
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinding characteristics of pyrrole-scaffold hepatitis B virus capsid inhibitors and identification of novel potent compounds†
Tanachote Ruengsatra a,
Arthitaya Meepraserta,
Eakkaphon Rattanangkoola,
Sirikan Deesiria,
Jakkrit Srisaa,
Udomsak Udomnilobola,
Wilasinee Dunkoksunga,
Natthaya Chuaypenab,
Rattanaporn Kiatbumrungab,
Pisit Tangkijvanichab,
Sornkanok Vimolmangkangc,
Khanitha Pudhomd and
Thomayant Prueksaritanont*a
a,
Arthitaya Meepraserta,
Eakkaphon Rattanangkoola,
Sirikan Deesiria,
Jakkrit Srisaa,
Udomsak Udomnilobola,
Wilasinee Dunkoksunga,
Natthaya Chuaypenab,
Rattanaporn Kiatbumrungab,
Pisit Tangkijvanichab,
Sornkanok Vimolmangkangc,
Khanitha Pudhomd and
Thomayant Prueksaritanont*a
aChulalongkorn University Drug Discovery and Drug Development Research Center (Chula4DR), Chulalongkorn University, 254 Phayathai Rd, Prathumwan, Bangkok 10330, Thailand. E-mail: thomayant.p@pharm.chula.ac.th
bCenter of Excellence in Hepatitis and Liver Cancer, Department of Biochemistry, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
cDepartment of Pharmacognosy and Pharmaceutical Botany, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Bangkok, Thailand
dDepartment of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
First published on 6th October 2023
Abstract
Hepatitis B virus (HBV) capsid assembly modulators (CAMs) are currently being evaluated in clinical trials as potential curative therapies for HBV. This study used in silico computational modeling to provide insights into the binding characteristics between the HBV core protein and two pyrrole-scaffold inhibitors, JNJ-6379 and GLP-26, both in the CAM-Normal (CAM-N) series. Molecular dynamics simulations showed that the pyrrole inhibitors displayed similar general binding-interaction patterns to NVR 3-778, another CAM-N, with hydrophobic interactions serving as the major driving force. However, consistent with their higher potency, the pyrrole inhibitors exhibited stronger nonpolar interactions with key residues in a solvent-accessible region as compared to NVR 3-778. This feature was facilitated by distinct hydrogen bond interactions of the pyrrole scaffold inhibitors with the residue 140 in chain B of the HBV core protein (L140B). Based on these findings, novel CAM-N compounds were designed to mimic the interaction with L140B residue while maximizing nonpolar interactions in the solvent-accessible region. Several 1H-pyrrole-2-carbonyl substituted pyrrolidine-based compounds with various hydrophobic side chains were synthesized and evaluated. Through analyses of the structure–activity and structure–druggability relations of a series of compounds, CU15 emerged as the most promising lead CAM-N compound, exhibiting sub-nanomolar potency and good pharmacokinetic profiles. Overall, the study demonstrated a practical approach to leverage computational methods for understanding key target binding features for rationale-based design, and for guiding the identification of novel compounds.
1 Introduction
Chronic hepatitis B virus (HBV) infection is a major public health concern that can lead to various liver-related diseases, including decompensated cirrhosis and hepatocellular carcinoma.1,2 HBV is a partially double-stranded DNA virus belonging to the Hepadnaviridae family. Upon infecting the liver, the viral replication cycle starts with the conversion of a 3.2 kb partially double-stranded, relaxed circular DNA (rcDNA) to covalently closed circular DNA (cccDNA), which serves as a nonreplicative minichromosome that persists throughout the lifespan of infected hepatocytes.3,4 The HBV nucleocapsid, also known as the HBV core protein (Cp), is generally formed by the aggregation of 120 Cp dimers, yielding an icosahedral structure (T = 4). The HBV capsid plays vital roles in several steps of the HBV life cycle, including genetic material protection, viral genome maturation, cccDNA stabilization, and interaction with host factors. Inhibiting the assembly of Cp dimers can reduce cccDNA formation, making it a viable target for drug development.5–7 Indeed, several capsid assembly modulators (CAMs) are currently in various stages of preclinical and clinical development,8 signifying a promising class of therapeutic compounds.CAMs can interfere with nucleocapsid formation through two modes of action:9,10 (i) CAM-Normal (CAM-N), represented by sulfamoyl benzamides (SBAs);11–13 sulfamoyl pyrrolamides (SPAs);14–16 and glyoxamide-substituted pyrrolamides,17,18 which accelerates the formation of empty morphologically normal capsid;19,20 and (ii) CAM-Aberrant (CAM-A), represented by heteroaryldihydropyrimidines (HAPs),21,22 induces the formation of a morphologically aberrant capsid.19,20 The first series of CAM-N compounds evaluated in clinical studies were SBAs,23,24 including NVR 3-778![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 and AB-423.12 Subsequently, several compounds with pyrrole scaffolds, such as SPAs (e.g., JNJ-6379)14,15 and the glyoxamide-substituted pyrrolamide series (e.g., GLP-26),18,25 have been developed and generally exhibit higher in vitro potency than SBAs.13,15 Understanding their key binding features holds the potential to discover novel chemotypes. While the bioactive conformation of SBA in complexation with the HBV core protein has been published in the protein data bank (PDB),26 no crystal structure studies of pyrrole-scaffold inhibitors bound with the protein are publicly available. Recently, the binding conformation of JNJ-6379 to the HBV core protein has been illustrated through molecular docking,27 but detailed analyzes on key potency-enhancing features were not provided.
11 and AB-423.12 Subsequently, several compounds with pyrrole scaffolds, such as SPAs (e.g., JNJ-6379)14,15 and the glyoxamide-substituted pyrrolamide series (e.g., GLP-26),18,25 have been developed and generally exhibit higher in vitro potency than SBAs.13,15 Understanding their key binding features holds the potential to discover novel chemotypes. While the bioactive conformation of SBA in complexation with the HBV core protein has been published in the protein data bank (PDB),26 no crystal structure studies of pyrrole-scaffold inhibitors bound with the protein are publicly available. Recently, the binding conformation of JNJ-6379 to the HBV core protein has been illustrated through molecular docking,27 but detailed analyzes on key potency-enhancing features were not provided.
Molecular modeling, including molecular docking and molecular dynamics (MD) simulation, is a widely adopted computational approach that enables the simulation of a ligand's bioactive conformation in its binding site, particularly when the crystal structures are unavailable.28–30 The available crystal structures of targeted protein complexes are commonly used as references for conformational and binding-efficiency predictions to assess the reliability of the computational techniques. Binding energy computations can provide insight into identifying the key binding features of ligand–protein interactions.31,32 Additionally, electrostatic complementarity (EC) analysis33 is also commonly performed to evaluate the compatibility of the electrostatic potential distribution on the molecular surface of the protein and the ligand for a more comprehensive understanding of the ligand–protein interactions.
This study has two primary objectives: (1) to provide insights into the binding characteristics of pyrrole-scaffold inhibitors, specifically the key binding features influencing their potency, and (2) to demonstrate the application of binding characteristic analysis to design and identify novel and potent CAMs. MD simulations and EC calculations were used to comprehensively analyze the binding characteristics of two potent, pyrrole-scaffold inhibitors, JNJ-6379 and GLP-26, with references to two compounds with available crystal structures: SBA NVR 3-778 and HAP, NVR 010-001-E2. GLS-4, which belongs to the HAP series and is expected to adopt a similar binding orientation to NVR 010-001-E2,21 was also included in the study. Novel inhibitors were designed based on the binding conformation insights obtained, and several compounds were selected to be synthesized and their anti-HBV activity and drug-like properties were characterized. The efforts resulted in the identification of novel potent CAMs with favorable pharmacokinetic properties.
2 Results and discussions
2.1 Molecular dynamics simulation of pyrrole-scaffold inhibitors with the HBV capsid protein
To investigate the binding characteristics of pyrrole-scaffold inhibitors in complexation with the HBV capsid protein, two independent all-atom MD simulations were performed for the representative compounds JNJ-6379, and GLP-26, and the three reference inhibitors. The HBV capsid structures, PDB 5T2P and 5E0I, were used as the representatives of the protein–ligand complex of CAM-N and CAM-A, respectively. The heavy atom root-mean-square deviation (RMSD) of the protein backbone and ligand was analyzed for 100 ns relative to an initial structure as a function of time to monitor the system stability and the overall convergence of simulations of the five MD systems (Fig. S1, left†). The global RMSDs of all complexes rapidly increased in the first 5 ns and remained relatively constant for the last 20 ns of simulation. Therefore, MD trajectories from 80 ns to 100 ns for each system were extracted for further structural and energetic analyses (Fig. 1). | ||
| Fig. 1 Binding conformations of five representative ligands, GLP-26, JNJ-6379, NVR 3-778, NVR 010-001-E2, and GLS-4, in the HBV capsid active site (chain B residue = green, chain C residue = cyan) as shown from the last MD snapshot. | ||
The root-mean-square fluctuation (RMSF) of the protein backbone was calculated during the last 20 ns of MD simulation. The results showed that all five complexes exhibited a similar pattern of backbone flexibility (Fig. S1, right†). Evidently, the binding residues that stabilized all inhibitors were in the same area. Specifically, these residues were found on chain B (P25, D29, L30, T33, W102, I105, T109, F110, Y118, S122, I139, and L140) and chain C (S121, V124, W125, T128, R133, and P134). The regions with high RMSF were residues 1–7, 40–50, 60–100, and 141–149, indicating that nonactive site residues exhibited greater flexibility compared to the residues in active sites.
To understand the ligand potency of the five CAMs against HBV capsid protein, the MM/PBSA approach was applied to investigate the binding free energy and its components relevant to the biological activities of five inhibitors toward HBV capsid protein binding. The results (Table 1) showed that the gas phase energy (ΔEMM) of GLP-26 and JNJ-6379 was larger than that of the three reference compounds. The contribution of the electrostatic energy (ΔEele) of pyrrole inhibitors was approximately twice as that of HAPs. However, the increase in ΔEMM for the pyrrole inhibitors was not only from ΔEele but also from the van der Waals energy (ΔEvdW) compared to NVR 3-778 (Table 1).
| Structural series | CAM-N | CAM-A | |||
|---|---|---|---|---|---|
| GLP-26 | JNJ-6379 | NVR 3-778 | NVR 010-001E2 | GLS-4 | |
| Pyrroles | Pyrroles | SBA | HAP | HAP | |
| ΔEele | −18.26 ± 0.08 | −18.92 ± 0.36 | −16.87 ± 0.16 | −8.49 ± 0.43 | −10.08 ± 1.03 |
| ΔEvdW | −57.78 ± 0.77 | −55.68 ± 0.31 | −52.91 ± 0.14 | −59.84 ± 1.50 | −61.39 ± 0.03 |
| ΔEMM | −76.05 ± 0.68 | −74.60 ± 0.05 | −69.78 ± 0.30 | −68.33 ± 1.07 | −71.47 ± 1.07 |
| ΔGele,sol | 26.56 ± 0.51 | 28.77 ± 0.19 | 26.31 ± 0.40 | 19.00 ± 0.01 | 20.64 ± 0.53 |
| ΔGnonpolar,sol | −6.37 ± 0.03 | −6.46 ± 0.02 | −6.51 ± 0.01 | −6.92 ± 0.05 | −7.02 ± 0.05 |
| ΔGsol | 20.18 ± 0.54 | 22.31 ± 0.17 | 19.80 ± 0.38 | 12.08 ± 0.05 | 13.62 ± 0.48 |
| ΔGele,sol + ΔEele | 8.29 ± 0.59 | 9.85 ± 0.41 | 9.44 ± 0.24 | 10.51 ± 0.44 | 10.56 ± 0.50 |
| ΔGnonpolar,sol + ΔEvdW | −64.16 ± 0.74 | −62.14 ± 0.31 | −59.42 ± 0.16 | −66.76 ± 1.55 | −68.41 ± 0.08 |
| ΔGtotal | −55.86 ± 0.15 | −52.29 ± 0.13 | −49.98 ± 0.08 | −56.25 ± 1.11 | −57.85 ± 0.59 |
| −TΔS | 18.31 ± 0.14 | 18.54 ± 0.18 | 20.53 ± 0.21 | 21.72 ± 0.03 | 19.87 ± 0.30 |
| ΔGbind | −37.55 ± 0.01 | −33.75 ± 0.04 | −29.45 ± 0.09 | −34.53 ± 0.76 | −37.98 ± 0.21 |
| pEC50 | 8.52 (ref. 17) | 7.22 (ref. 15) | 6.40 (ref. 11) | 7.95 (ref. 21) | 7.82 (ref. 21) |
Although the ΔEele of the two pyrrole inhibitors was remarkable, it was insufficient to compensate for the desolvation of the polar groups because of the positive value of the overall electrostatic term (ΔGele,sol + ΔEele). The overall electrostatic term of GLP-26 and JNJ-6379 was more negative, reflecting a higher polar interaction, compared to that of HAPs but comparable to that of NVR 3-778 (Table 1). For all five compounds, the contribution of nonpolar energy (ΔGnonpolar,sol + ΔEvdW) to ligand binding was greater than that of polar interaction (ΔGele,sol + ΔEele) (Table 1), suggesting that the major driving force of all inhibitors was the nonpolar interaction. Combining the total energetic term (ΔGtotal) with the entropic term (−TΔS), the overall binding free energies (ΔGbind) of the pyrrole inhibitors were larger than that of NVR 3-778 but comparable to that of HAPs (Table 1). These analyses and findings were consistent with experimental observations of potency (pEC50) (Table 1).
Decomposition energy calculations were performed to identify key residues involved in protein–ligand binding. The per-residue decomposition binding energy (ΔGresiduebind) calculations revealed similar essential binding residues (total binding energy ≤−1 kcal mol−1) at the capsid dimer interface for all five complexes (Fig. S2†). These residues were identified in chains B (P25, W102, I105, F110, I139, L140, S141, and T142) and C (V124, W125, S121, T128, R133, and P134). The interactions with these 14 essential residues were further analyzed separately based on the contributions of the polar (ΔEele + ΔGpolar) and nonpolar (ΔEvdW + ΔGnonpolar) energies (Fig. 2, left), as well as their backbones and side chains (Fig. 2, right). Consistent with the above findings that were common to all inhibitors, nonpolar interactions were observed to play an important role in the complexes (Fig. 2, left). Only W102B, L140B, T142B, and T128C residues contributed to some levels of polar energies. Interestingly, the residue T128C showed relatively high polar contributions for only the pyrrole inhibitors and NVR 3-778 (CAM-N series) but not for the two CAM-A compounds. Additionally, while high polar interactions facilitated by the residues T142B and L140B were observed for NVR 3-778 and the pyrrole inhibitors, respectively, such interactions were not observed for the 2 HAPs. In addition, it was found that for most residues, the main contribution to the binding energies was found to be from their side chains. However, both the backbone and the side chain of L140B played almost equal roles in contributing to the binding energies (Fig. 2, right).
 | ||
| Fig. 2 (Left) Energy contributions from electrostatic (ΔEele + ΔGpolar) and van der Waals (ΔEvdW + ΔGnonpolar) interactions, and (Right) energy contributions of residue side chains and backbones of the HBV Cp dimer complexed with (A) GLP-26, (B) JNJ-6379, (C) NVR 3-778, (D) NVR 010-001-E2, and (E) GLS-4. The results shown are the averaged values from two independent simulations. | ||
We next examined the ligand structural features that bind to the essential residues identified above. The ligand structures were divided into three different regions, for ease of the analysis, namely L1, L2 and L3, as shown in Fig. 3.
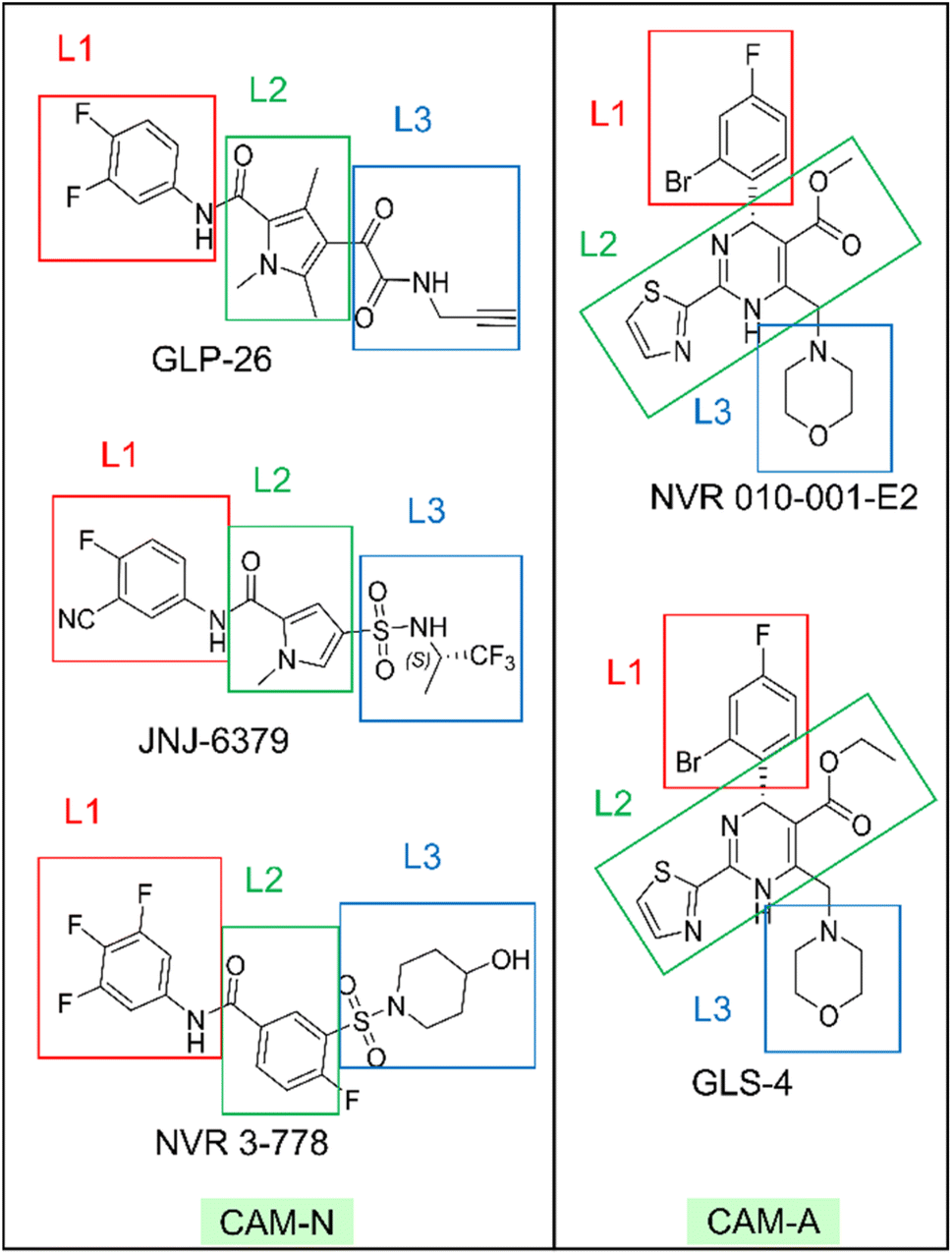 | ||
| Fig. 3 The molecular structures of all representative inhibitors are indicated with red, green, and blue rectangles for region assignment. | ||
The ligand structural features that formed polar interactions with the T128C residue was the NH amide between the L1 and L2 regions of the JNJ-6379, GLP-26 and NVR 3-778. Additionally, the hydroxyl group in the L3 region of NVR 3-778 facilitated a H-bond formation with the residue T142B. The presence of sulfonamide (–SO2NH–) and glyoxamide (–(CO)2NH–) moieties resided in the L3 region of the pyrrole inhibitors JNJ-6379 and GLP-26, respectively, enabled the formation of double H-bonds with residue L140B (Fig. 4). A similar finding has been reported previously for JNJ-6379.27 Notably, the polar contribution at T142B (−1.25 kcal mol−1 for NVR 3-778) and the double H-bonds at L140B residues (−1.62 and −1.16 kcal mol−1 for GLP-26 and JNJ-6379, respectively) were comparable (Table S1†). However, compared to the H-bond at T142B, the formation of the double H-bonds at L140B allowed for a closer distance between the α-carbon of the chain B residues (I139–T142) and the L3 nitrogen atom, and thus the adjacent L3 hydrophobic moieties (Fig. 4). The significance of these findings will be discussed in the next section.
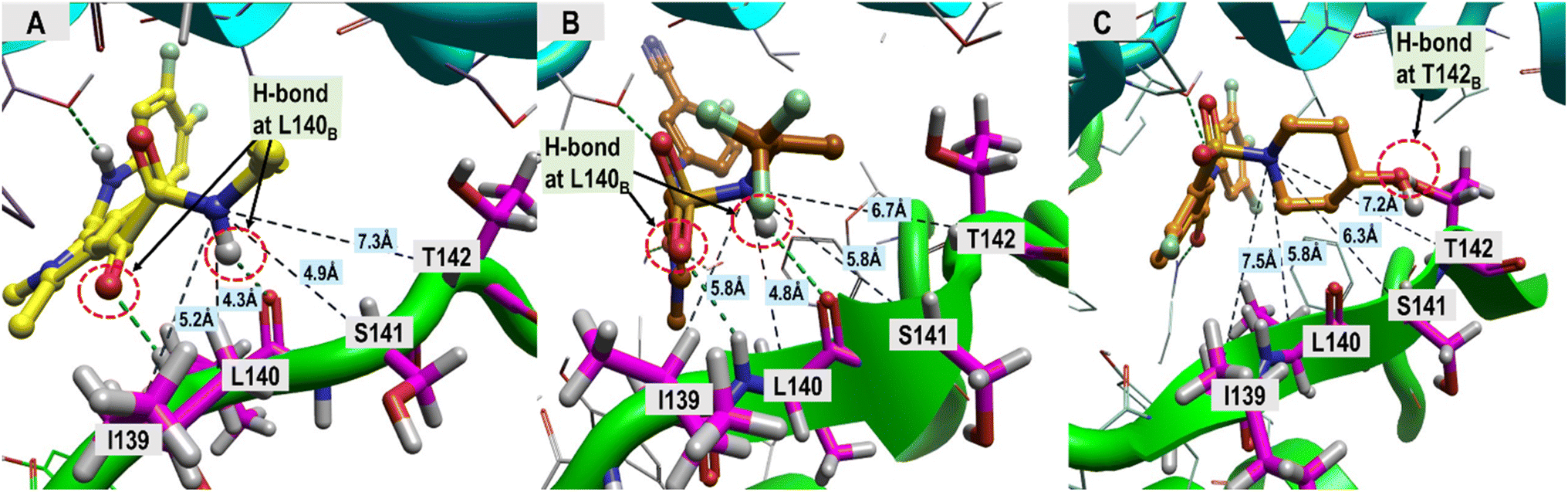 | ||
| Fig. 4 Distance measurements between the ligand's nitrogen atom, linked to an adjacent hydrophobic group, and HBV core protein residues I139–T142. (A) GLP-26, (B) JNJ-6379, (C) NVR 3-778. The ligand-binding regions and the associated structural features driving the hydrophobic interactions, which are the major contribution toward ligand binding, were analyzed, and the results are presented in Table 2. | ||
In the L1 region, the phenyl ring was identified as the key hydrophobic moiety contributing to the binding energies of all five inhibitors. It interacted with residues P25B, W102B, I105B, and V124C. The L1 nonpolar energy contribution of the pyrrole inhibitors was comparable to NVR 3-778 but lower than that of the HAPs (Table 2). In the L2 region, various structural features contributed to hydrophobic interactions with F110B, L140B, and T128C, with the pyrrole core being the primary contributor for the pyrrole inhibitors. Despite the structure diversity, the hydrophobic energies from this region appeared to be comparable across the inhibitors (Table 2).
| GLP-26 | JNJ-6379 | NVR 3-778 | NVR 010-001-E2 | GLS-4 | |
|---|---|---|---|---|---|
| L1 ΔGresiduebind | −5.91 | −5.78 | −6.10 | −8.45 | −8.91 |
| Binding residues: P25B, W102B, I105B, and V124C | |||||
| L2 ΔGresiduebind | −7.37 | −6.60 | −6.88 | −6.27 | −6.13 |
| Binding residues: F110B, L140B, and T128C | |||||
| L3 ΔGresiduebind | −7.67 | −7.15 | −6.66 | −4.88 | −4.61 |
| Overall binding residues: W125C, R133C, P134C, I139B, S141B, T142B, and S121C | |||||
| −3.79 | −3.35 | −2.82 | −1.70 | −1.33 | |
| Solvent-accessible binding residues: I139B, S141B, T142B, and S121C | |||||
In the L3 region, different hydrophobic functionalities of the five inhibitors interacted with the key residues W125C, R133C, P134C, S121C, I139B, S141B, and T142B. These hydrophobic interactions resulted in greater stabilization for the pyrroles inhibitors and NVR 3-778 binding compared to the two HAPs, indicating the potential significant role of the L3 hydrophobic moieties in designing CAM-N compounds. The residues W125C, R133C, and P134C formed a small hydrophobic pocket with a positive electrostatic potential surface (ESP) interacting with the electronegative sulfonamide (in NVR 3-778 and JNJ-6379) and glyoxamide moieties (in GLP-26) (Fig. S3†). JNJ-6379 (−3.81 kcal mol−1) and GLP-26 (−3.91 kcal mol−1) were bound to this pocket with a binding efficiency comparable to that of NVR 3-778 (−3.84 kcal mol−1) (Table S1†). Notably, the remaining four interacting residues (S121C, I139B, S141B, and T142B) were all in the solvent-accessible area. These residues interacted with the L3 hydrophobic moieties adjacent to the N atoms in the same region. As shown in Table 2, the L3 nonpolar binding energies of the pyrrole inhibitors were higher than those of the reference compounds (NVR 3-778, GLS-4, and NVR 010-001-E2). This strong hydrophobic interaction of the pyrrole-scaffold inhibitors is consistent with the proximity between the L3 hydrophobic moieties and the solvent-accessible residues, facilitated by the unique formation of the double H-bonds between their sulfonamide (–SO2NH–) or glyoxamide (–(CO)2NH–) groups with the L140B residue, as shown above in Fig. 4.
2.2 Electrostatic complementarity investigation of the L1 region
Our analysis of the structure–activity relationships (SARs) of available SPAs15 revealed that modification of the position and nature of substituents on the phenyl moiety at the L1 region could vary the potency of pyrrole-scaffold inhibitors by up to 10-fold. To further understand the changes in electrostatic contribution resulting from modification on the phenyl ring, we performed EC calculation using derivatives of JNJ-6379 (compound 094).15 To minimize conformational effects such as bond rotation, we manually modified the phenyl moiety and performed soft local minimization. The results of this analysis with six representative compounds (Fig. 5, row A) were visualized using EC and ESP surfaces (Fig. 5, rows B and C).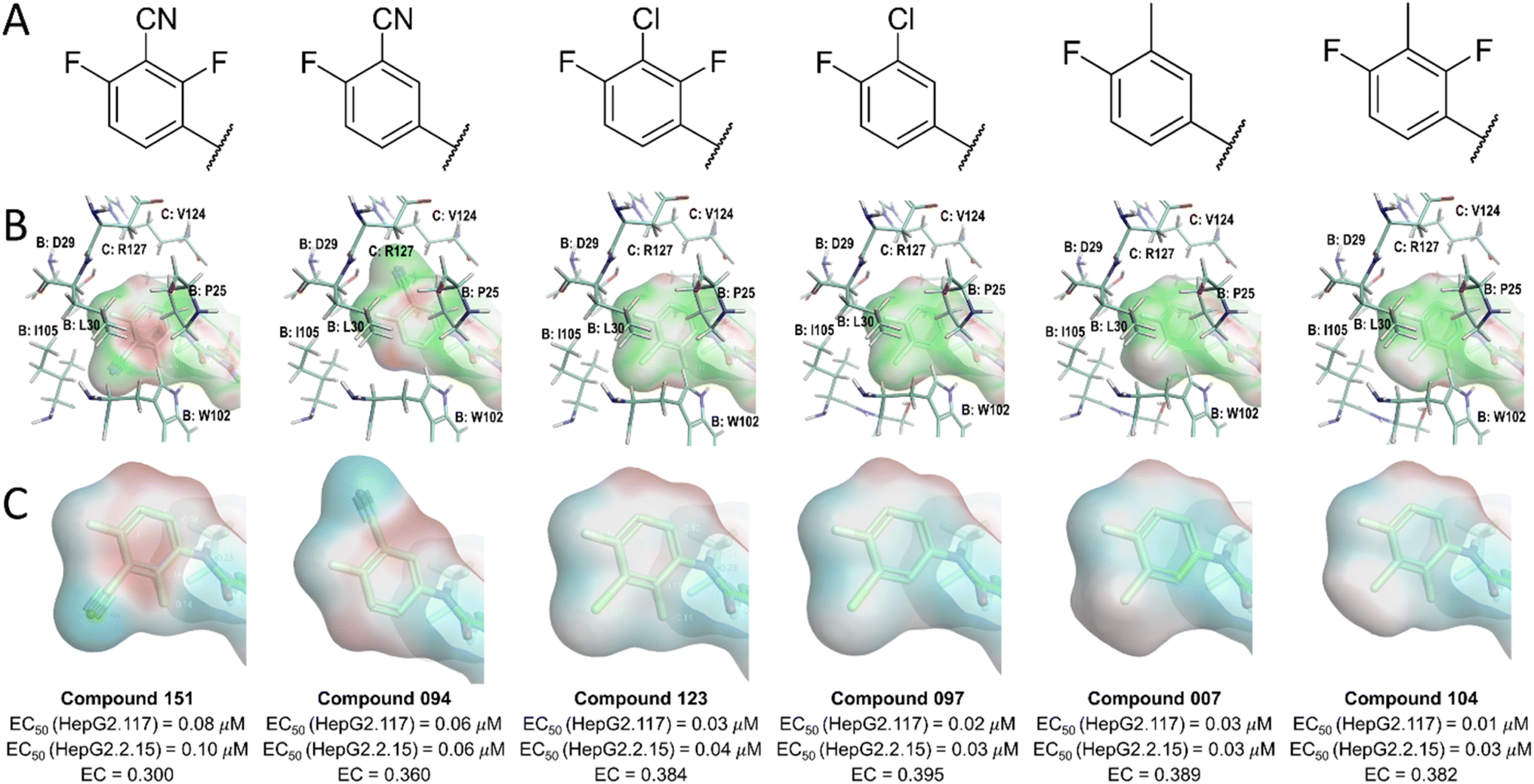 | ||
| Fig. 5 Structures (row A), EC surface (red = electrostatic crash, green = electrostatic complementarity, row B), ESP surface (red = positive ESP, blue = negative ESP, row C) of the ligand L1 moiety in the active site of the HBV Cp dimer. | ||
The results showed that the introduction of nitrile substitutions in compounds 094 and 151 reduced the negativity of π-clouds on the phenyl plane, leading to a stronger electrostatic crash. Moreover, replacing the nitrile group with either a chloro (compounds 097 and 123) or a methyl group (compounds 007 and 104) resulted in the phenyl ring gaining a more negative ESP, leading to a 3-to 8-fold increase in the anti-HBV activity in HepG2.117 and HepG2.2.15 cell lines (Fig. 5 row C). The chloro and methyl substituents enhanced the electrostatic interactions between the phenyl π-system and the side chain of residues P25B, W102B, and V124C, as shown by the green ligand EC surface (Fig. 5, row B). These findings suggest that the presence of strong electron-withdrawing groups reduces the negative potential of aromatic π-clouds, resulting in an increased electrostatic crash at the phenyl plane. However, the introduction of at least one of the electron-withdrawing groups may be necessary for interacting with the residues D29B, T33B, and I105B. Although these interactions may not appear significant, their absence could prevent the ligand from effectively binding the protein. Therefore, achieving a balance in the presence of electronegative groups on phenyl moiety is crucial to maximize ligand potency. An additional factor to consider is the potential steric hindrance effect of the substituents. The methyl substitution, for example, could increase potency due to its hydrophobic space-filling effect, while a nitrile substitution could lead to a decrease in ligand potency due to steric crash.
2.3 Incorporating the binding site analysis to design a potential lead compound
The insights gained from the MD analysis regarding the binding characteristics of pyrrole-scaffold inhibitors were used to guide our design strategy for novel CAM-N ligands. The design focus was on incorporating polar functionalities that can enable the formation of H-bonds with L140B, as well as incorporating hydrophobic groups that maximize nonpolar interactions with the key residues at the L3, and particularly the solvent-accessible region. Among possible core ligand structures considered, pyrrolidine was chosen as a viable alternative core to the pyrrole scaffold, given its comparable size and shape, as well as the potential for simplified reaction mechanisms during optimization. Considering the stronger hydrogen bonding interactions of the glyoxamide moiety with L140B compared to the sulfonamide moiety, we began our design with glyoxamide-substituted pyrrolidine-3-carboxamide derivatives.34 Subsequently, we replaced the glyoxamide moiety with a 1H-pyrrole-2-carbonyl functional group to mimic the hydrogen bonding interaction with L140B residue, resulting in a novel structural series (Fig. 6). | ||
| Fig. 6 Design strategy of the novel structural series. | ||
To investigate the impact of hydrophobic moieties in the L3 region on optimizing nonpolar interactions, we synthesized four derivatives (CU01–03) with different hydrophobic groups. As shown in Table 3, CU01 had the least potency among the compounds studied. This could be attributed to its hydrophobic group being smaller than those of other compounds, resulting in weaker nonpolar interactions. However, CU02 and CU03 showed comparable anti-HBV activity, indicating the importance of the hydrophobic interaction in the L3 region for enhancing ligand potency. Additionally, upon changing from the pyrrole core to the novel pyrrolidine-based series, the key interacting residues in the L3 region also changed from I139B S141B, T142B, and S121C to the newly identified residues: T109B, F110B, and T142B (Fig. S4†). These residues were taken into later design considerations to increase the potency of the next ligands.
|
|||||
|---|---|---|---|---|---|
| Code | R | % HBV DNA remaining | CLint (μL per min per mg protein) | % CYP inhibition at 10 μM | |
| 0.1 μM | 1 μM | ||||
| a NI is no inhibition; values were less than 20%. | |||||
| CU01 | 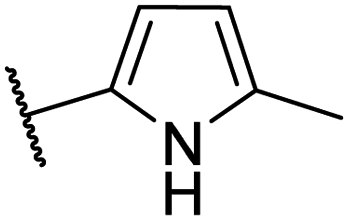 |
— | 59 | 14.4 | NIa |
| CU02 | 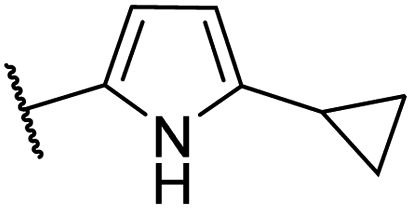 |
— | 48 | 14.5 | CYP2B6 (50%), CYP2C19 (53%) |
| CU03 | 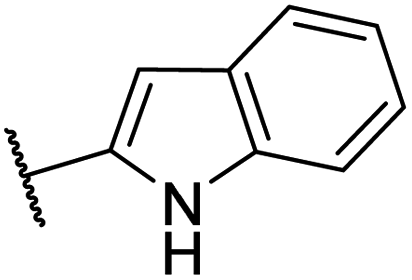 |
— | 44 | 9.2 | CYP2C9 (62%), CYP2C19 (63%), CYP2D6 (51%) |
| EC50 = 447 nM | |||||
| CU04 | 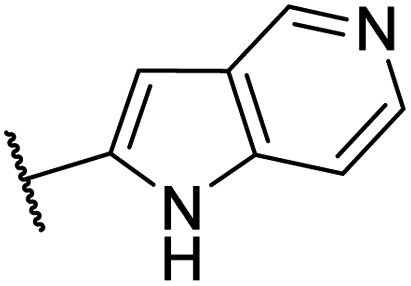 |
— | 67 | 1.1 | CYP2A6 (57%), CYP2C8 (55%), CYP2D6 (82%) |
| CU05 |  |
— | 47 | 2.9 | CYP2B6 (65%), CYP2C9 (87%) |
| CU06 | 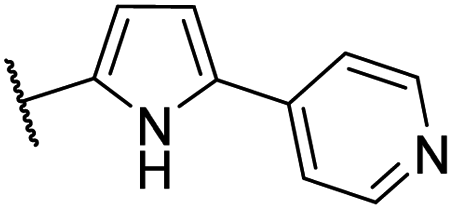 |
— | 28 | 4.6 | CYP2B6 (81%), CYP2C9 (67%), CYP2D6 (89%), CYP3A4/5 (76%) |
| CU07 |  |
— | 77 | 6.2 | NI |
| CU08 | 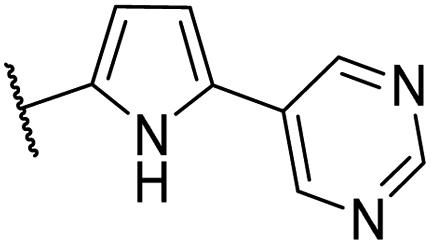 |
43 | 0.5 | CYP2B6 (61%), CYP2C9 (53%) | |
| CU09 | 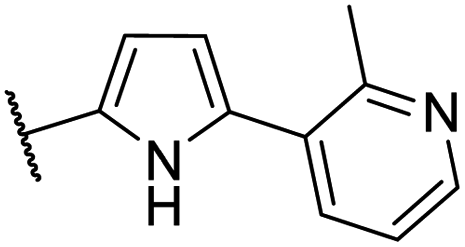 |
76 | 31 | 5.7 | NI |
| CU10 | 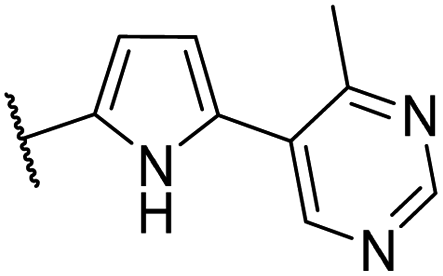 |
91 | 39 | 7.1 | CYP2C9 (90%) |
| CU11 | 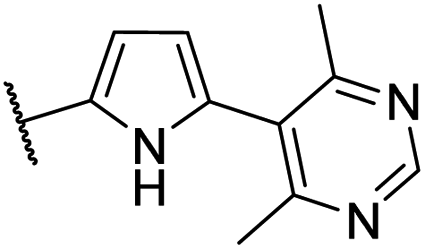 |
45 | 32 | 6.3 | NI |
| EC50 = 35 nM | |||||

A parallel effort was also made to study their drug-like properties. Specifically, the metabolic stability and potential for cytochrome P450 (CYP) inhibition were evaluated during the screening phase of this work. The results in Table 3 showed that CU01 and CU02 were less stable with higher CLint compared to CU03, while CU02 and CU03 showed inhibitory effects on multiple CYPs in human liver microsomes. The oxidative metabolic reactions primarily occurred on the R group of both CU02 and CU03 (ESI file S2†). To improve the metabolic stability, CU04 was synthesized by replacing an indole of CU03 with a more polar, azaindole moiety. However, CU04 showed a significant reduction in anti-HBV activity, suggesting that the polar nitrogen in the binding site interfered with hydrophobic interactions in the L3 region.
To further improve the ligand interactions, a pyridine moiety was incorporated into the R group, to give CU05–07 (Table 3). The presence of the moiety extended the aromatic nitrogen into the solvent-accessible area, allowing for interaction with water molecules. Moving the pyridine nitrogen from the meta position in CU05 to the para position in CU06, increased in potency. This improvement can be attributed to the favorable placement of the nitrogen atom, which enables interaction with water molecules in the solvent-accessible region. However, when the nitrogen atom was moved to the ortho position in CU07, a significant decrease in anti-HBV activity was observed. This could be due to the formation of an intramolecular H-bond between the pyridine nitrogen and the NH group of the pyrrole ring, resulting in the unavailability of the NH group for interaction with L140B.
Although the incorporation of the pyridine moiety improved metabolic stability, significant CYP inhibition was observed for CU05 and CU06, but not for CU07 (Table 3). A previous study suggested that an aromatic nitrogen lone pair of electrons could interact with CYP enzymes.35 Therefore, the CYP inhibitory effect could be associated with the existence of lone pairs of electrons on pyridine nitrogen. The decrease in CYP inhibition observed for CU07 was likely due to the intramolecular H-bond that can inhibit the interaction of pyridine with CYP enzymes. Furthermore, while addition of one aromatic nitrogen to CU08 did not affect its bioactivity compared to CU05, it appeared to slightly improve metabolic stability and CYP inhibition.
The pyridine moiety of CU05 was predicted to be close to and potentially in hydrophobic contact with the target residues (T109B, F110B, and T142B) (Fig. S5†). To enhance the anti-HBV activity while minimizing the CYP inhibition, CU09 and CU10 were designed with a methyl substitution adjacent to the aromatic nitrogen. This methyl group was expected to increase the anti-HBV activity by enhancing ligand binding to the target residues while reducing interactions between aromatic nitrogen and CYP enzymes via steric hindrance. However, a significant improvement in the CYP inhibition profile was only observed for CU09 (vs. CU05), but not for CU10 (vs. CU08). Additionally, their bioactivity did not improve as anticipated. We postulated that the introduction of the methyl substitution could lead to an asymmetrical structure, where 6-membered aromatic ring is arranged perpendicular to the pyrrole ring, resulting in two rotamers in the capsid binding site, with the methyl group pointing toward the target residues in one rotamer and not in another (Fig. 7A and B).
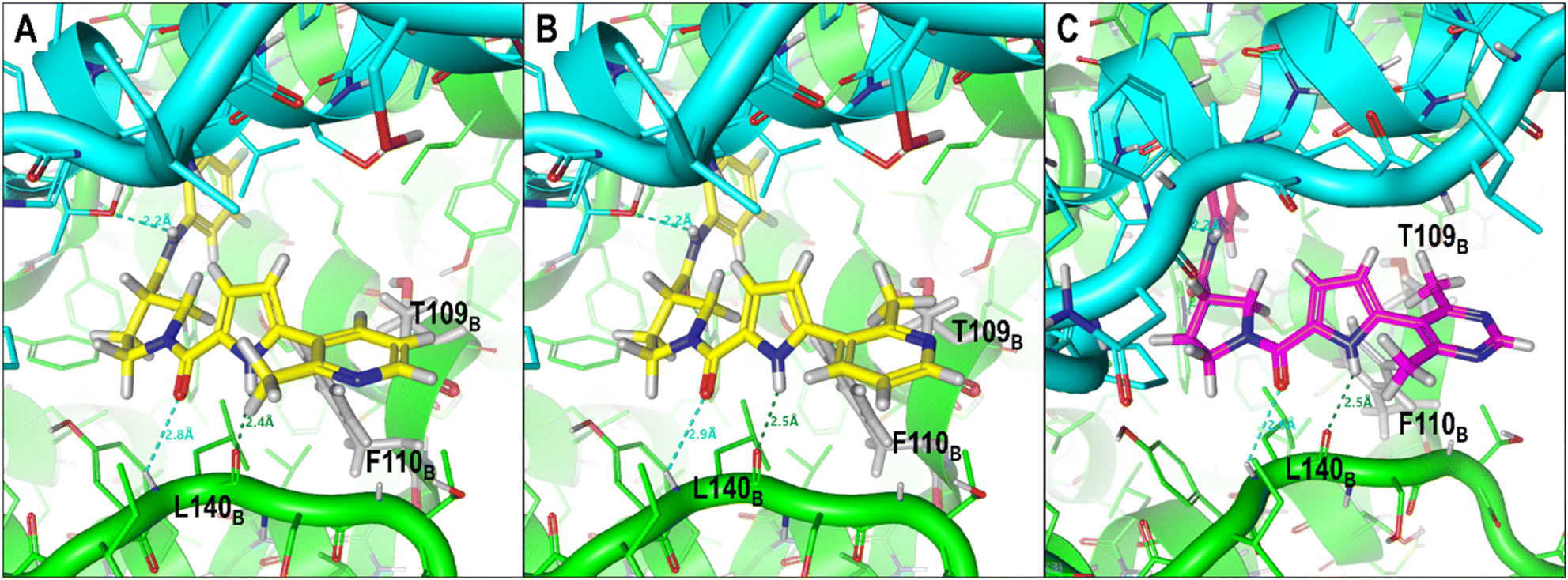 | ||
| Fig. 7 Predicted conformations of two compounds, CU09 and CU11, in the HBV capsid binding site (chain B residue = green, chain C residue = cyan). CU09 is depicted with the methyl group of the pyridine ring pointing outward from (A), and toward (B) the target residues. CU11 is depicted with the pyrimidine and pyrrole rings in a perpendicular conformation (C). | ||
To support the hypothesis that the methyl group can indeed interact with the target residues T109B, F110B, and T142B in the binding site, CU11 was designed with a symmetrical moiety of 4,6-dimethylpyrimidin-5-yl ring (Fig. 7C). Bioactivity studies showed that CU11 showed significantly higher anti-HBV activity than CU09, with an EC50 value of 35 nM (Table 3). Furthermore, CU11 showed acceptable metabolic stability, CYP inhibition and cytotoxicity profiles (CC50 > 10 μM). These results suggest that CU11 is a promising lead compound for further optimization of pyrrolidine-scaffold CAMs with favorable drug-like properties.
2.4 Optimization of the lead compounds
To improve the bioactivity of the lead compound, modifications were made to the substitution group on the phenyl moiety at the L1 region and the core structure at the L2 region. The EC analysis described earlier suggested that incorporating a less electronegative moiety in R1 and R2 could potentially enhance hydrogen–π interactions at L1, leading to improved potency. Therefore, the fluoro group in R1 was replaced with a less electronegative chloro group, while the fluorine in R2 was substituted with hydrogen. These modifications resulted in a twofold increase in potency for CU12 relative to CU11 (Table 3). It is noteworthy that we kept one fluoro functionality between R1 and R2 to maintain the interaction at D29B, T33B, and I105B (see 2.2 Electrostatic complementarity investigation of the L1 region).To improve the bioactivity further, we added an S-methyl group at position R3 of the pyrrolidine structure. This addition has been shown to enhance ligand bioactivity,34 potentially through increasing hydrophobic interactions at the L2 residues (L140B and F110B). This modification led to the discovery of a new structural series, designated as CU13–15. CU13 and CU14 exhibited superior potency compared to their predecessors, CU11 and CU12, respectively, confirming the potency-enhancing property of the methyl pyrrolidine structure.33 Substitution of chlorine in the R1 position of CU14 with a methyl group increased negative ESP to the phenyl ring, which led to the discovery of CU15. CU15 demonstrated a further increase in potency with an EC50 of less than 10 nM. These changes on R1, R2, and R3 had no effect on the metabolic stability (Table 4), CYP inhibition (IC50 > 10 μM for all tested CYPs) and cytotoxicity profile (CC50 > 10 μM) of the compounds. Further modification of CU15 by replacing the pyrimidine with a pyrazole ring resulted in CU16 with lower potency and less stability (Table 4). Other R4 modifications, including dimethyl-substituted pyridazine, isoxazole, and thiazole rings, as well as the use of a methoxy-substituted pyrimidine moiety, also did not result in improved bioactivity (data not shown).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Code | R1 | R2 | R3 | R4 | HBV DNA inhibition EC50 (nM) | CLint (μL per min per mg protein) | CLa (mL min−1 kg−1) | Fb (%) |
| a CL, in vivo rat clearance.b F, oral bioavailability in rats. | ||||||||
| CU11 | –F | –F | –H | 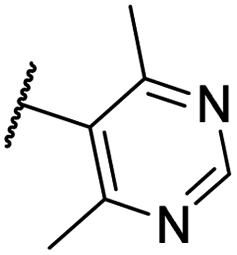 |
35.1 ± 3 | 6.3 | 15.2 | 79 |
| CU12 | –Cl | –H | –H |  |
18.2 ± 2 | 3.6 | — | — |
| CU13 | –F | –F | –Me | 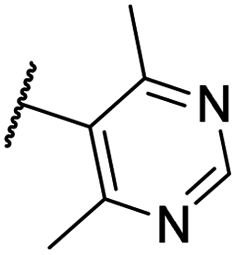 |
17.8 ± 3 | 2.1 | — | — |
| CU14 | –Cl | –H | –Me |  |
13.2 ± 2 | 2.9 | 12.3 | 66 |
| CU15 | –Me | –H | –Me |  |
8.6 ± 2 | 4.9 | 20.0 | 44 |
| CU16 | –Me | –H | –Me | 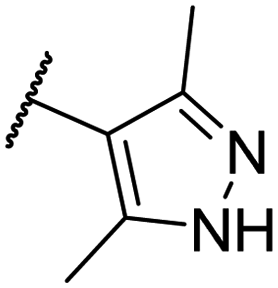 |
14.9 ± 3 | 11.7 | — | — |

Next, we evaluated the pharmacokinetics of our lead compounds following single oral dosing in rats. Table 4 shows that the lead compounds exhibited relatively low clearance and good oral bioavailability in rats. These findings, together with their in vitro potency, human liver microsomal stability and cytotoxicity results, suggested that CU15 was a promising lead compound worthy of further characterization.
2.5 Confirmation of CAM-N mechanism
To confirm that our leads belong to the CAM-N series, as was the case for the pyrroles, we employed size exclusion chromatography and transmission electron microscopy to study the effects of CU14, a representative compound, on HBV capsid assembly. The result was compared to the reference compounds NVR 3-778 (CAM-N compound) and BAY 41-4109 (CAM-A compound). In both assays, CU14 showed properties similar to those of NVR 3-778, but not BAY 41-4109, supporting the hypothesis that CU14, and likely all other compounds in Table 4, belongs to the CAM-N series (Fig. S5†).3 Conclusions
In this study, we employed MD simulations to investigate the binding characteristics of two representative pyrrole-scaffold inhibitors, JNJ-6379, and GLP-26, to the HBV capsid protein, and the results were compared to those of two other chemical series, SBAs (NVR 3-778) and HAPs (GLS-4 and NVR 010-001-E2). Our analysis revealed that all inhibitors displayed similar binding characteristics with essential binding residues at their capsid dimer interfaces (P25B, W102B, I105B, I139B, L140B, S141B, T142B, F110B, S121C, V124C, W125C, R133C, and P134C), and with hydrophobic interactions serving as the major ligand-binding component across all compounds. The hydrophobic interactions in the L3 region of the pyrroles and NVR 3-778 were higher compared with the HAPs. Distinct from NVR 3-778 and key to their potency, the pyrrole-scaffold inhibitors exhibited higher nonpolar interactions in the L3 solvent-accessible region. This strong hydrophobic interaction was associated with the proximity of their hydrophobic groups to the solvent-accessible residues enabled by the formation of the double H-bonds between the sulfonamide (–SO2NH–) or glyoxamide (–(CO)2NH–) moieties of the pyrrole inhibitors with the L140B residue. Furthermore, the electrostatic complementarity analysis of the L1 region revealed that increasing the negative ESP surface of phenyl moiety improved hydrogen–π interactions with W102B, P25B, and V124C. The insights into the binding features of pyrrole-scaffold inhibitors led to the design of novel CAM-N inhibitors by replacing the glyoxamide moiety with a 1H-pyrrole-2-carbonyl functional group to mimic the interaction at L140B, resulting in the development of a novel series of pyrrolidine-based inhibitors. We also optimized the hydrophobic groups of the compounds in the solvent-accessible region to increase their potency and metabolic stability and improve the CYP inhibition profile. Further optimization in other regions resulted in the identification of novel lead compounds with nanomolar potencies and favorable pharmacokinetic parameters. The CAM-N mechanism of lead compounds was also confirmed. Overall, this study demonstrates a practical approach for leveraging computational methods to guide the identification of novel potent CAMs, and potentially of modulators for other applicable target diseases.4 Experimental section
4.1 Molecular docking
System preparations were carried out using Flare (version 5.0).36 Considering that 3D structures of capsid protein complexes with GLP-26 and JNJ-6379 were unavailable, both inhibitors were individually docked into the binding site to identify ligand binding conformations and local minimum structures. The capsid protein (Cp) structure was obtained from the Protein Data Bank (PDB code 5T2P) to perform constrained docking.26 Only chains B and C were retained in the system before removing all water molecules. The complex was then prepared using a standard preparation procedure. Subsequently, the ligands were docked into the binding site using extra precision docking, and the number of runs was set to 15. Four hydrogen bonding interactions between the protein and ligand (atom Nε1 of W102B, N and O of L140B, and Oγ1 of T128C) were set as docking constraints. The N-phenyl formamide substructure of NVR 3-778 was used as a template. The best conformations of the ligand were then selected on the basis of the number of hydrogen bonds formed and binding energy. Ligand minimization was further applied to refine bond and ring conformation and reduce atomic crash. The docked complex was used as a starting structure for MD simulations.4.2 In silico system preparation
The initial structure of the wild-type (WT) capsid protein with NVR 010-001-E2 and NVR 3-778 bound was prepared on the basis of their crystal structures from the PDB entry codes 5E0I21 and 5T2P,26 respectively, which is a Y132A mutant. Only chains B and C were retained in the MD system. Residue 132 of chains B and C was edited from alanine (A) to tyrosine (Y) based on the Dunbrack rotamer library37 using a structural editing module implemented in Chimera 1.9 to build WT complexes.38 The WT capsid protein with GLS-4 bound was unavailable; thus, the methyl ester moiety of NVR 010-001-E2 in complexation with WT capsid protein was modified to ethyl ester moiety of GLS-4. Based on previous studies, the complex of JNJ-6379 and GLP-26 bound to HBV capsid protein was obtained from molecular docking. All system preparations and MD simulations were then performed using AMBER16 package program.39 The protonation state of all ionizable residues was characterized at pH = 7.0 by PDB2PQR.40 The missing hydrogen atoms were added using the LEAP module. The AMBER ff99SB force field was applied to proteins.41 Ligand structures were fully optimized using the HF/6-31G(d) method in Gaussian 09 to prepare ligand parameters,42 in which the electrostatic potential (ESP) was also obtained using the same method. The restrained electrostatic potential (RESP) charges were subsequently derived using RESP fitting.43 The AMBER ff99SB force field and general AMBER force field44 were applied to all ligands.Hydrogen atoms were minimized with 1500 steps of steepest descents (SD) and followed by 1500 steps of conjugate gradient (CG) using the SANDER module to decrease steric hindrance and atomic crash. All MD systems were solvated with TIP3P water45 in a 12 Å cubic box around the solute surface. Sodium counterions were also randomly added to neutralize the total negative charge of the system. Then, SD and CG minimizations were applied in three steps: (i) 500 steps of SD and CG were carried out to optimize the mutated residue; (ii) 1000 steps of SD and CG were subsequently applied to freely minimize all components, except for the protein backbone; (iii) 2500 steps of SD and CG were finally applied to minimize the whole complex.
4.3 Molecular dynamics simulations
Two independent MD simulations with different initial atomic velocities under a periodic boundary condition were carried out for each Cp/ligand complex (NVR 010-001-E2, GLS-4, NVR 3-778, GLP-26, and JNJ-6379). All covalent bonds involving hydrogen atoms were constrained using the SHAKE algorithm.46 The particle mesh Ewald summation approach was applied to address long-range electrostatic interactions, whereas the short-range cutoff of 10 Å was assigned for nonbonded interactions. The Langevin thermostat was also applied to control the temperature with a collision frequency of 5.0 ps−1 for the first 500 ps, after which it was decreased to 2.0 ps−1 for the remaining simulations. The simulation time step of 0.2 ps was used during simulation. Each simulated system was heated up from 0 K to 298.15 K under an NVT ensemble for 50 ps using positional restraints of 10.0 kcal mol−1·Å−2 to all heavy atoms of the Cp/ligand complex. Consequently, the whole system was simulated under the NPT ensemble at the target temperature and a pressure of 1 atm using the PMEMD.cuda module until the simulation time reached 100 ns. The MD system gradually decreased the restraint weights by 10.0, 5.0, 2.0, 1.0, 0.5, and 0.1 kcal mol−1 Å−2 for 50 ps of each stage. Afterward, the whole system was allowed to move freely. The MD trajectories were collected every 2 ps for further analysis. Notably, conformational analysis, including hydrogen bonding interaction, was investigated using CPPTRAJ.47 Protein–ligand binding interactions and associated binding free energies, including per-residue decomposition were evaluated using the MMPBSA.py module.48 Essential binding residues were identified based on their binding energy being less than −1.0 kcal mol−1. The molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) calculation has been widely applied to calculate binding energy.49,50 The entropic term (−TΔS) was carried out using normal mode analysis to evaluate the conformational entropy contribution upon ligand binding.51 The H-bond formation was evaluated based on the percentage of H-bond occupation recorded from the last 20 ns MD snapshots, based on the following criteria: (i) the distance between donor and acceptor atom ≤3.5 Å and (ii) the angle of H-bond ≥120°.4.4 Electrostatic complementarity investigation at the L1 region
All protein–ligand complexes and EC scores were obtained by Flare (version 5.0).36 The ligands, from the patent WO2014/184350,15 were prepared manually using the protein binding complex of JNJ-6379 (obtained from molecular docking) as the starting point. Structure editing (only at the phenyl moiety) and soft local minimization were performed, using ligand editing mode in Flare, to obtain the desired protein–ligand complex. If the substituents contain more than one rotamer (e.g., ortho or meta substituents on the phenyl ring), then the orientation with a high EC value was selected. The EC scores were used to determine EC.334.5 Chemistry
All reagents and solvents were used as purchased from commercial sources. Reactions were carried out under nitrogen or argon atmospheres. 1H and 13C spectra were recorded on a Bruker-400 or Jeol-500 NMR spectrometer (1H: 400 or 500 MHz, 13C: 100 or 125 MHz) using CD3OD, acetone-d6 or DMSO-d6 as a solvent and tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm) relative to TMS (δ = 0.00 ppm) and coupling constants (J) are reported in Hertz (Hz). Splitting patterns are indicated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; br s, broad singlet. Molecular weights of the final products were determined using high resolution mass spectrometry (HRMS) using AB Sciex X500B QTOF mass spectrometer. Products were purified by column chromatography (silica gel 60, Merck silica), or Sephadex-LH20. Analytical thin-layer chromatography (TLC) was performed with precoated Merck silica gel 60 F254 plates (thick layer, 0.25 mm) and visualized at 254 nm using an ultraviolet lamp or by staining with aqueous potassium permanganate solution as the detecting agent.4.5.11.1 Step 1. Methyl 5-bromo-1H-pyrrole-2-carboxylate (1.2 eq.) was dissolved in H2O
:1,4-dioxane (1:5). Then, pyridin-3-yl boronic acid (1.0 eq.) and K2CO3 (3.0 eq.) were added to the flask. The reaction was bubbled by the argon atmosphere for 15 min. Then 10 mol% Pd(PPh3)4 was added and heated the reaction under argon atmosphere at 110 °C for 3 hours. To achieve pure product, crude product was purified by liquid column chromatography with 20 to 50% EtOAc/Hexanes.
4.5.11.2 Step 2. Methyl 5-(pyridin-3-yl)-1H-pyrrole-2-carboxylate (1.0 eq.) was dissolved in THF. Then, LiOH (5.0 eq.) was dissolved in H2O and added to the THF solution. The reaction mixture was heated until it reached 90 °C and kept heating for an hour. The reaction was acidified by 2 N HCl until pH of solution equals to 2. The reaction mixture was concentrated by rotavapor. The solid was washed with 0 to 15% MeOH/EtOAc and transferred the solution into another flask and the crude solution was dried. Dried crude product was used in further synthesis.
4.5.11.3 Step 3. 5-(Pyridin-3-yl)-1H-pyrrole-2-carboxylic acid (1.0 eq.) was dissolved in DMF. Then HATU (1.5 eq.) and DIPEA (3.0 eq.) were added. The reaction mixture was stirred at room temperature for 10 min. Then, intermediate I-A (1.1 eq.) was added to the solution. The mixture was stirred overnight at room temperature. Then the mixture was diluted with water. The product was extracted with EtOAc 3 times. The organic layer was washed by brine and purified by column chromatography with 0 to 15% MeOH/EtOAc. The desired fractions were pooled and the solvent was removed under reduced pressure to provide CU05. 74% yield of white solid; 1H NMR (400 MHz, DMSO-d6) δ 11.89 (s, 1H), 10.48 (s, 1H), 9.03 (dd, J = 2.3, 0.6 Hz, 1H), 8.41 (dd, J = 4.7, 1.5 Hz, 1H), 8.21 (ddd, J = 8.0, 2.2, 1.6 Hz, 1H), 7.51 (dd, J = 10.1, 6.5 Hz, 4H), 7.37 (ddd, J = 8.1, 4.8, 0.7 Hz, 1H), 6.75 (dd, J = 3.8, 2.7 Hz, 1H), 6.72 (dd, J = 3.8, 2.3 Hz, 1H), 4.05–3.57 (m, 4H), 3.31–3.13 (m, 1H), 2.36–2.02 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 171.9, 171.6, 159.8, 150.2 (ddd, JCF = 242.8, 9.9, 5.3 Hz), 147.5, 146.3, 135.4 (td, JCF = 11.7, 3.5 Hz), 134.7 (dt, JCF = 243.1, 15.9 Hz), 131.9, 131.4, 127.8, 127.7, 123.7, 113.8, 108.3, 103.6 (d, JCF = 24.4 Hz, 2C), 50.2, 49.3 (isomer), 47.4, 46.3, 45.3 (isomer), 42.6 (isomer), 30.1, 27.5 (isomer); HRMS (ESI): m/z calcd for [C21H17F3N4O2 + H]+ 415.1382; found 415.1372.
4.5.13.1 Step 1. 2-Bromopyridine (1.2 eq.) was dissolved in H2O
:1,4-dioxane (1:5). Then, intermediate II (1.0 eq.) and K2CO3 (3.0 eq.) were added to the flask. The reaction mixture was bubbled by the argon atmosphere for 15 min. Then 10 mol% Pd(PPh3)4 was added to the reaction. Reaction was heated under argon atmosphere at 110 °C for 1 hour. To achieve pure product, crude product was purified by liquid column chromatography with 20 to 50% EtOAc/Hexanes.
4.5.13.2 Step 2:. Methyl 5-(pyridin-2-yl)-1H-pyrrole-2-carboxylate (1.0 eq.) was dissolved in THF. Then, LiOH (5.0 eq.) was dissolved in H2O and added to the THF solution. The reaction mixture was heated until it reached 90 °C and kept heating for an hour. The reaction was acidified by 2 N HCl until pH of solution equals to 2. The reaction mixture was concentrated by rotavapor. The solid was washed with 15% MeOH/EtOAc and transferred the solution into another flask and the crude solution was dried. Dried crude product was used in further synthesis.
4.5.13.3 Step 3. 5-(Pyridin-2-yl)-1H-pyrrole-2-carboxylic acid (1.0 eq.) was dissolved in DMF. Then, HATU (1.5 eq.) and DIPEA (3.0 eq.) were added. The reaction mixture was stirred at room temperature for 10 min. Then, intermediate I-A (1.1 eq.) was added to the solution. The mixture was stirred overnight at room temperature. Then the mixture was diluted with water. The product was extracted with EtOAc 3 times. The organic layer was washed by brine and purified by column chromatography with 0 to 15% MeOH/EtOAc. The desired fractions were pooled and the solvent was removed under reduced pressure to afford CU07. 80% yield of white solid; 1H NMR (400 MHz, DMSO-d6) d 11.43 (s, 1H), 10.51 (s, 1H), 8.55 (dd, J = 5.0, 1.8 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.79 (td, J = 8.0, 1.8 Hz, 1H), 7.54–7.50 (m, 2H), 7.23 (dd, J = 7.5, 4.9 Hz, 1H), 6.90 (dd, J = 4.0, 2.6 Hz, 1H), 6.74 (dd, J = 4.0, 2.4 Hz, 1H), 4.03–3.57 (m, 4H), 3.30–3.17 (m, 1H), 2.32–2.05 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 171.8, 171.5, 159.4, 150.1 (ddd, JCF = 242.7, 9.9, 4.9 Hz), 149.4, 149.3, 137.1, 135.4 (t, JCF = 11.5 Hz), 134.7 (dt, JCF = 243.1, 15.1 Hz), 133.8, 127.3, 121.9, 119.1, 113.7, 109.3, 103.6 (d, JCF = 24.6 Hz, 2C), 50.0, 49.3 (isomer), 47.3, 46.3, 45.2 (isomer), 42.5 (isomer), 30.0, 27.5 (isomer); HRMS (ESI): m/z calcd for [C21H17F3N4O2 + H]+ 415.1376; found 415.1357.
4.6 In vitro anti-HBV activity and cytotoxicity in HepG2.2.15 cell line
A stable HBV-expressed HepG2.2.15 cells were seeded at 5 × 105 cells per well in 24-well plate with high glucose Dulbecco's Modified Eagle Medium containing 10% heat inactivated fetal bovine serum, 300 mg L−1 G418, 1% non-essential amino acids, 100 units per ml antibiotic–antimycotic, and incubated for overnight at 37 °C and 5% CO2. The cells were treated with the medium containing solvent control (0.5% DMSO) or compounds at various concentrations for three days. Then, the medium was removed and replaced with fresh medium containing compounds for another 3 days. After 6 days of treatment, intracellular HBV DNA was extracted and determined by using quantitative real-time PCR (qPCR) technique with specific primers. For screening, the anti-HBV activity is reported as % HBV DNA remaining following compound treatment at 0.1 or 1 μM, compared to the vehicle control. For EC50 determination, the % HBV DNA remaining was plotted against compound concentration. The EC50 values were obtained from a 4-parameter nonlinear curve fitting using GraphPad Prism software.For cytotoxicity, the cells were seeded onto a 96-well plate and allowed to adhere for 24 h. Then, the cells were treated with various concentrations of the compounds for 6 days. After treatment, the cultured media were discarded and replaced with serum-free media containing 0.5 μg mL−1 of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) for 2 h. The resulting formazan crystals were dissolved in DMSO, and then measured its absorbance at 570 nm using a microplate reader. A concentration producing 50% cell death (CC50) was determined from a fitting of concentration–response curve (% cell viability vs. test concentration) to a four parameter Hill equation, using a GraphPad software (Prism, USA).
4.7 In vitro microsomal stability
Test compounds (1 μM) were incubated with pooled human liver microsome (0.4 mg mL−1) in potassium phosphate buffer (100 mM, pH 7.4) containing 3.3 mM of MgCl2 and 1.3 mM of β-NADPH. The organic solvent in reactions was kept not more than 1% in the incubations. The reactions were incubated at 37 °C. The reactions were quenched at various time points with ice-cold acetonitrile containing internal standard. Then, the supernatants of reaction mixtures were collected by centrifugation, and injected to LC-MS/MS for analysis. Data are expressed as intrinsic clearance (CLint) values.4.8 CYP450 inhibition
Test compounds (10 μM) were incubated with pooled human liver microsome (0.2 mg mL−1) and cocktail of 10 probe substrates [100 μM phenacetin for CYP1A2, 1.5 μM coumarin for CYP2A6, 12 μM bupropion for CYP2B6, 1 μM amodiaquine for CYP2C8, 100 μM tolbutamide for CYP2C9, 50 μM (S)-mephenytoin for CYP2C19, 2.5 μM dextromethorphan for CYP2D6, 15 μM chlorzoxazone for CYP2E1, and 2.5 μM midazolam and 50 μM testosterone for CYP3A4/5] in 100 mM potassium phosphate buffer (pH 7.4) and 3.3 mM of MgCl2 at 37 °C.52 The reaction was initiated by addition of β-NADPH (1.3 mM). After the incubation period of 10 minutes, the reaction was stopped with ice-cold 3% formic acid in 5% acetonitrile. The samples were centrifuged and collected for LC-MS/MS analysis. Data were expressed as % inhibition of each metabolite formed in samples compared to control.4.9 Size exclusion chromatography (SEC) and transmission electron microscopy (TEM)
HBV core protein (Cp149) protein was prepared based on a published method53 with slight modifications. A compound (50 μM) was incubated with 15 μM of Cp149 in 50 mM HEPES buffer (pH 7.5) containing 150 mM NaCl. The reaction mixture was incubated for 16 hours at 21 °C. After incubation, an aliquot of the reaction mixture was analyzed by SEC using a size exclusion column (YarraTM SEC-3000, 3 μm, 150 × 7.8 mm) with a running buffer (50 mM HEPES, 150 mM NaCl, adjusted to pH 7.5). The UV absorbance was monitored at 280 nm. Compound-induced capsid assembly was determined from the ratio of the retention time of the Cp149 dimer to that of the capsid fraction.For TEM study, another aliquot of the reaction mixture was negatively stained with 1% uranyl acetate and visualized on a JEOL JEM-1400 100 kV electron microscope. Images were acquired at a magnification of 100°000× to 300°000×.
4.10 Pharmacokinetic studies in rats
The study was approved by Pharmaron (Beijing) Institutional Animal Care and Use Committee (IACUC). Test compounds were administered to male Sprague-Dawley rats via intravenous bolus injection (0.5 mg kg−1) or oral gavage (2 mg kg−1). The formulations for intravenous and oral dosing are 10% dimethyl sulfoxide in 20% hydroxypropyl methyl β-cyclodextrin solution and 40% polyethylene glycol in water, respectively. Blood samples collected at pre-dose and at various time points up to 24 h post dosing, and centrifuged to obtain plasma. The concentration of test compounds in plasma were quantified using liquid chromatography-tandem mass spectrometric analysis. The pharmacokinetic parameters (i.e. CL, total body clearance; and F, oral bioavailability) were calculated via non-compartmental analysis using WinNonlin software (Phoenix™, version 8.3).Abbreviations
| CAM | Capsid assembly modulator |
| CAM-N | CAM-Normal |
| CAM-A | CAM-Aberrant |
| cccDNA | Covalently closed circular DNA |
| CLint | Intrinsic clearance |
| CL | Total body clearance |
| Cp | Core protein |
| EC | Electrostatic complementarity |
| ESP | Electrostatic potential |
| F | Oral bioavailability |
| H-bond | Hydrogen bond |
| HBV | Hepatitis B virus |
| MD | Molecular dynamic simulation |
| MM/PBSA | Molecular mechanics/Poisson–Boltzmann surface area |
| ns | Nanosecond |
| pgRNA | Pregenomic RNA |
| ps | Picosecond |
| rcDNA | Relaxed circular DNA |
| SBA | Sulfamoyl benzamide |
| SEC | Size exclusion chromatography |
| SI | Selectivity index |
| SPA | Sulfamoyl pyrrolamide |
| TEM | Transmission electron microscopy |
Data availability
(1) AMBER39 is a proprietary licensed software released by University of California, San Francisco. AMBER16 package program was used for MD simulation. (2) Chimera38 is an open-access software released by Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. Chimera 1.9 was used for protein editing. (3) Flare36 is a proprietary licensed software released by Cresset Biomolecular Discovery Ltd. Version 5.0.0 of the Flare was used for the molecular alignment, molecular docking, and EC calculation. (4) Gaussian 09 (ref. 42) is a proprietary licensed software released by Gaussian Inc., which was used for the preparation of ligand parameters. (5) All molecular data was manually obtained from the patents WO2014/184350 (ref. 15) which were validated and filtered through SwissADME54 and PAINS-Remover55 to eliminate the pan assay interference structures. (6) The protein structure data used in this experiment can be downloaded at https://www.rcsb.org/ (PDB code 5E0I21 and 5T2P26).Author contributions
Tanachote Ruengsatra was responsible for performing computational and in vitro ADMET experiments, strategizing compound design concept, and conceptualizing and writing manuscript. Arthitaya Meeprasert conducted compound modelling and docking and wrote manuscript. Eakkaphon Rattanangkool, Sirikan Deesiri, Jakkrit Srisa and Khanitha Pudhom were involved in chemistry related studies and manuscript writing. Udomsak Udomnilobol and Wilasinee Dunkoksung were involved in ADMET/PK studies and writing manuscript. Natthaya Chuaypen, Rattanaporn Kiatbumrung, and Pisit Tangkijvanich were responsible for cell biochemical experiments and analyses. Sornkanok Vimolmangkang was responsible for providing HBV capsid protein preparation and purification. Thomayant Prueksaritanont conceived the study project, obtained necessary fundings, and conceptualized/wrote manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported mainly by the Rachadapisek Sompoch Endowment Fund (Chulalongkorn University, Bangkok, Thailand), and partially by Thailand Center of Excellence for Life Sciences (TCELS, Thailand). The authors would like to thank Professor Dr Supa Hannongbua of the Faculty of Science, Kasetsart University, Professor Dr Supot Hannongbua of Faculty of Science, Chulalongkorn University, and Dr Mark Duggan of LifeSci Consulting LLC for their critical reviews of our manuscript. The Computational Chemistry Unit Cell, Faculty of Science; Center of Excellence in Hepatitis and Liver Cancer, Faculty of Medicine; and Faculty of Pharmaceutical Sciences, Chulalongkorn University are also gratefully acknowledged for some research facilities.References
- M. F. Yuen, D. S. Chen, G. M. Dusheiko, H. L. A. Janssen, D. T. Y. Lau, S. A. Locarnini, M. G. Peters and C. L. Lai, Hepatitis B virus infection, Nat. Rev. Dis. Prim., 2018, 4, 18035, DOI:10.1038/nrdp.2018.35.
- L. S. Y. Tang, E. Covert, E. Wilson and S. Kottilil, Chronic Hepatitis B Infection: A Review, JAMA, J. Am. Med. Assoc., 2018, 319, 1802–1813, DOI:10.1001/jama.2018.3795.
- J. Beck and M. Nassal, Hepatitis B virus replication, World J. Gastroenterol., 2007, 13, 48–64, DOI:10.3748/wjg.v13.i1.48.
- Center for Drug Evaluation and Research (CDER), Chronic Hepatitis B Virus Infection: Developing Drugs for Treatment Guidance for Industry [Online], https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chronic-hepatitis-b-virus-infection-developing-drugs-treatment, (accessed 20 April 2022) Search PubMed.
- T. Gerelsaikhan, J. E. Tavis and V. Bruss, Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis, J. Virol., 1996, 70, 4269–4274, DOI:10.1128/JVI.70.7.4269-4274.1996.
- A. Diab, A. Foca, F. Zoulim, D. Durantel and O. Andrisani, The diverse functions of the hepatitis B core/capsid protein (HBc) in the viral life cycle: Implications for the development of HBc-targeting antivirals, Antiviral Res., 2018, 149, 211–220, DOI:10.1016/j.antiviral.2017.11.015.
- A. Zlotnick, B. Venkatakrishnan, Z. Tan, E. Lewellyn, W. Turner and S. Francis, Core protein: A pleiotropic keystone in the HBV lifecycle, Antiviral Res., 2015, 121, 82–93, DOI:10.1016/j.antiviral.2015.06.020.
- V. Taverniti, G. Ligat, Y. Debing, D. B. Kum, T. F. Baumert and E. R. Verrier, Capsid Assembly Modulators as Antiviral Agents against HBV: Molecular Mechanisms and Clinical Perspectives, J. Clin. Med., 2022, 11, 1349, DOI:10.3390/jcm11051349.
- J. M. Berke, P. Dehertogh, K. Vergauwen, E. Van Damme, W. Mostmans, K. Vandyck and F. Pauwels, Capsid Assembly Modulators Have a Dual Mechanism of Action in Primary Human Hepatocytes Infected with Hepatitis B Virus, Antimicrob. Agents Chemother., 2017, 61, e00560, DOI:10.1128/AAC.00560-17.
- T. Lahlali, J. M. Berke, K. Vergauwen, A. Foca, K. Vandyck, F. Pauwels, F. Zoulim and D. Durantel, Novel Potent Capsid Assembly Modulators Regulate Multiple Steps of the Hepatitis B Virus Life Cycle, Antimicrob. Agents Chemother., 2018, 62, e00835, DOI:10.1128/AAC.00835-18.
- A. M. Lam, C. Espiritu, R. Vogel, S. Ren, V. Lau, M. Kelly, S. D. Kuduk, G. D. Hartman, O. A. Flores and K. Klumpp, Preclinical Characterization of NVR 3-778, a First-in-Class Capsid Assembly Modulator against Hepatitis B Virus, Antimicrob. Agents Chemother., 2019, 63, e01734, DOI:10.1128/AAC.01734-18.
- N. Mani, A. G. Cole, J. R. Phelps, A. Ardzinski, K. D. Cobarrubias, A. Cuconati, B. D. Dorsey, E. Evangelista, K. Fan and F. Guo, et al., Preclinical Profile of AB-423, an Inhibitor of Hepatitis B Virus Pregenomic RNA Encapsidation, Antimicrob. Agents Chemother., 2018, 62, e00082, DOI:10.1128/AAC.00082-18.
- S. J. Last, P. J.-M. B. Raboisson, G. Rombouts, K. Vandyck and W. G. Verschueren, Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis B, WIPO Pat., WO2014033170, 2014.
- J. M. Berke, P. Dehertogh, K. Vergauwen, W. Mostmans, K. Vandyck, P. Raboisson and F. Pauwels, Antiviral Properties and Mechanism of Action Studies of the Hepatitis B Virus Capsid Assembly Modulator JNJ-56136379, Antimicrob. Agents Chemother., 2020, 64, e02439, DOI:10.1128/AAC.02439-19.
- K. Vandyck, G. Y. P. Haché, S. J. Last, D. C. Mc Gowan, G. Rombouts, W. G. Verschueren and P. J.-M. B. Raboisson, Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B, WIPO Pat., WO2014184350, 2014.
- K. Vandyck, G. Y. P. Haché, S. J. Last, G. Rombouts, W. G. Verschueren and P. J.-M. B. Raboisson, Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B, WIPO Pat., WO2015118057, 2015.
- F. Amblard, S. Boucle, L. Bassit, B. Cox, O. Sari, S. Tao, Z. Chen, T. Ozturk, K. Verma and O. Russell, et al., Novel Hepatitis B Virus Capsid Assembly Modulator Induces Potent Antiviral Responses In vitro and in Humanized Mice, Antimicrob. Agents Chemother., 2020, 64, e01701–e01719, DOI:10.1128/AAC.01701-19.
- K. Vandyck, B. R. R. Kesteleyn, S. M. A. Pieters, G. Rombouts, W. G. Verschueren and P. J.-M. B. Raboisson, Glyoxamide substituted pyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B, WIPO Pat., WO2015011281, 2014.
- T. Vermes, M. Kielpinski, T. Henkel, M. A. Pericas, E. Alza, A. Corcuera, H. Buschmann, T. Goldner and A. Urban, An automated microfluidic platform for the screening and characterization of novel hepatitis B virus capsid assembly modulators, Anal. Methods, 2022, 14, 135–146, 10.1039/d1ay01227d.
- S. Wang, M. L. Fogeron, M. Schledorn, M. Dujardin, S. Penzel, D. Burdette, J. M. Berke, M. Nassal, L. Lecoq and B. H. Meier, et al., Combining Cell-Free Protein Synthesis and NMR Into a Tool to Study Capsid Assembly Modulation, Front. Mol. Biosci., 2019, 6, 67, DOI:10.3389/fmolb.2019.00067.
- K. Klumpp, A. M. Lam, C. Lukacs, R. Vogel, S. Ren, C. Espiritu, R. Baydo, K. Atkins, J. Abendroth and G. Liao, et al., High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15196–15201, DOI:10.1073/pnas.1513803112.
- G. Wu, B. Liu, Y. Zhang, J. Li, A. Arzumanyan, M. M. Clayton, R. F. Schinazi, Z. Wang, S. Goldmann and Q. Ren, et al., Preclinical characterization of GLS4, an inhibitor of hepatitis B virus core particle assembly, Antimicrob. Agents Chemother., 2013, 57, 5344–5354, DOI:10.1128/AAC.01091-13.
- E. Gane, C. Schwabe, K. Walker, L. Flores, G. Hartman, K. Klumpp, S. Liaw and N. Brown, Phase 1a Safety and Pharmacokinetics of NVR 3-778, a Potential First-In-Class HBV Core Inhibitor, Hepatology, 2014, 60, 1279A Search PubMed.
- K. Klumpp, T. Shimada, L. Allweiss, T. Volz, M. Lutgehetmann, G. Hartman, O. A. Flores, A. M. Lam and M. Dandri, Efficacy of NVR 3-778, Alone and In Combination With Pegylated Interferon, vs Entecavir In uPA/SCID Mice With Humanized Livers and HBV Infection, Gastroenterology, 2018, 154, 652–662, DOI:10.1053/j.gastro.2017.10.017.
- R. F. Schinazi, S. Boucle, F. Amblard, O. Sari and L. Bassit, Elimination of hepatitis B virus with antiviral agents, WIPO Pat., WO2017156255, 2017.
- Z. Zhou, T. Hu, X. Zhou, S. Wildum, F. Garcia-Alcalde, Z. Xu, D. Wu, Y. Mao, X. Tian and Y. Zhou, et al., Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms, Sci. Rep., 2017, 7, 42374, DOI:10.1038/srep42374.
- T. Verbinnen, Y. Tan, G. Wang, P. Dehertogh, K. Vergauwen, J. M. Neefs, E. Jacoby, O. Lenz and J. M. Berke, Anti-HBV activity of the HBV capsid assembly modulator JNJ-56136379 across full-length genotype A-H clinical isolates and core site-directed mutants in vitro, J. Antimicrob. Chemother., 2020, 75, 2526–2534, DOI:10.1093/jac/dkaa179.
- S. Y. Huang and X. Zou, Advances and challenges in protein–ligand docking, Int. J. Mol. Sci., 2010, 11, 3016–3034, DOI:10.3390/ijms11083016.
- X. Y. Meng, H. X. Zhang, M. Mezei and M. Cui, Molecular docking: a powerful approach for structure-based drug discovery, Curr. Comput.-Aided Drug Des., 2011, 7, 146–157, DOI:10.2174/157340911795677602.
- V. Salmaso and S. Moro, Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview, Front. Pharmacol, 2018, 9, 923, DOI:10.3389/fphar.2018.00923.
- J. Wang, T. Hou and X. Xu, Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models, Curr. Comput. Aided Drug Des., 2006, 2, 287–306, DOI:10.2174/157340906778226454.
- E. King, E. Aitchison, H. Li and R. Luo, Recent Developments in Free Energy Calculations for Drug Discovery, Front. Mol. Biosci., 2021, 8, 712085, DOI:10.3389/fmolb.2021.712085.
- M. R. Bauer and M. D. Mackey, Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein–ligand Complexes, J. Med. Chem., 2019, 62, 3036–3050, DOI:10.1021/acs.jmedchem.8b01925.
- K. Vandyck, G. Y. P. Haché, B. R. R. Kesteleyn and P. J.-M. B. Raboisson, Carboxamide derivatives and the use thereof as medicaments for the treatment of hepatitis b, WIPO Pat., WO2015059212, 2015.
- A. G. Leach and N. J. Kidley, Quantitatively Interpreted Enhanced Inhibition of Cytochrome P450s by Heteroaromatic Rings Containing Nitrogen, J. Chem. Inf. Model., 2011, 51, 1048–1063, DOI:10.1021/ci2000506.
- Flare Version 5.0, Cresset®, Litlington, Cambridgeshire, UK, 2021 Search PubMed.
- M. V. Shapovalov and R. L. Dunbrack Jr., A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions, Structure, 2011, 19, 844–858, DOI:10.1016/j.str.2011.03.019.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera-a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25, 1605–1612, DOI:10.1002/jcc.20084.
- D. A. Case, T. E. Cheatham 3rd, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, AMBER. Version 2016, University of California, San Francisco, San Francisco, CA, 2005 Search PubMed.
- T. J. Dolinsky, P. Czodrowski, H. Li, J. E. Nielsen, J. H. Jensen, G. Klebe and N. A. Baker, PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations, Nucleic Acids Res., 2007, 35, W522–W525, DOI:10.1093/nar/gkm276.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Comparison of multiple Amber force fields and development of improved protein backbone parameters, Proteins, 2006, 65, 712–725, DOI:10.1002/prot.21123.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09 (Revision A.2), Gaussian Inc., Wallingford, United States, 2009 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys. Chem., 2002, 97, 10269–10280, DOI:10.1021/j100142a004.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Chem., 2004, 25, 1157–1174, DOI:10.1002/jcc.20035.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935, DOI:10.1063/1.445869.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23, 327–341, DOI:10.1016/0021-9991(77)90098-5.
- D. R. Roe and T. E. Cheatham 3rd, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J. Chem. Theory Comput., 2013, 9, 3084–3095, DOI:10.1021/ct400341p.
- B. R. Miller 3rd, T. D. McGee Jr., J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, MMPBSA.py: An Efficient Program for End-State Free Energy Calculations, J. Chem. Theory Comput., 2012, 8, 3314–3321, DOI:10.1021/ct300418h.
- K. Huang, S. Luo, Y. Cong, S. Zhong, J. Z. H. Zhang and L. Duan, An accurate free energy estimator: based on MM/PBSA combined with interaction entropy for protein–ligand binding affinity, Nanoscale, 2020, 12, 10737–10750, 10.1039/c9nr10638c.
- T. Hou, J. Wang, Y. Li and W. Wang, Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations, J. Chem. Inf. Model., 2011, 51, 69–82, DOI:10.1021/ci100275a.
- B. Brooks and M. Karplus, Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 6571–6575, DOI:10.1073/pnas.80.21.6571.
- L. Guannan, H. Ke, N. Dejan and B. v. B. Richard, High-Throughput Cytochrome P450 Cocktail Inhibition Assay for Assessing Drug-Drug and Drug-Botanical Interactions, Drug Metab. Dispos., 2015, 43, 1670, DOI:10.1124/dmd.115.065987.
- A. Zlotnick, A. Lee, C. R. Bourne, J. M. Johnson, P. L. Domanico and S. J. Stray, In vitro screening for molecules that affect virus capsid assembly (and other protein association reactions), Nat. Protoc., 2007, 2, 490–498, DOI:10.1038/nprot.2007.60.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- J. B. Baell and G. A. Holloway, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays, J. Med. Chem., 2010, 53, 2719–2740, DOI:10.1021/jm901137j.
Footnote |
| † Electronic supplementary information (ESI) available: File S1 (Fig. S1–S5 and Table S1), File S2 (Metabolite identification of CU02–03), 1H-NMR and 13C-NMR spectrum of synthesized compounds. See DOI: https://doi.org/10.1039/d3ra04720b |
| This journal is © The Royal Society of Chemistry 2023 |