
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDensity functional theory study of the corrosion inhibition performance of 6-mercaptopurine and 6-thioguanine expired drugs toward the aluminium (111) surface†
Mahmoud A. A. Ibrahim *ab,
Nayra A. M. Moussaa,
Amna H. M. Mahmouda,
Shaban R. M. Sayedc,
Peter A. Sidhomd,
Mohamed K. Abd El-Rahmane,
Tamer Shoeib*f and
Lamiaa A. Mohameda
*ab,
Nayra A. M. Moussaa,
Amna H. M. Mahmouda,
Shaban R. M. Sayedc,
Peter A. Sidhomd,
Mohamed K. Abd El-Rahmane,
Tamer Shoeib*f and
Lamiaa A. Mohameda
aComputational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, Minia 61519, Egypt. E-mail: m.ibrahim@compchem.net
bSchool of Health Sciences, University of KwaZulu-Natal, Westville Campus, Durban 4000, South Africa
cDepartment of Botany and Microbiology, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
dDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Tanta University, Tanta 31527, Egypt
eDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
fDepartment of Chemistry, The American University in Cairo, New Cairo 11835, Egypt. E-mail: t.shoeib@aucegypt.edu
First published on 4th October 2023
Abstract
The potentiality of the 6-mercaptopurine (MP) and 6-thioguanine (TG) expired drugs toward the corrosion inhibition of the aluminium (Al) (111) surface was widely investigated using a series of density functional theory (DFT) calculations. A competition between the anti-corrosive features of the studied drugs in the gas and aqueous phases was conducted on both neutral and protonated forms by means of quantum mechanical descriptors. The results of the electrostatic potential analysis demonstrated the prominent nucleophilic nature of the sulfur and nitrogen atoms over the structures of the examined drugs. The frontier molecular orbital theory findings outlined the higher preferability of TG over MP as a corrosion inhibitor. Upon determining the most beneficial configurations of the MP/TG⋯Al (111) complexes, first-principles molecular dynamics simulations were executed. Interestingly, the competence of the TG drug in the corrosion inhibition process of Al (111) was more extensive than that of the MP one, which was confirmed by the interaction energy values of −1.79 and −1.64 eV, respectively. Upon obtaining the relaxed complexes, the effect of the presence of water solvent on the adsorption process was studied. These findings provide a foundation for developing green anti-corrosive inhibitors for the aluminium surface.
1 Introduction
Nowadays, aluminium (Al) and its alloys are used worldwide in versatile industrial applications, such as automotive, aerospace, electronics, and transportation, owing to their mechanical strength and economical manufacturing cost.1,2 It is noteworthy that Al and its alloys possess crucial resistance to atmospheric conditions due to a firm adherent metal oxide film.3 Nevertheless, Al-based alloys are susceptible to localized attack in corrosive environments, like media containing chloride ions.4–6 Hydrochloric acid (HCl) is a common solution used for chemical, pickling, and electrochemical etching of Al metal and its alloys.7,8 Further, the HCl solution is used to remove dust and rust in the metallurgical industries. Additionally, the surface of Al in an aqueous solution forms a compact and robust passive oxide coating. This film dissolves when submerged in HCl solution due to its amphoteric nature, leading to corrosion issues.8–11 Failures and losses resulting from the corrosion phenomena lead not only to economic problems but also environmental, security, social, and safety issues. Up to now, the investigation of corrosion inhibition of metallic surfaces has triggered much industrial and commercial interest. Indeed, corrosion could not be eliminated, but it could be only minimized by utilizing several methods comprising anodizing and coatings.12,13An innovative anti-corrosive approach is the incorporation of organic inhibitors,14–19 forming a protective barrier by getting adsorbed on the surface of the metals. Organic compounds containing heteroatoms (e.g., N, S, and O) in a conjugated system show high efficiency toward the corrosion inhibition of metals.20–22 Notwithstanding, the majority of the abovementioned compounds are highly expensive and toxic to the environment.23 In this regard, much scientific interest has been directed toward using pharmaceutical vehicles, more specifically expired drugs, as eco-friendly anti-corrosive inhibitors for metals and their corresponding alloys.24–27
The investigation of drug inhibitory properties has sparked much interest in recent years. A thorough review documented the potential efficiency of drugs as corrosion inhibitors with limited large-scale practical applications due to the high cost of active substances.28 In an attempt to avoid the drawback of the high cost of drugs, corrosion research was accordingly directed to explore the applicability of expired drugs as anti-corrosive inhibitors. It was unveiled that the utilization of expired drugs can solve two more crucial issues, including the reduction of drug disposal costs and limiting environmental pollution with drugs.29
As crucial N-heterocyclic pharmaceutical compounds, purines and their derivatives were deemed to be potential corrosion inhibitors for metals involving steel,30–33 aluminium,34,35 tin, indium, and their alloys.36,37 According to the literature, the 6-mercaptopurine (MP) drug showed promising corrosion inhibition efficiency for copper metal.38 Additionally, the bronze layer was protected by the purine derivative thioguanine (TG).39 The experimental findings revealed that the TG drug exhibited outstanding anodic and cathodic bifunctional corrosion inhibition performance.39 A literature survey unveiled that no solid investigation has been systematically proposed to assess the anti-corrosive efficiency of the 6-mercaptopurine (MP) and 6-thioguanine (TG) drugs on the Al (111) surface. In that spirit, the current work is devoted to elucidating the efficacy of the MP and TG drugs as corrosion inhibitors for the Al (111) surface (Fig. 1).
 | ||
| Fig. 1 (a) Illustrative representation for 6-mercaptopurine (MP) and 6-thioguanine (TG) drugs as corrosion inhibitors, and (b) side and top representations of the relaxed 6 × 6 supercell of aluminum Al (111). | ||
The correlation of the molecular structure with the potentiality of the investigated drugs as corrosion inhibitors was first examined by employing a series of quantum mechanical calculations. Henceforth, molecular dynamics (MD) simulations were conducted for the adsorption process of the MP and TG drugs on the Al (111) surface. Upon obtaining the preferable complexes, the interaction and binding energies were computed. Following that, charge analysis was executed to estimate the role of the charge transfer on the interactions between the investigated inhibitors and the metallic surface. The obtained findings would provide the foundation for utilizing eco-friendly drugs, especially thiopurines, as anti-corrosive inhibitors for the aluminium surface.
2 Computational Methods
2.1. Quantum chemical calculations
DFT computations were carried out for protonated and non-protonated forms of the 6-mercaptopurine (MP) and 6-thioguanine (TG) drugs in the gas and aqueous phases using the B3LYP/6-311G** level of theory with the help of Gaussian 09 software.40 The polarizable continuum model was utilized for the computations in the aqueous phase.41Optimization of the geometrical structures of the MP and TG drugs was first performed. Frequency computations were executed to confirm the true minima for their structures. To elucidate the nucleophilic and electrophilic regions over the surface of the chemical systems, electrostatic potential (ESP) analysis was executed on the optimized compounds. Upon earlier recommendations, an electron density envelope with a value of 0.002 au was devoted to extracting the ESP maps.42
To investigate the electronic parameters of the studied thiopurine drugs, FMOs theory was employed. Using FMOs, the electron density distribution of HOMO and LUMO were plotted, and their energies (i.e., EHOMO and ELUMO) were computed. Egap was calculated as the energy difference between ELUMO and EHOMO as follows:
| Egap = ELUMO − EHOMO | (1) |
Using Koopmans's approximation method, the IP and EA were computed as the negative values of the energies of HOMO and LUMO, respectively, as follows:
| Koopmans' IP ≈ −EHOMO | (2) |
| Koopmans' EA ≈ −ELUMO | (3) |
| Vertical's IP = ECationNeutral − ENeutral | (4) |
| Vertical's EA = ENeutral − EAnionNeutral | (5) |
Regarding the energy adiabatic method, the IP and EA were calculated upon obtaining the values of the total electronic energy of the optimized geometry of the neutral molecule (ENeutral), anion (EAnion) and cation (ECation) based on eqn (6) and (7).
| Adiabatic's IP = ECation − ENeutral | (6) |
| Adiabatic's EA = ENeutral − EAnion | (7) |
Upon calculating the values of IP and EA from the abovementioned methods, the global descriptors of reactivity, including η, σ, π, X, ΔEBack-donation, ω, and ΔN, were computed as per eqn (8)–(14).
 | (8) |
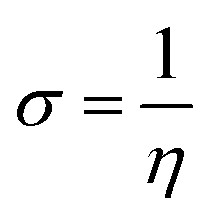 | (9) |
 | (10) |
| π = −χ | (11) |
 | (12) |
 | (13) |
 | (14) |
The metal work function (ΦMetal), inhibitor electronegativity (XInhibitor), and metal hardness (ηMetal), and inhibitor hardness (ηMetal) were involved in eqn (14). ΦMetal and ηMetal values were 4.26 eV and 0, respectively.43–45
The local reactivity of the investigated drugs was evaluated in terms of the Fukui function indices (i.e., fk+(r), fk−(r), and Δf(r)) upon eqn (15)–(17):46,47
| fk+(r) = ρk(N + 1) − ρk(N) | (15) |
| fk−(r) = ρk(N) − ρk(N − 1) | (16) |
| Δf(r) = fk+(r) − fk−(r) | (17) |
For atom k, the charge values of the neutral (ρk(N)), anionic (ρk(N + 1)), and cationic (ρk(N − 1)) forms were utilized to evaluate the aforesaid indices.
2.2. Molecular dynamics (MD) simulations
Using the Quantum ESPRESSO 6.4.1 package,48,49 molecular dynamics (MD) simulations in the NVT ensemble at 298 K were performed to investigate the interactions of the MP and TG drugs with the Al (111) surface. According to the experiments and theoretical studies, the Al (111) surface has been demonstrated to be the most thermodynamically stable surface.50 Therefore, the Al (111) surface was designed with three layers of aluminium atoms as a periodic slab, setting a 20 Å layer of vacuum along the z-axis of the Al (111) slab. A 6 × 6 × 1 supercell of Al (111), containing 108 atoms, was then constructed (Fig. 1). Afterwards, the MP and TG drugs were placed on the Al (111) surface to assess their ability toward corrosion inhibition (see Fig. 1).Generalized Gradient Approximation (GGA) within the Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional was utilized.51 The electron–ion interactions were described via the ultrasoft pseudopotential.52 The kinetic energy and charge density cutoffs were set to 50 and 500 Ry, respectively. The 10−4 eV and 10−3 eV Å−1 were set to be the energy and force convergences criteria, respectively. In order to sample the first Brillouin zone, the k-points of the 2 × 2 × 1 grid were utilized for the geometric optimization and charge transfer computations. The Al atoms in the bottom layer were set to be fixed through the relax calculations, while the two top layers of the slab were fully optimized with the inhibitors. For all calculations, the Marzari–Vanderbilt smearing technique was employed.53 Grimme's DFT-D2 correction method was considered to represent the long-range van der Waals interactions.54 The MD calculations were performed at a simulation time of 0.5 ps with a time step (dt) of 0.97 fs and steps number (nstep) of 1000 steps.
2.3. Interaction and binding energies calculations
Upon the MD simulations, the most stable configurations of the MP/TG⋯Al (111) complexes were selected and thoroughly studied. The interaction energies (Eint) of the selected complexes were calculated based on the following equation:| Eint = EMP/TG⋯Al (111) − (EAl (111) in complex + EMP/TG in complex) | (18) |
| Ebind = EMP/TG⋯Al (111) − (Eisolated Al (111) + Eisolated MP/TG) | (19) |
| Qt = Qcombined Al (111) − Qisolated Al (111) | (20) |
| Δρ = ρMP/TG⋯Al (111) − ρAl (111) − ρMP/TG | (21) |
For implicit water solvent calculations, the environ code58 of Quantum ESPRESSO was utilized with a dielectric constant of 78.3. The solvent effect on the interaction energy of the studied complexes was computed according to the following equation:
| Esolvent effectint = Ewaterint − Egasint | (22) |
3 Results and discussion
3.1. ESP analysis
Electrostatic potential (ESP) analysis is a dependable tool to provide pictorial evidence for either the nucleophilic or electrophilic nature of the chemical systems three-dimensionally.59,60 On the optimized structures of the neutral and protonated forms of the MP and TG drugs, molecular electrostatic potential (MEP) maps were portrayed in the gas and aqueous phases using an electron density envelope with a value of 0.002 au (Fig. 2). | ||
| Fig. 2 ESP maps of the 6-mercaptopurine (MP) and 6-thioguanine (TG) drugs. | ||
Looking at Fig. 2, electron-rich and -deficient centers were identified within the scouted drugs by red- and blue-colored regions, respectively. Evidently, the most negative potentials over the MEP map of the neutral drugs were observed above the sulfur and nitrogen atoms, reflecting their prominent nucleophilic nature. Notably, a negative ESP was found over the hexagonal ring of the drugs under study in the protonated form more than that in the neutral one. The observed nucleophilic character of the studied inhibitors predicted their potential versatility toward interacting with metallic surfaces (i.e., electrophilic surfaces).
3.2. FMOs analysis
The anti-corrosive efficacy of the MP and TG drugs was examined using the frontier molecular orbitals (FMOs) theory. From the FMOs, the distributions of the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) distributions were plotted for inhibitory compounds under investigation, and are shown in Fig. 3. Energies of the aforesaid molecular orbitals (i.e., EHOMO and ELUMO) along with their gap Egap were evaluated (Table 1). | ||
| Fig. 3 HOMO and LUMO distributions of the 6-mercaptopurine (MP) and 6-thioguanine (TG) drugs. | ||
| Compound | EHOMO | ELUMO | Egap |
|---|---|---|---|
| Gas (neutral) | |||
| MP | −5.74 | −2.06 | 3.68 |
| TG | −5.50 | −1.69 | 3.81 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Gas (protonated) | |||
| MP | −11.53 | −6.99 | 4.54 |
| TG | −10.96 | −6.31 | 4.64 |
|
|||
| Aqueous (neutral) | |||
| MP | −6.21 | −2.17 | 4.04 |
| TG | −6.07 | −1.82 | 4.25 |
|
|||
| Aqueous (protonated) | |||
| MP | −7.72 | −3.08 | 4.64 |
| TG | −7.26 | −2.64 | 4.62 |
As can be seen in Fig. 3, the HOMO levels of the two neutral thiopurine drugs in gas and aqueous phases were denoted surrounding the sulfur atom, while HOMO densities of the protonated thiopurine drugs were distributed around the whole molecules. The LUMO distributions were observed over the two rings of thiopurine drugs in the neutral and protonated molecules (Fig. 3).
According to the literature, EHOMO and ELUMO values describe the potentiality of the chemical systems to donate and accept electrons, respectively. From the literature, chemical systems with less negative EHOMO values showed a more prominent tendency toward giving electrons to the electrophiles that have low energy of unoccupied orbitals.61,62 It is apparent from Table 1 that the TG drug showed higher values of EHOMO (i.e., less negative) than the MP drug in all of the studied cases, demonstrating its prominent preferability toward donating electrons. Numerically, the EHOMO values of the neutral form of the TG and MP drugs in the gas phase were −5.50 and −5.74 eV, respectively. Comparatively, the neutral form of the investigated thiopurine drugs was denoted with higher EHOMO in the gas phase than in the aqueous phase, and the opposite is true in the protonated form. In both gas and aqueous phases, the studied drugs generally showed higher efficiency in the case of the neutral form than the protonated one. The TG drug showed less negative ELUMO values than the MP drug in the gas and aqueous phases, respectively.
Egap is a descriptive electronic parameter that depends on the reactivity of the inhibitory compound, unveiling its adsorption tendency toward the metallic surface. As Egap decreases (i.e., less positive), the molecular reactivity level is up. In this regard, as listed in Table 1, the TG and MP drugs had lower Egap values in the neutral form compared with the protonated one.
3.3. Global reactivity descriptors
To obtain deeper insight into the reactivity of corrosion inhibitors, a wide scope of global reactivity descriptors, including ionization potential (IP), electron affinity (EA), global hardness (η), chemical potential (π), global softness (σ), electronegativity (X), total energy change (ΔEBack-Donation), electrophilicity (ω), and a number of transferred electrons (ΔN), were evaluated by Koopmans, energy vertical and adiabatic methods (Table 2).| Compound | Method | IP (eV) | EA (eV) | η (eV) | σ (eV−1) | π (eV) | χ (eV) | ΔEBack-donation (eV) | ω (eV) | ΔN |
|---|---|---|---|---|---|---|---|---|---|---|
| Gas (neutral) | ||||||||||
| MP | Koopmans | 5.74 | 2.06 | 1.84 | 0.54 | −3.90 | 3.90 | −0.46 | 4.13 | 0.10 |
| Vertical | 7.83 | 0.25 | 3.79 | 0.26 | −4.04 | 4.04 | −0.95 | 2.15 | 0.03 | |
| Adiabatic | 7.64 | 0.39 | 3.63 | 0.28 | −4.01 | 4.01 | −0.91 | 2.22 | 0.03 | |
| TG | Koopmans | 5.50 | 1.69 | 1.90 | 0.52 | −3.59 | 3.59 | −0.48 | 3.39 | 0.17 |
| Vertical | 7.57 | 0.01 | 3.78 | 0.26 | −3.79 | 3.79 | −0.95 | 1.90 | 0.06 | |
| Adiabatic | 7.30 | 0.25 | 3.53 | 0.28 | −3.78 | 3.78 | −0.88 | 2.02 | 0.07 | |
|
||||||||||
| Gas (protonated) | ||||||||||
| MP | Koopmans | 11.53 | 6.99 | 2.27 | 0.44 | −9.26 | 9.26 | −0.57 | 18.88 | −1.10 |
| Vertical | 13.54 | 5.11 | 4.22 | 0.24 | −9.32 | 9.32 | −1.05 | 10.31 | −0.60 | |
| Adiabatic | 13.42 | 5.21 | 4.10 | 0.24 | −9.32 | 9.32 | −1.03 | 10.57 | −0.62 | |
| TG | Koopmans | 10.96 | 6.31 | 2.32 | 0.43 | −8.64 | 8.64 | −0.58 | 16.06 | −0.94 |
| Vertical | 12.82 | 4.5 | 4.16 | 0.24 | −8.66 | 8.66 | −1.04 | 9.01 | −0.53 | |
| Adiabatic | 12.65 | 4.67 | 3.99 | 0.25 | −8.66 | 8.66 | −1.00 | 9.40 | −0.55 | |
|
||||||||||
| Aqueous (neutral) | ||||||||||
| MP | Koopmans | 6.21 | 2.17 | 2.02 | 0.50 | −4.19 | 4.19 | −0.50 | 4.35 | 0.02 |
| Vertical | 6.07 | 2.33 | 1.87 | 0.53 | −4.20 | 4.20 | −0.47 | 4.71 | 0.02 | |
| Adiabatic | 5.96 | 2.45 | 1.76 | 0.57 | −4.20 | 4.20 | −0.44 | 5.03 | 0.02 | |
| TG | Koopmans | 6.07 | 1.82 | 2.13 | 0.47 | −3.94 | 3.94 | −0.53 | 3.66 | 0.07 |
| Vertical | 5.93 | 1.98 | 1.97 | 0.51 | −3.96 | 3.96 | −0.49 | 3.96 | 0.08 | |
| Adiabatic | 5.82 | 2.22 | 1.80 | 0.56 | −4.02 | 4.02 | −0.45 | 4.49 | 0.07 | |
|
||||||||||
| Aqueous (protonated) | ||||||||||
| MP | Koopmans | 7.72 | 3.08 | 2.32 | 0.43 | −5.40 | 5.40 | −0.58 | 6.28 | −0.25 |
| Vertical | 7.67 | 3.17 | 2.25 | 0.44 | −5.42 | 5.42 | −0.56 | 6.52 | −0.26 | |
| Adiabatic | 7.57 | 3.28 | 2.14 | 0.47 | −5.43 | 5.43 | −0.54 | 6.87 | −0.27 | |
| TG | Koopmans | 7.26 | 2.64 | 2.31 | 0.43 | −4.95 | 4.95 | −0.58 | 5.30 | −0.15 |
| Vertical | 7.21 | 2.75 | 2.23 | 0.45 | −4.98 | 4.98 | −0.56 | 5.56 | −0.16 | |
| Adiabatic | 7.03 | 3.07 | 1.98 | 0.51 | −5.05 | 5.05 | −0.50 | 6.44 | −0.20 | |
By employing Koopmans' theorem, IP and EA can be computed from the EHOMO and ELUMO energies using eqn (2) and (3), respectively. IP is a basic parameter for the reactivity of a molecule. A high IP means high stability and chemical inertness, and the opposite is true for small IP.63 As shown in Table 2, the TG drug showed lower IP and higher EA values than the MP drug. For example, the neutral form of the TG and MP drugs in the gas phase had Koopmans' IP values of 5.50 and 5.74 eV, respectively.
From the view of the hard and soft acid and bases (HSAB) theory, the inhibitory compound with high softness shows superb corrosion efficiency toward the metallic surfaces, and the opposite is true for the hardness property.64–66 The inhibitor could be adsorbed onto a metallic surface through the part of a molecule with the lowest hardness and greatest softness.67 In both gas and aqueous phases, the neutral form of the studied thiopurine drugs exhibited lower η and higher σ values than the protonated one, outlining their notable reactivity and, accordingly, their favorability in the corrosion inhibition process. Quantitatively, the neutral and protonated forms of the TG drug in the gas phase had Koopmans' η values of 1.90 and 2.32 eV, respectively. Generally, the drugs understudy in the gas phase showed lower η and higher σ values compared with the aqueous phase. As numerical evidence, the neutral form of the TG drug in the gas and aqueous phases had Koopmans' η values of 1.90 and 2.13 eV, respectively.
The HSAB theory documented the χ of a molecule as a crucial reactivity property. The χ difference can reveal the electron flow direction between the inhibitors and the surface of the metal.68 It has been reported that the χ value is inversely correlated with the inhibition effectiveness. From Table 2, further suitability of the TG drug over the MP drug toward the corrosion inhibition process was confirmed by the lower positive χ values of the former drug than the posterior one. Obviously, the lower positive χ values were ascribed to the neutral form of the investigated drugs more than the protonated ones, with prominent favorability in the gas phase.
ω allows an assessment of the global electrophilic nature of a chemical system. From the literature, the efficient electrophile and nucleophile are characterized by higher and lower values of ω,69 respectively. The listed ω values in Table 2 show the more eminent nucleophilic nature of the TG drug in all the studied forms and phases compared with the MP drug. Numerically, the neutral forms of the TG and MP drugs in the gas phase exhibited with Koopmans' ω values of 3.39 and 4.13 eV, respectively.
Regarding the ΔN descriptor, a high negative ΔN value indicates the superior preferable inhibition efficiency. For the studied thiopurine drugs, negative ΔN values were obtained, indicating the good inhibition efficiency of the examined drugs.70 ΔEBack-donation is another characteristic property for the chemical reactivity that was adopted to assess the received charge by each of the interacting species within the corrosion inhibition process. The two thiopurine drugs in all cases had negative values of ΔEBack-donation, outlining the occurrence of the back donation process, which strengthens the adhesion of the inhibitor to the aluminium surface.
Considering the evaluated global reactivity descriptors using Koopmans, energy vertical and adiabatic methods, the three methods exhibited a nearly similar pattern for the neutral and protonated forms of the studied drugs in the gas and aqueous phases.
3.4. Local reactivity: Fukui functions
Toward identifying the plausible nucleophilic and electrophilic regions, local reactivity of the investigated inhibitory compounds was estimated in terms of Fukui function indices. In this regard, nucleophilic (fk+(r)) and electrophilic (fk−(r)) Fukui function indices and their difference (Δf(r)) were computed, and are summarized in Table 3 for all atoms of the studied thiopurine drugs. Fig. S1† displays the plots of the nucleophilic and electrophilic Fukui function indices isosurfaces. As shown in Fig. S1,† the sulfur atom was observed with a higher positive purple-coded region than other atoms, reflecting its prominent suitability for the electrophilic attack. It is proven that the most appropriate regions for the nucleophilic and electrophilic attack were, in conjugation, the regions with the highest fk+(r) and fk−(r) values.71,72 High preferability for electrophilic and nucleophilic attacks was perceived for the atoms that possess values more and less than zero, respectively.73 According to the data gathered in Table 3, sulfur and nitrogen heteroatoms were identified as suitable sites for the electrophilic attack by their higher positive Δf(r) values compared with other atoms. In general, sulfur atoms of the MP and TG drugs were observed with the highest positive Δf(r) values, showing their eminent favorability for electrophilic attack (Table 3). For instance, sulfur atoms of the neutral form of the MP and TG drugs in the aqueous phase showed the highest Δf(r) values of 0.380 and 0.414, respectively.| Compound | Atom | Gas Phase | Aqueous Phase | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neutral | Protonated | Neutral | Protonated | ||||||||||
| fk+(r) | fk−(r) | Δf(r) | fk+(r) | fk−(r) | Δf(r) | fk+(r) | fk−(r) | Δf(r) | fk+(r) | fk−(r) | Δf(r) | ||
| MP | S | −0.368 | −0.578 | 0.209 | −0.223 | −0.387 | 0.165 | −0.344 | −0.724 | 0.380 | −0.196 | −0.435 | 0.239 |
| N | −0.006 | 0.017 | −0.023 | −0.007 | −0.018 | 0.011 | −0.010 | 0.009 | −0.019 | −0.011 | −0.022 | 0.011 | |
| N | −0.027 | −0.026 | −0.002 | −0.044 | −0.033 | −0.011 | −0.039 | −0.017 | −0.022 | −0.050 | −0.029 | −0.021 | |
| N | −0.042 | −0.044 | 0.002 | −0.042 | −0.051 | 0.009 | −0.028 | −0.020 | −0.008 | −0.032 | −0.032 | 0.000 | |
| N | −0.020 | 0.003 | −0.022 | −0.035 | −0.039 | 0.004 | −0.022 | −0.008 | −0.014 | −0.034 | −0.037 | 0.002 | |
| C | 0.003 | 0.004 | −0.002 | −0.009 | −0.069 | 0.060 | −0.015 | −0.007 | −0.008 | −0.025 | −0.083 | 0.057 | |
| C | −0.065 | −0.053 | −0.012 | −0.073 | −0.059 | −0.014 | −0.083 | −0.040 | −0.043 | −0.095 | −0.067 | −0.028 | |
| C | −0.037 | −0.046 | 0.009 | −0.086 | 0.026 | −0.112 | −0.090 | −0.044 | −0.046 | −0.130 | −0.006 | −0.124 | |
| C | −0.099 | −0.059 | −0.040 | −0.103 | −0.068 | −0.035 | −0.104 | −0.038 | −0.066 | −0.113 | −0.066 | −0.047 | |
| C | −0.093 | −0.054 | −0.040 | −0.094 | −0.051 | −0.043 | −0.106 | −0.038 | −0.068 | −0.110 | −0.048 | −0.062 | |
| TG | S | −0.355 | −0.582 | 0.227 | −0.212 | −0.246 | 0.035 | −0.328 | −0.742 | 0.414 | −0.190 | −0.190 | 0.000 |
| N | −0.027 | −0.023 | −0.003 | −0.036 | −0.037 | 0.001 | −0.031 | −0.013 | −0.018 | −0.038 | −0.037 | 0.000 | |
| N | −0.011 | 0.017 | −0.028 | −0.012 | −0.019 | 0.007 | −0.016 | 0.010 | −0.027 | −0.017 | −0.025 | 0.009 | |
| N | −0.038 | −0.042 | 0.004 | −0.038 | −0.050 | 0.013 | −0.025 | −0.018 | −0.006 | −0.028 | −0.040 | 0.012 | |
| N | −0.021 | 0.003 | −0.024 | −0.050 | −0.035 | −0.015 | −0.036 | −0.008 | −0.028 | −0.054 | −0.036 | −0.018 | |
| N | −0.033 | −0.036 | 0.003 | −0.041 | −0.086 | 0.045 | −0.029 | −0.015 | −0.014 | −0.034 | −0.099 | 0.065 | |
| C | 0.007 | 0.012 | −0.005 | 0.005 | −0.093 | 0.097 | −0.008 | −0.002 | −0.007 | −0.012 | −0.113 | 0.102 | |
| C | −0.071 | −0.052 | −0.019 | −0.081 | −0.058 | −0.022 | −0.091 | −0.037 | −0.054 | −0.102 | −0.079 | −0.023 | |
| C | −0.044 | −0.044 | 0.001 | −0.090 | 0.009 | −0.099 | −0.097 | −0.039 | −0.058 | −0.136 | −0.029 | −0.107 | |
| C | −0.061 | −0.039 | −0.022 | −0.058 | −0.029 | −0.029 | −0.073 | −0.033 | −0.040 | −0.078 | −0.052 | −0.026 | |
| C | −0.098 | −0.056 | −0.042 | −0.103 | −0.081 | −0.022 | −0.108 | −0.034 | −0.073 | −0.114 | −0.089 | −0.024 | |
3.5. Molecular dynamics (MD) simulations
The adsorption behavior of the MP and TG drugs toward the Al (111) surface as effective corrosion inhibitors was unveiled utilizing MD simulations. In this respect, the relationship between the total energy of the MP⋯ and TG⋯Al (111) complexes and the corresponding simulation time was elucidated, and is plotted in Fig. 4. | ||
| Fig. 4 Correlation between the total energy (in eV) and the molecular dynamics simulation time (in ps) for the 6-mercaptopurine (MP)⋯ and 6-thioguanine (TG)⋯aluminium (Al (111)) complexes. | ||
Based on the MD simulations, the most stable configurations of the MP⋯ and TG⋯Al (111) complexes with the highest negative energies were chosen (see Fig. 5). For the most stable configurations, the interaction (Eint) and binding (Ebind) energies were then computed and are presented in Table 4.
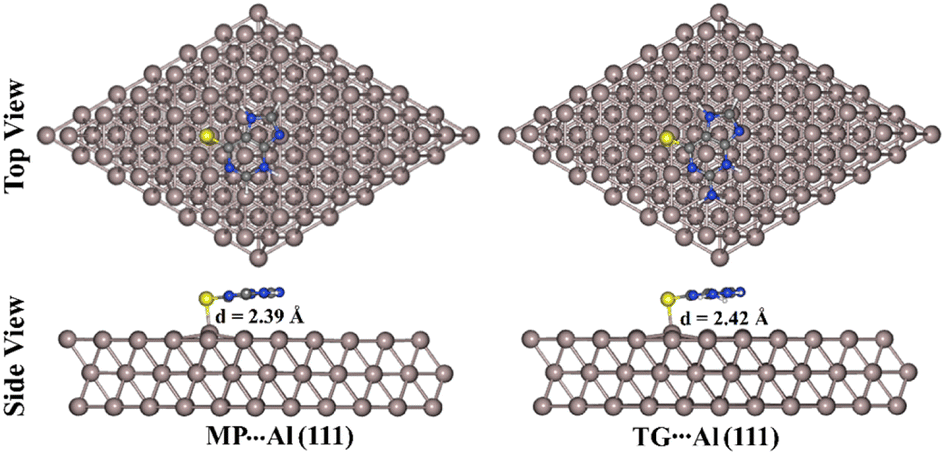 | ||
| Fig. 5 Top and side representations of the most stable 6-mercaptopurine (MP)⋯ and 6-thioguanine (TG)⋯ aluminium (Al (111)) complexes. The S⋯Al equilibrium distance (d) is in Å. | ||
As evident in Fig. 5, S⋯Al covalent bonds were noticed within the relaxed MP⋯ and TG⋯Al (111) complexes with lengths of 2.39 and 2.42 Å, respectively. These lengths were smaller than the entire summation of the vdW radii of the S and Al atoms. According to the data in Table 4, negative values of interaction energies (Eint) were observed for the MP⋯ and TG⋯Al (111) complexes, indicating that the adsorption processes were carried out. The TG⋯Al (111) complex had higher negative interaction energy than the MP⋯Al (111) one with values of −1.79 and −1.64 eV, respectively, demonstrating greater efficiency of the corrosion inhibition for the anterior one. The presence of an electron-donating group (i.e., NH2 group) within the TG structure could be the cause of the greater prominence of the molecular inhibitory potency of TG.74
Considering the structural deformation that occurred during the adsorption process, binding energies (Ebind) were estimated (see Table 4). In line with the Eint pattern, Ebind showed a higher negative value in the case of the TG⋯Al (111) complex in comparison with the MP⋯Al (111) counterpart. Numerically, the Ebind values of the optimized TG⋯ and MP⋯Al (111) complexes were −1.54 and −1.40 eV, respectively.
Toward a more reliable estimation of the inhibition efficiency, the structures of the most stable configurations of the MP⋯ and TG⋯Al (111) complexes were reoptimized utilizing five layers of Al (111) slab (Fig. S1†). Within the relax calculations, the Al atoms in the last two layers were fixed. At the same time, the three top layers of the slab were fully optimized with the inhibitors. Eint and Ebind were accordingly computed for the relaxed complexes and are listed in Table S1.† According to Fig. S1,† the relaxed MP⋯ and TG⋯Al (111) complexes employing five layers of Al (111) slab had interatomic distances that were nearly identical to those in the case of using three layers of Al (111) slab. For instance, S⋯Al bond lengths were observed with values of 2.39/2.42 and 2.40/2.42 Å within the relaxed MP⋯/TG⋯Al (111) complexes utilizing three and five layers of Al (111) slab, respectively.
From the negative Eint and Ebind values summarized in Table S1,† the occurrence of the adsorption process was confirmed. Compared to the adsorption process on the surface of the three layers of the Al (111) slab, five layers of the Al (111) slab showed slightly lower preferability by the energy difference ranging from −0.04 to −0.12 eV. Numerically, the Eint values of the adsorbed MP/TG molecule on three and five layers of the Al (111) slab were −1.64/−1.79 and −1.52/−1.75 eV, respectively.
3.6. Electronic properties calculations
The Bader charge analysis is an appropriate method to better understand the transfer of the charge between the adsorbent and the substrate.55,75 The charge transfer differences (Qt) of the most stable configurations of the MP and TG drugs on the surface of Al (111) were computed using Bader charge analysis, and the outcomes are summarized in Table 4. Based on the Bader charge findings, the negative values of the Qt indicate that the charge shifted from the inhibitors to the metallic surface, and the opposite is true for the positive values.It is clear from Table 4 that the MP and TG drugs transferred electrons to the Al (111) surface via the negative Qt values, indicating that the studied drugs had the potential to interact with the Al (111) surface. Such data consequently ensured the potential anti-corrosive activity of the investigated drugs toward the Al (111) surface. To gain a better understanding of the adsorption process, the maps of the charge density difference (Δρ) for the MP⋯ and TG⋯Al (111) complexes at the most stable configurations were constructed based on eqn (21). The extracted Δρ maps are illustrated in Fig. 6. In the Δρ maps, the sites with increasing and decreasing electron density were detected by colors of yellow and cyan, respectively.
 | ||
| Fig. 6 Top and side views of the charge density difference (Δρ) maps of the relaxed structures of the 6-mercaptopurine (MP)⋯ and 6-thioguanine (TG)⋯ aluminium (Al (111)) complexes at the most stable adsorption configurations obtained from the MD simulations. The charge accumulation and depletion were represented by yellow and cyan colors, respectively. The isosurfaces values are in e Å−3. | ||
From the data in Table 4, the charge transfer to the Al (111) surface from the investigated drugs was indicated by a negative sign of the Qt values. The amount of the distributed charge on the complexes was in line with the Eint findings, at which the TG⋯Al (111) complex had an Eint value that was larger than that of the MP⋯Al (111) complex.
Looking at Fig. 6, cyan- and yellow-colored regions in the Δρ maps were observed within the MP⋯ and TG⋯Al (111) complexes, indicating the electron gain and loss upon the adsorption process, respectively. Occurrence of charge accumulation (i.e., yellow-colored regions) confirmed the significant affinity of the studied drugs to interact with the surface of Al (111) as potential corrosion inhibitors.
To better understand the impact of the adsorption process on the electronic properties of the examined drugs, EHOMO, ELUMO, and Egap values were computed. The data for the studied drugs before and after the adsorption on Al (111) surface are listed in Table 5.
From the data in Table 5, notable differences in the EHOMO, ELUMO, and Egap values were observed for the studied drugs before and after the adsorption process. Moreover, the Egap values of MP and TG drugs were changed after their interaction with the Al (111) surface, confirming the occurrence of adsorption processes. For example, the isolated TG had an Egap value of 2.284 eV that changed to −0.341 eV after the adsorption process on the Al (111) surface (Table 5). Interestingly, the EHOMO values of the isolated drugs decreased after the adsorption on the Al (111) surface. For example, the isolated TG drug had an EHOMO value of −4.434 eV that decreased to −4.279 eV after the adsorption process on the Al (111) surface. The latter observation confirmed the electron-donating property of the TG drug, demonstrating that the FMO results were in line with the Bader charge findings (Table 4).
3.7. Solvent effect calculations
To understand the effect of the solvent on the adsorption process within the MP/TG⋯Al (111) complexes, the interaction energy was evaluated in the presence of a water solvent. Afterwards, the solvent effect (Esolvent effectint) energy was calculated for the most preferable configurations as the difference between the interaction energies of the water and gas media (see the Computational Methods section for details). The obtained (Ewaterint) and (Egasint) values are listed in Table 6.Based on the data in Table 5, the interaction energies of the MP/TG⋯Al (111) complexes in the water medium showed the same trend as those in the gas medium. For example, the Eint of the TG⋯Al (111) complex was larger than that for the MP⋯Al (111) complex in water, with values of −1.49 and −1.23 eV, respectively (Table 6). Subsequently, the negative values of Eint confirmed the occurrence of the adsorption process in the water medium.
4 Conclusions
Herein, detailed insight into the amplitude of the 6-mercaptopurine (MP) and 6-thioguanine (TG) drugs toward inhibiting the corrosion of the Al (111) surface was established using a series of DFT calculations. The ESP findings first showed the prominent nucleophilic nature of the S and N atoms of the examined drugs. Moreover, the FMO results revealed higher values of EHOMO (i.e., less negative) for the TG drug than the MP drug in all of the studied cases. These higher values demonstrated the greater prominent preferability of the former drug toward donating electrons than the latter one. Generally, the neutral form of the investigated thiopurine drugs had higher EHOMO in the gas phase than in the aqueous phase, and the opposite is true in the protonated form. Using Koopmans, energy vertical and adiabatic methods, the global reactivity descriptors were evaluated, and the higher inhibition efficiency of the TG drug over the MP analog was generally demonstrated. In summary, the obtained values of the QM descriptors indicated that the TG drug showed higher anti-corrosive activity than the MP one. In line with the global reactivity affirmations, the nucleophilic and electrophilic Fukui function indices documented a more noticeable preferability of donating electrons in the case of TG than MP. Consistent with QM descriptors, the interaction energies (Eint) confirmed the higher efficiency of the TG drug toward inhibiting the corrosion of the Al (111) surface than the MP one. Eint of TG⋯Al (111) and MP⋯Al (111) complexes showed values up to −1.79 and −1.64 eV, respectively. The energetic results of the solvent effect were in line with the interaction energies in the gas medium that demonstrated a more negative interaction energy for the TG⋯Al (111) complex compared with the MP⋯Al (111) complex. Overall, the above-discussed results are vital for using thiopurine-expired drugs as potential corrosion inhibitors, especially for the Al (111) surface.Author contributions
Conceptualization, Mahmoud A. A. Ibrahim, Tamer Shoeib, and Lamiaa A. Mohamed; methodology, Mahmoud A. A. Ibrahim, Nayra A. M. Moussa, Amna H. M. Mahmoud, Tamer Shoeib, and Lamiaa A. Mohamed; software, Mahmoud A. A. Ibrahim.; formal analysis, Lamiaa A. Mohamed; investigation, Nayra A. M. Moussa, Amna H. M. Mahmoud, and Lamiaa A. Mohamed; resources, Mahmoud A. A. Ibrahim, Shaban R. M. Sayed, and Tamer Shoeib; data curation, Lamiaa A. Mohamed; writing—original draft preparation, Lamiaa A. Mohamed; writing—review and editing, Mahmoud A. A. Ibrahim, Nayra A. M. Moussa, Amna H. M. Mahmoud, Shaban R. M. Sayed, Peter A. Sidhom, Mohamed K. Abd El-Rahman, Tamer Shoeib; visualization, Lamiaa A. Mohamed; supervision, Mahmoud A. A. Ibrahim; project administration, Mahmoud A. A. Ibrahim. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2023R743), King Saud University, Riyadh, Saudi Arabia, for funding this work. The computational work was performed with resources provided by the Science and Technology Development Fund (STDF-Egypt, Grants nos. 5480 and 7972), Bibliotheca Alexandrina (http://hpc.bibalex.org), and The American University in Cairo. Mahmoud A. A. Ibrahim thanks the Center for High-Performance Computing (CHPC, http://www.chpc.ac.za), Cape Town, South Africa, for providing computational resources.References
- Y. Otani and S. Sasaki, Effects of the addition of silicon to 7075 aluminum alloy on microstructure, mechanical properties, and selective laser melting processability, Mater. Sci. Eng., A, 2020, 777, 139079 CrossRef CAS.
- A. Heinz, A. Haszler, C. Keidel, S. Moldenhauer, R. Benedictus and W. S. Miller, Recent development in aluminium alloys for aerospace applications, Mater. Sci. Eng., A, 2000, 280(1), 102–107 CrossRef.
- V. Branzoi, F. Golgovici and F. Branzoi, Aluminium corrosion in hydrochloric acid solutions and the effect of some organic inhibitors, Mater. Chem. Phys., 2003, 78(1), 122–131 CrossRef.
- X. H. Liu, Y. Z. Li, L. Lei and X. Wang, The effect of nitrate on the corrosion behavior of 7075-T651 aluminum alloy in the acidic NaCl solution, Mater. Corros., 2021, 72(9), 1478–1487 CrossRef CAS.
- C. Luo, S. P. Albu, X. R. Zhou, Z. H. Sun, X. Y. Zhang, Z. H. Tang and G. E. Thompson, Continuous and discontinuous localized corrosion of a 2xxx aluminium-copper-lithium alloy in sodium chloride solution, J. Alloys Compd., 2016, 658, 61–70 CrossRef CAS.
- R. Ambat and E. S. Dwarakadasa, Studies on the influence of chloride ion and pH on the electrochemical behaviour of aluminium alloys 8090 and 2014, J. Appl. Electrochem., 1994, 24(9), 911–916 CrossRef CAS.
- A. S. Fouda, F. S. Mohamed and M. W. El-Sherbeni, Corrosion Inhibition of Aluminum–Silicon Alloy in Hydrochloric Acid Solutions Using Carbamidic Thioanhydride Derivatives, J. Bio- Tribo-Corros., 2016, 2(2), 11 CrossRef.
- U. Nazir, Z. Akhter, N. Z. Ali and F. U. Shah, Experimental and theoretical insights into the corrosion inhibition activity of novel Schiff bases for aluminum alloy in acidic medium, RSC Adv., 2019, 9(62), 36455–36470 RSC.
- I. B. Obot, N. O. Obi-Egbedi, S. A. Umoren and E. E. Ebenso, Adsorption and Kinetic Studies on the Inhibition Potential of Fluconazole for the Corrosion of Al in HCl Solution, Chem. Eng. Commun., 2011, 198(5), 711–725 CrossRef CAS.
- I. B. Obot and N. O. Obi-Egbedi, Fluconazole as an inhibitor for aluminium corrosion in 0.1M HCl, Colloids Surf., A, 2008, 330(2–3), 207–212 CrossRef CAS.
- E. E. Oguzie, G. N. Onuoha and E. N. Ejike, Effect of Gongronema latifolium extract on aluminium corrosion in acidic and alkaline media, Pigment. Resin Technol., 2007, 36(1), 44–49 CrossRef CAS.
- M. Paz Martínez-Viademonte, S. T. Abrahami, T. Hack, M. Burchardt and H. Terryn, A Review on Anodizing of Aerospace Aluminum Alloys for Corrosion Protection, Coatings, 2020, 10(11), 1106 CrossRef.
- K. Marcoen, P. Visser, G. F. Trindade, M. L. Abel, J. F. Watts, J. M. C. Mol, H. Terryn and T. Hauffman, Compositional study of a corrosion protective layer formed by leachable lithium salts in a coating defect on AA2024-T3 aluminium alloys, Prog. Org. Coat., 2018, 119, 65–75 CrossRef CAS.
- K. Hossam, F. Bouhlal, L. Hermouche, I. Merimi, H. Labjar, A. Chaouiki, N. Labjar, S.-I. Malika, A. Dahrouch, M. Chellouli, B. Hammouti and S. El Hajjaji, Understanding Corrosion Inhibition of C38 Steel in HCl Media by Omeprazole: Insights for Experimental and Computational Studies, J. Fail. Anal. Prev., 2020, 21(1), 213–227 CrossRef.
- K. F. Khaled, The inhibition of benzimidazole derivatives on corrosion of iron in 1 M HCl solutions, Electrochim. Acta, 2003, 48(17), 2493–2503 CrossRef.
- S. M. Elhadi, M. Bilel, B. Abdelmalek and C. Aissa, Experimental evaluation of quinolinium and isoquinolinium derivatives as corrosion inhibitors of mild steel in 0.5 M H2SO4 solution, Prot. Met. Phys. Chem. Surf., 2016, 52(4), 731–736 CrossRef CAS.
- H. Ashassi-Sorkhabi, B. Shabani, B. Aligholipour and D. Seifzadeh, The effect of some Schiff bases on the corrosion of aluminum in hydrochloric acid solution, Appl. Surf. Sci., 2006, 252(12), 4039–4047 CrossRef CAS.
- M. T. Saeed, M. Saleem, S. Usmani, I. A. Malik, F. A. Al-Shammari and K. M. Deen, Corrosion inhibition of mild steel in 1 M HCl by sweet melon peel extract, J. King Saud Univ., Sci., 2019, 31(4), 1344–1351 CrossRef CAS.
- P. B. Raja, M. Ismail, S. Ghoreishiamiri, J. Mirza, M. C. Ismail, S. Kakooei and A. A. Rahim, Reviews on Corrosion Inhibitors: A Short View, Chem. Eng. Commun., 2016, 203(9), 1145–1156 CrossRef CAS.
- M. H. O. Ahmed, A. A. Al-Amiery, Y. K. Al-Majedy, A. A. H. Kadhum, A. B. Mohamad and T. S. Gaaz, Synthesis and characterization of a novel organic corrosion inhibitor for mild steel in 1 M hydrochloric acid, Results Phys., 2018, 8, 728–733 CrossRef.
- S. K. Ahmed, W. B. Ali and A. A. Khadom, Synthesis and investigations of heterocyclic compounds as corrosion inhibitors for mild steel in hydrochloric acid, Int. J. Ind. Chem., 2019, 10(2), 159–173 CrossRef CAS.
- M. A. A. Ibrahim, N. A. M. Moussa, A. H. M. Mahmoud, A. M. Shawky and L. A. Mohamed, On the efficiency of barbituric acid and its thio derivatives as aluminium corrosion inhibitors: A computational study, J. Mol. Liq., 2023, 383, 122155 CrossRef CAS.
- K. Tamalmani and H. Husin, Review on Corrosion Inhibitors for Oil and Gas Corrosion Issues, Appl. Sci., 2020, 10(10), 3389 CrossRef CAS.
- G. Golestani, M. Shahidi and D. Ghazanfari, Electrochemical evaluation of antibacterial drugs as environment-friendly inhibitors for corrosion of carbon steel in HCl solution, Appl. Surf. Sci., 2014, 308, 347–362 CrossRef CAS.
- G. Gece, Drugs: A review of promising novel corrosion inhibitors, Corros. Sci., 2011, 53(12), 3873–3898 CrossRef CAS.
- A. A. Nazeer, H. M. El-Abbasy and A. S. Fouda, Antibacterial drugs as environmentally-friendly corrosion inhibitors for carbon steel in acid medium, Res. Chem. Intermed., 2013, 39(3), 921–939 CrossRef.
- A. S. Fouda, M. M. Farahat and M. Abdallah, Cephalosporin antibiotics as new corrosion inhibitors for nickel in HCl solution, Res. Chem. Intermed., 2014, 40(3), 1249–1266 CrossRef CAS.
- C. Verma, M. A. Quraishi and K. Y. Rhee, Present and emerging trends in using pharmaceutically active compounds as aqueous phase corrosion inhibitors, J. Mol. Liq., 2021, 328, 115395 CrossRef CAS.
- N. Vaszilcsin, V. Ordodi and A. Borza, Corrosion inhibitors from expired drugs, Int. J. Pharm., 2012, 431(1–2), 241–244 CrossRef CAS PubMed.
- M. Scendo, J. Trela and N. Radek, Purine as an effective corrosion inhibitor for stainless steel in chloride acid solutions, Corros. Rev., 2012, 30(1–2), 33–45 CAS.
- X. H. Li, S. D. Deng, H. Fu and T. H. Li, Adsorption and inhibition effect of 6-benzylaminopurine on cold rolled steel in 1.0 M HCl, Electrochim. Acta, 2009, 54(16), 4089–4098 CrossRef CAS.
- X. H. Li, S. D. Deng and H. Fu, Synergistic inhibition effect of 6-benzylaminopurine and iodide ion on the corrosion of cold rolled steel in H3PO4 solution, Corros. Sci., 2011, 53(11), 3704–3711 CrossRef CAS.
- Y. Yan, W. H. Li, L. K. Cai and B. R. Hou, Electrochemical and quantum chemical study of purines as corrosion inhibitors for mild steel in 1 M HCl solution, Electrochim. Acta, 2008, 53(20), 5953–5960 CrossRef CAS.
- M. A. Amin, Q. Mohsen and O. A. Hazzazi, Synergistic effect of I− ions on the corrosion inhibition of Al in 1.0M phosphoric acid solutions by purine, Mater. Chem. Phys., 2009, 114(2–3), 908–914 CrossRef CAS.
- N. O. Eddy, H. Momoh-Yahaya and E. E. Oguzie, Theoretical and experimental studies on the corrosion inhibition potentials of some purines for aluminum in 0.1 M HCl, J. Adv. Res., 2015, 6(2), 203–217 CrossRef CAS PubMed.
- A. R. El-Sayed, A. M. Shaker and H. M. A. El-Lateef, Corrosion inhibition of tin, indium and tin-indium alloys by adenine or adenosine in hydrochloric acid solution, Corros. Sci., 2010, 52(1), 72–81 CrossRef CAS.
- A.-R. El-Sayed, H. S. Mohran and H. M. Abd El-Lateef, Effect of some nitrogen-heterocyclic compounds on corrosion of tin, indium, and their alloys in HClO4, Monatsh. Chem., 2011, 143(1), 51–64 CrossRef.
- F. Alvarez, C. Grillo, P. Schilardi, A. Rubert, G. Benitez, C. Lorente and M. F. de Mele, Decrease in cytotoxicity of copper-based intrauterine devices (IUD) pretreated with 6-mercaptopurine and pterin as biocompatible corrosion inhibitors, ACS Appl. Mater. Interfaces, 2013, 5(2), 249–255 CrossRef CAS PubMed.
- X. Li, A. Feng, Y. Zu, P. Liu, F. Han and M. An, Electrochemical and Surface Analysis Investigation of Corrosion Inhibition Performance of 6-Thioguanine, Benzotriazole, and Phosphate Salt on Simulated Patinas of Bronze Relics, DOI:10.2139/ssrn.4435999.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. FoxGaussian 09, Revision E01; Gaussian09, Gaussian Inc., Wallingford CT, USA., 2009 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105(8), 2999–3094 CrossRef CAS PubMed.
- M. A. A. Ibrahim, Molecular mechanical perspective on halogen bonding, J. Mol. Model., 2012, 18(10), 4625–4638 CrossRef CAS PubMed.
- J. Hölzl, F. K. Schulte and H. Wagner, Solid Surface Physics, Springer, 1979, vol. 85 Search PubMed.
- H. B. Michaelson, The work function of the elements and its periodicity, J. Appl. Phys., 1977, 48(11), 4729–4733 CrossRef CAS.
- R. Oukhrib, B. El Ibrahimi, H. Abou Oualid, Y. Abdellaoui, S. El Issami, L. Bazzi, M. Hilali and H. Bourzi, In silico investigations of alginate biopolymer on the Fe (110), Cu (111), Al (111) and Sn (001) surfaces in acidic media: Quantum chemical and molecular mechanic calculations, J. Mol. Liq., 2020, 312, 113479 CrossRef CAS.
- W. Yang and W. J. Mortier, The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines, J. Am. Chem. Soc., 1986, 108(19), 5708–5711 CrossRef CAS PubMed.
- K. Fukui, Role of frontier orbitals in chemical reactions, Science, 1982, 218(4574), 747–754 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens.Matter, 2009, 21(39), 395502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio Jr., A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H. Y. Ko, A. Kokalj, E. Kucukbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Ponce, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens.Matter, 2017, 29(46), 465901 CrossRef CAS PubMed.
- Y. Kim and J. Choi, Oxide growth characteristics on Al (100), (110), and (111) surfaces: A chemo-mechanical evaluation, Mater. Today Commun., 2021, 26, 102006 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- D. Vanderbilt, Soft self-consistent pseudopotentials in a generalized eigenvalue formalism, Phys. Rev. B, 1990, 41(11), 7892–7895 CrossRef PubMed.
- N. Marzari, D. Vanderbilt, A. De Vita and M. C. Payne, Thermal contraction and disordering of the Al(110) surface, Phys. Rev. Lett., 1999, 82(16), 3296–3299 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jonsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36(3), 354–360 CrossRef.
- W. Kutzelnigg, Book Review: Atoms in Molecules. A Quantum Theory. (International Series Monographs on Chemistry, Vol. 22). By R. F. W. Bader, Angew. Chem., Int. Ed. Engl., 1993, 32(1), 128–129 CrossRef.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276 CrossRef CAS.
- O. Andreussi, I. Dabo and N. Marzari, Revised self-consistent continuum solvation in electronic-structure calculations, J. Chem. Phys., 2012, 136(6), 064102 CrossRef PubMed.
- A. J. Shusterman and L. M. Hoistad, Teaching Chemistry with Electron Density Models. 2. Can Atomic Charges Adequately Explain Electrostatic Potential Maps?, J. Chem. Educ., 2001, 6(1), 36–40 CrossRef CAS.
- F. J. Luque, J. M. Lopez and M. Orozco, Perspective on Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects - Miertus S, Scrocco E, Tomasi J (1981) Chem Phys 55 : 117, Theor. Chem. Acc., 2000, 103(3–4), 343–345 Search PubMed.
- K. F. Khaled, Electrochemical investigation and modeling of corrosion inhibition of aluminum in molar nitric acid using some sulphur-containing amines, Corros. Sci., 2010, 52(9), 2905–2916 CrossRef CAS.
- G. Gece, The use of quantum chemical methods in corrosion inhibitor studies, Corros. Sci., 2008, 50(11), 2981–2992 CrossRef CAS.
- Y. Abboud, A. Abourriche, T. Saffaj, M. Berrada, M. Charrouf, A. Bennamara and H. Hannache, A novel azo dye, 8-quinolinol-5-azoantipyrine as corrosion inhibitor for mild steel in acidic media, Desalination, 2009, 237(1–3), 175–189 CrossRef CAS.
- M. K. Awad, M. R. Mustafa and M. M. A. Elnga, Computational simulation of the molecular structure of some triazoles as inhibitors for the corrosion of metal surface, J. Mol. Struct.: THEOCHEM, 2010, 959(1–3), 66–74 CrossRef CAS.
- A. Kokalj, On the HSAB based estimate of charge transfer between adsorbates and metal surfaces, Chem. Phys., 2012, 393(1), 1–12 CrossRef CAS.
- N. Kovačević and A. Kokalj, Analysis of molecular electronic structure of imidazole- and benzimidazole-based inhibitors: A simple recipe for qualitative estimation of chemical hardness, Corros. Sci., 2011, 53(3), 909–921 CrossRef.
- H. Wang, X. Wang, H. Wang, L. Wang and A. Liu, DFT study of new bipyrazole derivatives and their potential activity as corrosion inhibitors, J. Mol. Model., 2007, 13(1), 147–153 CrossRef CAS PubMed.
- M. Abdallah, A. Al Bahir, H. M. Altass, A. Fawzy, N. El Guesmi, A. S. Al-Gorair, F. Benhiba, I. Warad and A. Zarrouk, Anticorrosion and adsorption performance of expired antibacterial drugs on Sabic iron corrosion in HCl solution: Chemical, electrochemical and theoretical approach, J. Mol. Liq., 2021, 330, 115702 CrossRef CAS.
- H. Bourzi, R. Oukhrib, B. El Ibrahimi, H. Abou Oualid, Y. Abdellaoui, B. Balkard, S. El Issami, M. Hilali, L. Bazzi and C. Len, Furfural Analogs as Sustainable Corrosion Inhibitors-Predictive Efficiency Using DFT and Monte Carlo Simulations on the Cu(111), Fe(110), Al(111) and Sn(111) Surfaces in Acid Media, Sustainability, 2020, 12(8), 3304 CrossRef.
- N. O. Obi-Egbedi, I. B. Obot and M. I. El-Khaiary, Quantum chemical investigation and statistical analysis of the relationship between corrosion inhibition efficiency and molecular structure of xanthene and its derivatives on mild steel in sulphuric acid, J. Mol. Struct., 2011, 1002(1–3), 86–96 CrossRef CAS.
- M. E. Belghiti, F. Benhiba, N. Benzbiria, C.-H. Lai, S. Echihi, M. Salah, A. Zeroual, Y. Karzazi, A. Tounsi, K. Abbiche, S. Belaaouad, H. Elalaoui-Elabdallaoui and Y. Naimi, Performance of triazole derivatives as potential corrosion in-hibitors for mild steel in a strong phosphoric acid medium: Combining experimental and computational (DFT, MDs & QSAR) approaches, J. Mol. Struct., 2022, 1256, 132515 CrossRef CAS.
- A. El Yaktini, A. Lachiri, M. El Faydy, F. Benhiba, H. Zarrok, M. El Azzouzi, M. Zertoubi, M. Azzi, B. Lakhrissi and A. Zarrouk, Inhibitor effect of new azomethine derivative containing an 8-hydroxyquinoline moiety on corrosion behavior of mild carbon steel in acidic media, Int. J. Corros. Scale Inhib., 2018, 7(4), 609–632 CAS.
- H. Allal, Y. Belhocine and E. Zouaoui, Computational study of some thiophene derivatives as aluminium corrosion inhibitors, J. Mol. Liq., 2018, 265, 668–678 CrossRef CAS.
- A. Singh, K. R. Ansari, M. A. Quraishi and H. Lgaz, Effect of Electron Donating Functional Groups on Corrosion Inhibition of J55 Steel in a Sweet Corrosive Environment: Experimental, Density Functional Theory, and Molecular Dynamic Simulation, Materials, 2019, 12(1), 1–17 CrossRef.
- R. F. W. Bader and T. T. Nguyen-Dang, Quantum Theory of Atoms in Molecules–Dalton Revisited, in Advances in Quantum Chemistry, ed. Löwdin, P.-O., Academic Press, 1981, vol. 14, pp. 63–124 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04954j |
| This journal is © The Royal Society of Chemistry 2023 |