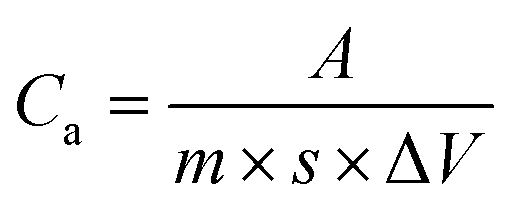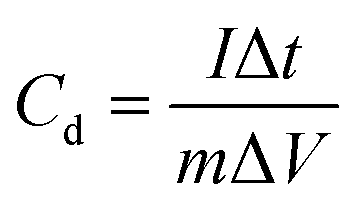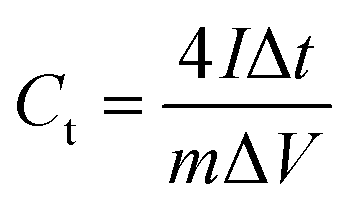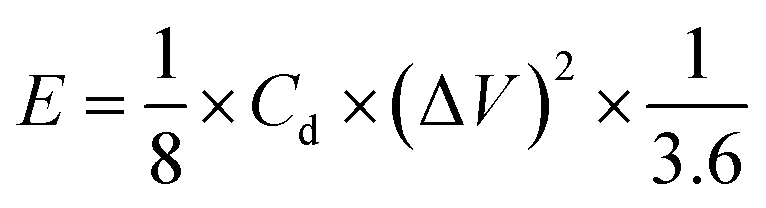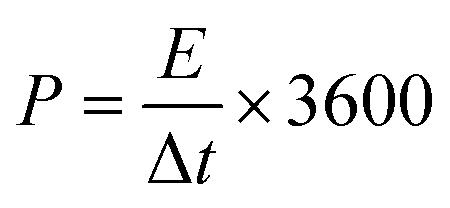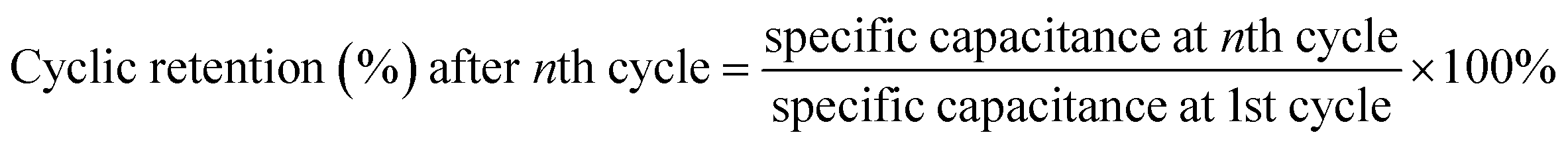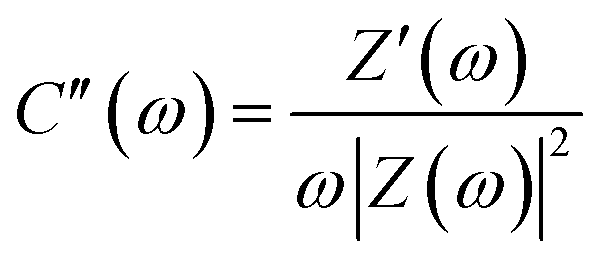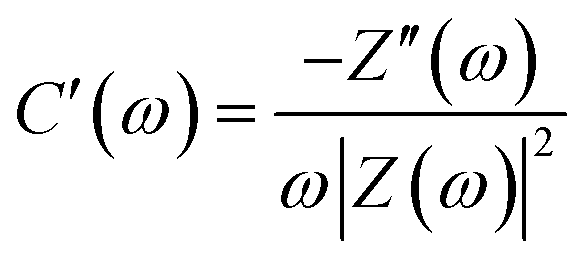DOI:
10.1039/D3RA04862D
(Paper)
RSC Adv., 2023,
13, 25877-25887
N/O Co-doped hierarchical nanoporous biochar derived from waste polypropylene nonwoven for high-performance supercapacitors†
Received
19th July 2023
, Accepted 10th August 2023
First published on 1st September 2023
Abstract
How to efficiently treat municipal solid waste (MSW) has become one of the critical solutions in response to the call for “carbon neutrality”. Here, the waste polypropylene nonwoven fabric of waste diapers was converted into hierarchical nanoporous biochar (HPBC) through pre-carbonization and activation processes as an ideal precursor for supercapacitors (SCs) with excellent performance. The prepared HPBC-750-4 with an ultrahigh specific surface area (3838.04 m2 g−1) and abundant heteroatomic oxygen (13.25%) and nitrogen (1.16%) codoped porous biochar structure. Given its structural advantages, HPBC-750-4 achieved a specific capacitance of 340.9 F g−1 at a current density of 1 A g−1 in a three-electrode system. Its capacitance retention rate was above 99.2% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at a current density of 10 A g−1, which indicated an excellent rate capability and long-term cycling stability. Furthermore, the HPBC-750-4//HPBC-750-4 symmetric SC exhibited a superb energy density of 10.02 W h kg−1 with a power density of 96.15 W kg−1 in a 6 M KOH electrolyte. This work not only demonstrates the enormous potential of waste polypropylene nonwoven fabric in the SC industry but also provides an economically feasible means of managing MSW.
000 cycles at a current density of 10 A g−1, which indicated an excellent rate capability and long-term cycling stability. Furthermore, the HPBC-750-4//HPBC-750-4 symmetric SC exhibited a superb energy density of 10.02 W h kg−1 with a power density of 96.15 W kg−1 in a 6 M KOH electrolyte. This work not only demonstrates the enormous potential of waste polypropylene nonwoven fabric in the SC industry but also provides an economically feasible means of managing MSW.
1. Introduction
In response to the “carbon neutrality” policy, renewable energy sources and corresponding energy storage technologies must be urgently developed to replace conventional fossil fuels.1,2 Numerous studies have been conducted on the development of efficient energy storage materials, among which supercapacitors (SCs) stand out as the most promising alternative devices.3,4 SCs are widely used in medical devices, portable electronics, and other renewable energy systems owing to their long service life, high specific capacity, fast charging and discharging capability, and environmental friendliness.5 Compared with conventional capacitors, the electrodes of SCs typically have a hierarchical porous structure. Depending on the electrode material and electrochemical charging and discharging mechanisms, SCs are subdivided into two categories: electric double-layer capacitors (EDLCs) and Faraday pseudocapacitors (PCs).6 The energy storage of PCs is generated by a fast and reversible redox reaction at the electrode surface,7 which can obtain a higher specific capacitance and energy density than EDLCs; however, the practical application of PCs is limited due to their poor cycling stability, unsatisfactory electrical conductivity, and high manufacturing cost.8 The energy storage of EDLCs stems from the formation of an electric double layer between the electrode and the electrolyte. By storing electrical energy on the electrode surface through physical absorption, EDLCs are characterized by high power density, high cycling stability, and good rate capability.9,10 The development of new robust electrode materials is crucial based on the considerable influence of electrode materials on EDLCs performance.
Typically, electrode materials with hierarchical porous structures and high specific surface area (SSA) can achieve a high energy density while maintaining a high power density. Carbon-based materials, transition metal oxides, and conductive polymers are basic electrode materials commonly used in SCs.11 Transition metal oxides and conductive polymers have relatively good electrical and mechanical properties, but their high cost, poor cycling performance, and thermal stability limit their applications. Carbon-based materials present high potential in SCs materials due to their structural stability, large SSA, well-developed pores, and low price.12 Carbon dots (zero-dimensional), carbon nanotubes and carbon fibers (one-dimensional), graphene (two-dimensional), and three-dimensional carbon frameworks obtained from the pyrolysis of waste biomass and organic polymers as precursors have gained considerable attention as electrode materials.13,14 Amongst them, functionalized porous biochar has gained much attention owing to it advantages of wide in raw materials, easy to modify, and high sustainability.15,16
Doping with heteroatoms (N, O, P, and S) can further improve the specific capacitance and cyclic stability of carbon materials.17,18 The presence of heteroatoms affects the electrochemical interfacial state and electric double-layer capacitive properties of carbon materials.2 As the most widely used dopant atom, N has a slightly smaller radius (0.74 Å) and larger electronegativity (3.04) than C atoms (0.77 Å and 2.55, respectively). The extra electrons in the nitrogen atom effectively alter the electronic band of carbon materials, which results in higher positive charge density and reduced lattice distortion.19–21 On the one hand, doping with N atoms can provide more active sites for energy storage, compensate for the poor electrical conductivity due to low graphitization, and maintain the stability of the original carbon skeleton due to the similar size of nitrogen and carbon atoms.22 On the other hand, the integration of nitrogen component increases the hydrophilicity of carbon materials and improves the lubricity of electrode materials to the electrolyte,23 which further improve the capacitance of EDLCs. Porous biochar materials are generally doped with nitrogen through two methods. The first method is in situ nitrogen doping, where the raw material itself contains nitrogen elements, and nitrogen-doped porous biochar is prepared directly by pyrolytic carbonization and activation without the addition of any nitrogen-containing dopant.24 The second method involves the addition of nitrogen-containing dopants, such as melamine, urea, polyacrylonitrile, and other chemical dopants,25 to introduce nitrogen to the precursor material for carbonization and activation. The second method has received increasing attention in the last decade due to its higher efficiency and generally better electrode performance compared with the first method.
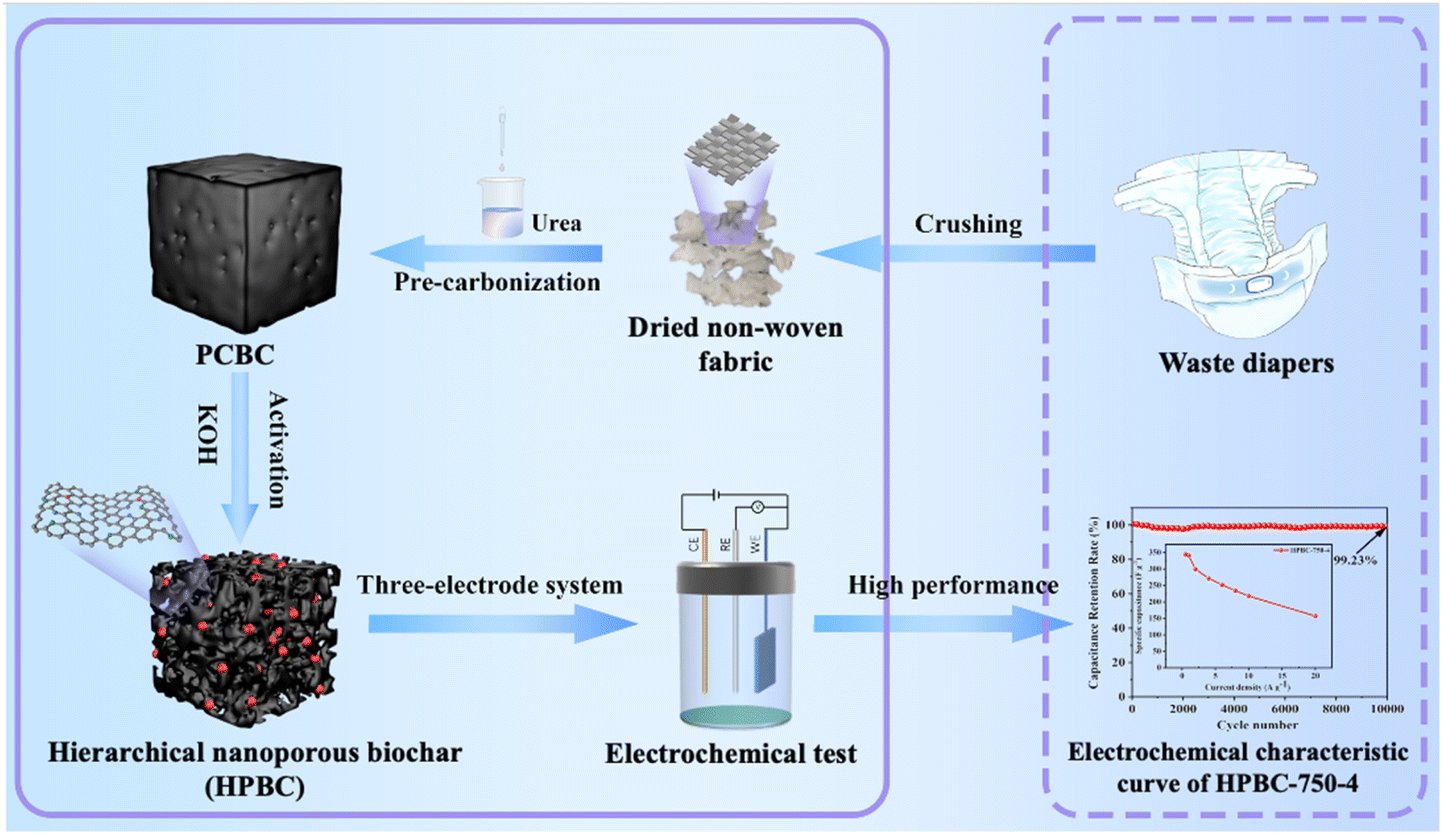
In this work, to alleviate the environmental burden caused by municipal disposable diapers,26 we selected waste polypropylene (PP) nonwoven fabric derived from diapers as the carbon source, and urea was used as the nitrogen source to prepare hierarchical nanoporous biochars (HPBC) through nitrogen doping, pre-carbonization, and chemical activation with KOH under a N2 inert atmosphere (Fig. 1). The specific capacitance, rate capability, and cycling stability of the porous biochar material were then measured to demonstrate its good potential as an electrode material in EDLCs.
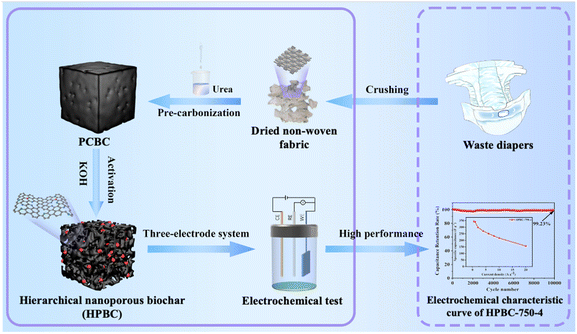 |
| | Fig. 1 Carbonization and activation processes of PP nonwovens fabrics. | |
2. Experimental section
2.1. Materials and reagents
Discarded baby diapers, which were all used by infants within one month of age, were collected from a postpartum care center near Henan University (Kaifeng, Henan). The equipment used in this study included a tubular furnace (CHY-1200, Henan Chengyi Equipment Technology Co., Ltd), a multifunctional crusher (QJ-08B, Shanghai Chaosun Technology Co., Ltd), and a press machine (YLJ-5T, Hefei Kejing Material Technology Co., Ltd). Nitrogen gas (N2, 99%, Xinyuan), anhydrous ethanol (CH3CH2OH, AR, Anhui Ante Food Co., Ltd), KOH (AR, Tianjin Kemio Chemical Reagent Co., Ltd) as the activating agent, urea (H2NCONH2, AR, Tianjin Kemio Chemical Reagent Co., Ltd), nickel foam, polytetrafluoroethylene emulsion (PTFE), and acetylene black (Force Chem) were used to prepare the working electrodes (Section 2.4). Deionized water (DI) was used throughout the experiment.
2.2. Preparation of the N-doped PCBC
The PP nonwoven fabric was removed from dried discarded baby diapers, cut into small pieces, and then crushed in a multifunctional crusher. Next, 10 g nonwoven fabric fragments were weighed and placed in a 500 ml beaker. Then, 200 ml urea solution with a concentration of 0.1 g ml−1 was added to the beaker and mixed thoroughly with a glass rod to ensure that the nonwoven fabric and urea were mixed well. The beakers were placed in an oven, and the temperature was set to 60 °C to completely impregnate both, followed by an increase in temperature to 80 °C to dry them, and then set aside. The impregnated material was then packed into two quartz boats of the same size and placed in a tubular furnace for pre-carbonization. After vacuuming, the temperature was raised to 500 °C at a rate of 2 °C min−1 under a N2 atmosphere. The material was maintained at 500 °C for 2 h to obtain precarbonized products doped with nitrogen and named PCBC.
2.3. Preparation of activated PCBC
The PCBC was rinsed with anhydrous ethanol and filtered using a Buchner funnel with an organic membrane to remove the grease and organic matter generated during the pre-carbonization process. After three rounds of filtration with anhydrous ethanol, the sample was further filtered using DI. The washed PCBC was then dried in an 80 °C oven. First, samples were prepared by mixing PCBC and the activating agent (KOH) at mass ratios of 1:1, 1:2, and 1:4. Then, the mixture was transferred to nickel crucibles. The crucibles were then placed in a tubular furnace, vacuumed, and heated to 750 °C at a rate of 5 °C min−1 under a N2 atmosphere. The temperature was maintained for 2 h, and the products obtained after cooling naturally to room temperature were named HPBC-750-1, HPBC-750-2, or HPBC-750-4 based on the activation ratio used. Second, samples with different activation temperatures were prepared by mixing PCBC and KOH at a mass ratio of 1:4 (where the electrochemical and physicochemical properties exhibited by the mass ratio of PCBC and KOH was 1:4 in Section 3 were deemed the best). The mixture was then placed in a tubular furnace and heated to 600 °C, 750 °C, or 900 °C at a rate of 5 °C min−1 under a N2 atmosphere. The material was maintained at each temperature for 2 h. After natural cooling to room temperature, the products obtained were named HPBC-600-4, HPBC-750-4, or HPBC-900-4.
2.4. Preparation of the HPBC electrodes
To prepare the working electrode, we mixed 80 wt% HPBC active substance, 10 wt% carbon black, and 10 wt% PTFE in a mortar using anhydrous ethanol as a solvent. The mixture was then ground for approximately 10 min until the solvent completely evaporated, and a roughly shaped electrode was formed. The small pieces on the mortar wall were collected with tweezers and attached to the electrode, which was then folded into a sheet-like electrode material. Then, the sheet electrode material was evenly coated on a nickel foam (1 × 1 cm2), compacted tightly using a sheet press under a pressure of 15–20 MPa for 1–2 min, and dried under vacuum at 60 °C for 12 h to obtain the final asymmetric working electrode for measurement of the electrochemical performance. For the two-electrode system, the working electrode was prepared by coating the active substance, which had been ground into thin slices, onto a circular nickel foam with a diameter of 1.1 cm. Cellulose membranes were then used for the assembly of the symmetric capacitor.
2.5. Electrochemical measurement
In this section, the electrochemical properties of electrode sheets were measured by cyclic voltammetry (CV), constant-current charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) reactions at room temperature on an electrochemical workstation (CHI760E). In the three-electrode system, the calomel electrode, platinum wire electrode, and prepared electrode sheet were used as the reference, counter, and working electrodes, respectively, with 6 mol L−1 KOH solution as the electrolyte. EIS was conducted continuously on an electrochemical workstation at an open-circuit voltage with an AC amplitude of 5 mV and a frequency range of 0.1 Hz to 100 kHz. The voltage operating window of CV was set to −1 to 0 V at different scan rates (5–200 mV s−1), and the specific capacitance (Ca, F g−1) can be calculated from the CV curves:| |
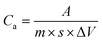 | (1) |
where Ca is the mass specific capacitance, ΔV (V) is the voltage window of the working electrode, A represents half of the integrated area of the CV curve, m (g) is the mass of the active material on the electrode, and s (mV s−1) is the scan rate.
The voltage working window of GCD was set to −1 to 0 V, and charge and discharge were performed at different current densities (0.5–20 A g−1). The specific capacitance of a single electrode (Cd, F g−1) can also be calculated according to the following equation:27
| |
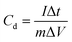 | (2) |
where
Cd is the specific capacitance,
I (A) is the current during discharge, Δ
t (s) is the discharge time, Δ
V (V) is the discharge voltage window, and
m (g) is the mass of the electrode active material.
Second, to further investigate the electrode materials for practical applications, we assembled a symmetric SC from circular electrode sheets in a two-electrode system. The voltage working window for CV and GCD was set to 0–1.3 V (Section 3.3), with the same scanning rate and current density as in the three-electrode system. The specific capacitance of the symmetric capacitor can be calculated using eqn (3).28 The energy density (E, W h kg−1) and power density (P, W kg−1) of the capacitor can be obtained from the discharge curve of GCD using eqn (4) and (5), respectively:
| |
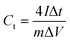 | (3) |
| |
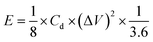 | (4) |
| |
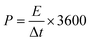 | (5) |
where
Ct is the specific capacitance of the entire symmetrical capacitor, and
I (A), Δ
t (s), and Δ
V (V) are the same as in
eqn (2).
M (g) is the total mass of active material on both electrodes.
The cycle capacitance retention of SCs was obtained from eqn (6):29
| |
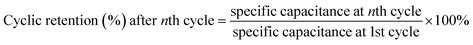 | (6) |
For the further analysis of the capacitance, the real (C′) and imaginary (C′′) parts of the capacitance can be obtained from the following equations:
| |
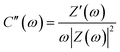 | (7) |
| |
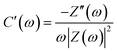 | (8) |
where
Z(
ω) represents the complex impedance,
Z′(
ω) denotes the real part of
Z(
ω), and
Z′′(
ω) refers to the imaginary part of
Z(
ω) (
ω = 2πf). With the use of the peak position of the imaginary part of the complex capacitance (
C′′(
ω)), the relaxation time constant (
τ0) can be calculated using the equation
τ0 = 1/
f0, where
f0 is the frequency corresponding to the peak position.
2.6. Physicochemical characterization
The thermal decomposition process of raw materials and the amount of residual carbon at specific temperatures were examined by thermogravimetric analysis (TGA) using a TGA/DSC3+ type simultaneous thermal analyzer (Mettler-Toledo, Switzerland) to study the stability of synthesized electrode materials at a temperature increase rate of 10 °C min−1 in an air environment. The stability of the synthesized electrode materials was investigated by field-emission scanning electron microscopy (SEM, JSM-7610F) to study the morphology of microscopic surfaces of the analyzed samples, and energy diffraction spectroscopy (EDS) was used to investigate the elemental composition. The crystallinity of samples was analyzed by X-ray diffraction (XRD, Bruker D8 Advance) with a scan rate of 2° min−1 in the range of 5°–60° to obtain diffractograms and determine the crystal structure of all samples. Raman spectra were collected on a Raman spectrometer (Renishaw inVia Reflex) with a laser wavelength of 532 nm to detect defects in the samples.30 The composition and chemical state of the surface elements of the activated samples were further characterized by X-ray photoelectron spectroscopy (XPS). The surface area and pore size distribution of the samples were analyzed by an automatic SSA and pore size analyzer (ASAP 2020 Particle Science), via N2 adsorption and desorption measurements. The monolayer adsorption and multilayer adsorption SSA were calculated by the Langmuir and Brunauer–Emmett–Teller (BET) methods, respectively. The micropore volume, micropore surface area, and external surface area were calculated using the t-plot method. The analysis by the Barrett–Joyner–Halenda method gives the average pore size, pore volume, and pore size distribution of mesopores of a material. In addition, the pore size distribution of microporous and mesoporous multistage pore materials was calculated using the nonlocal density functional theory model.
3. Results and discussion
3.1. Physical properties
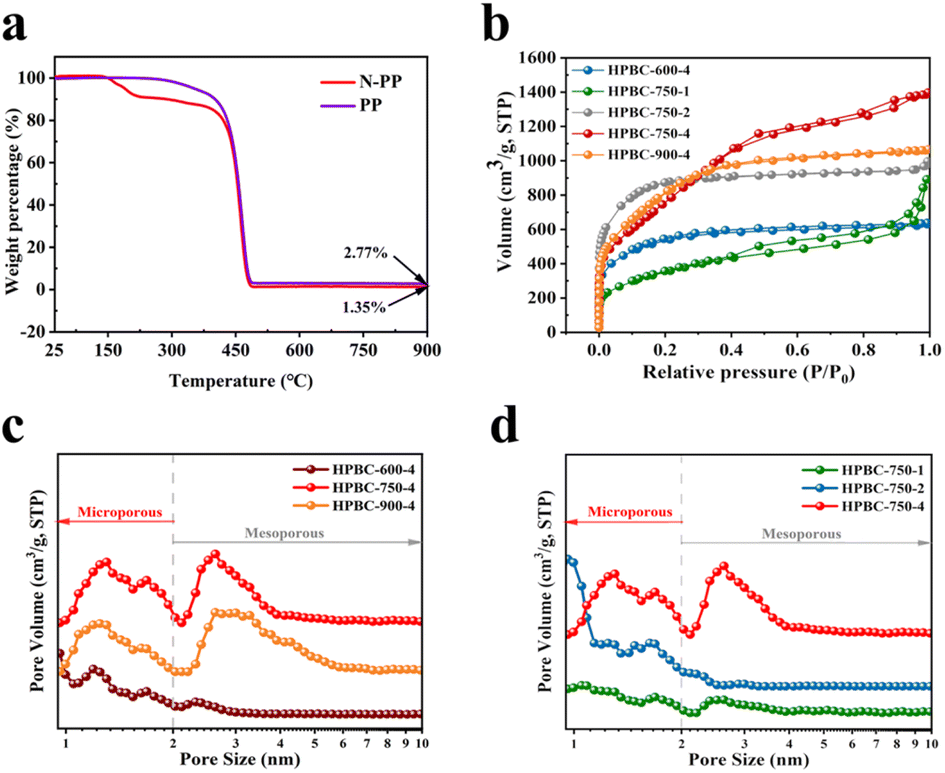
3.1.1. TGA. To investigate the formation process of nanoporous biochars and determine its pyrolysis temperature, we performed TGA (Fig. 3a). Compared with the nonwoven precursor without urea impregnation, the urea-impregnated precursor (N-PP) started to lose weight at approximately 141.9 °C. The first stage was the evaporation of surface and adsorbed water in the N-PP material, with approximately 15.62 wt% water loss. The second stage of weight loss occurred at approximately 401 °C due to the decomposition of organic polymers and urea to release volatiles of mass loss, with approximately 83.03 wt% weight loss. Finally, 1.35 wt% N/O atoms co-doped with carbon were produced at 900 °C.
3.1.2. SEM-EDS. Fig. 2 shows the SEM images of HPBC-T-4 (T = 600, 750, 900) and HPBC-750-x (x = 1, 2, 4). When the pyrolysis temperature was 600 °C (Fig. 2a), HPBC-600-4 showed the morphology of a dense bulk material and a smooth surface and no evident pores. When the pyrolysis reaction temperature increased to 750 °C, an increasingly developed pore structure of HPBC-750-x was observed with the increase in KOH ratios (Fig. 2b–d). The formation of pore structure and carbon fragments was attributed to the addition of KOH during activation. The specific pore-forming mechanism was further explained in terms of chemical reactions involved in the active agent (reactions (9)–(12)):| | |
6KOH + 2C ⇒ 2K + 3H2 + 2K2CO3
| (9) |
| | |
K2CO3 + 2C ⇒ 2K + 3CO
| (12) |
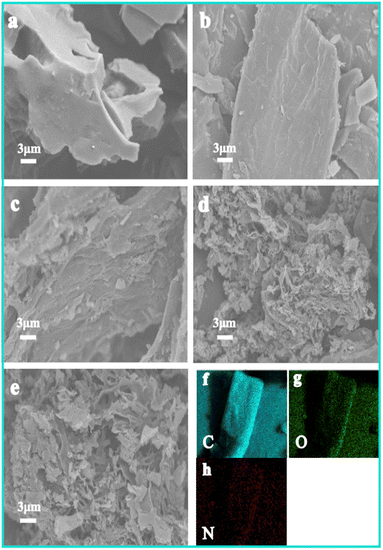 |
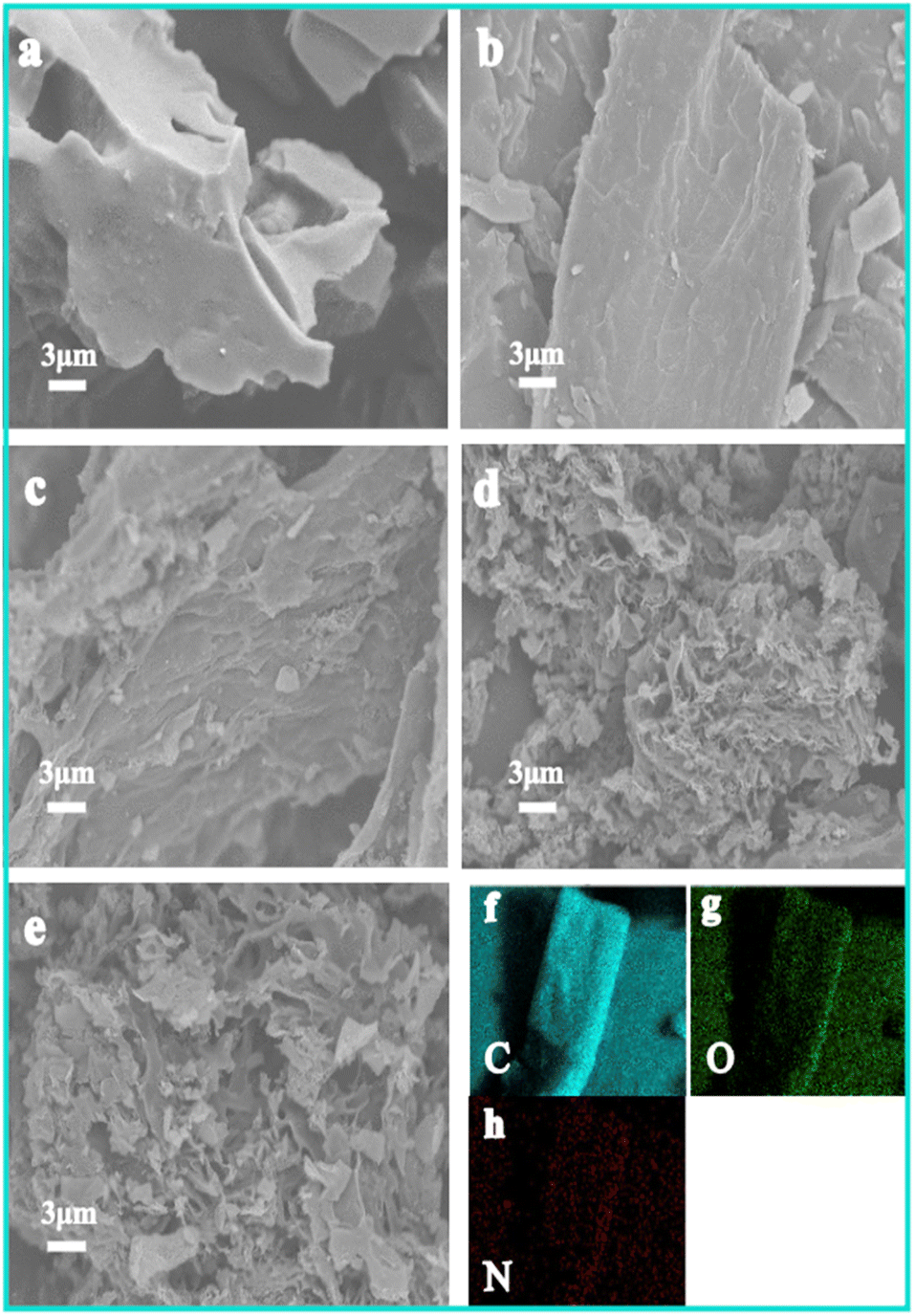
| | Fig. 2 SEM images of samples: (a) HPBC-600-4, (b) HPBC-750-1, (c) HPBC-750-2, (d) HPBC-750-4, (e) HPBC-900-4. (f)–(h) EDS element mapping of HPBC-750-4. | |
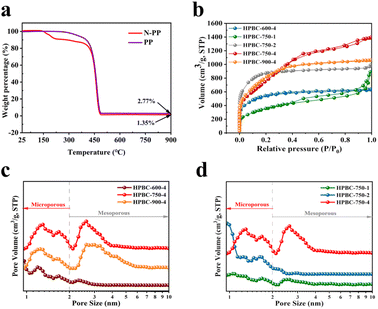 |
| | Fig. 3 (a) TGA curves of N-PP and PP, (b) N2 adsorption/desorption isotherm diagram, and (b) and (c) are the pore size distribution of the samples. | |
The decomposition of urea at high temperatures produces NH3 and CO2, which both contribute to the development of micro/mesopores.31 HPBC-750-4 formed a hierarchical nanoporous structure, which resulted in abundant in micropores and mesopores and provided high-speed channels that promoted the rapid diffusion of ions. With the further increase in the pyrolysis temperature to 900 °C (Fig. 2e), the alkaline activator intensified the erosion of the carbon framework, which resulted in a fragile carbon skeleton and collapse phenomenon. As a result, the reduction of the SSA and porosity of HPBC-900-4 were observed. In addition to the randomly distributed pores, heteroatoms are key to improving capacitive performance.32 As shown in Fig. 2f–h, the EDS analysis of HPBC-750-4 showed a uniform distribution of C, O, and N, which indicates the successful self-doping of HPBC-750-4.
3.1.3. N2 adsorption and desorption. The BET SSA and pore structure distribution of the samples were detected by the N2 adsorption and desorption curves shown in Fig. 3b. All samples had hierarchical microporous (1–2 nm) and mesoporous (2–50 nm) structures (Fig. 3c and d), where micropores can provide a large SSA and pore capacity, which increased the number of active sites, and mesopores can promote the rapid diffusion of ions and rapid transfer of substances.31 As shown in Fig. 3b, HPBC-750-1 and HPBC-750-4 exhibited typical type IV isotherms (0.4 < P/P0 < 1), and HPBC-600-4, HPBC-750-2, and HPBC-900-4 showed a combination of types I and IV isotherms. At low relative pressures (P/P0 < 0.05), the isotherms exhibited a rapid increase in adsorption capacity, which indicated the presence of abundant micropores.33 By contrast, at higher relative pressures (P/P0 > 0.4), adsorption and desorption were not fully reversible, and a hysteresis effect occurred, which demonstrated an H4-type hysteresis loop and the presence of a typical mesoporous structure.34 In addition, the steep increase in HPBC-750-1 and HPBC-750-2 at approximately P/P0 = 1 proved the presence of macropores in the sample.35 The gradually increase in the SSA (1259.47, 2820.86, and 3838.04 m2 g−1) of HPBC-750-1, HPBC-750-2, and HPBC-750-4, respectively (Table 1), can be attributed to the addition of more KOH. The reaction of KOH with carbon can produce H2, which led to the formation of microporous structures (reaction (9)); in addition, the etchants K2O and K2CO3 can react with carbon to generate CO and CO2, which contributed to the generation of micropores and mesopores,34 respectively. Given the lower activation temperature of HPBC-600-4, the volatile components generated by the reaction were not released, and a poor pore structure was observed. With increasing temperature, the number of micropores and mesopores of HPBC-750-4 and HPBC-900-4 increased, but the high temperature caused the carbon skeleton to be destroyed by KOH erosion and collapse to form larger pores, which resulted in a decreased SSA of HPBC-900-4. HPBC-750-4 showed the highest SSA of 3838.04 m2 g−1 and a total pore volume of 3.03 cm3 g−1, which indicate its excellent hierarchical porosity.
Table 1 Porosity parameters, specific capacitance at 1 A g−1 and the capacitance retention rate at 10 A g−1 of the HPBCs
| Sample |
SBETa (m2 g−1) |
Smicro (m2 g−1) |
Smeso (m2 g−1) |
Vporeb (cm3 g−1) |
Vmicroc (cm3 g−1) |
Vmesod (cm3 g−1) |
Capacity (F g−1) |
Retention rate (%) |
| SBET is specific surface area by the BET method at P/P0 = 0.001–0.05. Vpore is total pore volume at P/P0 = 0.99. Vmicro is micropore volume and is evaluated by the t-plot method. Vmeso is mesopore volume and is evaluated by the BJH method. |
| HPBC-600-4 |
1873.46 |
972.65 |
900.8 |
0.97 |
0.42 |
0.55 |
450.92 |
32.2 |
| HPBC-750-1 |
1259.47 |
171.72 |
1087.75 |
1.37 |
0.51 |
0.86 |
222.76 |
78.4 |
| HPBC-750-2 |
2820.86 |
2053.02 |
767.84 |
1.54 |
1.00 |
0.54 |
290.73 |
72.6 |
| HPBC-750-4 |
3838.04 |
2634.07 |
1203.97 |
3.03 |
1.37 |
1.66 |
340.92 |
63.9 |
| HPBC-900-4 |
3474.10 |
3299.78 |
174.32 |
3.43 |
1.27 |
2.16 |
205.76 |
60.6 |
3.2. Chemical properties
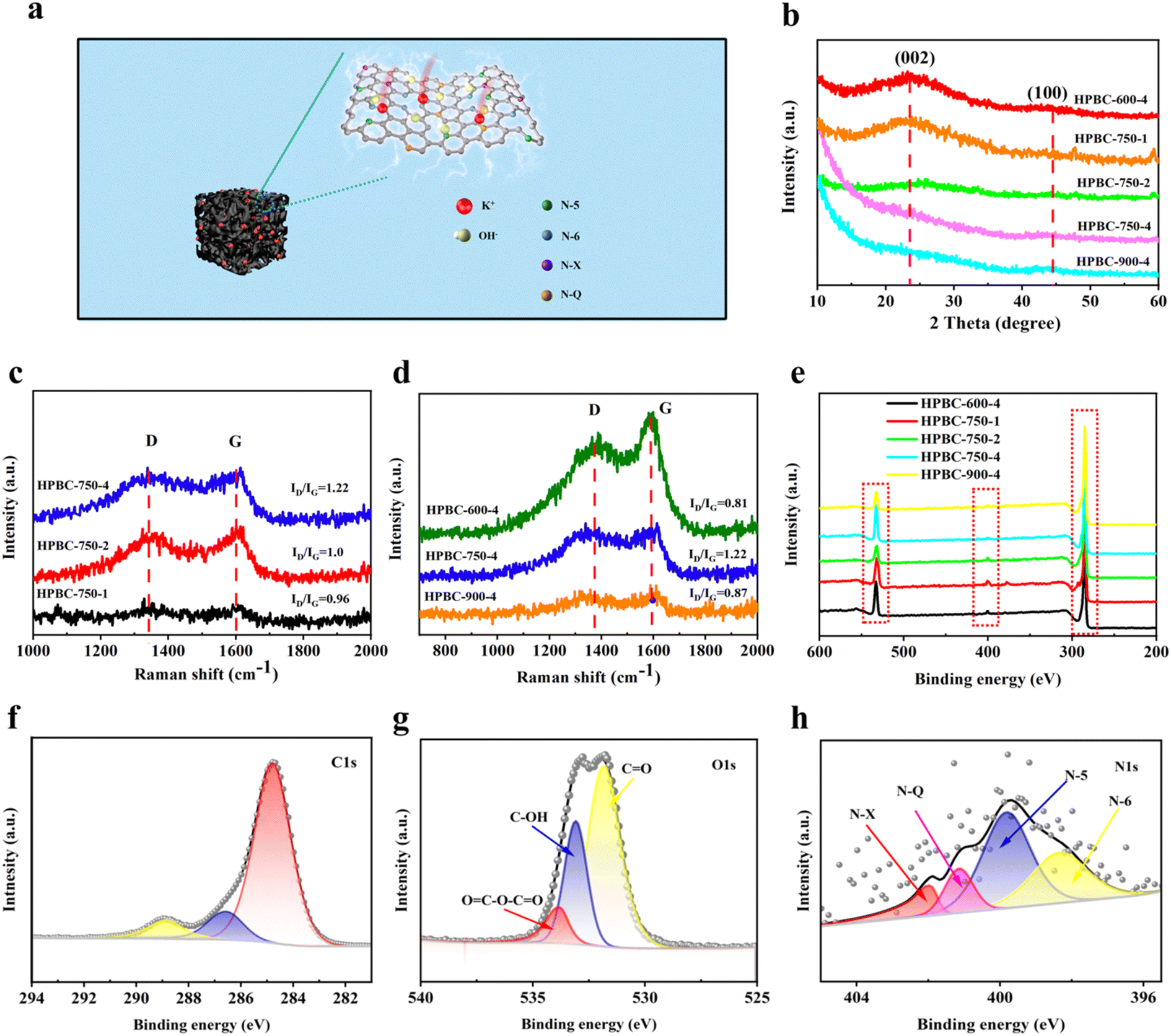
3.2.1. XRD. The crystallinity of carbon materials is a critical factor affecting the performance of the SCs. Fig. 4b shows the crystal structures of all samples. The broad diffraction peaks near 25° and weak peaks near 45° were attributed to the interlayer spacing (002) and planar spacing (100) lattice planes, respectively, which indicated the presence of certain amorphous carbon and graphitized structures in the samples.36 These weak and broad peaks indicated that all samples were composed of graphitized carbon atoms and mixed-layer stacking with a well-developed pore structure, but the carbon material exhibited a relatively poorly crystalline structure.37 The broad peaks reflected by the (002) and (100) crystalline surfaces exhibited graphitic carbon and interlayer condensation, respectively, and no additional peaks were found in all samples; thus, the materials were completely carbonized, and no other impurities were introduced.38 In addition, HPBC-750-4 showed a distinct high-intensity peak in the small-angle region (2θ < 20°), which indicated the presence of high-density micropores, consistent with the results of SEM and N2 adsorption–desorption isotherm analysis.
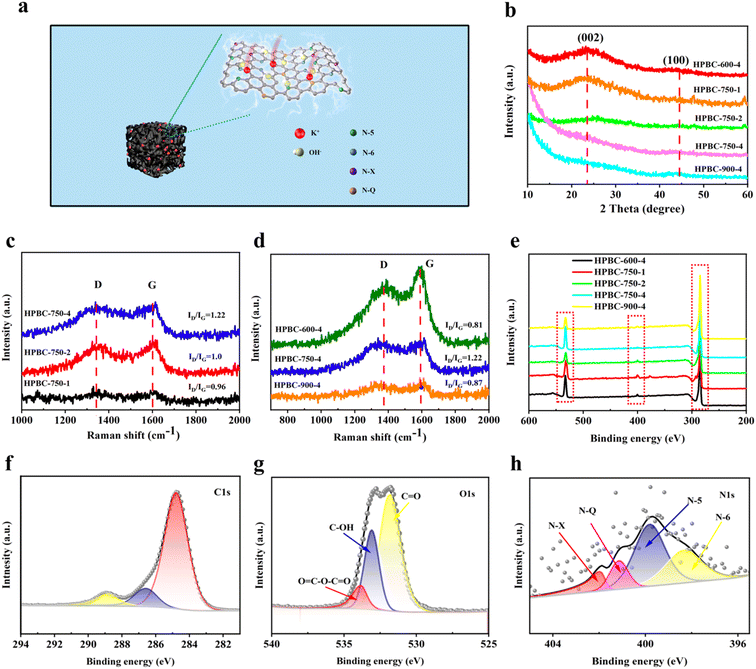 |
| | Fig. 4 (a) Microscopic schematic of nanoporous biochar, (b) XRD spectra of all samples, (c) and (d) Raman spectra, (e) XPS spectra of all samples, (f)–(h) are the C 1s, O 1s and N 1s XPS spectra of HPBC-750-4. | |
3.2.2. Raman spectra. The defects and graphitization of HPBCs were further characterized through Raman analysis (Fig. 4c and d). Two distinctive characteristic peaks were observed in the Raman spectra of the activated carbon materials, and they represented the D-band (1367 cm−1) caused by structural defects in graphite and the G-band (1605 cm−1) generated by in-plane stretching vibration of sp2 hybridized carbon in graphite crystals. Fig. 4c shows the Raman spectra of the samples with the different mass ratios of PCBC and KOH. The intensity ratios (ID/IG) of the G- and D-band can be used to evaluate the degree of graphitization, and the calculated ID/IG values of 0.96, 1.0, and 1.22 for HPBC-750-1, HPBC-750-2, and HPBC-750-4, respectively, indicated that the increase in the KOH mass ratio can produce more defect vacancies due to the deep etching of the carbon skeleton by KOH; as a result, a gradual increase in the disordered and a decrease in graphitization of the prepared carbon materials was observed.39 As shown in Fig. 4d, the ID/IG values of HPBC-600-4, HPBC-750-4, and HPBC-900-4 were 0.81, 1.22, and 0.87, respectively, and a slight increase in ID/IG was observed with the increase activation temperature, which was related to the increased defect rate of the prepared carbon materials.40 Compared with those of other HPBC materials, the higher ID/IG values and defect degree of HPBC-750-4 resulted in a higher SSA and fine pore structure, which allowed the activated carbon material to adsorb more electrolyte ions that increased its specific electric capacity.
3.2.3. XPS. The X-ray photoelectron spectra of the relevant HPBCs were measured to further investigate the elemental composition and bonding states of the doped heteroatoms (Fig. 4e). Typical C 1s, O 1s, and N 1s characteristic peak signals were observed from the measured XPS spectrum of HPBC-750-4, which implied the presence of C atoms (85.59%) and abundant heteroatoms oxygen (13.25%) and nitrogen (1.16%). Table S2 (ESI†) summarizes the elemental contents of carbon, oxygen, and nitrogen. The peaks at 284.7, 286.47, and 288.79 eV in the high-resolution C 1s spectra of HPBC-750-4 (Fig. 4f) corresponded to those caused by C–C (sp2 hybridization) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (79.63%), C–N (11.07%), and C–O (9.3%) groups,41 respectively. The presence of C–N groups indicated the successful self-doping of nitrogen in the materials. The high-resolution O 1s spectrum (Fig. 4g) showed that the oxygen-based component was mainly present in the carbon skeleton in the form of CO (531.79 eV), C–OH (533.08 eV), and OC–O–CO (533.81 eV) bonds. The spectrum of N 1s (Fig. 4h) can be decomposed into four different peaks, namely, 397.96, 399.32, 401.15, and 402.42 eV, which corresponded to pyridine nitrogen (N-6), pyrrole nitrogen (N-5), graphite nitrogen (N-Q), and nitrogen oxide (N-X), respectively. The microscopic molecular structure of nanoporous biochar is represented in Fig. 4a. N-6 and N-5, which can promote energy storage,42 presented relatively high proportions of 33.26% and 29.03%, respectively (Fig. S1 (ESI†)). The nitrogen atoms in N-6 can act as electron-absorbing groups to produce localized electron defects in carbon materials, which can be used to store charges and increase the capacitance of the material. N-5 can function as an electron donor-group to generate “off-domain electrons” in carbon materials, and the excess electrons produced can make the material more conductive and reduce the charge transfer resistance.43 The presence of N-Q (7.39%) implied the formation of graphitized frameworks, which accelerated the transfer of electrons. Overall, the results indicated that PP nonwoven fabrics have been successfully transformed into functionalized nanoporous biochar co-doped with N and O.
C (79.63%), C–N (11.07%), and C–O (9.3%) groups,41 respectively. The presence of C–N groups indicated the successful self-doping of nitrogen in the materials. The high-resolution O 1s spectrum (Fig. 4g) showed that the oxygen-based component was mainly present in the carbon skeleton in the form of CO (531.79 eV), C–OH (533.08 eV), and OC–O–CO (533.81 eV) bonds. The spectrum of N 1s (Fig. 4h) can be decomposed into four different peaks, namely, 397.96, 399.32, 401.15, and 402.42 eV, which corresponded to pyridine nitrogen (N-6), pyrrole nitrogen (N-5), graphite nitrogen (N-Q), and nitrogen oxide (N-X), respectively. The microscopic molecular structure of nanoporous biochar is represented in Fig. 4a. N-6 and N-5, which can promote energy storage,42 presented relatively high proportions of 33.26% and 29.03%, respectively (Fig. S1 (ESI†)). The nitrogen atoms in N-6 can act as electron-absorbing groups to produce localized electron defects in carbon materials, which can be used to store charges and increase the capacitance of the material. N-5 can function as an electron donor-group to generate “off-domain electrons” in carbon materials, and the excess electrons produced can make the material more conductive and reduce the charge transfer resistance.43 The presence of N-Q (7.39%) implied the formation of graphitized frameworks, which accelerated the transfer of electrons. Overall, the results indicated that PP nonwoven fabrics have been successfully transformed into functionalized nanoporous biochar co-doped with N and O.
3.3. Electrochemical studies
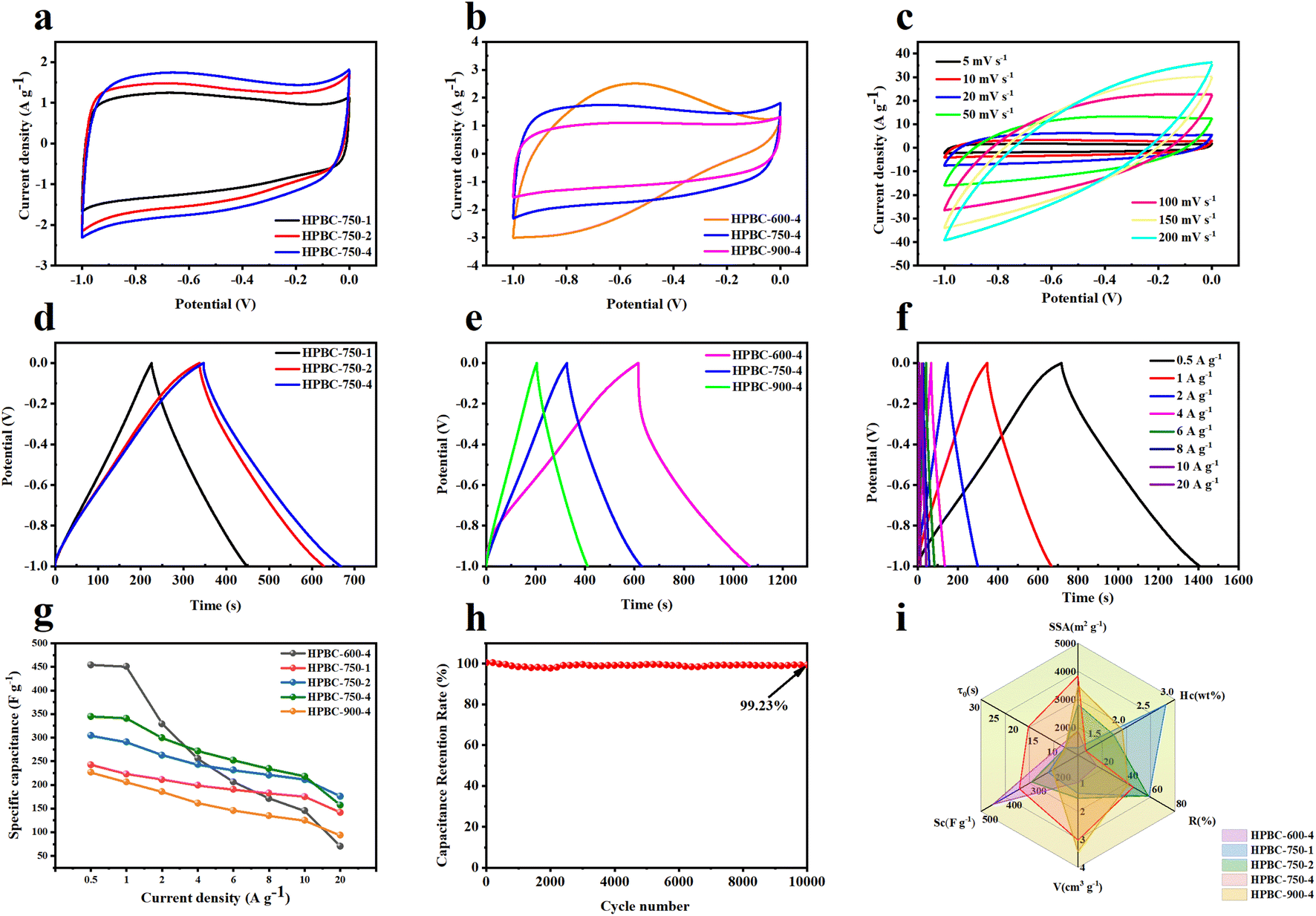
3.3.1. Three-electrode test. The electrochemical performance of HPBC-T-4 and HPBC-750-x as electrode materials was first evaluated in a three-electrode system with 6 M KOH as the electrolyte. Fig. 5a shows the CV curves of the samples with different KOH ratios at a scan rate of 5 mV s−1. The CV curve showed a quasirectangular shape, which indicates a fast electrochemical reaction and the behavior of dominant EDLCs.44 Compared with HPBC-750-1 and HPBC-750-2, HPBC-750-4 had the largest rectangular area and therefore exhibited the best capacitive performance. In addition, a small hump in the CV curve was observed at −0.8 V, which possibly resulted from the reversible redox reaction of N atom doping. No bending was observed, and it indicated charge diffusion dynamics and good reversibility.45 Fig. 5b displays the CV curves of the activated carbon materials at different activation temperatures. HPBC-750-4 had a larger area of the CV curve than HPBC-900-4, which implied its better double-layer capacitance. The CV curves of HPBC-750-4 all presented desirable rectangular shapes at different scan rates (Fig. 5c), which indicated fast ion transport rates and excellent rate capability.46
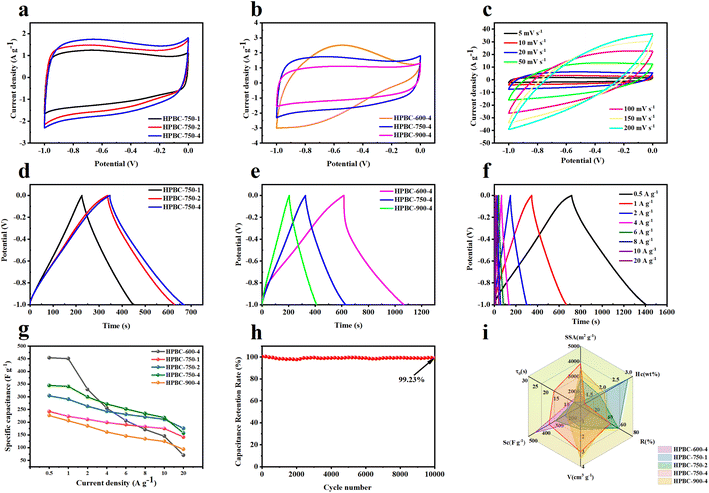 |
| | Fig. 5 Electrochemical performance test in the three-electrode system: (a) CV curves of HPBC-750-x at a scan rate of 5 mV s−1, (b) CV curves of HPBC-T-4 at a scan rate of 5 mV s−1, and (c) CV curves of HPBC-750-4 at different scan rates. (d) GCD curves of HPBC-750-x at a current density of 1 A g−1, (e) GCD curves of HPBC-T-4 at a current density of 1 A g−1, and (f) GCD curves of HPBC-750-4 at different current densities. (g) Specific capacitance of all samples at different current densities, (h) cycling stability of HPBC-750-4 at 20 A g−1; (i) comparison chart of parameters, including SSA, porous volume (V), heteroatom contents (Hc), current density at 1 A g−1 (Rc), ion transport time (τ0), and rate capability (R). | |
The GCD curves of the charge–discharge process (Fig. 5d and e) were almost straight and roughly symmetric isosceles triangles and further demonstrated the typical double-layer capacitor behavior and good charge–discharge reversibility of the SCs.30 HPBC-600-4 had the longest charge–discharge time, which implied its maximum specific capacitance; however, its charge–discharge curves were slightly asymmetric, which suggests that the HPBC-600-4 electrode was less reversible.47 Notably, the HPBC-750-4 curve showed less distortion (Fig. 5f), where all GCD curves had similar triangles as the current density increased. Fig. 5g (eqn (2)) present the comprehensive specific capacitance performance of the electrodes calculated from the GCD curves. Table 1 shows the specific capacitance values of all the samples at a current density of 1 A g−1. It was observed that HPBC-750-4 had a notably high specific capacitance of 340.9 F g−1 at 1 A g−1. However, when the current density was increased from 1 A g−1 to 10 A g−1, HPBC-750-4 exhibited a specific capacitance of 217.9 F g−1, with a capacitance retention of 63.9%. This indicates that HPBC-750-4 can maintain a significant portion of its capacitance even at higher current densities, making it a promising candidate for high-performance energy storage applications. During the test of the cycling stability of HPBC-750-4, after 10000 cycles at a current density of 10 A g−1 (Fig. 5h), the capacitance retention was 99.23% (derived from eqn (7)) with negligible capacity decay, which indicated excellent cycling stability and a reversible and efficient charging and discharging behavior. As expected, the HPBC-750-4 electrode exhibited high current density and rate capability (Fig. 5i). The high specific capacitance of HPBC-750-4 can be attributed to several reasons: (1) the ultrahigh SSA (3838.04 m2 g−1) and total pore volume (3.03 cm3 g−1) provided more adsorption sites and improved the diffusion rate of electrolyte ions. The unique hierarchical porous structure formed favored the volume changes during charge and discharge.48 (2) Heteroatom doping increased the number of active sites and thus stored more charge accordingly. The improved wettability of the electrode promoted sufficient contact between the electrode material and electrolyte ions.
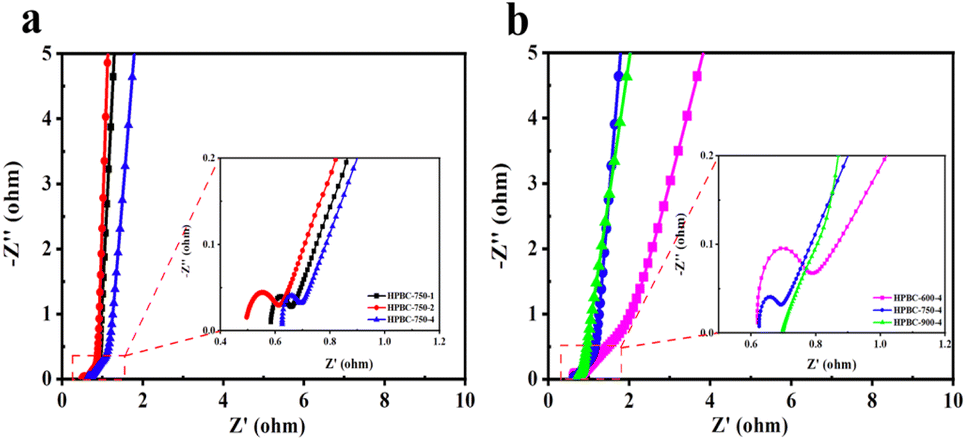
Furthermore, EIS was used to study the ion diffusion and charge transfer of different electrode materials. Fig. 6a and b show the Nyquist plots of the HPBC-750-4 electrode in the frequency range of 0.01 Hz to 100 kHz. The Nyquist plots consisted of a semicircular part at high frequencies and a linear part at low frequencies. The diameter of the semicircle corresponds to the charge transfer resistance (Rct). The intercept of the semicircle with the X-axis yielded the diffusion resistance (Rs) of the active substance with the electrolyte.49 Based on the equivalent circuit diagram fitting calculations in Fig. S2 (ESI†), Rct corresponding to those of HPBC-750-1, HPBC-750-2, and HPBC-750-4 samples initially increased and then decreased as the activator ratio increased (Fig. 6a), with values of 0.059 Ω, 0.119 Ω, and 0.045 Ω, respectively. Thus, the electrode prepared with an activator ratio of 4 had the lowest Rct. For activation temperatures, the Rcts at 600 °C and 750 °C were 0.102 Ω and 0.045 Ω, respectively. The straight line on the EIS curve in the low-frequency region corresponds to the Rs of the electrolyte in the pores of the electrode active material. A large slope was considered to indicate a low Rs.50 The slope of the straight line approximation in the frequency region for the three samples with different activator ratios was approximately 45°, which indicates that the prepared carbon material had ideal EDLCs characteristics. Among the three samples with different pyrolysis temperatures, a smaller semicircle and a steeper straight line were observed in the Nyquist plot of HPBC-750-4, which implied its higher electrical conductivity. Combined with the characteristics of the internal resistance and slope listed in Table S1 (ESI†), the HPBC-750-4 electrode possessed a lower Rct and Rs values, resulting in improved rate capability and capacitive performance. In addition, the Bode curves of the electrode materials (Fig. S3 (ESI†)) showed that the phase angle of HPBC-600-4 was 54.8°, which deviated from the ideal state. The phase angles of HPBC-750-1, HPBC-750-2, HPBC-750-4, and HPBC-900-4 were 83.5°, 82.5°, 76.9°, and 74.2°, respectively, which were all close to the ideal capacitor phase angle of 90°. Thus, these samples exhibited a good capacitive behavior.
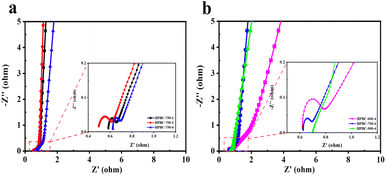 |
| | Fig. 6 (a) AC impedance spectra of HPBC-750-x, (b) AC impedance spectra of HPBC-T-4. | |
Based on EIS data, the relationship between capacitance and frequency was analyzed by the complex model of capacitance, C(ω) = C′(ω) + jC′′(ω).51 The relaxation time constant (τ0) was used to evaluate the charge/discharge rate, which can be obtained from the characteristic frequency (f0) and maximum C′′ (eqn (7) and (8)) (Fig. S4 (ESI†)). For the HPBC-600-4 electrode, a distinct peak was not observed, which rendered the determination of the value of τ0 impossible. The values of τ0 for the HPBC-750-1, HPBC-750-2, HPBC-750-4, and HPBC-900-4 electrodes were 8.3, 8.3, 17.8, and 8.3 s, respectively. The value of real part (C′(ω)) at low frequencies represents the maximum capacity of the electrode during constant current discharge. A small slope allowed HPBC-750-4 to attain a fast ion diffusion rate.
3.3.2. Symmetric electrode test. To evaluate the performance of HPBC-750-4 in practical applications, we assembled a symmetric SC HPBC-750-4//HPBC-750-4 (Fig. 7a) using 6 M KOH as the electrolyte. CV test was performed to verify the rate performance of the symmetric SC in the voltage window of 0–1.8 V at a scan rate of 50 mV s−1 (Fig. 7b). The anode current appeared in the CV curve when the voltage window was increased to 1.3 V, which indicates that the symmetric SC can work stably in the voltage window of 0–1.3 V. Fig. 7c and d show the results of CV and constant GCD tests, respectively. The CV curves maintained a rectangular-like shape at different scan rates, which proved the good capacitive performance of the material. The GCD curves showed an isosceles triangle shape at different current densities, which indicated the good multiplicative and fast reversible charging and discharging performances of the symmetric SC.52 The specific capacitance of the symmetrical SC reached 259 F g−1 at a current density of 1 A g−1 and maintained over 100 F g−1 at a current density of 20 A g−1 (Fig. 7e). To observe the overall performance of HPBC-750-4//HPBC-750-4, we calculated the relationship between energy density and power density based on the GCD curves using eqn (4) and (5), respectively (Fig. 7f). The energy density reached 10.02 W h kg−1 at a power density of 96.15 W kg−1 and remained at 4.36 W h kg−1 when the power density changed to 2000 W kg−1, which proved its good electrochemical performance and application prospects.
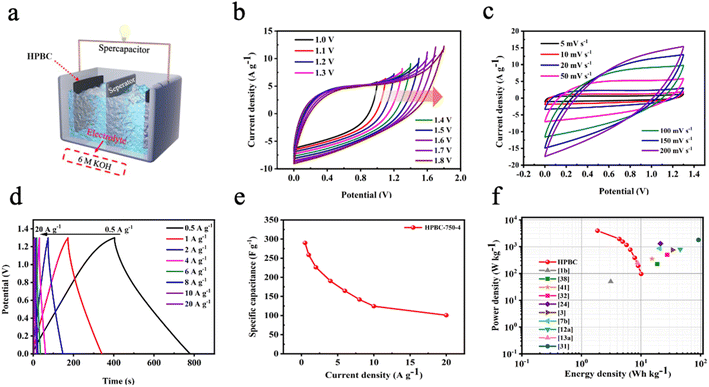 |
| | Fig. 7 (a) HPBC-750-4//HPBC-750-4 symmetric supercapacitor. (b) CV of HPBC-750-4 at different open circuit voltages, (c) CV of HPBC-750-4 at 1.3 V at open-circuit voltage; (d) GCD of HPBC-750-4 at different current densities in the voltage window range of 0–1.3 V, (e) specific capacitance of HPBC-750-4 at different scan rates in the voltage window range of 0–1.3 V, and (f) relationship between power density and energy density of HPBC-750-4. | |
4. Conclusions
In summary, a series of nitrogen-doped nanoporous biochar materials was prepared from waste nonwoven fabrics through pre-carbonization coupled with pyrolytic activation using urea as the nitrogen source and KOH as the activator. The effect of pyrolysis temperature and KOH on the physicochemical properties and electrochemical performance of the obtained nanoporous biochars was investigated. The specific capacitance of HPBC-750-4 reached 340.9 F g−1 at a current density of 1 A g−1, and its retention rate was higher than 99.23% after 10000 cycles at a current density of 10 A g−1. In addition, the assembled HPBC-750-4//HPBC-750-4 symmetrical SC also exhibited an energy density of 10.02 W h kg−1 when the power density was 96.15 W kg−1 in 6 M KOH electrolyte, which indicated a good overall SC performance. This work not only provides a simple and environmentally friendly pathway for the synthesis of highly active and low-cost SCs using waste nonwoven fabrics as precursors but also opens up a new circular economy for recycling waste diaper components and reducing the burden of urban environmental pollution.
Author contributions
Yihao Geng: methodology, conceptualization, data curation, writing – original draft. Jieni Wang: conceptualization, writing – review & editing. Qizhao Wang: investigation, validation. Xuanyu Chen: methodology, visualization. Suinan Sun: data curation and editing. Shuqin Zhang & Chenxiao Liu: software, data curation. Yijun Tian: supervision, writing – review & editing. Lin Wang: conceptualization, supervision, resources. Zhangdong Wei: writing – review & editing, language editing. Leichang Cao: project administration, conceptualization, supervision, language editing. Jinglai Zhang: investigation, writing – review & editing. Shicheng Zhang: supervision.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (No. 52100164), the China Postdoctoral Science Foundation (No. 2023M731169), the Key Scientific Research Projects of Universities in Henan Province (No. 23A610006), the Key Science and Technology Department Project of Henan Province (No. 222102320252), the Science and Technology Development Plan of Kaifeng City – Science and Technology Research Project, and Yellow River Scholar Program of Henan University. The authors are also grateful for the provision of a scholarship to Leichang Cao by Shanghai Tongji Gao Tingyao Environmental Science & Technology Development.
References
- F. Mao and S.-H. Son, Chem. Eng. Sci., 2023, 276, 118702 CrossRef CAS.
- L. Gao, Y. Wang, Y. Liu and L. Xu, Colloids Surf., A, 2023, 663, 131056 CrossRef CAS.
- Y. Ding, Y. Li, L. Wang, X. Han, L. Zhu and S. Wang, Fuel, 2021, 304, 121449 CrossRef CAS.
- D. R. Lobato-Peralta, D. M. Arias and P. U. Okoye, J. Energy Storage, 2021, 40, 102760 CrossRef.
- W. He, G. Zhao, P. Sun, P. Hou, L. Zhu, T. Wang, L. Li, X. Xu and T. Zhai, Nano Energy, 2019, 56, 207–215 CrossRef CAS.
- M. Cakici, R. R. Kakarla and F. Alonso-Marroquin, Chem. Eng. J., 2017, 309, 151–158 CrossRef CAS.
- J. Yin, W. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Small Methods, 2020, 4, 1900853 CrossRef CAS.
- L. Sinha and P. M. Shirage, J. Electrochem. Soc., 2019, 166, A3496 CrossRef CAS.
- A. C. Forse, C. Merlet, J. M. Griffin and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 5731–5744 CrossRef CAS PubMed.
- M. Song, Y. Zhou, X. Ren, J. Wan, Y. Du, G. Wu and F. Ma, J. Colloid Interface Sci., 2019, 535, 276–286 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, H. Hou, W. Xu, G. Duan, S. He, K. Liu and S. Jiang, J. Mater. Sci., 2020, 56, 173–200 CrossRef CAS.
- X. Zhang, R. Han, Y. Liu, H. Li, W. Shi, X. Yan, X. Zhao, Y. Li and B. Liu, Chem. Eng. J., 2023, 460, 141607 CrossRef CAS.
- R. Kumar, E. Joanni, S. Sahoo, J.-J. Shim, W. K. Tan, A. Matsuda and R. K. Singh, Carbon, 2022, 193, 298–338 CrossRef CAS.
- L. Wan, E. Shamsaei, C. D. Easton, D. Yu, Y. Liang, X. Chen, Z. Abbasi, A. Akbari, X. Zhang and H. Wang, Carbon, 2017, 121, 330–336 CrossRef CAS.
- Y. Ding, S. Huang, Y. Sun, Y. Li, L. Zhu and S. Wang, ChemElectroChem, 2021, 8, 3745–3754 CrossRef CAS.
- A. P. Khedulkar, B. Pandit, V. D. Dang and R.-a. Doong, Sci. Total Environ., 2023, 869, 161441 CrossRef CAS PubMed.
- D. Khalafallah, X. Quan, C. Ouyang, M. Zhi and Z. Hong, Renewable Energy, 2021, 170, 60–71 CrossRef CAS.
- X. Li, X. Du, Y. Li, X. Tian, H. Zheng and X. Li, J. Mater. Sci., 2022, 57, 9357–9369 CrossRef CAS.
- D. Guo, R. Xin, Y. Wang, W. Jiang, Q. Gao, G. Hu and M. Fan, Microporous Mesoporous Mater., 2019, 279, 323–333 CrossRef CAS.
- K. Zou, Y. Deng, J. Chen, Y. Qian, Y. Yang, Y. Li and G. Chen, J. Power Sources, 2018, 378, 579–588 CrossRef CAS.
- Y. Zheng, K. Chen, K. Jiang, F. Zhang, G. Zhu and H. Xu, J. Energy Storage, 2022, 56, 105995 CrossRef.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- Y. Wang, H. Li, W. Yang, S. Jian, C. Zhang and G. Duan, Diamond Relat. Mater., 2022, 130, 109526 CrossRef CAS.
- Y. Shu, J. Maruyama, S. Iwasaki, S. Maruyama, Y. Shen and H. Uyama, J. Power Sources, 2017, 364, 374–382 CrossRef CAS.
- A. Gopalakrishnan and S. Badhulika, J. Power Sources, 2020, 480, 228830 CrossRef CAS.
- J. Wang, Y. Liang, S. Wang, P. U. Okoye, H. Chen, Y. Zhou, J. Xu, Z. Meng, L. Wang, S. Li and U. Maschke, Int. J. Polym. Sci., 2020, 2020, 1–9 CAS.
- M. Karnan, K. Subramani, N. Sudhan, N. Ilayaraja and M. Sathish, ACS Appl. Mater. Interfaces, 2016, 8, 35191–35202 CrossRef CAS PubMed.
- P. Yang and W. Mai, Nano Energy, 2014, 8, 274–290 CrossRef CAS.
- M. M. Rahman, M. R. Hossen, I. Alam, M. H. Rahman, O. Faruk, M. Nurbas, M. M. Rahman and M. M. R. Khan, J. Alloys Compd., 2023, 947, 169471 CrossRef CAS.
- Z. Shang, X. An, H. Zhang, M. Shen, F. Baker, Y. Liu, L. Liu, J. Yang, H. Cao, Q. Xu, H. Liu and Y. Ni, Carbon, 2020, 161, 62–70 CrossRef CAS.
- R. Wang, S. Khoshk Rish, J.-M. Lee, R. Bahadur, A. Vinu, A. Tahmasebi and J. Yu, J. Anal. Appl. Pyrolysis, 2022, 168, 105785 CrossRef CAS.
- J. Mu, S. I. Wong, Q. Li, P. Zhou, J. Zhou, Y. Zhao, J. Sunarso and S. Zhuo, J. Alloys Compd., 2020, 832, 154950 CrossRef CAS.
- L. Hou, B. Xing, H. Zeng, W. Kang, H. Guo, S. Cheng, G. Huang, Y. Cao, Z. Chen and C. Zhang, J. Alloys Compd., 2022, 922, 166134 CrossRef CAS.
- X. Wang, Q. Li, Y. Zhang, Y. Yang, Z. Cao and S. Xiong, Appl. Surf. Sci., 2018, 442, 565–574 CrossRef CAS.
- X. Zheng, L. Miao, Z. Song, W. Du, D. Zhu, Y. Lv, L. Li, L. Gan and M. Liu, J. Mater. Chem. A, 2022, 10, 611–621 RSC.
- Y. Qin, Z. Song, L. Miao, C. Hu, Y. Chen, P. Liu, Y. Lv, L. Gan and M. Liu, Chem. Eng. J., 2023, 470, 144256 CrossRef CAS.
- X. Xiao, L. Song, Q. Wang, Z. Wang, H. Wang, J. Chu, J. Liu, X. Liu, Z. Bian and X. Zhao, RSC Adv., 2022, 12, 16257–16266 RSC.
- M. Pang, S. Jiang, J. Zhao, S. Zhang, R. Wang, N. Li, R. Liu, Q. Pan, W. Qu and B. Xing, RSC Adv., 2020, 10, 35545–35556 RSC.
- G. Zhao, D. Yu, C. Chen, L. Sun, C. Yang, H. Zhang, B. Du, F. Sun, Y. Sun and M. Yu, J. Electroanal. Chem., 2020, 878, 114551 CrossRef CAS.
- R. Srinivasan, E. Elaiyappillai, H. P. Pandian, R. Vengudusamy, P. M. Johnson, S.-M. Chen and R. Karvembu, J. Electroanal. Chem., 2019, 849, 113382 CrossRef CAS.
- T. Yuan, W. He, G. Yin and S. Xu, Fuel, 2020, 261, 116450 CrossRef CAS.
- L. Cui, Y. Huan, J. Shan, B. Liu, J. Liu, H. Xie, F. Zhou, P. Gao, Y. Zhang and Z. Liu, ACS Nano, 2020, 14, 15327–15335 CrossRef CAS PubMed.
- M. S. Khan, D. Jhankal, P. Shakya, A. K. Sharma, M. K. Banerjee and K. Sachdev, Carbon, 2023, 208, 227–237 CrossRef CAS.
- C. Costentin, T. R. Porter and J.-M. Savéant, ACS Appl. Mater. Interfaces, 2017, 9, 8649–8658 CrossRef CAS PubMed.
- K. R. Thines, E. C. Abdullah, M. Ruthiraan, N. M. Mubarak and M. Tripathi, J. Anal. Appl. Pyrolysis, 2016, 121, 240–257 CrossRef CAS.
- Y. Xu, H. Lei, S. Qi, F. Ren, H. Peng, F. Wang, L. Li, W. Zhang and G. Ma, J. Energy Storage, 2020, 32, 101970 CrossRef.
- A. Ramadoss, K.-Y. Yoon, M.-J. Kwak, S.-I. Kim, S.-T. Ryu and J.-H. Jang, J. Power Sources, 2017, 337, 159–165 CrossRef CAS.
- K. R. Thines, E. C. Abdullah, N. M. Mubarak and M. Ruthiraan, Biomass Bioenergy, 2017, 98, 95–111 CrossRef CAS.
- Y. Jiang, L. Chen, H. Zhang, Q. Zhang, W. Chen, J. Zhu and D. Song, Chem. Eng. J., 2016, 292, 1–12 CrossRef CAS.
- L. Ma, W. Zhang, R. Zhang, H. Niu, Q. Yang, F. Li, M. Zhou, L. Zhang and Y. Huang, Appl. Surf. Sci., 2023, 626, 157148 CrossRef CAS.
- Y. Zhang, Z. Song, L. Miao, Y. Lv, L. Li, L. Gan and M. Liu, Chem. Eng. J., 2023, 467, 143497 CrossRef CAS.
- X. Du, W. Zhang, M. Zhang, K. Su and Z. Li, Electrochim. Acta, 2023, 441, 141803 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Jinglai Zhang
*ab,
Jinglai Zhang