
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen-doped antimonene monolayer as a promising anchoring material for lithium–sulfur batteries: a first-principles study
Victor Zhu and
Xuan Luo*
and
Xuan Luo*
National Graphene Research and Development Center, Springfield, Virginia 22151, USA. E-mail: xluo@ngrd.org
First published on 16th October 2023
Abstract
To effectively mitigate the dissolution of lithium polysulfides (Li2Sx) in the electrolyte, the search for an effective anchoring material is crucial. In this study, we employed density functional theory (DFT) computations to investigate the adsorption behavior of long-chain Li2Sx species on an O-doped antimonene monolayer. Our results demonstrate that the O-doped antimonene mono-layer exhibits stronger adsorption for long-chain Li2Sx species compared to the pristine antimonene monolayer, resulting in enhanced adsorption energies. This improved adsorption effectively curtails the dissolution of lithium polysulfides and preserves the structural integrity of the Li2Sx species. The charge transfer analysis also revealed the strong chemical interactions between the Li2Sx species and the O-doped antimonene monolayer. These findings suggest that the O-doped anti-monene monolayer holds promise as an effective anchoring material for enhancing the performance of lithium–sulfur batteries.
I. Introduction
To meet the ever-increasing global energy consumption1,2 and accommodate the grow-ing use of electric devices, electric vehicles, and renewable energy sources,3–5 the develop-ment of high-performance rechargeable batteries is desirable. Currently, lithium-ion batter-ies (LIBs) are the optimal rechargeable battery, featuring excellent environmental compat-ibility, high energy density, and long cycle life.6,7 However, LIBs have limited applications in electric vehicles due to safety, durability, and cost considerations.4,6,8 In fact, LIBs are approaching their theoretical energy density limit.9,10 As a potential alternative, lithium–sulfur (Li–S) batteries have gathered great interest due to their higher energy density and theoretical capacity.11,12 Furthermore, the natural abundance, low cost, and non-toxicity of sulfur make the development of Li–S batteries more attractive.13,14 Yet the practical ap-plication of lithium–sulfur batteries is impeded by multiple major obstacles.15 One critical issue is the dissolution of soluble long-chain lithium polysulfides (Li2Sx, x = 4, 6, 8) in the electrolyte during the charge/discharge process. The phenomenon referred to as the shuttle effect results in the loss of active materials, rapid capacity fade, and self-discharge.16–18Extensive research has been performed to inhibit the shuttle effect, and various strategies have been proposed to suppress it. One effective strategy is to use anchoring materials to bind lithium polysulfides onto their surface through physical/chemical interactions.19 Many anchoring materials have been studied, including various carbon materials such as carbon composites,20 carbon heterostructures21 because of their high conductivity and large surface area. However, these carbon materials exhibited weak interaction with lithium polysulfides, making them ineffective in fully suppressing the shuttle effect.22,23 Many other functional materials have also been studied, including polymers,24,25 metal oxides and sulfides,26 metal organic frameworks,27,28 and other metal compounds.29 Although these functional materi-als have demonstrated strong chemical adsorption strength with lithium polysulfides, their reversible capacity and cycle stability remain unsatisfactory.30,31 In light of this, previous studies have demonstrated that two-dimensional,32–34 with their unique electronic properties,35,36 high surface-volume ratio,37 and multiple adsorption sites38 are promising anchoring materials.
Various two-dimensional materials have been investigated to anchor lithium polysul-fides, including transition metal sulfides (e.g. TiS2,32 VS2,33 WS2 (ref. 34)), other metal com-pounds (e.g. SiC2,39 V2CS2,40 Ti2C41) and monoelemental two-dimensional materials (e.g. borophene,42 phosphorene,43 arsenene,44 and bismuthene44). Many of them exhibit weak interactions with lithium polysulfides. Therefore, further studies have explored several ap-proaches to enhance the adsorption strength of lithium polysulfides onto two-dimensional materials. With vacancies, substitution doping of atoms, and surface functionalizations with atom and molecules, these methods not only improved the adsorption capability of two-dimensional materials but also exposed more adsorption sites.34,45,46 For example, N-doping of graphene,47 transition metal doping of C2N,48 and S-termination of Ti2C Mxene41 have all improved their adsorption capabilities. As such, the use of these strategies are of great interest towards strengthening the performance of anchoring materials.
One promising anchoring material for Li–S batteries is antimonene. Due to its moderate band gap,49 high carrier mobility,50,51 and high structural stability at ambient temperatures,46,52 antimonene is promising for application in energy storage. While pre-vious research has demonstrated its effectiveness as an electrode material for LIBs53 and sodium-ion batteries,54 recent studies have explored its potential for Li–S batteries.44,49 However, pristine antimonene exhibits only weak to moderate adsorption capabilities of anchoring lithium polysulfides in Li–S batteries.44 To overcome this limitation, researchers have turned to doping strategies, with vanadium, tin, and selenium dopants showing promis-ing results through atom substitution.49 Nevertheless, concerns remain about the strength of adsorption and potential detachment of the adsorbed polysulfides from the doped an-choring material.55 To address this challenge, oxygen doping has shown promise. Studies involving oxygen doping, such as carbon nitride tubes, revealed improved adsorption of lithium polysulfides through chemical interactions upon substantial oxygen doping.56–58 Furthermore, doping can decrease the band gap of the monolayer and enhance the intrinsic conductivity, facilitating better lithium diffusion.59 Thus, we aim to study the adsorption of lithium polysulfides on oxygen-doped antimonene monolayer, potentially improving the performance and stability of Li–S batteries.
We performed first-principle calculations based on Density Functional Theory (DFT) to study the structural and electronic properties of lithium polysulfides adsorbed on pure and doped antimonene. The adsorption energies of the lithium polysulfides adsorbed on pure and oxygen-doped antimonene were calculated to study the suppression of the shuttle effect. In addition to adsorption energies, the band structure, and charge transfer were calculated.
II. Methods
A. Computational details
We performed first-principle calculations based on Density Functional Theory (DFT) within the Perdew–Burke–Ernzerhof (PBE) Generalized Gradient Approximation (GGA) implemented in the ABINIT60 code. We used the Projected Augmented Wave (PAW) method61 with projectors generated with the ATOM code.62 The cut-off radii are 1.0, 1.5, 1.4, 2.4, 1.6, and 1.9 Bohr, and the electrons configurations are 1s1, [He] 2s2 2p2, [He] 2s2 2p4, [Kr] 5s2 5p3 4d10, 1s2 2s1, and [Ne] 3s2 3p4 for H, C, O, Sb, Li, and S, respectively.Convergence was carried out to determine the appropriate converged values for the kinetic energy cutoff, Monkhorst–Pack k point grids, and vacuum. The values were considered converged when the difference in total energy was less than 1.0 × 10−4 Hartree twice consecutively.63 During the convergence calculations, the self-consistent field (SCF) total energy calculations were considered complete when the total energy difference was less than 1.0 × 10−10 Hartree for the second time.63
The Broyden–Fletcher–Goldfarb–Shanno64 (BFGS) method was used for the relax-ation of the lattice parameters and atomic structure. During relaxation calculations, SCF iterations were completed when the total difference in forces was less than 2.0 × 10−5 Hartree Bohr−1 twice consecutively. The relaxation calculations were considered complete when all of the forces were less than 6.0 × 10−4 Hartree Bohr−1 (around 0.03 eV Å−1).65
B. Atomic structure
Monolayer antimonene exists in several allotropes, differentiated by their prefixes, α, β, γ, and others.46 It has been predicted by phonon dispersion spectra, mechanically, and thermally that the α- and β-phases are stable and semiconducting and β-phase is the most stable allotrope.66Previous studies demonstrated β-phase antimonene has nonplanar structure and hexagonal arrangement.66 In this study, a 4 × 4 × 1 supercell of β-phase antimonene with 32 Sb atoms was used for calculations. We will be substitutionally doping the monolayer with O to enhance its effects on the adsorption of long-chain lithium polysulfides Li2S4, Li2S6, and Li2S8. The defect formation energy (Eform)67 is defined by
| Eform = EOSbML − ESbML − EO + ESb | (1) |
C. Li2Sx adsorption
To demonstrate the adsorption capabilities of the antimonene monolayer, the ad-sorption energies of Li2Sx (x = 1, 2, 4, 6, 8) with the typical electrolytes dimethyl ether (DME)/1,3-dioxolane (DOL) were calculated with the following equation| Ebind = ELiPS+electro − ELiPS − Eelectro | (2) |
Alongside this, the adsorption energies Eads of long-chain lithium polysulfides Li2Sx (x = 4, 6, 8) on the pristine and O-doped antimonene monolayers were calculated by the following equation
| Eads = ELiPS+ML − EML − ELiPS | (3) |
D. Electronic structure
The band structure of the antimonene system was calculated before and after the adsorption of Li2Sx (x = 4, 6, 8) polysulfides, and it was plotted using the high symmetry k-points Γ (0, 0, 0) M (1/2, 0, 0) K (2/3, 1/3, 0) and Γ (1, 1, 1).The interaction between adsorbed Li2Sx (x = 4, 6, 8) and O-doped antimonene was further confirmed by calculating the charge transfer. The charge transfer Δρ(r) is defined by
| Δρ(r) = ρLiPS/OSbML(r) − ρOSbML(r) − ρLiPS(r) | (4) |
III. Results and discussion
We carried out first-principle calculations to investigate the adsorption of lithium poly-sulfides species on commonly used electrolytes DME and DOL molecules, and both pristine and oxygen-doped antimonene monolayers. We fully relaxed the atomic structures and cal-culated the adsorption energy, band structure, as well as charge transfer.A. Adsorption of Li2Sx species on typical electrolytes
 | ||
| Fig. 1 Optimized atomic structures of Li2Sx (x = 1, 2, 4, 6, 8) and electrolytes DME/DOL: (a) Li2S, (b) Li2S2, (c) Li2S4, (d) Li2S6, (e) Li2S8 (f) DOL, and (g) DME. H, O, C, Li and S atoms are represented by white, red, blue, green, and yellow, respectively. | ||
| Species | Li2S | Li2S2 | Li2S4 | Li2S6 | Li2S8 |
|---|---|---|---|---|---|
| a Ref. 71.b Ref. 72.c Ref. 73. | |||||
| dS–S (Å) | — | 2.19 | 2.09 | 2.04 | 2.08 |
| dS–S (Å) | — | 2.19a | 2.19a | 2.08a | 2.07a |
| dS–S (Å) | — | — | 2.141b | 2.261b | 2.087b |
| dLi–S (Å) | 2.09 | 2.22 | 2.34 | 2.38 | 2.38 |
| dLi–S (Å) | 2.09a | 2.22a | 2.48a | 2.55a | 2.42a |
| dLi–S (Å) | 2.073b | 2.227b | 2.377b | 2.407b | 2.412b |
| dLi–S (Å) | 2.09c | 2.23c | 2.36/2.40c | 2.35/2.41c | 2.38/2.39c |
| θLi–S–Li (deg) | 109.3 | 95.32 | 77.38 | 68.65 | 66.25 |
| θLi–S–Li (deg) | 131.8c | 96.8c | 73.5c | 69.1c | 66.3c |
| dLi–Li (Å) | 3.41 | 3.28 | 2.82 | 2.67 | 2.59 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of dimethyl ether (DME) and 1,3-dioxolane (DOL). This combination exhibits superior reactivity with polysulfides, ensuring enhanced stability and improved electrochemical performance in Li–S batteries.68,79,80
:1 mixture of dimethyl ether (DME) and 1,3-dioxolane (DOL). This combination exhibits superior reactivity with polysulfides, ensuring enhanced stability and improved electrochemical performance in Li–S batteries.68,79,80The optimized structure of DME and DOL are shown in Fig. 1. DME consists of a central ethane backbone with a methyl (–CH3) group attached to each of the carbon atoms. The calculated C–O–C bond angle after relaxation was measured to be 112.13°, indicating a bent molecular geometry, which is in good agreement with the experimentally calculated angle of approximately 111.43°.81 DOL adopts a puckered five-member ring, where the ring is not perfectly planar but instead exhibits slight deviation from planarity. The bond lengths between the carbon and oxygen atoms in DOL are relatively equal about 2.7 Å.
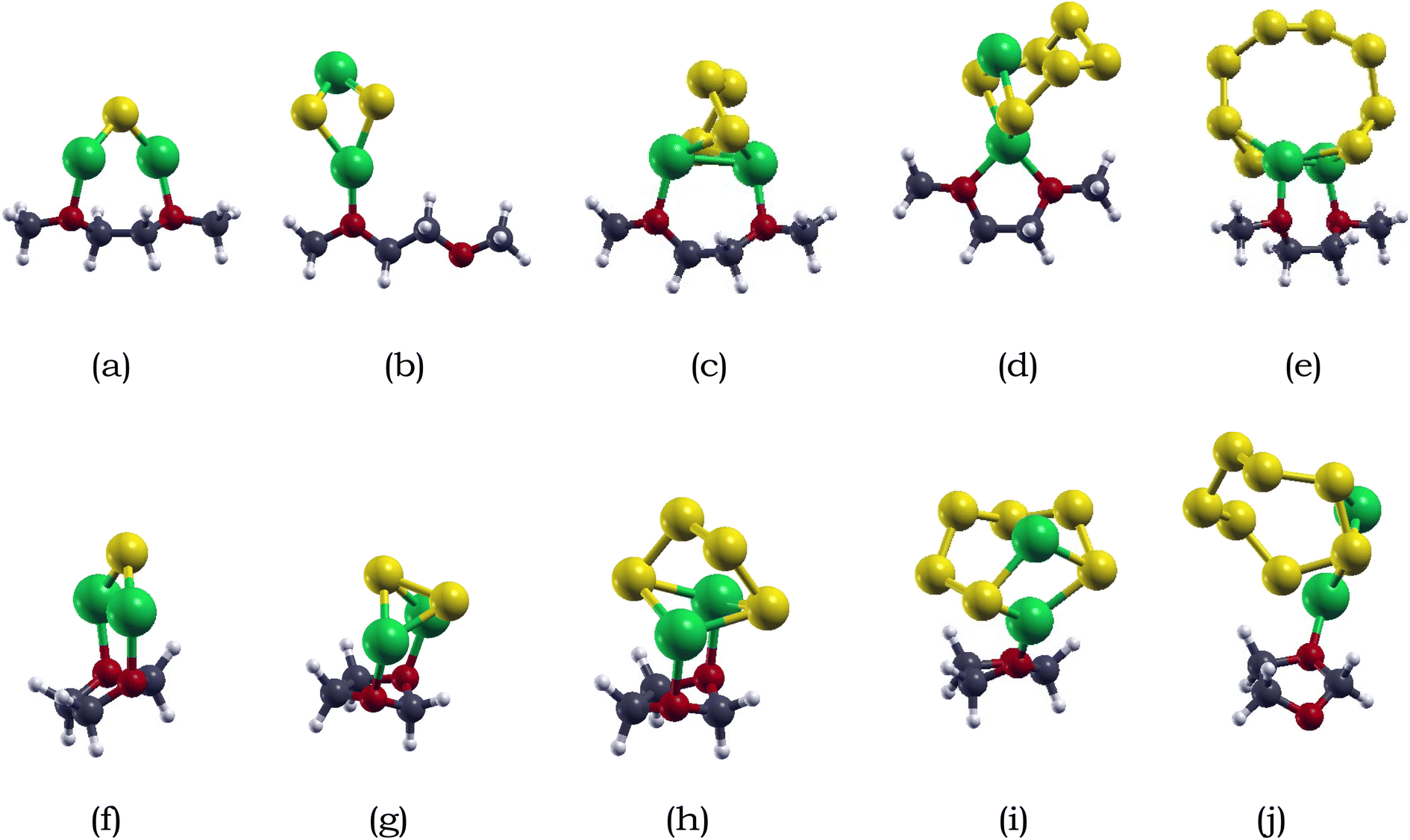 | ||
| Fig. 2 Optimized atomic structures of Li2Sx (x = 1, 2, 4, 6, 8) adsorbed with electrolytes DME/DOL: (a) Li2S + DME, (b) Li2S2 + DME, (c) Li2S4 + DME, (d) Li2S6 + DME, (e) Li2S8 + DME, (f) Li2S + DOL, (g) Li2S2 + DOL, (h) Li2S4 + DOL, (i) Li2S6 + DOL, (j) Li2S8 + DOL. H, O, C, Li, and S atoms are represented by white, red, blue, green and yellow. | ||
The adsorption energies of lithium polysulfides on DME and DOL are summarized in Table 2. The adsorption energies of Li2S, Li2S2, Li2S4, Li2S6, and Li2S8 on DOL are −0.83, −0.94, −0.78, −0.69, and −1.05 eV respectively. For DME, the adsoprtion energies of Li2S, Li2S2, Li2S4, Li2S6, and Li2S8 are −0.94, −0.73, −0.62, −1.05, and −0.77 eV, respectively. The adsorption energies between the lithium polysulfides and electrolytes range of −0.69 to −1.05 eV for DOL, and −0.62 to −1.05 eV for DME, showing that the adsorption of lithium polysulfides on DME and DOL do not significantly differ. Therefore, the preferable range of adsorption energies would be from around −1.00 eV to −2.00 eV, as extreme adsorption can hinder the detachment of adsorbed lithium polysulfide from the anchoring material.82
| Li2S | Li2S2 | Li2S4 | Li2S6 | Li2S8 | |
|---|---|---|---|---|---|
| Ebind-DME (eV) | −0.94 | −0.73 | −0.62 | −1.05 | −0.77 |
| Ebind-DOL (eV) | −0.83 | −0.94 | −0.78 | −0.69 | −1.05 |
B. Adsorption of Li2Sx species on pristine antimonene monolayer
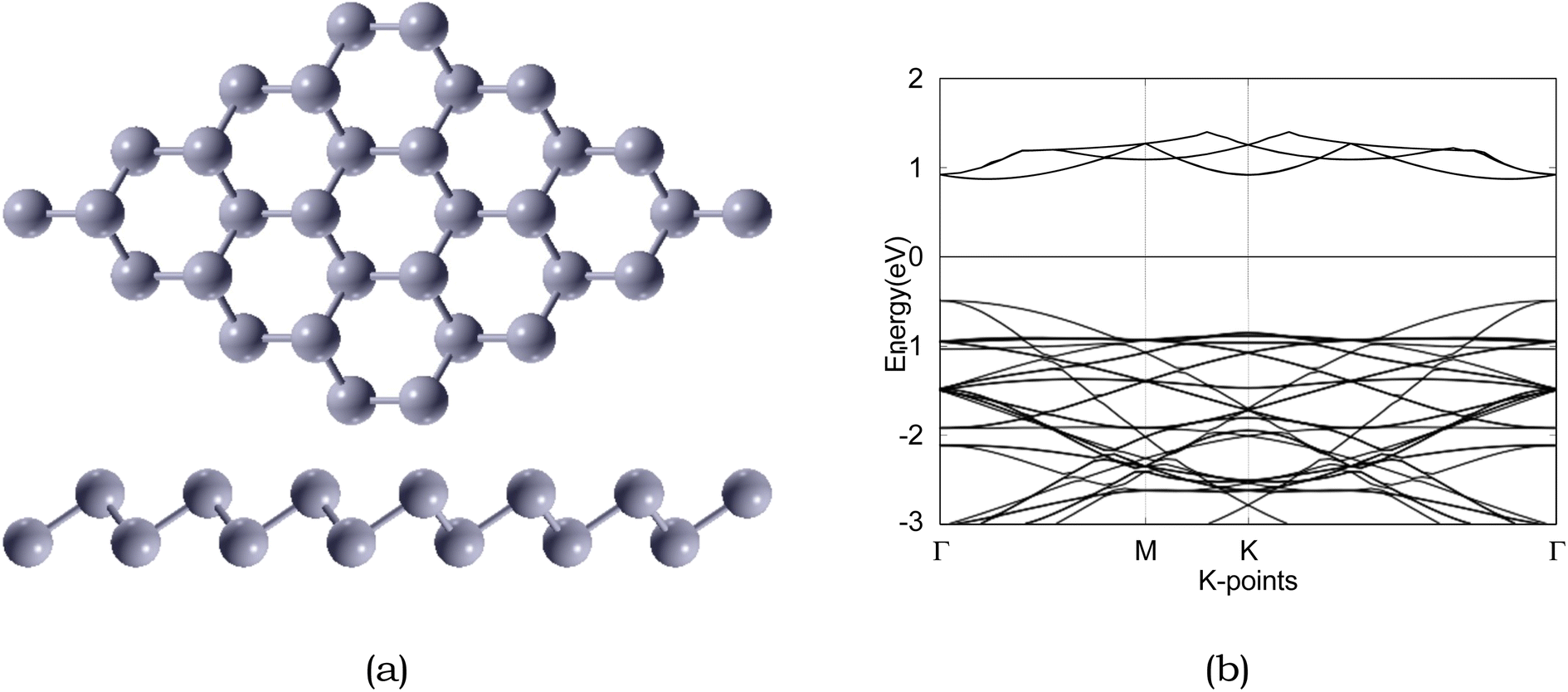 | ||
| Fig. 3 Atomic structure and band structure of 4 × 4 × 1 pristine. The Fermi level is set to 0. Sb is represented by silver, respectively. | ||
Due to the insulating nature of sulfur, an ideal anchoring material for the Li–S battery should possess excellent electronic conductivity, which will greatly affect the performance and operability of the battery. Therefore, we have computed the electronic band structure to understand the electronic properties of antimonene. The monolayer exhibits a band gap of 1.52 eV, as presented in Table 5. The monolayer is an indirect band gap semiconductor between the Γ and M points, as presented in Fig. 3.
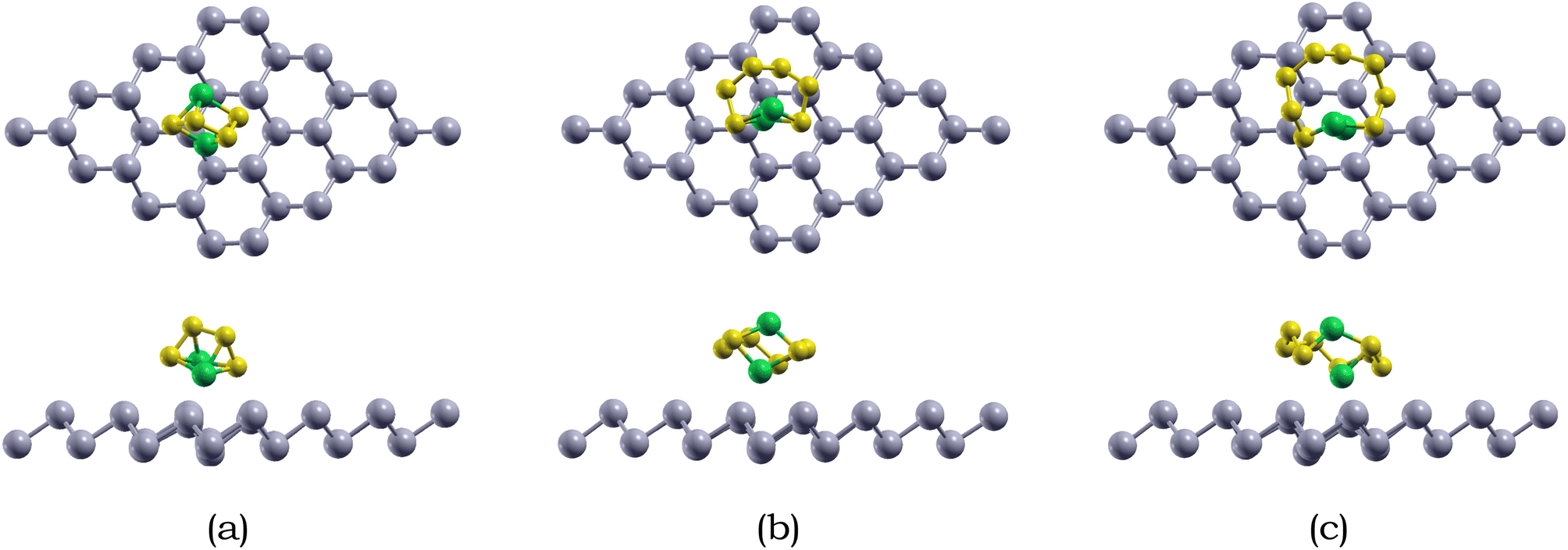 | ||
| Fig. 4 Top and side views of the optimized atomic structures Li2Sx (x = 4, 6, 8) adsorbed on 4 × 4 × 1 pristine antimonene monolayer: (a) Li2S4, (b) Li2S6, and (c) Li2S8. Sb, Li, and S are represented by silver, green, and yellow, respectively. | ||
For Li2S4 adsorption, the Li atoms face downwards towards the monolayer, while for Li2S6 and Li2S8 adsorption, the S chain is parallel to the monolayer surface. The shortest intermolecular distance between Li2S6, Li2S6, and Li2S8 and the antimonene monolayer is 2.93, 2.98, and 3.16 Å, respectively. Specifically, the Li atoms are closer than the S atoms when interacting with the antimonene monolayer, and the two Li atoms prefer to adsorb around the hexagonal edges of antimonene through Li–Sb interactions.
We also calculated the variances of the corresponding structural parameters for both Li2Sx and pristine antimonene as summarized in Table 3. The average change of the Li–S bond in Li2S4, Li2S6, and Li2S8 is 0.04, 0.02, and 0.07 Å. Not only this, for the antimonene monolayer, the nearby Sb–Sb bonds increased only around 0.01 Å. Therefore, little structural deformation is observed for both the adsorbed long-chain lithium polysulfides and the antimonene monolayer, which is preferable as severe deformation away from the stable configuration is unfavorable.83
| Species | dSb–Li (Å) | ΔdLi–S (Å) | ΔdSb–Sb (Å) |
|---|---|---|---|
| Li2S4 | 2.93 | 0.04 | 0.01 |
| Li2S6 | 2.98 | 0.02 | 0.01 |
| Li2S8 | 3.16 | 0.07 | 0.01 |
The adsorption energies of the Li2Sx (x = 4, 6, 8) species on antimonene are listed in Table 4. The adsorption energies of Li2S4, Li2S6, and Li2S8 on the antiomonene monolayer are −0.90, −0.82, and −0.70 eV, respectively. The overall adsorption energies of antimonene to the Li2Sx (x = 4, 6, 8) species range from −0.70 to −0.90 eV, which are similar to previous results.49
C. Adsorption of Li2Sx species on O-doped antimonene monolayer
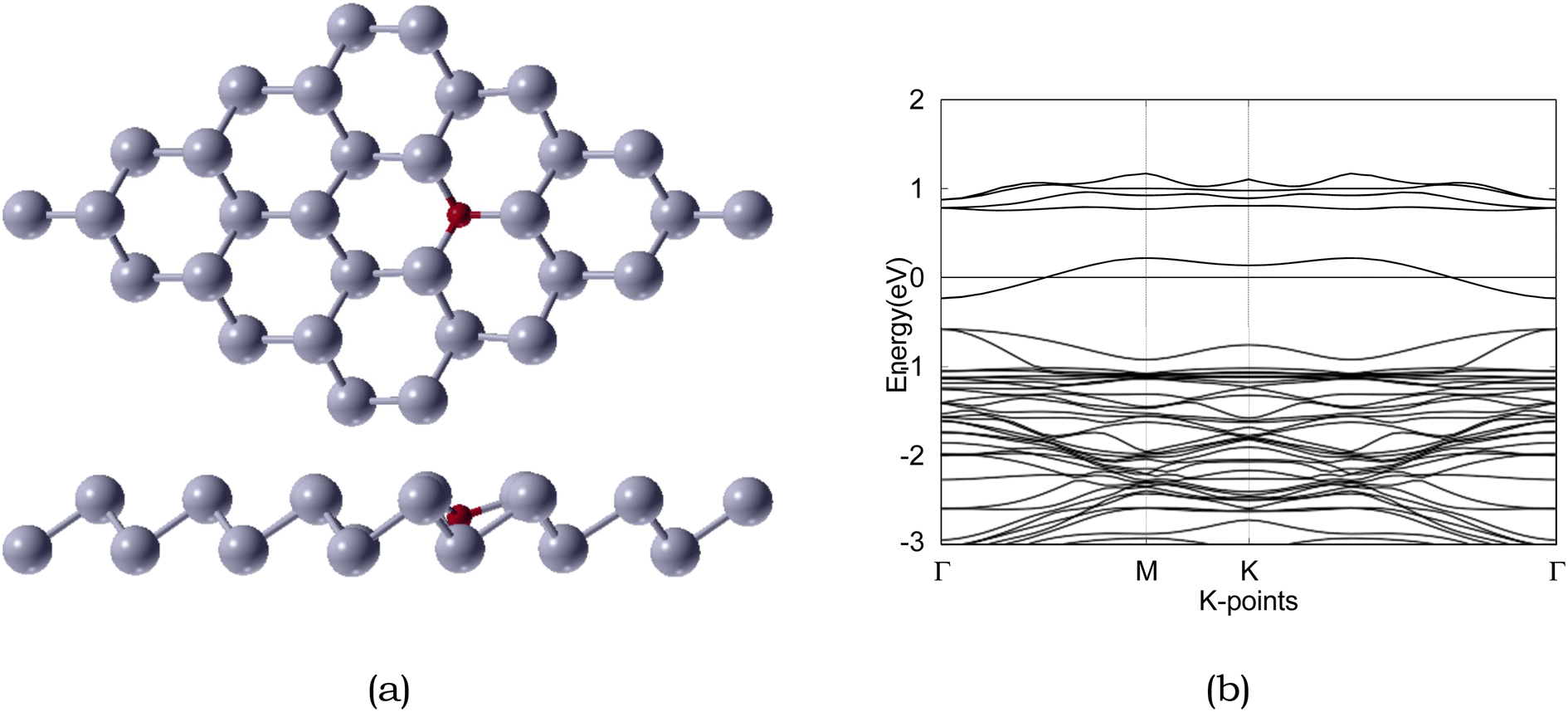 | ||
| Fig. 5 Atomic structure and band structure of 4 × 4 × 1 O-doped antimonene monolayer. The Fermi level is set to 0. Sb and O are represented by silver and red, respectively. | ||
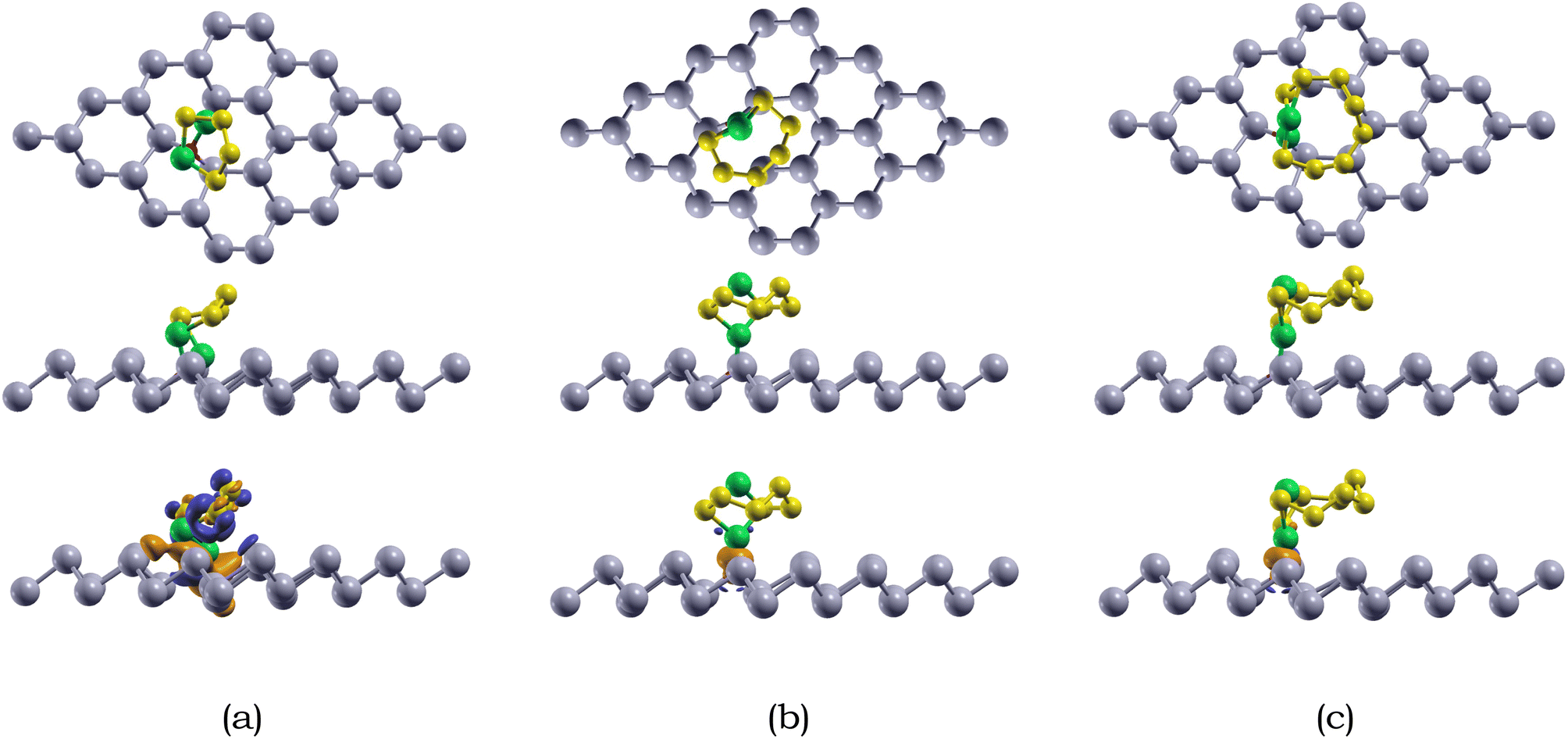 | ||
| Fig. 6 Top view (1st row) and side view (2nd row) of the optimized atomic structure and charge transfer (3rd row) of Li2Sx (x = 4, 6, 8) species on the O-doped antimonene monolayer (OSbML) (a) Li2S4/OSbML (b) Li2S6/OSbML, and (c) Li2S8/OSbML. The orange and dark blue bubbles represent charge accumulation and depletion, respectively. The isosurface is 0.002 e per Å3. Sb, O, Li, and S are represented by silver, red, green, and yellow, respectively. | ||
We also calculated the variances of the corresponding structural parameters for both Li2Sx (x = 4, 6, 8) and the O-doped antimonene monolayer, as summarized in Table 6. The average change of the Li–S bond in Li2S4, Li2S6, and Li2S8 is 0.44, 0.04, and 0.07 Å. Not only this, for the antimonene monolayer, the nearby O–Sb bonds increased around 0.57, 0.43, and 0.47 Å for the adsorption of Li2S4, Li2S6, and Li2S8. The considerable change in the substrate is observed due to the attraction between the Li and O atoms, resulting in the O atom moving away from one of the adjacent Sb atoms to bond with the Li atoms. The variances of the structural parameters of both adsorbates and substrates are considerably larger, indicating that the Li2Sx species experienced some deformation, but are nearly intact.
| Species | dO–Li (Å) | ΔdLi–S (Å) | ΔdSb–O (Å) |
|---|---|---|---|
| Li2S4 | 1.95 | 0.44 | 0.57 |
| Li2S6 | 1.87 | 0.04 | 0.43 |
| Li2S8 | 1.88 | 0.07 | 0.47 |
The adsorption energies of the Li2Sx (x = 4, 6, 8) species on O-doped antimonene are listed in Table 4. The adsorption energies of Li2S4, Li2S6, and Li2S8 on the doped monolayer is −1.24, −1.21, and −1.12 eV, respectively. The overall adsorption energies Li2Sx (x = 4, 6, 8) species adsorbed on O-doped antimonene range from −1.12 to −1.24 eV, stronger than those of pristine antimonene. The higher chemical reactivity between the Li2Sx species and O-doped antimonene than pristine antimonene is due to the electronegativity difference between Li and O.
Moreover, we compared the energy gain for Li2Sx species to form large Li–S interconnected clusters (or networks) and the adsorption energies of the Li2Sx (x = 4, 6, 8) on O-doped antimonene. The energy gain to create interconnected Li2Sx clusters is less than around 0.40 eV for Li2Sx (x = 4, 6, 8),69 which is smaller than the Li2Sx and O-doped anti-monene interactions (−1.24, −1.21, −1.12 eV, respectively). Consequently, the three long-chain lithium polysulfides generally prefer anchoring on O-doped antiomonene than nucleation into larger Li2Sx clusters. In addition, we compared the binding energy for Li2Sx species with DME and DOL and the adsorption energies of Li2Sx (x = 4, 6, 8) on O-doped antimonene. The binding energies of the Li2Sx species with the electrolytes are smaller than those with the O-doped antimonene monolayer. Therefore, it can be seen that the Li2Sx species would prefer to anchor on the O-doped antimonene monolayer and not dissolve in the electrolyte.
Overall, the adsorption energies of the soluble Li2Sx species are moderate (−1.00 to −2.00 eV), the adsorbed Li2Sx species and the O-doped antimonene are nearly intact. Therefore, we expect that O-doped antimonene is suitable as an anchoring material for Li–S batteries.
IV. Conclusion
By using first-principles calculations based on DFT, the adsorption behavior of Li2Sx (x = 4, 6, 8) species on the pristine and O-doped antimonene monolayers was investigated. The Li2Sx species were weakly adsorbed on the pristine antimonene monolayer while mod-erately adsorbed on the O-doped antimonene monolayer. Therefore, the Li2Sx species are adsorbed on the O-doped antimonene monolayer and the dissolution of the Li2Sx species into the electrolyte is prevented from an energetic point of view. Furthermore, the charge transfer from the Li2Sx species to the O-doped antimonene monolayer revealed strong chem–ical interactions between the Li2Sx species and O-doped antimonene monolayer. Therefore, the O-doped antimonene monolayer is a promising anchoring material for high-performance Li–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Geifei Qian for his technical support throughout our research.References
- L. Pérez-Lombard, J. Ortiz and C. Pout, Energy Buildings, 2008, 40, 394 CrossRef.
- K. Karpov, Studies on Russian Economic Development, 2019, 30, 38 CrossRef.
- A. Bloom, U. Helman, H. Holttinen, K. Summers, J. Bakke, G. Brinkman and A. Lopez, IEEE Power Energy Mag., 2017, 15, 22 Search PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854 RSC.
- Y. Wang, B. Liu, Q. Li, S. Cartmell, S. Ferrara, Z. D. Deng and J. Xiao, J. Power Sources, 2015, 286, 330 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419 CrossRef CAS.
- M.-Y. Yan, G. Li, J. Zhang, Y.-F. Tian, Y.-X. Yin, C.-J. Zhang, K.-C. Jiang, Q. Xu, H.-L. Li and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2020, 12, 27202 CrossRef CAS PubMed.
- L. Lu, X. Han, J. Li, J. Hua and M. Ouyang, J. Power Sources, 2013, 226, 272 CrossRef CAS.
- T. C. Wanger, Conserv. Lett., 2011, 4, 202 CrossRef.
- K. Abraham, J. Phys. Chem. Lett., 2015, 6, 830 CrossRef CAS PubMed.
- K. Divya and J. Østergaard, Electr. Power Syst. Res., 2009, 79, 511 CrossRef.
- J. Cho, S. Jeong and Y. Kim, Prog. Energy Combust. Sci., 2015, 48, 84 CrossRef.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125 CrossRef CAS PubMed.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018 RSC.
- J. Sun, T. Wang, Y. Gao, Z. Pan, R. Hu and J. Wang, InfoMat, 2022, 4, e12359 CrossRef CAS.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q.-H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed.
- J. Yan, X. Liu and B. Li, Advanced Science, 2016, 3, 1600101 CrossRef PubMed.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969 CrossRef CAS.
- W. Ren, W. Ma, S. Zhang and B. Tang, Energy Storage Mater., 2019, 23, 707 CrossRef.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 1 CrossRef.
- C. Zhang, Q. He, W. Chu and Y. Zhao, Appl. Surf. Sci., 2020, 534, 147575 CrossRef CAS.
- L. Chen, H. Zhou, C. Fu, Z. Chen, C. Xu and Y. Kuang, Int. J. Hydrogen Energy, 2016, 41, 21850 CrossRef CAS.
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021 CrossRef CAS PubMed.
- Y. Fu, Y.-S. Su and A. Manthiram, J. Electrochem. Soc., 2012, 159, A1420 CrossRef CAS.
- X. Zhao, C. Wang, Z. Li, X. Hu, A. A. Razzaq and Z. Deng, J. Mater. Chem. A, 2021, 9, 19282 RSC.
- X. Liu, J.-Q. Huang, Q. Zhang and L. Mai, Adv. Mater., 2017, 29, 1601759 CrossRef PubMed.
- J. Zheng, J. Tian, D. Wu, M. Gu, W. Xu, C. Wang, F. Gao, M. H. Engelhard, J.-G. Zhang and J. Liu, et al., Nano Lett., 2014, 14, 2345 CrossRef CAS PubMed.
- Y. Hou, H. Mao and L. Xu, Nano Res., 2017, 10, 344 CrossRef CAS.
- J. Xu, T. Lawson, H. Fan, D. Su and G. Wang, Adv. Energy Mater., 2018, 8, 1702607 CrossRef.
- C. Li, Z. Xi, D. Guo, X. Chen and L. Yin, Small, 2018, 14, 1701986 CrossRef PubMed.
- Y. Zhu, S. Wang, Z. Miao, Y. Liu and S.-L. Chou, Small, 2018, 14, 1801987 CrossRef PubMed.
- W. Zhao, L.-C. Xu, R. Li, Y. Guo, Z. Yang, R. Liu and X. Li, Mater. Today Commun., 2022, 30, 103196 CrossRef CAS.
- Y. Wang, Z. Ma, N. Song, T. Zhang, Q. Zhang, D. Yang and F. Wang, Chem. Phys. Lett., 2020, 741, 137121 CrossRef CAS.
- R. Jayan and M. M. Islam, J. Phys. Chem. C, 2020, 124, 27323 CrossRef CAS.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutíerrez, T. F. Heinz, S. S. Hong, J. Huang and A. F. Ismach, et al., ACS Nano, 2013, 7, 2898 CrossRef CAS PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699 CrossRef CAS PubMed.
- D. Chimene, D. L. Alge and A. K. Gaharwar, Adv. Mater., 2015, 27, 7261 CrossRef CAS PubMed.
- H. Xu, Z. Kong, J. Siegenthaler, B. Zheng, Y. Tong, J. Li, T. Schuelke, Q. H. Fan, K. Wang and H. Xu, et al., EcoMat, 2023, 5, e12286 CrossRef CAS.
- Y. Zhao, J. Zhao and Q. Cai, Appl. Surf. Sci., 2018, 440, 889 CrossRef CAS.
- Y. Wang, J. Shen, L.-C. Xu, Z. Yang, R. Li, R. Liu and X. Li, Phys. Chem. Chem. Phys., 2019, 21, 18559 RSC.
- X. Liu, X. Shao, F. Li and M. Zhao, Appl. Surf. Sci., 2018, 455, 522 CrossRef CAS.
- L. Zhang, P. Liang, H.-b. Shu, X.-l. Man, F. Li, J. Huang, Q.-m. Dong and D.-l. Chao, J. Phys. Chem. C, 2017, 121, 15549 CrossRef CAS.
- H. H. Haseeb, Y. Li, S. Ayub, Q. Fang, L. Yu, K. Xu and F. Ma, J. Phys. Chem. C, 2020, 124, 2739 CrossRef CAS.
- X. Mao, L. Zhu and A. Fu, Int. J. Quantum Chem., 2021, 121, e26661 CrossRef CAS.
- N. Yamsang, J. Sittiwong, P. Srifa, B. Boekfa, M. Sawangphruk, T. Maihom and J. Limtrakul, Appl. Surf. Sci., 2021, 565, 150378 CrossRef CAS.
- G. Wang, R. Pandey and S. P. Karna, ACS Appl. Mater. Interfaces, 2015, 7, 11490 CrossRef CAS PubMed.
- G. S. Yi, E. S. Sim and Y.-C. Chung, Phys. Chem. Chem. Phys., 2017, 19, 28189 RSC.
- H. Lin, R. Jin, A. Wang, S. Zhu and H. Li, Ceram. Int., 2019, 45, 17996 CrossRef CAS.
- D. Singh, S. K. Gupta, T. Hussain, Y. Sonvane, P. Gajjar and R. Ahuja, Energy Fuels, 2021, 35, 9001 CrossRef CAS.
- S. Zhang, M. Xie, F. Li, Z. Yan, Y. Li, E. Kan, W. Liu, Z. Chen and H. Zeng, Angew. Chem., 2016, 128, 1698 CrossRef.
- X. Wang, J. Song and J. Qu, Angew. Chem., Int. Ed., 2019, 58, 1574 CrossRef CAS PubMed.
- D. R. Kripalani, A. A. Kistanov, Y. Cai, M. Xue and K. Zhou, Phys. Rev. B, 2018, 98, 085410 CrossRef CAS.
- J. Su, T. Duan, W. Li, B. Xiao, G. Zhou, Y. Pei and X. Wang, Appl. Surf. Sci., 2018, 462, 270 CrossRef CAS.
- S. Upadhyay and P. Srivastava, Mater. Chem. Phys., 2020, 241, 122381 CrossRef CAS.
- L. Zhang, P. Liang, H. B. Shu, X. L. Man, X. Q. Du, D. L. Chao, Z. G. Liu, Y. P. Sun, H. Z. Wan and H. Wang, J. Colloid Interface Sci., 2018, 529, 426 CrossRef CAS PubMed.
- L. Zhang, W. Zhao, S. Yuan, F. Jiang, X. Chen, Y. Yang, P. Ge, W. Sun and X. Ji, J. Energy Chem., 2021, 60, 531 CrossRef CAS.
- L. Ding, Q. Lu, A. D. C. Permana, S. Oswald, M. Hantusch, K. Nielsch and D. Mikhailova, Energy Technol., 2021, 9, 2001057 CrossRef CAS.
- F. Hu, H. Peng, T. Zhang, W. Shao, S. Liu, J. Wang, C. Wang and X. Jian, J. Energy Chem., 2021, 58, 115 CrossRef CAS.
- J. Zhao, Y. Yang, R. S. Katiyar and Z. Chen, J. Mater. Chem. A, 2016, 4, 6124 RSC.
- X. Gonze, B. Amadon, P.-M. Anglade, J.-M. Beuken, F. Bottin, P. Boulanger, F. Bruneval, D. Caliste, R. Caracas and M. Cote, et al., Comput. Phys. Commun., 2009, 180, 2582 CrossRef CAS.
- P. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- N. A. W. Holzwarth, A. R. Tackett and G. E. Matthews, Comput. Phys. Commun., 2001, 135, 329 CrossRef CAS.
- C. Liu and X. Luo, J. Mater. Chem. B, 2021, 9, 2736 RSC.
- J. D. Head and M. C. Zerner, Chem. Phys. Lett., 1985, 122, 264 CrossRef CAS.
- W. Lee and X. Yao, Comput. Mater. Sci., 2015, 106, 76 CrossRef CAS.
- B. Zhang, H. Zhang, J. Lin and X. Cheng, Phys. Chem. Chem. Phys., 2018, 20, 30257 RSC.
- A. Zhang and X. Luo, Mater. Adv., 2022, 3, 5845 RSC.
- M. R. Kaiser, S. Chou, H.-K. Liu, S.-X. Dou, C. Wang and J. Wang, Adv. Mater., 2017, 29, 1700449 CrossRef PubMed.
- B. Wang, S. M. Alhassan and S. T. Pantelides, Phys. Rev. Appl., 2014, 2, 034004 CrossRef CAS.
- C. Barchasz, F. Molton, C. Duboc, J.-C. Leprêtre, S. Patoux and F. Alloin, Anal. Chem., 2012, 84, 3973 CrossRef CAS PubMed.
- Q. Zhang, X. Zhang, Y. Xiao, C. Li, H. H. Tan, J. Liu and Y. Wu, ACS Omega, 2020, 5, 29272 CrossRef CAS PubMed.
- T.-T. Yu, P.-F. Gao, Y. Zhang and S.-L. Zhang, Appl. Surf. Sci., 2019, 486, 281 CrossRef CAS.
- T. Li, C. He and W. Zhang, J. Mater. Chem. A, 2019, 7, 4134 RSC.
- L.-C. Yin, J. Liang, G.-M. Zhou, F. Li, R. Saito and H.-M. Cheng, Nano Energy, 2016, 25, 203 CrossRef CAS.
- X. Liu, N. Xu, T. Qian, J. Liu, X. Shen and C. Yan, Nano Energy, 2017, 41, 758 CrossRef CAS.
- V. Kolosnitsyn and E. Karaseva, Russ. J. Electrochem., 2008, 44, 506 CrossRef CAS.
- W. Wang, Y. Wang, Y. Huang, C. Huang, Z. Yu, H. Zhang, A. Wang and K. Yuan, J. Appl. Electrochem., 2010, 40, 321 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89, 206 CrossRef CAS.
- R. Rauh, K. Abraham, G. Pearson, J. Surprenant and S. Brummer, J. Electrochem. Soc., 1979, 126, 523 CrossRef CAS.
- L. Lodovico, PhD thesis, dissertation, Karlsruher Institut für Technologie (KIT), Karlsruhe, 2019.
- U. Blukis, P. H. Kasai and R. J. Myers, J. Chem. Phys., 1963, 38, 2753 CrossRef CAS.
- Q. Zhang, Y. Wang, Z. W. Seh, Z. Fu, R. Zhang and Y. Cui, Nano Lett., 2015, 15, 3780 CrossRef CAS PubMed.
- R. A. Jarrin, et al., PhD thesis, Johns Hopkins University, 2023.
- T.-Z. Hou, X. Chen, H.-J. Peng, J.-Q. Huang, B.-Q. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283 CrossRef CAS PubMed.
- C. Tang, H.-F. Wang, X. Chen, B.-Q. Li, T.-Z. Hou, B. Zhang, Q. Zhang, M.-M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845 CrossRef CAS PubMed.
- K. C. Wasalathilake, M. Roknuzzaman, K. K. Ostrikov, G. A. Ayoko and C. Yan, RSC Adv., 2018, 8, 2271 RSC.
| This journal is © The Royal Society of Chemistry 2023 |