DOI:
10.1039/D3RA05897B
(Paper)
RSC Adv., 2023,
13, 34610-34617
A polyacrylamide gel containing an engineered hexameric hemoprotein as a cross-linking unit toward redox-responsive materials†
Received
29th August 2023
, Accepted 20th November 2023
First published on 27th November 2023
Abstract
Hydrogels containing synthetic polymers and supramolecular cross-linking units are expected to exhibit unique functions and properties. The heme–heme pocket interaction in hemeproteins may be useful for development of a cross-linking unit because heme binding depends on the redox states of the iron center. In this work, hexameric tyrosine-coordinated hemoprotein (HTHP) is employed as a cross-linking unit in a polyacrylamide gel to create redox-responsive hydrogels. First, redox-dependent stability of the heme–heme pocket interaction in HTHP was evaluated, and it was found that the heme affinity dramatically decreases in the Fe(II) state. Second, the polymerization of acrylamide and engineered HTHP possessing acryloyl group-tethering heme moieties provided a polyacrylamide gel containing HTHP as a cross-linking unit. A reduction-triggered gel–sol transition in the presence of apomyoglobin was observed. Furthermore, the mechanical properties of the gels containing the engineered HTHP and methylene bisacrylamide were evaluated by a tensile test, and the Young's modulus value was determined to be 14 kPa, which is higher than that of the control gel containing only methylene bisacrylamide (8.5 kPa). Compression tests of the gels revealed redox-responsive mechanical behavior, resulting in a decrease in the compressive modulus upon the addition of a reductant. This behavior is qualitatively consistent with the redox-responsive heme binding of HTHP in a solution state. This finding is expected to contribute to the development of redox-responsive materials for biomedical and biological applications.
Introduction
Stimuli-responsive hydrogels demonstrate changes in the physicochemical properties, volumes, and/or shapes triggered by external stimuli such as temperature, chemicals, pH, ionic strength, electric field/voltage/current, and light intensity.1–3 These properties have been widely expected to be harnessed in fabrication of smart biomaterials for use in chemical sensors, actuators, and other applications.4 Efforts to develop stimuli-responsive materials have focused on rational design of composites of functional molecules and polymeric matrices.5–7 In the polymer network, several functional molecules provide non-covalent connections including hydrophobic,8 hydrogen bonding,9,10 π–π,11,12 electrostatic,13 coordination,14–17 and host-guest interactions.18–21 In particular, a series of stimuli-responsive hydrogels containing cyclodextrin-based host-guest systems have been demonstrated by Harada and co-workers. Various combinations with guest molecules such as azobenzene22 and ferrocene23 have produced photo- and redox-responsive hydrogels. Furthermore, other host molecules, crown ethers,24,25 cucurbit[n]urils,26–28 calix[n]arenes,29–31 and pillar[n]arenes,32–34 have been employed to provide stimuli responsive interactions, leading to the development of various stimuli-responsive gels.35
Proteins are also potent building blocks due to their attractive properties. Protein-based hydrogels and elastomers have been prepared by taking advantage of covalent connections as well as supramolecular linkages.36–40 Folding and denaturation properties in protein units provide unique mechanical properties and responsiveness to stimuli. Miyata and co-workers utilized the supramolecular interaction between a protein and small molecules to generate a cross-linking unit of a gel.41–47 A polyacrylamide gel formed by strong and specific interaction between antibody and antigen has been demonstrated to display swelling behavior upon the addition of free-antigen.41 In another example, a biotin-attached four-armed poly(ethylene glycol) was reacted with avidin to demonstrate supramolecular gelation via the biotin-avidin interaction.45 Further addition of free biotin induces its dissociation to a sol state. Similarly, the glucose–lectin concanavalin A interaction46,47 was found to serve as a cross linkage for gelation as well as providing peptide–protein interactions.48–50 The specific molecular binding behavior of proteins provides gel systems with useful properties for applications in chemical sensors and drug delivery systems.51,52
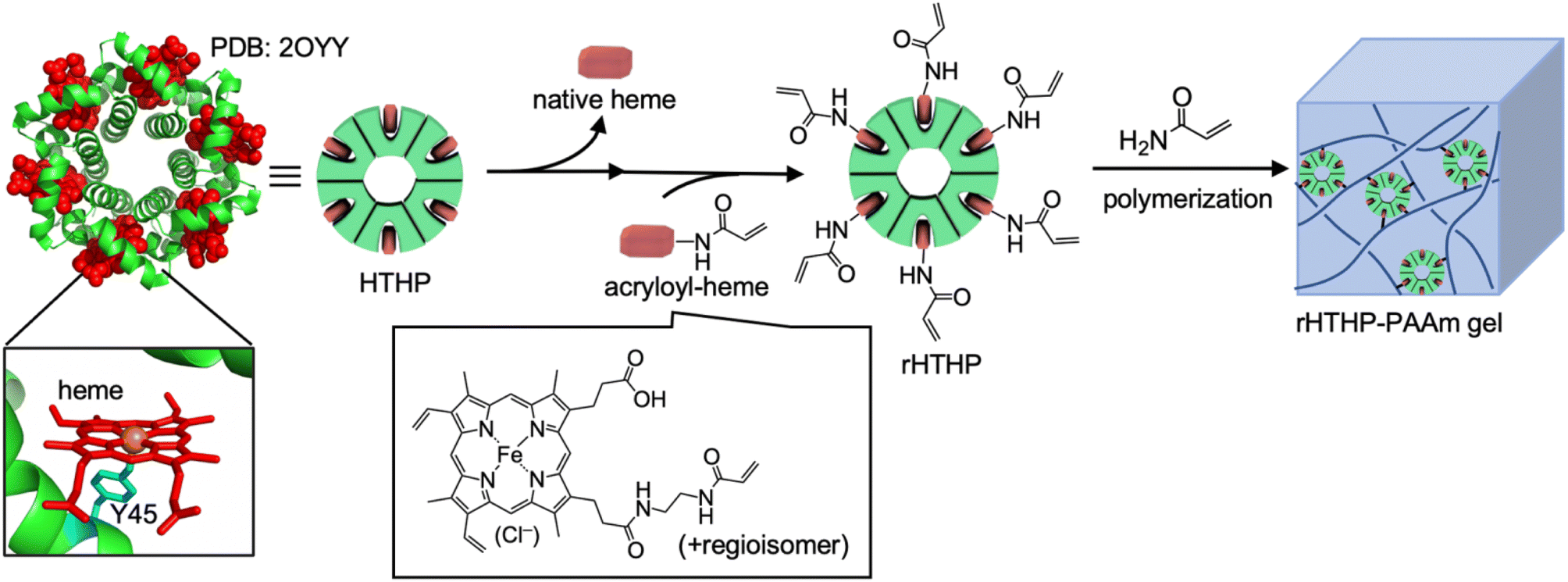
The heme–heme pocket interaction in hemoproteins is known as one of the most stable supramolecular interactions in biological systems.53–55 Unique stimuli–responsive properties are expected to be provided by materials that have a heme–heme pocket interaction because the affinity depends on the properties of the sixth ligand and the redox states of the iron center.53,54,56,57 Our group has reported a series of supramolecular hemoprotein assembling systems driven by successive heme–heme pocket interactions.58–62 In particular, the supramolecular assembly of myoglobin (Mb), a dioxygen binding hemoprotein, demonstrates stimuli-responsive stability.62 The interaction was also utilized to provide a connection between a gel containing the apo-form of heme-dependent peroxidase and a gel containing heme moieties, indicating that an enzymatic reaction can occur at the gel interface.63 The heme–heme pocket interaction has been recognized as an attractive cross linkage of the polymer network in hydrogels. In our previous work, supramolecular hydrogels were prepared by heme-tethering of polyacrylic acid and the apo-form of the Mb dimer.64 However, the low solubility of the heme-tethering polymer and the intrinsically monomeric structure of Mb have imposed limits on further investigations. In the present work, we focus on hexameric tyrosine coordinated heme protein (HTHP),65 an intrinsically oligomeric hemoprotein, as a cross-linking unit to develop a redox-responsive polyacrylamide hydrogel (Fig. 1). The axial ligand of the heme cofactor in each monomer of HTHP is a tyrosine residue located at position 45 of the protein. The Fe(III) state is highly stabilized in tyrosine-coordinated hemoproteins,66 inspiring significantly redox-dependent heme-binding behavior. Furthermore, the remarkably high thermal stability (Tm > 130 °C) and the ability to replace heme with synthetic cofactors are useful features for development of a cross-linking unit.65,67,68 In this study, we demonstrate the preparation of a gel by copolymerization of acrylamide with an engineered HTHP as a cross-linking unit and evaluate its redox-responsive behavior.
 |
| | Fig. 1 Schematic representation for preparation of HTHP reconstituted with acryloyl-heme (rHTHP) and a polyacrylamide gel containing rHTHP (rHTHP-PAAm gel). | |
Results and discussion
Evaluation of heme–heme pocket interaction in Fe(II) and Fe(III) states of HTHP
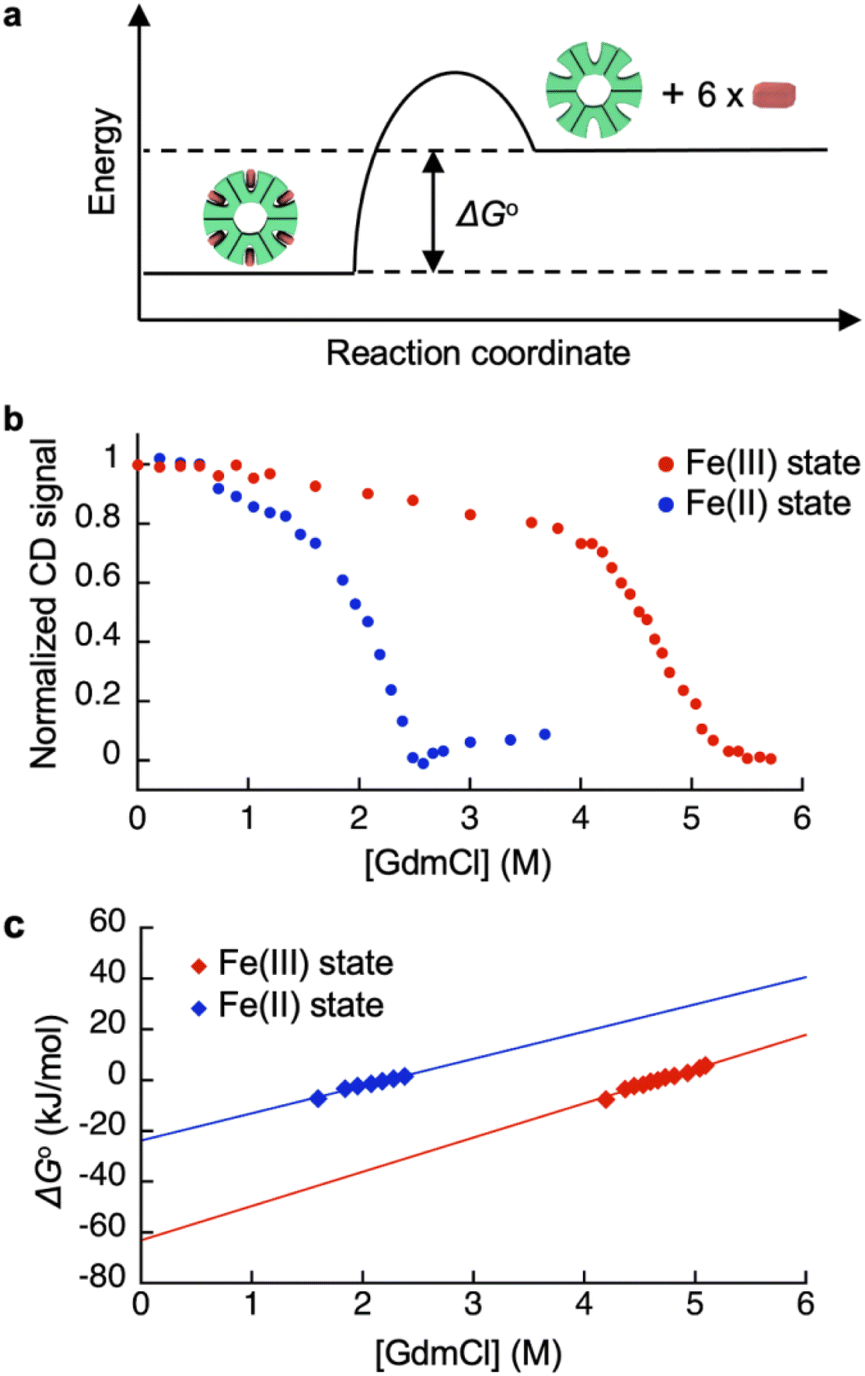
Redox-dependent heme-binding behavior of HTHP was first investigated. The iron center of HTHP is in the Fe(III) state under ambient conditions, whereas the Fe(II) state is obtained upon the addition of dithionite as a reductant. The UV-vis spectrum of the Fe(III) state of HTHP displays a strong absorption Soret band at 402 nm and several Q band peaks near 550 and 620 nm. Upon conversion to the Fe(II) state of HTHP, the Soret band shifts to 420 nm and the Q bands shift to 559 and 590 nm.65 Circular dichroism (CD) spectra of the Fe(III) and Fe(II) states of HTHP were also measured. In the Fe(III) state, an obvious CD signal at 418 nm is observed due to the chiral heme-binding sites of HTHP (Fig. S5†).66 Upon addition of dithionite, this CD signal shifts to 438 nm, indicating that the Fe(II) states of heme molecules are also bound in the protein matrix of HTHP (Fig. S6†). Next, ΔG°, the energy gap in heme dissociation from the HTHP matrix, was evaluated to assess the stability of the heme–heme pocket interaction in HTHP (Fig. 2a), because the ΔG° values are known to represent a thermodynamic parameter indicating the stability of folding in proteins as well as heme-binding in hemoproteins.57,69–71 The ΔG° values were determined by titration of guanidium chloride (GdmCl), a chemical denaturant, into a solution of wild-type HTHP in potassium phosphate buffer at pH 7.0.72
 |
| | Fig. 2 (a) Schematic energy diagram of heme binding in HTHP. (b) CD signal changes of the Fe(III) and Fe(II) states of HTHP upon addition of various concentration of GdmCl. (c) Plots of ΔG° values of the Fe(III) and Fe(II) states of HTHP against concentration of GdmCl. Conditions: 100 mM potassium phosphate buffer, pH 7.0, at 90 °C (Fe(III) state) or 25 °C (Fe(II) state). | |
Due to HTHP's high thermodynamic stability, we used severe denaturing conditions at a high temperature of 90 °C. Fig. 2b shows plots of normalized CD signal changes of the Fe(III) state of HTHP in the presence of various concentrations of GdmCl. The two-state equilibrium transition is considered as below:
| |
 | (1) |
In Fig. 2b, a clear decrease of the CD intensity in the Fe(III) state was observed in the concentration range of 4.0 M to 5.8 M GdmCl. This indicates that the heme-bound state at 4.0 M GdmCl converts to the unbound state at 5.8 M GdmCl. The equilibrium constants at each GdmCl concentration were calculated using the following equation:
| | |
FB(C) = 1 − FU (C) = (yx(C) − yU(C))/(yB(C) − yU(C))
| (2) |
where
FB(
C) and
FU(
C) represent fractions of the heme-bound and unbound states, respectively, at a given GdmCl concentration (
C).
yx(
C) is an observed CD signal intensity at a GdmCl concentration (
C).
yB(
C) and
yU(
C) are the dependencies of the assumed CD signal intensities on denaturant concentrations for the fully heme-bound and unbound states, respectively. Subsequently, the equilibrium constants,
Keq, were determined as:
| | |
Keq (C) = FB(C)/FU(C) = (yx(C) − yu(C))/(yB(C) − yx(C))
| (3) |
The Keq(C) values were converted into ΔG°(C) values using eqn (4), as follows:
| |
ΔG° (C) = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKeq(C) lnKeq(C)
| (4) |
The ΔG° value in the absence of chemical denaturant was determined as the y-intercept of the linear extrapolation model,72 as shown in Fig. 2c. The ΔG° value for heme binding in the Fe(III) state of HTHP was determined to be −57 kJ mol−1. In the presence of an excess amount of dithionite at 25 °C, ΔG° for the Fe(II) state of HTHP was also determined to be −25 kJ mol−1. For reference, the ΔG° values for the Fe(II) and Fe(III) states of Mb were evaluated using UV-vis spectral changes which occur upon addition of GdmCl. The ΔG° values are summarized in Table 1. The ΔG° value of HTHP in the Fe(III) state is notably lower than that of other hemoproteins such as Mb (−45.7 kJ mol−1)70 or cytochrome b562 (−46 kJ mol−1).71 In the case of Mb, which has a histidine residue as an axial ligand of heme, the Fe(II) state is more stable than the Fe(III) state. In contrast, the Fe(II) state of HTHP exhibits weakened heme binding, possibly because the heme cofactors are coordinated by phenolate moieties derived from tyrosine residues in HTHP. The high energy difference of heme binding between the Fe(II) and Fe(III) states corresponds to the Fe(II)/Fe(III) redox potential of HTHP at −0.54 V (vs. Ag/AgCl).73
Table 1 The ΔG° values of HTHP and Mb in different redox states
| Hemoprotein |
Redox state |
ΔG° (kJ mol−1) |
| The ΔG° values were determined by the CD signal changes at 90 °C. The ΔG° values were determined the CD signal changes at 25 °C in the presence of excess ditihonite. The ΔG° values were determined by UV-vis absorbance changes at 25 °C. The ΔG° values were determined by UV-vis absorbance changes in the presence of 100 mM dithionite at 25 °C. |
| HTHP |
Fe(III) |
−57 ± 6a |
| HTHP |
Fe(II) |
−25 ± 2b |
| Mb |
Fe(III) |
−46 ± 4c |
| Mb |
Fe(II) |
−49 ± 2d |
Preparation of a polyacrylamide gel containing an engineered HTHP as a cross-linking unit
Reactive moieties were introduced into HTHP to convert it to a cross-linking unit for a polyacrylamide gel and HTHP was reconstituted with an artificial cofactor. In this context, acryloyl-heme was designed and synthesized, in which heme b has an acryloyl group attached to one of the heme-propionate side chains via an ethylene diamine linker unit (see ESI†). This freebase precursor was characterized by 1H-NMR and ESI MS, and the acryloyl-heme unit was confirmed by ESI-MS and UV-vis spectroscopy. Preparation of apoHTHP and its reconstitution were conducted using conventional methods (Fig. 1).67 Acryloyl-heme dissolved in DMSO was added to the apo-form of HTHP (apo-HTHP) in a 100 mM potassium phosphate buffer, pH 7.0, containing 1 M NaCl, potentially enhancing hydrophobic interaction between acryloyl-heme and the heme pocket of HTHP. The obtained protein, reconstituted HTHP (rHTHP), was characterized using UV-vis spectroscopy. The Rz value (ratio of absorbances at 402 nm and 280 nm) is similar to that of the holo-form of HTHP (Fig. 3a). The CD spectrum in the far-UV region indicates the typical α-helical structure of HTHP (Fig. S7†). Analytical size exclusion chromatography of rHTHP shows a similar elution volume as wild type HTHP (Fig. S8†), indicating that the hexameric structure is maintained.
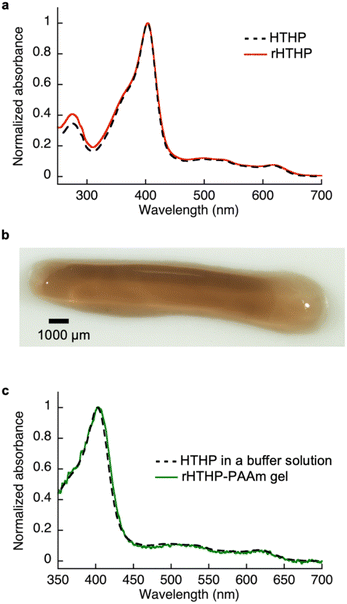 |
| | Fig. 3 (a) UV-vis absorption spectra of the Fe(III) states of wild type and rHTHP in 100 mM potassium phosphate buffer, pH 7.0, containing 1 M NaCl. (b) Photograph of rHTHP-PAAm gel, (c) UV-vis absorption spectra of the Fe(III) states of rHTHP-PAAm gel and HTHP in solution. | |
Copolymerization of acrylamide with rHTHP as a cross-linking unit was performed using conventional radical polymerization in the presence of ammonium persulfate and N,N,N′,N′-tetramethylenediamine. Interestingly, gelation occurred within a few hours without the support of other covalent cross-linkers such as methylene bis-acrylamide. Fig. 3b displays a representative image of the polyacrylamide gel containing rHTHP (rHTHP-PAAm gel). A control experiment using HTHP instead of rHTHP did not produce a gel. This observation confirms that rHTHP acts as a cross-linking unit. The UV-vis spectrum of the gel has a Soret band near 400 nm and Q-bands near 500 nm (Fig. 3c), which are typical bands for the heme cofactors bound into each HTHP matrix. This suggests that the radical polymerization reaction does not significantly affect the environment of the heme cofactor in rHTHP.
Gel–sol transition in rHTHP-PAAm gel induced by a heme-transfer event using the apo-form of Mb
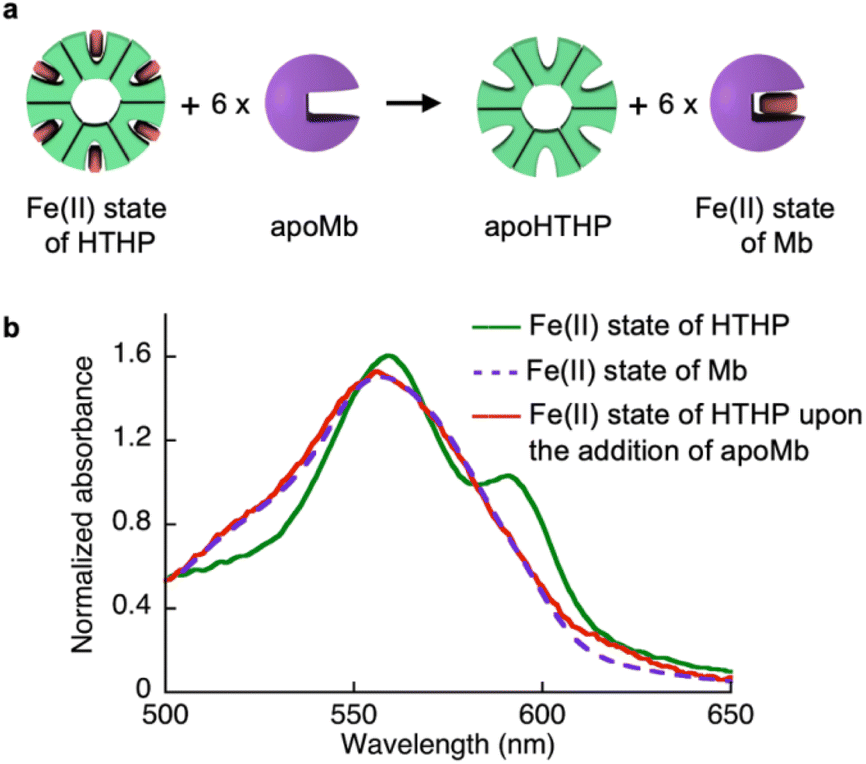
As shown in Table 1, we characterized the cross linkage provided by the heme–heme pocket interaction in the gel, noting the high energy gap of the heme binding between Mb and HTHP in the Fe(II) states, as shown in Table 1. This observation encouraged us to reconstitute the apo-form of Mb (apoMb) with heme in the Fe(II) state, as depicted in Fig. 4a. This type of heme-transfer event was previously utilized for removal of heme in cytochrome P450-BM3 upon the addition of apoMb in the Fe(II) state.74 First, the heme transfer reaction from HTHP to apoMb was attempted in solution. The UV-vis spectral changes of the Fe(II) state of HTHP were monitored upon the addition of excess apo-Mb at room temperature. The characteristic spectrum of the Q-band of the Fe(II) state of HTHP was converted within one hour, revealing a spectrum consistent with that of the Fe(II) state of Mb (Fig. 4b). This finding clearly indicates that the Fe(II) state of the heme cofactor was transferred from HTHP to apoMb, following the determined energy difference in the heme-binding process. Next, this experiment was applied to the rHTHP-PAAm gel (Fig. 5a). The gel with a height of 3 mm and a diameter of 6 mm was prepared in a small vial, as shown in Fig. 5b. When the vial was inverted, the rHTHP-PAAm gel was located at the bottom of the vial. A 0.1 mM apoMb solution was then added to the vial. After 3 hours, apoMb diffused into the gel and the gel state was maintained and located at the bottom of the inverted vial. Into this mixture, an equal volume of a 200 mM dithionite solution was added. Interestingly, the gel was observed to change to the sol state upon gentle shaking. These behaviors were caused by reduction-triggered heme transfer from HTHP to apo-Mb, leading to the decomposition of the cross linkage in the polymer network. Furthermore, the UV-vis spectrum of the resulting solution is consistent with that of Fe(II) state of Mb (Fig. S9†). This type of gel–sol transition is typical for hydrogels containing supramolecular interactions as cross linkages, demonstrating that the heme–heme pocket interaction in rHTHP is the principal cross linkage formed in the gelation process.
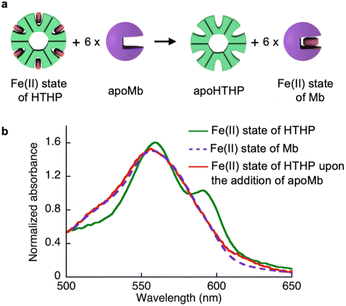 |
| | Fig. 4 (a) Scheme of a heme-transfer event from Fe(II) state of HTHP to apoMb. (b) Absorption spectra of the Fe(II) state of HTHP (green solid line), the Fe(II) state of Mb (purple broken line) and the Fe(II) state of HTHP upon the addition of apoMb (red solid line) in 100 mM potassium phosphate buffer, pH 7.0. | |
 |
| | Fig. 5 (a) Scheme of gel–sol transition by the reduction-triggered heme transfer in the presence of apoMb. (b) Photographs of the gel–sol transition experiment. The left-side photographs show the Fe(III) state of rHTHP-PAAm gel immersed in apoMb solution. The right-side photographs show the solution obtained by liquefaction of rHTHP-PAAm gel containing apo Mb upon addition of 200 mM dithionite solution. | |
Mechanical properties of the gel containing rHTHP and the redox responsiveness
The mechanical properties of the gels were investigated. Initially, an evaluation of the polyacrylamide gel containing rHTHP as a cross-linking unit was attempted, but the gel was too fragile to quantify the properties. Therefore, a dual cross-linked gel was prepared by radical polymerization of 1.0 M acrylamide with 1.5 mM rHTHP and 5.0 mM N,N′-methylene bis-acrylamide as cross-linking units (Fig. S10†). The obtained gel, which we designate “rHTHP-MBA-PAAm gel,” exhibits sufficient toughness to conduct a mechanical evaluation. As a control sample, the gel prepared without rHTHP, MBA-PAAm gel, was also investigated. The tensile tests of the gels were carried out at a strain speed of 10 mm min−1. The gels were swollen in a buffer solution overnight at room temperature to reach the equilibrium state before obtaining measurements. Fig. 6a shows the stress–strain curves of the gels. The Young's modulus value was determined as a slope of the curve from 0% to 10% of the rupture points. The rupture energy was calculated as the area under the curve representing the gel deformation until rupture. The Young's modulus and rupture energy values are summarized in Fig. 6b and c, respectively. The Young's modulus values of MBA-PAAm gel and rHTHP-MBA-PAAm gel were determined to be 8.5 kPa and 14 kPa, respectively. The elastic property of the rHTHP-MBA-PAAm gel is obviously higher than that of the MBA-PAAm gel. The rupture energies of MBA-PAAm gel and rHTHP-MBA-PAAm gel were determined to be 8.8 kJ m−3 and 11 kJ m−3, respectively. The heme–heme pocket interaction of rHTHP which provides the cross linkage in the gel contributes to enhancement of the mechanical property of the gel due to increased cross-linking density.75,76
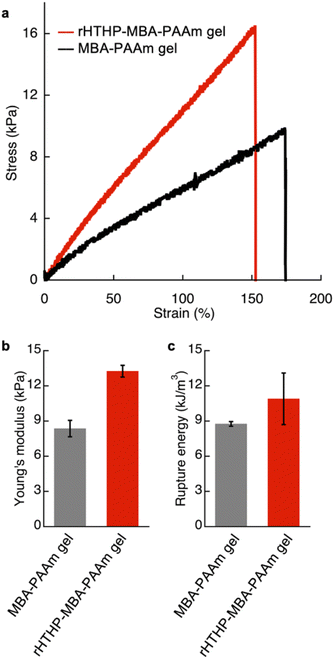 |
| | Fig. 6 (a) Stress–strain curves of the Fe(III) state of rHTHP-MBA-PAAm gel (red) and MBA-PAAm gel (black). (b) Young's modulus of rHTHP-MBA-PAAm gel and MBA-PAAm. (c) Rupture energy of rHTHP-MBA-PAAm gel and MBA-PAAm gel. | |
Since the heme binding in HTHP strongly depends on the redox states of the iron center, the mechanical properties of the gel containing rHTHP were evaluated in the Fe(II) and Fe(III) states. Due to the easily oxidized feature of the Fe(II) states of HTHP in air, the gel sample must be rapidly evaluated. Thus, a compression test was employed. First, reduction of the iron center in the rHTHP-MBA-PAAm gel was carried out by soaking a cylindrical gel into a buffer solution, pH 7.0, containing excess dithionite as shown in Fig. 7a and b. No significant differences in size were observed with or without dithionite. The reduction of the iron center was confirmed by UV-vis spectra of the gel: the peaks near 500 nm are consistent with the peaks of the characteristic Q bands of the Fe(II) state HTHP in a solution (Fig. 7c). Compression tests of the gels with or without dithionite were conducted at a 0.05 mm s−1 loading rate up to 60% height changes (Fig. S11†). The compressive modulus values were determined by slopes during the decrease in height ranging from 0% to 40%. Fig. 7d summarizes the compressive modulus values. The compressive modulus values of the rHTHP-MBA-PAAm gels containing the Fe(III) and Fe(II) states of heme are 11 kPa and 8.5 kPa, respectively. The reduction of the iron center significantly decreases the compressive modulus without changes in size. This finding ruled out changes in modulus due to differences in water content. The decrease of compressive modulus due to reduction appears to correspond to the redox-dependent heme binding in HTHP. Although mechanical properties of the gel are typically regulated by swelling of the gels and/or interactions between main-chain polymers, the present result is mainly caused by a cross-linking unit of the gel. This indicates that the heme–heme pocket interaction in HTHP provides a sufficiently stable cross linkage to affect the mechanical property. To the best of our knowledge, this is the first demonstration of a stimuli-responsive hydrogel triggered by a redox-dependent heme–heme pocket interaction.
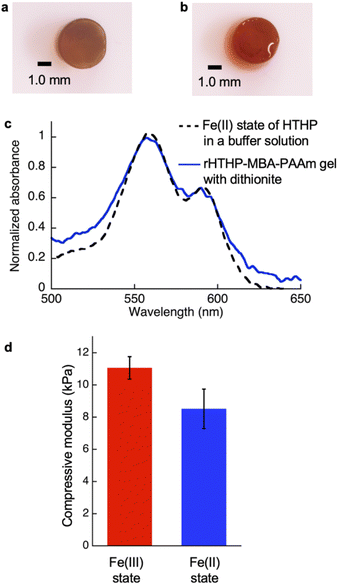 |
| | Fig. 7 (a) Photograph of the Fe(III) state of the rHTHP-MBA-PAAm gel. (b) Photograph of the rHTHP-MBA-PAAm gel soaked in 50 mM dithionite solution for 2 h. (c) UV-vis spectra of the Fe(II) state of HTHP in a solution (black broken line) and rHTHP-MBA-PAAm gel with dithionite (blue solid line). (d) Compressive modulus of the Fe(III) and Fe(II) states of rHTHP-MBA-PAAm gels. | |
Experimental procedures
Instruments
UV-vis spectral measurements of aqueous solution samples were carried out with a Shimadzu UV-2700 or a Shimadzu UV-3600 plus spectrophotometer. UV-vis spectra of the gels were recorded using a Shimadzu UV-3600 plus spectrophotometer equipped with an integrating sphere attachment. CD spectra were recorded on a JASCO (Model J-820). 1H NMR spectra were recorded on a Bruker BioSpin AVANCE III HD NMR spectrometer (400 MHz). The chemical shifts were calibrated by residual solvent signals. ESI-TOF MS analyses were performed with a Bruker Daltonics micrOTOF-II mass spectrometer. The pH values were monitored with a Horiba F-52 pH meter. Tensile tests were conducted using an EZ Graph with a 10 N EZ Graph load cell. Compression analysis was carried out on an HAAKE RheoStress 6000 rheometer equipped with a P20 TiL plate.
Materials
Ultrapure water (Milli-Q) was prepared by a Millipore Integral 3 apparatus. All reagents were of the highest commercially available guaranteed grade and were used as received unless otherwise indicated. HTHP and apoHTHP were prepared according our previous report.67 Mb was purchased from Sigma Aldrich and purified by a CM-Sepharose column.
CD spectra for ΔG° evaluation
CD spectra were measured in a solution containing wild-type HTHP in 100 mM potassium phosphate buffer at pH 7.0, with or without 100 mM dithionite. In the case of the Fe(III) state, the temperature was maintained at 90 °C by an equipped thermal controller. In the case of the Fe(II) state, CD signals were recorded at 25 °C. The optical path length was 1 cm, and the bandwidth was 2 μm.
Preparation of rHTHP
Five equivalents of acryloyl-heme in DMSO (stock solution: 5 mM) were added into a solution of apoHTHP (10 μM in 100 mM potassium phosphate buffer containing 1 M NaCl) at room temperature, and the reaction mixture was gently shaken overnight. Dialysis was carried out to remove the DMSO against 100 mM potassium phosphate buffer, pH 7.0, at 4 °C. The excess acryloyl-heme was removed by a HiTrap Desalting column. The obtained rHTHP was characterized by UV-vis spectrum. Purity was confirmed by an Rz value > 2.3.
Preparation of polyacrylamide gel containing rHTHP
Reconstituted HTHP was concentrated to 5 mM by ultrafiltration using a 30 kDa cut-off Amicon Ultra tube via centrifugation at 6000 rpm. Polymerization of 1.0 mM rHTHP and 1 M acrylamide in 100 mM potassium phosphate buffer, pH 7.0, containing 1 M NaCl was carried out with 0.25% ammonium sulfate and 0.38% N,N,N′,N′-tetramethylenediamine as radical initiators. Then, a dual cross-linked hydrogel was prepared in the presence of 5 mM N,N′-methylene bis-acrylamide and 1.5 mM rHTHP under the same radical polymerization conditions.
Mechanical properties evaluation
Samples were prepared in cuboid shapes with ca. 14 mm × ca. 1.0 mm × ca. 1.0 mm in equilibrium swelling states. During tensile tests, samples were attached to a metal sheet using the adhesive reagent Aron Alpha®, within 8 mm. The Young's modulus was determined by the slope in the strain ranging from 0% to 10% of the rupture point. The rupture energy was determined by the area sum in the strain–stress curve. Compression analysis was performed using a HAAKE RheoStress 6000 instrument in compression mode using P20 TiL as a plate. The compression test was carried out with a 0.05 mm s−1 stroke at 20 °C.
Conclusions
The hexameric hemoprotein reconstituted with acryloyl group-tethered heme derivatives functions as a cross-linking unit to provide a functional polyacrylamide hydrogel. The obtained hydrogels clearly exhibit redox-responsive mechanical properties without significant changes in volume, and the responsiveness arises from a large decrease of the heme–protein affinities generated by reduction of the iron center. The elastic modulus of the hydrogel depends on the supramolecular heme-binding behavior in the cross-linking unit. This finding suggests that the process of heme binding of hemoproteins in cross-linking units is useful for generating redox-responsive mechanical properties. Further details will be investigated to clarify the relationship between heme binding and mechanical properties of gels. Compared to previous redox-responsive hydrogels containing simple host–guest systems such as ferrocene–cyclodextrin interaction,23 the present hemoprotein-based hydrogels have two potential advantages: (i) redox potentials and/or binding constants for the heme moieties can be fine-tuned by protein mutation as well as metal substitution of the heme derivative, and (ii) specific heme-binding to the protein can create unique hydrogels in combination with various other supramolecular systems. Processes to control the gel properties by modulation of heme-binding behavior such as small molecule ligation and protein matrix mutation will be investigated.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was funded by Grants-in-Aid for Scientific Research provided by JSPS KAKENHI Grant Numbers JP15H05804, JP18KK0156, JP20H02755, JP20H00403, JP20KK0315, JP22H05364, JP22K21348, JP23H03832 and JST PPRESTO JPMJPR15S2 and JPMJPR22A3. K. K. appreciates the establishment of university fellowships towards the creation of science technology innovation, Grant Number JPMJFS2125, supported by JST. We acknowledge Prof. Hiroshi Uyama and Dr Akihide Sugawara (Osaka University) for tensile tests and compression analysis. K. O. thanks the financial supports by the Tokuyama Science Foundation, the Takeda Science Foundation, and the Noguchi Institute.
Notes and references
- M. Annaka and T. Tanaka, Nature, 1992, 355, 430–432 CrossRef CAS.
- H. Katono, A. Maruyama, K. Sanui, N. Ogata, T. Okano and Y. Sakurai, J. Controlled Release, 1991, 16, 215–227 CrossRef CAS.
- T. Tanaka, Sci. Am., 1981, 244, 124–138 CrossRef CAS PubMed.
- L. Lonov, Mater. Today, 2014, 17, 494–503 CrossRef.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345–347 CrossRef CAS.
- H. Yang, H. Liu, H. Kang and W. Tan, J. Am. Chem. Soc., 2008, 130, 6320–6321 CrossRef CAS PubMed.
- D. C. Tuncaboylu, M. Sari, W. Oppermann and O. Okay, Macromolecules, 2011, 44, 4997–5005 CrossRef CAS.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- S. Burattini, H. M. Colquhoun, J. D. Fox, D. Friedmann, B. W. Greenland, P. J. F. Harris, W. Hayes, M. E. Mackay and S. J. Rowan, Chem. Commun., 2009, 6717–6719 RSC.
- J. Fox, J. J. Wie, B. W. Greenland, S. Burattini, W. Hayes, H. M. Colquhoun, M. E. Mackay and S. J. Rowan, J. Am. Chem. Soc., 2012, 134, 5362–5368 CrossRef CAS PubMed.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2010, 10, 14–27 CrossRef PubMed.
- P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- C. Li, J. Tan, Z. Guan and Q. Zhang, Macromol. Rapid Commun., 2019, 40, 1800909 CrossRef PubMed.
- G. Liu, Q. Yuan, G. Hollett, W. Zhao, Y. Kang and J. Wu, Polym. Chem., 2018, 9, 3436–3449 RSC.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- Y. Yang, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Commun., 2018, 9, 1906 CrossRef PubMed.
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 RSC.
- K. Iwaso, Y. Takashima and A. Harada, Nat.
Chem., 2016, 8, 625–632 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, A. Hashidzume and A. Harada, Angew. Chem., Int. Ed., 2013, 52, 5731–5735 CrossRef CAS PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 51, 7011–7015 CrossRef CAS PubMed.
- D. Liu, D. Wang, M. Wang, Y. Zheng, K. Koynov, G. K. Auernhammer, H.-J. Butt and T. Ikeda, Macromolecules, 2013, 46, 4617–4625 CrossRef CAS.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Z. Yu, N. Li, C. Abell and O. A. Scherman, Adv. Mater., 2017, 29, 1605325 CrossRef PubMed.
- C. S. Y. Tan, J. Liu, A. S. Groombridge, S. J. Barrow, C. A. Dreiss and O. A. Scherman, Adv. Funct. Mater., 2018, 28, 1702994 CrossRef.
- K.-P. Wang, Y. Chen and Y. Liu, Chem. Commun., 2015, 51, 1647–1649 RSC.
- Q. Zhao, Y. Chen, S. H. Li and Y. Liu, Chem. Commun., 2018, 54, 200–203 RSC.
- H. Yang, J. Tang, C. Shang, R. Miao, S. Zhang, K. Liu and Y. Fang, Macromol. Rapid Commun., 2018, 39, 1700679 CrossRef PubMed.
- S. Wang, Z. Xu, T. Wang, T. Xiao, X.-Y. Hu, Y.-Z. Shen and L. Wang, Nat. Commun., 2018, 9, 1737 CrossRef PubMed.
- H. Ju, F. Zhu, H. Xing, Z. L. Wu and F. Huang, Macromol. Rapid Commun., 2017, 38, 1700232 CrossRef PubMed.
- S. Wang, Z. Xu, T. Wang, X. Liu, Y. Lin, Y.-Z. Shen, C. Lin and L. Wang, J. Photochem. Photobiol., A, 2018, 355, 60–66 CrossRef CAS.
- T. Xiao, L. Xu, L. Zhou, X.-Q. Sun, C. Lin and L. Wang, J. Mater. Chem. B, 2019, 7, 1526–1540 RSC.
- J. Fang, A. Mehlich, N. Koga, J. Huang, R. Koga, X. Gao, C. Hu, C. Jin, M. Rief, J. Kast, D. Baker and H. Li, Nat. Commun., 2013, 4, 2974 CrossRef PubMed.
- Y. Cao and H. Li, Chem. Commun., 2008, 4144–4146 RSC.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS PubMed.
- H. Li, N. Kong, B. Laver and J. Liu, Small, 2016, 12, 973–987 CrossRef CAS PubMed.
- S. Lyu, J. Fang, T. Duan, L. Fu, J. Liu and H. Li, Chem. Commun., 2017, 53, 13375–13378 RSC.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS PubMed.
- T. Miyata, M. Jige, T. Nakaminami and T. Uragami, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 1190–1193 CrossRef CAS PubMed.
- T. Miyata, N. Asami and T. Uragami, Macromolecules, 1999, 32, 2082–2084 CrossRef CAS.
- C. Norioka, K. Okita, M. Mukada, A. Kawamura and T. Miyata, Polym. Chem., 2017, 8, 6378–6385 RSC.
- T. Miyata, N. Asami and T. Uragami, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2144–2155 CrossRef CAS.
- T. Miyata, A. Jikihara, K. Nakamae and A. S. Hoffman, Macromol. Chem. Phys., 1996, 197, 1135–1146 CrossRef CAS.
- T. Miyata, A. Jikihara, K. Nakamae and A. S. Hoffman, J. Biomater. Sci., Polym. Ed., 2004, 15, 1085–1098 CrossRef CAS PubMed.
- Y. Liang and K. L. Kiick, Acta Biomater., 2014, 10, 1588–1600 CrossRef CAS PubMed.
- N. Yamaguchi, L. Zhang, B. Chae, C. S. Palla, E. M. Furst and K. L. Kiick, J. Am. Chem. Soc., 2007, 129, 3040–3041 CrossRef CAS PubMed.
- K. L. Kiick, Soft Matter, 2008, 4, 29–37 RSC.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- T. Miyata, T. Uragami and K. Nakamae, Adv. Drug Delivery Rev., 2002, 54, 79–98 CrossRef CAS PubMed.
- C. J. Reedy and B. R. Gibney, Chem. Rev., 2004, 104, 617–650 CrossRef CAS PubMed.
- S. Schneider, J. Marles-Wright, K. H. Sharp and M. Paoli, Nat. Prod. Rep., 2007, 24, 621–630 RSC.
- K. Oohora and T. Hayashi, Dalton Trans., 2021, 50, 1940–1949 RSC.
- B. A. Springer, S. G. Sligar, J. S. Olson and G. N. Phillips Jr, Chem. Rev., 1994, 94, 699–714 CrossRef CAS.
- M. S. Hargrove and J. S. Olson, Biochemistry, 1996, 35, 11310–11318 CrossRef CAS PubMed.
- K. Oohora, A. Onoda and T. Hayashi, Chem. Commun., 2012, 48, 11714–11726 RSC.
- K. Oohora, N. Fujimaki, R. Kajihara, H. Watanabe, T. Uchihashi and T. Hayashi, J. Am. Chem. Soc., 2018, 140, 10145–10148 CrossRef CAS PubMed.
- K. Oohora, R. Kajihara, M. Jiromaru, H. Kitagishi and T. Hayashi, Chem. Lett., 2019, 48, 295–298 CrossRef CAS.
- K. Oohora, Y. Onuma, Y. Tanaka, A. Onoda and T. Hayashi, Chem. Commun., 2017, 53, 6879–6882 RSC.
- K. Oohora, A. Onoda, H. Kitagishi, H. Yamaguchi, A. Harada and T. Hayashi, Chem. Sci., 2011, 2, 1033–1038 RSC.
- Y. Kobayashi, Y. Takashima, A. Hashidzume, H. Yamaguchi and A. Harada, Sci. Rep., 2015, 5, 16254 CrossRef CAS PubMed.
- T. Ono, Y. Hisaoka, A. Onoda, K. Oohora and T. Hayashi, Chem.–Asian J., 2016, 11, 1036–1042 CrossRef CAS PubMed.
- J.-H. Jeoung, D. A. Pippig, B. M. Martins, N. Wagener and H. Dobbek, J. Mol. Biol., 2007, 368, 1122–1131 CrossRef CAS PubMed.
- T. Mashima, K. Oohora and T. Hayashi, J. Porphyrins Phthalocyanines, 2017, 21, 824–831 CrossRef CAS.
- K. Oohora, T. Mashima, K. Ohkubo, S. Fukuzumi and T. Hayashi, Chem. Commun., 2015, 51, 11138–11140 RSC.
- S. Hirayama, K. Oohora, T. Uchihashi and T. Hayashi, J. Am. Chem. Soc., 2020, 142, 1822–1831 CrossRef CAS PubMed.
- M. S. Hargrove, A. J. Wilkinson and J. S. Olson, Biochemistry, 1996, 35, 11300–11309 CrossRef CAS PubMed.
- P. A. Sykes, H.-C. Shiue, J. R. Walker and R. C. Bateman Jr, J. Chem. Educ., 1999, 76, 1283–1284 CrossRef CAS.
- C. R. Robinson, Y. Liu, J. A. Thomson, J. M. Sturtevant and S. G. Sligar, Biochemistry, 1997, 36, 16141–16146 CrossRef CAS PubMed.
- G. I. Makhatadze, J. Phys. Chem. B, 1999, 103, 4781–4785 CrossRef CAS.
- P. Kielb, T. Utesch, J. Kozuch, J.-H. Jeoung, H. Dobbek, M. A. Mroginski, P. Hildebrandt and I. Weidinger, J. Phys. Chem. B, 2017, 121, 3955–3964 CrossRef CAS PubMed.
- S.-C. Chien, O. Shoji, Y. Morimoto and Y. Watanabe, New J. Chem., 2017, 41, 302–307 RSC.
- T. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, Md. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Takashi Hayashi
* and
Takashi Hayashi