DOI:
10.1039/D3RA06109D
(Paper)
RSC Adv., 2023,
13, 35537-35550
Fabrication and characterization of g-C3N4 supported Cu-single atom catalysts for the photocatalytic degradation of dyes
Received
7th September 2023
, Accepted 28th November 2023
First published on 7th December 2023
Abstract
Methylene blue and Congo red are widely used organic dyes in biomedical laboratories and textile industries. The abundant use of these dyes has led to their emission in wastewaters, which causes major health hazards to exposed populations. Therefore, it is necessary to properly treat the dye effluents before being discharged into the water bodies. The present study presents Cu–g-C3N4 as an efficient and cost-effective catalyst for the photocatalytic degradation of these dyes. The single-atom catalyst was prepared by a simple co-precipitation method and the composition, structure, morphologies, and electronic state were determined by FT-IR SEM, XRD, XPS, PL, and TGA analyses. The photocatalytic activity of the catalyst was studied by optimizing various parameters i.e. concentration of dye, time, catalyst dose, and pH under UV irradiation and dark conditions. The results evidenced that Cu–g-C3N4 is an excellent catalyst as it achieved 100% degradation of the methylene blue and Congo red dyes in only 5 and 30 minutes respectively. The kinetics of photocatalytic degradation revealed that the half-life of methylene blue and Congo red was reduced significantly. The stability of the catalyst was determined by using it for five consecutive cycles and the results proved that Cu–g-C3N4 is a highly stable catalyst. Thus, Cu–g-C3N4 proved itself to be a highly active, stable, and cost-effective catalyst for the degradation of dyes with minimum resources. This material is also believed to have great potential to degrade other environmental pollutants too.
Introduction
Water contamination is becoming a crucial environmental and health issue with time. Industrial development and a massive increase in population are playing a significant role in aggravating the water contamination. Various techniques are being used to remove contaminants from water including membrane filtration, electro-dialysis, adsorption, precipitation, electro-deionization, electrochemical reduction, etc. But there are several disadvantages associated with these techniques i.e. high energy consumption, lower efficiency, and production of various by-products. Thus, there is a need to explore new methods for mitigating the problem of water contamination that are energy efficient, low cost and easy to execute. In this context, the photocatalytic degradation has emerged as an alternative method with many advantages over other conventional techniques including excellent performance, low cost, no waste formation, and also that it is functional at ambient temperature and pressure.1–3
Semiconductor photocatalysts have been considered as the most efficient and environment-friendly catalysts for the photocatalytic degradation of contaminants.4,5 Their activity can be further increased by converting them to single-atom catalysts. In the field of photocatalysis, single-atom catalysts have received a lot of attention owing to their fascinating properties in comparison to their bulk counterparts or clusters. They offer exceptionally high selectivity and activity, reduced use of metals due to atomic dispersion, and easy-to-follow mechanism due to single-atom reactive sites.6 Single-atom catalysts also offer high surface energy owing to their small size, but they sometimes agglomerate during the reaction process. This issue of aggregation can be resolved by selecting appropriate support and developing strong bonding between the support material and single atoms. This bonding and interaction affect the efficiency and stability of the catalysts.7 The popular approaches for increasing the stabilization of single-atom catalysts also include the reduction in mobility of single atoms by their confinement into a limited space.
Considering the above mentioned factors, graphitic carbon nitride has presented itself to be an ideal candidate for the support of single-atom catalysts. It is a polymeric semiconductor consisting of two-dimensional (2D) layered structure. It can be synthesized easily from some precursors i.e. urea, melamine, thiourea etc.8 The combined effect of the high catalytic activity of single atoms and the photo-response characteristics of graphitic carbon nitride makes them ideal candidates for photocatalytic reactions.9,10 The rich surface loading sites are achieved owing to the 2D layered structure of graphitic carbon nitride in which the layers are connected by weak van der Waals forces. The carbon and nitrogen atoms in the plane are sp2 hybridized forming a highly delocalized π-conjugated system. An opportunity for coordinating metal atoms is provided by the lone-pair of electrons in two-coordinated nitrogen atoms. The rich lone-pair electrons of nitrogen atoms in the carbon–nitrogen heterocycle confine the highly active single metal atoms that lead to the formation of stabilized graphitic carbon nitride-based single-atom photocatalysts.11,12
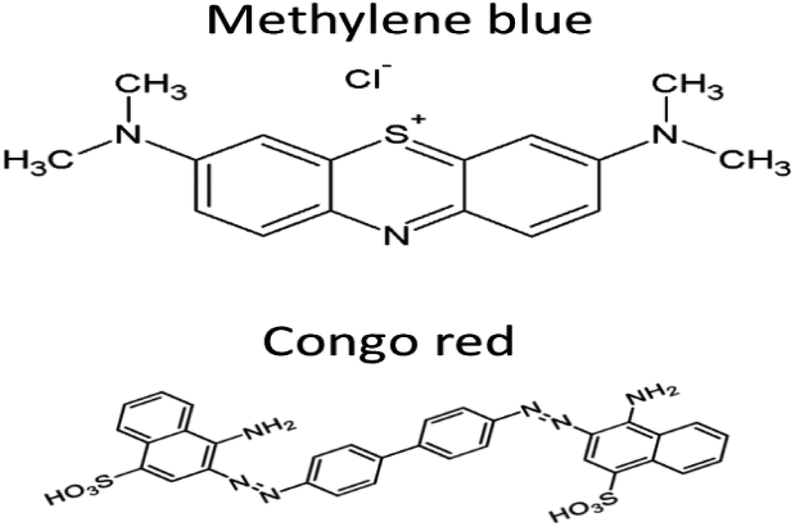
The present work reports the synthesis of graphitic carbon nitride-supported copper single-atom (Cu–g-C3N4) catalyst. The commonly used dyes (methylene blue and Congo red) contaminating the water systems were used as model compounds to study the potential of synthesized catalyst for their degradation. These dyes were selected because of their different properties. Methylene blue is cationic dye, basic in nature, and belongs to the phenothiazines family and Congo red is anionic, acidic dye and belongs to the group of azo dyes.13,14 Their structures are shown in Fig. 1. The synthesized catalyst exhibited excellent catalytic activity both under UV and dark conditions.
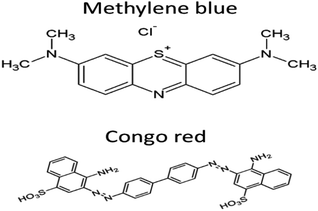 |
| | Fig. 1 Structure of dyes. | |
Experimental methodology
The chemicals used for present research work were of analytical grade with a purity of 99.8%. Deionized distilled water was used throughout the study for making various solutions. The glassware used was of acid resistant borosilicate glass that was unaffected by leaching effect of strong acidic or alkaline solutions. All the volumetric apparatus used was calibrated prior to use. The graphitic carbon nitride supported copper single-atom catalyst was synthesized and used for the photocatalytic degradation of both dyes by optimizing various parameters. The Focus degradation kinetic software was used to determine the kinetics of photocatalytic degradation and half life for photodegradation.15
Preparation of graphitic carbon nitride supported single atom catalyst
The Cu–g-C3N4 catalyst was synthesized by adopting the co-precipitation method. Firstly, the support was prepared by heating the urea in a covered crucible at 550 °C in a muffle furnace set at the heating rate of 10 °C min−1. Then it was washed with nitric acid and dried at 80 °C. To prepare the single-atom catalyst, 0.1341 g CuCl2·2H2O was added to 5 g g-C3N4 solution. Under continuous stirring, the solution was heated up to 80 °C and maintained at this temperature till further processing. The pH of the solution was set at 8 by dropwise addition of sodium hydroxide solution. The heating was then stopped and the solution was left to age for 4 hours. Afterward, the precipitates were carefully filtered, thoroughly washed with hot distilled water and finally dried at 80 °C.16 The synthesized catalyst was characterized by FTIR, XRD, EDX, SEM, TGA, XPS, and PL analyses before being used for the photodegradation of dyes.
Photocatalytic degradation of methylene blue and Congo red dyes
Methylene blue and Congo red dyes are widely used in the textile industry. Their unwise use is increasing day by day that has led to serious environmental concerns. They enter water bodies through runoff or leaching which poses serious health risks to aquatic and other living organisms. Therefore, these dyes should be properly treated before being disposed of. The photocatalytic degradation of the dyes is one of the most convenient and efficient methods to treat the dye's waste. The use of photocatalysts such as graphitic carbon nitride supported copper single-atom can accelerate the degradation process, converting the dyes to simpler and non-toxic compounds. However, the optimization of catalyst dose, irradiation time, and pH is necessary to achieve controlled and desirable results.
To study the photocatalytic degradation of dyes, their solutions of different concentrations (15, 25, and 35 mg L−1) were prepared. These solutions were stirred in the presence of Cu–g-C3N4 catalyst under UV light for specific intervals of time. A UVC light of 8 W was used during the reaction with maximum emission at 230 nm. These samples were then analyzed by a UV-visible spectrophotometer (LABOMED, INC, Model No. “UVD-3200”) at 665 and 497 nm respectively to determine the leftover concentration of dyes after degradation. The same procedure was followed for the reactions performed in the dark.
The degradation efficiency was calculated by using the following equation:
| (%) degradation = (Co − Ct)/Co × 100 |
where
Co represents the initial concentration of dye and
Ct represents the remaining concentration of dye after degradation, at a specific interval of time.
The effect of different parameters on the degradation of dyes was also studied. The parameters included: the effect of the irradiation time, the concentration of dyes, the dose of the catalyst, and pH. To study the effect of UV irradiation time, the dye solutions were stirred for specific intervals of time i.e., from 5 to 45 minutes under UV light. The similar control experiments were carried out under dark in the presence of the catalyst. The solutions of different concentrations i.e., 15 mg L−1, 25 mg L−1, and 35 mg L−1 were prepared to study the effect of the initial concentration of dyes. A 0.025 g portion of the catalyst per 100 mL volume of dye solution was added to each of these solutions. These solutions were then continuously stirred for a specific interval of time in the presence of UV light. The samples were then analyzed for the remaining dye concentration. Another set of experiments was performed in the dark with similar conditions. To study the effect of catalyst dose, different specific amounts of Cu–g-C3N4 catalyst i.e., 0.025 g, 0.05 g, and 0.075 g were added to 100 mL volumes of the dye solutions under UV light and in the dark for a specific interval of time. The pH of the dye solutions was maintained at 3, 5, 7, 9, and 11 by the addition of NaOH or HCl solutions, in the presence of 0.075 g of catalyst with continuous stirring to study the effect of pH on photocatalytic degradation. The process was repeated in the dark as well.
Results and discussion
Characterization of graphitic carbon nitride supported copper single-atom catalyst
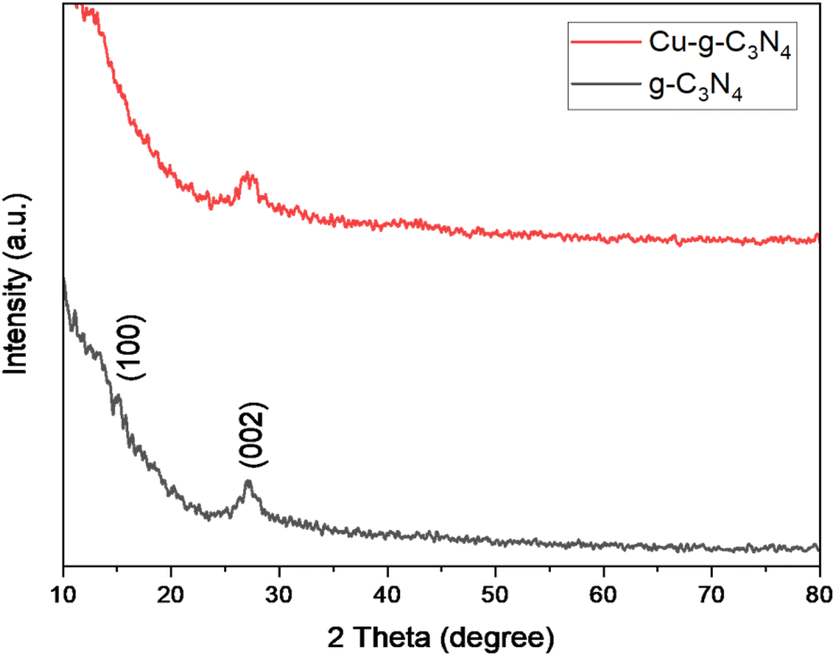
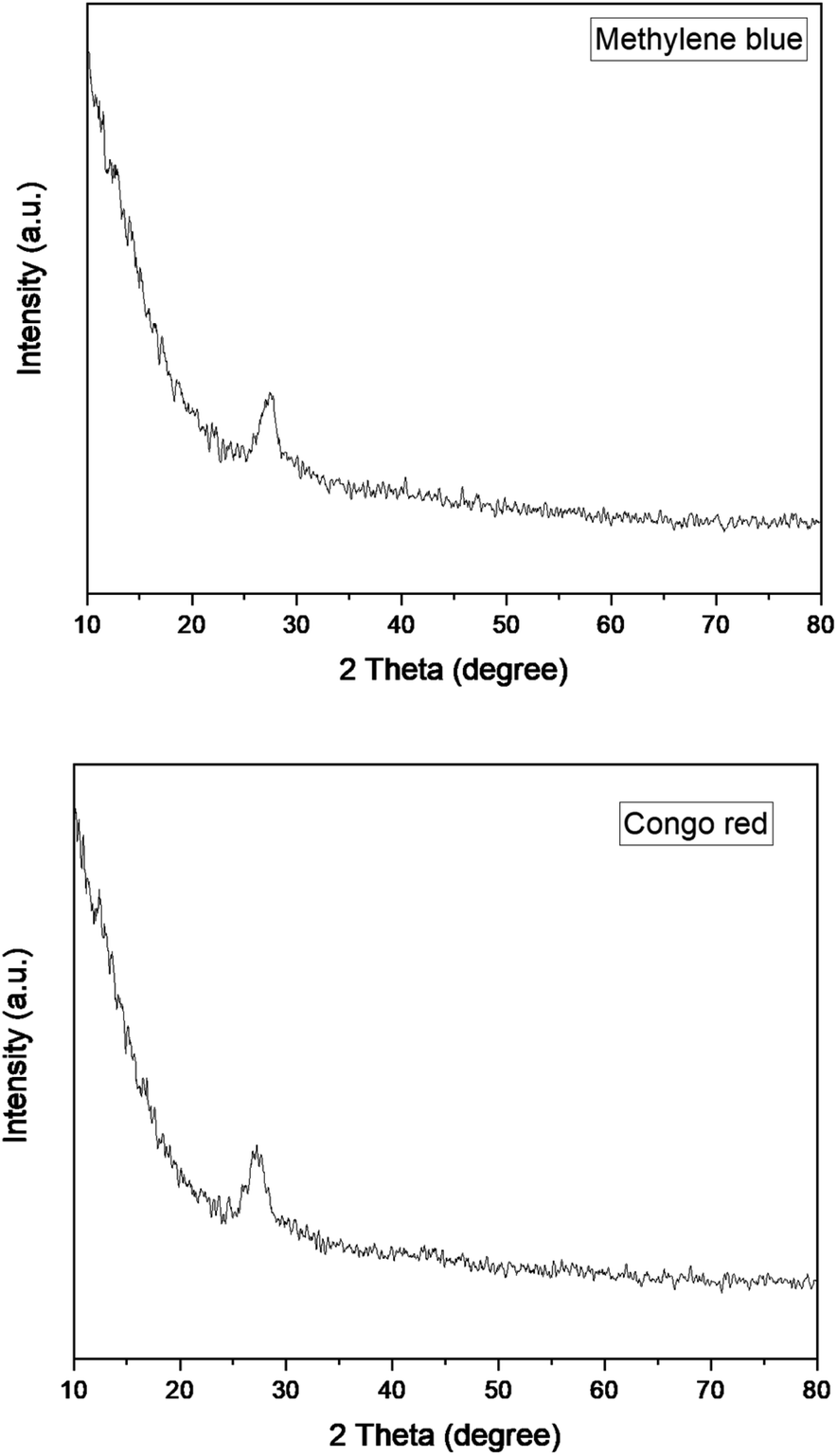
The XRD patterns of graphitic carbon nitride (g-C3N4) and graphitic carbon nitride supported single-atom catalyst (Cu–g-C3N4) were recorded in the range of 2θ = 10–80° as shown in Fig. 2. The XRD pattern clearly showed two diffraction peaks at 2θ values of 13.1° and 27.3° indexed to (100) and (002) planes respectively. The strong diffraction peak (002) is due to the interlayer stacking of the conjugated aromatic system of graphitic carbon nitride. While the other comparatively weaker peak (100) is associated with the in-plane repeat units. The observed peaks matched well with the JCPDS card no. 01-087-1526.8,17 No characteristic peak for copper was observed as the metal loading was quite low.
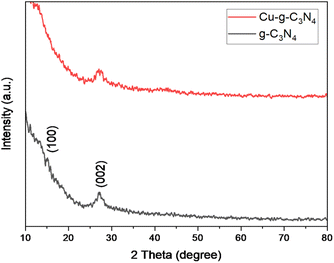 |
| | Fig. 2 XRD patterns of g-C3N4 and Cu–g-C3N4. | |
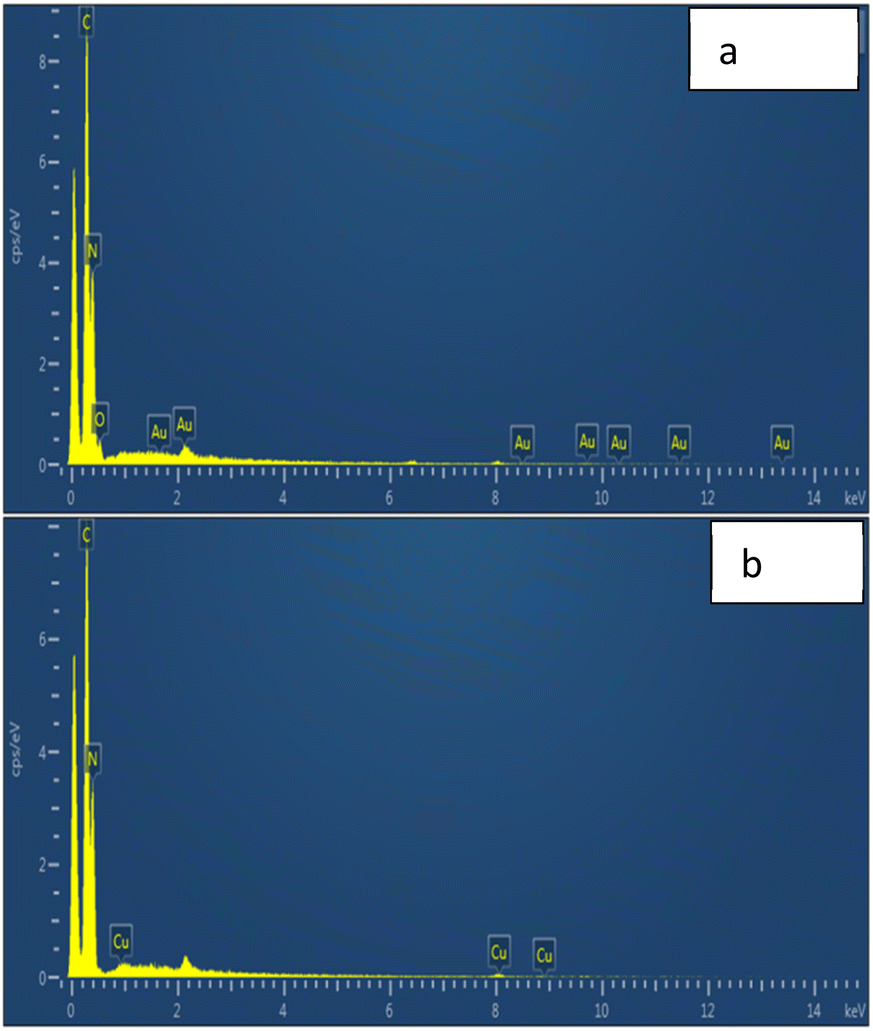
The scanning electron microscopy images of graphitic carbon nitride supported copper single-atom catalyst are shown in Fig. 3. The figure clearly showed the nano-sized particles of graphitic carbon nitride. The single atoms or nanoclusters of copper cannot be seen at this magnification. However, the presence of copper single atoms was evidenced by energy dispersive X-ray (EDX) analysis (Fig. 4). The EDX spectra for Cu–g-C3N4 clearly showed that graphitic carbon nitride supported copper single atom catalyst was successfully prepared and there was no impurity present in it. The presence of copper is confirmed by the peaks in EDX spectra followed by the presence of carbon and nitrogen.
 |
| | Fig. 3 SEM images of Cu–g-C3N4. | |
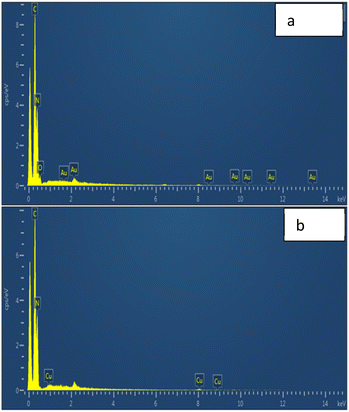 |
| | Fig. 4 EDX spectra of (a) g-C3N4 and (b) Cu–g-C3N4. | |
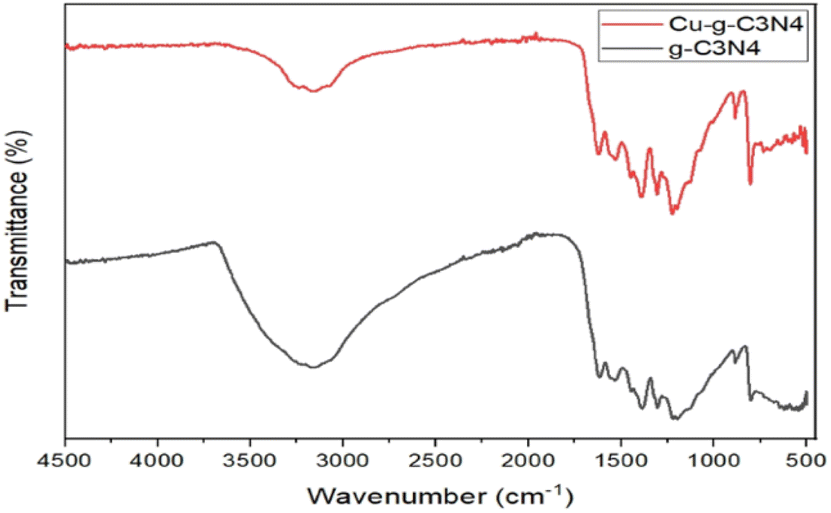
The FTIR spectrum of g-C3N4 and Cu–g-C3N4 is presented in Fig. 5. Several bands present in the range of 1000 cm−1 to 1700 cm−1 were ascribed to the stretching vibrations of C–N heterocycles. The vibrations observed at 1231, 1313, and 1397 cm−1 were characteristic of aromatic C–N stretching. The band observed at 806 cm−1 was ascribed to the tris-s-triazine rings of graphitic carbon nitride, while the band observed at 3185 cm−1 was ascribed to the stretching of the N–H group of residual uncondensed amino groups.18
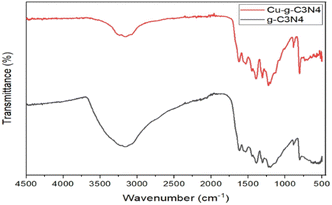 |
| | Fig. 5 FTIR spectra of g-C3N4 and Cu–g-C3N4. | |
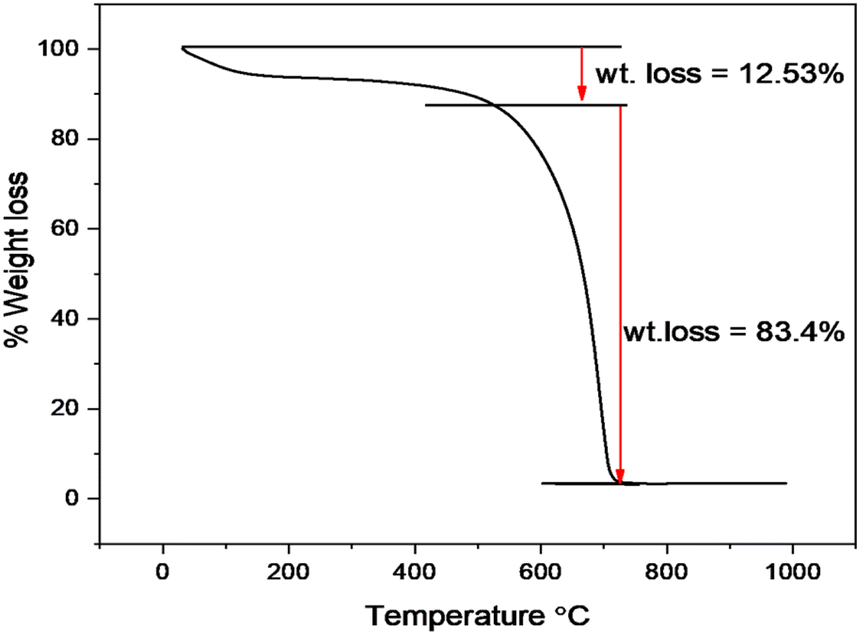
The thermal stability of Cu–g-C3N4 was investigated by thermo-gravimetric analysis (TGA) as depicted in Fig. 6. The TGA was performed under a nitrogen atmosphere at the heating rate of 10 °C min−1. Up to 550 °C, a small weight loss occurred that involved the removal of moisture adsorbed on the surface. After 550 °C, the catalyst starts to decompose up to 700 °C. This sharp weight loss is attributed to the oxidation of the g-C3N4 to form nitrogen and graphite.19 This curve indicated the typical behavior of polymeric graphitic carbon nitride.
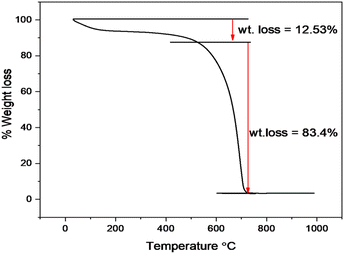 |
| | Fig. 6 TGA curve of Cu–g-C3N4. | |
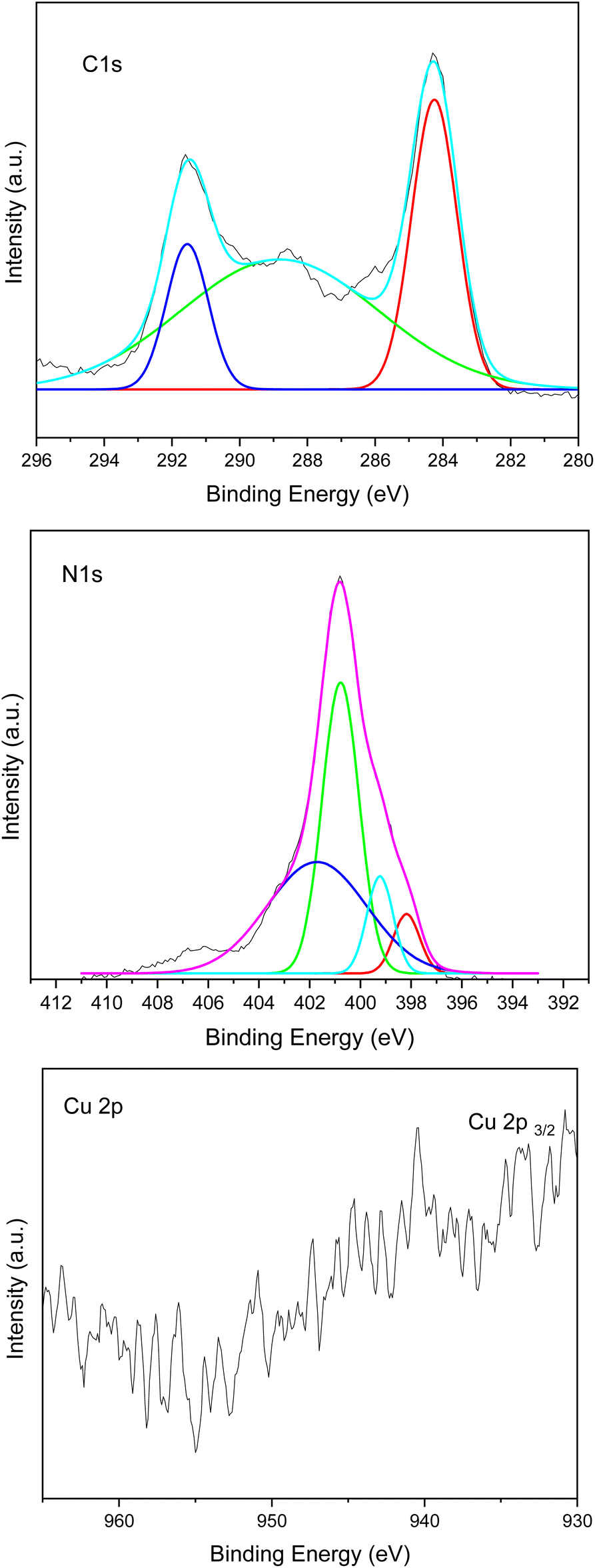
The chemical composition and chemical state of the Cu–g-C3N4 catalyst were determined by X-ray photoelectron spectroscopy (XPS). The spectrum for C 1s, N 1s, and Cu 2p is shown in Fig. 7. The C 1s region showed peaks around 284.6, 288.7, and 291.6 eV. The peak at 288.6 eV is attributed to adventitious carbon. The adventitious carbon peak is visible because of a very thin film of the sample deposited in the sample holder to avoid charging effects. The peak at 288.7 eV is characteristic of graphitic carbon nitride and is attributed to C–(N)3. The peak at 291.6 eV is associated with π–π* satellite structure. For the N 1s region, the broad spectrum was deconvoluted into four peaks at 398.1, 399.2, 400.7, and 401.8 as shown in Fig. 6. These peaks were assigned to C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N–H2, (C)2–N–H, and N–(C)3 species respectively. It is evident from the fit of the Cu 2p3/2 region that copper is present in different oxidation states. The first component centered at 933.3 eV is assigned to the presence of both Cu+ (933.7 eV) and metallic Cu (933.2 eV). While the second component at 934.5 eV in the Cu 2p3/2 peak, and the intense satellite peak at 940.4 eV, are associated with the presence of Cu2+, which confirmed the presence of copper in two different oxidation states.8,20,21 A distinct shift in binding energy compared with references for pure metal may elucidate the oxidation state of isolated metal atoms and exclude the existence of nanoparticles.22 The characteristic peak of single copper atom was shifted to 933.7 eV as compared to pure metal peaks generally observed at 932.3 eV.23 So it may provide evidence for the presence of single atom.
C, C–N–H2, (C)2–N–H, and N–(C)3 species respectively. It is evident from the fit of the Cu 2p3/2 region that copper is present in different oxidation states. The first component centered at 933.3 eV is assigned to the presence of both Cu+ (933.7 eV) and metallic Cu (933.2 eV). While the second component at 934.5 eV in the Cu 2p3/2 peak, and the intense satellite peak at 940.4 eV, are associated with the presence of Cu2+, which confirmed the presence of copper in two different oxidation states.8,20,21 A distinct shift in binding energy compared with references for pure metal may elucidate the oxidation state of isolated metal atoms and exclude the existence of nanoparticles.22 The characteristic peak of single copper atom was shifted to 933.7 eV as compared to pure metal peaks generally observed at 932.3 eV.23 So it may provide evidence for the presence of single atom.
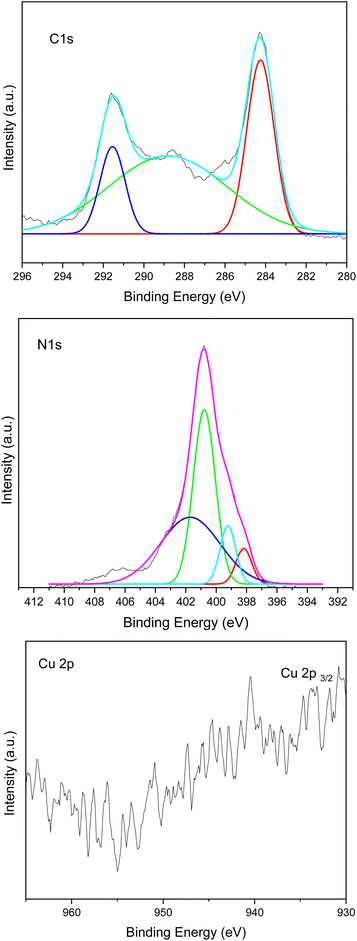 |
| | Fig. 7 XPS spectra of Cu–g-C3N4. | |
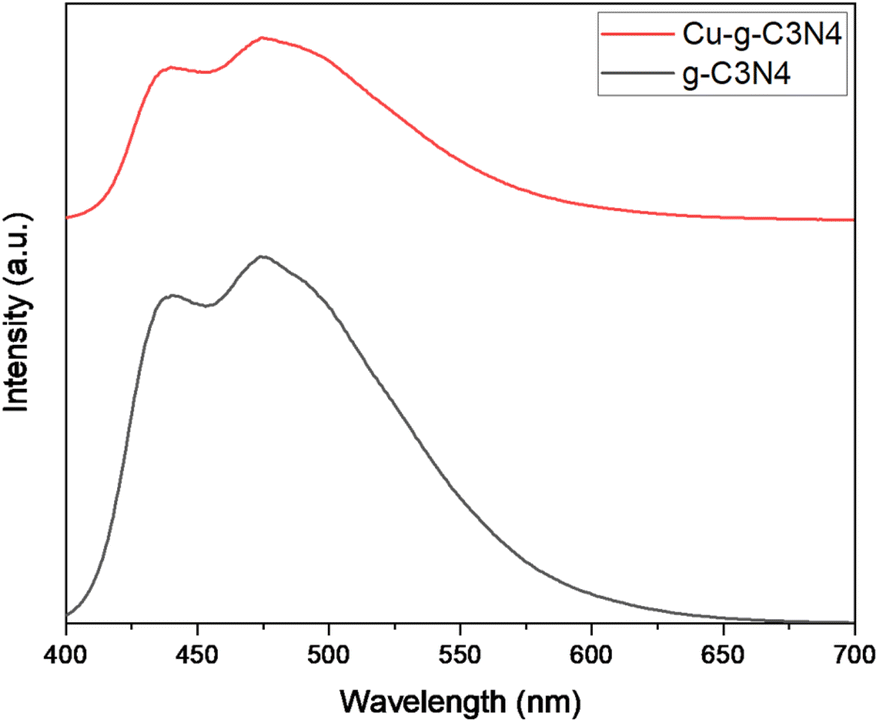
The Fig. 8 shows the photo-luminescence spectra of g-C3N4 and Cu–g-C3N4. For g-C3N4 and Cu–g-C3N4, the emission bands were centered at 437 and 474 nm that were attributed to blue emission produced from electronic transitions. The intense emission peaks are assigned to the higher recombination rate of the photogenerated electrons and holes. In case of Cu–g-C3N4, the decrease in the intensity of emission peaks is observed. The decrease in emission peak intensity is due to inhibited recombination of photogenerated electron–hole pairs in Cu–g-C3N4 which indicates a successful charge separation. However, PL study confirmed the increased lifetime of the photogenerated carrier in Cu–g-C3N4 as compared to pristine g-C3N4.24,25
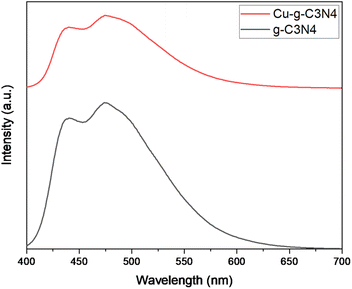 |
| | Fig. 8 Photo luminescence spectra of g-C3N4 and Cu–g-C3N4. | |
Photocatalytic degradation of methylene blue by graphitic carbon nitride supported single atom catalyst
Various parameters i.e. irradiation time, catalyst dose, concentration of dye, and pH were optimized in the presence of Cu–g-C3N4 catalyst to study the photocatalytic degradation of methylene blue. The UV-visible spectra of methylene blue (Fig. 9) depicted its λmax to be 665 nm.26 This value of λmax was used to determine the concentration of methylene blue throughout the experiment.
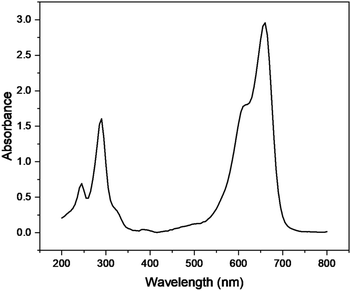 |
| | Fig. 9 UV-vis absorption spectra of methylene blue. | |
Effect of time on degradation of methylene blue
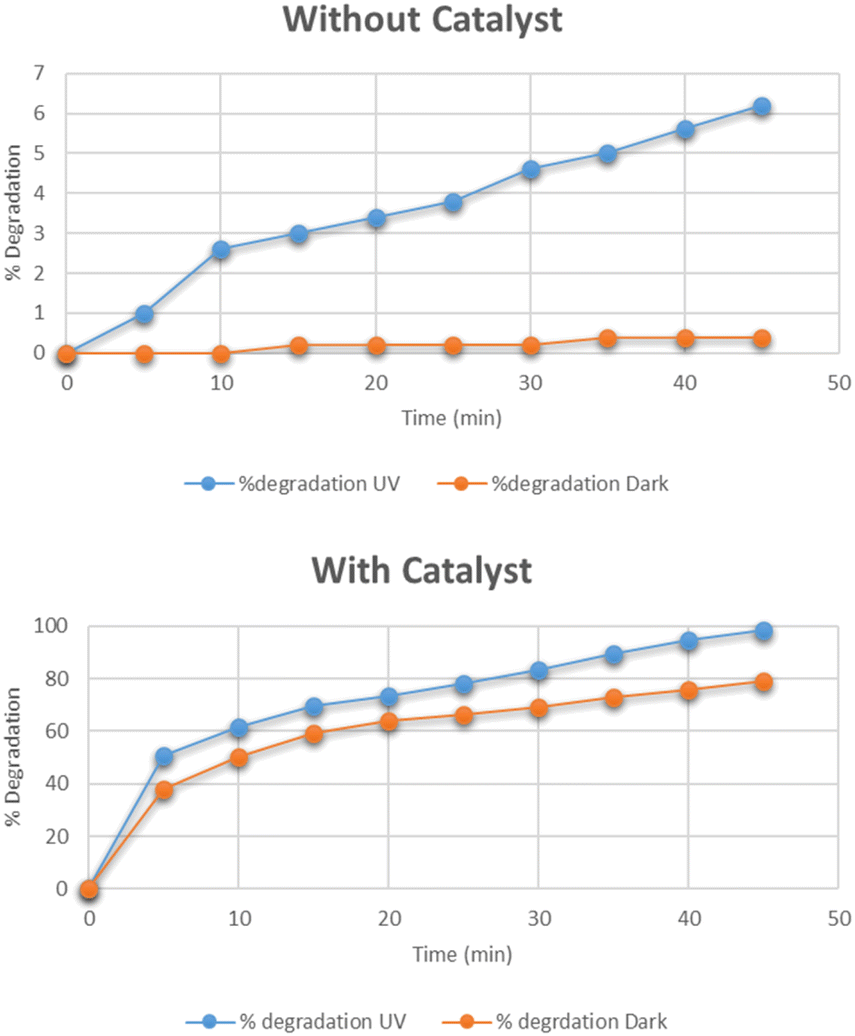
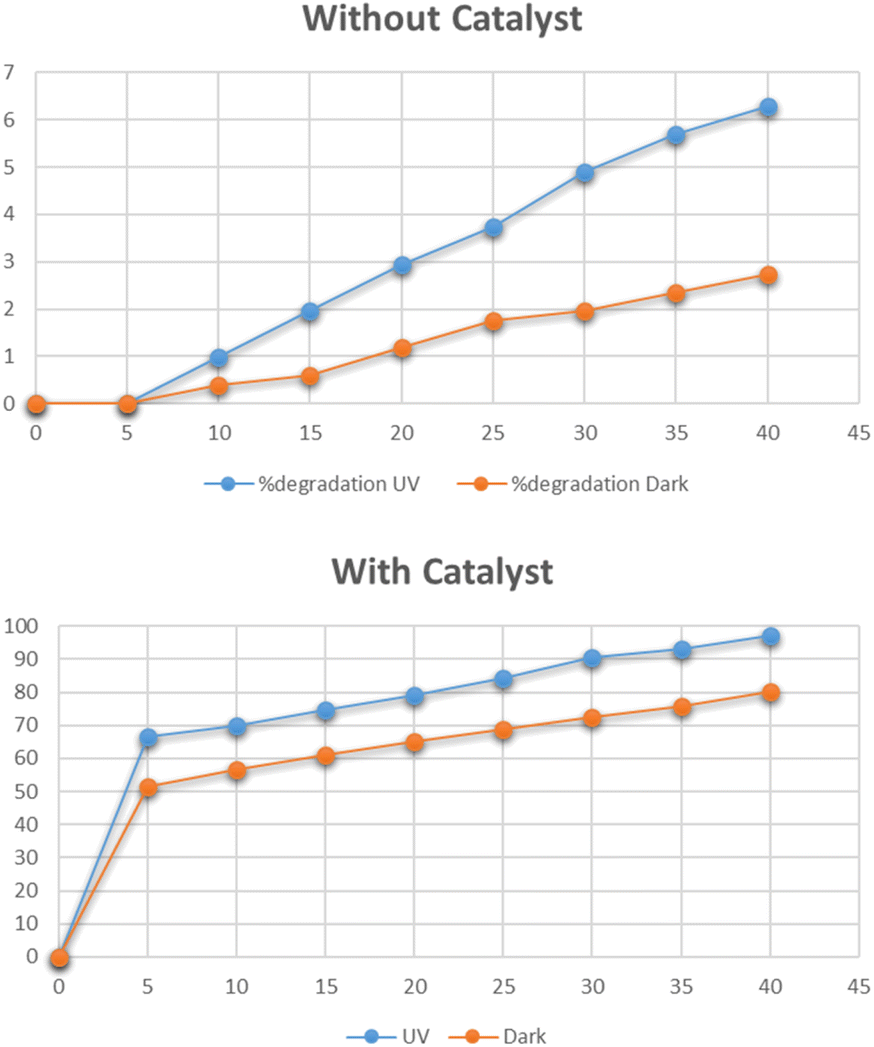
The optimum time for the photocatalytic degradation of methylene blue was determined by the addition of 0.025 g of Cu–g-C3N4 catalyst to 100 mL solutions of 15 mg per L dye with continuous stirring. The data for the photocatalytic degradation of methylene blue in the absence of the catalyst under UV light and dark is depicted in Fig. 10. The data for the degradation of methylene blue in the presence of the catalyst is also depicted in this figure. In the first case, the data clearly showed that the degradation of methylene blue reached only 0.4% in the dark, and under UV light it reached up to 6.2% in 45 minutes. The UV light slightly improved the degradation process. But the Cu–g-C3N4 catalyst significantly enhanced the degradation and reached up to 98% under UV which was only 79% under the dark condition in just 45 minutes. It was worth noting that in the presence of the catalyst, the degradation rapidly increased after adding the catalyst for up to 5 minutes leading to maximum degradation in 45 minutes. The degradation of dye is dependent on contact time. As the contact time is increased, more electron–hole pairs are generated which lead to higher degradation of dye.27 The degradation rate was higher in the first five minutes due to the combined effect of adsorption of dye molecules on the catalyst surface and subsequent photocatalytic degradation. It can be considered that firstly the dye molecules are adsorbed onto the catalyst which are then photodegraded so, adsorption is an important factor for dye removal under dark conditions. After that UV light was majorly responsible for the degradation of adsorbed dye molecules creating vacancies for more molecules to be mineralized.28
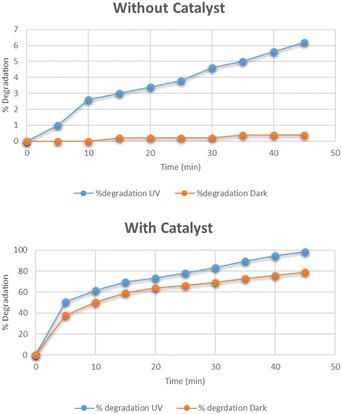 |
| | Fig. 10 Effect of time on degradation of methylene blue. | |
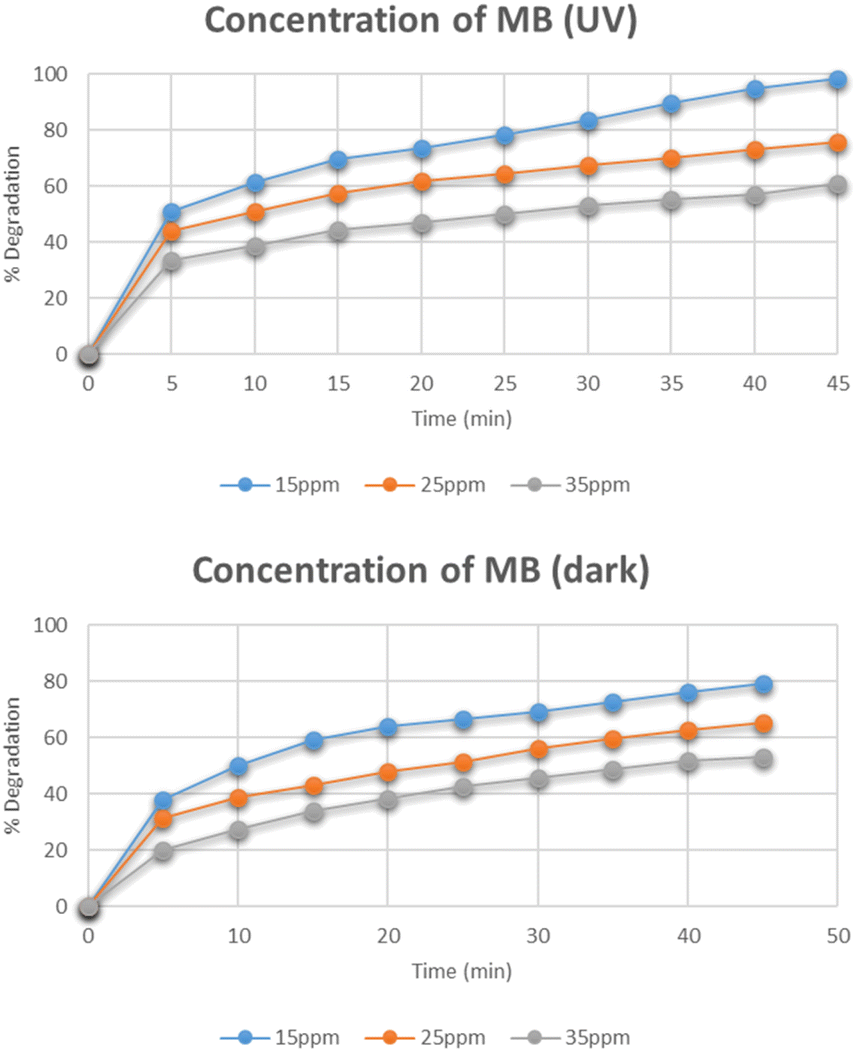
Effect of concentration of methylene blue
Fig. 11 represents the degradation of different concentrations of methylene blue i.e. 15 mg L−1, 25 mg L−1, and 35 mg L−1. The maximum degradation was recorded for the 15 mg L−1 solution which degraded to 98% under UV light while in dark, its degradation was only 79%. As the concentration of methylene blue was increased, the degradation was found to be decreased. The reason for the decrease in degradation was the increased adsorption of methylene blue molecules on the active catalytic sites and decreased adsorption of OH− which led to reduced production of OH˙ radicals. The degradation was also decreased with an increase in concentration that caused reduced number of photons reaching the dye molecules and hence leading to decreased concentration of light-triggered active sites.29,30
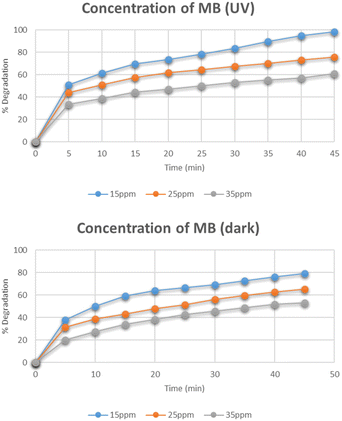 |
| | Fig. 11 Effect of concentration of methylene blue on its degradation. | |
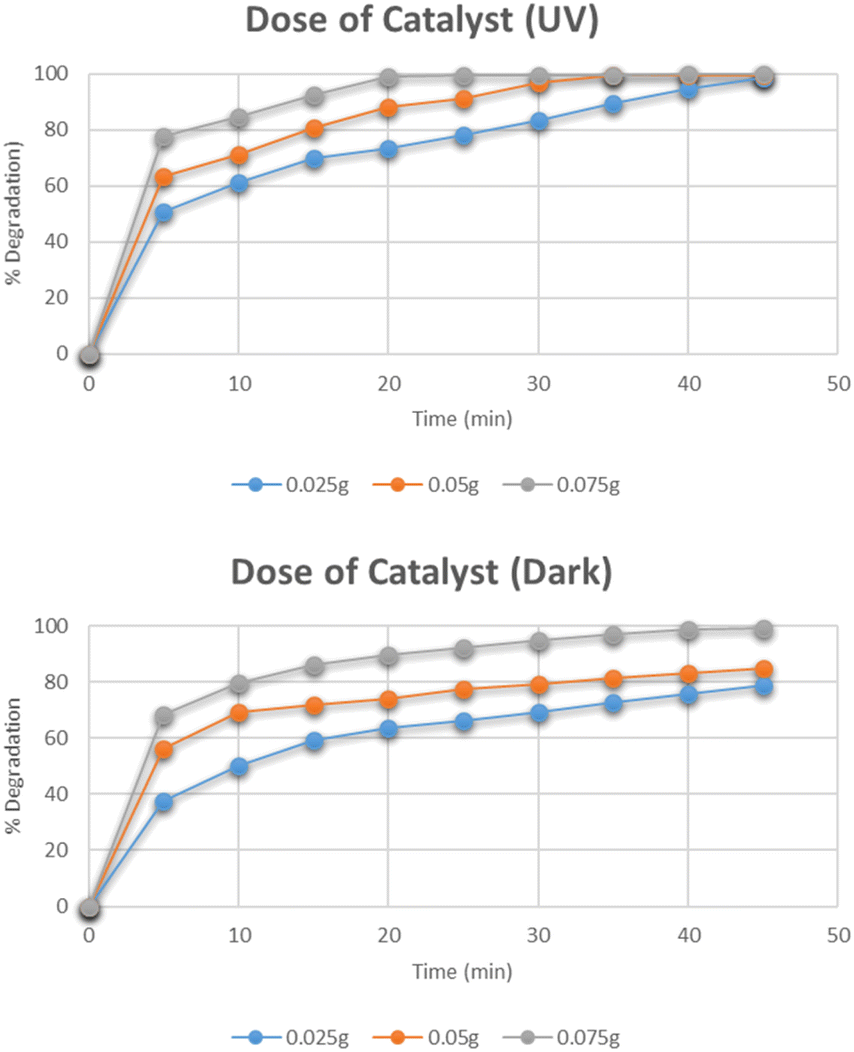
Effect of dose of catalyst
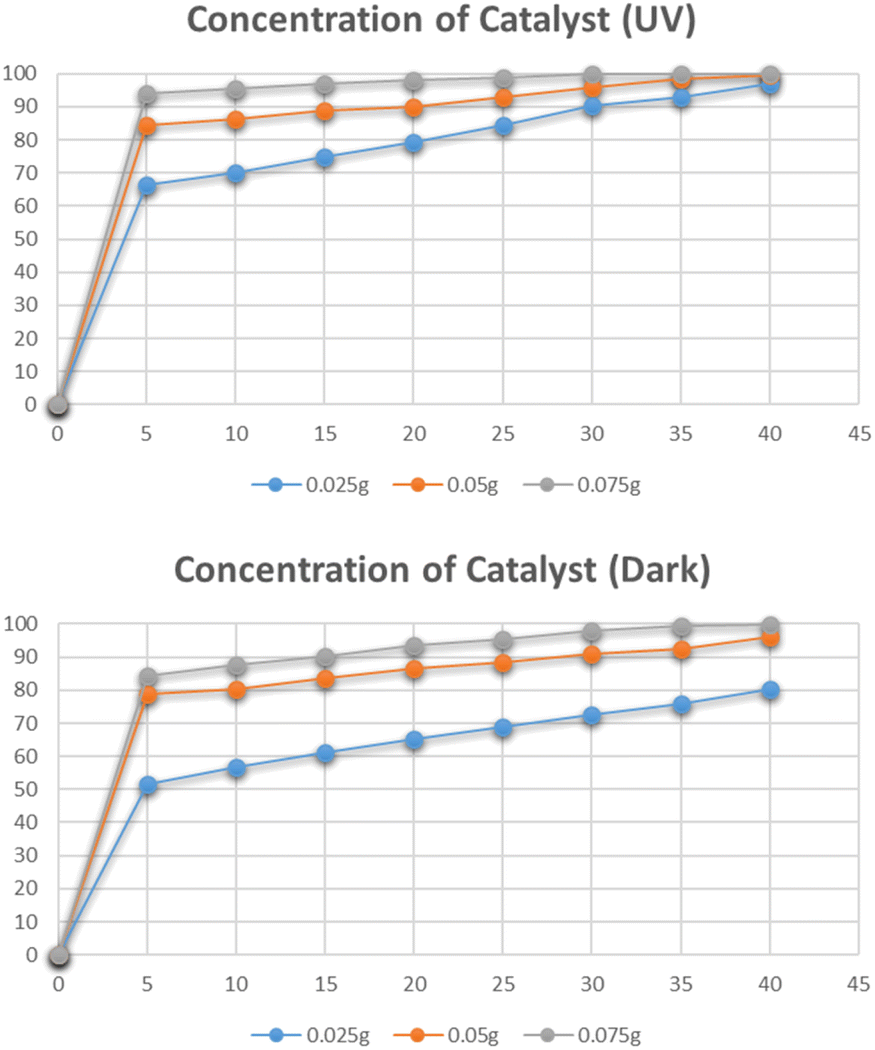
The effect of the dose of catalyst on the degradation of methylene blue was studied by varying the amounts of catalyst i.e. 0.025 g, 0.05 g, and 0.075 g. The data for the effect of the catalyst dose on degradation is shown in Fig. 12. The highest percentage degradation was 99% observed in just 20 minutes for a 0.075 g catalyst in the presence of UV light. The same effect was observed for the solutions degraded under dark. The maximum degradation efficiency was recorded for the 0.075 g catalyst. The increase in degradation efficiency is related to the increased availability of active catalytic sites due to the uniform dispersion of catalyst particles.31
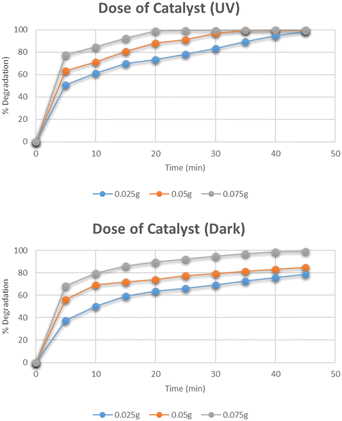 |
| | Fig. 12 Effect of dose of catalyst on degradation of methylene blue. | |
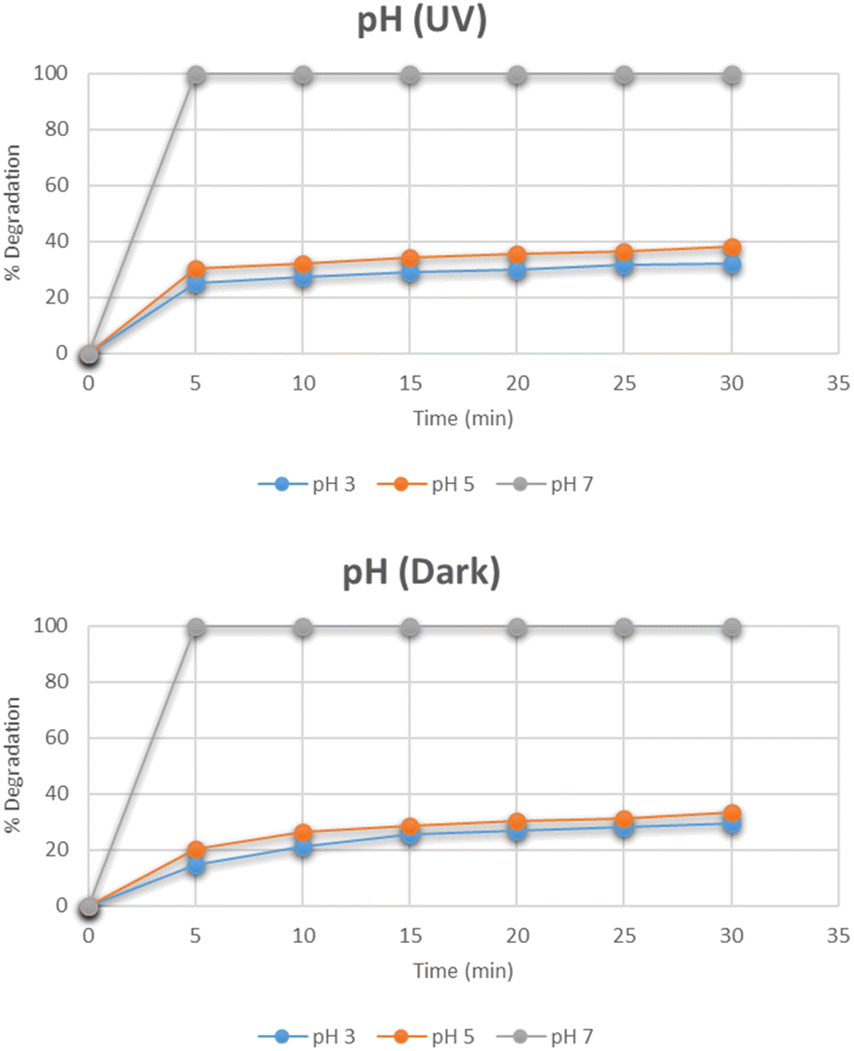
Effect of pH
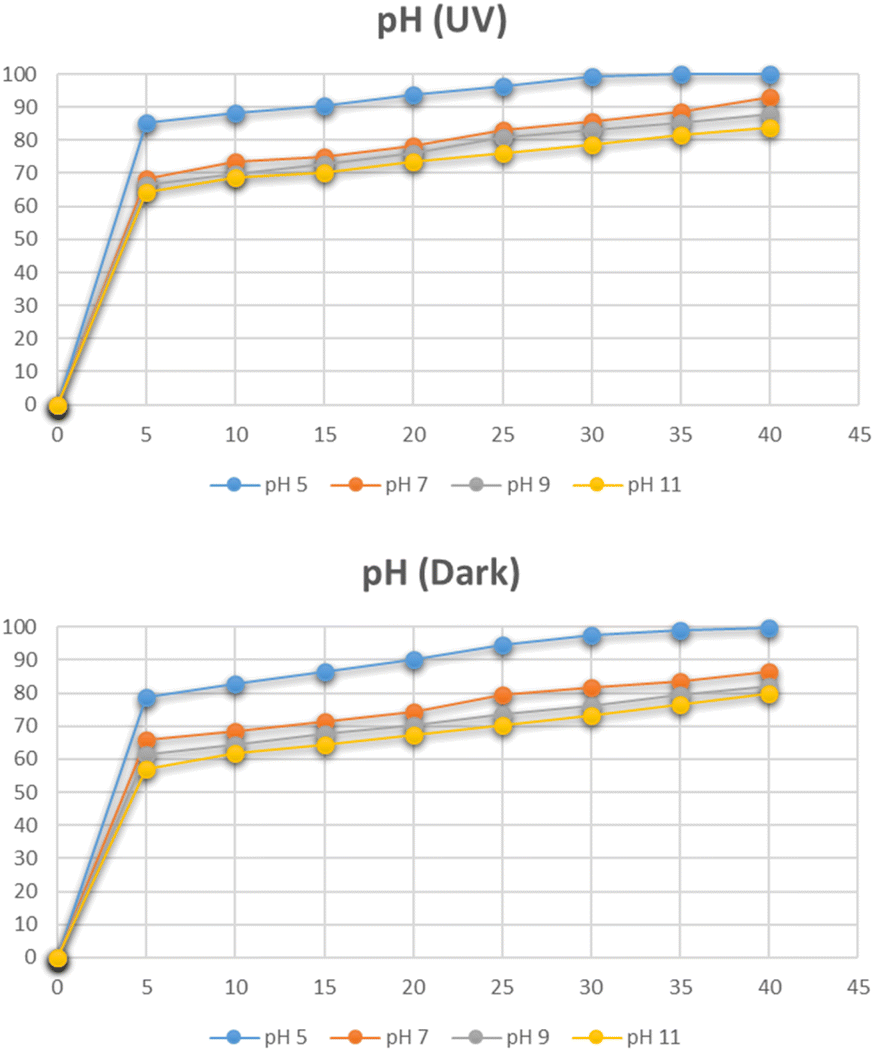
The effect of pH on the degradation pattern for methylene blue under the optimum conditions of dye concentration and the catalyst dose was studied by maintaining different pH i.e. 3, 5, 7, 9, and 11 as depicted in Fig. 13. At pH 7, the degradation reached 100% in only 5 minutes. By further increasing the pH to 9 and 11, the complete degradation was observed even before 5 minutes. However, the degradation was reduced when moving towards acidic side. The same pattern was observed under dark conditions as well. The increase in degradation with the increase in pH is related to the cationic nature of methylene blue. At a basic pH, the increased number of OH− ions are adsorbed rapidly on the surface of the catalyst. They further lead to the subsequent degradation of methylene blue in several intermediates and finally into simplest molecules.32
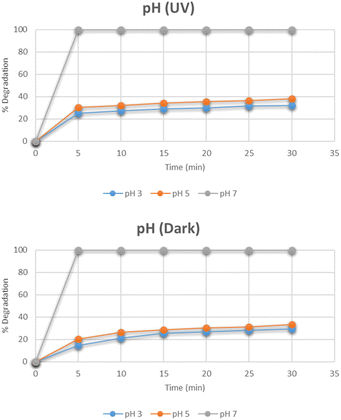 |
| | Fig. 13 Effect of pH on degradation of methylene blue. | |
Kinetics of photocatalytic degradation of methylene blue
The degradation kinetics of methylene blue was evaluated by using three different models, i.e., single first order rate model (SFO), indeterminate order rate equation model (IORE), and double first-order in parallel (DFOP) model. First-order kinetics is followed in most photocatalytic degradation reactions, but it is not compulsory for all cases. It can vary according to the conditions and nature of the dye. Thus, three different models were applied to study the kinetics of photocatalytic degradation of methylene blue. Table 1 depicts the parameters for all the models studied. The degradation kinetics of methylene blue carried out in the absence of the catalyst in the dark is depicted in Fig. 14(a). The degradation data exhibited a Sc value less than SSFO. Thus, the single first-order model cannot be applied to represent degradation data according to the NAFTA guidelines. This can be described based on initial degradation of dye as the degradation process was very slow at the initial stages. Thus, the degradation data may follow either of the other two models. The TIORE for the IORE model and DT50 corresponding to the slower rate constant were compared to get the representative half-life. The model to be followed will be decided based on these values. The model with the smaller value will be followed. And for this data set TIORE was greater than 2DT50, so, the DFOP model was the most suitable model to determine the half-life value of methylene blue.
Table 1 Kinetic Parameters of different degradation models
| Model |
Parameters |
Without catalyst |
With catalyst |
| Dark |
UV |
Dark |
UV |
| SFO |
K |
0.0009 |
0.001 |
0.054 |
0.10 |
| Ssfo |
0.97 |
0.38 |
24.64 |
22.45 |
| DT50 |
770.2 |
620.4 |
12.83 |
6.932 |
| DT90 |
2558 |
2061 |
42.62 |
23.03 |
| IORE |
Kiore |
2.29 × 10−6 |
6.10 × 10−6 |
3.60 × 10−4 |
7.01 × 10−4 |
| Niore |
2.00 |
3.10 |
3.50 |
3.43 |
| Siore |
0.02 |
0.04 |
1.16 |
18.40 |
| DT50 |
29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 239 239 |
869.8 |
5.895 |
3.59 |
| DT90 |
262575 |
33051 |
400.9 |
218.8 |
| Tiore |
79043 |
9949 |
120.7 |
65.86 |
| DFOP |
K1 |
0.00 |
0.00 |
0.21 |
1.00 |
| K2 |
1.00 × 10−3 |
2.00 × 10−3 |
8.50 × 10−3 |
1.00 × 10−2 |
| Sdfop |
0.09 |
1.18 |
1.83 |
34.96 |
| 1DT50 |
62386 |
6932 |
3.32 |
0.69 |
| 2DT50 |
693.1 |
346.6 |
81.55 |
69.31 |
| g |
0.5 |
0.5 |
0.5 |
0.5 |
| Sc |
|
0.03 |
0.06 |
1.60 |
25.27 |
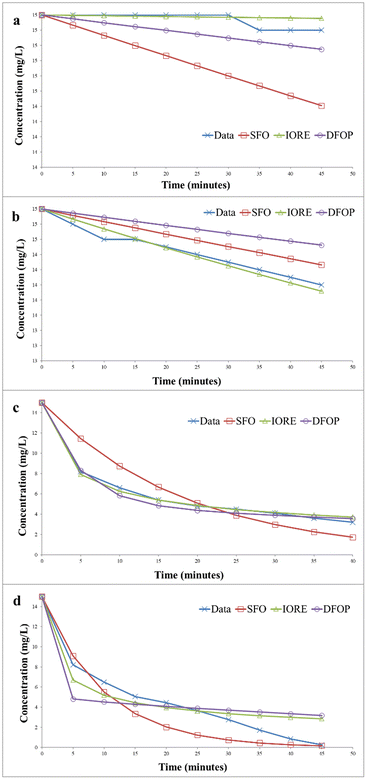 |
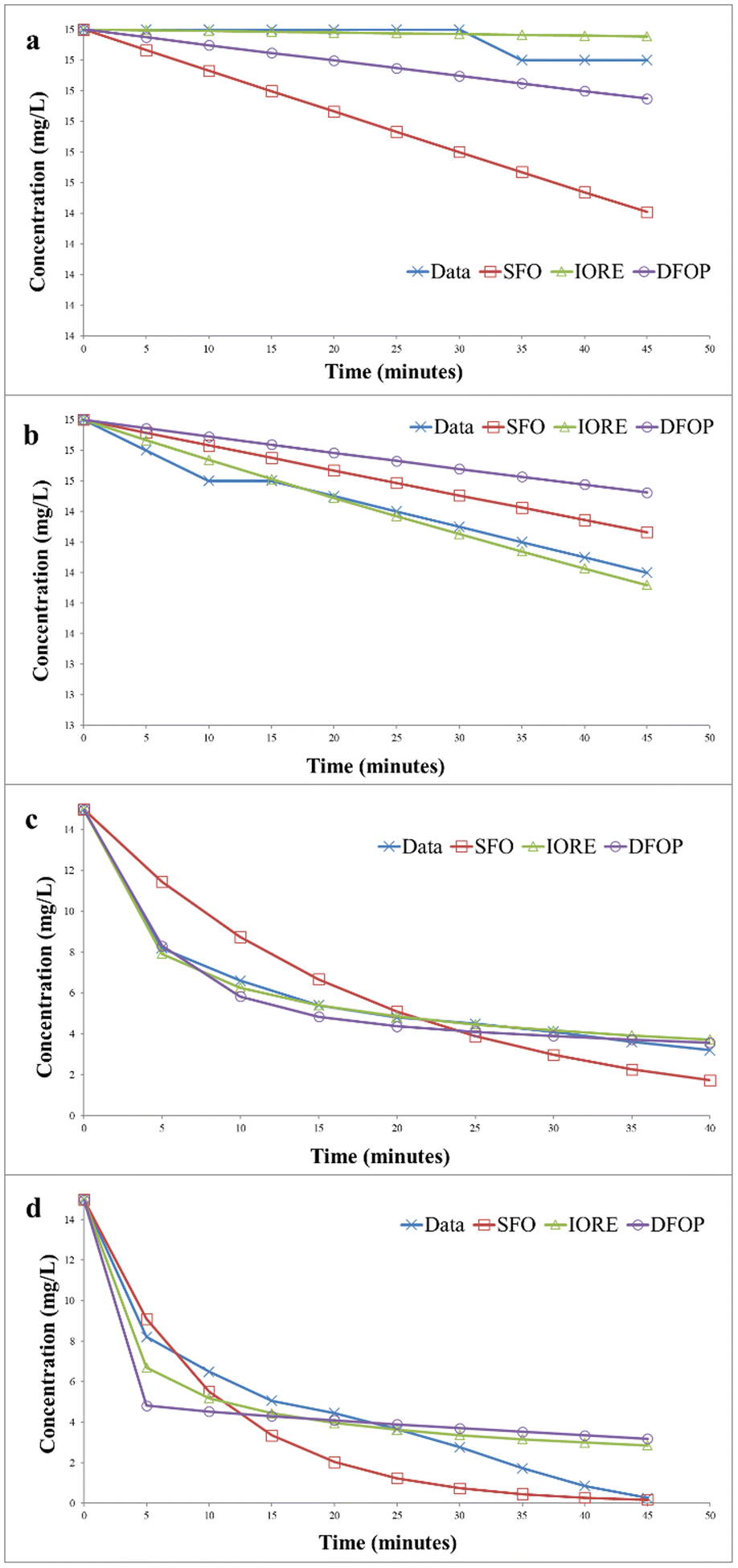
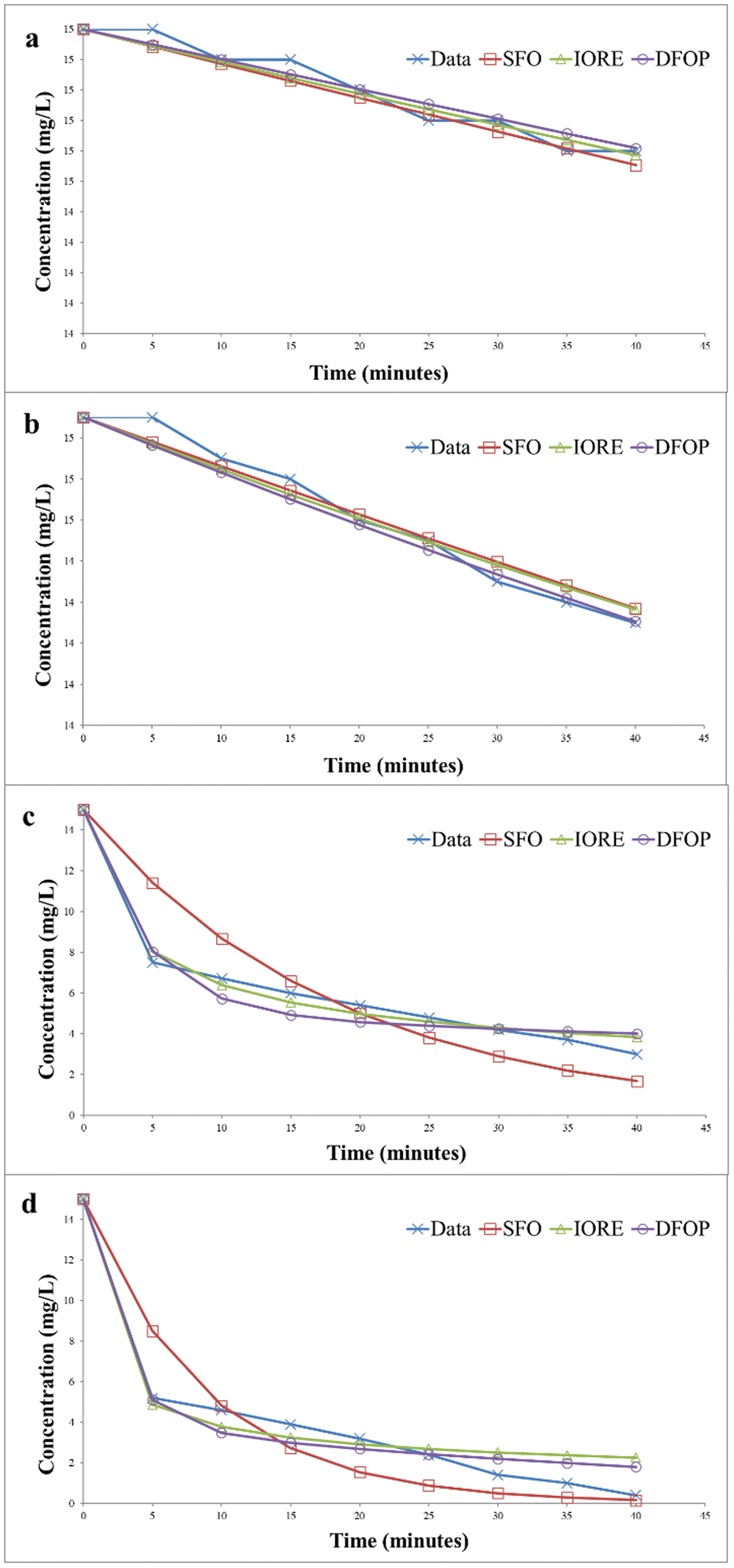
| | Fig. 14 Kinetics of photocatalytic degradation of methylene blue (a) without catalyst under dark (b) without catalyst under UV irradiation (c) with catalyst under dark (d) with catalyst under UV irradiation. | |
Fig. 14(b) represents the data for the degradation of methylene blue in the absence of the catalyst under UV irradiation. In this case, SSFO was greater than Sc, so the single first-order model cannot be applied here. Moreover, the 2DT50 value was much lower than TIORE. So, the DFOP model is most suitable for the determination of the half-life of methylene blue under UV in the absence of the catalyst. The half-life was reduced from 693 to 346 minutes in the presence of UV irradiations.
The degradation kinetics of methylene blue in the presence of the catalyst in the dark is depicted in Fig. 14(c). The data depicted that SSFO was greater than Sc, so the SFO model cannot be applied here. The DFOP model was found to be the most suitable model for determining the half-life of the dye as the value of 2DT50 was lower than TIORE. According to the calculations based on this model, the half-life of the dye was reduced to 81 min only even in the dark, thus depicting that the prepared catalyst is even effective in reducing the half-life of the dye in the dark. The data for UV irradiation in the presence of the catalyst is depicted in Fig. 14(d). In this case, the Sc is greater than SSFO, so the single first-order model best is the best choice to describe the degradation kinetics. The half-life of the dye was further reduced to only 6 min, showing that the catalyst more efficiently degraded the dye under UV irradiation.33,34
A comparison of the degradation efficiency of methylene blue in the presence of various catalysts is presented in Table 2.
Table 2 Comparison of photocatalytic degradation of methylene blue in the presence of different catalysts
| Sr no. |
Catalyst |
Time |
Degradation (%) |
Reference |
| 1 |
g-C3N4/ZnO |
120 |
91 |
33 |
| 2 |
CuO–ZnO |
120 |
95.6 |
34 |
| 3 |
CuO–ZnO |
150 |
98.5 |
35 |
| 4 |
Cr2O3/ZnO |
90 |
85 |
36 |
| 5 |
Cu–g-C3N4 |
5 |
100 |
This work |
Photocatalytic degradation of Congo red by graphitic carbon nitride supported single atom catalyst
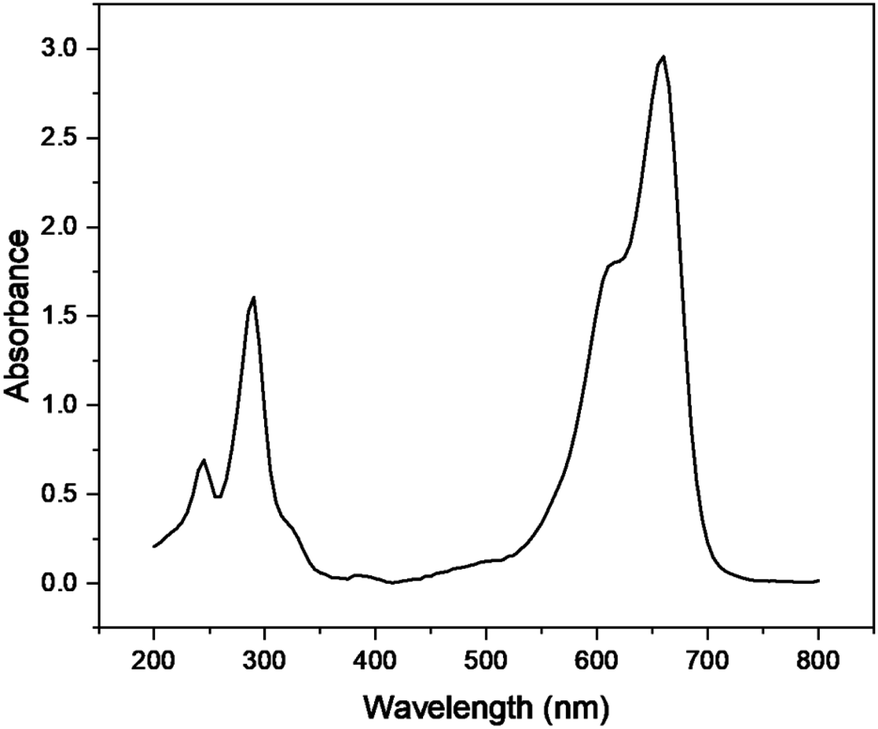
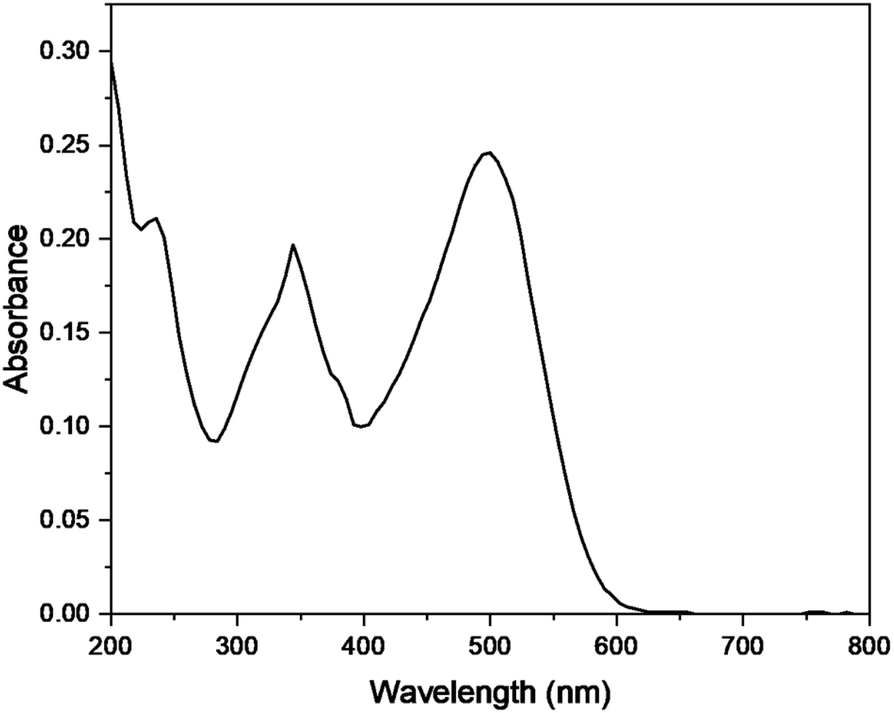
The photocatalytic degradation of Congo red was also studied by optimizing various parameters such as catalyst dose, irradiation time, pH, and concentration of dye in the presence of Cu–g-C3N4 catalyst. The UV-visible spectra of Congo red (Fig. 15) depicted its λmax to be 497 nm.37 This value of λmax was used throughout the study to determine the concentration of Congo red during different sets of experiments.
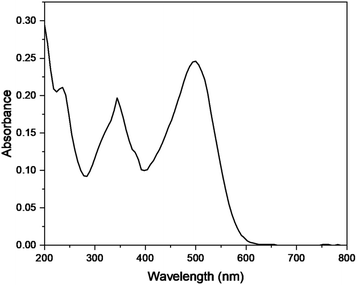 |
| | Fig. 15 UV spectra of Congo red. | |
Effect of time on degradation of Congo red
The optimum time for photocatalytic degradation of Congo red was determined by the addition of 0.025 g of Cu–g-C3N4 catalyst to 100 mL solutions of 15 mg per L Congo red with continuous stirring. The data for the degradation of methylene blue under UV light and dark in the presence and absence of the catalyst is shown in Fig. 16. In the absence of the catalyst, the degradation of Congo red reached only 2% in the dark and 6.2% under UV light in 40 minutes as shown in Fig. 16. The UV irradiation has a slight effect on the degradation process. But the degradation was improved significantly in the presence of Cu–g-C3N4 catalyst and reached up to 97% under UV light and 80% in the dark in just 40 minutes. As in the case of methylene blue, the degradation rapidly increased after adding the catalyst for up to 5 minutes and then it slowly progressed to maximum degradation in 40 minutes. The degradation of dye is affected by the contact time. By increasing the contact time, the production of electron–hole pairs increases leading to improved degradation of the dye.38
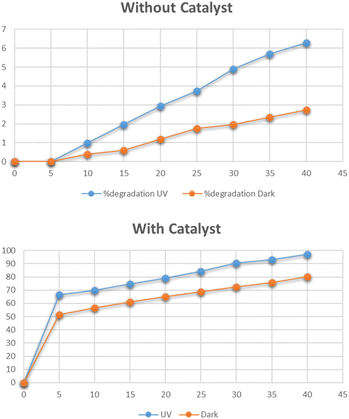 |
| | Fig. 16 Effect of time on degradation of Congo red. | |
Effect of concentration of Congo red
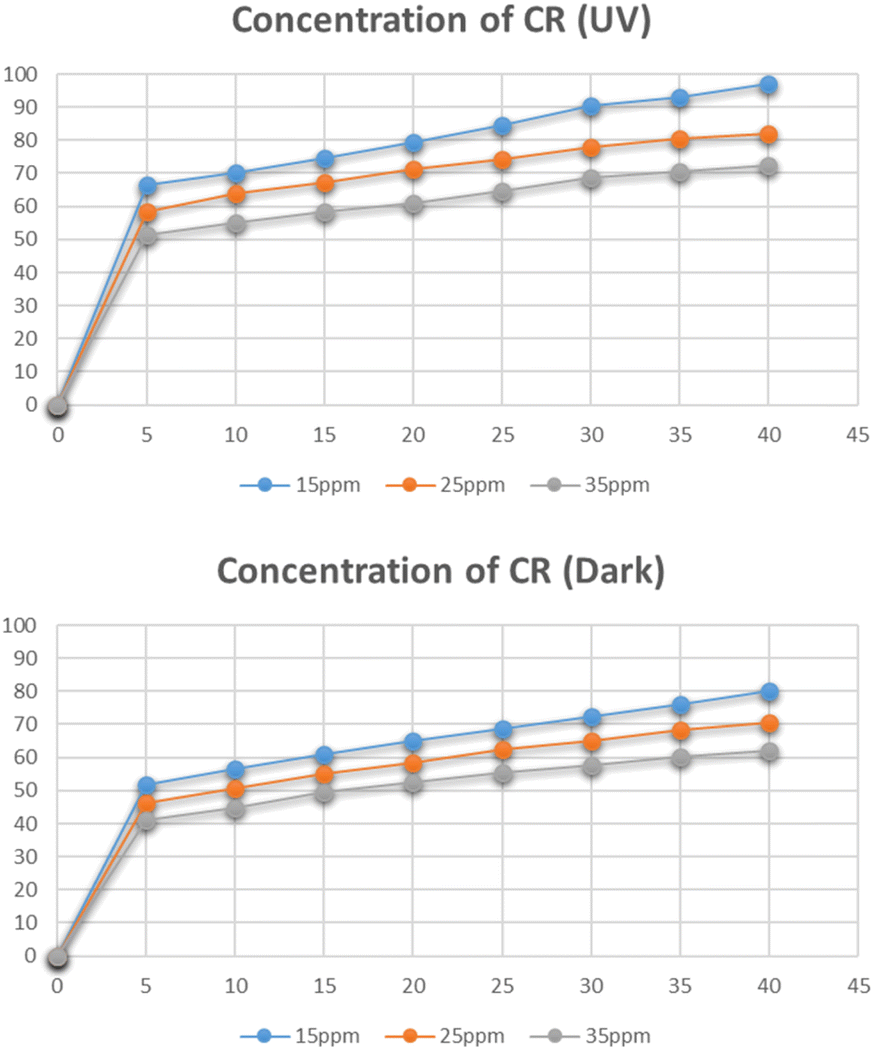
Fig. 17 represents the degradation of different concentrations of Congo red i.e. 15 mg L−1, 25 mg L−1, and 35 mg L−1. The maximum degradation was recorded for the 15 mg L−1 solution which was 97% in the presence of UV light and 80% in dark. With an increase in the concentration of Congo red the degradation was reduced. As the increased concentration of the dye blocks the radiation and lets only a small amount of radiation to reach the catalyst, hence reducing the number of radicals generated and hence leading to a decrease in degradation percentage.39
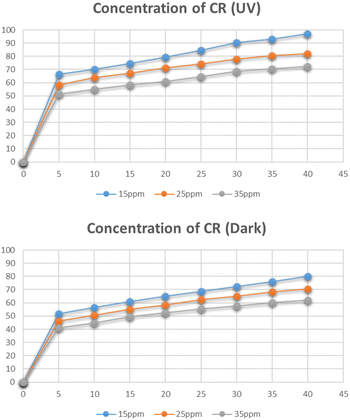 |
| | Fig. 17 Effect of concentration of Congo red on its degradation. | |
Effect of dose of catalyst
The effect of the catalyst dose on the photocatalytic degradation of Congo red was studied by varying the amounts of catalyst i.e. 0.025 g, 0.05 g, and 0.075 g. The data for the effect of the dose of catalyst on degradation is shown in Fig. 18. The highest degradation was 99% in just 30 minutes for a 0.075 g catalyst under UV irradiation. The same effect was observed for the solutions degraded under dark. The maximum degradation efficiency was recorded for the 0.075 g catalyst. By increasing the catalyst dose, a higher number of catalytic sites were available that efficiently degraded the dye molecules.40
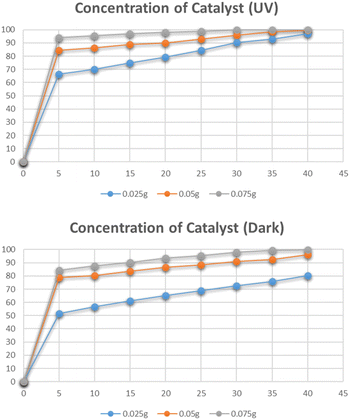 |
| | Fig. 18 Effect of dose catalyst on degradation of Congo red. | |
Effect of pH
The effect of pH on the degradation of Congo red dye at the optimum concentration and the catalyst dose was studied by varying pH between 5 and 11. The pH had a detrimental effect on the degradation of the dye as depicted in Fig. 19. At pH 5, the degradation reached 99% in 30 minutes. At pH less than 5, CR is chemically unstable, it protonates and takes on a blue color. Therefore, tests were not carried out below pH 5.37 The degradation is increased at acidic pH due to the anionic nature of Congo red dye which contains two sulphonic acid groups and these groups are easily ionized in an acidic medium to produce soluble Congo red anion.41
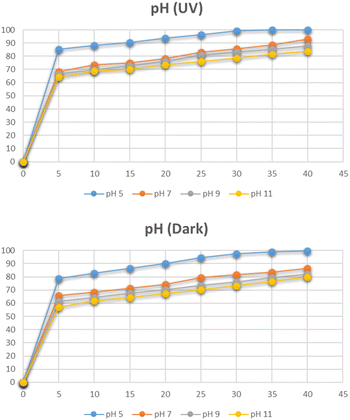 |
| | Fig. 19 Effect of pH on degradation of Congo red. | |
Kinetics of photocatalytic degradation of Congo red
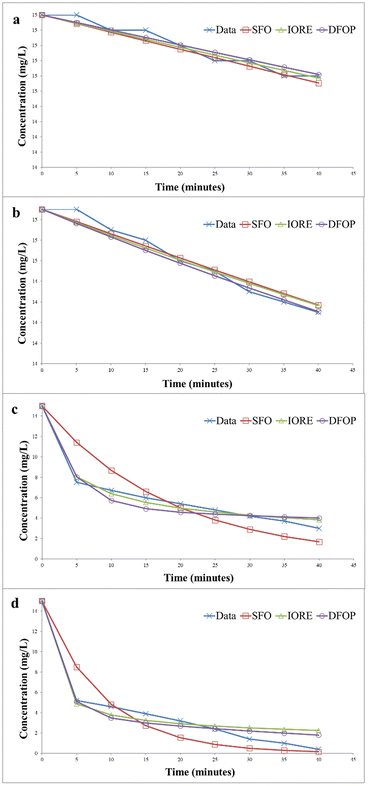
The photocatalytic degradation kinetics of Congo red was also determined by IORE, SFO, and DFOP model. Table 3 provides the parameters for the all three models. The degradation kinetics of Congo red carried out in the dark and in the absence of catalyst are depicted in Fig. 20(a). According to the degradation data the Sc value was less than SSFO. Thus, the single first-order model cannot be applied to represent degradation data. Thus, either the IORE or DFOP model will be followed. The TIORE for the IORE model and DT50 were compared to determine the half-life. And for this data set TIORE was greater than 2DT50, so, the DFOP model was followed to determine the half-life value of Congo red. Fig. 20(b) represents the data for the degradation of Congo red in the presence of UV light without catalyst. In this case, SSFO was less than Sc, so the single first-order model is followed to determine the half-life of the dye. In the presence of UV light, the half-life was reduced from 693 to 433 minutes. Fig. 20(c) shows the degradation kinetics of Congo red in the dark and in the presence of the catalyst. The data depicted that SSFO was greater than Sc, so the SFO model cannot be applied here. The DFOP model was found to be most suitable for determining the half-life of the dye as the value of 2DT50 was lower than TIORE. The half-life of the dye was reduced to only 125 min even in the dark in the presence of the catalyst. It depicts that the prepared catalyst is efficient enough to reduce the half-life of the dye in the dark. The data for the degradation in the presence of the catalyst under UV is provided in Fig. 20(d).
Table 3 Parameters of different kinetic degradation models
| Model |
Parameters |
Without catalyst |
With catalyst |
| Dark |
UV |
Dark |
UV |
| SFO |
K |
0.0010 |
0.0016 |
0.0548 |
0.11 |
| Ssfo |
0.11 |
0.04 |
24.15 |
18.75 |
| DT50 |
692.9 |
433.2 |
12.65 |
6.11 |
| DT90 |
2302 |
1439 |
42.01 |
20.23 |
| IORE |
Kiore |
1.02 × 10−5 |
2.00 × 10−6 |
3.00 × 10−4 |
1.09 × 10−3 |
| Niore |
2.60 |
3.50 |
3.56 |
3.65 |
| Siore |
0.01 |
0.04 |
1.66 |
7.91 |
| DT50 |
1647 |
1069 |
6.21 |
1.41 |
| DT90 |
31342 |
72347 |
459.5 |
117.9 |
| Tiore |
9435 |
21779 |
138.3 |
35.49 |
| DFOP |
K1 |
0.00 |
0.00 |
0.23 |
0.40 |
| K2 |
1.00 × 10−3 |
5.50 × 10−3 |
5.51 × 10−3 |
2.00 × 10−2 |
| Sdfop |
0.13 |
0.04 |
4.46 |
5.92 |
| 1DT50 |
13472 |
60533 |
3.01 |
1.73 |
| 2DT50 |
693.2 |
126.0 |
125.8 |
34.66 |
| g |
0.5 |
0.5 |
0.5 |
0.636 |
| Sc |
|
0.02 |
0.06 |
2.40 |
11.42 |
 |
| | Fig. 20 Kinetics of photocatalytic degradation of Congo red (a) without catalyst under dark, (b) without catalyst under UV irradiation, (c) with catalyst under dark (d) with catalyst under UV irradiation. | |
In this case, the single first-order model cannot be applied here because the Sc is lower than SSFO. The DFOP model was found to be most suitable for determining the half-life of the dye as the value of 2DT50 was lower than TIORE. The catalyst more efficiently degraded the dye under UV irradiation as the half-life of the dye was further reduced to only 34 min.44,45 A comparison of the degradation efficiency of Congo red in the presence of different catalysts is presented in Table 4.
Table 4 Comparison of percentage degradation of Congo red in the presence of different catalysts
| Sr no. |
Catalyst |
Time |
% degradation |
Reference |
| 1 |
CoFe2O4 |
90 |
83 |
42 |
| 2 |
CeO2–chitosan |
90 |
86.2 |
43 |
| 3 |
Cocatalyst loaded Al–SrTiO3 |
90 |
81 |
40 |
| 4 |
SnO2–Fe3O4 |
90 |
50.76 |
38 |
| 5 |
Cu–g-C3N4 |
30 |
99 |
This work |
Reusability of the catalyst
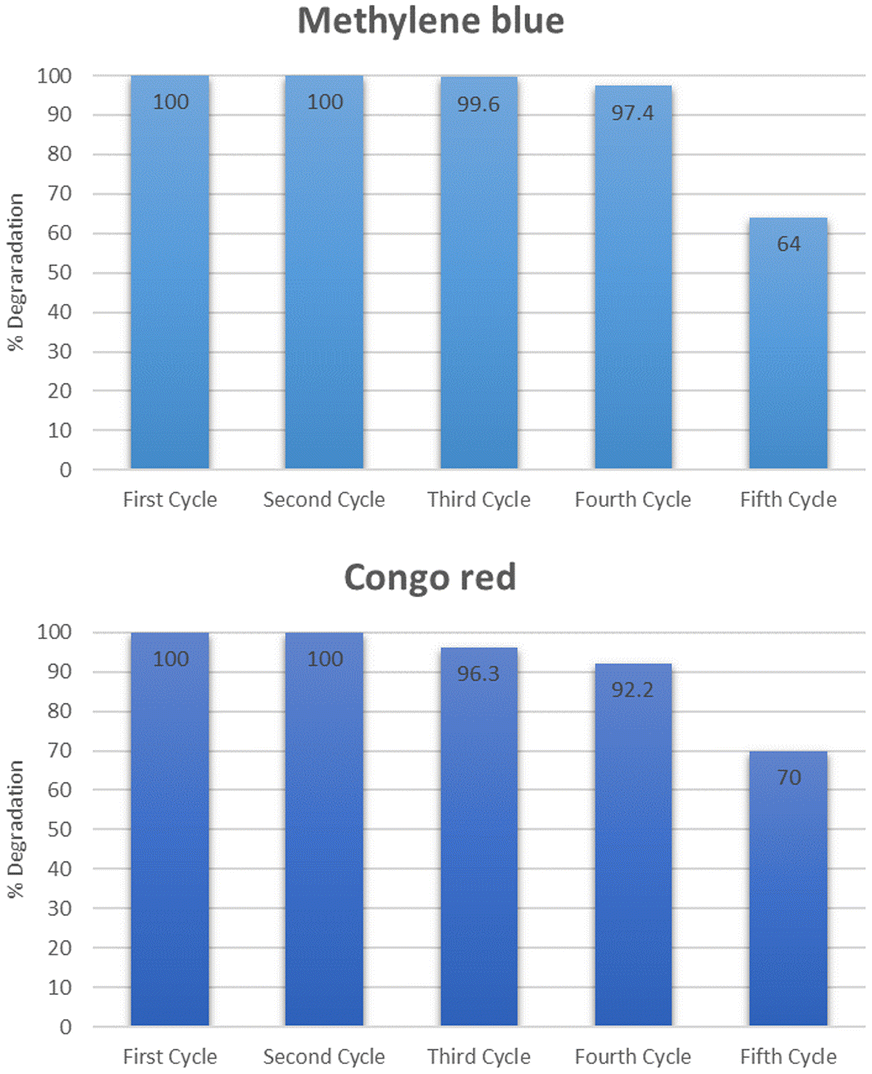
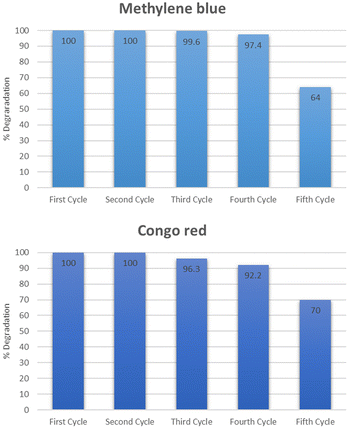
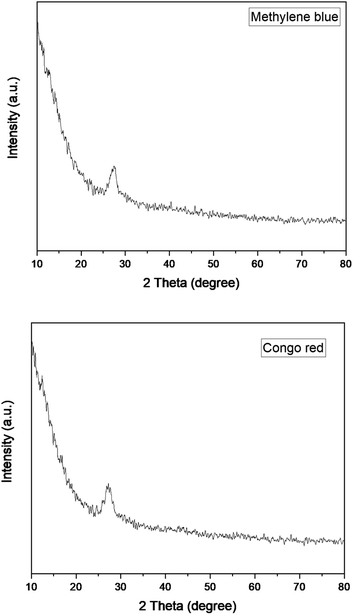
The reusability and stability of the catalyst were determined by using it for five consecutive cycles. The catalyst was first used to degrade methylene blue at optimum conditions exhibiting maximum degradation of 100% in 5 minutes for methylene blue. After that, the catalyst was regenerated by thoroughly washing with distilled water followed by drying at 100 °C. The dried catalyst was then used again for the second cycle at optimum conditions. The second cycle also exhibited 100% degradation of methylene blue. The catalyst was similarly reused for the third, fourth, and fifth cycles after activation. The degradation observed for the third, fourth, and fifth cycles was 99.6, 97.4, and 64% respectively as shown in Fig. 21. The same set of cycles was conducted for Congo red dye. The percentage degradation observed was 100, 100, 96.3, 92.2, and 70% for the cycles starting from 1 to 5. After five consecutive cycles, the XRD spectra of the catalyst was recorded which is shown in Fig. 22. The structural stability of the catalyst is evident from the XRD patterns. The XRD patterns confirm that there is no significant change in the structure of the catalyst even after five reaction cycles. The reduced activity after four cycles might be due to the agglomeration of particles which leads to reduced surface area.46 After using the catalyst for five consecutive cycles it was disposed of according to the SDGs practiced worldwide. The catalyst was disposed of properly in waste bins specified for solid chemicals.
 |
| | Fig. 21 Reusability of the catalyst. | |
 |
| | Fig. 22 XRD pattern of Cu–g-C3N4 catalyst after five reaction cycles. | |
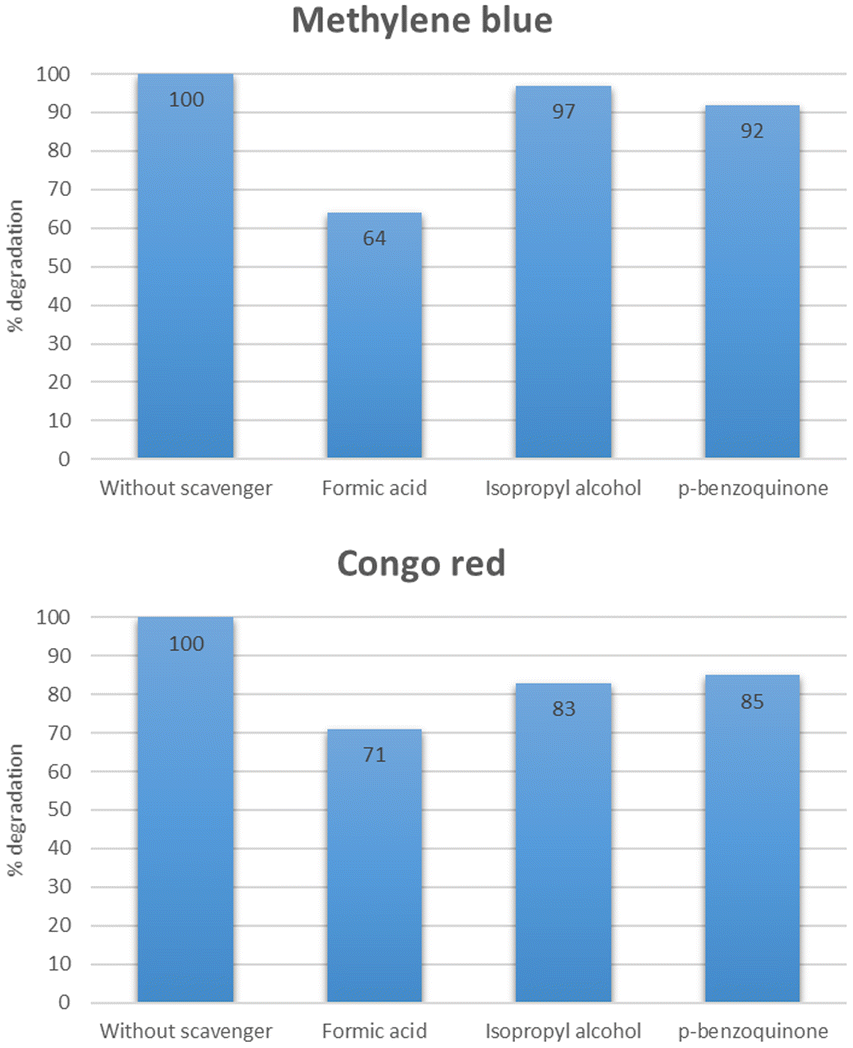
Scavenger studies
To study the role of different reactive species, a series of experiments were performed using different scavengers such as isopropyl alcohol, p-benzoquinone, and formic acid which acted as the source of HO˙, O2˙−, and h+ respectively. As shown in Fig. 23, the % degradation of methylene blue was slightly decreased in the case of isopropyl alcohol and p-benzoquinone. While the % degradation was significantly reduced in the case of formic acid. However, 100% degradation was observed when there was no scavenger added. This study indicated that HO˙ plays a more critical role than O2˙−, and holes.47,48 In the case of Congo red, it was deduced from Fig. 23 that maximum degradation was observed when no scavenger was added. Other than that the O2˙− exhibited an essential role in the degradation of Congo red.49,50
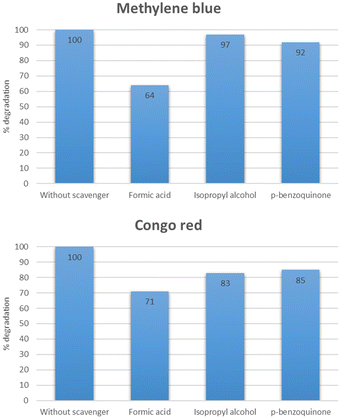 |
| | Fig. 23 Effect of different scavenger on the degradation of methylene blue and Congo red. | |
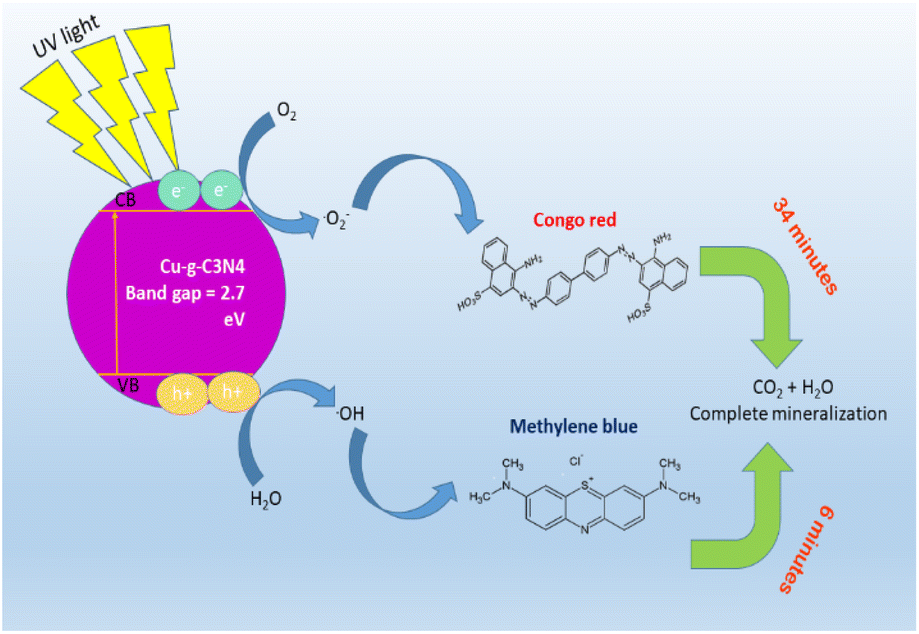
The mechanism of degradation of dyes in the presence of the Cu–g-C3N4 catalyst is elaborated in Fig. 24. The bandgap of the catalyst was calculated to be 2.7 eV. The catalyst is activated by the incident UV light, and electrons and holes are generated. These photo-generated electron and hole pairs react with oxygen and water to produce reactive oxygen species, including hydroxyl radicals and superoxide radical anions. These radicals interact with dye molecules, resulting in the complete degradation of dyes.51 To determine the possible degradation products of dyes, GC-MS analysis was carried out. Before analysis with the GC-MS system, the products were extracted using trichloromethane in a separating funnel for three times. Remaining water content from the sample was removed using Na2SO4. The metabolites extracted were identified using Thermo Scientific ISQ Single Quadrupole GC-MS with Trace TR-35 GC Column. Ionization voltage was set at 70 eV and helium gas flow was set at 1 mL min−1 for 35 minutes of run time. Starting temperature was set at 80 °C for two minutes and increased gradually by 10 °C min−1 until temperature reached 280 °C and held for seven minutes.52,53 In the case of both dyes, no metabolites were observed. Hence, it can be deduced that the single-atom catalyst was able to completely mineralize the dye solutions.
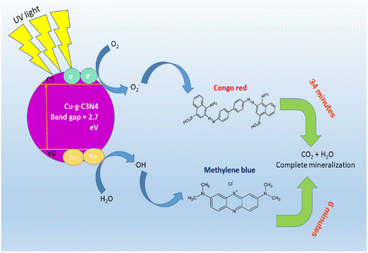 |
| | Fig. 24 Schematic diagram for photocatalytic degradation of dyes using Cu–g-C3N4 catalyst. | |
Conclusions
This work focused on the synthesis of an efficient and cost-effective single-atom catalyst that was used for the photocatalytic degradation of methylene blue and Congo red. The maximum degradation for methylene blue and Congo red was observed for 15 mg L−1 concentration in 5 and 30 minutes respectively, under UV irradiation. The prepared catalyst was able to efficiently reduce the half-life of both dyes. The half life of methylene blue was reduced from 346 to 6 minutes and the half life of Congo red was reduced from 433 to 34 minutes in the presence of supported single atom catalyst under UV irradiation. The catalyst was consecutively used for five cycles and showed excellent stability with a little reduction in activity after the fifth cycle. The catalyst's structural stability after five cycles was also depicted by XRD patterns which were not changed after reuse. Thus, Cu–g-C3N4 can be used as a cost-effective, stable, and highly active catalyst for the removal of pollutants like organic dyes from contaminated industrial effluents.
Author contributions
Saadia Rashid Tariq planned, supervised, edited and finalized the whole project. Zunaira Niaz performed experimental work and initial write up, formatting and editing. Ghayoor Abbas provided the instrumental facilities for material characterization and testing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial assistance provided by Lahore College for Women University, Lahore to carry out this research work as a part of PhD research project. We are also thankful to Mr Zajif from LUMS, Lahore for carrying out the XRD and SEM-EDX analyses.
References
- M. A. Al-Nuaim, A. A. Alwasiti and Z. Y. Shnain, Chem. Pap., 2023, 77, 677–701 CrossRef CAS PubMed.
- G. Ren, H. Han, Y. Wang, S. Liu, J. Zhao, X. Meng and Z. Li, Nanomaterials, 2021, 11, 1804 CrossRef CAS PubMed.
- V. Ruta, A. Sivo, L. Bonetti, M. A. Bajada and G. Vilé, ACS Appl. Nano Mater., 2022, 5, 14520–14528 CrossRef CAS PubMed.
- J. Hong, K. H. Cho, V. Presser and X. Su, Curr. Opin. Green Sustainable Chem., 2022, 36, 100644 CrossRef CAS.
- P. Klara, F. M. dela Rosa, M. Kovačić, H. Kušić, U. L. Štangar, F. Fresno, D. D. Dionysiou and A. L. Bozic, Materials, 2020, 13, 1338 CrossRef PubMed.
- B. Xia, Y. Zhang, J. Ran, M. Jaroniec and S. Z. Qiao, ACS Cent. Sci., 2021, 7, 39–54 CrossRef CAS PubMed.
- S. Hejazi, M. S. Killian, A. Mazare and S. Mohajernia, Catalysts, 2022, 12, 905 CrossRef CAS.
- C. Cometto, A. Ugolotti, E. Grazietti, A. Moretto, G. Bottaro, L. Armelao, C. Di Valentin, L. Calvillo and G. Granozzi, npj 2D Mater. Appl., 2021, 5, 63 CrossRef CAS.
- G. Gentile, M. Marchi, M. Melchionna, P. Fornasiero, M. Prato and G. Filippini, Eur. J. Org. Chem., 2022, 2022(37), e202200944 CrossRef CAS.
- H. Lin, J. Wu, F. Zhou, X. Zhao, P. Lu, G. Sun, Y. Song, Y. Li, X. Liu and H. Dai, J. Environ. Sci., 2023, 124, 570–590 CrossRef CAS PubMed.
- J. Fu, S. Wang, Z. Wang, K. Liu, H. Li, H. Liu, J. Hu, X. Xu, H. Li and M. Liu, Front. Phys., 2020, 15, 33201 CrossRef.
- X. Mao, R. Guo, Q. Chen, H. Zhu, H. Li, Z. Yan, Z. Guo and T. Wu, Molecules, 2023, 28, 3292 CrossRef CAS PubMed.
- I. Khan, K. Saeed, I. Zekker, B. Zhang, A. H. Hendi, A. Ahmad, S. Ahmad, N. Zada, H. Ahmad, L. A. Shah, T. Shah and I. Khan, Water, 2022, 14, 242 CrossRef CAS.
- I. M. Alarifi, Y. O. Al-Ghamdi, R. Darwesh, M. O. Ansari and M. K. Uddin, J. Mater. Res. Technol., 2021, 13, 1169–1180 CrossRef CAS.
- J. J. T. I. Boesten, K. Aden, C. Beigel, S. Beulke, M. Dust, J. S. Dyson, S. Fomsgaard, R. L. Jones, S. Karlsson, A. M. A. Van Der Linden, O. Richter, J. O. Magrans and G. Soulas, Guidance Document on Estimating Persistence and Degradation Kinetics from Environmental Fate Studies on Pesticides in EU Registration. The Final Report of the Work Group on Degradation Kinetics of FOCUS (Forum for the Co-ordination of Pesticide Fate Models and Their Use), p. 1005 Search PubMed.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- E. H. Kim, M. H. Lee, J. Kim, E. C. Ra, J. H. Lee and J. S. Lee, Chin. J. Catal., 2023, 47, 214–221 CrossRef CAS.
- L. Jurado, J. Esvan, L. A. Luque-Álvarez, L. F. Bobadilla, J. A. Odriozola, S. Posada-Pérez, A. Poater, A. Comas-Vives and M. R. Axet, Catal. Sci. Technol., 2023, 13, 1425–1436 RSC.
- M. Kaur and K. Pal, J. Mater. Sci.: Mater. Electron., 2021, 32, 12475–12489 CrossRef CAS.
- C. Yang, X. Hu, Y. Bai, B. Cai and Y. Li, Catalysts, 2023, 13, 619 CrossRef CAS.
- E. J. Lin, Y. B. Huang, P. K. Chen, J. W. Chang, S. Y. Chang, W. T. Ou, C. C. Lin, Y. H. Wu, J. L. Chen, C. W. Pao, C. J. Su, C. H. Wang, U. S. Jeng and Y. H. Lai, Appl. Surf. Sci., 2023, 615, 156372 CrossRef CAS.
- Z. H. Xue, D. Luan, H. Zhang and X. W. D. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- T. Zhang, X. Nie, W. Yu, X. Guo, C. Song, R. Si, Y. Liu and Z. Zhao, iScience, 2019, 22, 97–108 CrossRef CAS PubMed.
- J. Bian, C. Huang and R. Q. Zhang, ChemSusChem, 2016, 9, 2723–2735 CrossRef CAS PubMed.
- Z. Song, Z. Li, L. Lin, Y. Zhang, T. Lin, L. Chen, Z. Cai, S. Lin, L. Guo, F. Fu and X. Wang, Nanoscale, 2017, 9, 17737–17742 RSC.
- N. Alsubaie, R. Alshamrani, D. Domyati, N. Alahmadi and F. Bannani, Open J. Phys. Chem., 2021, 11, 106–127 CrossRef CAS.
- D. Chahar, D. Kumar, P. Thakur and A. Thakur, Mater. Res. Bull., 2023, 162, 112205 CrossRef CAS.
- W. Chaisorn, P. Nuengmatcha, A. Noypha, R. Pimsen, P. Porrawatkul, A. Kuyyogsuy, Y. Thepchuay, P. Sricharoen, N. Limchoowong, S. Chanthai and P. Nuengmatcha, Environ. Sci. Pollut. Res., 2023, 30, 96840–96859 CrossRef CAS PubMed.
- Z. Z. Vasiljevic, M. P. Dojcinovic, J. D. Vujancevic, I. Jankovic-Castvan, M. Ognjanovic, N. B. Tadic, S. Stojadinovic, G. O. Brankovic and M. V. Nikolic, R. Soc. Open Sci., 2020, 7, 200708 CrossRef CAS PubMed.
- S. Alkaykh, A. Mbarek and E. E. Ali-Shattle, Heliyon, 2020, 6, e03663 CrossRef PubMed.
- A. A. Abd El Khalk, M. A. Betiha, A. S. Mansour, M. G. Abd El Wahed and A. M. Al-Sabagh, ACS Omega, 2021, 6, 26210–26220 CrossRef CAS PubMed.
- M. Awais, S. Khursheed, R. Tehreem, S. Uddin, Y. S. Mok and G. U. Siddiqui, SSRN Electron. J., 2022, 643, 118764 CAS.
- S. R. Tariq, G. A. Chotana and A. Rashid, Int. J. Environ. Sci. Technol., 2022, 19, 2583–2598 CrossRef CAS.
- G. O. Aliyu, C. U. Anyanwu, C. I. Nnamchi and C. O. Onwosi, Soil Sediment Contam., 2023, 32, 105–124 CrossRef CAS.
- S. Merrad, M. Abbas, R. Brahimi and M. Trari, J. Mol. Struct., 2022, 1265, 133349 CrossRef CAS.
- R. C. Ngullie, S. O. Alaswad, K. Bhuvaneswari, P. Shanmugam, T. Pazhanivel and P. Arunachalam, Coatings, 2020, 10, 500 CrossRef CAS.
- H. Ullah, L. Mushtaq, Z. Ullah, A. Fazal and A. M. Khan, Inorg. Nano-Met. Chem., 2021, 51, 963–975 CrossRef CAS.
- P. Shanmugam, R. C. Ngullie, S. M. Smith, S. Boonyuen, R. Boddula and R. Pothu, Mater. Sci. Energy Technol., 2023, 6, 359–367 CAS.
- O. A. Zelekew, P. A. Fufa, F. K. Sabir and A. D. Duma, Heliyon, 2021, 7, e07652 CrossRef CAS PubMed.
- M. Said, W. T. Rizki, W. R. Asri, D. Desnelli, A. Rachmat and P. L. Hariani, Heliyon, 2022, 8, e09204 CrossRef CAS PubMed.
- P. L. Hariani, M. Said, A. Rachmat, A. Nurmansyah and R. H. T. Amallia, Int. J. Environ. Sci. Dev., 2022, 13, 57–62 CrossRef CAS.
- M. Abd Elkodous, A. M. El-Khawaga, M. M. Abouelela and M. I. A. A. Maksoud, Sci. Rep., 2023, 13, 6331 CrossRef CAS PubMed.
- U. O. Bhagwat, J. J. Wu, A. M. Asiri and S. Anandan, ChemistrySelect, 2018, 3, 11851–11858 CrossRef CAS.
- N. Ali, A. Said, F. Ali, F. Raziq, Z. Ali, M. Bilal, L. Reinert, T. Begum and H. M. N. Iqbal, Water, Air, Soil Pollut., 2020, 231, 50 CrossRef CAS.
- W. A. Al-Onazi and M. H. H. Ali, J. Mater. Sci.: Mater. Electron., 2021, 32, 12017–12030 CrossRef CAS.
- A. K. Sibhatu, G. K. Weldegebrieal, S. Sagadevan, N. N. Tran and V. Hessel, Chemosphere, 2022, 300, 134623 CrossRef CAS PubMed.
- D. Gogoi, P. Makkar and N. N. Ghosh, ACS Omega, 2021, 6, 4831–4841 CrossRef CAS PubMed.
- H. Ashrafi, M. Akhond and G. Absalan, J. Photochem. Photobiol., A, 2020, 396, 112533 CrossRef CAS.
- S. A. Al-Zahrani, M. B. Patil, S. N. Mathad, A. Y. Patil, A. Al Otaibi, N. Masood, D. Mansour, A. Khan, V. Gupta, N. S. Topare, A. Somya and M. Ayyar, Crystals, 2023, 13, 577 CrossRef CAS.
- S. P. Pattnaik, A. Behera, R. Acharya and K. Parida, J. Environ. Chem. Eng., 2019, 7, 103456 CrossRef CAS.
- M. Saeed, M. Muneer, A. Ul Haq and N. Akram, Environ. Sci. Pollut. Res., 2022, 29, 293–311 CrossRef CAS PubMed.
- X. Q. Wang, S. F. Han, Q. W. Zhang, N. Zhang and D. D. Zhao, in MATEC Web of Conferences, EDP Sciences, 2018, 238, p. 03006 Search PubMed.
- H. Victor, V. Ganda, B. Kiranadi and R. Pinontoan, KnE Life Sci., 2020, 2020, 102–110 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Ghayoor Abbas Chotana
*a and
Ghayoor Abbas Chotana