in the model compounds having piperidino group (I and i). The energy differences decreased (J and K) when the carbon atom of a piperidino group was replaced with an electronegative atom, such as S and O. These results indicate that the presence of electronegative atoms in a cyclic amino group accelerate the tautomerization to phosphite forms, which agrees with our hypothesis. It was found that heterocyclic amino groups having electronegative atoms are recommended for the activation using acidic activators.
Table 4 Total energies of the model compounds calculated at the B3LYP/6-31G* level
|
| Model compounds |
X |
Total energy (au) |
Δa (kcal mol−1) |
| H-Phosphonate |
Phosphite |
| (Phosphite-form)-(H-phosphonate-form) × 627.51. |
| I or i |
CH2 |
−783.33553 |
−783.32123 |
8.98 |
| J or j |
S |
−1142.17342 |
−1142.16018 |
8.31 |
| K or k |
O |
−819.22236 |
−819.21000 |
7.76 |
Investigation of acidic activators in pyridine
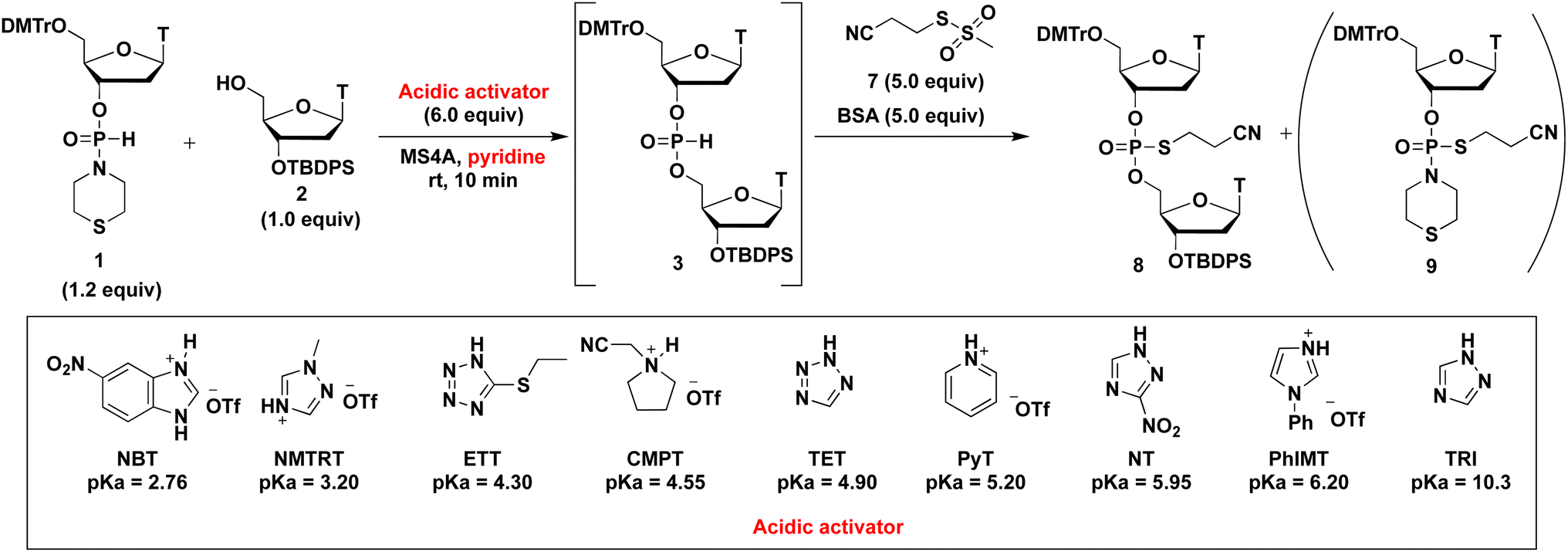
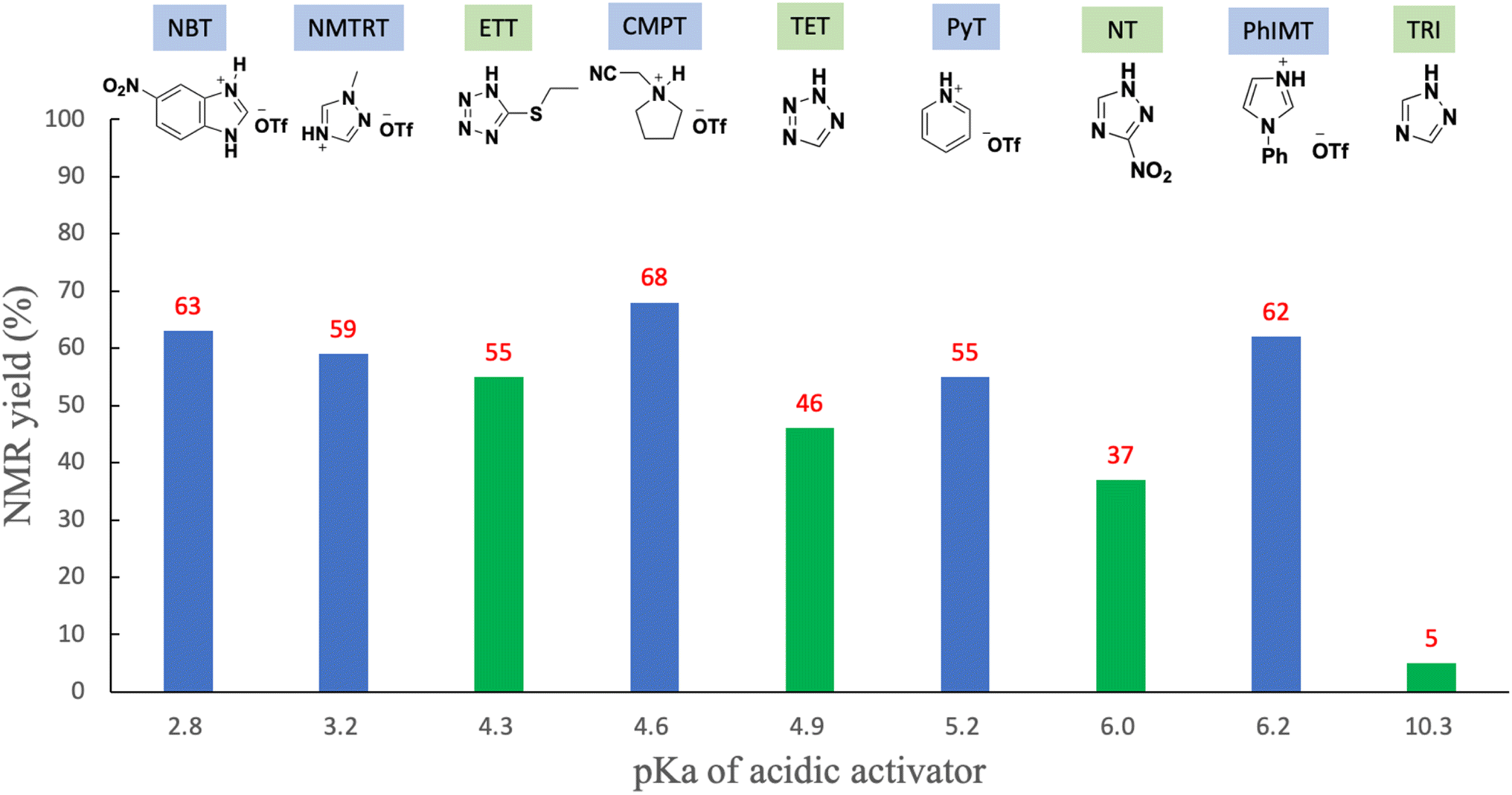
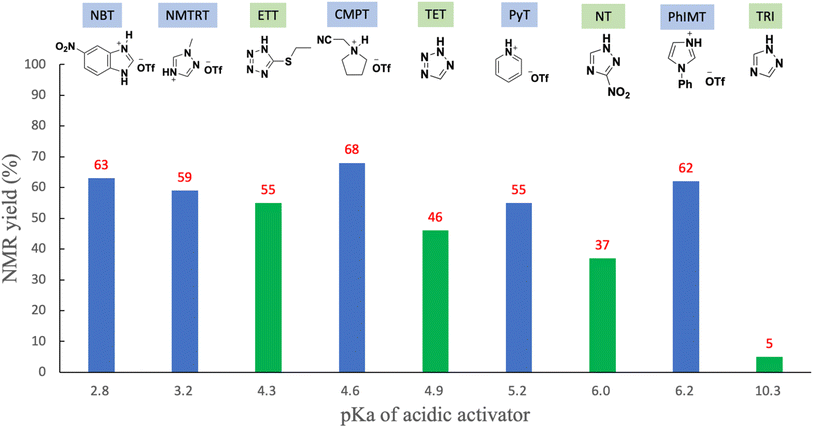
The results mentioned above reveal that the optimized solvent and leaving group are pyridine and the thiomorpholino group, respectively. Next, we aimed to determine an optimized acidic activator in pyridine. The 1.2 equivalents of H-phosphonamidate 1 were condensed using 2 bearing the 5′-OH group using acidic activators. The condensation reaction was allowed to proceed in pyridine at rt. In this experiment, the reaction time was set to 10 min to evaluate the differences in condensation efficacy. The condensation reaction was quenched by the addition of a sulfurizing reagent 7 (ref. 16) and N,O-bis(trimethylsilyl)acetamide (BSA)17,18 as a silylating reagent to the reaction mixture. Our previous study revealed that this sulfurization reaction proceeded instantly to yield the stable phosphorothioate diester 8 and phosphorothioamidate monoester derivative 9 from H-phosphonate diester 3 and H-phosphonamidate monomer 1, respectively.9 Therefore, the condensation efficacy can be determined via the 31P NMR analysis of the reaction mixture after sulfurization. The results are shown in Table 5 and Fig. 2.
Table 5 Investigation of acidic activators in pyridine
|
| Entry |
Acidic activator |
pKa of acidic activator |
NMR yields (%) |
| 1 |
TRI |
10.3 |
5 |
| 2 |
PhIMT |
6.20 |
62 |
| 3 |
NT |
5.95 |
37 |
| 4 |
PyT |
5.20 |
55 |
| 5 |
TET |
4.90 |
46 |
| 6 |
CMPT |
4.55 |
68 |
| 7 |
ETT |
4.30 |
55 |
| 8 |
NMTRT |
3.20 |
59 |
| 9 |
NBT |
2.76 |
63 |
 |
| | Fig. 2 Relationships between the pKa of acidic activators and NMR yields. | |
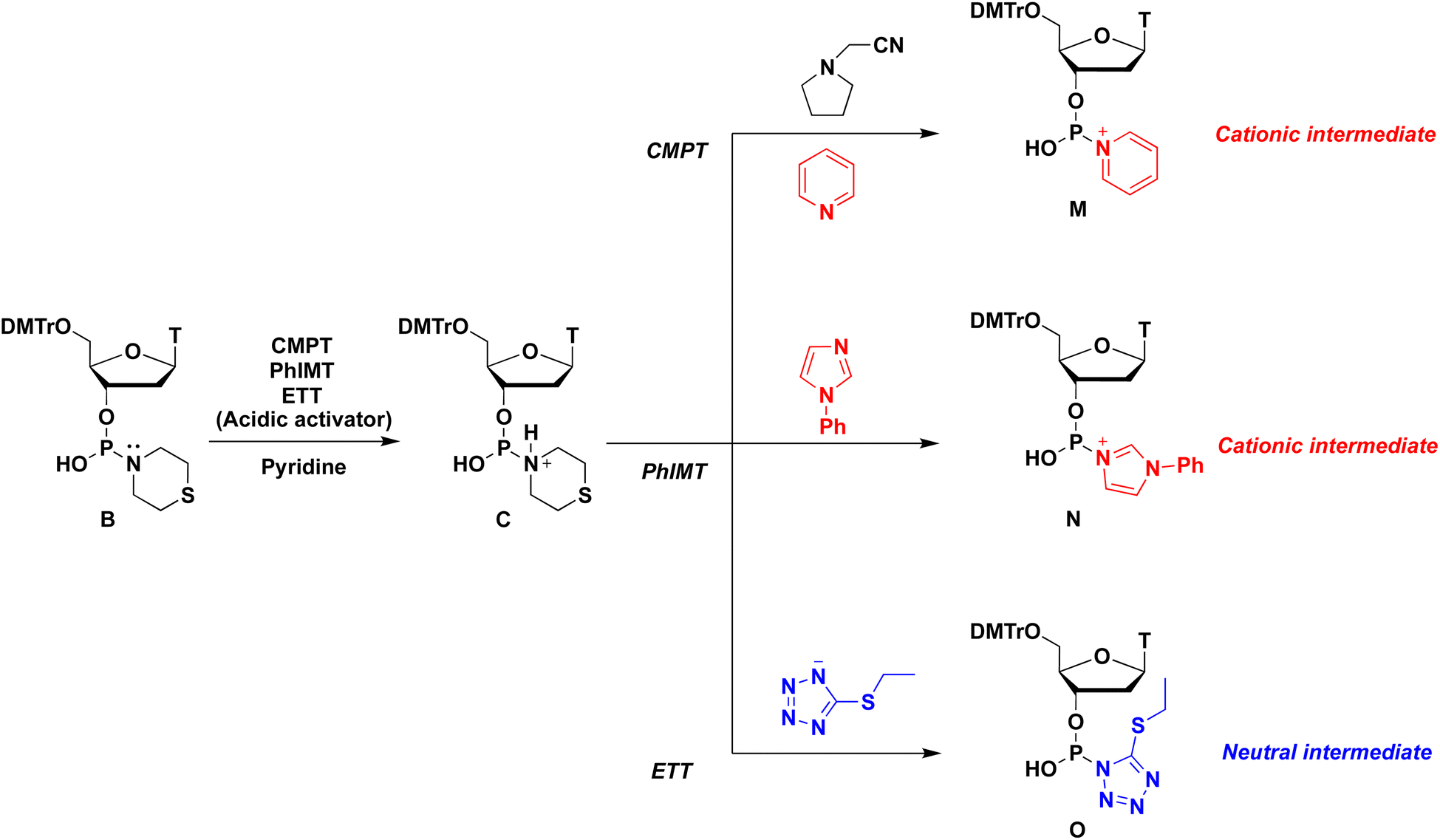
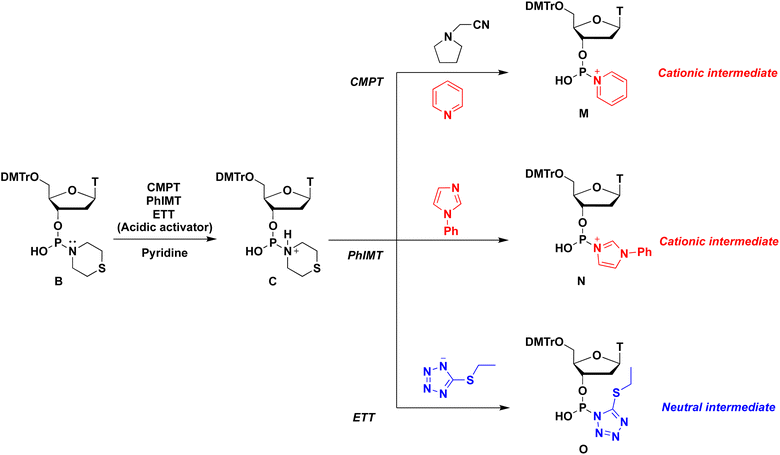
Various acidic activators whose pKa values ranged from 2.76 to 10.3 were investigated in pyridine. Fig. 2 shows that when the neutral acidic activators were used, more acidic activators attained improved condensation efficacies (TRI, NT,19 TET, and ETT; entries 1, 3, 5, and 7, respectively). On the other hand, almost the same results were observed for CMPT (pKa = 4.55), NMTRT (pKa = 3.2),20 and NBT (pKa = 2.76)21(entries 6, 8, and 9, respectively). These findings indicate that the acidity effect of acidic activators on the condensation reaction plateaued at around pKa 4.50. Although CMPT gave the poorest result in CH3CN (Table 1, entry 2), it showed the best result in pyridine. No nucleophilic catalysts were present when CMPT was used in CH3CN. Meanwhile, when other acidic activators, such as TET, were used in CH3CN, their conjugated bases acted as nucleophilic catalysts and accelerated the condensation reaction. By contrast, pyridine itself acted as a nucleophilic catalyst and accelerated the reaction even when nonnucleophilic CMPT was used (Scheme 3).
 |
| | Scheme 3 Formation of various intermediates. | |
Specific results were observed with the use of PhIMT and ETT. Despite its lower acidity, PhIMT exhibited a relatively good result (62% in entry 2). Meanwhile, ETT (pKa = 4.3), despite its higher acidity, yielded a poorer result than CMPT (pKa = 4.55). We hypothesized that these results were due to the intermediates formed by the conjugated base of PhIMT and ETT. The conjugated base of PhIMT formed the cationic intermediate N, which has a high activity. Therefore, relatively good results were obtained with the use of PhIMT. On the other hand, the conjugated base of ETT formed the neutral intermediate O, which is less reactive compared with cationic intermediates M and N. These results indicate that CMPT, which presents acidity to some extent and gives the cationic intermediate M, is the most optimal for the activation of H-phosphonamidate derivatives.
DFT calculations for the reaction intermediate
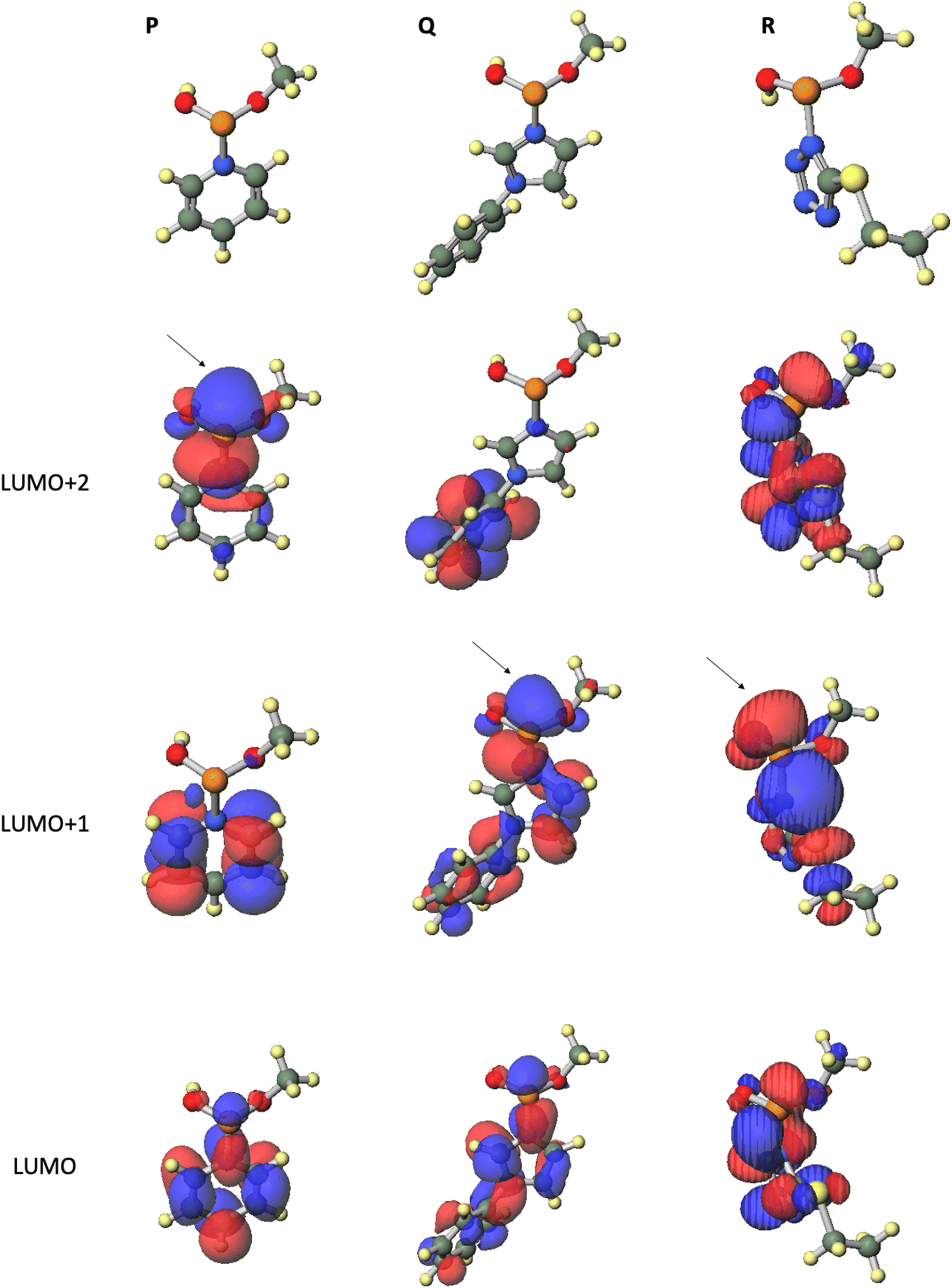
To elucidate the leaving group-reactivity relationship of the intermediates, we conducted DFT calculations for the reaction intermediates. Calculations for the model compounds P, Q, and R, which are models of pyridinium intermediate M, phenylimidazolium intermediate N, and ethylthiotetrazolide intermediate O, respectively, were carried out at the B3LYP/6-31G* level.15 In these calculations, the nucleoside residue was substituted for the methyl group. Table 6 and Fig. 3 provide the orbital energies, optimized geometries, and molecular orbitals (MOs) of these compounds. For the condensation reaction, an orbital, which exists on the back side of a leaving group of the phosphorus atom, is important. In the structure of pyridinium intermediate P, a large lowest unoccupied molecular orbital (LUMO)+2 is located on the back side of the leaving group of the phosphorus atom. By contrast, in the structures of phenylimidazolium intermediate Q and ethylthiotetrazolide intermediate R, a large LUMO+1 is located on the back side of the leaving group of phosphorus atom (Fig. 3). Compared with that of ethylthiotetrazolide intermediate R (LUMO+1: −0.4626 eV), the orbital energy levels of pyridinium intermediate P (LUMO+2: −4.1769 eV) and phenylimidazolium intermediate Q (LUMO+1: −3.8123 eV) were considerably lower. Thus, intermediates P and Q are more reactive to nucleophiles than intermediate R (Table 6). These calculation results were in good agreement with the experimental findings. To summarize, acidic activators, which have a suitable acidity and yield cationic intermediates such as a pyridinium intermediate, effectively enable the condensation reaction of H-phosphonamidate monomers.
Table 6 Orbital energies of the model compounds calculated at the B3LYP/6-31G* level
| |
Orbital energy (eV) |
| Pyridinium intermediate (P) |
Phenylimidazolium intermediate (Q) |
Ethylthiotetrazolide intermediate (R) |
| LUMO+2 |
−4.1769 |
−3.5130 |
0.5116 |
| LUMO+1 |
−5.1420 |
−3.8123 |
−0.4626 |
| LUMO |
−6.1226 |
−4.7620 |
−0.7538 |
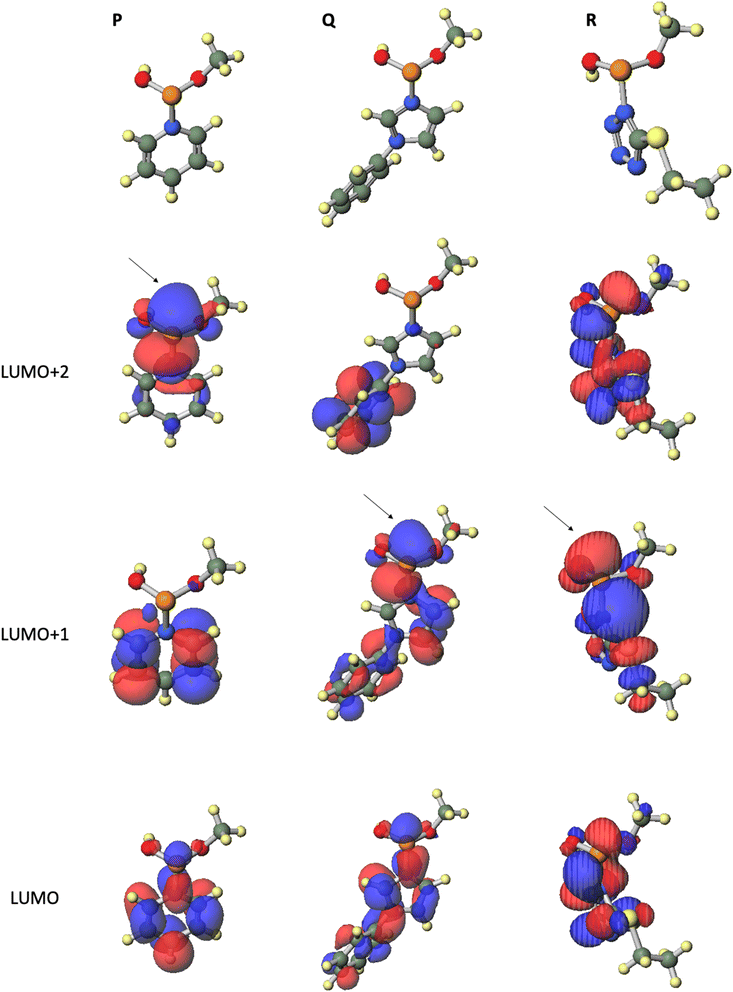 |
| | Fig. 3 Optimized geometries and MOs of the model compounds calculated at the B3LYP/6-31G* level. | |
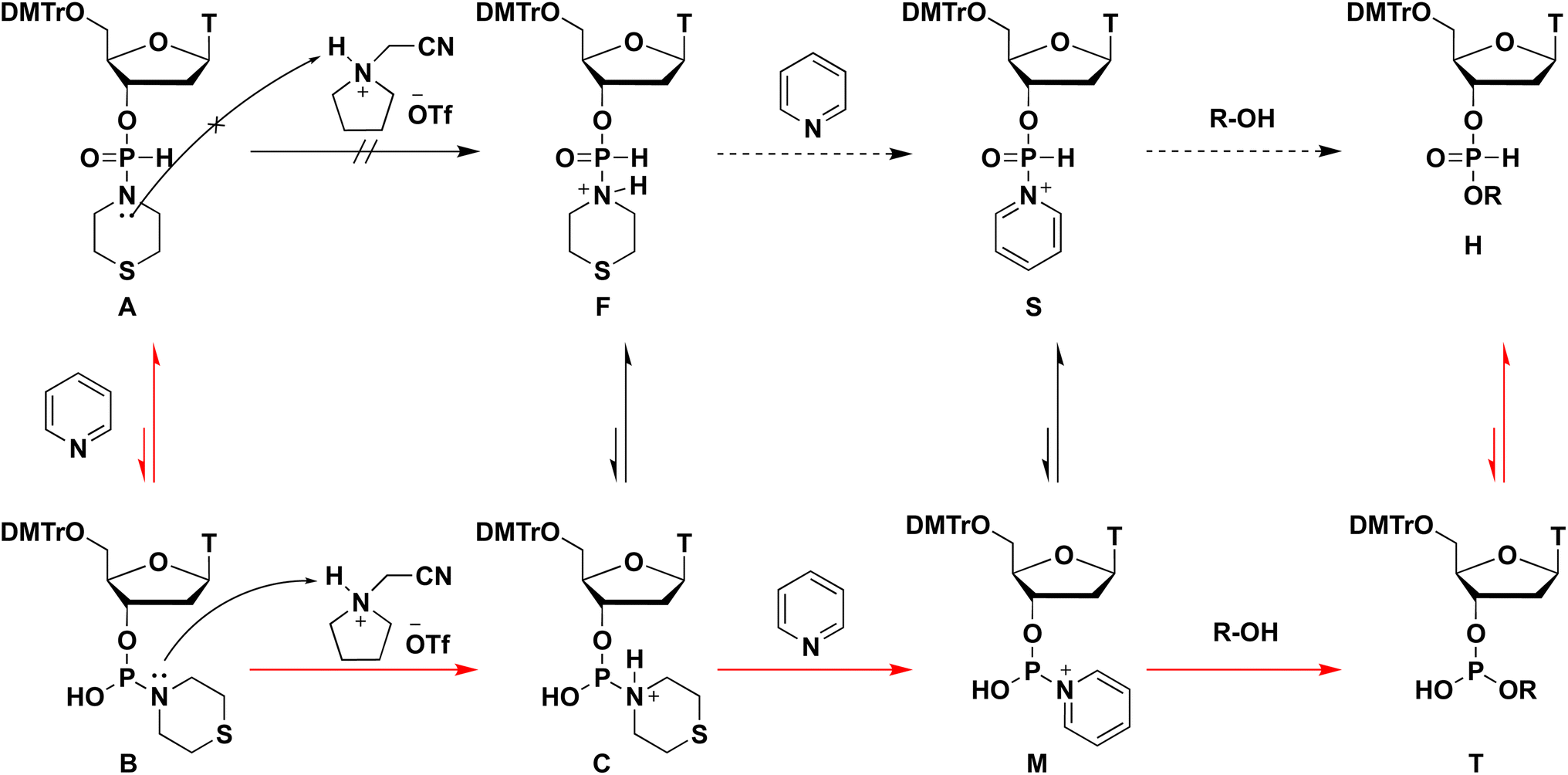
Plausible mechanism of the condensation reaction
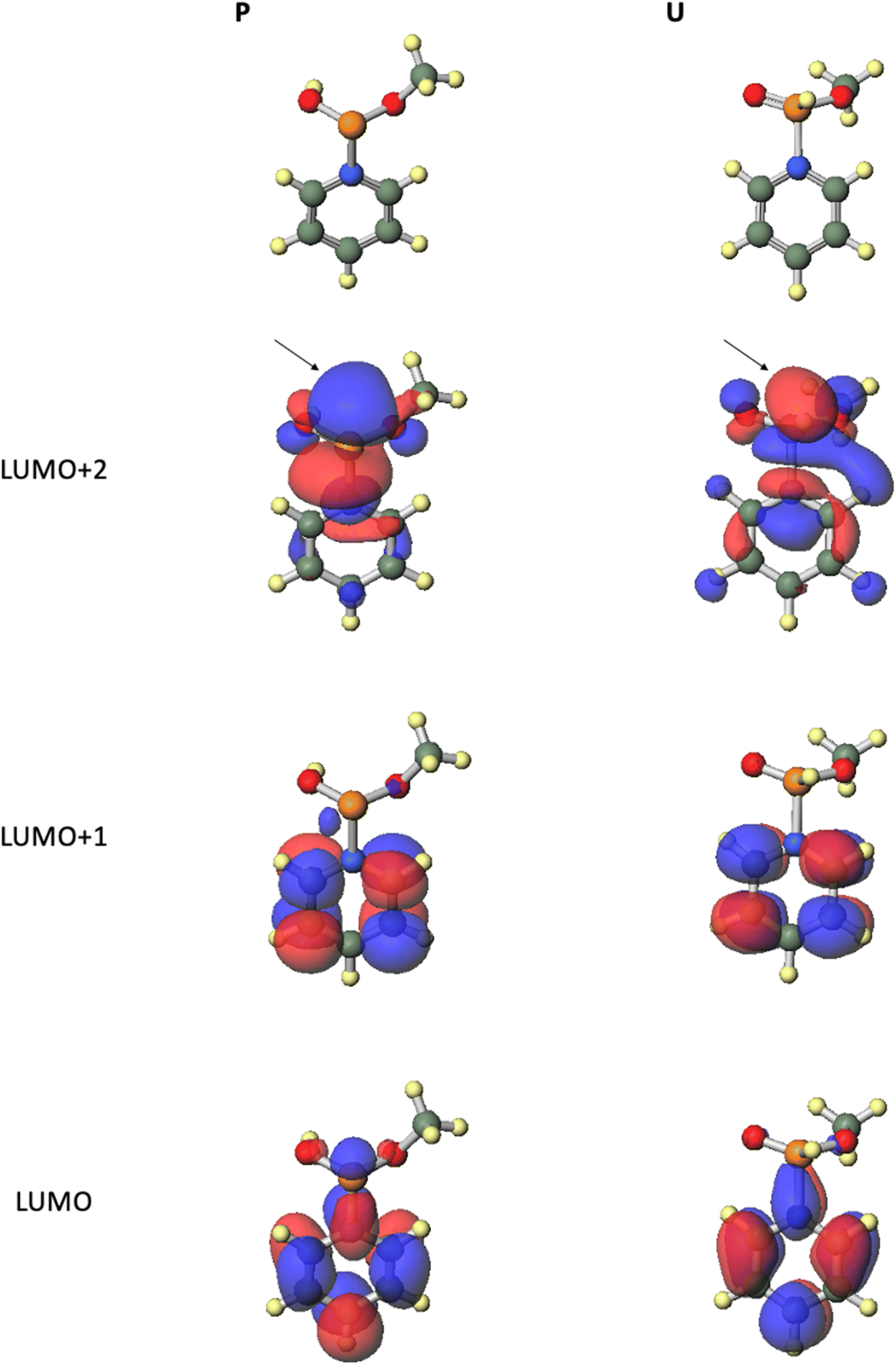
From all the results of experiments and calculations, we concluded the mechanism of the condensation reaction when CMPT was used as an acidic activator (Scheme 4). First, the tautomerization of H-phosphonate-form A of the H-phosphonamidate monomer to phosphite-form B occurred, and this step was accelerated by pyridine. Then, CMPT activated the thiomorpholino group of B to afford an intermediate C, which was converted to pyridinium intermediate M. Finally, pyridinium intermediate M was reacted with alcohol to give an H-phosphonate diester H. However, in this mechanism, pyridinium intermediate S also reacted with alcohol. Therefore, we performed DFT calculations for the reaction intermediates to elucidate their reactivities. The calculations for model compounds P and U, which are models of phosphite-form intermediate M and H-phosphonate-form intermediate S, respectively, were carried out at the B3LYP/6-31G* level. In the calculations, the nucleoside residue was substituted for the methyl group. Table 7 and Fig. 4 show the orbital energies, optimized geometries, and MOs of these compounds. In the structure of pyridinium intermediates P and U, a large LUMO+2 is located on the back side of the leaving group of the phosphorus atom (Fig. 4). Compared with the orbital energy level of H-phosphonate-form intermediate U (LUMO+2: −3.0722 eV), that of phosphite-form intermediate P (LUMO+2: −4.1769 eV) was substantially lower, which indicates that phosphite-form P is more reactive to nucleophiles than H-phosphonate-form U (Table 7). Thus, pyridinium intermediate M is the real active species, and it reacts with alcohol.
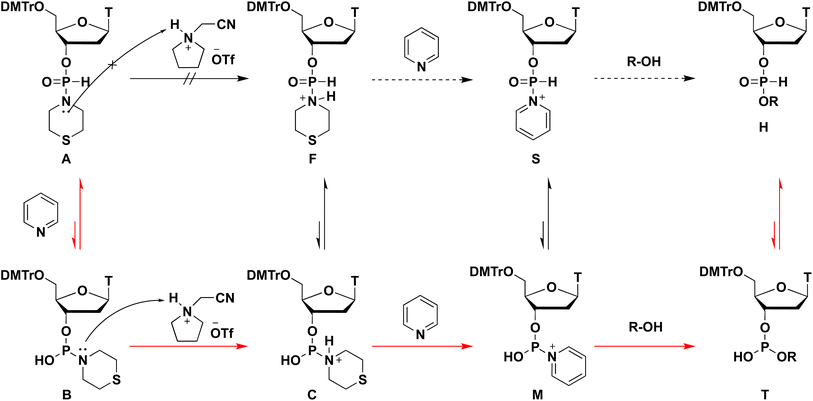 |
| | Scheme 4 Plausible mechanism of the condensation reaction. | |
Table 7 Orbital energies (au) of the model compounds calculated at the B3LYP/6-31G* level
| |
Orbital energy (eV) |
| Phosphite-form (P) |
H-Phosphonate-form (U) |
| LUMO+2 |
−4.1769 |
−3.0722 |
| LUMO+1 |
−5.1402 |
−5.4831 |
| LUMO |
−6.1226 |
−6.6124 |
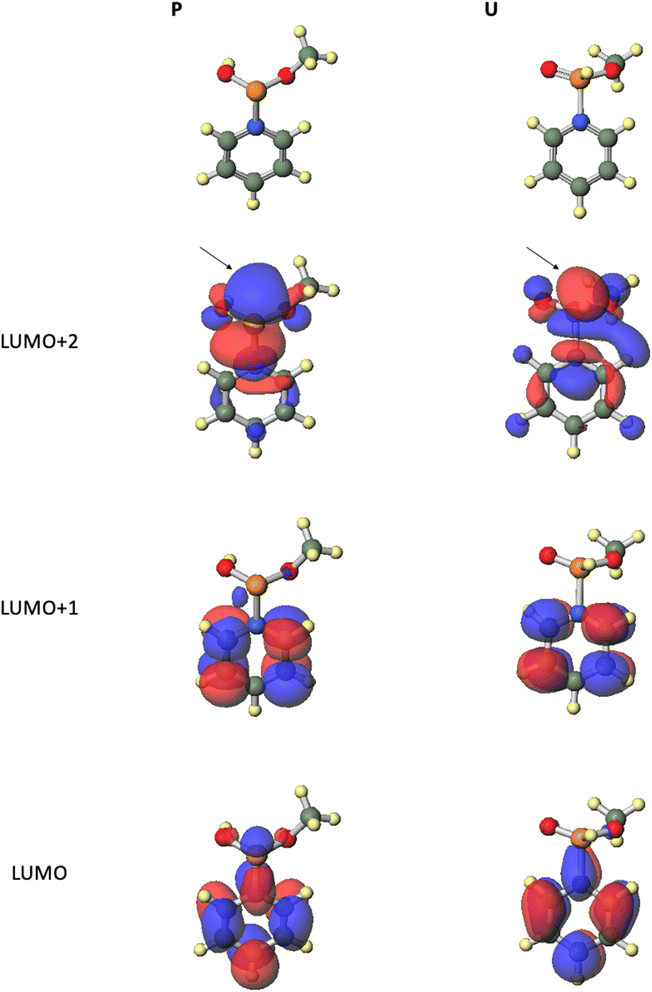 |
| | Fig. 4 Optimized geometries and MOs of the model compounds calculated at the B3LYP/6-31G* level. | |
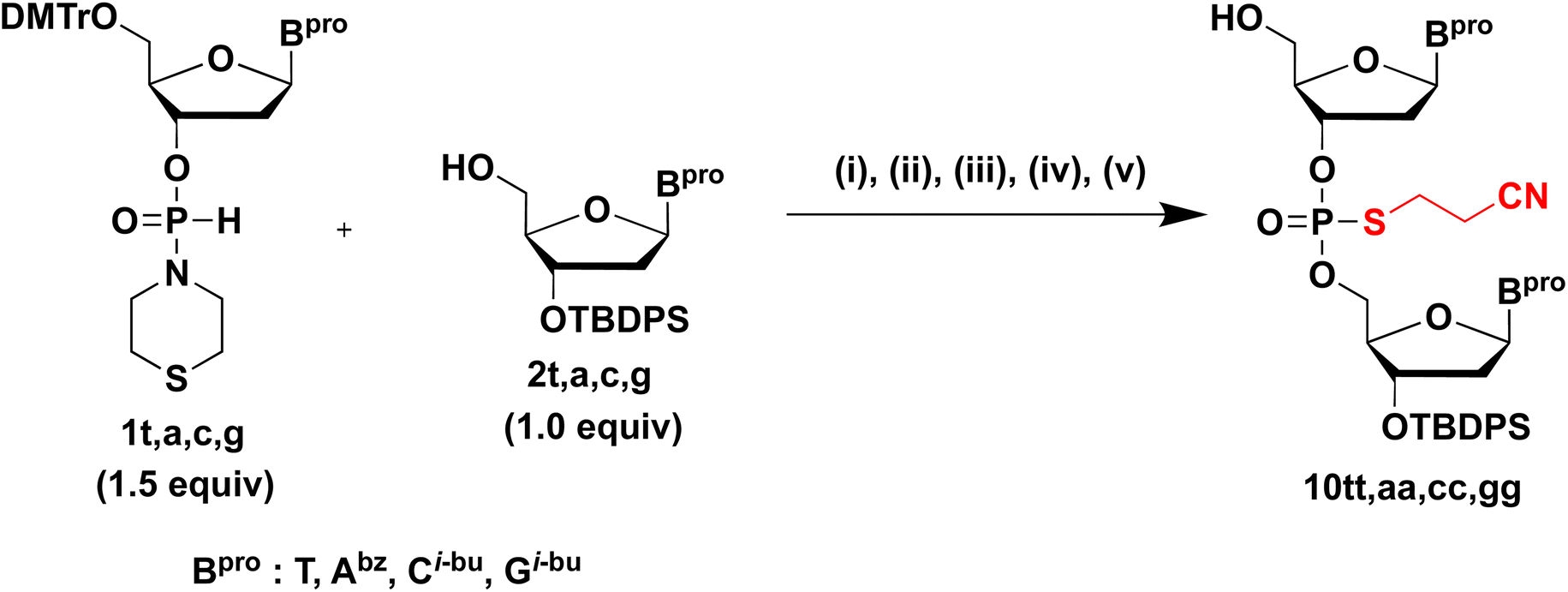
Synthesis of dinucleoside S-cyanoethyl phosphorothioate diester (NPSCEN). Finally, under optimized conditions, NPSCEN dimers bearing all nucleobases were synthesized using H-phosphonamidate derivatives 1t, 1a, 1c, or 1g and 5′-OH derivatives 2t, 2a, 2c, or 2g, respectively (Table 8). H-Phosphonamidate 1 (1.5 equivalents) was condensed with 2 bearing the 5′-OH group using 6.0 equivalents of CMPT in pyridine at rt for 30 min. Thereafter, sulfurization of the H-phosphonate diester linkage was conducted using sulfurizing reagent 7. These steps were conducted as a one-pot reaction. After the mixture was concentrated, 10 was produced by detritylation using 1% TFA in CHCl3 and simple extraction. The excess amount of 1 was removed completely via simple extraction because H-phosphonamidate monomer 1 was converted into a water-soluble derivative by a detritylation reaction.9 The sulfurization reaction was accelerated by the addition of BSA as a silylating reagent; BSA promoted the conversion of H-phosphonamidate monomer 1 to phosphorothioamidate monoester derivative 9, which was not removed by extraction after detritylation.9 It has been shown that an H-phosphonate diester linkage shows a higher reactivity to electrophiles than an H-phosphonamidate linkage. Owing to the differences in the reactivities to sulfurizing reagents, sulfurization without a silylating reagent was only successful on the H-phosphonate diester linkage.9 After simple extraction, the crude mixtures were purified by silica-gel column chromatography. All dimers were obtained in 74–78% isolated yields, which indicates the possible application of this strategy to all nucleobases. We verified the completion of condensation and sulfurization reactions through thin-layer chromatography monitoring. Consequently, minor losses during purification steps, such as extraction and silica-gel column chromatography, afforded modest isolated yields. Compared with our previous approach that used mild basic conditions (40 °C, 2 h), this novel strategy is more efficient and practical (rt, 30 min).
Table 8 Synthesis NPSCEN 10 a b
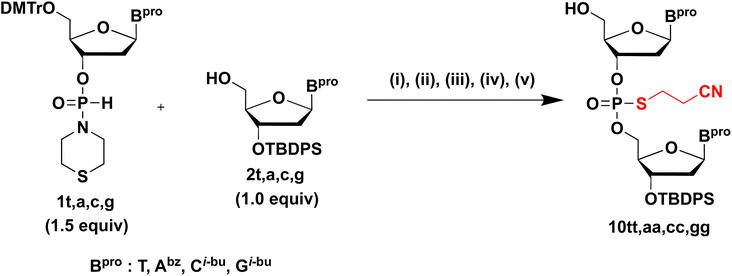
|
| Entry |
H-Phosphonamidate monomer |
5′-OH nucleoside |
Product |
Isolated yield (%) |
| Reagents and conditions: (i) CMPT (6.0 equiv.), pyridine, MS4A, rt, 30 min; (ii) sulfurizing reagent 7 (1.2 equiv.), pyridine, rt, 1 h; (iii) 1% TFA in CHCl3, rt, 10 min; (iv) extraction with a saturated aqueous solution of NaHCO3; (v) silica-gel column chromatography. 1t, 2t, 10tt: Bpro = thymine, 1a, 2a, 10aa: Bpro = N6-benzoyladenine, 1c, 2c, 10cc: Bpro = N4-isobutyrylcytosine, 1g, 2g, 10gg: Bpro = N2-isobutyrylguanine. |
| 1 |
1t |
2t |
10tt |
78 |
| 2 |
1a |
2a |
10aa |
76 |
| 3 |
1c |
2c |
10cc |
75 |
| 4 |
1g |
2g |
10gg |
74 |
Conclusion
This study focused on the investigation of the acidic activator-mediated internucleotidic bond formation reaction using H-phosphonamidate derivatives bearing a heterocyclic amine. H-Phosphonamidate derivatives having a thiomorpholino group affords an internucleotidic linkage in the presence of acidic activators such as 1H-tetrazole. This study is the first to report the activation of H-phosphonamidate derivatives by acidic activators. Pyridine as a solvent effectively accelerated the reaction. In addition, an efficient reaction necessitates the introduction of an electron negative atom, such as O and S atom, to the cyclic amine structure. From these results, the tautomerization of H-phosphoramidates to their phosphite-form is an important process for the investigated reaction. In addition, exploration of a variety of acidic activators revealed the good condensation efficacy obtained with the use of acidic activators giving a cationic intermediate. Our experimental results are supported by MO calculation, which also revealed pyridinium intermediate as the most reactive one. Superior results were obtained with the combination of a thiomorpholino group as a leaving group, pyridine as a solvent, and N-cyanomethyl pyrrolidinium triflate (CMPT) as an acidic activator. Under the optimized conditions, four kinds of dinucleoside phosphorothioate derivatives were synthesized in three steps, namely, condensation, sulfurization, and detritylation, in good yields. In our previous approach, the condensation reaction cannot proceed at rt. However, in this study, the condensation reaction was completed at rt under the optimized conditions. The synthesis of longer ODNs using this strategy is in progress.
Author contributions
T. T. established the research plans. T. T. conducted the experiments. All authors analyzed the data. T. T. wrote the manuscript. K. S. and T. W. revised the manuscript. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Dr Yayoi Yoshimura (Tokyo University of Science) for the mass spectrum measurements. We would like to thank Enago (https://www.enago.jp) for the English language review. This work was supported by JST, the establishment of university fellowships toward the creation of science technology innovation, Grant Number JPMJFS2144 and AMED under Grant Number JP21ae0121025.
References
- S. T. Crooke, J. L. Witztum, C. F. Bennett and B. F. Baker, Cell Metab., 2018, 27, 714–739 CrossRef CAS PubMed.
- J. Marugg, A. Burik, M. Tromp, G. Van der Marel and J. Van Boom, Tetrahedron Lett., 1986, 27, 2271–2274 CrossRef CAS.
- A. Sobkowska, M. Sobkowski, J. Cieślak, A. Kraszewski, I. Kers and J. Stawiński, J. Org. Chem., 1997, 62, 4791–4794 CrossRef CAS.
- A. Sobkowska, M. Sobkowski, J. Stawi ski and A. Kraszewski, Nucleosides, Nucleotides Nucleic Acids, 1995, 14, 703–706 CrossRef CAS.
- I. Kers, J. Stawiński and A. Kraszewski, Tetrahedron, 1999, 55, 11579–11588 CrossRef CAS.
- A. Kraszewski, M. Sobkowskia and J. Stawiński, J. Chem. Soc., Perkin Trans. 1, 1993, 1699–1704 RSC.
- T. Wada, K. Ishikawa and T. Hata, Tetrahedron Lett., 1990, 31, 6363–6366 CrossRef CAS.
- T. Wada, K. Ishikawa and T. Hata, Tetrahedron, 1993, 49, 2043–2054 CrossRef CAS.
- T. Tsurusaki, K. Sato and T. Wada, Org. Biomol. Chem., 2023, 21, 2486–2492 RSC.
- F. R. Benson, Chem. Rev., 1947, 41, 1–61 CrossRef CAS PubMed.
- E. Lieber and T. Enkoji, J. Org. Chem., 1961, 26, 4472–4479 CrossRef CAS.
- Y. Hayakawa, R. Kawai, A. Hirata, J.-i. Sugimoto, M. Kataoka, A. Sakakura, M. Hirose and R. Noyori, J. Am. Chem. Soc., 2001, 123, 8165–8176 CrossRef CAS PubMed.
- N. Oka, T. Wada and K. Saigo, J. Am. Chem. Soc., 2003, 125, 8307–8317 CrossRef CAS PubMed.
- E. Nurminen and H. Loennberg, J. Phys. Org. Chem., 2004, 17, 1–17 CrossRef CAS.
- G. M. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina and E. Guidez, J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed.
- N. Iwamoto, D. C. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Sah, S. M. Standley, G. Lu, L. H. Apponi and M. Frank-Kamenetsky, Nat. Biotechnol., 2017, 35, 845–851 CrossRef CAS PubMed.
- T. Wada, Y. Sato, F. Honda, S.-i. Kawahara and M. Sekine, J. Am. Chem. Soc., 1997, 119, 12710–12721 CrossRef CAS.
- E. De Vroom, C. Dreef, H. Van den Elst, G. Van der Marel and J. Van Boom, Recl. Trav. Chim. Pays-Bas, 1988, 107, 592–595 CrossRef CAS.
- T. Kofman, Russ. J. Org. Chem., 2002, 38, 1231–1243 CrossRef CAS.
- R. M. Claramunt, D. Sanz, G. Boyer, J. Catalän, J. L. G. de Paz and J. Elguero, Magn. Reson. Chem., 1993, 31, 791–800 CrossRef CAS.
- M. Sekine, A. Ohkubo and K. Seio, J. Org. Chem., 2003, 68, 5478–5492 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Kazuki Sato
,
Kazuki Sato