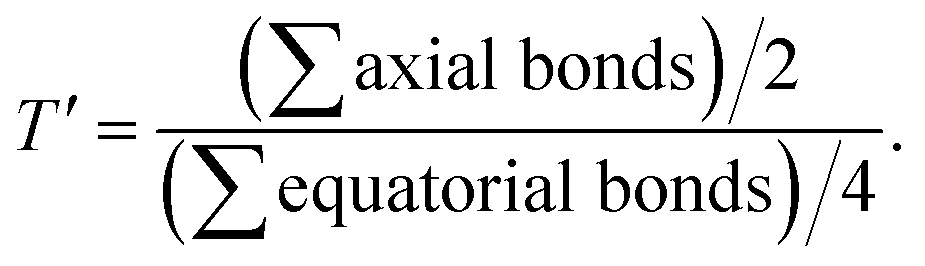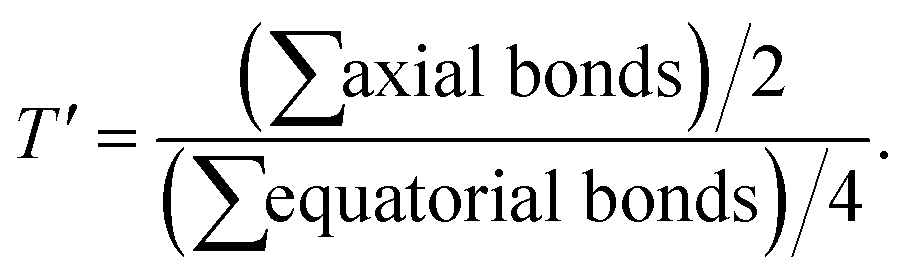DOI:
10.1039/D3RA04834A
(Paper)
RSC Adv., 2023,
13, 24674-24690
Mononuclear nickel(II)–flavonolate complexes of tetradentate tripodal 4N ligands as structural and functional models for quercetin 2,4-dioxygenase: structures, spectra, redox and dioxygenase activity†
Received
18th July 2023
, Accepted 1st August 2023
First published on 18th August 2023
Abstract
Three new nickel(II)-flavonolate complexes of the type [Ni(L)(fla)](ClO4) 1–3, where L is the tripodal 4N ligand tris(pyrid-2-ylmethyl)amine (tpa, L1) or (pyrid-2-ylmethyl)bis(6-methylpyrid-2-ylmethyl)amine (6-Me2-tpa, L2) or tris(N-Et-benzimidazol-2-ylmethyl)amine (Et-ntb, L3), have been isolated as functional models for Ni(II)-containing quercetin 2,4-dioxygenase. Single crystal X-ray structures of 1 and 3 reveal that Ni(II) is involved in π-back bonding with flavonolate (fla−), as evident from enhancement in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length upon coordination [H(fla), 1.232(3); 1, 1.245(7); 3, 1.262(8) Å]. More asymmetric chelation of fla− in 3 than in 1 [Δd = (Ni–Ocarbonyl − Ni–Oenolate): 1, 0.126; 3, 0.182 Å] corresponds to lower π-delocalization in 3 with electron-releasing N-Et substituent. The optimized structures of 1–3 and their geometrical isomers have been computed by DFT methods. The HOMO and LUMO, both localized on Ni(II)-bound fla−, are highly conjugated bonding π- and antibonding π*-orbitals respectively. They are located higher in energy than the Ni(II)-based MOs (HOMO–1, dx2–y2; HOMO–2/6, dz2), revealing that the Ni(II)-bound fla− rather than Ni(II) would undergo oxidation upon exposure to dioxygen. The results of computational studies, in combination with spectral and electrochemical studies, support the involvement of redox-inactive Ni(II) in π-back bonding with fla−, tuning the π-delocalization in fla− and hence its activation. Upon exposure to dioxygen, all the flavonolate adducts in DMF solution decompose to produce CO and depside, which then is hydrolyzed to give the corresponding acids at 70 °C. The highest rate of dioxygenase reactivity of 3 (kO2: 3 (29.10 ± 0.16) > 1 (16.67 ± 0.70) > 2 (1.81 ± 0.04 × 10−1 M−1 s−1)), determined by monitoring the disappearance of the LMCT band in the range 440–450 nm, is ascribed to the electron-releasing N-Et substituent on bzim ring, which decreases the π-delocalization in fla− and enhances its activation.
O bond length upon coordination [H(fla), 1.232(3); 1, 1.245(7); 3, 1.262(8) Å]. More asymmetric chelation of fla− in 3 than in 1 [Δd = (Ni–Ocarbonyl − Ni–Oenolate): 1, 0.126; 3, 0.182 Å] corresponds to lower π-delocalization in 3 with electron-releasing N-Et substituent. The optimized structures of 1–3 and their geometrical isomers have been computed by DFT methods. The HOMO and LUMO, both localized on Ni(II)-bound fla−, are highly conjugated bonding π- and antibonding π*-orbitals respectively. They are located higher in energy than the Ni(II)-based MOs (HOMO–1, dx2–y2; HOMO–2/6, dz2), revealing that the Ni(II)-bound fla− rather than Ni(II) would undergo oxidation upon exposure to dioxygen. The results of computational studies, in combination with spectral and electrochemical studies, support the involvement of redox-inactive Ni(II) in π-back bonding with fla−, tuning the π-delocalization in fla− and hence its activation. Upon exposure to dioxygen, all the flavonolate adducts in DMF solution decompose to produce CO and depside, which then is hydrolyzed to give the corresponding acids at 70 °C. The highest rate of dioxygenase reactivity of 3 (kO2: 3 (29.10 ± 0.16) > 1 (16.67 ± 0.70) > 2 (1.81 ± 0.04 × 10−1 M−1 s−1)), determined by monitoring the disappearance of the LMCT band in the range 440–450 nm, is ascribed to the electron-releasing N-Et substituent on bzim ring, which decreases the π-delocalization in fla− and enhances its activation.
Introduction
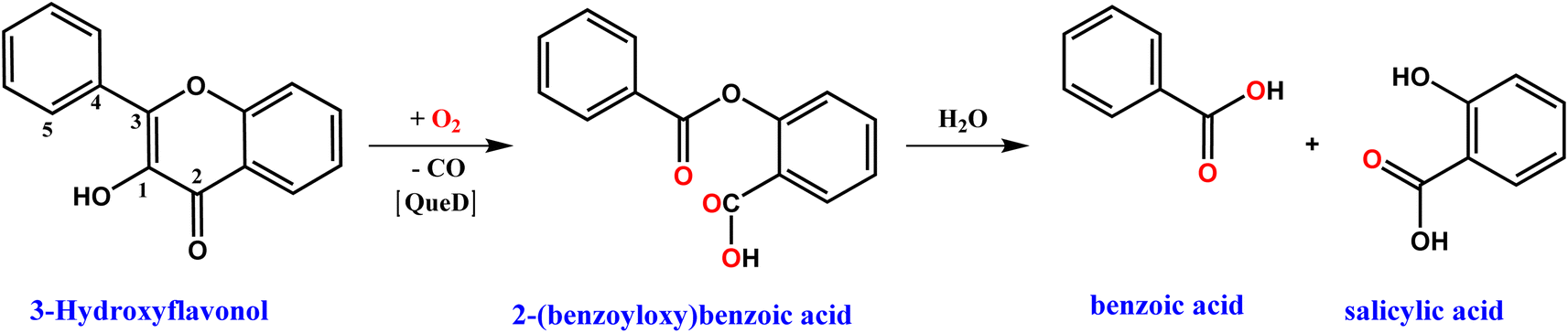
The Quercetin 2,4-Dioxygenase (2,4-QueD) enzymes, found in soil microorganisms, catalyze the aerobic degradation of quercetin (3,5,7,3′,4′-pentahydroxyflavone), which is mainly derived from the decomposition of plant materials and exudates of roots and leaves, via activation of molecular oxygen.1 The oxygenative ring-opening and cleavage of two carbon–carbon bonds in the substrate to form the phenolic carboxylic acid esters (depside), which is further hydrolyzed to benzoic acid and salicylic acid, with concomitant release of carbon monoxide, involves a fascinating enzyme mechanism.2 Although many experimental advances and theoretical analyses concerning these enzymes have been reported,3,4 there are still some issues regarding their biosynthesis and catalytic activity due to variation in their active sites. Like many other metal-based enzymes, which play the most important roles in dictating the biological functions like synthesis of biomolecules, metabolic reactions and conversions and oxidative detoxifications, the native QueD enzymes use mainly type II copper5,6 and iron7,8 as cofactors. Also, they use many other metal ion cofactors like Mn, Co, Ni and Zn for the catalytic degradation of quercetin, and differ by the variation in percentage of metals in the active sites.9
In very recent times, the enzyme QueD has engrossed the attention of bioinorganic chemists, and many synthetic models have been isolated and studied to understand their enzymatic reaction mechanism, which is still imprecise and vague. The most investigated models for the dioxygenases include copper(II),10,11 Mn(II)12 and Fe(III)13 flavonolate complexes. Many synthetic models for Co, Ni, Cd, Hg and Zn containing QueD enzymes14–18 have been isolated and studied as structural and functional models of QueDs enzyme. Very recently, Grubel et al. have synthesized a series of metal complexes of Mn(II), Co(II), Ni(II), Cu(II) and Zn(II) with N,N-bis(6-phenyl-pyrid-2-ylmethyl)-N-(pyrid-2-ylmethyl)amine (6-Ph2TPA) as QueDs models to understand the chemistry behind the native enzymes.15 Sun et al. have reported metal complexes of certain dipicolylamine-based ligands with the first row transition metals like Mn, Fe, Co, Ni, Cu and Zn as QueD models, and studied the catalytic dioxygenation of flavonol.16 Metal complexes of N-propanoate-N,N-bis(2-pyridylmethyl)amine (HPBMPA) and N-methylpropanoate-N,N-bis(2-pyridylmethyl)amine (MPBMPA) with the first row transition metals Mn, Fe, Co, Ni, and Cu and 3-hydroxyflavoneas substrate have been studied.17 Very recently, a series of metal complexes with the ligand hydrotris(3-mesityl)pyrazolylborate and 3-hydroxyflavone has been found to show better QueD-like dioxygenation activity.18
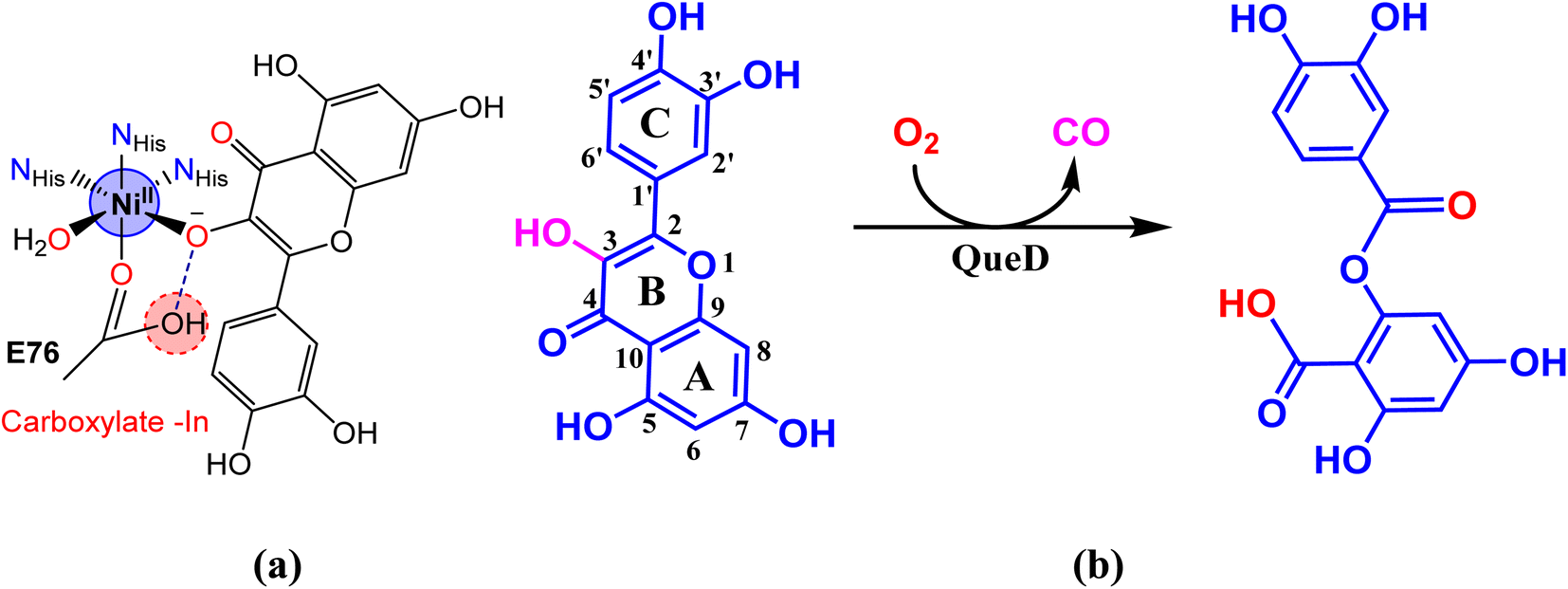
Interestingly, there is one form of nickel containing 2,4-QueD isolated from Streptomyces sp. FLA, that shows the highest reactivity9 and it is similar to that found in other nickel-containing enzymes such as acireductone dioxygenase containing nickel.19,20 In every part of Ni(II)-QueDs, the metal is bound to three histidine-derived imidazoles and a glutamate-derived carboxylic acid in the active sites (Scheme 1).5c,21,22
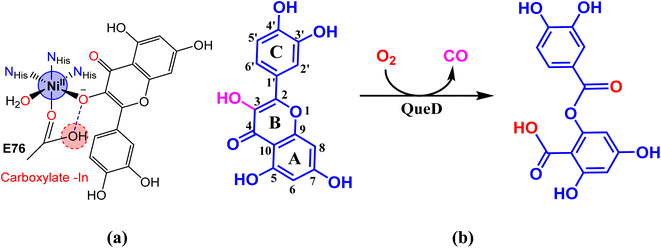 |
| | Scheme 1 (a) Active site structure of nickel(II)-derived flavonol 2,4-dioxygenase enzyme. (b) Degradation of quercetin catalyzed by the enzyme to produce depside. | |
Although many biomimetic studies on 2,4-QueD have been made, not much attention has been paid to the biomimetic study of Ni(II)-QueD which exhibits high dioxygenase activity in spite of the redox-inactivity of the metal. The X-ray structures of only a few Ni(II)-flavonolate model complexes are known but only one of the complexes shows dioxygenation activity upon exposure to UV radiation.12,13 Very recently, a few nickel(II) complexes of the tripodal ligand 2-([bis(pyridin-2-ylmethyl)amino]methyl)-p/m-R-benzoic acid (R = p-OMe, p-Me, m-Br and m-NO2) and 3-hydroxyflavone, that show structural features related to the native enzyme, have been isolated and their ability to degrade coordinated flavonolate correlates with the Lewis acidity of the metal.23 As QueDs model, a metastable organo peroxide complex of Ni(II) has been shown to exhibit ring cleavage of flavonols.24 Nickel(II) complexes of hydrotris(3,5-dimethyl)pyrazolylborate and 3-hydroxyflavone (HFla) as substrate analogue have been isolated and the structural changes that occur upon modification of CO function in these complexes to obtain the corresponding CS and CSe analogues as QueD models have been studied and, interestingly, the complexes are found to retain dioxygenase activity.25 Also, theoretical aspects of many model complexes have been investigated with an to better understand the enzyme activity of nickel containing QueDs.26,27 During the preparation of this article, the Ni(II)complex of 1,4,7-trimethyl-1,4,7-triazacyclononane (Me3TACN) and 3-hydroxyflavonol, which acts as a structural and functional model for the Ni(II)-QueD enzyme, has been reported.28
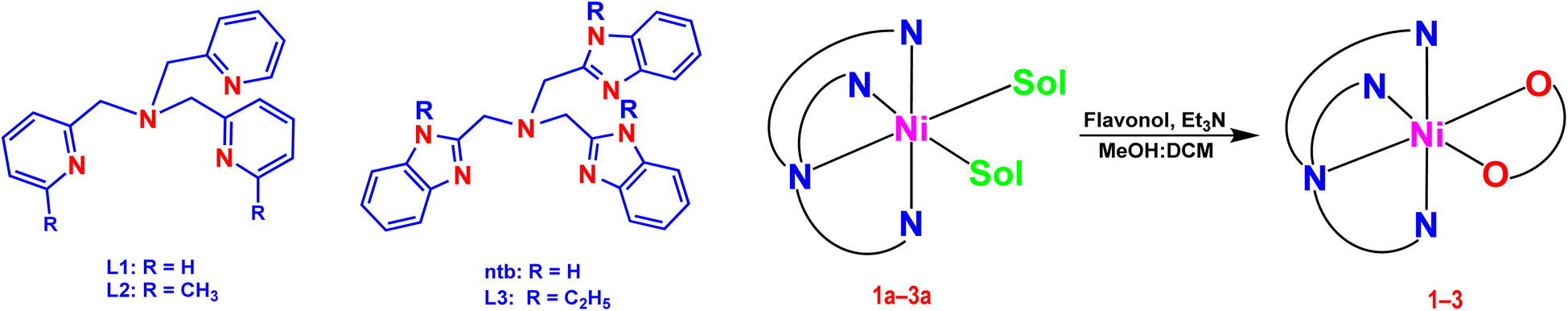
In our laboratory, we have successfully isolated and investigated diiron(III)29 and nickel(II) complexes,20c,30,31 which show the potential to act as bio-mimetic and bio-inspired functional models for sMMO enzymes. Now, we have synthesized and characterized a few nickel(II) complexes of tripodal 4N ligands like tris(pyridylmethyl)amine (tpa, L1; 6-Me2-tpa, L2) and tris(N-Et-benzimidazol-2-yl-methyl)amine (N-Et-ntb, L3) and 3-hydroxyflavone (H(fla)), as structural and functional models for Ni(II)-containing 2,4-QueD enzyme (Scheme 2). While H(fla) would act as the substrate analogue for quercetin, the pyridyl and benzimidazolyl nitrogen donors of the primary ligands would mimic the three histidyl imidazole nitrogen atoms of the enzyme active site. The incorporation of electron-releasing N-Et substituent on the bzim ring and sterically hindering 6-methyl substituent on the pyridyl ring is expected to fine tune the electronic properties of Ni(II), and hence the dioxygenase activity. And this study is expected to throw light on the function of Glu-73 carboxylate group bound to Ni(II) in the enzyme active site. Interestingly, the X-ray structures of two of our Ni(II)-flavonolate adducts reveal that Ni(II) exhibits π-back bonding with fla− and tunes the π-delocalization in, and activation of fla− towards reaction with dioxygen. Density functional theory (DFT) computations reveal that the HOMO and LUMO, localized on the Ni(II)-bound flavonolate, possess energies higher than the metal-based MOs (HOMO–1, dx2–y2; HOMO–2/6, dz2) so that the coordinated fla− rather than Ni(II) is oxidized during dioxygenation. While the incorporation of N-Et substituent on bzim moiety enhances the rate of quercetinase activity the sterically hindering 6-Me substituent on the pyridyl ring decreases it, which is consistent with the inversion in energies of dx2–y2 and dz2 orbitals.
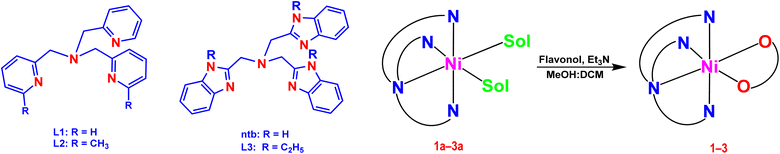 |
| | Scheme 2 Chemical structures of the ligands L1, L2 and L3 and their Ni(II) complexes [Ni(L)(Sol)2](ClO4)2 1a–3a and [Ni(L)(fla)](ClO4) 1–3. | |
Experimental
Materials
Pyridine-2-carboxaldehyde, 6-methylpyridine-2-carboxaldehyde, 2-(aminomethyl)pyridine, nickel(II) perchlorate hexahydrate, nitrilotriacetic acid, iodoethane (Sigma-Aldrich), sodium triacetoxyborohydride, 1,2-diaminobenzene, 3-hydroxyflavone (Alfa Aesar), ethyl acetate, triethylamine, sodium sulphate (Merck, India), were used as received. Dichloromethane, diethylether, tetrahydrofuran, acetonitrile (Merck, India) and methanol, acetone (Sisco Research Laboratory, Mumbai) were distilled before use. The supporting electrolyte tetra-N-butylammonium perchlorate (Aldrich) was recrystallized and used.
Synthesis of ligands
The ligands tris(pyrid-2-yl-methyl)amine (tpa, L1),30,32 (pyrid-2-yl-methyl)bis(6-methyl-pyrid-2-ylmethyl)amine (6-Me2-tpa, L2),30,32 and tris(benzimidazol-2-ylmethyl)amine (ntb)30,33 were prepared by using the previously reported procedures with slight modifications. The ligand tris(N-et-benzimidazol-2-ylmethyl)amine (Et-ntb, L3) was prepared by N-ethylating ntb.
Tris(2-pyridylmethyl)amine (L1)30,32
The compound L1 is a yellow solid (1.21 g, 75%). 1H NMR (400 MHz, CDCl3): δ = 8.43 (d, 3H), 7.54 (t, 3H), 7.50 (d, 3H), 7.04 (d), 3.79 (s, 6H) (Fig. S13 and 14†). Anal. calcd for C18H19N4: C, 74.46; H, 6.25; N, 19.30. Found: C, 74.55; H, 6.30; N, 19.58. LC-MS (ethyl acetate) displays a peak at m/z 291.25 [C18H18N4+] (calc. 291.16, Fig. S17†).
(Pyrid-2-ylmethyl)bis(6-methyl-pyrid-2-ylmethyl)amine (L2)30,32
The ligand L2 is a brown solid (1.33 g, 73%). 1H NMR (400 MHz, CDCl3): δ = 8.60 (d, 1H), 7.74 (t, 3H), 7.24 (m, 4H), 7.04 (d, 2H), 3.79 (s, 6H), 2.45 (s, 6H). Anal. calcd for C20H22N4: C, 75.44; H, 6.96; N, 17.60. Found: C, 75.60; H, 6.70; N, 17.78.
Tris(N-Et-benzimidazol-2-ylmethyl)amine (L3)
Nitrilotriacetic acid (5 g, 26.2 mmol) was ground to a fine powder and intimately mixed with 1,2-diaminobenzene (8.483 g, 78.5 mmol). The mixture was heated at 170–180 °C for 1 h at which stage effervescence had ceased. The mixture was cooled to room temperature, and the resulting red glass was taken up in dilute (∼4 M) hydrochloric acid (150 mL) to obtain a greyish blue precipitate. The precipitate was filtered off and washed by slurrying it in acetone several times. The precipitate was then dissolved in water and neutralized with dilute ammonia. The off-white precipitate was collected by filtration, recrystallized from acetone, and ground to a fine powder prior to vacuum drying to give ntb (yield, 11.85 g, 87%). After thorough drying, the powder was suspended in dry tetrahydrofuran (50 mL) and stirred overnight with NaOH (3.494 g, 87.31 mmol). Iodoethane (13.62 g, 87.31 mmol) was added to this solution, and the solution left stirring for 2 days. The solvent was then stripped off to dryness and the resulting powder dissolved in chloroform. The insoluble NaBr was removed by filtration, and the filtrate was rotaevaporated to a very small volume. A little acetone was added, and upon standing crystalline reddish-brown compound L3 was obtained (14.24 g, 86%). The compound was recrystallized and the crystals were collected and dried in vacuum. Yield, 1H NMR (400 MHz, CDC13): δ = 7.68 (m, 3H), 7.16 (m, 9H), 4.21 (s, 6H), 3.37 (m, 6H), 0.67 (t, 9H) (Fig. S15 and S16†). Anal. calcd for C30H33N7: C, 73.29; H, 6.77; N, 19.94. Found: C, 73.33; H, 6.80; N, 19.98. HR-MS (ethyl acetate) displays a peak at m/z 492.2870 [C30H34N7+] (calc. 492.2870, Fig. S18†).
Isolation of Ni(II) complexes
The nickel(II) complexes were prepared by using the previously reported procedures with slight modifications.30,31,34
[Ni(L1)(CH3CN)2](ClO4)2 1a30. A methanol solution (5 mL) of Ni(ClO4)2·6H2O (0.365 g, 1 mmol) was added to a methanol solution (5 mL) of L1 (0.290 g, 1 mmol) under stirring at room temperature. The solution turned green in color and was stirred for an additional 30 min when light green precipitate was obtained. The precipitate was filtered off, washed with small quantities of ice-cold methanol and then dried. The precipitate was used as such for isolation of nickel–flavonolate complexes. Yield, 0.41 g, 63%. λmax/nm, in ACN (εmax/dm3 mol−1): 858 (280), 523 (410), 384sh. Anal. calcd for C22H24N6Cl2NiO8: C, 41.94; H, 3.84; N, 13.34. Found: C, 41.77; H, 3.70; N, 13.48. HR-MS: (ACN) displays a peak at m/z 421.1179 [M + CH3OH – CH3CN – 2ClO4]2+ (calc. 421.1413, Fig. S19†).
[Ni(L2)(CH3CN)(H2O)](ClO4)2 2a34. This was prepared by using the procedure used for preparing complex 1a but by using L2 ligand instead of L1. Yield, 0.48 g, 70%. λmax/nm, in ACN (εmax/dm3 mol−1): 936 (270), 545 (290), 428sh. Anal. calcd for C22H27N5Cl2NiO9: C, 41.61; H, 4.29; N, 11.03. Found: C, 41.52; H, 4.32; N, 11.12. HR-MS: (ACN) displays a peak at m/z 575.2894 [M + 2CH3CN + H2O – ClO4]+ (calc. 575.1320), 603.1600 [M + CH3OH + 2H2O – CH3CN – ClO4]+ (calc. 603.3206) and 447.0368 [M + 3H2O – CH3CN – 2ClO4]2+ (calc. 447.1537) (Fig. S20†).
[Ni(L3)(CH3CN)2](ClO4)2 3a30,35. This was also prepared by using the procedure used for preparing complex 1a but by using L2 ligand instead of L1. Yield, 0.62 g, 72%. λmax/nm, in ACN (εmax/dm3 mol−1): 932 (110), 571 (180), 361sh. Anal. calcd for C34H39N9Cl2NiO8: C, 49.12; H, 4.73; N, 15.16. Found: C, 49.22; H, 4.62; N, 15.27. HR-MS: (ACN) displays a peak at m/z 648.1625 [M – 2CH3CN – ClO4]+ (calc. 648.1636, Fig. S21†).
[Ni(L1)(fla)]ClO4 1. The complex 1a (0.211 g, 0.5 mmol) and 3-hydroxyflavone (0.11 g, 0.5 mmol) were suspended in methanol/DCM (15 mL) and Et3N (0.05 g, 0.5 mmol) was added with stirring. The reaction mixture was stirred for 2 h at room temperature and filtered. The residue was washed twice with MeCN and all volatiles were removed in vacuo to give 1 as a brown crystalline solid. Crystals of 1 suitable for single-crystal X-ray diffraction studies were obtained by slowly evaporating the volatiles from a saturated solution of 1 in acetonitrile. Yield, 0.210 g, 66%. ATR (cm−1): ν(CO), 1536; ν(CN), 1598. λmax/nm, in ACN (εmax/dm3 mol−1): 929 (180), 528 (370), 441sh. Anal. calcd for C33H27N4ClNiO7: C, 57.80; H, 3.97; N, 8.17. Found: C, 57.68; H, 3.80; N, 8.30. HR-MS: (ACN) displays a peak at m/z 585.1432 [M – ClO4]+ (calc. 585.1437, Fig. S22†).
[Ni(L2)(fla)]ClO4 2. This complex was prepared as a brown solid by using the procedure employed for obtaining 1 and using 2a. Yield, 0.27 g, 81%. ATR (cm−1): ν(CO), 1544; ν(CN), 1595. λmax/nm, in ACN (εmax/dm3 mol−1): 1007 (170), 582 (260), 441sh. Anal. calcd for C35H31N4ClNiO7: C, 58.89; H, 4.38; N, 7.85. Found: C, 58.78; H, 4.47; N, 7.77. HR-MS: (ACN) displays a peak at m/z 613.1752 [M – ClO4]+ (calc. 613.1750, Fig. S23†).
[Ni(L3)(fla)]ClO4 3. This complex was prepared as a green crystalline solid by using the procedure employed for obtaining 1 and using 3a. Crystals of 3 suitable for single-crystal X-ray diffraction studies were obtained by slowly evaporating the volatiles from a saturated solution of 3 in acetonitrile. Yield, 0.322 g, 74%. ATR (cm−1): ν(CO), 1495; ν(CN), 1543. λmax/nm, in ACN (εmax/dm3 mol−1): 1022 (210), 580 (510), 446sh. Anal. calcd for C45H42N7ClNiO11: C, 54.79; H, 4.29; N, 9.94. Found: C, 54.66; H, 4.40; N, 9.82. HR-MS: (ACN) displays a peak at m/z 786.2687 [M – ClO4]+ (calc. 786.2703, Fig. S24†).
Caution!. Perchlorate salts of the compounds are potentially explosive. Only small quantities of these compounds should be prepared and suitable precautions should be taken when they are handled.
Physical methods
1H and 13C NMR spectra were recorded (400 and 100 MHz, respectively) on a Bruker Avance DPX 400 MHz using CDCl3. Chemical shifts for proton and carbon resonances are reported for the major isomer in parts per million (d) relative to tetramethylsilane (d 0.00), chloroform (d 77.23) respectively. Multiplicities are indicated by singlet (s), doublet (d), triplet (t) and multiplet (m). FT-IR spectra of solid samples were obtained using a Bruker ATR-Alpha (Diamond) spectrophotometer. Electronic absorbance spectra were recorded in a 1 cm cuvette on a Varian Cary 300 UV-visible spectrophotometer. Kinetic measurements were recorded using a 1 cm cuvette on an Agilent diode array spectrometer (Agilent 8453). Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) on glassy carbon electrode were performed in DMF at 25 °C. The voltammograms were generated using CH instruments 620C electrochemical analyzer. A three-electrode system was used to study the electrochemical behavior of complexes (0.001 M) consists of a glassy carbon working electrode (A, 0.0707 cm2), a platinum wire auxiliary electrode and a saturated calomel reference electrode and TBAP (0.1 M) was used as the supporting electrolyte. Solutions were deoxygenated by purging with nitrogen gas for 15 min prior to the measurement. Microanalyses (C, H and N) were carried out using PR 2400 Series II PerkinElmer equipment. ESI-mass spectra were recorded using a Thermo LC-MS instrument. High resolution mass analyses were performed using electron spray ionization (ESI) technique on a Thermo Scientific Exactive plus EMR instrument. GC-MS and GC analysis were performed on Agilent 5977E GCMSD using HP-5 MS ultra-inert (30 m × 250 μm × 0.25 μm) capillary column.
Crystallographic data collection, refinement and structure solution
The molecular structures of the complexes 1 and 3 were unambiguously determined by measuring X-ray intensity data on a Bruker SMART APEX II single crystal X-ray CCD diffractometer at ambient temperature having graphite-monochromatized Mo-Kα(λ = 0.71073 Å) radiation. The data were solved using direct methods with SHELXS-97 and refined using SHELXL-97, SHELXTL and SHELXL-2014, SHELXTL.36 The graphics interface package used was PLATON, and the figures were generated using the ORTEP 3.07 generation package.37 The positions on all the atoms were obtained by direct methods. Metal atoms in each complex were located from the E-maps and non-hydrogen atoms were refined anisotropically. The hydrogen atoms bound to the carbon were placed in geometrically constrained positions and refined with isotropic temperature factors, generally1.2 Ueq of their parent atoms. Crystallographic data are listed in Tables 1, S1 and S2.†
Computational details
The coordination geometries of nickel(II) complexes with triplet spin in the ground state were fully optimized by using Density Functional Theory (DFT) at U-B3LYP level of theory by employing the Gaussian 09 program package.38 The calculations were carried out with a mixed basis set of LANL2DZ for the nickel center, which has a relativistic effective core potential with a valence basis set, and 6-31G* for the remaining atoms.39,40 The solvation of the complexes was carried out by employing conductor-like polarizable continuum model (CPCM) method using acetonitrile as the solvent. The normal mode analyses were performed to check the minimal energy nature of the geometry in solution state.
Kinetic measurements
Reactivity studies. Quercetin 2,3-dioxygenase activity
The reactions of the flavonol substrate adducts [Ni(L)(fla)]+ with O2 in DMF at desired temperature were performed in a 10 mm path length UV-vis cell that was held in a Unisoku thermostated cell holder (Osaka, Japan). The solutions of the flavonolate complexes [Ni(L)(fla)](ClO4) 1–3 (1 × 10−4 M in 3 mL DMF) were maintained at 70 °C under N2 for several minutes, and then N2 was replaced by bubbling with O2. The time course of the reactions was followed by monitoring the absorption spectral changes at 444–450 nm. The kinetic parameters for the catalytic dioxygenation reactions were obtained from the ln{(At − A∞)/(A0 − A∞)} vs. time plot (Table 5, Fig. 7, S9–S11†) at 70 °C.
Identification of dioxygenated products23
In a typical experiment, deaerated 10 mL DMF solution of the flavonolate substrate adducts [Ni(L)(fla)](ClO4) 1–3 (1.0 × 10−3 M) was taken in a 50 mL RB flask, tightly closed with a rubber septum and the reaction vessel maintained at 70 °C under N2 for several minutes by continuous bubbling of N2 using a balloon with syringe. Then N2 was replaced with a desired amount of dioxygen to start the reaction. The reaction mixture was taken by using a syringe periodically (ca. every 2 min), and the time course of the reaction was followed by monitoring the absorption spectral changes at 444–450 nm. The O2 concentrations in DMF were calculated from literature data taking into account the partial pressure of DMF.41
Results and discussion
Synthesis and characterization of ligands and complexes
The ligands L1 and L2 were prepared by reductive amination7b,30 which involves treating one equivalent of 2-(aminomethyl)pyridine with two equivalents of the corresponding pyridine aldehydes and using sodium triacetoxyborohydride as the reducing agent. And the benzimidazole ligand ntb was prepared according to a known Philips condensation reaction,30,33 by refluxing o-phenylenediamine with nitrilotriacetic acid in the presence of dilute hydrochloric acid. The N-ethylation of ntb to get L3 was carried out in dry tetrahydrofuran as a solvent by using iodoethane in the presence of NaOH.5a,b All the ligands were characterized by 1H, 13C NMR, elemental analysis and mass spectrometry. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 nickel(II) complexes 1a–3a of the tripodal ligands L1–L3 were prepared by adding one equivalent of the ligand to a methanol solution of an equivalent amount of Ni(ClO4)2·6H2O. The complexes 1–3 were prepared by adding one equivalent of 3-hydroxyflavonol in MeOH:DCM (3:1 v/v) solution to the complexes 1a–3a dissolved in methanol and then adding Et3N as base. Single crystals of 1 and 3 suitable for X-ray crystallographic analysis were obtained by slow evaporation of a CH3CN:CH3OH solution of the complexes. The flavonolate complexes are formulated as [Ni(L)(fla)](ClO4) 1–3, which is supported by HR-MS and the X-ray crystal structures of 1 and 3. All the complexes with pyridyl/benzimidazolyl nitrogen donors are expected to mimic the active site environment of the substrate-bound enzyme QueDs and so they have been chosen to model the catalytic degradation reactions of the enzyme. The identity of the complexes in solution were examined by HR(ESI)-MS. All the complexes exhibit a main peak cluster at m/z (pos.) = 585.1432 for [Ni(L1)(fla)]+ 1, 613.1752 for [Ni(L2)(fla)]+ 2 and 786.2687 for [Ni(L3)(fla)]+ 3. The m/z values and the isotope distribution pattern of each peak agree well with the calculated values, indicating that the complexes maintain their mononuclear structure and Ni(II) oxidation state in solution, which is consistent with other spectroscopic results. The ν(CN) vibration observed for the pyridyl complexes 1 and 2 absorb as a strong peak around 1597 cm−1, which is higher than that for the benzimidazolyl complex 3 (∼1545 cm−1, Fig. S2†), which is expected.42 All the complexes show the ν(CO) vibration of flavonolate (fla−) as a strong peak in the range 1536–1495 cm−1, which is lower than that (1602 cm−1) of free flavonol, revealing that the CO of fla− is coordinated to Ni(II). The values of ν(CO) varies in the order 3 < 1 < 2, revealing that Ni(II) is involved in π-back bonding with CO in 3 more strongly than in 1 and 2.11–13,18,23 Also, the decrease in ν(CO) observed for 1 is higher than that observed for 2, indicating that the π-back bonding of Ni(II) with CO in the former is stronger than that in 2. A similar decrease in ν(CO) observed for Cu(II)-fla− complexes has been ascribed to π-back bonding of Cu(II) with CO of fla− (cf. above).43
:1 nickel(II) complexes 1a–3a of the tripodal ligands L1–L3 were prepared by adding one equivalent of the ligand to a methanol solution of an equivalent amount of Ni(ClO4)2·6H2O. The complexes 1–3 were prepared by adding one equivalent of 3-hydroxyflavonol in MeOH:DCM (3:1 v/v) solution to the complexes 1a–3a dissolved in methanol and then adding Et3N as base. Single crystals of 1 and 3 suitable for X-ray crystallographic analysis were obtained by slow evaporation of a CH3CN:CH3OH solution of the complexes. The flavonolate complexes are formulated as [Ni(L)(fla)](ClO4) 1–3, which is supported by HR-MS and the X-ray crystal structures of 1 and 3. All the complexes with pyridyl/benzimidazolyl nitrogen donors are expected to mimic the active site environment of the substrate-bound enzyme QueDs and so they have been chosen to model the catalytic degradation reactions of the enzyme. The identity of the complexes in solution were examined by HR(ESI)-MS. All the complexes exhibit a main peak cluster at m/z (pos.) = 585.1432 for [Ni(L1)(fla)]+ 1, 613.1752 for [Ni(L2)(fla)]+ 2 and 786.2687 for [Ni(L3)(fla)]+ 3. The m/z values and the isotope distribution pattern of each peak agree well with the calculated values, indicating that the complexes maintain their mononuclear structure and Ni(II) oxidation state in solution, which is consistent with other spectroscopic results. The ν(CN) vibration observed for the pyridyl complexes 1 and 2 absorb as a strong peak around 1597 cm−1, which is higher than that for the benzimidazolyl complex 3 (∼1545 cm−1, Fig. S2†), which is expected.42 All the complexes show the ν(CO) vibration of flavonolate (fla−) as a strong peak in the range 1536–1495 cm−1, which is lower than that (1602 cm−1) of free flavonol, revealing that the CO of fla− is coordinated to Ni(II). The values of ν(CO) varies in the order 3 < 1 < 2, revealing that Ni(II) is involved in π-back bonding with CO in 3 more strongly than in 1 and 2.11–13,18,23 Also, the decrease in ν(CO) observed for 1 is higher than that observed for 2, indicating that the π-back bonding of Ni(II) with CO in the former is stronger than that in 2. A similar decrease in ν(CO) observed for Cu(II)-fla− complexes has been ascribed to π-back bonding of Cu(II) with CO of fla− (cf. above).43
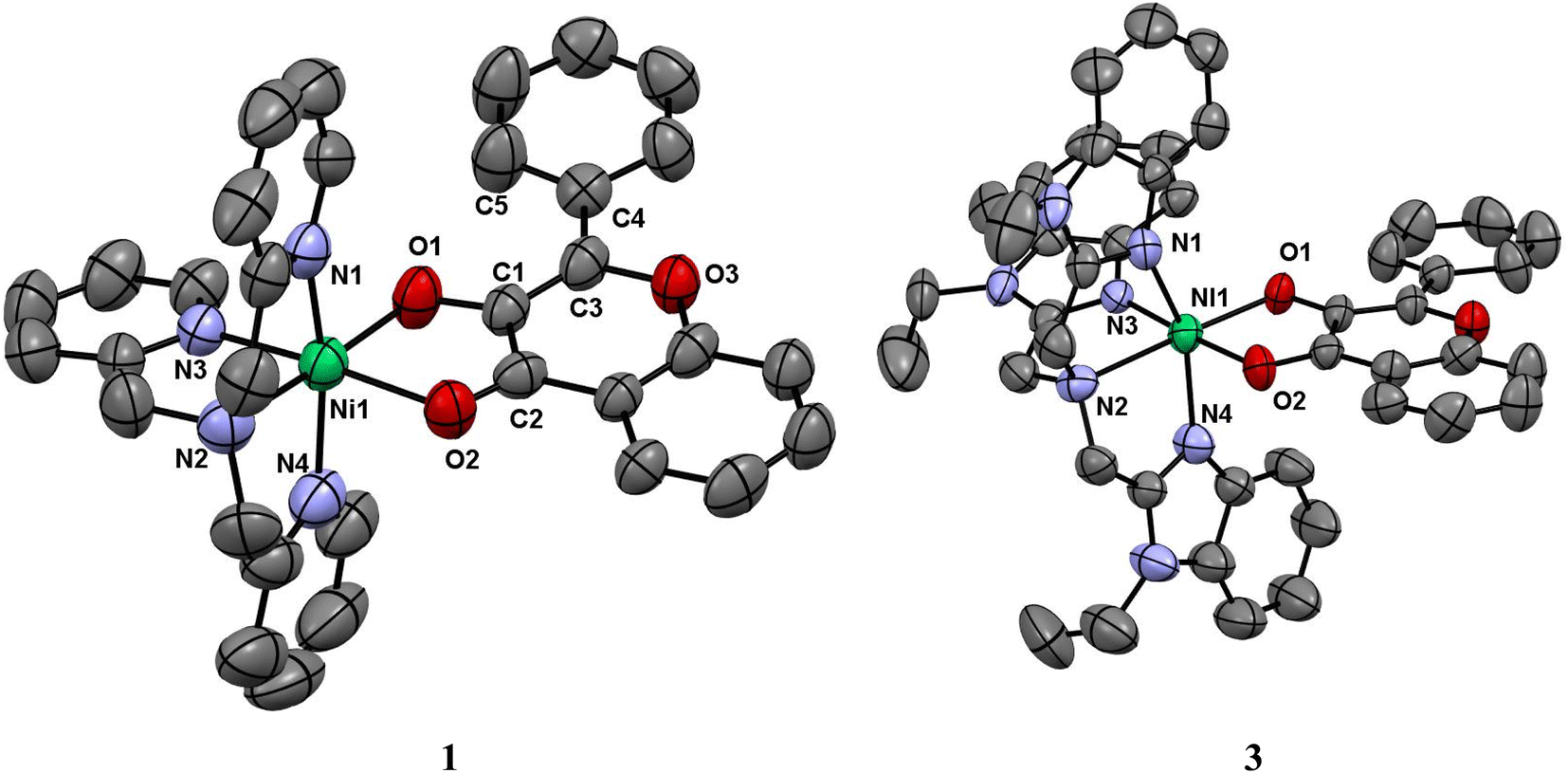
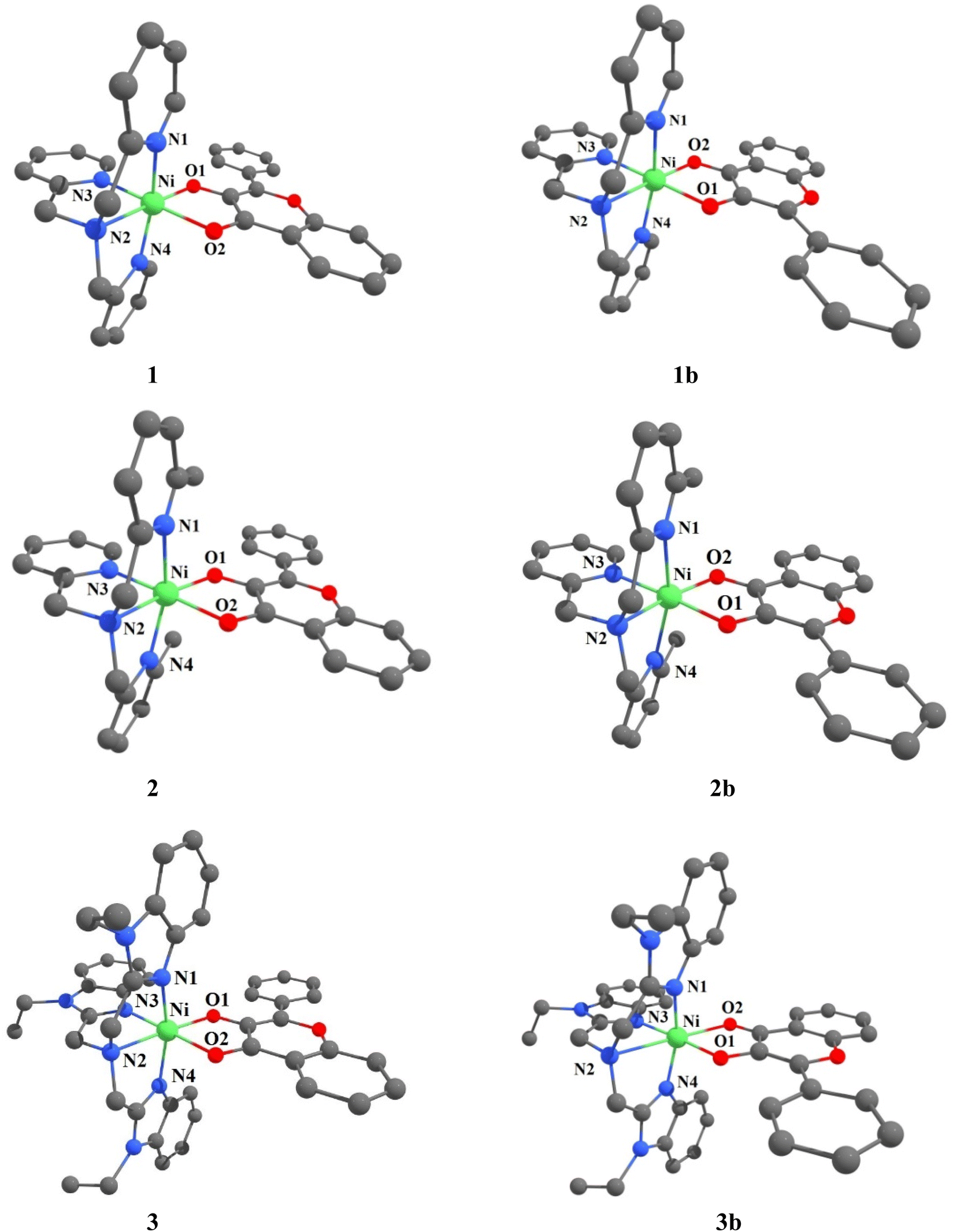
Description of X-ray crystal structures of [Ni(L1)(fla)](ClO4) 1 and [Ni(L3)(fla)](ClO4) 3
The molecular structures of complex cations of [Ni(L1)(fla)](ClO4) 1 and [Ni(L3)(fla)](ClO4) 3 are shown in Fig. 1, together with the atom numbering scheme. The principal bond lengths and bond angles of 1 and 3 are collected in Table 1. The complex cation of 1 possesses a NiN4O2 coordination sphere with distorted octahedral geometry constituted by all the four nitrogen atoms of the tripodal tetradentate ligand L1. One of the three pyridyl nitrogens (N3) and the tertiary amine nitrogen N2 of L1 and O1 and O2 atoms of deprotonated 3-hydroxyflavonone (fla−) occupy the corners of equatorial plane of the octahedral geometry while the other two py nitrogen atoms (N1, N4) occupy the trans axial positions. The O1 atom is coordinated trans to N2 amine nitrogen, and O2 is weakly coordinated trans to the strongly bound pyridyl N3 nitrogen. All the Ni–Npy (Ni–N1, 2.080(5); Ni–N3, 2.050(4); Ni–N4, 2.073(6)Å) bond lengths fall in the range observed for similar complexes (Ni–N1, 2.076(10); Ni–N3, 2.056(8); Ni–N4, 2.057(9) Å).20c,44 The Ni–N2amine (2.087(5) Å) bond is longer than all the three Ni–Npy bonds, which is expected of sp3 and sp2 hybridizations respectively of amine and py nitrogen donors. The Ni–O1enolate (1.984(4) Å) bond is stronger than the Ni–O2carbonyl (2.110(4) Å) bond, as expected, and the bond lengths fall within the same range as those observed for similar Ni(II)-flavonolate complexes.16,18,23 Interestingly, the CO bond in 1 (1.245(7) Å) is longer than that (1.232(3) Å) in the free ligand H(L1).45 This reveals that while the filled dxz and dyz orbitals of Ni(II) located perpendicular to CO bond are involved in π-back bonding with the antibonding π* orbital of CO (cf. below), leading to elongation of the CO bond, and supports the IR spectral results.
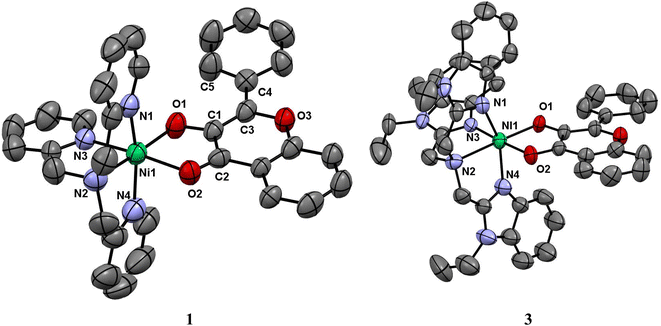 |
| | Fig. 1 ORTEP diagrams of [Ni(L1)(fla)]ClO4 1 and [Ni(L3)(fla)]ClO4 3 showing 50% probability thermal ellipsoids and labeling scheme for selected atoms. All the hydrogen atoms and perchlorate counter ions are omitted for clarity. | |
Table 1 Selected bond lengths [Å] for 1 and 3
| Bond lengths/Å |
1 |
3 |
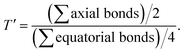 |
| Ni–N1 |
2.080(5) |
2.062(7) |
| Ni–N2 |
2.087(5) |
2.221(7) |
| Ni–N3 |
2.050(4) |
2.039(7) |
| Ni–N4 |
2.073(6) |
2.092(7) |
| Ni–O1 |
1.984(4) |
1.962(5) |
| Ni–O2 |
2.110(4) |
2.144(6) |
| O1–C1 |
1.314(6) |
1.302(9) |
| O2–C2 |
1.245(7) |
1.262(8) |
| C1–C2 |
1.464(7) |
1.498(10) |
| Axial donors |
Equatorial donors |
T′a |
T′a |
| N1N4 |
N2N3O1O2 |
1.009 |
0.993 |
| N3O2 |
N1N2N4O1 |
1.012 |
1.003 |
| N2O1 |
N1N3N4O2 |
0.979 |
1.003 |
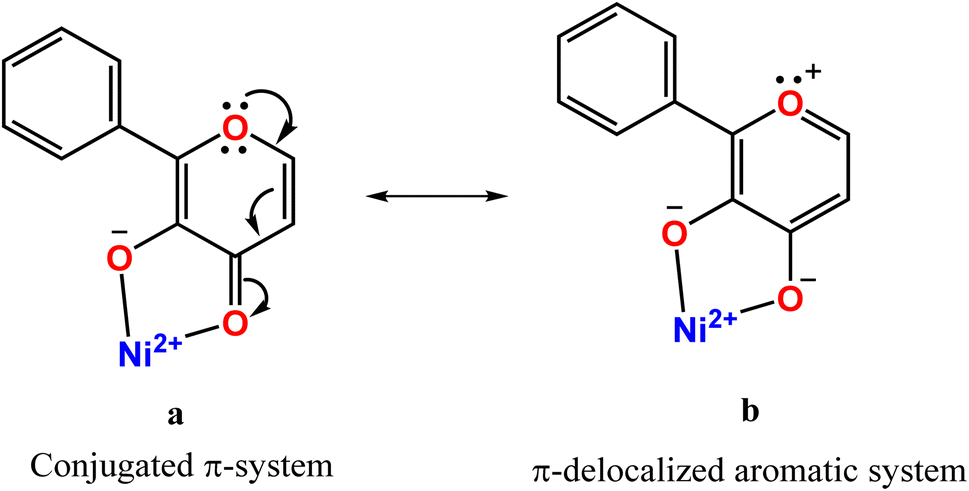
The complex cation of 3, like its py analogue 1, possesses a NiN4O2 coordination sphere with distorted octahedral geometry. The tertiary amine nitrogen atom N2 and one of the three benzimidazole (bzim) nitrogens (N3) of L3 and O1 and O2 atoms of fla− occupy the equatorial positions of the octahedron while the other two bzim nitrogen atoms (N1, N4) occupy the trans axial positions. All the Ni–Nbzim (Ni–N1, 2.062(7); Ni–N3, 2.039(7); Ni–N4, 2.092(7) Å) bond lengths are close to those observed for a similar Ni(II) complex of Me-ntb ligand.46 The Ni–Namine (2.221(7) Å) bond is longer than the Ni–Nbzim bonds, which is expected of sp3 and sp2 hybridizations respectively of amine and bzim nitrogen donors. As in 1, the Ni–O1enolate (1.962(5) Å) bond is shorter than the Ni–O2carbonyl bond (2.144(6) Å). The Ni–Nbzim bonds (2.039–2.092 Å) are shorter than the corresponding Ni–Npy (2.050–2.080 Å) bonds in 1, which is expected of the higher basicity of bzim nitrogen donor (pKa: pyH+, 5.25; 6-Me-pyH+, 5.5; MeImH+, 7.8).47 The strongly σ-bonding bzim N-donor is expected to enhance the electron density on Ni(II), causing elongation of both Ni–O1enolate and Ni–O2carbonyl bonds. However, the former is shorter (1.962(5) Å) than that (1.984(4) Å) in the py analogue 1 while the Ni–O2carbonyl bond is longer, as expected. It is evident that the CO bond oriented along the z-axis is involved in σ-bonding with Ni(II) (t2g6eg2) while the antibonding π* orbital of CO is involved in π-back bonding from the filled dxz and dyz orbitals of Ni(II), more strongly than those in 1 (cf. below), causing the CO bond in 3 (1.262(8) Å) to be longer than that in 1 (1.245(7) Å). Consequently, the π-back bonding renders the Ni–O2carbonyl bond (2.144(4) Å) weaker than that (2.110(4) Å) in 1, the Ni–O1enolate bond stronger synergistically, and the C2–C1 bond weaker (3: 1.498(10) Å; 1: 1.464(7) Å). Also, interestingly, the difference in Ni–O1enolate and Ni–O2carbonyl bonds in 3 (Δd, 0.182 Å) is higher than that in 1 (0.126 Å), revealing that the –CO–C(OH)– group in 3 is coordinated more asymmetrically, and the π-conjugated carbonyl form is stabilized more than the π-delocalized aromatic form of fla− (Scheme 3a and b, cf. below) or simply the π-delocalization in fla− decreases. Hence, the shorter the Ni–O1enolate bond and longer the Ni–O2carbonyl bond or higher the asymmetry in coordination of flavonolate (fla−), as given by the difference in Ni–O1 and Ni–O2 bond lengths (Δd), the lower the π-delocalization (decrease in aromatic character) in fla−, and hence the activation of Ni(II)-bound flavonone. Similar involvement of Ni(II) in π-back bonding is evident in the X-ray crystal structures of the complex [Ni(Me3TACN)(fla)(NO3)] with two geometric isomers a and b. The COcarbonyl and C–Oenolate bond lengths of both the isomers a (COcarbonyl, 1.261(18); C–Oenolate, 1.391(14) Å) and b (COcarbonyl, 1.280(20); C–Oenolate, 1.423(17) Å) are lower than those of free H(fla).28 Also, in the X-ray crystal structures of [NiII(L)(fla)],23 where LH is the tripodal ligand 2-([bis(pyridin-2-yl-methyl)amino]methyl)-p/m-R-benzoic acid [R = p-OMe and m-NO2], we find that the CO bond in the p-OMe complex is elongated, and the Ni–Oenolate bond is shortened, more than the respective bonds in the parent complex. The π-back bonding of Ni(II) with CO and synergistic σ-bonding of the enolate group to Ni(II) (CO: –OMe, 1.253(7); –NO2, 1.231(5); Ni–O: –OMe, 2.070(4), –NO2, 2.062(4); Ni–O(enolate): –OMe, 2.004(4), –NO2, 2.062(5) Å) facilitates the dioxygenation reaction of the p-OMe complex, rendering it more reactive. So, the highest asymmetry in coordination of fla− and π-back bonding in 3 would be expected to lead to confer the highest rate of dioxygenation (cf. below).
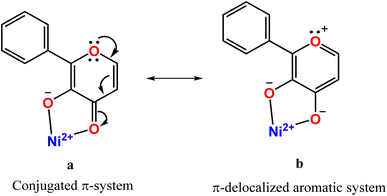 |
| | Scheme 3 Structures of π-conjugated carbonyl (a) and π-delocalized aromatic forms bound to nickel(II) (b). | |
Computational studies
A Density Functional Theory (DFT) study has been performed at the computational level of U-B3LYP/6-31G* exchange correlation functional on the Ni(II)-flavonolate complexes 1–3 to throw light on their unique structural features and to illustrate their reactivity towards dioxygen. The initial coordinates of 1 and 3 were taken from their single crystal X-ray structures and the calculated structures subjected to optimization. The structure of [Ni(ntb)(fla)]+ 4 has been also computed for comparison with 3. And the computed structural parameters (Tables 2, S4–S6, Fig. 2, S3, S4†) are in reasonably good agreement with those of the X-ray structures. So, they are valid and can be used to discuss the trends in bonding and reactivity of the model complexes. Also, we have calculated the structures of the other geometric isomers 1b–3b (Fig. 2, Tables S3 and S7†) for S = 0 and S = 1 spin states. We found that the S = 1 states have energies lower than the former, which is supported by the observation of three ligand field bands (cf. below), and the orientation of fla− in the distorted octahedral geometries of 1b–3b is different from that in 1–3. It may be noted that both such geometric isomers have been found in the X-ray structures of [Ni(Me3TACN)(fla)(NO3)].28 The incorporation of electron-releasing N-Et substituent on bzim ring in 4 to get 3 (cf. above) renders the coordination of bzim nitrogen slightly stronger,48 and the Ni–O2 bond in 3 (2.119 Å) is slightly longer than that in 4 (2.112 Å). This reveals that the stronger π-back bonding of Ni(II) with CO in 3 results in decrease in π-delocalization in fla− ring. In contrast, the Ni–Npy bonds 2 formed by the sterically hindered pyridyl nitrogen are longer than those in 1, which results in weaker π-back bonding of Ni(II) with CO and hence the higher π-delocalization in fla− ring (Scheme 3b). The asymmetry in coordination of fla−, as given by the difference in Ni–O1 and Ni–O2 bond lengths (Δd), varies as 3 (0.070) ≥ 1 (0.069) > 2 (0.048 Å), revealing that the fla− in 1 is expected to be activated more than in 2 and to almost the same extent as in 3 (cf. below).
Table 2 Computed structural parameters and HOMO and LUMO energies and band gap energies for complexes 1–3 and [Ni(ntb)(fla)]+ 4
| Bond lengths/Å |
1 |
2 |
3 |
[Ni(ntb)(fla)]+ 4 |
 |
| Ni–N1 |
2.095 |
2.216 |
2.084 |
2.091 |
| Ni–N2 |
2.149 |
2.114 |
2.260 |
2.279 |
| Ni–N3 |
2.060 |
2.053 |
2.063 |
2.065 |
| Ni–N4 |
2.095 |
2.222 |
2.087 |
2.088 |
| Ni–O1 |
2.044 |
2.051 |
2.049 |
2.049 |
| Ni–O2 |
2.113 |
2.099 |
2.119 |
2.112 |
| O1–C1 |
1.342 |
1.343 |
1.341 |
1.342 |
| O2–C2 |
1.297 |
1.297 |
1.295 |
1.296 |
| C1–C2 |
1.471 (1.464) |
1.467 |
1.473 (1.498) |
1.472 |
| Total energy (eV) |
−5.136 × 104 |
−5.350 × 104 |
−6.852 × 104 |
−6.210 × 104 |
| HOMO (eV) |
−5.582 |
−5.613 |
−5.508 |
−5.528 |
| LUMO (eV) |
−2.526 |
−2.567 |
−2.480 |
−2.495 |
| Band gap (eV) |
3.056 |
3.045 |
3.029 |
3.033 |
| Axial donors |
Equatorial donors |
T′a |
|
|
|
| N1N4 |
N2N3O1O2 |
1.002 |
1.067 |
0.983 |
0.982 |
| N3O2 |
N1N2N4O1 |
0.996 |
0.965 |
0.982 |
0.986 |
| N2O1 |
N1N3N4O2 |
1.003 |
0.970 |
1.036 |
1.032 |
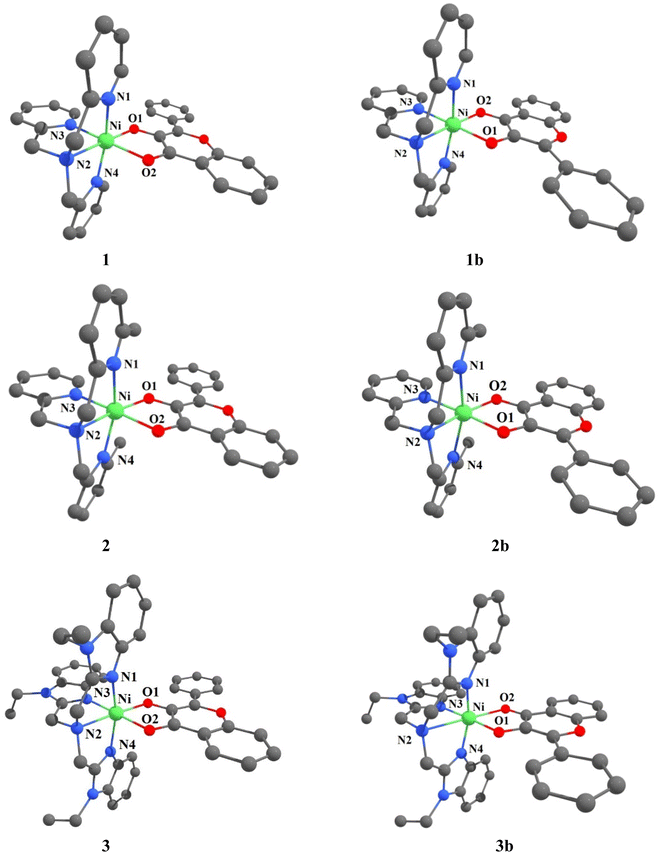 |
| | Fig. 2 Computed structures of [Ni(L)(fla)]ClO4 1–3 and its geometric isomer [Ni(L)(fla)]ClO4 1b–3b were calculated at U-B3LYP combined correlation function and basis sets for metal atom LANL2DZ and 6-31G* basis sets for other non-metal atoms and acetonitrile as a solvent by the CPCM method. | |
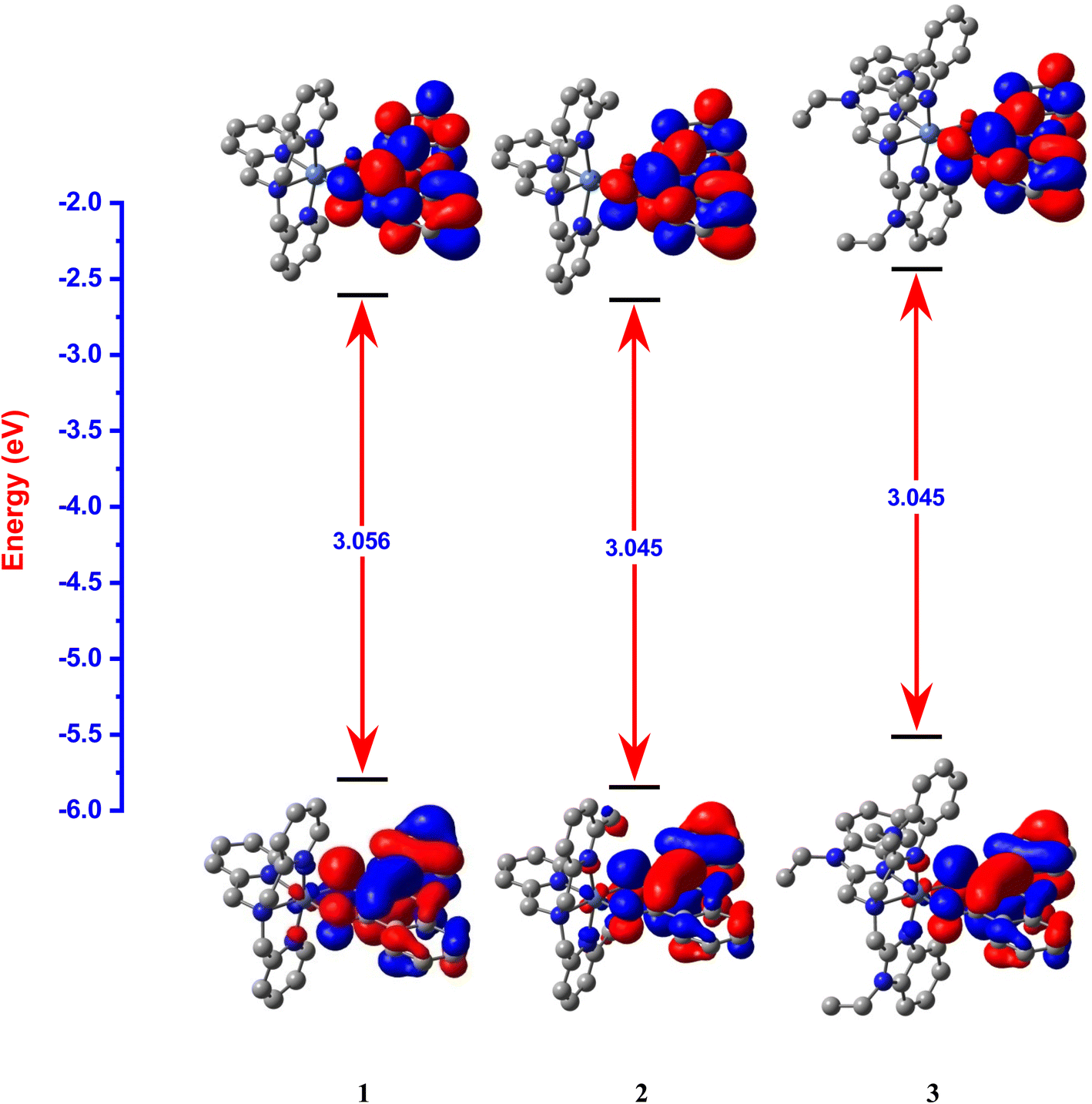
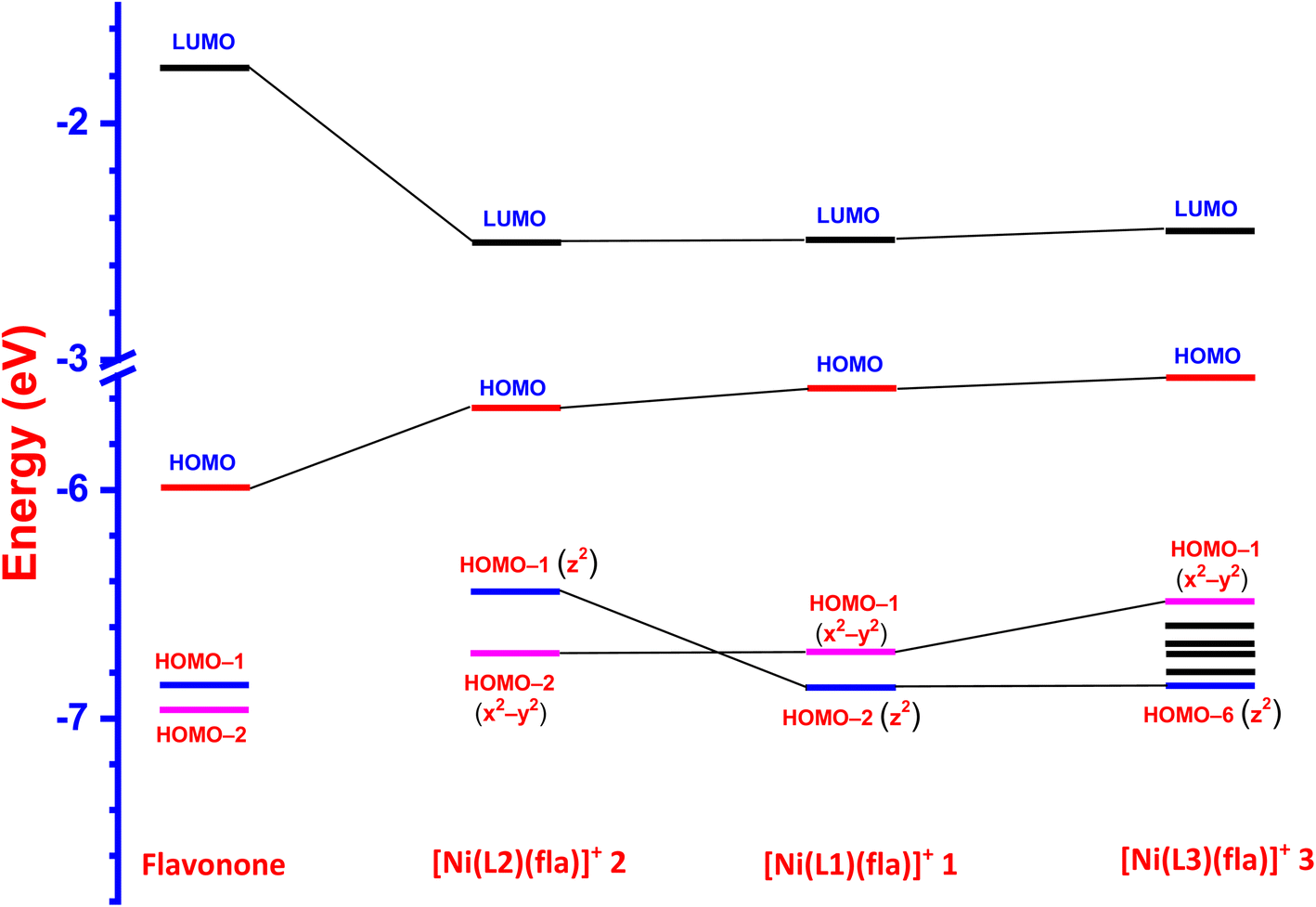
Interestingly, the doubly filled HOMO, localized on the coordinated fla−, is higher in energy than the metal based HOMO-1, and HOMO-2/6 (Table 2, Fig. 3, 4).49,50 This non-Aufbau electronic configuration is expected of π-conjugated molecules like H(fla), conferring enhanced stabilities on them, and such HOMO-1–HOMO inversion has been observed previously.51 The HOMO is a highly conjugated π-MO of fla− while the LUMO is the unoccupied highly conjugated π* orbital. The HOMO-1 and HOMO-2 are made up respectively of dx2–y2 (σ*) and dz2 (σ*) orbitals of Ni(II) (Fig. S3 and S4†). The observed trend in HOMO energy, H(fla) (−6.002) < 2 (−5.613) < 1 (−5.582) < 3 (−5.508 eV), is the same as that in LUMO energy, 2 (−2.567) < 1 (−2.526) < 3 (−2.480 eV), and a plot of the two energies is linear, reflecting that the 4N ligands tune the π-delocalization in fla− (Scheme 3b). The replacement of pyridyl nitrogen donor in 1 by the more basic N-Et-bzim nitrogen donor to obtain 3 raises the energies of both HOMO and LUMO, by enhancing the π-back bonding of Ni(II) with fla− and decreasing the π-delocalization in fla− in 3 (cf. above). On the other hand, interestingly, the inclusion of sterically hindering 6-Me group in 1 to obtain 2 increases the π-delocalization in fla− ring in 2, leading to a significant decrease in energies of both HOMO and LUMO, so that the HOMO-1 (dx2–y2) and HOMO–2 (dz2) in 2 become higher and hence are inverted in energies. It is expected that the higher the HOMO energy the easier is the removal of electron from it and hence more facile is the oxidation of fla−. It is seen that in 1 and 3, the dx2–y2 orbital is oriented towards N1N2N3O2 donor set while the dz2 orbital is oriented towards N3O2 donor atoms. On the other hand, the dz2 orbital in 2 is oriented towards N2O1 donors while the dx2–y2 orbital is oriented towards N1N3N4O1 donor set, which is in accordance with the calculated values of T'.52
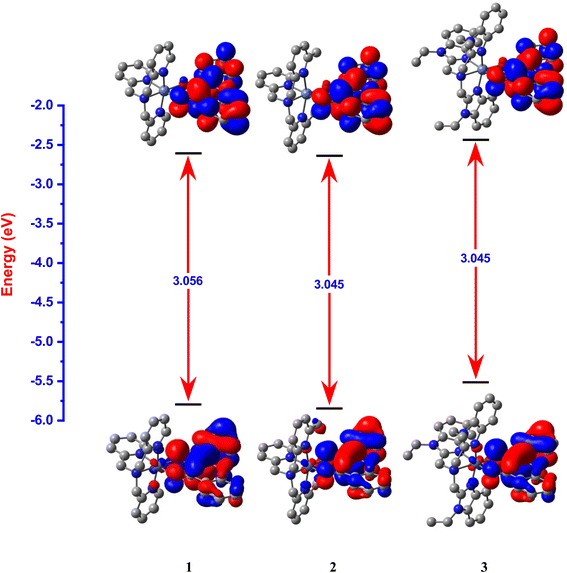 |
| | Fig. 3 HOMO and LUMO band gap energy profile diagram for [Ni(L1)(fla)]ClO4 1, [Ni(L2)(fla)]ClO4 2 and [Ni(L3)(fla)]ClO4 3 were calculated at U-B3LYP combined correlation function and basis sets for metal atom LANL2DZ and 6-31G* basis sets for other non-metal atoms and acetonitrile as a solvent by the CPCM method. | |
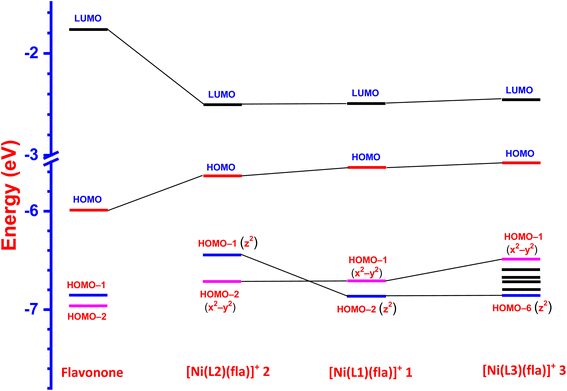 |
| | Fig. 4 LUMO and HOMO energy profile diagram for the ligand 3-hydroxyflavone and complexes 1–3. | |
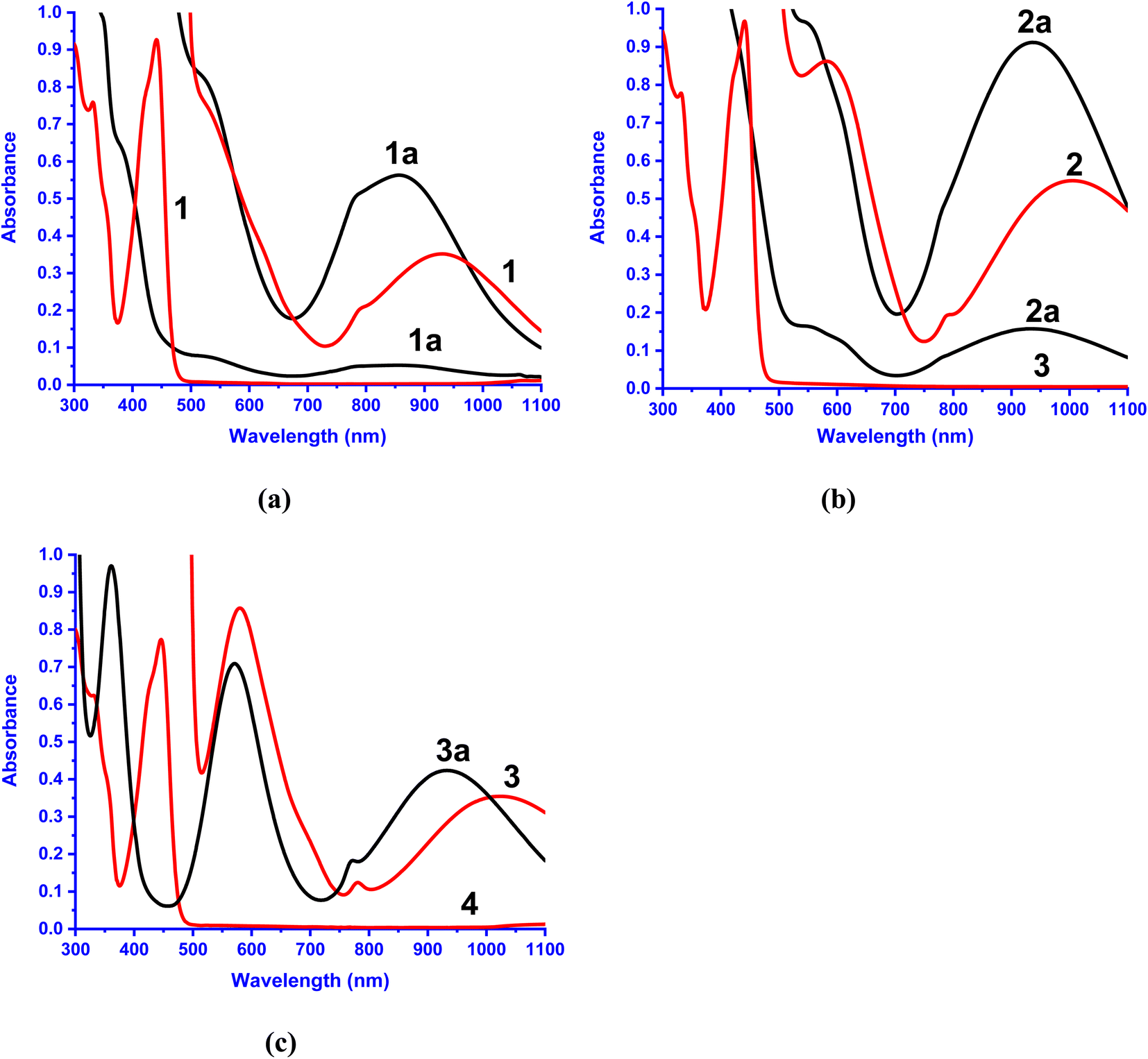
Electronic absorption spectral properties
The electronic absorption spectral data of all the Ni(II) complexes obtained in CH3CN solvent are summarized in Table 3 and typical spectra of [Ni(L)(CH3CN)2]2+ 1a–3a and the flavonolate adducts [Ni(L)(fla)]+ 1–3 are shown in Fig. 5. The complexes 1–3 exhibit an intense absorption band in the range 440–450 nm (ε, 3.1–5.6 × 104 M−1 cm−1, Table 3, Fig. 5), which is assigned to the π → π* transition within the coordinated flavonolate.53 All these assignments are in good agreement with those observed for the previously reported octahedral Ni(II) complexes with NiIIN6 (ref. 30 and 31) and similar NiIIN4O2 chromophores.54 The band energy observed for 3 (446 nm) is lower than those for 1 and 2 (441 nm), which correlates with the trend in the HOMO (π) − LUMO (π*) energy gap, 1 (3.056) > 2 (3.045) > 3 (3.029 eV) (Table 2, Fig. 3), revealing the influence of primary ligands on the coordinated fla−. The shift in π → π* band from that (458 nm) observed15a for free flavonolate anion of Me4N[fla] is higher for 3 (12 nm) than that (17 nm) for 1 and 2, confirming the influence of primary ligands on π-delocalization within fla−. The lower shift observed for 3, which is consistent with the higher asymmetry in fla− coordination (cf. above), corresponds to higher π-delocalization within fla− ring.18 Hence, it is evident that the lower the shift in π → π* band position the higher the disruption of π-delocalization within fla−. All the complexes exhibit two well-defined ligand field (LF) absorption bands together with an absorption spectral feature in the low energy region in 1–3. The lowest energy band observed in the range 929–1022 nm for 1–3 is assigned to 3A2g → 3T2g(F) (ν1) transition in Ni(II) located in an octahedral environment. The higher energy band observed (528–582 nm) is assigned to 3A2g → 3T1g(F) (ν2) transition while the feature located on the low energy side of ν2 band is assigned to 3A2g → 1E1g(D) spin-forbidden transition.30,31,44,54a Upon adding fla− to complexes 1a–3a, the ν1 and ν2 bands are red-shifted (Table 3, Fig. 5), suggesting coordination of flavonolate to Ni(II).30,31 Also, the trend in ν1 band position reveals that the LF imposed on Ni(II) is weaker in 2 and 3, which corresponds to the weaker coordination of L2 in 2, and the ligand steric crowding around Ni(II) in 3, which renders the Ni–Nbzim bonds weaker in solution.
Table 3 Electronic absorption spectral data for complexes in acetonitrile solutiona at 25 °C
| Complex |
Electronic absorption spectra |
| Wavelength (nm) |
ε (M−1 cm−1) |
| Conc. 1a, 1: 2.0 × 10−3 M; 2a: 3.4 × 10−3 M; 2: 3.3 × 10−3 M; 3a: 3.9 × 10−3 M; 3: 1.7 × 10−3 M. Conc. 1a, 1: 1.3 × 10−4 M; 2a, 2, 3: 1.4 × 10−4 M; 3a: 3.9 × 10−3 M. |
| [Ni(L1)(CH3CN)2](ClO4)2 1a |
858(ν1) |
280 |
| 523(ν2) |
410 sh |
| 384(ν3)b |
| [Ni(L2)(CH3CN)(H2O)](ClO4)2 2a |
936(ν1) |
270 |
| 545(ν2) |
290 sh |
| 428(ν3)b |
| [Ni(L3)(CH3CN)2](ClO4)2 3a |
932(ν1) |
110 |
| 571(ν2) |
180 sh |
| 361(ν3)b |
| [Ni(L1)(fla)](ClO4) 1 |
929(ν1) |
180 |
| 528(ν2) |
370 sh |
| 441b |
| [Ni(L2)(fla)](ClO4) 2 |
1007(ν1) |
170 |
| 582(ν2) |
264 sh |
| 441b |
|
| [Ni(L3)(fla)](ClO4) 3 |
1022(ν1) |
210 |
| 580(ν2) |
510 sh |
| 446b |
|
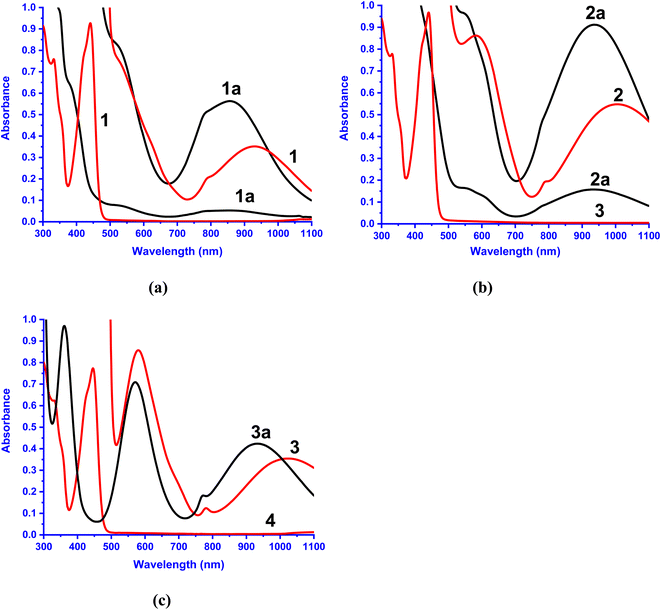 |
| | Fig. 5 Electronic absorption spectra of 1a–3a and 1–3. Conc. (a) 1a, 1: 2.0 × 10−3 M, 1.3 × 10−4 M; (b) 2a: 3.4 × 10−3 M, 1.4 × 10−4 M, 1.3 × 10−4 M; 2: 3.3 × 10−3 M, 1.4 × 10−4 M and (c) 3a: 3.9 × 10−3 M; 3: 1.7 × 10−3 M, 1.4 × 10−4 M in acetonitrile solution at 25 °C. | |
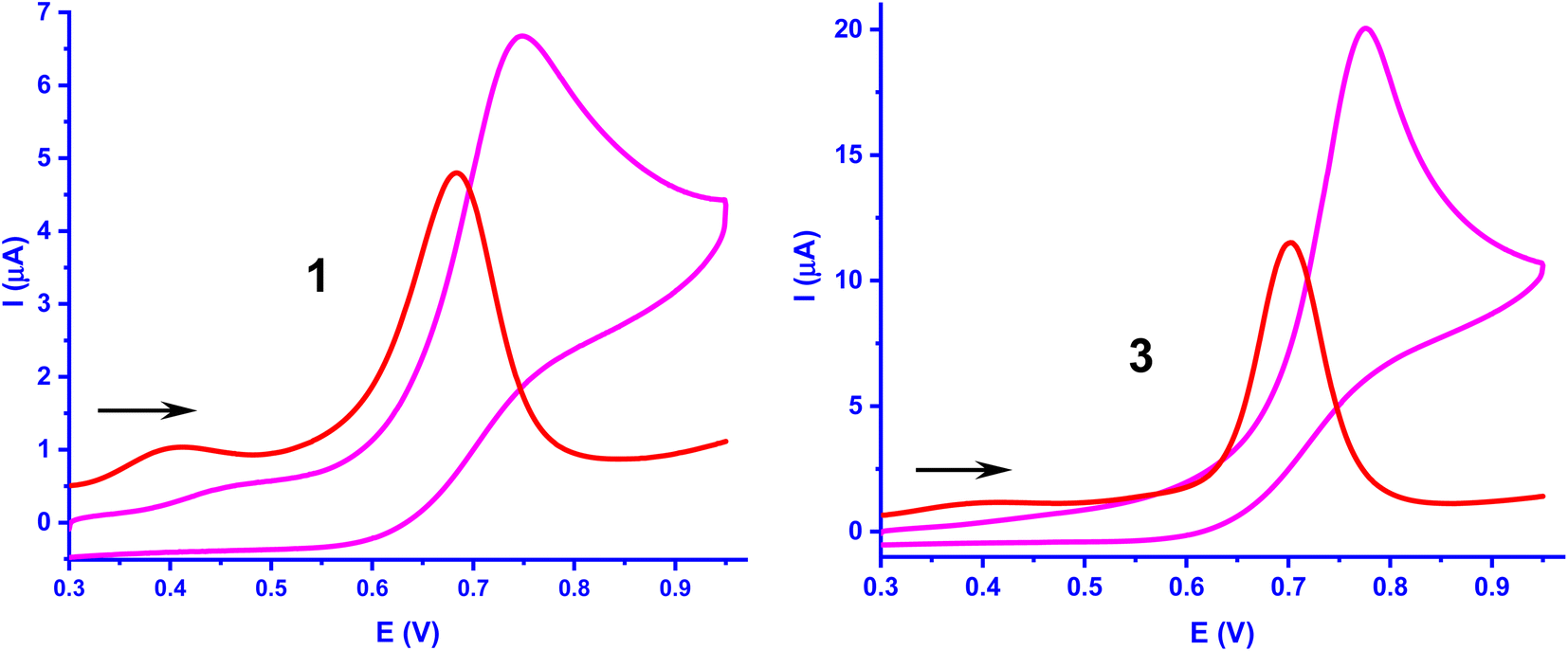
Redox studies
The electrochemical oxidation of the complexes 1–3 was investigated by using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) (Table 4, Fig. 6, S1†). The irreversible oxidation process observed in the range, 0.770–0.800 V is assigned to oxidation of bound flavonolate (fla−) to fla˙– (fla–/fla˙ redox couple), as free H(fla) is found to exhibit an oxidation wave at 0.715 V (vs. SCE), and as the Ni(II)/Ni(III) redox couple is expected to occur at more positive potentials.31b The E1/2 values from DPV follow the trend, H(fla) (0.715 V) > 3 (0.683 V) > 1 (0.657 V) < 2 (0.679 V vs. SCE). The E1/2 values observed for fla− are lower than that for H(fla), revealing that fla− is destabilized towards oxidation to give fla˙, upon coordination to Ni(II). The incorporation of Me group in 1 to obtain 2 sterically hinders the coordination of pyridyl nitrogen, lowers the energy of HOMO (cf. above), and renders the removal of electron from the HOMO more difficult and stabilizes fla− towards oxidation more. Similarly, the bzim nitrogens in 3, which are coordinated to Ni(II) more strongly than the py nitrogens in 1, and raise the energy of HOMO (cf. above), are expected to facilitate the removal of electron from HOMO, and lower the oxidation potential. However, the observed oxidation potential of 3 is higher than that for 1. The three bulky bzim nitrogens sterically crowd around Ni(II) in solution, as evident from the lower LF strength (cf. above), and possibly one of the weak Ni–Nbzim bonds is substituted by ClO4− available in high concentration, which lowers the HOMO energy and renders the removal of electron from HOMO more difficult. Thus, the removal of electron to form the radical fla˙ intermediate and reactive O2˙− is facilitated by the strong σ-bonding of primary ligands. This observation is reminiscent of Nature choosing carboxylate donor to coordinate to redox-inactive Ni(II), increase the electron density on it, and hence enhance the π-back bonding of Ni(II) with fla−.
Table 4 Electrochemical dataa of complexes 1–3 in DMF solution at 25 °C
| Complex |
Cyclic voltammetry |
DPV |
Redox process |
| Epc (V) |
E1/2 (V) |
E1/2 (V) |
| Add 0.252 V to convert NHE. Scan rate, 50 mV s−1. Working electrode: glassy carbon, reference electrode: calomel electrode. Pulse width (ΔE), 0.050 V, DPV: E1/2 = Ep + ΔE/2. |
| Flavonone |
0.795 |
0.725 |
0.715 |
HFla/Fla˙ |
| [Ni(L1)(fla)]+ 1 |
0.747 |
0.674 |
0.657 |
Fla−/Fla˙ |
| [Ni(L2)(fla)]+ 2 |
0.773 |
0.695 |
0.679 |
Fla−/Fla˙ |
| [Ni(L3)(fla)]+ 3 |
0.775 |
0.719 |
0.683 |
Fla−/Fla˙ |
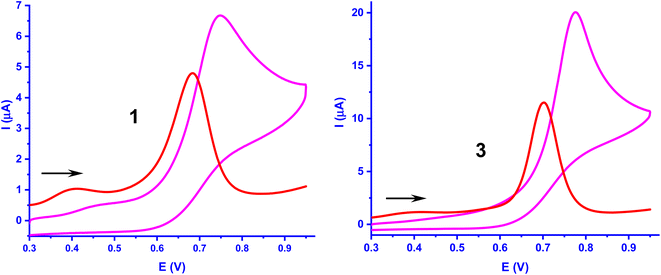 |
| | Fig. 6 Cyclic voltammogram and DPV of complexes 1 and 3 (1.0 × 10−3 M) in DMF at 25 °C. Conditions: supporting electrolyte, 0.1 M TBAP; scan rate, CV, 50 mV s−1 and DPV, 2 mV s−1 for all complexes, reference electrode, calomel electrode; working electrode, glassy carbon; counter electrode, platinum wire. | |
Study of degradation of Ni(II)-flavonolate adducts
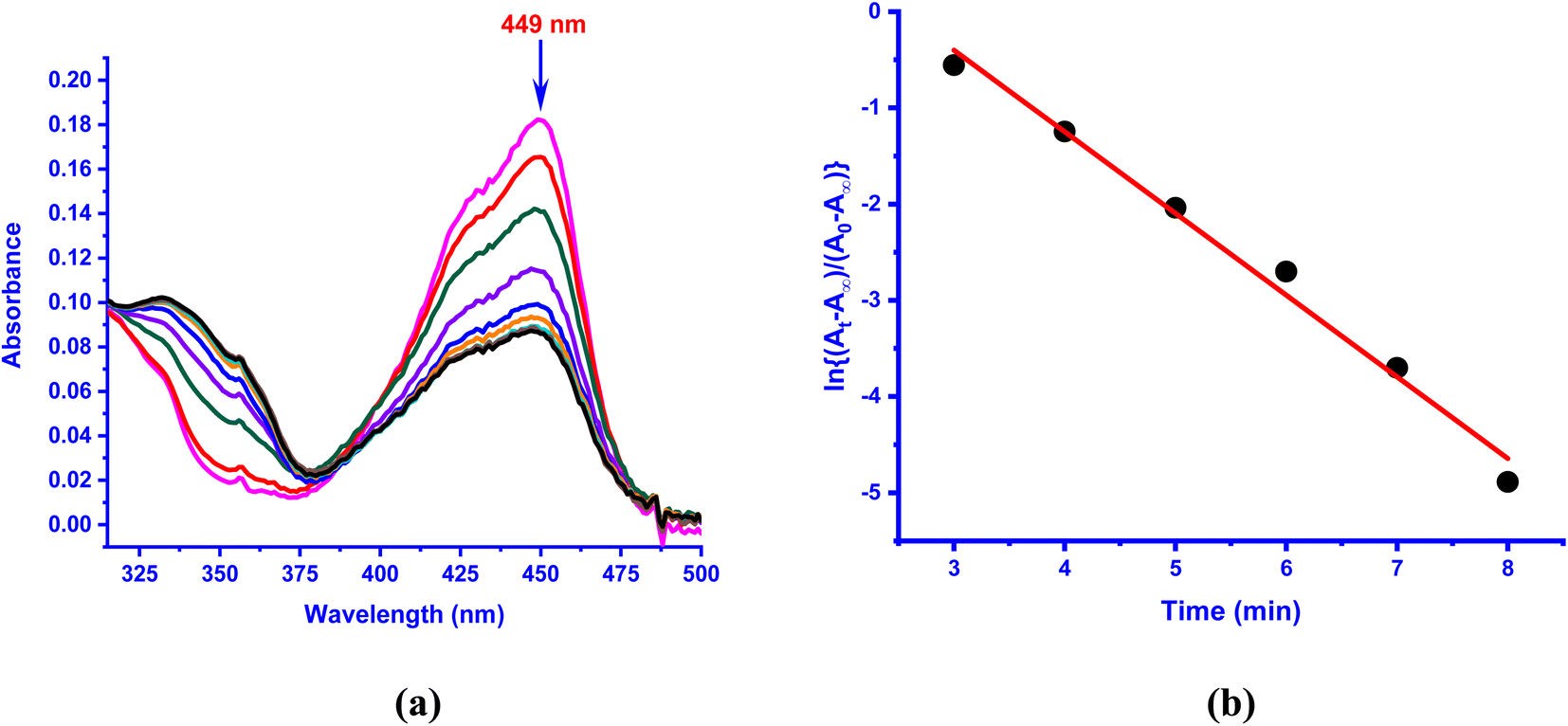
The kinetics of reaction of the enzyme–substrate model complexes [Ni(L)(fla)](ClO4) 1–3 in DMF solution with dioxygen was monitored by following the decrease in absorption intensity of the π → π* band (440–450 nm) upon exposure to atmospheric air or pure dioxygen. At room temperature and under atmospheric air, 1–3 did not show any measurable decrease in absorption intensity; however, when dioxygen was bubbled through the solution, the reaction proceeds slowly, with 1 and 3 reacting at almost the same rate. At a higher temperature (70 °C) and in the presence of atmospheric dioxygen, the reaction proceeds faster than at room temperature. The absorption intensity decreases to reach a minimum within 4–6 min for 1 and 3 (Fig. 7a and S10a†), and decreases slowly and then fast for 2 (Fig. S11a†). The decrease in intensity resembles that observed for the enzyme–substrate adduct,55 and corresponds to degradation of Ni(II)-bound flavonolate. But, when dioxygen is bubbled through the solutions at 70 °C, the reaction proceeds faster, confirming that the reaction rate is dependent upon dioxygen concentration. Under the reaction conditions with excess dioxygen, the pseudo-first order rate constant kobs was calculated from the slope of the plot of ln{(At-A∞)/(A0-A∞)} vs. time (Fig. 7 and S9–S11†). The kinetic data and the values of kobs and kO2 (= kobs/[O2]) are collected in Table 5. At 70 °C (pure O2), the rate of dioxygenolysis (kO2) calculated based on the initial decrease in intensity varies as: 1 (16.67 ± 0.70) > 2 (1.81 ± 0.04) < 3 (29.10 ± 0.16 × 10−1 M−1 s−1). Thus, 3 reacts almost twice faster than 1, and nearly 25 times faster than 2. The GC-MS analysis of the final colorless reaction mixture obtained for 1–3 (Fig. S12, Table S12†) reveals the ultimate products (Scheme 4) as salicylic acid [m/z (neg): 136, [(M – 2H)−] (20–60%) and benzoic acid (m/z (neg): 122 [(M)+] (21–43%).
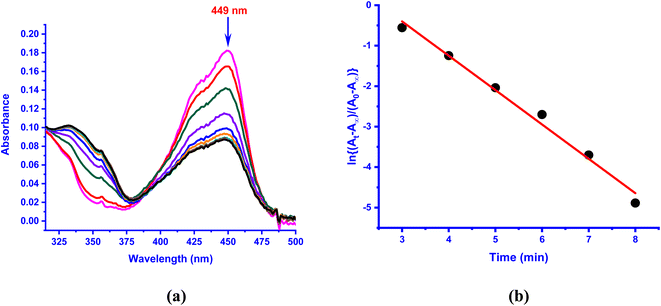 |
| | Fig. 7 (a) UV-vis spectral changes observed in the presence of O2 in DMF solution of [Ni(L3)(fla)]ClO4 3 (1 × 10−4 M) at 70 °C; (b) plot of ln{(At − A∞)/(A0 − A∞)} vs. time for the band observed at 449 nm. | |
Table 5 The kinetic dataa,b for dioxygenation of Ni(II)-bound flavonol in DMF as solvent using dioxygen at 70 °C
| Compound |
kobs (×10−3 s−1) |
kO2 (×10−1 M−1 s−1) |
| Dioxygen filled in a balloon as an external dioxygen source. Conc. of dioxygen in DMF solution,41 4.86 × 10−3 M. Calculated rate for the changes obtained after 33 min. |
| [Ni(L1)(fla)]ClO4 1 |
8.10 ± 0.40 |
16.67 ± 0.70 |
| [Ni(L2)(fla)]ClO4 2 |
0.88 ± 0.02 |
1.81 ± 0.04 |
| 3.27 ± 0.27c |
6.73 ± 0.56c |
| [Ni(L3)(fla)]ClO4 3 |
14.14 ± 0.20 |
29.10 ± 0.16 |
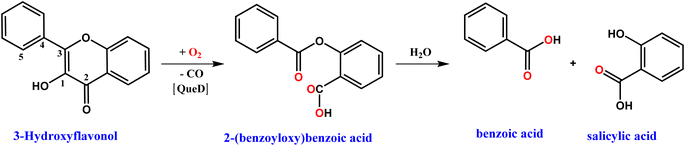 |
| | Scheme 4 Dioxygenation of 3-hydroxyflavonol and subsequent hydrolysis of depside to products, catalyzed by complexes 1–3. | |
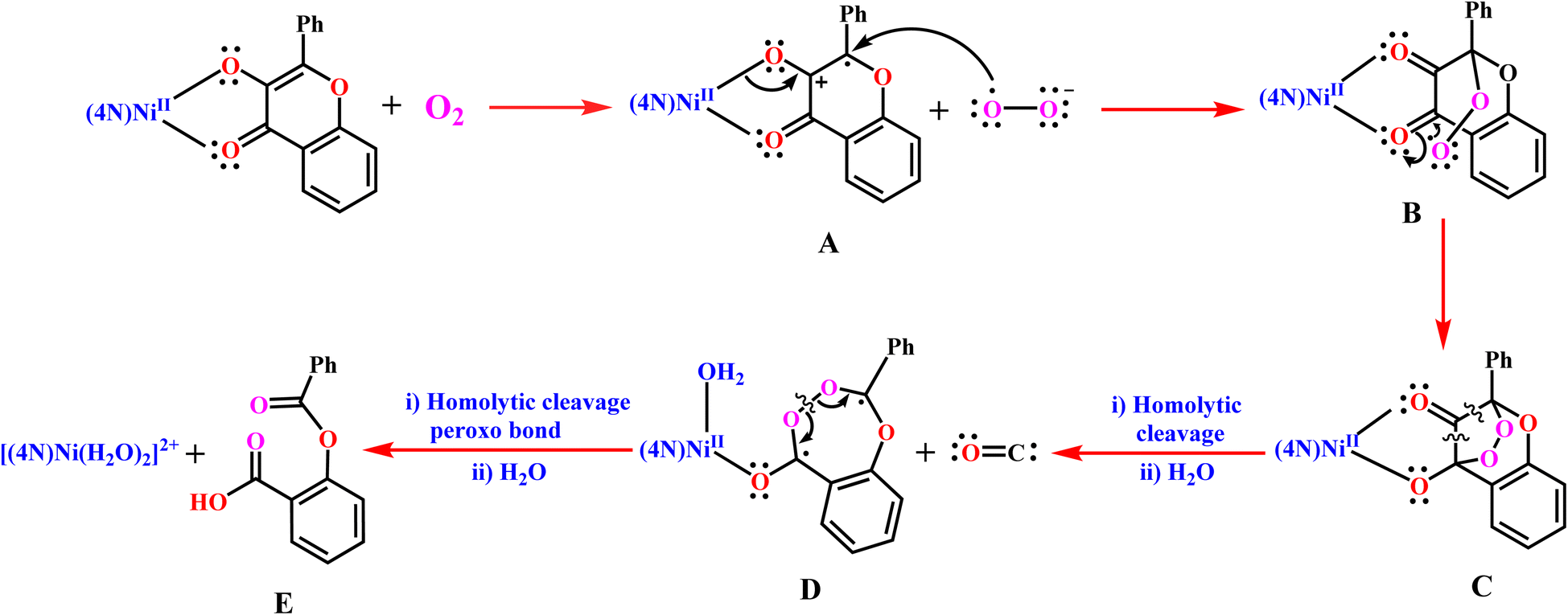
The observed trend in rates of dioxygenolysis can be illustrated by invoking the reaction mechanism proposed in Scheme 5, which involves formation of the intermediates A–D. A similar mechanism has been already proposed for certain Ni(II)-flavonolate complexes.18,28 The first step in the reaction mechanism involves dioxygen abstracting an electron from the π-orbital on C1 carbon bearing electron-rich enolate to form the intermediate [Ni(L)(fla˙)]2+ A and O2˙– radical. This step is considered the rate-determining step as it involves activation of dioxygen. The unpaired electron on the benzylic C3 carbon (Scheme 3) in A is significantly stabilized by delocalization into the electron-sink phenyl group and on to C1 and C2 atoms (cf. below). The electron density in the Ni(II)–O1(enolate) oxygen in A is then transferred to C1 carbon to form the Ni(II)-bound carbonyl group. In the next step, the reactive O2˙– immediately attacks the C3 carbon carrying the unpaired electron to form the peroxide radical B. Then the peroxide radical immediately attacks the C2 carbon in B to form the bridging peroxide intermediate C with Ni(II) weakly bound to C–O(enolate). In the third step, the unstable bicyclic species C undergoes homolysis at the two weaker C–C bonds adjacent to the carbonyl group, generating the diradical D and releasing the CO. The diradical D is stabilized by delocalization of electron density into the phenyl ring and the Ni(II)-bound enolate oxygen as well. In the penultimate step, the peroxo group undergoes homolysis to form substituted (benzoyloxy)benzoic acid E as the desired product and regenerate the Ni(II) complex. In the presence of traces of water, the depside E readily undergoes hydrolysis to produce benzoic acid and salicylic acid.
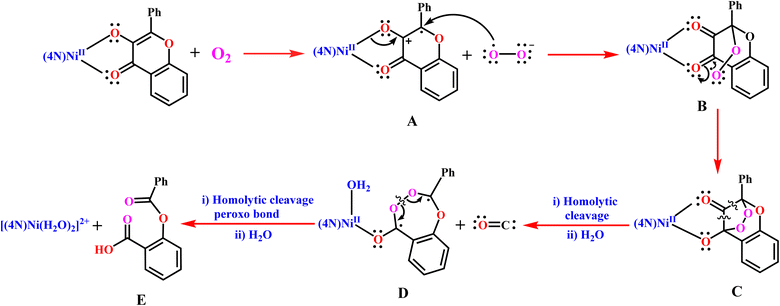 |
| | Scheme 5 Proposed reaction mechanism for Ni(II)-flavonolate adducts 1–3 as quercetin 2,4-dioxygenase enzyme models. | |
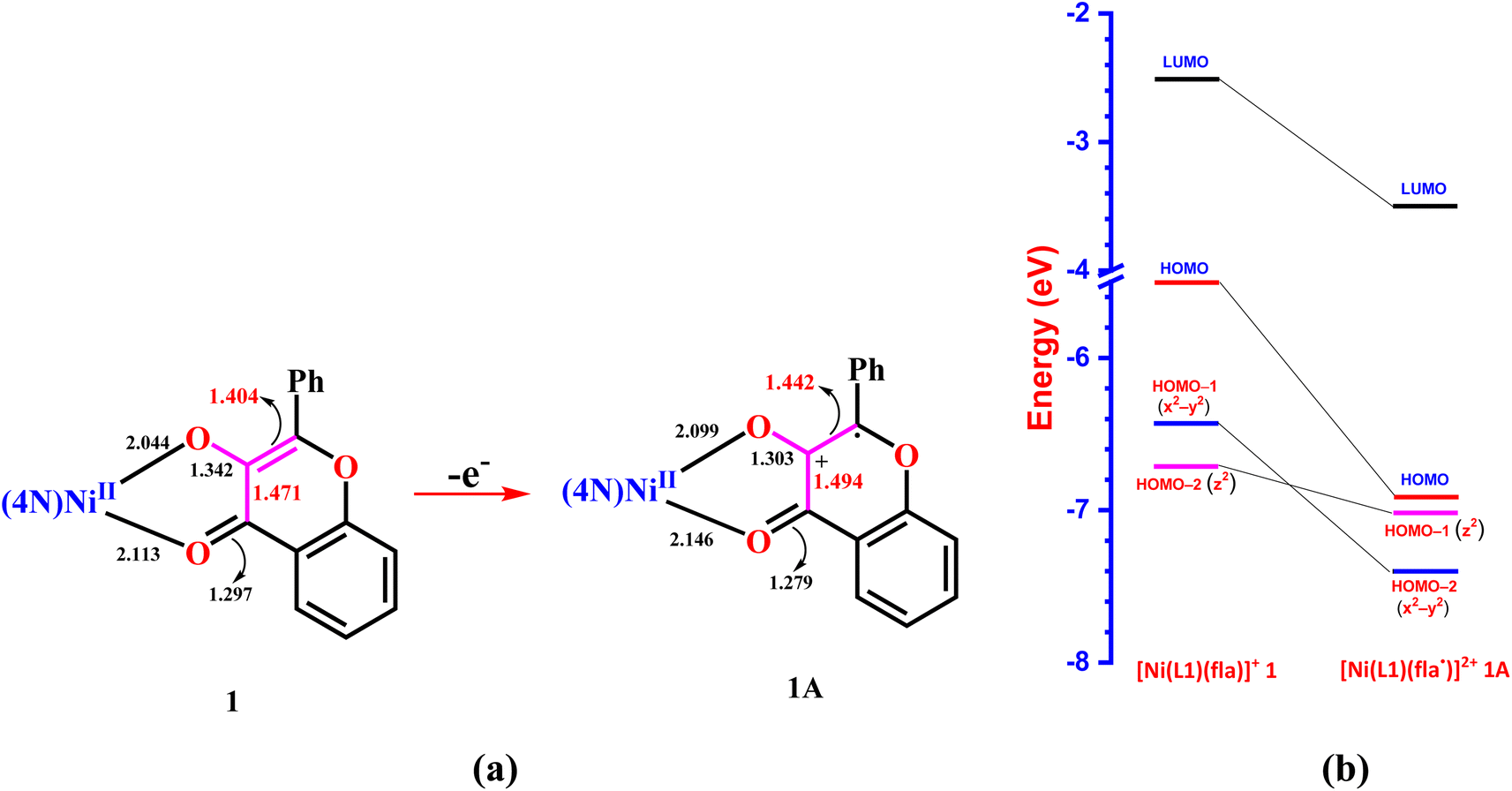
The formation of superoxide ion during the reaction has been demonstrated by performing the nitro blue tetrazolium (NBT2+) test which involves treating one equivalent of 3 with two equivalents of NBT2+ under nitrogen and then exposing the reaction mixture to dioxygen at 70 °C. The appearance of a band at 520 nm (Fig. S5†) characteristic of monoformazan species (MF+) reveals the in situ production of superoxide radical which reduces NBT2+ to give MF+.56 Also, when the reaction was performed in the presence of small amount of the free radical scavenger DMSO the reaction did not proceed,57 lending strong support to the involvement of superoxide in the reaction mechanism proposed. To understand the extent of delocalization of π-electrons into Ni(II)-bound fla˙, the electronic structures of the intermediate radicals 1A–3A have been computed and optimized using DFT method (Fig. S8, Tables S10 and S11†). Upon radical formation, the Ni–O1 (2.047–2.102 Å) and Ni–O2 (2.105–2.135 Å) coordinate bonds become weaker, and the C1–O1 and CO2 bonds become stronger, revealing that fla• is weakly bound and has become susceptible to undergo homolytic cleavage at the weaker C1–C2 and C1–C3 bonds (Scheme 6, S1†). Also, the HOMOs of the intermediates have become significantly lower in energy (1.246–1.356 eV) than those of the reactants, but remain still higher in energy than the MOs. This reveals significant stabilization of the oxidized radical species (Scheme 6b, S1, Fig. S6, S7, Tables S8 and S9†), possibly due to delocalization of unpaired electron density over C1 and C2 carbons and the phenyl ring (Scheme S1†). Also, the trend in HOMO energy, viz., 3A > 1A ≈ 2A is similar to that of the reactants, viz., 3 > 1 > 2.
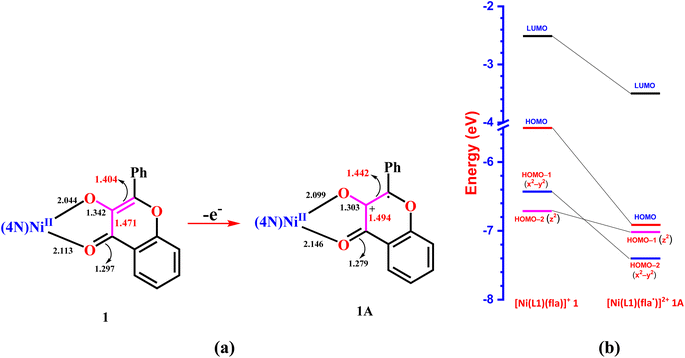 |
| | Scheme 6 Changes in bond lengths (a) and energies of molecular orbitals (b) upon removal of an electron from Ni(II)-bound flavonolate in 1 to form the intermediate radical 1A. | |
The trend in the rate of dioxygenation observed for 1–3 can be illustrated by invoking the above reaction mechanism. Thus, the increase in electron density on Ni(II) by the more basic bzim nitrogens and the electron-releasing N-Et substituent in 3 enhance the π-back bonding of Ni(II) with the antibonding π* orbital of CO of fla− (cf. above), raise the energy of HOMO and facilitate the rate-determining step of electron abstraction by dioxygen. In other words, the higher asymmetric coordination of fla− (cf. above) causes decrease in π-delocalization in fla− ring, activates it towards dioxygen and accounts for the highest dioxygenation rate of 3. Also, the weaker C1–C2 bond in 3, arising from the stronger π-back bonding of Ni(II) and the weaker C1–C2 and C1–C3 bonds in 3A, facilitate the homolytic cleavage of the peroxide intermediate C in 3 more than that in 1. A similar enhancement in reaction rate has been observed23 for [NiII(L)(fla)], due to the π-back bonding of Ni(II) with CO and synergistic σ-bonding of the enolate group to Ni(II) when electron-releasing p-OMe group is incorporated in the tripodal pyridyl ligand (cf. above). Also, it may be noted that the incorporation of –OMe group on the phenyl ring of the flavonolate increases the rate of degradation of [K(fla)] while that of –CN group decreases it.45 The sterically hindering 6-Me groups in 2 weaken the coordination of L2 to Ni(II), resulting in its HOMO to be more stable than that of 1 (cf. above), causing the abstraction of electron from 2 to obtain 2A more difficult; this illustrates that 2 undergoes degradation at a rate lower than 1, which is consistent with the lower asymmetry in bonding of flavonolate (cf. above). Very recently, Mandal et al.58 and Sun et al.16,23 have illustrated the trends in reactivity of certain Cu(II)-flavonolate adducts and the substituent effects on the basis of the torsion angles calculated from solid state X-ray structures.58 However, for the present complexes, the trends in the low values of twist angles obtained from X-ray structures of 1 and 3, and those calculated for 1–3, fail to correlate well with the dioxygenation rates.
Thus, both the computational and experimental results illustrate the trend in rates of the enzyme-type dioxygenation of the present functional models designed for Ni(II)-containing 2,4-QueD, and support the proposed reaction mechanism, particularly, the radical formation by abstraction of an electron from the carbon carrying the coordinated hydroxyl group, and the stabilization of benzylic radical by the electron-sink phenyl ring. Also, the X-ray structures of 1 and 3 lend support to the enolate-one to one-enolate shift in bonding in the flavonolate during dioxygenation, as tuned by the redox-inactive Ni(II) involved in π-back bonding with the CO group and the synergistic σ-bonding of the enolate group of flavonolate to Ni(II). This is similar to the bonding in carbonyls like [Ni(CO)4]. Thus, a metal carbonyl like [Ni(CO)4] owes its strong metal coordination to π-back bonding and synergistic σ-bonding through the same carbon atom while the flavonolate owes its decreased π-delocalization in the aromatic ring and hence activation to involvement of the conjugated carbonyl group in π-back bonding from Ni(II) and that of enolate oxygen atom in synergistic σ-bonding to Ni(II). Thus, the strong σ-bonding of more basic bzim nitrogen and electron-releasing N-Et-bzim decrease the π-delocalization in the aromatic flavonolate ring, leading to higher rates of degradation. This observation is reminiscent of the higher rate of degradation of enzyme–substrate complex on account of electron-releasing effect of the N-Glu carboxylate bound to Ni(II) in the active site. This supports the role of redox-inactive Ni(II) in the enzyme–substrate model complex by involving in π-back bonding, delocalizing π-electron density into fla− and activating it towards oxidation.
Conclusions
To conclude, three new Ni(II)-flavonolate complexes of the type [Ni(L)(fla)]+, where L is a tripodal 4N ligand with three pyridyl/benzimidazolyl nitrogen donors, have been isolated as structural and functional models for the Ni(II)-containing quercetin 2,4-dioxygenase (2,4-QueD) enzymes. Single crystal X-ray structures of two flavonolate adducts contain a NiN4O2 coordination sphere with distorted octahedral geometry. They reveal that Ni(II) is involved in π-back bonding with flavonolate and the latter in synergistic σ-bonding with Ni(II). The DFT optimized structures of the complexes and their geometric isomers have been obtained. The HOMO and LUMO orbitals localized on Ni(II)-bound flavonolate are highly conjugated π-bonding and antibonding π*-orbitals respectively. They are located higher in energy than the Ni(II)-based MOs (HOMO–1, dx2–y2; HOMO–2/6, dz2), revealing that Ni(II)-bound flavonolate rather than Ni(II) undergoes oxidation upon exposure to dioxygen. The energies of HOMO, in combination with spectral and electrochemical studies, illustrate the potential of the tripodal ligands to coordinate to Ni(II) and facilitate the conversion of Ni-enolate to Ni-one bond enabling release of CO from the activated substrate. The highest dioxygenase reactivity of the flavonolate adduct with N-Et-bzim moiety is on account of the more basic bzim and electron-releasing N-Et substituent on it, which leads to enhanced π-back bonding of Ni(II) with flavonolate, causing decreased π-delocalization in the substrate and hence its activation. This observation is reminiscent of Ni(II)-carboxylate coordination in the enzyme active site, which promotes the reaction remarkably by electron-release into the π-delocalized flavonolate substrate through π-back bonding with Ni(II).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the financial support by SERB, New Delhi (EMR/2015/002222) and INSA, New Delhi, for the INSA Senior Scientist position to M. P. HRMS facility was sanctioned to School of Chemistry, Bharathidasan University through DST-FIST. And, we thank Professor Dr Nashreen S. Islam and Dr Ms Mitu Sharma, Tezpur University, Tezpur for collecting the X-ray data and Professor M. Velusamy, NEHU, Shillong for his help in solving the X-ray structures.
References
- P. Pietta, C. Gardana and A. Pietta, Oxid. Stress, 2003, 9, 43–70 CAS.
- S. Tranchimand, P. Brouant and G. Iacazio, Biodegradation, 2010, 21, 833–859 CrossRef CAS PubMed.
-
(a) K. C. Ryan, O. E. Johnson, D. E. Cabelli, T. C. Brunold and M. J. Maroney, J. Biol. Inorg. Chem., 2010, 15, 795–807 CrossRef CAS PubMed;
(b) M. Sparta, C. E. Valdez and A. N. Alexandrova, J. Mol. Biol., 2013, 425, 3007–3018 CrossRef CAS;
(c) B. Desguin, P. Goffin, E. Viaene, M. Kleerebezem, V. Martin-Diaconescu, M. J. Maroney, J. P. Declercq, P. Soumillion and P. Hols, Nat. Commun., 2014, 5, 3615 CrossRef.
-
(a) S. L. Chen, M. R. Blomberg and P. E. Siegbahn, Phys. Chem. Chem. Phys., 2014, 16, 14029–14035 RSC;
(b) K. C. Ryan, A. I. Guce, O. E. Johnson, T. C. Brunold, D. E. Cabelli, S. C. Garman and M. J. Maroney, Biochemistry, 2015, 54, 1016–1027 CrossRef CAS.
-
(a) E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS;
(b) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1046 CrossRef CAS PubMed;
(c) F. Fusetti, K. H. Schröter, R. A. Steiner, P. I. van Noort, T. Pijning, H. J. Rozeboom, K. H. Kalk, M. R. Egmond and B. W. Dijkstra, Structure, 2002, 10, 259–268 CrossRef CAS PubMed;
(d) R. A. Steiner, K. H. Kalk and B. W. Dijkstra, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16625–16630 CrossRef CAS PubMed.
-
(a) I. M. Kooter, R. A. Steiner, B. W. Dijkstra, P. I. van Noort, M. R. Egmond and M. Huber, Eur. J. Biochem., 2002, 269, 2971–2979 CrossRef CAS;
(b) R. A. Steiner, I. M. Kooter and B. W. Dijkstra, Biochemistry, 2002, 41, 7955–7962 CrossRef CAS.
-
(a) E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed;
(b) R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS PubMed;
(c) J. B. Vincent, G. L. Olivier-Lilley and B. A. Averill, Chem. Rev., 1990, 90, 1447–1467 CrossRef CAS;
(d) J. D. Lipscomb, Annu. Rev. Microbiol., 1994, 48, 371–399 CrossRef CAS PubMed;
(e) A. L. Feig and S. J. Lippard, Chem. Rev., 1994, 94, 759–805 CrossRef CAS;
(f) B. J. Wallar and J. D. Lipscomb, Chem. Rev., 1996, 96, 2625–2658 CrossRef CAS PubMed;
(g) M. Costas, K. Chen and L. Que, Coord. Chem. Rev., 2000, 200–202, 517–544 CrossRef CAS;
(h) M. H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem. Rev., 2003, 103, 2385–2419 CrossRef CAS PubMed.
-
(a) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed;
(b) E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef CAS PubMed;
(c) S. V. Kryatov, E. V. Rybak-Akimova and S. Schindler, Chem. Rev., 2005, 105, 2175–2226 CrossRef CAS PubMed;
(d) L. Que Jr, Acc. Chem. Res., 2007, 40, 493–500 CrossRef PubMed;
(e) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS;
(f) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS;
(g) K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73–96 CrossRef CAS;
(h) M. Sallmann and C. Limberg, Acc. Chem. Res., 2015, 48, 2734–2743 CrossRef CAS PubMed;
(i) A. J. Jasniewski and L. Que Jr, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed;
(j) O. Y. Lyakin, K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2019, 384, 126–139 CrossRef CAS.
- D. Nianios, S. Thierbach, L. Steimer, P. Lulchev, D. Klostermeier and S. Fetzner, BMC Biochem., 2015, 16, 10 CrossRef PubMed.
- J. Kaizer, É. Balogh-Hergovich, M. Czaun, T. Csay and G. Speier, Coord. Chem. Rev., 2006, 250, 2222–2233 CrossRef CAS.
- J. S. Pap, J. Kaizer and G. Speier, Coord. Chem. Rev., 2010, 254, 781–793 CrossRef CAS.
- J. Kaizer, G. Baráth, J. Pap, G. Speier, M. Giorgi and M. Réglier, Chem. Commun., 2007, 5235–5237, 10.1039/b711864c.
- G. Baráth, J. Kaizer, G. Speier, L. Párkányi, E. Kuzmann and A. Vértes, Chem. Commun., 2009, 3630–3632, 10.1039/b903224j.
-
(a) A. Nishinaga, N. Numada and K. Maruyama, Tetrahedron Lett., 1989, 30, 2257–2258 CrossRef CAS;
(b) A. Nishinaga, T. Tojo and T. Matsurra, J. Chem. Soc., Chem. Commun., 1974, 952, 896–897 RSC.
-
(a) K. Grubel, K. Rudzka, A. M. Arif, K. L. Klotz, J. A. Halfen and L. M. Berreau, Inorg. Chem., 2010, 49, 82–96 CrossRef CAS;
(b) K. Grubel, A. R. Marts, S. M. Greer, D. L. Tierney, C. J. Allpress, S. N. Anderson, B. J. Laughlin, R. C. Smith, A. M. Arif and L. M. Berreau, Eur. J. Inorg. Chem., 2012, 2012, 4750–4757 CrossRef CAS.
-
(a) Y. J. Sun, Q. Q. Huang, T. Tano and S. Itoh, Inorg. Chem., 2013, 52, 10936–10948 CrossRef CAS;
(b) Y. J. Sun, Q. Q. Huang, P. Li and J. J. Zhang, Dalton Trans., 2015, 44, 13926–13938 RSC.
- A. Matuz, M. Giorgi, G. Speier and J. Kaizer, Polyhedron, 2013, 63, 41–49 CrossRef CAS.
- S. Hoof and C. Limberg, Inorg. Chem., 2019, 58, 12843–12853 CrossRef CAS PubMed.
- Y. Dai, T. C. Pochapsky and R. H. Abeles, Biochemistry, 2001, 40, 6379–6387 CrossRef CAS PubMed.
-
(a) S. W. Ragsdale, J. Biol. Chem., 2009, 284, 18571–18575 CrossRef CAS PubMed;
(b) J. L. Boer, S. B. Mulrooney and R. P. Hausinger, Arch. Biochem. Biophys., 2014, 544, 142–152 CrossRef CAS PubMed;
(c) M. Sankaralingam, M. Balamurugan and M. Palaniandavar, Coord. Chem. Rev., 2020, 403, 213085 CrossRef.
- B. Gopal, L. L. Madan, S. F. Betz and A. A. Kossiakoff, Biochemistry, 2005, 44, 193–201 CrossRef CAS PubMed.
- J. H. Jeoung, D. Nianios, S. Fetzner and H. Dobbek, Angew. Chem., Int. Ed., 2016, 55, 3281–3284 CrossRef CAS PubMed.
- Y. J. Sun, Q. Q. Huang and J. J. Zhang, Dalton Trans., 2014, 43, 6480–6489 RSC.
- P. Holze, T. Corona, N. Frank, B. Braun-Cula, C. Herwig, A. Company and C. Limberg, Angew. Chem., Int. Ed., 2017, 56, 2307–2311 CrossRef CAS.
- S. Hoof and C. Limberg, Z. Anorg. Allg. Chem., 2019, 645, 170–174 CrossRef CAS.
- W. J. Wang, W. J. Wei and R. Z. Liao, Phys. Chem. Chem. Phys., 2018, 20, 15784–15794 RSC.
- H. Li, X. Wang, G. Tian and Y. Liu, Catal. Sci. Technol., 2018, 8, 2340–2351 RSC.
- D. Jeong, S. Sun, D. Moon and J. Cho, J. Inorg. Biochem., 2022, 226, 111632 CrossRef CAS PubMed.
-
(a) M. Balamurugan, E. Suresh and M. Palaniandavar, Dalton Trans., 2016, 45, 11422–11436 RSC;
(b) M. Sankaralingam and M. Palaniandavar, Polyhedron, 2014, 67, 171–180 CrossRef CAS;
(c) R. Viswanathan, M. Palaniandavar, P. Prabakaran and P. T. Muthiah, Inorg. Chem., 1998, 37, 3881–3884 CrossRef CAS PubMed.
- M. Balamurugan, R. Mayilmurugan, E. Suresh and M. Palaniandavar, Dalton Trans., 2011, 40, 9413–9424 RSC.
-
(a) M. Sankaralingam, P. Vadivelu, E. Suresh and M. Palaniandavar, Inorg. Chim. Acta, 2013, 407, 98–107 CrossRef CAS;
(b) M. Sankaralingam, M. Balamurugan, M. Palaniandavar, P. Vadivelu and C. H. Suresh, Chem.–Eur. J., 2014, 20, 11346–11361 CrossRef CAS PubMed;
(c) M. Sankaralingam, P. Vadivelu and M. Palaniandavar, Dalton Trans., 2017, 46, 7181–7193 RSC.
-
(a) G. J. Britovsek, J. England and A. J. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed;
(b) A. G. Blackman, Eur. J. Inorg. Chem., 2008, 2008, 2633–2647 CrossRef.
- V. McKee, M. Zvagulis, J. V. Dagdigian, M. G. Patch and C. A. Reed, J. Am. Chem. Soc., 1984, 106, 4765–4772 CrossRef CAS.
- M. Inosako, A. Kunishita, M. Kubo, T. Ogura, H. Sugimoto and S. Itoh, Dalton Trans., 2009, 9410–9417, 10.1039/b910237j.
- J.-W. Wang, H.-H. Huang, J.-K. Sun, D.-C. Zhong and T.-B. Lu, ACS Catal, 2018, 8, 7612–7620 CrossRef CAS.
- G. M. Sheldrick, Acta. Crystallogr. A, 2015, 71, 3–8 CrossRef PubMed.
- M. N. Burnett and C. K. Johnson, ORTEP-III: Oak Ridge thermal ellipsoid plot program for crystal structure illustrations, Oak ridge national laboratory report ORNL-6895, Tennessee, 1996 Search PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009, 121, pp. 150–166–.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
-
(a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS;
(b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- M. Velusamy, R. Mayilmurugan and M. Palaniandavar, Inorg. Chem., 2004, 43, 6284–6293 CrossRef CAS PubMed.
-
(a) L. K. Thompson, B. S. Ramaswamy and R. D. Dawe, Can. J. Chem., 1978, 56, 1311–1318 CrossRef CAS;
(b) J.-L. Tian, M.-J. Xie, Z.-Q. Liu, S.-P. Yan, D.-Z. Liao and Z.-H. Jiang, J. Coord. Chem., 2005, 58, 833–840 CrossRef CAS.
- É. Balogh-Hergovich, J. Kaizer, J. Pap, G. Speier, G. Huttner and L. Zsolnai, Eur. J. Inorg. Chem., 2002, 2002, 2287–2295 CrossRef.
- M. Velusamy, M. Palaniandavar and K. R. Justin Thomas, Polyhedron, 1998, 17, 2179–2186 CrossRef CAS.
- L. Barhács, J. Kaizer and G. Speier, J. Org. Chem., 2000, 65, 3449–3452 CrossRef PubMed.
- H. Wu, K. Li, T. Sun, B. Liu, F. Jia, J. Yuan and G. Chen, Z. Naturforsch. B, 2010, 65, 1341–1348 CrossRef CAS.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa prediction for organic acids and bases, Springer, 1981 Search PubMed.
- T. Ajaykamal, M. Sharma, N. S. Islam and M. Palaniandavar, Dalton Trans., 2021, 50, 8045–8056 RSC.
-
(a) T. Kusamoto, S. Kume and H. Nishihara, J. Am. Chem. Soc., 2008, 130, 13844–13845 CrossRef CAS PubMed;
(b) Y. Wang, H. Zhang, M. Pink, A. Olankitwanit, S. Rajca and A. Rajca, J. Am. Chem. Soc., 2016, 138, 7298–7304 CrossRef CAS PubMed;
(c) A. Kumar and M. D. Sevilla, J. Phys. Chem. B, 2018, 122, 98–105 CrossRef CAS PubMed;
(d) S. Kasemthaveechok, L. Abella, M. Jean, M. Cordier, T. Roisnel, N. Vanthuyne, T. Guizouarn, O. Cador, J. Autschbach, J. Crassous and L. Favereau, J. Am. Chem. Soc., 2020, 142, 20409–20418 CrossRef CAS PubMed.
-
(a) H. Guo, Q. Peng, X. K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J. L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed;
(b) L. Abella, J. Crassous, L. Favereau and J. Autschbach, Chem. Mater., 2021, 33, 3678–3691 CrossRef CAS;
(c) Z. Wang, R. Murata and M. Abe, ACS Omega, 2021, 6, 22773–22779 CrossRef CAS PubMed.
- B. L. Westcott, N. E. Gruhn, L. J. Michelsen and D. L. Lichtenberger, J. Am. Chem. Soc., 2000, 122, 8083–8084 CrossRef CAS.
- N. Saravanan, T. Ajaykamal and M. Palaniandavar, Communicated, 2023 Search PubMed.
- L. Jurd and T. A. Geissman, J. Org. Chem., 1956, 21, 1395–1401 CrossRef CAS.
-
(a) A. P. Lever, Studies in physical theoretical chem, 1984, p. 33 Search PubMed;
(b) M. S. Kryatova, O. V. Makhlynets, A. Y. Nazarenko and E. V. Rybak-Akimova, Inorg. Chim. Acta, 2012, 387, 74–80 CrossRef CAS;
(c) T. R. Holman, M. P. Hendrich and L. Que, Inorg. Chem., 1992, 31, 937–939 CrossRef CAS.
- D. W. S. Westlake, J. M. Roxburgh and G. Talbot, Nature, 1961, 189, 510–511 CrossRef CAS.
- B. H. Bielski, G. G. Shiue and S. Bajuk, J. Phys. Chem., 1980, 84, 830–833 CrossRef CAS.
- R. Jangir, M. Ansari, D. Kaleeswaran, G. Rajaraman, M. Palaniandavar and R. Murugavel, ACS Catal., 2019, 9, 10940–10950 CrossRef CAS.
- N. Podder, S. Dey, A. Anoop and S. Mandal, Dalton Trans., 2022, 51, 4338–4353 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Mallayan Palaniandavar
and
Mallayan Palaniandavar