
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multicomponent synthesis of pyrido[2,3-b]pyrazine derivatives: electrochemical DNA sensing, nonlinear optical properties and biological activity†
Muhammad Rashida,
Muhammad Khalid*bc,
Abida Ashrafad,
Tahira Saleema,
Iqra Shafiqbc,
Muhammad Azeem Shakila,
Briha Zainaba,
Attalla F. El-kottef,
Muhammad Yaqub*a and
Zahid Shafiq *a
*a
aInstitute of Chemical Sciences, Bahauddin Zakariya University, Multan 60800, Pakistan. E-mail: mayaqub2@yahoo.com; zahidshafiq@bzu.edu.pk; Tel: +92-3006559811
bInstitute of Chemistry, Khwaja Fareed University of Engineering & Information Technology, Rahim Yar Khan, 64200, Pakistan. E-mail: muhammad.khalid@kfueit.edu.pk
cCentre for Theoretical and Computational Research, Khwaja Fareed University of Engineering & Information Technology, Rahim Yar Khan, 64200, Pakistan
dDepartment of Chemistry, Govt. Graduate College Shah Rukne-Alam Multan, Pakistan
eDepartment of Biology, College of Science, King Khalid University, Abha, Saudi Arabia
fDepartment of Zoology, College of Science, Damanhour University, Egypt
First published on 1st November 2023
Abstract
We synthesized novel pyrido[2,3-b]pyrazin based heterocyclic compounds (4–7) and their chemical structures were ascertained by spectral techniques (NMR, FT-IR). Besides experimental investigation, density functional theory (DFT) computations with B3LYP/6-31G(d,p) level of theory were executed to obtain spectroscopic and electronic properties. Nonlinear optical (NLO) properties, frontier molecular orbitals (FMOs), UV-visible, vibrational analysis, natural bond orbitals (NBOs), transition density matrix (TDM) and density of states (DOS) analyses of molecules (4–7) were accomplished at B3LYP/6-31G (d,p) level. Global reactivity parameters (GRPs) were correlated with the band gap (Egap) values; compound 7 with lower Egap (3.444 eV), exhibited smaller value of hardness (1.722 eV) with greater softness value (0.290 eV−1). The dipole moment (μ), average polarizability 〈α〉, first (βtot) and second 〈γ〉 hyper-polarizabilities were calculated for compounds (4–7). Compound 7 showed less Egap, highest absorption wavelength and remarkable NLO response. The highest 〈α〉, βtot and 〈γ〉 values for compound 7 were observed as 3.90 × 10−23, 15.6 × 10−30 and 6.63 × 10−35 esu, respectively. High NLO response revealed that pyrido[2,3-b]pyrazin based heterocyclic compounds had very remarkable contributions towards NLO technological applications. Further compounds (4–7) are utilized for the first time in electrochemical sensing of DNA, in vitro antioxidant and antiurease activity.
1. Introduction
A type of sensor uses DNA as a recognition element to detect specific molecules or analytes in a sample is called DNA electrochemical biosensors.1,2 The sensor is typically made of a conductive material that is coated with a layer of DNA or other biomolecules.3 When the DNA in the sample encounters the sensor, it can bind to the DNA on the sensor surface, causing a change in the electrical properties of the sensor. This change can be measured and used to determine the presence or concentration of the DNA in the sample. Several types of DNA electrochemical biosensors exist, including hybridization-based sensors, DNA-enzyme sensors, and DNA-modified electrode sensors.A label-free electrochemical DNA biosensor uses an electrochemical sensor to detect and measure the presence of DNA in a sample without the need for a detectable label.4 It offers several advantages over traditional DNA biosensors, including high sensitivity, specificity, and the ability to detect small amounts of DNA. They are also relatively simple and inexpensive to manufacture and operate, making them an attractive option for many applications.5 One of the most promising applications of DNA electrochemical biosensors is in the field of medical diagnosis.6 These sensors have the potential to quickly and accurately detect the presence of disease-associated biomarkers,7 such as proteins or nucleic acids, in body fluids. This could potentially allow for the early detection of diseases, such as cancer,8 infectious diseases, and genetic disorders, leading to more timely and effective treatment.9 In addition to their medical applications, DNA electrochemical biosensors also have potential uses in environmental monitoring for example, these sensors could be used to detect the presence of harmful chemicals or contaminants in water, soil, or air.10
DNA electrochemical biosensors can be used to detect and measure the presence of DNA from pathogens or other contaminants in food products. This can assist to ensure the quality and safety of the food supply.11 DNA electrochemical biosensors can be used in forensic science to identify and quantify DNA from crime scenes or other forensic samples.12 Biosecurity is another field in which DNA electrochemical biosensors can be used to detect specific microorganisms or toxins that could be used in bioterrorism or other forms of sabotage.13
DNA electrochemical biosensors have revolutionized the field of agriculture. This can be used to monitor crop health and assess the effectiveness of agricultural practices.14 One of the main challenges in designing DNA electrochemical biosensors is the stability of the DNA recognition element. Researchers are working on ways to stabilize the DNA recognition element to improve the performance and longevity of DNA electrochemical biosensors.15 In conclusion, DNA electrochemical biosensors offer a sensitive, specific, and cost-effective means of detecting and measuring DNA in a wide range of applications.16–19 Out of different techniques employed for DNA biosensing, cyclic voltammetry is frequently used to detect the presence of specific DNA sequences or to measure the concentration of DNA in a sample.20,21 Cyclic voltammetry (CV) is an electrochemical analysing technique that includes applying a voltage to an electrode and measuring the resulting current. CV has several advantages over other DNA detection methods, including the capacity to detect extremely low DNA concentrations, great sensitivity to small fluctuations in DNA concentration, and simultaneous detection of different DNA sequences. Furthermore, it is relatively simple and does not require expensive or specialized equipment.22
The heterocyclic compounds with a pyrido[2,3-b]pyrazine core have been shown in the scientific literature to exhibit a wide range of biological applications, including selective inhibition of PI3K isozymes and, for treating myocardial infarction.23 They have also been explored as dipeptidyl peptidase IV inhibitors for treatment of type 2 diabetes mellitus,24 inhibitors of Mycobacterium tuberculosis growth25,26 antagonists for TRPV1 in pain management,27 fungicides,28 inhibitors of the Wnt/b-catenin pathway,29 inhibitors of PKB,30 ALK inhibitors,31 antagonists for the human neurokinin-3 (hNK-3) receptor and GnRH antagonist, as well as inhibitors of p38aMAP kinase and BRAF in cancer treatment.31,32 DFT calculations have attracted significant attention to predict structural parameters of novel synthesized compounds (4–7) such as non-covalent interactions, electronic properties, chemical reactivity, and stability. These results propose that the compounds under consideration have potential NLO uses. So overall, pyrido[2,3-b]pyrazine based compounds have shown potentials in various fields, including display technology, pharmaceuticals, and materials science.
Keeping in view the importance of DNA electrochemical biosensing and material sciences in various fields, our research group endeavors to report derivatives of indeno[2′,1′:5,6]pyrido[2,3-b]pyrazin (4–7), and anticipating that these molecules may acquire electrochemical DNA sensing and non-linear properties which have not yet been reported in the literature. The interaction between DNA and synthesized compounds is the fundamental biological tool for improving the therapeutic uses of organic compounds and we for the first time, determine application of compounds (4–7) in in vitro antioxidant and antiurease activity.
2. Experimental section
2.1 Materials and methods
Substituted aromatic aldehydes, 1,3-Indane dione, 2-aminopyrazine, p-TSA, salmon sperm DNA (SS-DNA), and different solvents were acquired from Merck/Sigma Aldrich and used as received. The melting points of synthesized compounds with fused ring heterocyclic structures were determined using an electro-thermal Gallenkamp apparatus. Thin layer chromatography (TLC) on a Merck aluminium sheet with coated silica gel was used to monitor the progress of the synthesis. The TLC plate was visualized using locating agents such as UV light with a wavelength of 366/254 nm or iodine. The FT-IR spectrometer was used to measure infrared spectra, and the 400 MHz Brauker Avance III HD 1H-NMR spectrometer was used to record 1H-NMR spectra, the solvent used was DMSO-d6 and internal standard was TMS. Chemical shift and coupling constant values were measured in ppm and Hz, respectively. The redox activity of DNA was measured using cyclic voltammetry (AUT50296, Autolab Potentiostat) in an electrochemical biosensor with a three-electrode system in a glass cell of 100 mL having a platinum wire, an Ag/AgCl reference electrode, and a glassy carbon electrode (GCE) with some modification along with ss-DNA as the working electrode. The working electrode passed through a cleaning process involving sonication, soap washing, triple rinsing with distilled water, and drying in an oven. A paste was prepared by blending our synthesized heterocyclic compound and ss-DNA and the in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (w/w) in 2 mL of ethanol. Solutions of varying concentrations were prepared in PBS (pH 7), and all the readings were taken at room temperature.
:1 ratio (w/w) in 2 mL of ethanol. Solutions of varying concentrations were prepared in PBS (pH 7), and all the readings were taken at room temperature.
2.2 Computational procedure
To investigate the electronic properties, absorption spectra, and NLO characteristics of the heterocyclic compounds, we conducted DFT simulations. In a recent study, we performed quantum chemical analyses of compounds 4 to 7 at the B3LYP/6-31G(d,p) level using Gaussian 09 software packages.33 We employed the same level and basis set in conjunction with the Gaussian 09 embedded NBO 3.1 program package to examine natural bond orbitals (NBO) and natural populations (NPA). Similarly, we utilized time-dependent density functional theory (TD-DFT) at the B3LYP/6-31G(d,p) level for the frontier molecular orbital (FMO) analysis. GaussView 5.0 was employed for organizing input files.34 Additionally, Gauss View 5.0, Avogadro35 and Chemcraft36 were utilized for the elucidation of outcomes. Dipole moment was calculated by eqn (1).37| μ = (μx2 + μy2 + μz2)1/2 | (1) |
Average polarizability was calculated with eqn (2).38
| 〈α〉 = (axx + ayy + azz)/3 | (2) |
Gaussian outcome analysis file contains ten hyper-polarizability tensors oriented in the x, y, and z directions: βxxx, βxyy, βxzz, βyyy, βxxy, βyzz, βzzz, βxxz, βyyz, βxyz. First hyper-polarizability (βtot) is calculated with the help of eqn (3).39
| βtot= (βx2 + βy2 + βz2)1/2 | (3) |
The second hyper-polarizability was calculated with eqn (4).38
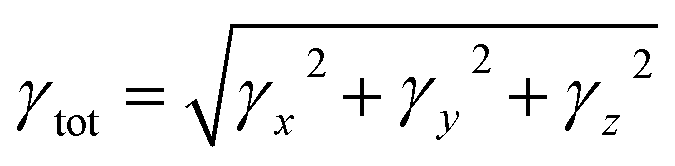 | (4) |
2.3 Electrochemical DNA sensing
2.4 Biological activity assay
| DPPH˙ + RH → DPPH + R˙ |
1 mg of a chemical compound was solubilized in 1 mL of ethanol to generate initial stock solutions. Subsequently, this substance was dissolved in 3 mL of ethanol, resulting in solutions with varying concentrations of 10, 50, and 100 g. Just before the UV measurements, a DPPH solution in ethanol was prepared. Following that, 3 mL of the sample and 1 mL of the DPPH solution were mixed and left in the dark for 30 minutes at room temperature before measuring the decrease in absorption. A blank sample containing the same amount of ethanol and DPPH solution was used to test the control absorption. Ascorbic acid was employed as the reference. The experiment was done three times. The scavenging activity increased as the concentration of the test samples increases.
The radical scavenging activity was calculated by the following formula:
| % Inhibition = [Ab − Aa/Ab] × 100 |
Percentage inhibition was determined using the following formula:
| % Inhibition = [Control OD − (Sample OD/Control OD)] × 100 |
In the realm of scientific investigation, thiourea functions as a benchmark inhibitor. The determination of the IC50 values entails subjecting both standard and synthesized compounds to varying concentrations under identical experimental conditions.43,44
2.5 Synthetic procedure
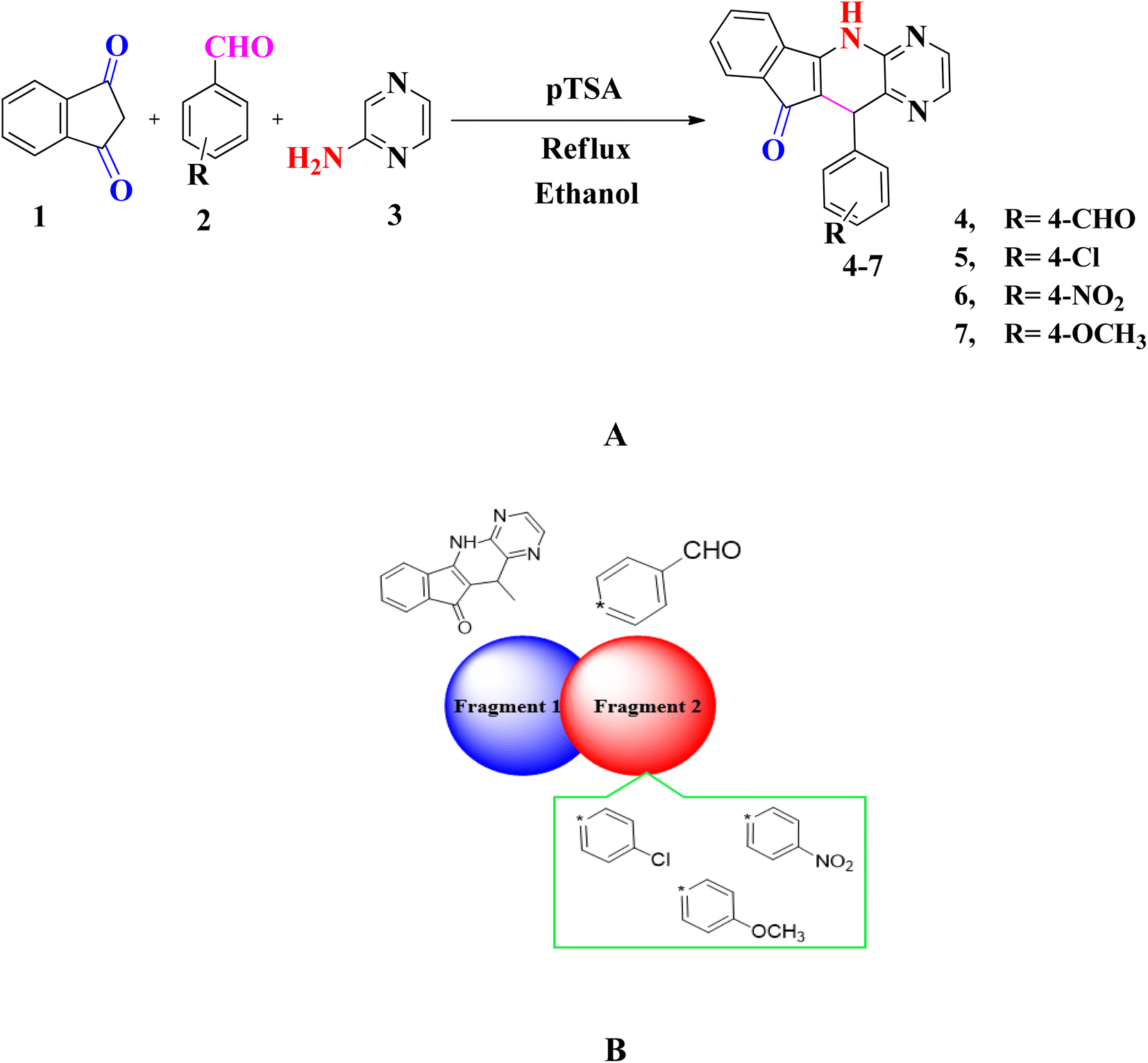 | ||
| Scheme 1 (A) Synthesis of indeno[2′,1′:5,6]pyrido[2,3-b]pyrazin (4–7) (B) sketch map of studied compounds (4–7). | ||
3. Results and discussion
3.1 Chemistry
A series of substituted pyrido[2,3-b]pyrazine based heterocyclic compounds (4–7) have been synthesized to explore the DNA electrochemical sensing, NLO properties and biological screening.These compounds are prepared by reacting a mixture of indane 1,3 dione (1), substituted aromatic aldehyde (2) and 2-aminopyrazine (3) with 20 mol% p-TSA in ethanol. The optimization of reaction conditions is done for model compound 7 by treating 4-methoxybenzaldehyde, 2-aminopyrazine and indane 1,3 dione in equimolar amounts by using solvents of different polarity i.e. H2O, ethanol, DCM, THF, CH3CN and DMF. Furthermore, the reaction is also carried out in the presence and absence of catalyst. Thus after several attempts the reaction conditions defined to obtain pyrido[2,3-b]pyrazine based heterocyclic compounds (4–7) is shown in entry 8 (20 mol% p-TSA, 9 h) with yield of 89% (Table S1†). The substrate scope of reaction is extended vide reacting indane 1,3 dione (1), different aromatic aldehydes (2) and 2-aminopyrazine (3). The targeted compounds (4–7) are obtained in good to excellent yield (82–89%) (Table S2†) (Scheme 1A).
The structure of novel pyrido[2,3-b]pyrazine based heterocyclic compounds (4–7) are established by using FTIR, NMR (1H & 13C) and CHN analysis. The NH stretching in FTIR appear in the range of 3193–3459 cm−1 while the peaks in range of 1566–1661 cm−1 showed the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group. The 1H NMR spectra of compounds (4–7) show the presence of singlet for (CH) non-aromatic proton in the range of 5.46–5.64 ppm while the singlet peak in the range of 8.95–9.62 ppm unveiled the presence of NH proton. Total count of proton in aromatic region also justifies the structural requirement of compounds. The 13C NMR spectra of compounds (4–7) supports the FTIR and 1H NMR data. The proposed mechanism of reaction is shown in Scheme S1a.†
O group. The 1H NMR spectra of compounds (4–7) show the presence of singlet for (CH) non-aromatic proton in the range of 5.46–5.64 ppm while the singlet peak in the range of 8.95–9.62 ppm unveiled the presence of NH proton. Total count of proton in aromatic region also justifies the structural requirement of compounds. The 13C NMR spectra of compounds (4–7) supports the FTIR and 1H NMR data. The proposed mechanism of reaction is shown in Scheme S1a.†
3.2 Electrochemical DNA sensing study
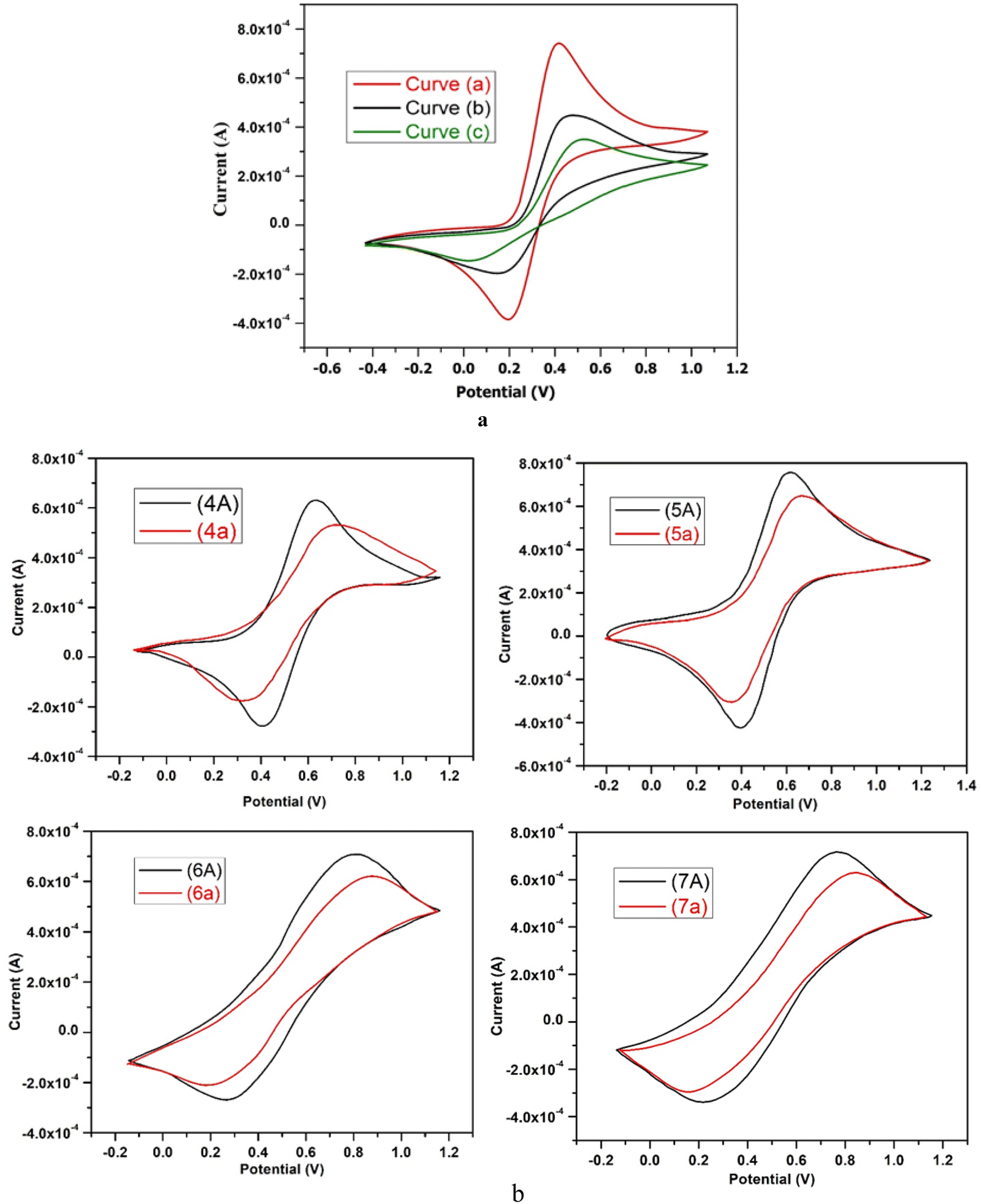 | ||
| Fig. 1 (a) CV curves of (a) glassy carbon electrode (GCE) (b) bare electrode and (c) after immobilization of ss-DNA on glassy carbon electrode (GCE) in 0.1 M PBS (pH 7), 0.1 M KCl, 0.001 M K3Fe(CN)6, and scan rate 100V s−1 with scanning range from −0.7 V to 1.2 V. (b) CV curves in the absence of ss-DNA (curve, 7A) and in the presence of 10 mg L−1 of ss-DNA (curve, 7a) in 0.1 M PBS (pH 7), 0.1 M KCl, 0.001 M K3Fe(CN)6, and scan rate 100V s−1, scanning range from −0.3 V to 1.3 V. | ||
When ss-DNA is immobilized on the surface of a glassy carbon electrode (GCE), it causes a decrease in the electrochemical reversibility of the Fe(CN)63−/Fe (CN)64− redox couple, and an increase is shown by ΔEp to 470 mV (curve “c” in Fig. 1a). Further, an increase in charge transfer resistance and a decrease in the peak current of the redox probe [Fe(CN)6]3−/4− occur as a result of contact between the amine groups of ss-DNA bases and the carboxylic groups of the modified GCE surface.46 Additionally, the immobilization of ss-DNA on the GCE surface results in an enhancement of the specific surface area and electrical conductivity of the sensing platform, leading to a lower peak potential for the electrooxidation of guanine residues in ssDNA.47 Due to electrostatic repulsion, the phosphate groups which are negatively charged on the ss-DNA may inhibit the Fe(CN)63−/Fe (CN)64− redox pair from accessing the electrode surface. The difference in CV profiles between both electrodes can be exploited in this investigation to detect ss-DNA immobilisation on the electrode surface.41
We found two distinct anodic and cathodic peaks with peak potentials of 505 mV and 400 mV, respectively, when using cyclic voltammetry to analyse the results of our synthesised heterocyclic compound 5. The black coloured curves 5A in Fig. 1b depict this peak-to-peak potential separation (Ep), which was 105 mV. When ss-DNA was added, the current of both peaks reduced. Peak-to-peak potential increased, and anodic and cathodic peak potentials both were improved towards more positive and more negative values, respectively. The spacing between these peak-to-peak potentials (Ep) has now increased to 290 mV, as illustrated by curve 5a in Fig. 1b, and the anodic and cathodic peaks potentials are now 650 mV and 360 mV, respectively.
When discussing the cyclic voltammetry data of our synthesised compound 6, we found two distinct anodic and cathodic peaks with peak potentials of 800 mV and 290 mV, respectively, in the absence of ss-DNA. The black coloured curves 6A in Fig. 1b reflect this peak-to-peak potential separation (Ep), which was 510 mV. With the addition of ss-DNA, the current of both peaks reduced. Along with an increase in peak-to-peak potential, the anodic and cathodic peak potentials were both increased towards more positive and more negative values, respectively. As indicated by curve 6a in Fig. 1b, the anodic and cathodic peaks potentials now change to 870 mV and 210 mV, respectively, with an increased peak-to-peak potential separation (Ep) of 660 mV.
The effect of ss-DNA on the electrochemical behavior of (synthesized compound, 7) in 0.1 M PBS (pH 7) was examined using cyclic voltammetry and the results are shown in Fig. 1b. While talking about the cyclic voltammetry results of our synthesized compound 7, in the absence of ss-DNA we noticed two distinct anodic and cathodic peaks with peak potential of 780 mV and 210 mV respectively. The separation between these peak-to-peak potentials (ΔEp) was 570 mV, represented by the black colored curves 7A, in Fig. 1b. The current of both peaks decreased upon the addition of ss-DNA. Anodic and cathodic peak potentials both were enhanced towards more positive and more negative values, respectively along with increase of peak-to-peak potential. Now the anodic and cathodic peaks potentials become 820 mV and 170 mV respectively, with the separation between these peak-to-peak potentials (ΔEp) increased to 650 mV, as shown by curve 7a in Fig. 1b.
The formation of a DNA-bound complex with synthesized compounds lead to a decrease in the concentration of unbound synthesized compound and thus a drop in the anodic current, is observed due to the presence of ss-DNA.48 The decrease in anodic current is mainly because of the lower rate of diffusion of the DNA-bound species and the decrease in the concentration of unbound synthesized compound as the synthesized compound–DNA complex formed upon addition of DNA. Interaction with ss-DNA also resulted in a shift of the peak potential to a more positive value. According to literature a rise in peak potential suggests an interaction in intercalative mode.49 So, while observing the positive shift in the peak potential of the synthesized compound, it is suggested that the synthesized compound is intercalating into the DNA double helix. Next the decrease in peak current of the synthesized compound after the addition of ss-DNA is likely caused by the intercalation of the synthesized compound into the large, slowly diffusing DNA, which results in a significant decreasing in the apparent diffusion coefficient, as per observations.40
The term “strong interactions with ss-DNA for biosensor applications” refers to a chemical or material's strong affinity for binding to ss-DNA and its possible effects in the field of biosensor technology. These implications include a number of important points.
Firstly, these interactions can increase the sensitivity of biosensors, allowing for the detection and measurement of specific DNA sequences or chemicals even at low concentrations.50 Secondly, strong interactions can improve the selectivity of biosensors, reducing the likelihood of false positives or false negatives by enhancing the ability to differentiate between different DNA sequences or targets.51 Additionally, strong interactions can lead to faster response times in biosensors, enabling quick and accurate detection of biological analytes or modifications to DNA structure.52 Finally, biosensors with strong interactions with ss-DNA may find a wider range of applications in industries such as biotechnology, environmental monitoring, and medical diagnostics, due to their enhanced performance features.53
3.3 Computational analysis
In this research paper, configuration of investigated compounds (4–7) has two different portions: fragment 1 and fragment 2. In compounds 4, 5 as well as 6 and 7 (5,11-dihydro-10H-indeno[2′,1′:5,6]pyrido[2,3-b]pyrazin-10-one) acts as a fragment 1 whereas terephthalaldehyde, 4-chlorobenzaldehyde, 4-nitrobenzaldehyde and 4-methoxybenzaldehyde respectively act as a fragment 2. In the context of examined molecular entities, the regions responsible for charge transfer phenomena are depicted in Scheme 1B, whereas the structural representations of the investigated compounds, as generated using ChemDraw, are elucidated in Fig. 2a. Furthermore, their optimized structures are epitomized in Fig. 2b. DFT and TDDFT calculations were performed to investigate the impact of two distinct molecular fragments on their NLO characteristics and the energy gap between molecular orbitals. This study is expected to provide valuable insights for the development of NLO materials with superior performance.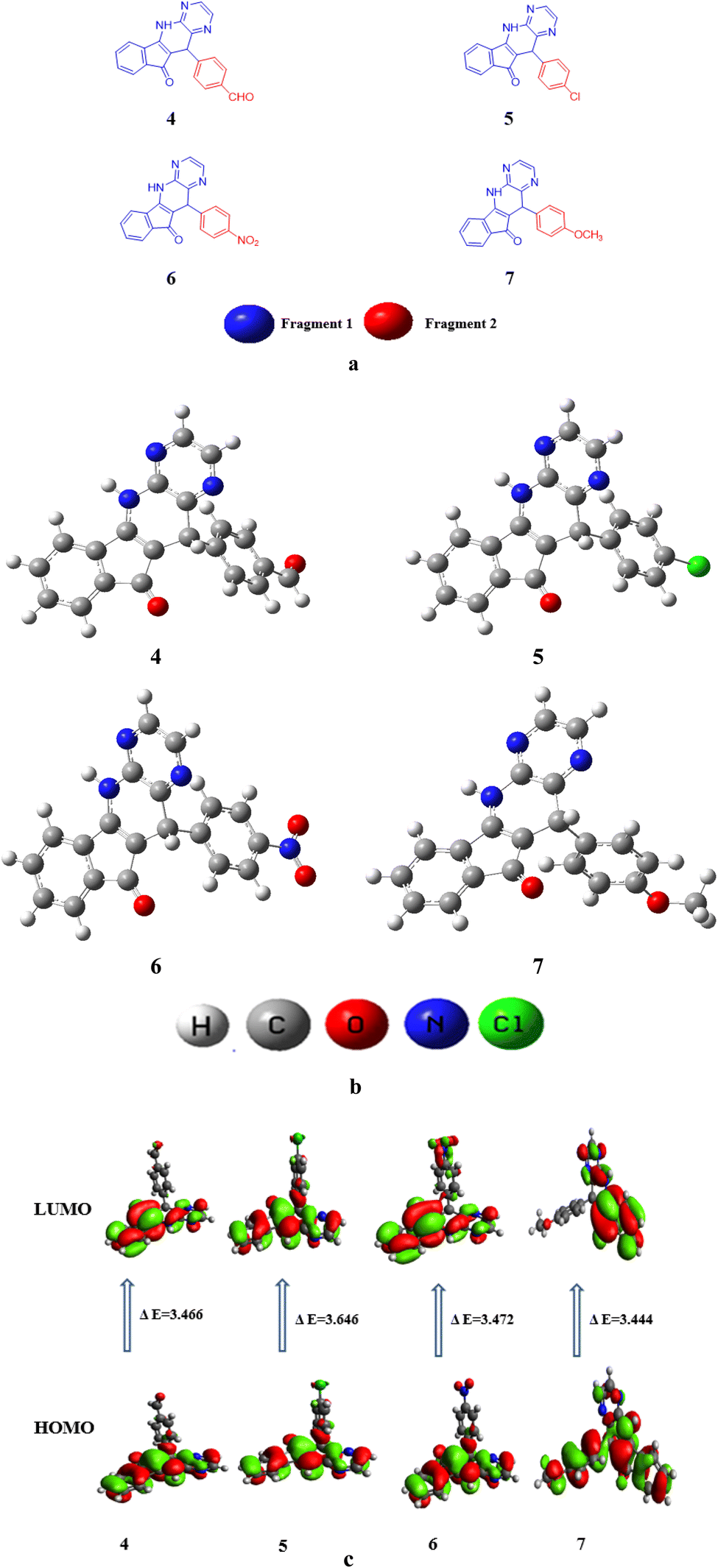 | ||
| Fig. 2 (a) Structures of studied compounds (4–7) (b) optimized structures of investigated compounds (4–7) (c) contour surfaces of FMOs of the studied compounds (4–7). | ||
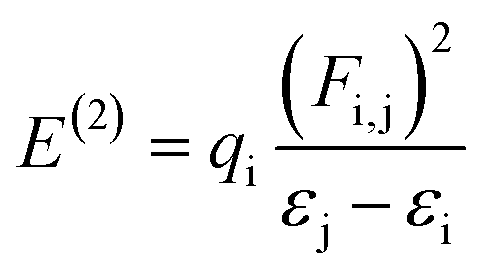
F(i,j) indicates the diagonal, qi donor orbital occupancy, and εj and εi are off-diagonal NBO Fock or Kohn–Sham medium elements. E(2) specifies the stabilization energy56 When E(2) has a high value and the system as a whole has more conjugation, there is a significant interaction between i and j. The findings show that substituent's capacity to withdraw or donate electrons plays a significant part in causing the atomic charge on the carbon atom connected with substituents. Electronegativity plays a significant part in stabilization because highly electronegative groups, such as nitrogen, chloro, and methoxy group insertion, can promote hyper-conjugation and therefore stabilize the lone pair of atoms in transition states.57 The specific nominated values for compounds 4 to 7 in the second-order perturbation approach are listed in Table 1.
| Compounds | Donor(i) | Type | Acceptor(j) | Type | E(2) [kcal mol−1] | E(j) − E(i) [a.u.] | F(i,j) [a.u.] |
|---|---|---|---|---|---|---|---|
| 4 | C14–C15 | π | C26–O27 | π* | 25.77 | 0.3 | 0.079 |
| C16–C18 | π | C14–C15 | π* | 0.79 | 0.28 | 0.014 | |
| C15–C26 | σ | C14–N34 | σ* | 6.54 | 1.1 | 0.076 | |
| C28–H29 | σ | C30–H31 | σ* | 0.5 | 0.97 | 0.02 | |
| N34 | LP(1) | C14–C15 | π* | 43.81 | 0.32 | 0.108 | |
| O38 | LP(2) | C37–H39 | σ* | 23.26 | 0.63 | 0.11 | |
| 5 | C14–C15 | π | C26–O27 | π* | 25.78 | 0.3 | 0.079 |
| C16–C17 | π | C14–C15 | π* | 0.86 | 0.28 | 0.014 | |
| C5–C6 | σ | C4–C14 | σ* | 5.81 | 1.15 | 0.073 | |
| C28–H29 | σ | C30–H31 | σ* | 0.5 | 0.97 | 0.02 | |
| N34 | LP(1) | C14–C15 | π* | 43.3 | 0.32 | 0.107 | |
| O27 | LP(2) | C3–C26 | σ* | 21.58 | 0.67 | 0.109 | |
| 6 | C19–C23 | π | N37–O39 | π* | 27.57 | 0.15 | 0.061 |
| C14–C15 | π | C16–C17 | π* | 0.52 | 0.31 | 0.012 | |
| C5–C6 | σ | C4–C14 | σ* | 5.83 | 1.15 | 0.073 | |
| C28–H29 | σ | C30–H31 | σ* | 0.5 | 0.97 | 0.02 | |
| O38 | LP(3) | N37–O39 | π* | 164.39 | 0.14 | 0.14 | |
| O27 | LP(2) | C3–C26 | σ* | 21.52 | 0.67 | 0.109 | |
| 7 | C14–C15 | π | C26–O27 | π* | 25.06 | 0.31 | 0.078 |
| C12–N33 | π | C16–C18 | π* | 0.62 | 0.37 | 0.014 | |
| C15–C26 | σ | C14–N34 | σ* | 6.66 | 1.1 | 0.076 | |
| C3–C26 | σ | C14–N34 | σ* | 0.51 | 1.08 | 0.021 | |
| N34 | LP(1) | C14–C15 | π* | 43.45 | 0.32 | 0.107 | |
| O27 | LP(2) | C3–C26 | σ* | 21.9 | 0.67 | 0.109 |
NBO study depicts four types of transition i.e., σ → σ*,π → π*, LP → σ* and LP → π*. These transitions are vital for the intramolecular charge transferal and results in the stabilization of system. In π → π* transitions, the charge transference takes place within the molecule is more prominent as compared to σ → σ*. A few transitions are presented in Tables S3–S6.†
In compound 6, the significant π → π* electronic transition which showed the highest stabilization energy value (27.57 kcal mol−1) is for π(C19–C23) → π*(N37–O39). While π(C14–C15) → π*(C16–C17) was of the lowermost energy transition having 0.52 kcal mol−1 energy. Moreover, σ → σ* transition as σ(C5–C6) → σ*(C4–C14) had 5.83 kcal mol−1 as maximum energy and σ(C28–H29) → σ*(C30–H31) having 0.51 kcal mol−1 lowest energy transitions. Moreover, LP → π* shows the highest value of 164.39 kcal mol−1 for LP3(O38) → π*(N37–O39) transition and LP → σ* had the stabilization energy value of 21.52 kcal mol−1 for LP2(O27) → σ*(C3–C26) transitions.
For compound 4, π → π* electronic transition with the maximum value of stabilization energy (25.77 kcal mol−1) is shown by π(C14–C15) → π*(C26–O27). Whereas, π(C16–C18) → π*(C14–C15) shows lowest energy transition of 0.79 kcal mol−1 energy. In addition, σ → σ* transition like σ(C15–C26) → σ*(C14–N34) has 6.54 kcal mol−1 as uppermost energy and σ(C28–H29) → σ*(C30–H31) depicts 0.50 kcal mol−1 lowest energy. The LP → π* is perceived at 43.81 kcal mol−1 by LP1(N34) → π*(C14–C15), while the σ* value perceived in LP2(O38) to σ*(C37–H39) transition is 23.26 kcal mol−1.
For compound 5, the π → π* transition shows the utmost value of stabilization energy at 25.78 kcal mol−1 by π(C14–C15) → π*(C26–O27) whereas, π(C16–C17) → π*(C14–C15) consisting of the lowest energy as 0.86 kcal mol−1. The σ → σ* transition: σ(C5–C6) → σ*(C4–C14) had 5.81 kcal mol−1 highest and σ(C28–H29) → σ*(C30–H31) comprising of the lowest energy as 0.5 kcal mol−1. Moreover, LP → π* had the maximum value at 43.3 kcal mol−1 for LP1(N34) → π*(C14–C15) while the σ* value (21.58 kcal mol−1) is observed for LP2(O27) → σ*(C3–C26) transition. For compound 7, π → π* transition contained highest value of stabilization energy 25.06 kcal mol−1 for π(C14–C15) → π*(C26–O27). However, lowest energy (0.62 kcal mol−1) was disclosed for π(C12–N33) → π*(C16–C18). Further, σ → σ* transition such as σ(C15–C26) → σ*(C14–N34) comprising highest energy of 6.66 kcal mol−1. The σ(C3–C26) → σ*(C14–N34) is found with lowest energy of 0.51 kcal mol−1. Moreover, LP → π* has the highest value of 43.45 kcal mol−1 due to LP1(N34) → π*(C14–C15) while the σ* value noticed in LP2(O27) → σ*(C3–C26) was 21.9 kcal mol−1 transition. Among all the entitled compounds, compound 6 shows the maximum stability due to extended hyper-conjugation with 27.57 kcal mol−1. The overall stability order is 6 > 4 > 5 > 7. Therefore, NBO study of these compounds shows that the extended hyper conjugation and strong ICT plays a significant role in stabilizing these compounds and signifies charge transmission properties.
Table S7† shows that the EHOMO/ELUMO values for compounds (4–7) are obtained as −6.131/−2.665, −6.068/−2.604, −6.261/−2.789, and −5.858/−2.414 eV, correspondingly. The ELUMO − EHOMO values of titled compounds are calculated as 3.477, 3.646, 3.472 and 3.444 eV, respectively. Table S7† describes that the compound 7 has lowest band gap value than all other derivatives. This might be due to the presence of methoxy group at the para position of benzene ring that have positive inductive effect and improve the charge transference in molecule. The Egap is indispensable for charge transfer process, lesser the band gap greater will be the transference of charge. The contour surface diagrams of HOMO and LUMO are presented in Fig. 2c. It represents that HOMO is mainly located over the [5,11-dihydro-10H-indeno[2′,1′:5,6]pyrido[2,3-b]pyrazin-10-one] part of compound 4. While, in LUMO the charge density is majorly concentrated on [5,11-dihydro-10H-indeno[2′,1′:5,6]pyrido[2,3-b]pyrazin-10-one] moiety and some effects are on oxygen atom of benzaldehyde moiety. In 5 compound the charge density for HOMO is positioned over 5,11-dihydro-10H-indeno[2′,1′:5,6]pyrido[2,3-b] pyrazin-10-one and small effect on chlorine atom of chlorobenzen moiety, Whereas, in LUMO the charge density is concentrated on the same portion like HOMO.
In compound 6 the charge density for HOMO is situated on 5,11-dihydro-10H-indeno [2′,1′:5,6] pyrido[2,3-b]pyrazin-10-one, while at LUMO the charge density additionally observed at nitro group of nitrobenzene. Similarly, for compound 7 charge density for HOMO is located over 3-amino-2-(4-methoxybenzyl)-1H-inden-1-one part and small effect are observed at pyrazine part, while at LUMO charge density observed at 3-amino-2-(4-methoxybenzyl)-1H-inden-1-one part are displayed in Fig. 2c. The values of HOMO−1, LUMO+1, HOMO−2 and LUMO+2 is exhibited in Table S7† and their FMOs diagrams are provided in Fig. S1.†
| IP = −EHOMO | (5) |
| EA = −ELUMO | (6) |
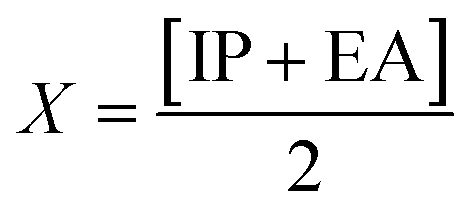 | (7) |
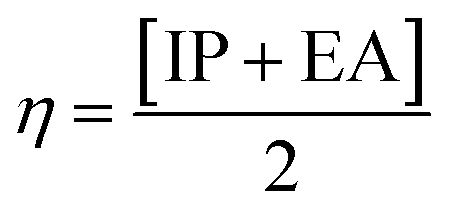 | (8) |
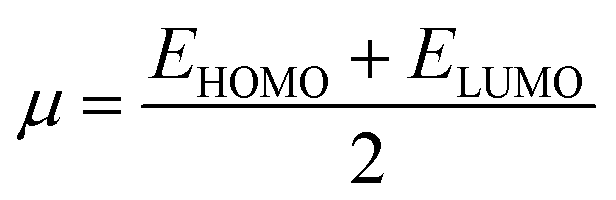 | (9) |
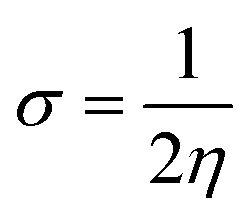 | (10) |
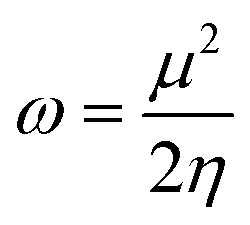 | (11) |
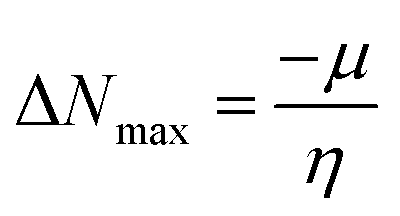 | (12) |
The movement of electrons within a compound is designated as the value of μ,76 whereas the chemical reactivity and stability is determined by η.77 When a molecule gains an extra electronic charge from an external source,78 this determines its stabilisation energy ω. The date in Table S9† shows the GRPs of titled chromophores at B3LYP functional. The energy gap is directly affected by the chemical potential (μ) and hardness (η) parameters. The negative findings of chemical potential (μ) indicates that the compound under study is sufficiently stable. The Egap, hardness, stability and chemical potential exhibit direct relation with each other, whereas inverse relation with reactivity.57 The η values of these compounds are 1.733, 1.732, 1.736 and 1.722 eV. Compound 6 exhibits the highest stability owing to the greatest hardness value (1.736 eV). Whereas σ values are 0.288, 0.288, 0.288 and 0.290 eV−1. The smaller hardness and larger softness values of compound 7 in comparison to others show its higher reactivity and less stability due to lower Egap of compound 7. The outcomes revealed that compound 7 is highly polarizable and demonstrates exceptional NLO response.
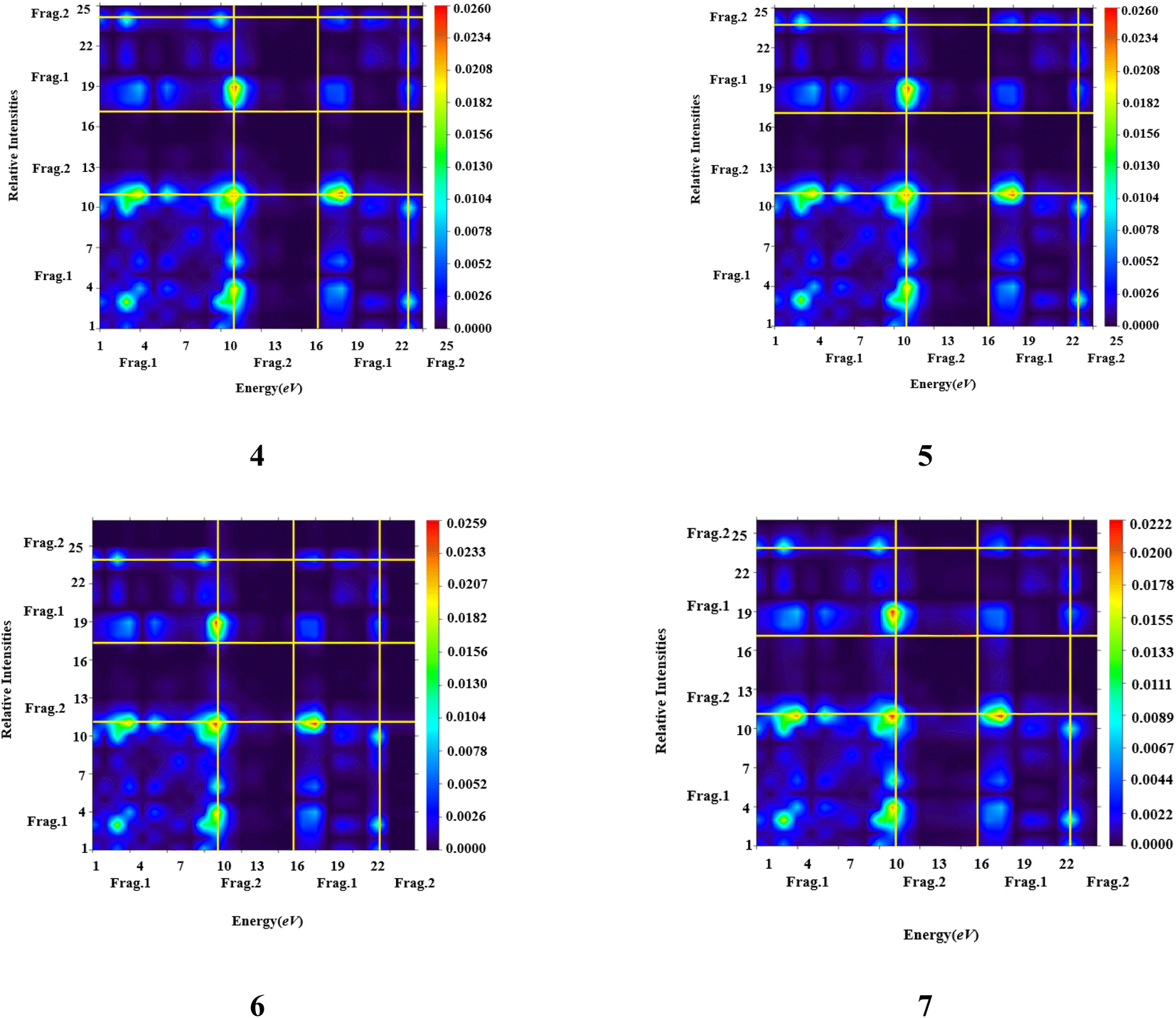 | ||
| Fig. 3 TDM heat maps of compound (4–7). | ||
It is apparent from the data presented in Table S11† that there exists an inverse correlation between transition energy outcomes and the maximum absorption wavelengths (λmax). As the transition energy of molecules escalates λmax decreases. The computed values of λmax for all synthesised compounds are in the range of 310.131–443.704 nm. The lowest value of λmax is examined in 5 (310.131 nm) due to poor charge transference as chloro has electron withdrawing effect it withdraws electrons from fragment 1. Charge transference was improved in 6 (315.417 nm) due to nitro group which withdraws electrons. Compound 7 with λmax (443.704 nm) exhibits maximum absorption wavelength due to methoxy group as it donates electron to fragment 1 and charge is efficiently transferred to fragment 1 (Tables S12–S15†). In summary, the comprehensive discourse supports the notion that all chemical compounds can be regarded as adept candidates for NLO materials. However, it is noteworthy that compound 7 exhibits the potential for heightened proficiency in this context, attributable to its diminished band gap energy (Egap) and augmented values for the maximum wavelength (λmax).
3.3.7.1 C–H vibrations. In the aromatic ring stretching bands related to C–H vibrational are present at 3100–3000(cm−1).81 In the current study, we got computational calculations for C–H of compound 4 at 3194–3186 cm−1, which were enough close to the observed vibrational band 3214 cm−1. Similarly, compounds 5, 6 and 7 were obtained at range at 3193–3160, 3224–3174 and 3203–3130 cm−1 which were very close to the experimental vibrational bands 3193, 3347 and 4359 cm−1. A reasonable consistency has been observed between DFT based and experimentally determined FT-IR values. Further, the calculated in-plane bending vibrational wavenumbers for C–H bond appeared 1474 to 892 cm−1 for all studied compounds as reported in Tables S16–S19.†
3.3.7.2 C–C vibrations. The stretching frequencies of C
C are shown at 1650–1400 (cm−1).82 The measured C–C vibrational modes are observed at 1665–1433 cm−1 for compounds 4 to 7. Absorption bands which are measured clearly matching to experimental bands of 1660, 1661, 1659 and 1658 cm−1 for 4–7 compounds respectively as can be seen in Tables S16–S19.†
3.3.7.3 C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) N vibrations. The values for the CN vibrations were mentioned at 1585, 1545, 1544 and 1544 cm−1 for compounds 4–7 respectively. However, some other vibrations are observed at 1600 cm−1 range for all studied compounds between the carbon and nitrogen of pyridine ring.
N vibrations. The values for the CN vibrations were mentioned at 1585, 1545, 1544 and 1544 cm−1 for compounds 4–7 respectively. However, some other vibrations are observed at 1600 cm−1 range for all studied compounds between the carbon and nitrogen of pyridine ring.
3.3.7.4 N–H vibrations. Usually, the N–H wavenumbers lie in the span of 3450–3250 (cm−1).83 In the current work, the N–H stretching bands simulated, are observed at 3614, 3614, 3613 and 3615 cm−1 in compounds 4–7 respectively. However, some other vibrations in-plane bending vibrations are observed at 892 cm−1 for all investigate compounds can be seen in Tables S16–S19.†
3.3.7.5 Other vibrations. In our studied compounds 4–7 different substituents are attached with fragment 2 showing different vibrations. However, some other calculated vibrations of CHO, C–Cl, NO2 and OCH3 were observed at 892, 770, 771 and 772 cm−1 which were very close to that of experimental vibrational band 843, 759, 765 and 720 cm−1 as represented in Tables S16–S19.†
Among all the entitle compounds, the greatest value of μtot is observed in 6 which may be due to the existence of highly electronegative nitro group. The decreasing order of dipole moments of computed molecules is: 6 > 4 > 5 > 7. Thus, it is evident from the results that ICT and electron transport rate increase with increasing μtot value. The results of 〈α〉 of all the compounds are found comparable with one another. The compound 7 unveiled the highest value of 〈α〉 which is 3.90 × 10−23 esu which may be owing to the inductive effect of methoxy group at para position while, 5, shows the lowermost 〈α〉 value of 3.78 × 10−23 esu. Among these newly synthesize compounds, the order of 〈α〉 for all studied compounds is as: 7 > 4 > 6 >5.
The charge transfer within the molecule is associated with the first hyper-polarizability. All of the titled compounds (4–7) exhibit the strongest ICT. The greater the ICT, greater the NLO reaction would be. The βtot value of 6 is examined to be 9.20 × 10−30 esu which is the lowermost observed value, while 7is found to have the utmost value (15.6 × 10−30 esu) of βtot among all the studied molecules. The βtot values of 4–7 is observed to be 7.88 × 10−30, 9.74 × 10−30, 9.20 × 10−30, and 15.6 × 10−30 esu, respectively. The βtot values were found to be in the declining order as: 7 > 5 > 6 > 4. For the investigation of hyper-polarizability characteristics, Para-nitro aniline (PNA) is used as the reference molecule (βtot = 0.3610 × 10−30 esu).88 By comparing our results with PNA molecule, we determined that the hyper-polarizability values of 4 to 7 are 21, 26, 25 and 43 times larger than PNA molecule, respectively.
Two-photon absorption (TPA) phenomena in NLO materials are thought to be the basis for the second hyper-polarizability of the compounds. As shown in Tables S21–S24,† a substantial second-order hyper polarizability response was perceived along the x-axis in all the studied compounds, with 7 having the largest value of 〈γ〉 among the derivatives. The decreasing order of 〈γ〉 of the above-mentioned compounds is as follows: 7 > 6 > 4 > 5. From the above explanation, it should be clear that different kinds of substituents played a crucial role in exhibiting amazing NLO amplitude.89
3.4 Biological evaluation
The empirical findings pertaining to the antioxidative efficacy of the tested compounds, in conjunction with the reference pharmaceutical agent, are displayed within Table S25† for reference. The purpose of this work is to identify the potential pyrido[2,3-b]pyrazine based heterocyclic compound as antioxidant agent. The results given therein indicate that all the synthesized compounds (4–7) demonstrated potent to moderate free radical scavenging activity or inhibition. Compounds 5 proved to be potent DPPH scavengers, as it exhibited antioxidant activity with IC50 values 5.92 ± 0.11 μM in comparison to the standard antioxidant, which showed IC50 values of 5.31 ± 1.22. While the compounds 4, 6 and 7 showed moderate activity ranging 5.98 ± 1.95 to 7.76 ± 1.76 μM (Fig. 4). The order of decreasing antioxidant activity is 5 > 4 > 6 > 7. Further the analysis of Table S25† indicated that radical scavenging activity in DPPH method increases with increasing concentration.
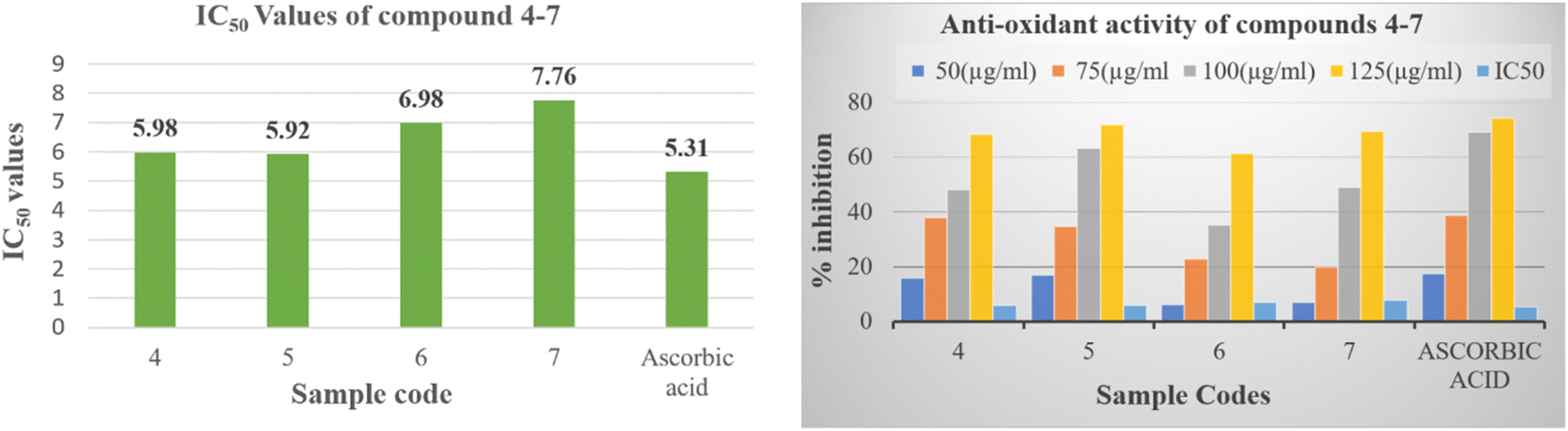 | ||
| Fig. 4 In vitro antioxidant activity of compounds (4–7) and ascorbic acid reference. | ||
4. Conclusion
Four Pyrido[2,3-b]pyrazine based heterocyclic compounds (4–7) were synthesized and evaluated for their electrochemical DNA sensing, computational studies and biological activities. Cyclic voltammetry is used to detect electrochemical DNA sensing, showing that interaction of compound (4–7) with ssDNA is in intercalative mode. These results indicated that Pyrido[2,3-b]pyrazine based heterocyclic compounds (4–7) demonstrated significant potential for DNA sensing under optimised conditions and can be employed for sensing applications of different biomolecules with minor modifications. In DFT study, NBO findings revealed that effective CT is possible within the molecules. The FMOs exploration designates the phenomena of charge transfer and chemical reactivity in 4–7 compounds. With the assistance of FMO energies, GRP has been also calculated that showed that compound 7 with lower band gap (3.444 eV), exhibited smaller hardness (1.722 eV) with larger value of softness which indicated its larger polarizability. Additionally, compound 7 showed less energy gap and remarkable NLO response with highest 〈α〉, βtot and 〈γ〉 values observed as 3.90 × 10−23, 15.6 × 10−30 and 6.63 × 10−35 esu, respectively. Comparative analysis with P-NA molecule showed that the fused ring heterocyclic pyrazin based heterocyclic compounds act as efficient NLO materials and can be used in progressive photonic applications. Furthermore, the novel compound (4–7) showed encouraging antioxidant and urease inhibitory properties, highlighting their potential as therapeutic agents.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors express their appreciation to the Deanship of Scientific Research at King Khalid University, Saudi Arabia, for funding this work through research group program under grant number (RGP. 2/232/44). Dr Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (project no. 20-14703/NRPU/R&D/HEC/2021).References
- C. Dincer, R. Bruch, E. Costa-Rama, M. T. Fernández-Abedul, A. Merkoçi, A. Manz, G. A. Urban and F. Güder, Adv. Mater., 2019, 31, 1806739 CrossRef PubMed.
- A. Khalilian, M. R. R. Khan and S.-W. Kang, Sens. Actuators, B, 2017, 249, 700–707 CrossRef CAS.
- H. Kaur and M. Shorie, Nanoscale Adv., 2019, 1, 2123–2138 RSC.
- Y. Ye, J. Xie, Y. Ye, X. Cao, H. Zheng, X. Xu and Q. Zhang, Carbon, 2018, 129, 730–737 CrossRef CAS.
- H. A. M. Faria and V. Zucolotto, Biosens. Bioelectron., 2019, 131, 149–155 CrossRef CAS PubMed.
- Z. Shabaninejad, F. Yousefi, A. Movahedpour, Y. Ghasemi, S. Dokanehiifard, S. Rezaei, R. Aryan, A. Savardashtaki and H. Mirzaei, Anal. Biochem., 2019, 581, 113349 CrossRef CAS PubMed.
- X. Fan, S. Wang, H. Liu, Z. Li, Q. Sun, Y. Wang and X. Fan, Talanta, 2022, 236, 122830 CrossRef CAS PubMed.
- D. Kwong Hong Tsang, T. J. Lieberthal, C. Watts, I. E. Dunlop, S. Ramadan, A. E. del Rio Hernandez and N. Klein, Sci. Rep., 2019, 9, 13946 CrossRef PubMed.
- S. Eissa, S. Alkhaldi, R. Chinnappan, A. Siddiqua, M. Abduljabbar, A. M. A. Rahman, M. Dasouki and M. Zourob, Talanta, 2020, 216, 120951 CrossRef CAS PubMed.
- A. Zubiarrain-Laserna and P. Kruse, J. Electrochem. Soc., 2020, 167, 037539 CrossRef CAS.
- C. McVey, F. Huang, C. Elliott and C. Cao, Biosens. Bioelectron., 2017, 92, 502–508 CrossRef CAS PubMed.
- D. Rawtani, M. Tharmavaram, G. Pandey and C. M. Hussain, TrAC, Trends Anal. Chem., 2019, 120, 115661 CrossRef CAS.
- M. Ibrahim, T.-C. Liang, K. Scott, K. Chakrabarty and R. Karri, IEEE Trans. Inf. Forensics Secur., 2020, 15, 3595–3609 Search PubMed.
- M. Kundu, P. Krishnan, R. Kotnala and G. Sumana, Trends Food Sci. Technol., 2019, 88, 157–178 CrossRef CAS.
- A. Singh, A. Sharma, A. Ahmed, A. K. Sundramoorthy, H. Furukawa, S. Arya and A. Khosla, Biosensors, 2021, 11, 336 CrossRef CAS PubMed.
- P. Liu, X. Qian, X. Li, L. Fan, X. Li, D. Cui and Y. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 45648–45656 CrossRef CAS PubMed.
- Z. Izadi, M. Sheikh-Zeinoddin, A. A. Ensafi and S. Soleimanian-Zad, Biosens. Bioelectron., 2016, 80, 582–589 CrossRef CAS PubMed.
- J. Wang, P. Xue, Y. Jiang, Y. Huo and X. Zhan, Nat. Rev. Chem, 2022, 6, 614–634 CrossRef PubMed.
- M. K. Yadav, T. Prasad, R. Rathi, V. K. Kushwaha, V. Kumar, S. K. Yadav and S. Rai, International Journal of Pharma Professional’s Research (IJPPR), 2023, 14, 36–54 Search PubMed.
- D. M. Kim and S. M. Yoo, Crit. Rev. Biotechnol., 2020, 40, 787–803 CrossRef CAS PubMed.
- Z. Zhang, Q. Li, X. Du and M. Liu, Thorac. Cancer, 2020, 11, 840–850 CrossRef PubMed.
- M. Manzano, S. Viezzi, S. Mazerat, R. S. Marks and J. Vidic, Biosens. Bioelectron., 2018, 100, 89–95 CrossRef CAS PubMed.
- M. S. Palanki, E. Dneprovskaia, J. Doukas, R. M. Fine, J. Hood, X. Kang, D. Lohse, M. Martin, G. Noronha and R. M. Soll, J. Med. Chem., 2007, 50, 4279–4294 CrossRef CAS PubMed.
- J. M. Cox, B. Harper, A. Mastracchio, B. Leiting, R. S. Roy, R. A. Patel, J. K. Wu, K. A. Lyons, H. He and S. Xu, Bioorg. Med. Chem. Lett., 2007, 17, 4579–4583 CrossRef CAS PubMed.
- E. L. White, W. J. Suling, L. J. Ross, L. E. Seitz and R. C. Reynolds, J. Antimicrob. Chemother., 2002, 50, 111–114 CrossRef CAS PubMed.
- B. Mathew, S. Srivastava, L. J. Ross, W. J. Suling, E. L. White, L. K. Woolhiser, A. J. Lenaerts and R. C. Reynolds, Bioorg. Med. Chem., 2011, 19, 7120–7128 CrossRef CAS PubMed.
- C. A. Blum, T. Caldwell, X. Zheng, R. Bakthavatchalam, S. Capitosti, H. Brielmann, S. De Lombaert, M. T. Kershaw, D. Matson and J. E. Krause, J. Med. Chem., 2010, 53, 3330–3348 CrossRef CAS PubMed.
- P. J. Crowley, C. Lamberth, U. Müller, S. Wendeborn, K. Nebel, J. Williams, O. A. Sageot, N. Carter, T. Mathie and H. J. Kempf, Pest Manag. Sci., 2010, 66, 178–185 CrossRef CAS PubMed.
- Y.-D. Gong, M.-S. Dong, S.-B. Lee, N. Kim, M.-S. Bae and N.-S. Kang, Bioorg. Med. Chem., 2011, 19, 5639–5647 CrossRef CAS PubMed.
- L. Kékesi, A. Sipos, G. Németh, J. Pató, N. Breza, F. Baska, L. Őrfi and G. Kéri, Bioorg. Med. Chem. Lett., 2013, 23, 6152–6155 CrossRef PubMed.
- T. R. Webb, J. Slavish, R. E. George, A. T. Look, L. Xue, Q. Jiang, X. Cui, W. B. Rentrop and S. W. Morris, Expert Rev. Anticancer Ther., 2009, 9, 331–356 CrossRef CAS PubMed.
- A. Zambon, D. Ménard, B. M. Suijkerbuijk, I. Niculescu-Duvaz, S. Whittaker, D. Niculescu-Duvaz, A. Nourry, L. Davies, H. A. Manne and F. Lopes, J. Med. Chem., 2010, 53, 5639–5655 CrossRef CAS PubMed.
- J. Ashburner, Magn. Reson. Imaging, 2009, 27, 1163–1174 CrossRef PubMed.
- N. M. O'boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- M. Khalid, I. Shafiq, K. Mahmood, R. Hussain, M. F. ur Rehman, M. A. Assiri, M. Imran and M. S. Akram, Sci. Rep., 2023, 13, 1395 CrossRef CAS PubMed.
- C. Valverde, Acta Chim. Slov., 2018, 65, 739–774 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- B. König, I. Tammer, V. Sollich and W. König, J. Clin. Microbiol., 2005, 43, 3500–3503 CrossRef PubMed.
- W. Qamar, M. R. Khan and A. Arafah, Saudi J. Biol. Sci., 2017, 24, 1465–1469 CrossRef CAS PubMed.
- M. Mohamadi, A. Mostafavi and M. Torkzadeh-Mahani, Biosens. Bioelectron., 2014, 54, 211–216 CrossRef CAS PubMed.
- S. V. Chitreddy and S. Shanmugam, J. Mol. Liq., 2017, 243, 494–502 CrossRef CAS.
- N. Baltaş, F. YILMAZ and E. Menteşe, Hacettepe J. Biol. Chem., 2016, 44, 293–305 CrossRef.
- H. Bektaş, Ş. Ceylan, N. Demirbaş, Ş. Alpay-Karaoğlu and B. B. Sökmen, Med. Chem. Res., 2013, 22, 3629–3639 CrossRef PubMed.
- M. Mohamadi, A. Mostafavi and M. Torkzadeh-Mahani, J. Electrochem. Soc., 2015, 162, B344 CrossRef CAS.
- M. Moshari, D. Koirala and P. B. Allen, J. Solid State Electrochem., 2021, 25, 1667–1678 CrossRef CAS.
- A. Mohammadpour-Haratbar, S. B. A. Boraei, Y. Zare, K. Y. Rhee and S.-J. Park, Biosensors, 2023, 13, 80 CrossRef CAS PubMed.
- H. Mahmoudi-Moghaddam, S. Tajik and H. Beitollahi, Microchem. J., 2019, 150, 104085 CrossRef CAS.
- S. Ponkarpagam, G. Mahalakshmi, K. N. Vennila and K. P. Elango, J. Biomol. Struct. Dyn., 2022, 40, 8394–8404 CrossRef CAS PubMed.
- D. S. Pisetsky, R. Shaffer, D. D. Armstrong and D. M. Spencer, Clin. Immunol., 2021, 231, 108848 CrossRef CAS PubMed.
- A. C. Carpenter, I. T. Paulsen and T. C. Williams, Genes, 2018, 9, 375 CrossRef PubMed.
- F. Li, X. Mao, F. Li, M. Li, J. Shen, Z. Ge, C. Fan and X. Zuo, J. Am. Chem. Soc., 2020, 142, 9975–9981 CrossRef CAS PubMed.
- J. Ali, J. Najeeb, M. A. Ali, M. F. Aslam and A. Raza, J. Biosens. Bioelectron., 2017, 8, 1–9 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. Szafran, A. Komasa and E. Bartoszak-Adamska, J. Mol. Struct., 2007, 827, 101–107 CrossRef CAS.
- R. Hussain, M. U. Khan, M. Y. Mehboob, M. Khalid, J. Iqbal, K. Ayub, M. Adnan, M. Ahmed, K. Atiq and K. Mahmood, ChemistrySelect, 2020, 5, 5022–5034 CrossRef CAS.
- S. He, Y. Tan, X. Xiao, L. Zhu, Y. Guo, M. Li, A. Tian, X. Pu and N.-B. Wong, J. Mol. Struct.: THEOCHEM, 2010, 951, 7–13 CrossRef CAS.
- M. Wielopolski, J.-H. Kim, Y.-S. Jung, Y.-J. Yu, K.-Y. Kay, T. W. Holcombe, S. M. Zakeeruddin, M. Grätzel and J.-E. Moser, J. Phys. Chem. C, 2013, 117, 13805–13815 CrossRef CAS.
- I. Tammer, S. Brandt, R. Hartig, W. König and S. Backert, Gastroenterology, 2007, 132, 1309–1319 CrossRef CAS PubMed.
- R. D. Fonseca, M. G. Vivas, D. L. Silva, G. Eucat, Y. Bretonnière, C. Andraud, L. De Boni and C. R. Mendonca, J. Phys. Chem. C, 2018, 122, 1770–1778 CrossRef CAS.
- M. R. S. A. Janjua, M. U. Khan, B. Bashir, M. A. Iqbal, Y. Song, S. A. R. Naqvi and Z. A. Khan, Comput. Theor. Chem., 2012, 994, 34–40 CrossRef CAS.
- M. R. S. A. Janjua, Inorg. Chem., 2012, 51, 11306–11314 CrossRef CAS PubMed.
- P. K. Chattaraj, S. Giri and S. Duley, Chem. Rev., 2011, 111, PR43–PR75 CrossRef PubMed.
- A. J. Garza, O. I. Osman, N. A. Wazzan, S. B. Khan, A. M. Asiri and G. E. Scuseria, Theor. Chem. Acc., 2014, 133, 1–8 Search PubMed.
- M. I. Nan, E. Lakatos, G.-I. Giurgi, L. Szolga, R. Po, A. Terec, S. Jungsuttiwong, I. Grosu and J. Roncali, Dyes Pigm., 2020, 181, 108527 CrossRef CAS.
- W. Rahmalia, J.-F. Fabre, T. Usman and Z. Mouloungui, Spectrochim. Acta, Part A, 2014, 131, 455–460 CrossRef CAS PubMed.
- P. Goszczycki, K. Stadnicka, M. Z. Brela, J. Grolik and K. Ostrowska, J. Mol. Struct., 2017, 1146, 337–346 CrossRef CAS.
- M. Ans, J. Iqbal, Z. Ahmad, S. Muhammad, R. Hussain, B. Eliasson and K. Ayub, ChemistrySelect, 2018, 3, 12797–12804 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, Chem. Phys., 1978, 68, 3801–3807 CAS.
- N. Kovačević and A. Kokalj, Corros. Sci., 2011, 53, 909–921 CrossRef.
- N. Sheela, S. Muthu and S. Sampathkrishnan, Spectrochim. Acta, Part A, 2014, 120, 237–251 CrossRef CAS PubMed.
- T. J. Seiders, K. K. Baldridge, J. S. Siegel and R. Gleiter, Tetrahedron Lett., 2000, 41, 4519–4522 CrossRef CAS.
- W. D. Jones, Science, 2000, 287, 1942–1943 CrossRef CAS.
- A. R. Pasha, M. Khalid, Z. Shafiq, M. U. Khan, M. M. Naseer, M. N. Tahir, R. Hussain, A. A. C. Braga and R. Jawaria, J. Mol. Struct., 2021, 1230, 129852 CrossRef CAS.
- M. Khalid, S. Naseer, M. S. Tahir, I. Shafiq, K. S. Munawar, S. F. de Alcântara Morais and A. A. Braga, Opt. Quantum Electron., 2023, 55, 258 CrossRef CAS.
- P. K. Chattaraj and D. R. Roy, Chem. Rev., 2007, 107, PR46–PR74 CrossRef CAS.
- Y. Li and C. Ullrich, Chem. Phys., 2011, 391, 157–163 CrossRef CAS.
- M. Khalid, M. U. Khan, I. Shafiq, R. Hussain, A. Ali, M. Imran, A. A. Braga, M. Fayyaz ur Rehman and M. S. Akram, R. Soc. Open Sci., 2021, 8, 210570 CrossRef CAS PubMed.
- J. E. Lisman and N. A. Otmakhova, Hippocampus, 2001, 11, 551–568 CrossRef CAS PubMed.
- U. Slotta, M. Tammer, F. Kremer, P. Koelsch and T. Scheibel, Supramol. Chem., 2006, 18, 465–471 CrossRef CAS.
- M. Subramanian, P. Anbarasan and S. Manimegalai, Spectrochim. Acta, Part A, 2009, 73, 642–649 CrossRef CAS PubMed.
- S. M. B. Dhas and S. Natarajan, Opt. Commun., 2007, 278, 434–438 CrossRef CAS.
- M. Shkir, S. Muhammad, S. AlFaify, A. R. Chaudhry and A. G. Al-Sehemi, Arab. J. Chem., 2019, 12, 4612–4626 CrossRef CAS.
- W. Sun, M. M. Bader and T. Carvalho, Opt. Commun., 2003, 215, 185–190 CrossRef CAS.
- T. Zhang, X. Wei, Y. Zuo and J. Chao, Optik, 2019, 182, 295–302 CrossRef CAS.
- S. Muhammad, R. A. Shehzad, J. Iqbal, A. G. Al-Sehemi, M. Saravanabhavan and M. Khalid, J. Theor. Comput. Chem., 2019, 18, 1950030 CrossRef CAS.
- A. Saeed, S. Muhammad, S.-u. Rehman, S. Bibi, A. G. Al-Sehemi and M. Khalid, J. Mol. Graphics Modell., 2020, 100, 107665 CrossRef CAS PubMed.
- C. V. Subbareddy, R. Subashini and S. Sumathi, Mol. Diversity, 2017, 21, 841–848 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05365b |
| This journal is © The Royal Society of Chemistry 2023 |