
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise synthesis route to access bioactive natural products-dihydrocurcumins/1,7-diarylheptanoids†
Datendra Nath Tripathiab and
Saravanakumar Rajendran *a
*a
aChemistry Division, School of Advanced Sciences, Vellore Institute of Technology Chennai campus, Vandalur-Kelambakkam Road, Chennai – 600127, Tamil Nadu, India. E-mail: saravanakumar.r@vit.ac.in; sar.org@gmail.com; Fax: +91 44 3993 2555; Tel: +91 44 3993 1479
bAnthem Biosciences Private Limited, No 49, Canara Bank Road, Hosur Rd, Electronics City Phase 1, Bommasandra Industrial Area, Bengaluru, Karnataka 560099, India
First published on 1st September 2023
Abstract
Dihydrocurcumin (DHCUR), a natural product and reductive metabolite of curcumin (CUR), is limitedly explored owing to its low bioavailability and limited accessibility due to lack of straightforward synthetic routes and poor yields with known methods. Herein, we report a concise and straightforward route to synthesize DHCUR and its analogs in excellent yields. Dihydroferuloylacetone is condensed with aldehydes to obtain desired DHCURs/1,7-diarylheptanoids in 81–90% yields. The developed protocol facilitates easy access to bioactive natural products, 1,7-diarylheptanoids and DHCUR, for therapeutic study.
Introduction
Dihydrocurcumin (DHCUR, 1) and tetrahydrocurcumin (THCUR) are reduced metabolites of curcumin (CUR, Fig. 1), a yellow pigment found in the rhizome of Curcuma longa, which has been traditionally and widely used as a food coloring additive.1–3 In recent years, there has been a significant increase in the demand for naturally occurring pigments in the health sector owing to their lesser adverse effects and superior binding capacity than curcumin.4 There are few biotransformation5 and plant extraction procedures6 to prepare DHCUR (1) from CUR (2) with low yields. In the reported biotransformation process,5 CUR was incubated with Marchantia polymorpha, which resulted in the formation of THCUR (90% yield) and DHCUR (5% yield). Similarly, the extraction of DHCUR (1) from Curcuma longa rhizome was reported6 with low yield (400 mg of DHCUR extracted from 7 kg of turmeric powder). The synthetic methodology to produce DHCUR is quite limited and difficult to produce in bulk.7–9 Notably, the current reaction conditions provided significantly improved reaction yield when compared to the reported procedures.7–9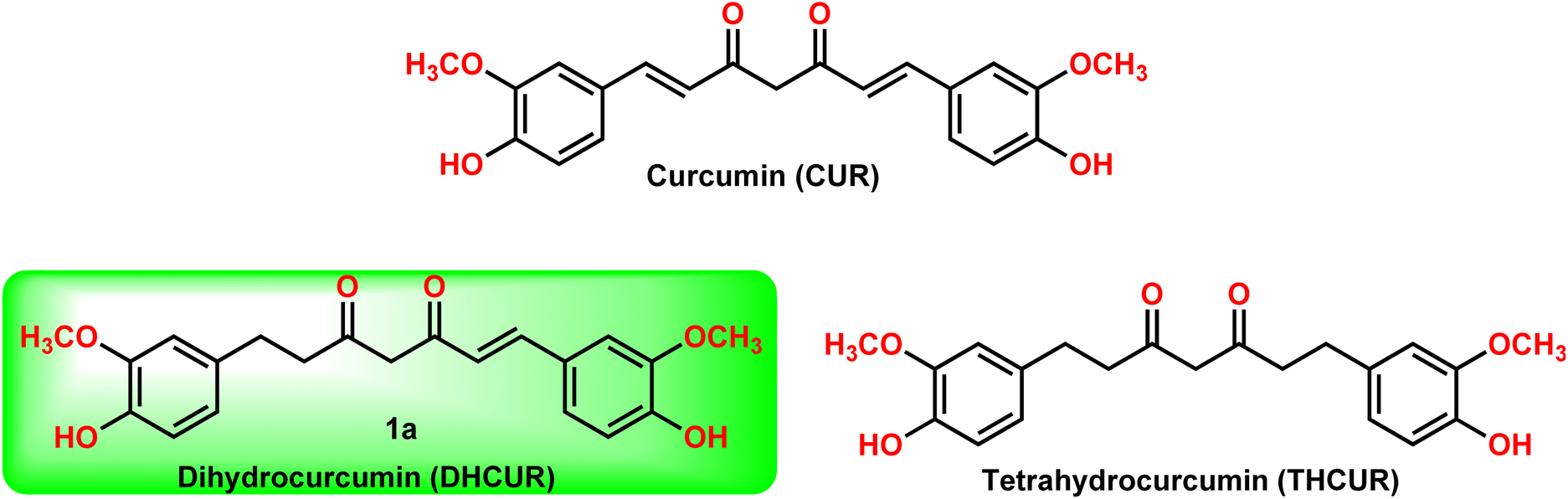 | ||
| Fig. 1 Curcumin and its reductive metabolites. | ||
The synthetic procedures in literature8,9 for the synthesis of DHCUR (1) involve multi-step process and/or require harsh reaction conditions (Fig. 2). For example, S. Venkateswarlu et al. reported8 a newly developed multi-step approach for the synthesis of DHCUR from cinnamic acid. However, the overall yield of this method is limited to only 19%. Furthermore, there is a direct synthetic transformation of CUR to DHCUR using Zn–AcOH,7 however DHCUR was obtained in poor yield (30%). Therefore, a more concise and efficient method is deemed essential for the synthesis of DHCURs. In this study, we report that DHCURs can be readily synthesized by condensing dihydroferuloylacetone (2, Scheme 1) with aldehyde using the Pabon method,10,11 which can be adopted from literature (Scheme 2).12
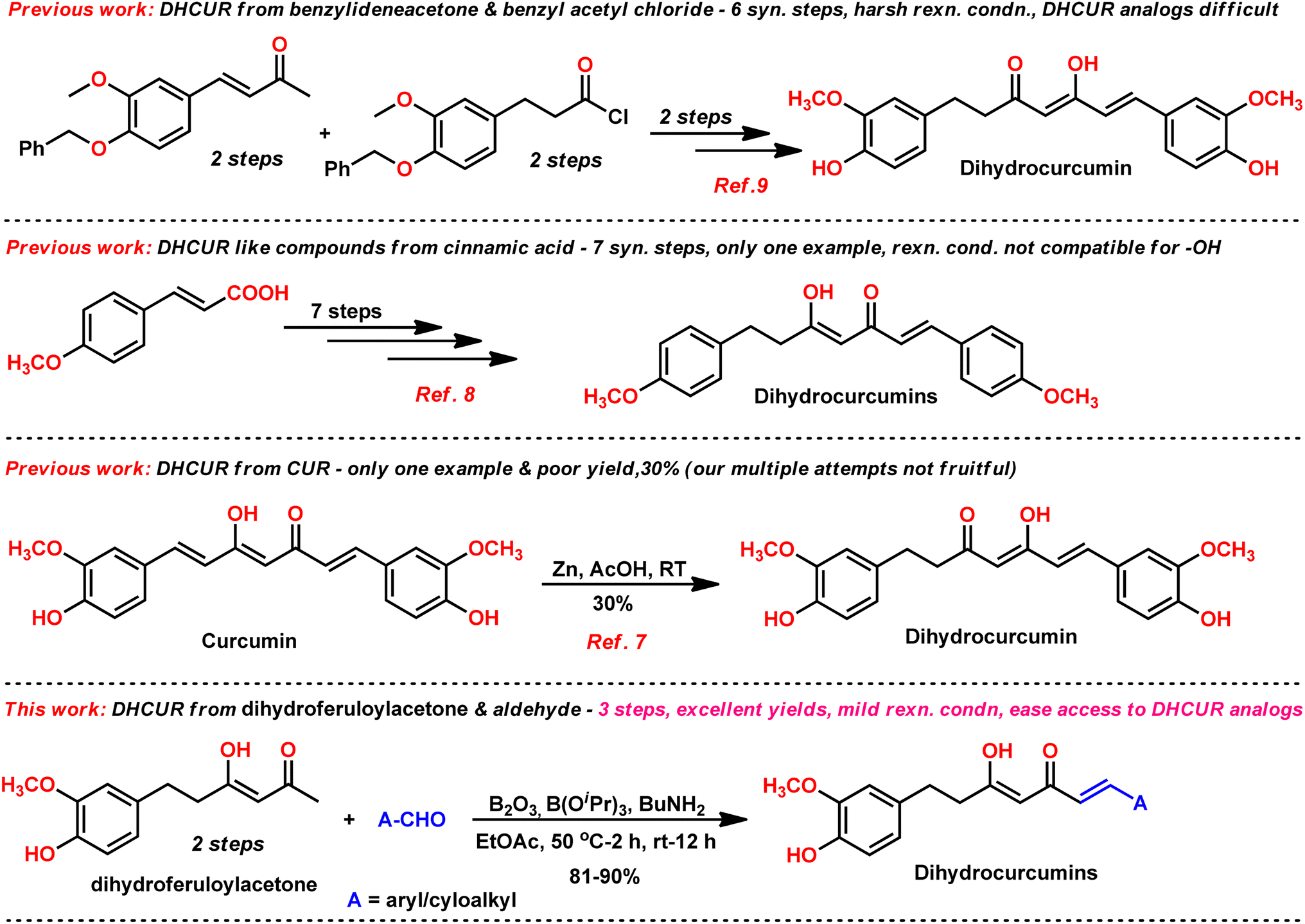 | ||
| Fig. 2 Comparison of the present work with previous works. | ||
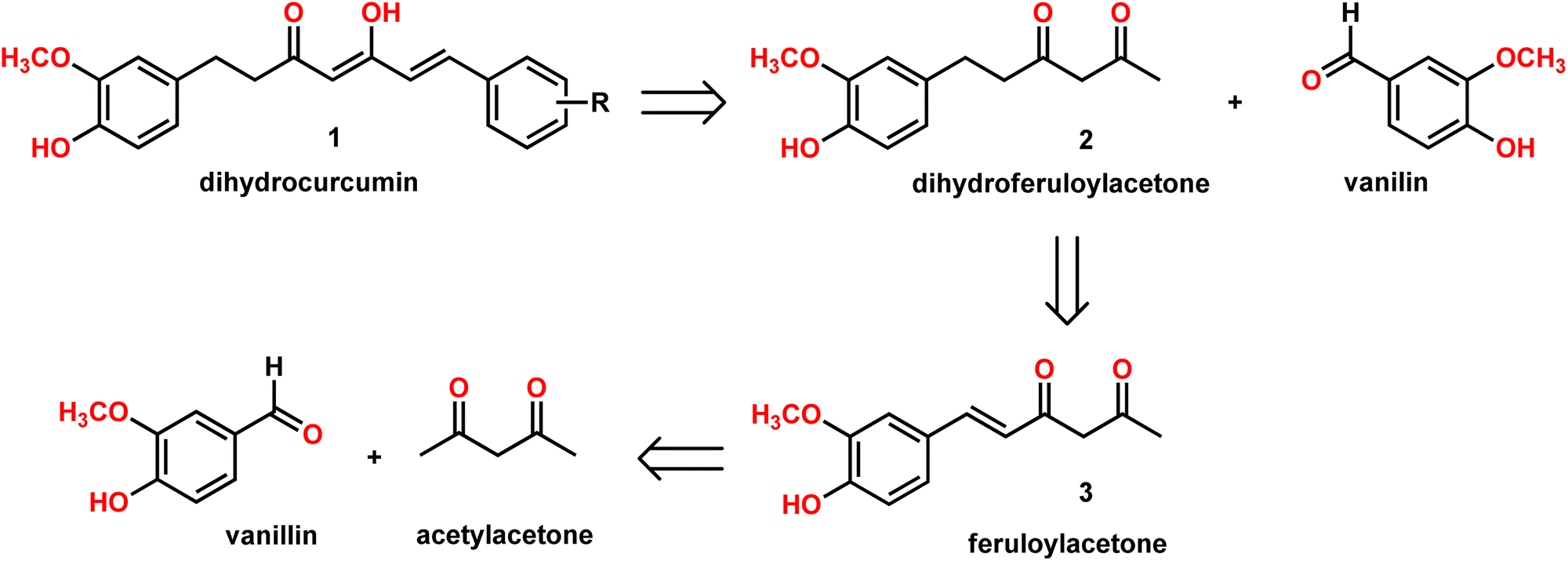 | ||
| Scheme 1 Proposed retrosynthesis for dihydrocurcumins (DHCURs). | ||
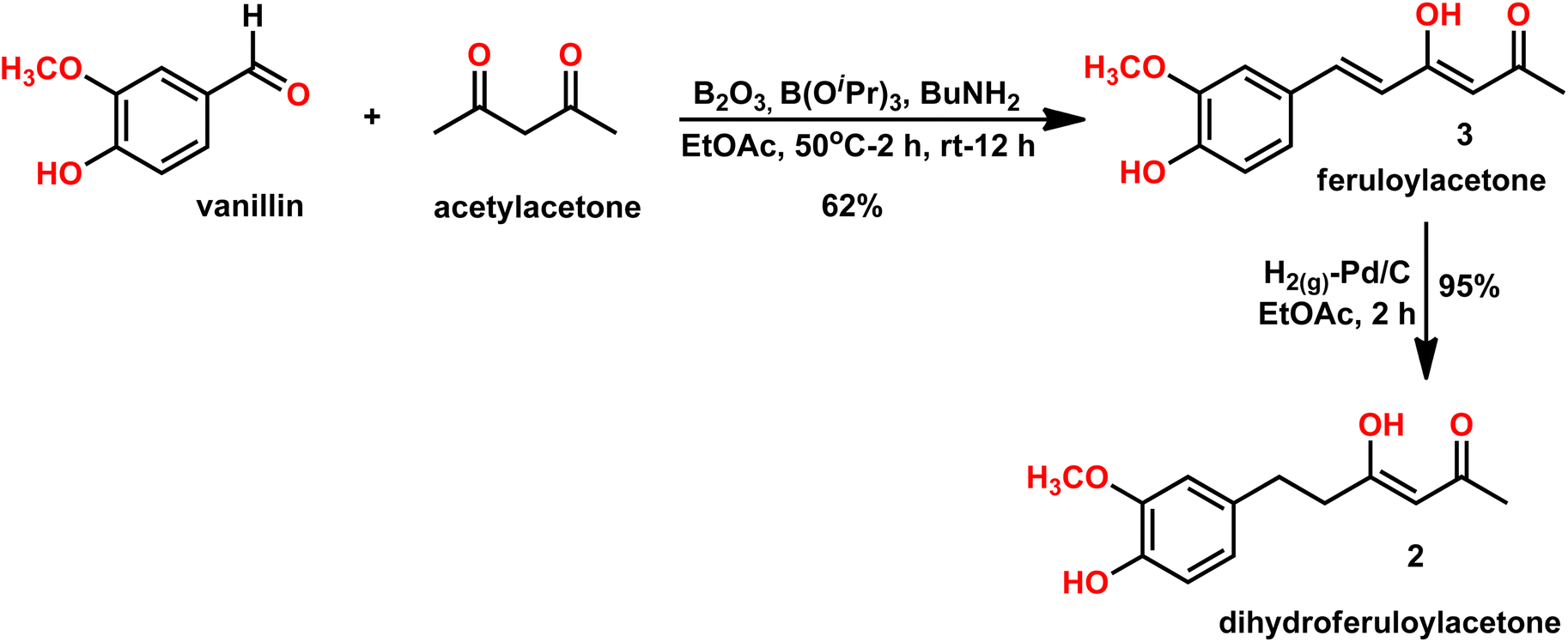 | ||
| Scheme 2 Synthesis of dihydroferuloylacetone (2). | ||
Results and discussion
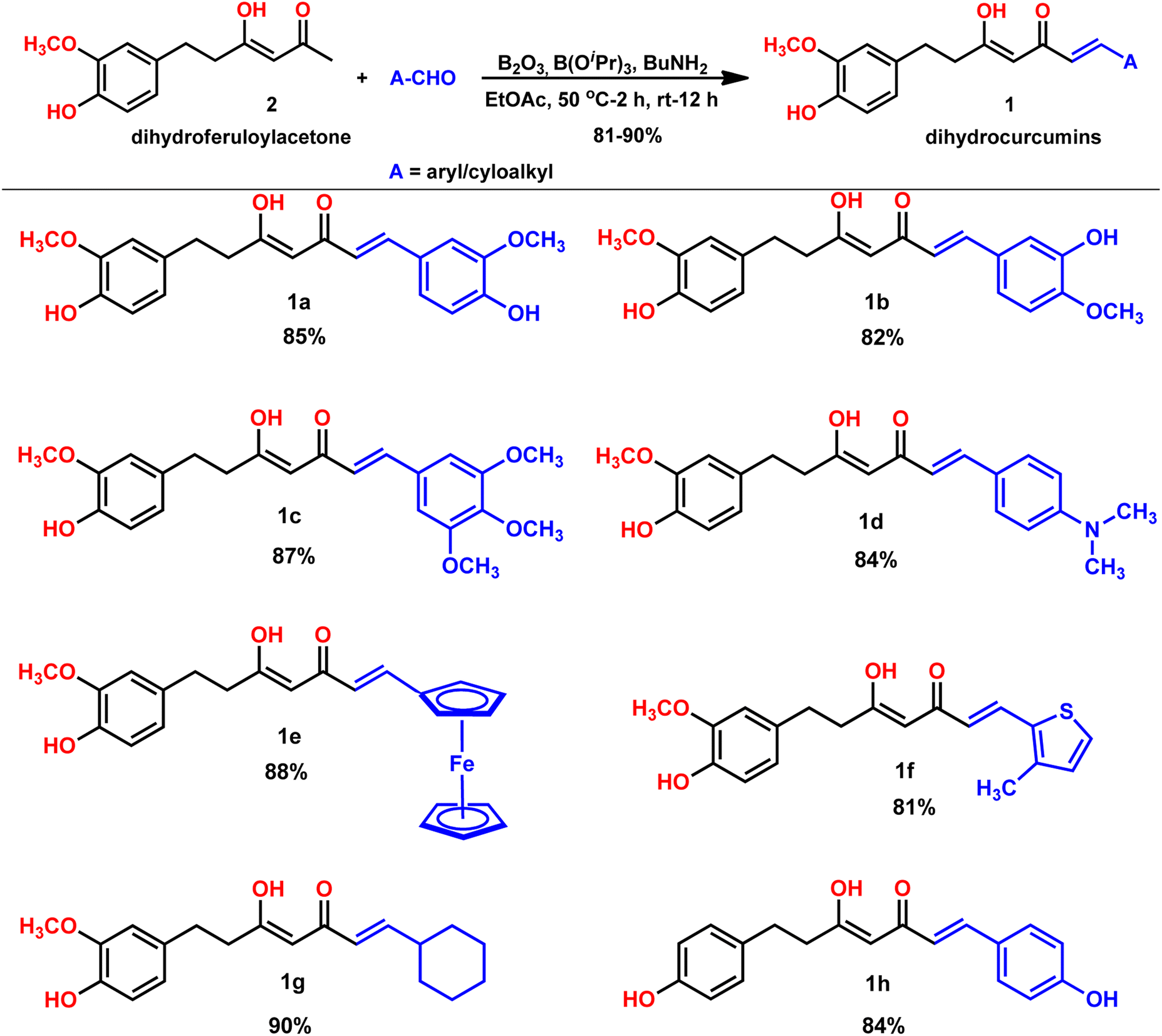
Our group is actively involved in the synthesis of curcumin hybrids for anticancer and anti-inflammatory studies. In our efforts to synthesize curcumin hybrids, we sought to synthesize DHCUR as an intermediate. Literature survey indicated the existence of only three synthetic methods to prepare DHCUR. Two were multi-step approaches,8,9 while the third was a direct reduction of CUR to DHCUR in the presence of Zn–AcOH,8 whereby the best yield reported was 30%.7 Thus, we decided to follow the latter procedure. However, our multiple attempts to reduce CUR using the Zn–AcOH system failed. The starting material (CUR) was the only one recovered. We then attempted partial hydrogenation using the Pd/C catalyst system at various time scales, and the results always ended up with THCUR. Also, we tried this reaction with Pd/C/HCOONH4 (in molar equivalents), but only the formation of THCUR was observed. M. Soriano-Garcia et al. reported the crystal structure of DHCUR,13 whereby they mentioned the synthesis of DHCUR; however, no detailed synthetic procedure or reference was provided. So, we decided to develop a new and better synthetic route to access DHCUR and its analogs with better yield. Our retrosynthetic (Scheme 1) analysis revealed that dihydroferuloylacetone (2) could be a potential starting material for the synthesis of DHCUR (1) and its analogs. The desired dihydroferuloylacetone (2) could be readily prepared from feruloylacetone (3), which could be synthesized easily from acetyl acetone and vanillin according to the procedure in literature.12Synthesis of dihydroferuloylacetone (2)
A previously reported procedure in the literature was adopted for the synthesis of dihydroferuloylacetone (2).12 The synthesis started with vanillin and acetylacetone, using the Pabon10,11 method, whereby the vanillin was condensed with acetylacetone. In this method, initially, acetylacetone reacted with boric anhydride at 50 °C to form an acetylacetone–boron complex (Scheme 2), which is then condensed with vanillin in the presence of butylamine to produce 62% yield of feruloylacetone (3). The latter compound was hydrogenated (in an autoclave at 1 kg cm−2 pressure for 2 h) with 10% Pd/C catalyst at room temperature to form dihydroferuloylacetone (2) in 95% yield.12 The formation of feruloylacetone (3) and dihydroferuloylacetone (2) was confirmed by m.pt, 1H NMR, 13C NMR, FT-IR spectroscopy, and LC-MS. Dihydroferuloylacetone (2) is a dihydro congener of feruloylacetone (3) but has a significant melting point difference. Feruloylacetone (3) melts at 145–147 °C, whereas dihydroferuloylacetone (2) melts at a much lower temperature of 39–41 °C. 1H NMR clearly showed the absence of alkene protons in dihydroferuloylacetone (2), which appeared at 7.54 ppm and 6.33 ppm in feruloylacetone (3), and further verified by mass spectrometry. The corresponding M + 1 peak was observed for feruloylacetone (3) and dihydroferuloylacetone (2), while the melting point and spectral attributes were as per the literature-reported values.12After synthesizing the potential precursor, dihydroferuloylacetone (2), we set out to condense various benzaldehydes/cyclohexyl aldehydes to achieve the desired target compounds, dihydrocurcumins. The Pabon10,11 method was adopted for the condensation reaction. As per the method, dihydroferuloylacetone (2) was reacted with boric anhydride at 50 °C to form the dihydroferuloylacetone–boron complex. It was then condensed with vanillin and other aldehydes (Table 1) in the presence of butylamine to achieve the desired target compounds, dihydrocurcumins of 81–90% yield. Electron donating, heterocyclic, organometallic, and cyclohexyl aldehydes were found to be compatible under the reaction. Electronic effect on the reaction rate and yield were undetected. The structure of synthesized dihydrocurcumins was confirmed by m.pt, 1H NMR, 13C NMR, FT-IR spectroscopy, LC-MS, and elemental analysis. 1H and 13C NMR spectral attributes, atomic and mass analysis were as per the structure of dihydrocurcumins. To highlight the broad applicability of our protocol, the synthesis of DHCUR (1a) was demonstrated in multi-gram scale (see Experimental section).
 |
The structural elucidation was performed by using 1a as the sample. In the 1H NMR spectrum, the singlet at 15.67 ppm was assigned to the enolic proton. Similarly, two separate singlets at around 9.63 and 8.71 ppm were assigned to two phenolic protons. To further confirm this, D2O exchange experiment was conducted. The enolic and phenolic proton peaks eventually disappeared (refer ESI† for the spectrum) as expected. Surprisingly, the active methylene protons were exchanged to greater extent with D2O. It is only integrated to 0.2. A doublet at 7.45 ppm is assigned to the β-alkene proton, and its coupling constant (J) value of 16 Hz indicated that it is an E/trans alkene. The α-alkene proton peak appeared along with aromatic protons and hence it is non-differentiable. One of the aromatic proton peaks appeared as a sharp singlet at 7.29 ppm. A doublet at 7.10 ppm with coupling constant (J) value of 8 Hz was assigned to one of the aromatic proton. The other aromatic proton peaks appeared as two separate multiplets, one around 6.81–6.79 ppm and the other around 6.67–6.59 ppm. A sharp singlet at 5.86 ppm was assigned to the methine proton (Fig. 3). Due to the unsymmetrical structure, the two methoxy protons appeared as two separate singlets, one at 3.82 and the other at 3.73 ppm. Benzylic and methylene protons appeared as two separate triplets at 2.79 ppm and 2.66 ppm, respectively.
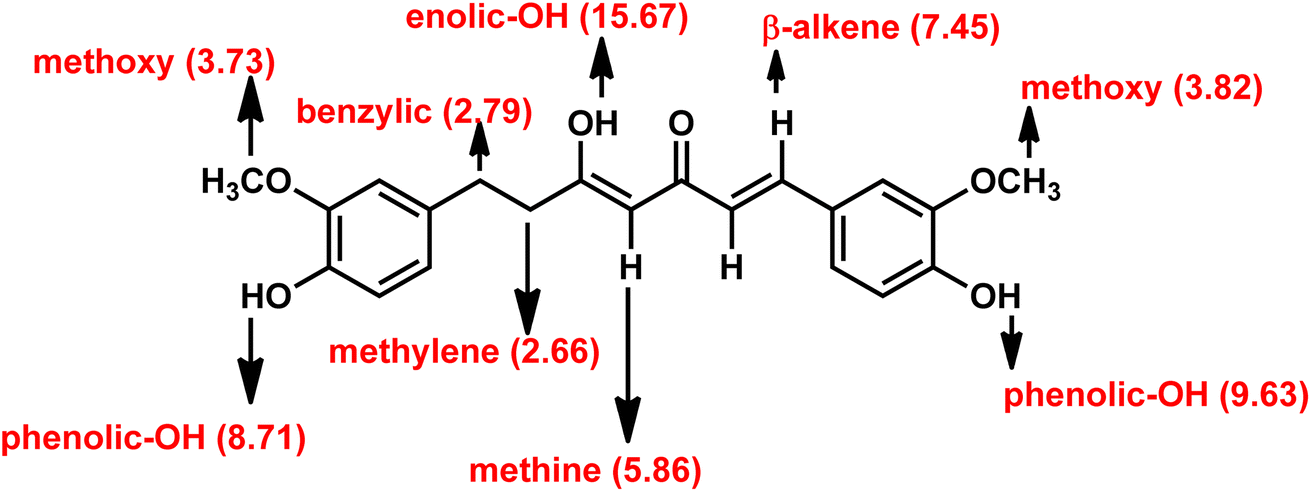 | ||
| Fig. 3 Proton label of compound 1a. In the bracket 1H NMR (DMSO-d6) δ value in ppm. | ||
The 13C NMR spectrum of 1a showed a characteristic negative signal for keto and enolic carbon at 198.9 and 177.9 ppm, respectively. Positive signals at 41.1 and 30.3 ppm were assigned to the benzylic and methylene carbons. The aromatic carbon signals appeared around 149.1–111.1 ppm. A positive signal at 100.0 ppm was assigned to methine carbon. FT-IR spectrum of 1a featured a characteristic –OH absorption band at 3414 cm−1 and a keto carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) absorption band at 1630 cm−1. LC-MS spectrum showed a characteristic M + 1 peak at 371.9 for 1a. The obtained elemental analysis value of 1a is in accord with theoretical data.
O) absorption band at 1630 cm−1. LC-MS spectrum showed a characteristic M + 1 peak at 371.9 for 1a. The obtained elemental analysis value of 1a is in accord with theoretical data.
To highlight the broad applicability of our protocol, we demonstrated the synthesis of dihydro-bisdemethoxycurcumin (1h, Table 1) using the above protocol with 84% yield. This compound had been isolated as an antiplatelet diarylheptanoid from the Chinese medicinal plant, Alpina blepharocalyx K. Schum (Zingeberaceae). The formation and structure of dihydro-bisdemethoxycurcumin (1h) is analyzed and verified by various spectroscopic techniques.
Conclusions
A simple and concise protocol was developed for the synthesis of dihydrocurcumins/1,7-diarylheptanoids, a small group of bioactive natural products. Dihydroferuloylacetone was used as a precursor for the synthesis of dihydrocurcumins/1,7-diarylheptanoids. The condensation of dihydroferuloylacetone with various aromatic and cyclohexyl aldehydes using the Pabon method produced dihydrocurcumins with 81–90% yield. In particular, the developed protocol offers a new pathway to generate dihydrocurcumins/1,7-diarylheptanoids with significantly improved reaction yield when compared to the previously reported procedures. Thus, our protocol enables easy access to dihydrocurcumins/1,7-diarylheptanoids on a multi-gram scale to explore the bioactivity potential of these compounds.Experimental section
Synthesis of feruloylacetone (3)
A mixture of acetylacetone (50 g, 1 eq.) and boric anhydride (17.5 g, 0.5 eq.) was refluxed in ethyl acetate (500 mL) for 30 min followed by vanillin (19.0 g, 0.25 eq.) in ethyl acetate (60 mL) was added to the acetyl acetone–boron complex mixture at 50 °C together with triisopropyl borate (230.5 mL, 2.0 eq.). The reaction mixture was stirred vigorously for 30 min at 50 °C, followed by the dropwise addition of n-butyl amine (19.74 mL, 0.2 eq.) in ethyl acetate (100 mL) over 2 h at 50 °C. Then, the reaction mixture was cooled to room temperature with continuous stirring overnight. Upon completion of the reaction (followed by TLC), the reaction mass was neutralized with 1 N HCl (300 mL) and the mixture was stirred for another 30 min. The organic layer was separated, and the aqueous fraction was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were washed with water, and the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to get the crude product. The crude product was purified by column chromatography using gradient hexane-ethyl acetate (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) to afford feruloylacetone (3) as a pale yellow solid in 18.12 g (62% yield).
:3) to afford feruloylacetone (3) as a pale yellow solid in 18.12 g (62% yield).
Pale yellowish solid, m.pt 145–147 °C (lit. 146–147 °C), IR (KBr): 3263 (–O–H), 1634 (–CO), 1571 (–CC–CO), 1515 (–CC–), 1030 (–C–O) cm−1. 1H NMR: (400 MHz, CDCl3) δ: 7.54 (d, J = 16 Hz, 1H, Ar–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) CH–), 7.11–7.09 (m, 1H, Ar–), 7.03 (d, J = 4 Hz, 1H, Ar–), 6.93 (d, J = 8 Hz, 1H, Ar–), 6.33 (d, J = 16 Hz, 1H, Ar–CHC–C(O)–), 5.86 (bs, 1H, Ar–O), 5.64 (s, 1H, methine–), 3.95 (s, 3H, –OC3), 2.17 (s, 3H, (O)CC3). 13C NMR: (100 MHz, CDCl3) δ: 197.1 (–
CH–), 7.11–7.09 (m, 1H, Ar–), 7.03 (d, J = 4 Hz, 1H, Ar–), 6.93 (d, J = 8 Hz, 1H, Ar–), 6.33 (d, J = 16 Hz, 1H, Ar–CHC–C(O)–), 5.86 (bs, 1H, Ar–O), 5.64 (s, 1H, methine–), 3.95 (s, 3H, –OC3), 2.17 (s, 3H, (O)CC3). 13C NMR: (100 MHz, CDCl3) δ: 197.1 (–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O), 177.9 (–OH), 147.7 (Ar–C–), 146.8 (Ar), 140.1 (Ar), 127.7 (Ar), 122.6 (Ar), 120.3 (Ar), 114.8 (Ar), 109.5 (Ar), 100.7 (methine–), 55.9 (–OH3), 26.8 (H3–C(O)). MS: (LC-MS): m/z [M + 1] 235.6.
O), 177.9 (–OH), 147.7 (Ar–C–), 146.8 (Ar), 140.1 (Ar), 127.7 (Ar), 122.6 (Ar), 120.3 (Ar), 114.8 (Ar), 109.5 (Ar), 100.7 (methine–), 55.9 (–OH3), 26.8 (H3–C(O)). MS: (LC-MS): m/z [M + 1] 235.6.
Synthesis of dihydroferuloylacetone (2)
To a stirred solution of feruloylacetone (25 g) in ethyl acetate (300 mL), 2.5 g of 10% Pd/C was added, and the resulting mixture was hydrogenated in an autoclave at 1 kg cm−2 pressure for 2 h. Upon the completion of the reaction (followed by TLC), the reaction mass was filtered through Celite bed to remove solid residues. The filtrate solvent was evaporated under reduced pressure to obtain the crude product as thick viscous oil, which was further purified via column chromatography (60–120 mesh silica) using hexane-ethyl acetate (8:2) mixture as the eluent to get dihydroferuloylacetone (2) as a white solid in 23.95 g with 95% yield.
Off white solid, m.pt 39–41 °C (lit. 40–41 °C), IR (KBr): 3447 (–O–H), 1723(–CO), 1571 (–CC–CO), 1517 (–CC–), 1084 (–C–O) cm−1. 1H NMR: (400 MHz, CDCl3) δ: 15.49 (s, 1H, enolic–O), 6.84–6.81 (m, 1H, Ar–), 6.71–6.65 (m, 2H, Ar–), 5.49 (s, 1H, Ar–O), 5.47 (s, 1H, methine–H), 3.87 (s, 3H, –OC3), 2.88–2.83 (m, 2H, Ar–C2-), 2.58–2.54 (m, 2H, Ar–CH2–C2–), 2.04 (s, 3H, C3–C(O)–). 13C NMR: (100 MHz, CDCl3) δ: 193.4 (–O), 191.2 (–OH), 146.5 (Ar), 144.1 (Ar), 132.7 (Ar), 120.9 (Ar), 114.4 (Ar), 111.1 (Ar), 100.2 (methine–), 56.0 (–OH3), 40.5 (Ar–H2–), 31.4 (Ar–CH2–H2–), 24.9 (H3–C(O)). MS: (LC-MS): m/z [M + 1] 237.2.
General procedure for the synthesis of dihydrocurcumins (DHCURs)
A mixture of dihydroferuloylacetone (2.0 g, 1 eq.) and boric anhydride (0.5 eq.) was heated at 50 °C in ethyl acetate (8 mL) for 45 min. Then, the corresponding aldehyde (0.9 eq.) in ethyl acetate (5 mL) was added to the dihydroferuloylacetone–boron complex mixture at 50 °C along with triisopropyl borate (2.0 eq.). The reaction mixture was stirred vigorously for 30 min at 50 °C until the solution became milky. Next, n-butyl amine (0.2 eq.) in ethyl acetate (5 mL) was added dropwise over 30 min. The reaction mixture was cooled to room temperature and stirred overnight. The reaction mass was neutralized with 1 N HCl (15 mL) and the mixture was stirred for another 30 min. The organic layer was separated and the aqueous fraction was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were washed with water and the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain the crude product. The crude product was purified by recrystallization in ethanol to get dihydrocurcumin (DHCUR, 1) and its analogs with 81 to 90% yield.Multi-gram scale synthesis of dihydrocurcumin (DHCUR, 1a)
A mixture of dihydroferuloylacetone (10.0 g, 1 eq.) and boric anhydride (1.49 g, 0.5 eq.) was heated at 50 °C in ethyl acetate (80 mL) for 45 min. Then, vanillin (5.85 g, 0.9 eq.) in ethyl acetate (30 mL) was added to the dihydroferuloylacetone–boron complex mixture at 50 °C together with triisopropyl borate (19.7 mL, 2.0 eq.). The reaction mass was stirred vigorously for 30 min at 50 °C until the solution became milky. Next, n-butyl amine (0.84 mL, 0.2 eq.) in ethyl acetate (25 mL) was added dropwise over 30 min. The reaction mixture was cooled to room temperature and stirred overnight. The reaction mass was neutralized with 1 N HCl (75 mL), and the mixture was stirred for another 30 min. The organic layer was separated, and the aqueous fraction was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were washed with water and the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain the crude product. The crude dihydrocurcumin was crystallized from ethanol (70 mL) at room temperature. The precipitated solid was filtered, washed with ethanol (10 mL), and dried under reduced pressure at 50 °C to obtain the yellow crystalline powder of dihydrocurcumin (DHCUR, 1a) (13.4 g, 85% yield).Pale yellowish solid, m.pt 113–116 °C, IR (KBr): 3447 (–O–H), 1630 (–CO), 1593 (–CC–CO), 1509 (–CC–), 1131 (–C–O) cm−1. 1H NMR: (400 MHz, DMSO-d6) δ: 15.43 (s, 1H, enolic–O), 9.63 (CH2–Ar–O), 8.71 (Ar–O), 7.48 (d, J = 16 Hz, 1H, Ar–C), 7.29 (s, 1H, Ar–), 7.11–7.09 (m, 1H, Ar–), 6.81–6.79 (m, 2H, Ar–), 6.67–6.59 (m, 3H, Ar–), 5.86 (s, 1H, methine–), 3.82 (s, 3H, –OC3), 3.73 (s, 3H, –OC3), 2.81–2.77 (m, 2H, Ar–C2–), 2.68–2.65 (m, 2H, Ar–CH2–C2–). 13C NMR: (100 MHz, DMSO-d6) δ: 198.9 (–O), 177.9 (–C–OH), 149.1 (Ar–HCH–), 147.9 (Ar–), 147.3 (Ar–), 144.6(Ar–), 140.1 (Ar–), 131.5 (Ar–CHH–), 126.3 (Ar–), 122.8 (Ar–), 120.2 (Ar–), 119.6 (Ar–), 115.6 (Ar–), 115.2 (Ar–), 112.5 (Ar–), 111.1 (Ar–), 100.0 (methine–H–), 55.6 (–OH3), 55.5 (–OH3), 41.1 (Ar–H2–), 30.3 (Ar–CH2–H2–). MS: (LC-MS): m/z [M + 1] 371.9. Anal. calculated for C21H22O6: C, 68.10: H, 5.99: O, 25.92. Found C, 67.96: H, 5.97.
Data availability
The full experimental data related to this article are given in the ESI File.† It includes the analytical data, 1H NMR, and 13C NMR spectra of dihydrocurcumins and their precursors.Conflicts of interest
Authors have no conflict of interest to declare.Acknowledgements
D. N. T. and R. S. K. thank the Chemistry Division and VIT Chennai for providing the infrastructure facility and instrumentation facility. D. N. T. gratefully acknowledges Dr Ganesh Sambasivam, CSO of Anthem Biosciences Pvt. Ltd, for his constant support to conduct the reactions and for allowing the use of the instrumentation facilities, including the 1H and 13C NMR, FT-IR, LC-MS and elemental analysis to characterize the synthesized compounds.References
- S. Fuloria, J. Mehta, A. Chandel, M. Sekar, N. N. I. M. Rani, M. Y. Begum, V. Subramaniyan, K. Chidambaram, L. Thangavelu, R. Nordin, Y. S. Wu, K. V. Sathasivam, P. T. Lum, D. U. Meenakshi, V. Kumarasamy, A. K. Azad and N. K. Fuloria, Front. Pharmacol, 2022, 13, 820806 CrossRef CAS PubMed.
- A. Pandey, M. Chaturvedi, S. Mishra, P. Kumar, P. Somvanshi and R. Chaturvedi, Heliyon, 2020, 6, e05469 CrossRef CAS PubMed.
- M. Heger, R. F. Van Golen, M. Broekgaarden and M. C. Michel, Pharmacol. Rev., 2014, 66, 222–307 CrossRef PubMed.
- K. V. Dileep, I. Tintu and C. Sadasivan, Interdiscip. Sci.: Comput. Life Sci., 2011, 3, 189–197 CrossRef CAS PubMed.
- K. Shimoda, N. Kubota, H. Hirano, M. Matsumoto, H. Hamada and H. Hamada, Nat. Prod. Commun., 2012, 7, 1934578X1200700 Search PubMed.
- V. Ravindranath and M. N. Satyanarayana, Phytochem, 1980, 19, 2031–2032 CrossRef CAS.
- C. Changtam, P. Hongmanee and A. Suksamrarn, Eur. J. Med. Chem., 2010, 45, 4446–4457 CrossRef CAS PubMed.
- S. Venkateswarlu, M. S. Ramachandra, M. Rambabu and G. V. Subbaraju, J. Asian Nat. Prod. Res., 2000, 2, 111–120 CrossRef CAS PubMed.
- H. G. Krishnamurty and S. Ghosh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1986, 25B, 411–412 CAS.
- H. J. J. Pabon, Recl. Trav. Chim. Pays-Bas, 1964, 83, 379–386 CrossRef CAS.
- P. J. Roughley and D. A. Whiting, J. Chem. Soc., Perkin Trans. 1, 1973, 2379 RSC.
- J.-Y. Feng and Z.-Q. Liu, Eur. J. Med. Chem., 2011, 46, 1198–1206 CrossRef CAS PubMed.
- M. C. Lozada, C. E. Lobato, R. G. Enríquez, B. Ortíz, D. Gnecco, W. F. Reynolds and M. Soriano-García, Anal. Sci.: X-Ray Struct. Anal. Online, 2005, 21, X59–X60 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra05049a |
| This journal is © The Royal Society of Chemistry 2023 |