
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessing the evolution of oxygenated functional groups on the graphene oxide surface upon mild thermal annealing in water†
Francesco Amato a,
Irene Ferraria,
Alessandro Mottaab,
Robertino Zanonia,
Enrique A. Dalchielec and
Andrea Giacomo Marrani*a
a,
Irene Ferraria,
Alessandro Mottaab,
Robertino Zanonia,
Enrique A. Dalchielec and
Andrea Giacomo Marrani*a
aDipartimento di Chimica, Università di Roma “La Sapienza”, p.le A. Moro 5, Rome I-00185, Italy. E-mail: andrea.marrani@uniroma1.it
bConsorzio INSTM, UdR Roma “La Sapienza”, Italy
cInstituto de Física & CINQUIFIMA, Facultad de Ingeniería, Julio Herrera y Reissig 565, C.C. 30, Montevideo 11000, Uruguay
First published on 6th October 2023
Abstract
Graphene oxide (GO) is known to be a 2D metastable nanomaterial that can be reconstructed under thermal annealing into distinct oxidized and graphitic phases. Up to now, such phase transformation, mainly related to epoxide and hydroxyl functional groups, has been usually achieved by thermally treating layers of GO in the solid state. Here, we present the mild annealing of GO dispersed in an aqueous medium, performed at two temperatures, 50 °C and 80 °C, for different intervals of time. We show experimental evidences of the epoxide instability in the presence of water by means of XPS, cyclic voltammetry and Raman spectroscopy, demonstrating the reorganization of epoxide and hydroxyl moieties initiated by water molecules. In fact, at 50 °C an increase in oxygen content is detected in all annealed samples compared to untreated GO, with a transformation of epoxide groups into vicinal diols. On the other hand, at 80 °C the oxygen content decreases towards the initial value since the vicinal diols, previously formed, transform into single hydroxyls and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Moreover, the higher temperature annealing likely favours oxygenated functional groups rearrangements and clustering, in accordance with the literature, leading to a higher electron affinity and conductivity of the graphenic network.
C bonds. Moreover, the higher temperature annealing likely favours oxygenated functional groups rearrangements and clustering, in accordance with the literature, leading to a higher electron affinity and conductivity of the graphenic network.
Introduction
Graphene oxide (GO) is a bidimensional nanomaterial obtained from exfoliation and oxidation of graphite, a process which results in mixed sp2 and sp3 areas, with an irregular presence of oxygenated functional groups (OFG).1–3 In particular, both hydroxyl and epoxide groups are mainly localized in the basal plane of the layer, differently to the less abundant carboxyl-functionalities, which are distributed at the edges.4,5The growing popularity of GO as a unique substrate in a wide range of applications, from biomedicine to electronics, can be traced back to its versatility in terms of lateral dimensions, chemical groups, structural defectivity, and extent of chemical/physical treatments leading to reduced GO (RGO).6–8 GO is a temperature-sensitive nanomaterial, whose structure can be altered up to a reductive process by means of extensive thermal treatments as several studies have already demonstrated. Acik et al. reported the formation of oxygenated free radicals in the presence of trapped water in GO upon annealing in the 60–850 °C range under moderate vacuum.9 Kim et al. evidenced GO metastability, with a spontaneous evolution of multilayer GO films towards chemical modification and reduction at room temperature.10 Mild thermal annealing in the 50–80 °C range has emerged as a route towards scalable enhancement of GO properties via structural rearrangement of GO oxygen-containing functional groups. A phase transformation of GO was first reported by Kumar et al., showing a transition of mixed sp2–sp3 hybridized domains into distinct oxidized and graphitic phases, with preservation of oxygen functionalities and large improvements in the optical and electronic properties of the annealed GO.11 This phase transformation of GO triggered by temperature and already reported in other works is known as “oxygen clustering”.11–13 The possible reason for such preservation upon mild annealing, notwithstanding a gradual decrease of chemically bonded oxygen atoms found on explored graphene membranes, was assigned by Zhu et al. to the strongly physisorbed oxygen species trapped within the GO lattice.14 Furthermore, Foller et al. reported evidence by TEM measurements of a size enhancement of graphitic domains from ∼40 nm2 to >200 nm2, evidenced from high and low contrast regions, respectively interpreted as disordered areas with oxygen functionalities and ordered areas of graphitic domains. They also confirmed the preservation of the functional groups during annealing by a comprehensive chemical analysis.15 Sun et al. addressed the question of the oxygen content upon mild annealing of GO membranes, in consideration of the metastable nature of GO, commented above. They reported significant transformation among diverse oxygen functionalities in addition to phase separation, concluding that a slight reduction of GO occurs.14 Moreover, the oxygen clustering of GO for the interaction with biomolecules was also exploited by Chen et al. who investigated the effect of the temperature on the structure of GO and in particular on a GO-coated glass substrate heated in an oven at 90 °C for 9 days. As proved by XPS analyses, in the reported experimental conditions, the extent of the π-conjugated domains increases whilst the oxygen content and the morphology, investigated by means of SEM and AFM microscopies, remain almost the same, compared to the starting GO material.12 Finally, Singh et al. have demonstrated very recently that heat treatment of GO at 200 °C is the most effective option for developing a porous structure.16
In this contest, the present study focuses on the modifications undergone by GO upon thermal treatment of its aqueous dispersions conducted at 50 °C up to 168 h and at 80 °C up to 48 h. These temperatures were chosen to investigate the mild thermal annealing of GO that takes place in an aqueous acidic medium, where the presence of the dispersing medium could have a role in the dynamic evolution of the GO structure. Mild thermal annealing of GO was followed by an ample set of experimental techniques, namely XPS, cyclic voltammetry and Raman spectroscopy, combined with DFT modeling. In particular, the time evolution of hydroxyl and epoxide groups was monitored, demonstrating that even at these relatively low temperatures chemical transformations occur on the GO plane, with a critical role exerted by water molecules.
Experimental
All the chemicals used here were of analytical reagent grade and were used as received. GO was purchased from ACS Materials, USA, and consists of single-layered graphene oxide from modified Hummers oxidation method, dispersed in water at a concentration of 5 mg mL−1. This dispersion was then diluted to a 0.06 mg mL−1 concentration with distilled water.17,18 See ESI† for further details of the following methods.Preparation of graphene oxide samples
10 mL of GO samples diluted in water (0.06 mg mL−1) were collected in sealed vials and placed in an oven at two temperatures, 50 and 80 °C, for different times, ranging from few hours to one week. These samples were then extracted from the oven and subject to electrochemical and spectroscopical analysis. According to the temperature used and the time of heating, the GO samples will henceforth be referred to using the following general nomenclature: GO-temperature (°C)-time (h). For example, the sample GO-80-48 was heated at 80 °C for 48 h.After thermal treatment, GO thin films were obtained by drop-casting 50 μL of the GO solution onto the surface of a glassy graphite disc, for the electrochemical characterization, and onto H-terminated Si(111) surfaces for the spectroscopical analysis.17,18
Characterization
Results and discussion
A first series of experiments was carried out on films obtained from GO solutions heated in an oven at 50 °C (GO-50 dataset) for few hours up to 168 h; a second series focused on the effects of heating at 80 °C (GO-80 dataset) for few hours up to 48 h. The longer time scale adopted for the GO-50 dataset was necessary to make appreciable the chemical and structural modification of GO starting materials and, in turn, for a better comparison with the modification observed in the GO-80 dataset. A GO solution with the same dilution was left at room temperature for a week as control sample, showing negligible chemical and structural modifications compared to the reference pristine GO (GO-0).Heating at 50 °C (GO-50 dataset)
The cyclic voltammograms of GO films from GO solutions annealed at 50 °C for heating times of 24, 48 and 168 h (samples GO-50-24, GO-50-48, GO-50-168) are reported in Fig. 1 along with the reference pristine GO (GO-0). GO and its derivatives typically present a multiplicity of voltammetric features, related to the reduction of the various oxygenated functional groups located at the edges and on the basal plane of the carbon nanosheets. Here, four main features (I, II, II and IV, Fig. 1) can be recognized in response to the reductive potential stimulus of the cyclic voltammetry, as we previously described.17,19–21 | ||
| Fig. 1 Cyclic voltammograms of GO deposits from corresponding solutions thermally annealed at 50 °C for 24 h (GO-50-24, red), 48 h (GO-50-48, blue) and 168 h (GO-50-168, green). Pristine GO (GO-0, black) is also shown. Potential scan rate 20 mV s−1. | ||
In pristine GO (GO-0, black curve in Fig. 1) the well-behaved component I at −0.93 V is assigned to the reduction of epoxide groups, which leads to the restoration of sp2 graphitic network in a bi-electronic reduction path.19 Component II is accounted for by a shapeless current wave enveloped between component I and III, and can be assigned to the one-electron carbonyl reduction, leading to C–OH moieties,19 yet with a possible partial contribution from a further epoxy reduction.17 The well definite component III at −1.22 eV is compatible with the reduction of basal hydroxyl groups leading to restoration of CC bonds, as we recently reported according to a mechanism where hydroxyl islands are considered.17 A further component (IV) appears in the range −1.45 ÷ −1.5 V, immediately followed by bulk water reduction onset at −1.53 V. This feature can be tentatively associated to water reduction in an initial adsorptive stage on GO, with water molecules probably intercalated or H-bound to the GO layers.22–25 Since the intensity of this feature is strongly dependent on the adsorbed water amount, which can sensibly vary among different samples, its voltammetric response can hardly be correlated to the experimental conditions, therefore it will not be commented further in the text.
Stepping to the GO-50-24 sample (red curve, Fig. 1), which underwent thermal annealing in water for 24 h, a shape modification of the voltammetric response can be seen. Component I decreases its intensity, together with a parallel increase of component III, which becomes broader and extends to more negative potentials, gaining intensity towards component IV. Upon extension of the thermal annealing time, samples GO-50-48 and GO-50-168 (blue and green curves in Fig. 1, respectively) present only slight variations on the position and intensity of component I, whereas features II and III appear depressed compared to GO-50-24, yet maintaining a significant intensity in the range between −1.25 and −1.4 V, prior to component IV. An intermediate annealing time between 48 and 168 h (96 h) was also investigated with CV, but no significant difference was found with the GO-50-48 sample (see Fig. S1†).
After thermal annealing selected samples were investigated with X-ray photoelectron spectroscopy (XPS), with particular attention to the C 1s ionization region, which is diagnostic of the chemically inequivalent carbon species present within the GO nanosheets. The raw C 1s XPS spectra (dots in Fig. 2), are composed by a low energy feature peaked at 284.8 eV binding energy (BE) related to unoxidized C atoms, and a component tailed towards higher BE accounting for oxygenated carbon moieties around 287 eV. This second main feature appears to decrease from pristine GO to the GO-50-168 sample. Since the nanoscale structure of GO is intrinsically chemically heterogeneous, mainly consisting of graphitic islands embedded within highly disordered oxidized areas,26 it is very likely that the above mentioned C 1s features represent an envelope of more specific chemically shifted contributions, which, therefore, require a curve-fitting analysis for their unravelling. To this aim, the curve-fitting results for the C 1s XPS regions are reported in Fig. 2a–d for each thermally annealed sample, together with the pristine GO. This latter (Fig. 2a) can be fitted to a unique component at 284.85 eV BE, associated to the aromatic CC network (blue curve, Fig. 2), plus a multiplicity of components in the oxidized area.20,27,28 This region can be fitted to several components, according to the expected oxygenated functional groups in GO. In fact, it is well known that the main functionalities appended to the basal plane of GO are epoxy and hydroxyl groups, while carboxylic, carboxylate, carbonyl (ketones, quinones), phenol, ester and lactone groups decorate edges and defects of the nanosheets.29 Therefore, the proposed curve-fitting involves all the expected functional groups (see Fig. 2 with assignments and Table 1 for quantitative details), as we already reported elsewhere.17,18,27 The first two contributions to the oxidized area are due to hydroxylated C atoms (286.35 eV, magenta curve) and epoxide groups (286.85 eV, green curve), whereas the tail at higher BE can be fitted to two further components at 288.0 eV and 289.0 eV. The former can be assigned to carbonyl (ketones, quinones) and carboxylate and the latter to carboxyl (esters, lactones, carboxylic acids) groups.17
 | ||
| Fig. 2 XPS C 1s spectrum of GO samples belonging to the GO-50 dataset: (a) pristine GO (GO-0), (b) GO-50-2, (c) GO-50-24, (d) GO-50-168. Raw data are reported in dots, while curve fitting results in coloured curves, with chemical assignments reported in (a). | ||
| Sample | Annealing time (h) | CC |
C–OH | C–O–C | CO/COO− |
COOH | RO/C | ΔGO-0 | ΔGO-0 |
|---|---|---|---|---|---|---|---|---|---|
| C–OH | C–O–C | ||||||||
a Associated error is ±10%.b As estimated by the following equation, where Px represents the area of peak x in the C 1s spectrum: 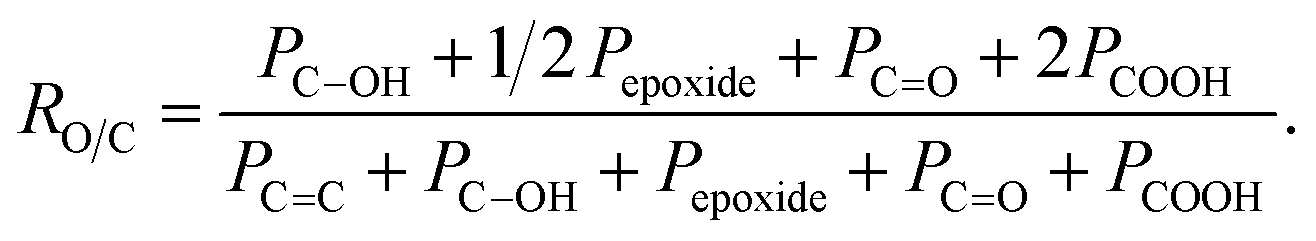 c ΔGO-0 values represent the variation in percent area related to selected functional groups with respect to GO-0 sample. c ΔGO-0 values represent the variation in percent area related to selected functional groups with respect to GO-0 sample. |
|||||||||
| GO-0 | — | 45.6 | 10.2 | 34.1 | 7.6 | 2.4 | 0.40 | — | — |
| GO-50-2 | 2 | 44.6 | 24.4 | 19.1 | 10.5 | 1.4 | 0.47 | +14.2 | −15.0 |
| GO-50-24 | 24 | 44.1 | 27.5 | 18.2 | 8.1 | 2.0 | 0.49 | +17.3 | −15.9 |
| GO-50-168 | 168 | 50.9 | 23.8 | 16.4 | 5.7 | 3.2 | 0.44 | +13.6 | −17.7 |
As reported in Table 1, the percent contribution of each carbon species in GO can be extracted from the area of each component resulting from the curve-fitting. The main outcome of this evaluation is that the contribution from C–OH groups increases upon thermal annealing at 50 °C, and it is paralleled by an almost equivalent decrease in the epoxide concentration. In order to evidence this finding, the variations in percent area related to C–OH and epoxide C atoms in all annealed samples with respect to the pristine GO-0 sample were determined as ΔGO-0 values and listed in Table 1. These differences show that, to a fair approximation, for each epoxide C atom disappearing, a corresponding hydroxylated C atom is formed, strongly suggesting that thermal treatment in these conditions leads to a transformation of a fraction of epoxide groups into vicinal diols. Furthermore, this hypothesized transformation would increase the number of O atoms bound to GO, which nicely matches the trend of O/C ratio (RO/C) reported in Table 1 (see ESI† for further details on the O/C evaluation). The increase in RO/C supports the chemical evolution of epoxide groups into vicinal diols, suggesting that this reaction might possibly be exerted by water molecules through an epoxide ring-opening path (see the Theoretical modeling section).
As to the trend of variations in intensity of the various chemical components among the different samples, one can see that RO/C slightly increases up to GO-50-24 (RO/C = 0.49), apparently due to an enrichment in C–OH groups. On the other hand, upon extending the duration of thermal annealing up to 168 h, a drop in RO/C is detected, probably due to a more pronounced elimination of epoxide groups. This latter decrease of epoxide at 168 h is also accompanied by a relevant increase in the percent intensity of CC functions (50.9%), calling for a partial restoration of C sp2 network.
The XPS findings are consistent with the results from the cyclic voltammetry, where, upon passing from pristine GO-0 to thermally treated samples, a displacement of intensity from the epoxide reduction wave (I) to the hydroxyl one (III) is detected, with an increase in current signal between feature III and IV, which may account for the freshly generated diol groups and the increased O/C ratio determined by XPS.
Heating at 80 °C (GO-80 dataset)
The cyclic voltammograms of GO films from GO solutions annealed at 80 °C for heating times of 2, 8, 24, and 48 h (samples GO-80-2, GO-80-8, GO-80-24 and GO-80-48) are reported in Fig. 3 along with the reference pristine GO (GO-0). As discussed in the previous case of the GO-50 samples, four main reductive features (I, II, II and IV, Fig. 3) can also be recognized in this case, and their evolution upon extension of thermal annealing duration is described. | ||
| Fig. 3 Cyclic voltammograms of GO deposits from corresponding solutions thermally annealed at 80 °C for 2 h (GO-80-2, red), 8 h (GO-80-8, blue), 24 h (GO-80-24, magenta) and 48 h (GO-80-48, green). Pristine GO (GO-0, black) is also shown. Potential scan rate 20 mV s−1. | ||
Compatibly with the results obtained from GO samples annealed at 50 °C, an intensity decrease of feature I, related to epoxide electrochemical reduction, is also detected, with a parallel increase in the intensity of feature III (C–OH groups), at least up to 8 h annealing (blue curve). A further annealing up to 48 h (green curve) leads to a sizeable decrease of the overall signal, pointing at a partial loss of electroactivity of the GO nanosheets. Two main differences can further be seen in these CVs with respect to those related to GO-50 samples. The first is a potential shift to less negative values of both features I and III, which respectively move of −0.04 and −0.06 V in the GO-80-2 sample compared to pristine GO, and of further −0.015 and −0.03 V in the GO-80-8 sample. In the samples annealed for longer times, i.e., GO-80-24 and GO-80-48, feature III becomes smoother and broader, while feature I is still detectable and is found to further shift down to −0.78 V in GO-80-48 (green curve in Fig. 3). This potential backshift of the voltammetric reduction features of GO was already recently detected by us in samples treated with mild chemical reducing agents,27 and associated to an extension of C sp2 islands or to their coalescence into larger and interconnected domains, with a consequent increase in electron affinity and conductivity. The mild thermal treatment at 80 °C is therefore also found to modify the overall electronic properties of GO, and eventually facilitate the electrochemical reduction of its oxygenated functional groups.27
Another difference between the GO-50 and GO-80 CV datasets is the absence in these latter samples of a current signal in the potential range between features III and IV, which remains very similar in all samples to pristine GO.
The C 1s XPS spectra of GO-80 samples are reported in Fig. 4, with the corresponding curve fitting results and a comparison with pristine GO.
 | ||
| Fig. 4 XPS C 1s spectrum of GO samples belonging to the GO-80 dataset: (a) pristine GO (GO-0), (b) GO-80-2, (c) GO-80-8, (d) GO-80-24, (e) GO-80-48. Raw data are reported in dots, while curve fitting results in coloured curves, with chemical assignments reported in (a). | ||
In this set of samples, the contributions from the various chemically inequivalent C atoms in GO can also be found, and, similarly to the GO-50 samples, upon heating a decrease in the epoxide contribution (green curve) is detected, parallel to an increase in the C–OH signal (magenta curve). On the other hand, as reported in Table 2, the ΔGO-0 values related to C–O–C (epoxide) groups in each of the GO-80 samples are found to be roughly twice as those related to C–OH. Considering that the contribution of epoxide C atoms to the XPS signal is double compared to that coming from hydroxylated C atoms, this relative variation points at the formation of a single C–OH group after the ring opening of an epoxide group. According to this hypothesis, the number of O atoms in GO is expected to be maintained constant, in accord, within the ±10% associated error with the RO/C values computed for these samples, which vary in a narrow range between 0.39 and 0.43 (Table 2).
| Sample | Annealing time (h) | CC |
C–OH | C–O–C | CO/COO− |
COOH | RO/C | ΔGO-0 | ΔGO-0 |
|---|---|---|---|---|---|---|---|---|---|
| C–OH | C–O–C | ||||||||
a Associated error is ±10%.b As estimated by the following equation, where Px represents the area of peak x in the C 1s spectrum:  c ΔGO-0 values represent the variation in percent area related to selected functional groups with respect to GO-0 sample. c ΔGO-0 values represent the variation in percent area related to selected functional groups with respect to GO-0 sample. |
|||||||||
| GO-0 | — | 45.6 | 10.2 | 34.1 | 7.6 | 2.4 | 0.40 | — | — |
| GO-80-2 | 2 | 50.5 | 16.2 | 22.5 | 9.7 | 1.1 | 0.39 | +6.0 | −11.6 |
| GO-80-8 | 8 | 50.2 | 19.4 | 20.0 | 8.9 | 1.5 | 0.41 | +9.2 | −14.1 |
| GO-80-24 | 24 | 53.8 | 22.3 | 13.3 | 8.0 | 2.6 | 0.42 | +12.1 | −20.8 |
| GO-80-48 | 48 | 54.5 | 22.9 | 11.8 | 7.6 | 3.2 | 0.43 | +12.7 | −22.3 |
As to the variations among the different GO-80 samples, the effect of longer annealing times seems to increase the extent of the conversion of epoxide groups into C–OH, with the deepest ΔGO-0 variations found in the GO-80-48 sample.
The resulting invariance of the RO/C value compared to pristine GO is coherent with the CV results, where, at least in the samples heated up to 8 h, the decrease in feature I (epoxide reduction) seems to be counterbalanced by an increase in feature III (C–OH reduction), with no increased current between III and IV. At the same time, the increased percent area related to the CC signal (Table 2) in GO-80 samples compared to pristine GO supports the enhanced electron affinity experienced by all GO-80 samples, and resulting in the potential backshift of the voltammetric features (Fig. 3). Overall, these results are compatible with a reaction path reported by Kim et al.10 In this case, temperature affects the mobility of basal C–H groups, which are required for an intramolecular redox process occurring with a vicinal hydroxyl group (see the Theoretical modeling section). Therefore, we hypothesize that at 80 °C a C–H group migrates in close vicinity to a C–OH moiety. These two groups condense together, restoring a CC bond with a loss of a water molecule (see Fig. 6b). The proposed path is compatible with both the increasing of the CC XPS signal and the ΔGO-0 values found for the epoxide and the hydroxyl groups (Table 2).
Raman analysis
Fig. 5 reports the experimental Raman spectra taken in the D and G regions for the investigated samples. It is evident, from visual inspection of Fig. 5a and b, that already after 24 h of thermal treatment at both 50 and 80 °C a structural modification occurs on the pristine GO material. | ||
| Fig. 5 Raman spectra of the D and G regions of the GO samples annealed at (a) 50 °C and (b) 80 °C for the reported time intervals. (c) Comparison between Raman spectra of GO samples annealed at 50 and 80 °C for the respective largest time intervals tested. | ||
The Raman spectrum of pristine GO displays the D and G bands localized at ∼1354 cm−1 and ∼1598 cm−1, respectively.5,21,26,30 The ratio ID/IG increases from 0.71 in pristine GO to 0.87 in the GO-50-24 sample (Fig. 5a, red curve), indicating a partial introduction of surface defects in the basal plane of GO, probably due to chemical transformations initiated by water molecules, as pointed out by both CV and XPS analyses. Furthermore, the spectrum of GO-50-24 is markedly comparable to that of GO-50-168 (Fig. 5a, blue curve), suggesting a negligible structural evolution of the GO layers despite the prolonged time of thermal treatment. The same trend of variation can be found in the GO-80 samples (Fig. 5b), where a closely comparable increase of the ID/IG ratio is observed after 24 h (ID/IG = 0.86, Fig. 5b, red curve), and is maintained constant up to 48 h (Fig. 5b, blue curve). The comparison between Raman spectra of GO samples annealed at 50 and 80 °C for the respective largest time intervals is reported in Fig. 5c and shows that the two samples display a very similar defectivity. It is worth noting that, since the oxygen content after thermal treatments for both GO-50 and GO-80 series presents changes which are not sizeable (see O/C ratios in Tables 1 and 2), the observed modifications of the Raman spectra as a consequence of the mild annealing process are most likely related to morphological changes due to clustering processes13 rather than to a variation in the oxygen content.
According to the ID/IG values found, the mean distance among defects in the graphene layer, LD, can be approximated to decrease from ∼12 nm in GO to ∼9 nm on passing from GO-0 to GO-50 and GO-80 samples.31,32
Theoretical modeling
DFT calculations were performed to get insights into the mechanistic aspects of the thermal treatment. GO was modelled within a cluster approach using a B3LYP hybrid functional and a triple basis set plus polarization (6-311g**). See ESI† for computational details. At 50 °C, water molecules, approaching the GO basal epoxide groups, can promote the epoxy ring opening and, in turn, the formation of vicinal diols species (Fig. 6a). In fact, the corresponding process is computed to be isoergonic (ΔG = 1.8 kcal mol−1), but the huge amount of solvent water molecules around the GO sheets suggests that the equilibrium is strongly shifted toward the formation of diols. | ||
| Fig. 6 Proposed modification paths of GO after mild annealing at (a) 50 °C and (b) 80 °C. Values of ΔG refer to Gibbs free energy (kcal mol−1). | ||
At 80 °C (Fig. 6b), temperature can activate the proton migration toward the vicinal hydroxyl groups on the GO basal plane. This step requires about 40 kcal mol−1 to occur. Once the hydroxyl and the proton are close enough, a proton transfer process leads to the formation of a new CC double bond with the elimination of water. This step requires 30 kcal mol−1 to occur and it is strongly exorgonic (ΔG = −25.8 kcal mol−1). As a matter of fact, heating at 50 °C promotes an equilibrium process pushed to products formation by an excess of reactant, while at 80 °C a GO reduction path is activated, kinetically driven by the proton migration upon the GO surface.
Conclusions
In this work, the mild thermal annealing of GO dispersions in water has been investigated. The layers annealed at 50 °C display an increased O concentration correlated to a transformation of the epoxide functional groups into vicinal diols, as revealed by the combined characterization through XPS and cyclic voltammetry. This reaction is most likely the consequence of a near-equilibrium epoxide-rings aperture exerted by water molecules shifted toward the formation of diols (Fig. 6a). The longest duration of thermal treatment, 168 h, leads to a partial reduction of GO owing to the prominent depletion of epoxide groups and an increase of CC up to about 50.9%, as proved by XPS analyses. Conversely, in the samples heated at 80 °C, the XPS spectra clearly show that an epoxide-to-hydroxyl conversion occurs, but with an O concentration maintained constant with respect to pristine GO. Cyclic voltammetry is coherent with this picture within the first hours of thermal annealing, wherein an intensity gain is detected in the reduction wave assigned to hydroxyl groups at the expenses of that related to epoxide groups. These findings call for a reaction path at T = 80 °C where a further step is added compared to the lower T case. In this case, the higher T enhances the mobility of C–H groups throughout the GO basal plane, eventually driving the thermodynamically favoured restoration of a CC bond via loss of water and leaving a single hydroxyl group on the layer. All these modifications in the chemical composition of oxygen-based functional groups were also supported by Raman analyses, which revealed that the main modification to the D and G band lineshape and intensity occurs in the first hours of the annealing treatment. Overall, these experimental results shed light on the chemical transformations that occur on GO at mild temperatures, with a specific significance assigned to the role of water molecules. Particular attention and accuracy should then be taken when GO is handled in aqueous media at high temperatures.
Author contributions
I. Ferrari: investigation, data curation, formal analysis. A. Motta: conceptualization, methodology, software, writing – original draft, writing – review &. R. Zanoni: conceptualization, supervision, writing – original draft. F. Amato: visualization, data curation, writing – review & editing. E. A. Dalchiele: visualization, writing – review & editing. A. G. Marrani: conceptualization, methodology, investigation, data curation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Sapienza Università di Roma: grant numbers RM120172B80059DA (R. Z.), RM11916B88D8E044 (A. G. M.), AR2221814CBEA5BC (F. A.), and Visiting Professor Grant 2022 assignation to E. A. D. E. A. D. acknowledges CSIC (Comisión Sectorial de Investigación Científica), Universidad de la República, Montevideo, Uruguay, PEDECIBA – Física. Prof D. Gazzoli is kindly acknowledged for assistance in Raman spectroscopy. Computational resources were provided by CINECA Computing Cluster under the ISCRA initiative (award no. HP10C0T7WT 2022). SNN-Lab-Sapienza Nanoscience & Nanotechnology Lab is kindly acknowledged for assistance in the FE-SEM images acquisition.References
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- F. Amato, A. Motta, L. Giaccari, R. Di Pasquale, F. A. Scaramuzzo, R. Zanoni and A. G. Marrani, Nanoscale Adv., 2023, 5, 893–906 RSC.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761–19781 CrossRef CAS.
- S. Kim, S. Zhou, Y. Hu, M. Acik, Y. J. Chabal, C. Berger, W. De Heer, A. Bongiorno and E. Riedo, Nat. Mater., 2012, 11, 544–549 CrossRef CAS PubMed.
- P. V. Kumar, N. M. Bardhan, S. Tongay, J. Wu, A. M. Belcher and J. C. Grossman, Nat. Chem., 2014, 6, 151–158 CrossRef CAS PubMed.
- C. Y. Chen, H. Y. Chiu, S. J. Chang, N. L. Yeh, C. H. Chan, C. C. Shih, S. L. Chen, J. W. Yang, C. Y. Huang and G. Y. Chen, ACS Biomater. Sci. Eng., 2023, 9, 2148–2155 CrossRef CAS PubMed.
- P. V. Kumar, N. M. Bardhan, G.-Y. Y. Chen, Z. Li, A. M. Belcher and J. C. Grossman, Carbon, 2016, 100, 90–98 CrossRef CAS.
- P. Sun, Y. Wang, H. Liu, K. Wang, D. Wu, Z. Xu and H. Zhu, PLoS One, 2014, 9, e111908 CrossRef PubMed.
- T. Foller, R. Daiyan, X. Jin, J. Leverett, H. Kim, R. Webster, J. E. Yap, X. Wen, A. Rawal, K. K. H. De Silva, M. Yoshimura, H. Bustamante, S. L. Y. Chang, P. Kumar, Y. You, G. H. Lee, R. Amal and R. Joshi, Mater. Today, 2021, 50, 44–54 CrossRef CAS.
- S. B. Singh and S. A. Dastgheib, Carbon Trends, 2023, 10, 100251 CrossRef CAS.
- I. Ferrari, A. Motta, R. Zanoni, F. A. Scaramuzzo, F. Amato, E. A. Dalchiele and A. G. Marrani, Carbon, 2023, 203, 29–38 CrossRef CAS.
- A. G. Marrani, A. Motta, F. Amato, R. Schrebler, R. Zanoni and E. A. Dalchiele, Nanomaterials, 2022, 12, 1–21 Search PubMed.
- A. G. Marrani, A. Motta, R. Schrebler, R. Zanoni and E. A. Dalchiele, Electrochim. Acta, 2019, 304, 231–238 CrossRef CAS.
- A. G. Marrani, R. Zanoni, R. Schrebler and E. A. Dalchiele, J. Phys. Chem. C, 2017, 121, 5675–5683 CrossRef CAS.
- A. G. Marrani, A. C. Coico, D. Giacco, R. Zanoni, F. A. Scaramuzzo, R. Schrebler, D. Dini, M. Bonomo and E. A. Dalchiele, Appl. Surf. Sci., 2018, 445, 404–414 CrossRef CAS.
- T. AL-Gahouari, P. Sayyad, G. Bodkhe, N. Ingle, M. Mahadik, S. Shirsat and M. Shirsat, Appl. Phys. A: Mater. Sci. Process., 2021, 127, 170 CrossRef CAS.
- S. Shit, P. Samanta, S. Bolar, N. C. Murmu and T. Kuila, Electrochim. Acta, 2020, 62, 137146 CrossRef.
- V. V. Neklyudov, N. R. Khafizov, I. A. Sedov and A. M. Dimiev, Phys. Chem. Chem. Phys., 2017, 19, 17000–17008 RSC.
- V. H. Pham, T. Gebre and J. H. Dickerson, Nanoscale, 2015, 7, 5947–5950 RSC.
- S. Guo, S. Garaj, A. Bianco and C. Ménard-Moyon, Nat. Rev. Phys., 2022, 4, 247–262 CrossRef CAS.
- A. G. Marrani, A. Motta, V. Palmieri, G. Perini, M. Papi, E. A. Dalchiele, R. Schrebler and R. Zanoni, Mater. Adv., 2020, 1, 2745–2754 RSC.
- M. P. Briggs and D. Seah, Practical Surface Analysis, J. Wiley & Sons, Chichester, 2nd edn., 1990 Search PubMed.
- P. P. Brisebois and M. Siaj, J. Mater. Chem. C, 2020, 8, 1517–1547 RSC.
- J. Bin Wu, M. L. Lin, X. Cong, H. N. Liu and P. H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- S. Eigler, C. Dotzer and A. Hirsch, Carbon, 2012, 50, 3666–3673 CrossRef CAS.
- L. G. Cançado, A. Jorio, E. H. M. Ferreira, F. Stavale, C. A. Achete, R. B. Capaz, M. V. O. Moutinho, A. Lombardo, T. S. Kulmala and A. C. Ferrari, Nano Lett., 2011, 11, 3190–3196 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational methods, O/C ratio calculation from XPS, FE-SEM micrographs. See DOI: https://doi.org/10.1039/d3ra05083a |
| This journal is © The Royal Society of Chemistry 2023 |