
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, in silico studies, and biological evaluation of novel pyrimidine-5-carbonitrile derivatives as potential anti-proliferative agents, VEGFR-2 inhibitors and apoptotic inducers†
Abdulrahman M. Saleh ab,
Hazem A. Mahdy*a,
Mohamed Ayman El-Zahabia,
Ahmed B. M. Mehanyb,
Mohamed M. Khalifa*a and
Ibrahim H. Eissa*a
ab,
Hazem A. Mahdy*a,
Mohamed Ayman El-Zahabia,
Ahmed B. M. Mehanyb,
Mohamed M. Khalifa*a and
Ibrahim H. Eissa*a
aPharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt. E-mail: Ibrahimeissa@azhar.edu.eg; Hazem_hady2001@azhar.edu.eg; MohamedKhalifa2321.el@azhar.edu.eg
bZoology Department, Faculty of Science (Boys), Al-Azhar University, Cairo 11884, Egypt
First published on 24th July 2023
Abstract
A novel series of pyrimidine-5-carbonitrile derivatives bearing benzylidene and hydrazone moieties with different linkers (spacers) were designed and synthesized as possible inhibitors of the vascular endothelial growth factor receptor-2 (VEGFR-2). The newly synthesized compounds were evaluated in vitro for their cytotoxic activities against two human cancer cell lines namely colon cancer (HCT-116) and breast cancer (MCF-7) using sorafenib as a standard anticancer drug. Compounds 9d, 11e, 12b, and 12d showed higher cytotoxic activities than sorafenib with IC50 values ranging from 1.14 to 10.33 μM. In particular, compound 11e exhibited excellent activities against HCT-116 and MCF-7 with IC50 values of 1.14 and 1.54 μM, respectively. Moreover, compound 11e exhibited about 47.32-fold cytotoxic activity against normal human fibroblast (WI-38) cells, lower than the cytotoxicity against the cancer cells. Compounds 11e and 12b were the most potent VEGFR-2 inhibitors with IC50 values of 0.61 and 0.53 μM, respectively, compared to sorafenib. Bedsides, compound 11e arrested the HCT-116 cell growth at S and sub-G1 phases, induced a significant increase in the apoptotic cells, and caused remarkable decrease in the levels of TNF-α, IL-6, and caspase-3. Finally, the binding patterns of the target derivatives were investigated through the docking study against the proposed molecular target (VEGFR-2, PDB ID 1YWN). The results of molecular docking studies showed similar binding modes to sorafenib against VEGFR-2. In addition, molecular dynamic simulations revealed the stability of compound 11e in the active site for 100 ns.
1. Introduction
Cancer, a disease characterized by uncontrollable cell growth, is still one of the most serious diseases to threaten human life.1 Although there are wide advances dealing with its biochemistry and trying to understand its progression, the complete cure of cancer has not been achieved yet.2–4 The increased incidence of cancer cases returns primarily to the resistance of the cancer cells to the known medications, the reason that opens the door to research to either find new targets or to construct new chemical scaffolds to act on the previously identified targets.Vascular Endothelial Growth Factor (VEGF) has been nominated as one of the most prevalent tumor angiogenesis mediators. VEGF initiates endothelial cell activation and proliferation.5 However, it promotes vascular permeability in solid tumors.6 These processes are all mediated via stimulation of a specific type of tyrosine kinase receptors, VEGFR-2 receptor.7 However, VEGF/VEGFR-2 pathway contributes to the progress of numerous cancer types including breast, colon, gastric and lung via migration, metastasis and stimulation of angiogenesis.8 Therefor hindering the VEGF/VEGFR-2 pathway appears crucial in stopping angiogenesis.9 Accordingly, VEGFR-2 gained an increasing importance in the anti-angiogenic therapy against cancer. Unfortunately, treatment with the currently FDA approved VEGFR-2 inhibitors faced several drawbacks represented in their serious adverse reactions and development of a secondary resistance.10 There drawbacks motivated the medicinal chemists to continue searching for new small molecules aiming to overcome the adverse effects and minimize resistance.11–14
Observing the structure–activity relationship of the FDA approved VEGFR-2 inhibitors revealed that they almost shared four common features namely, (a) a heterocyclic head with minimum one nitrogen atom to occupy the hinge region of the active site, (b) a spacer to occupy gatekeeper region, (c) a hydrogen bonding moiety or a pharmacophore that makes essential H-bonds with the DFG amino acids, and (d) a hydrophobic tail that fills the receptor's allosteric site15–20 (Fig. 1A). However, Fig. 1B illustrated the role of sorafenib in inhibiting the VEGFR-2-mediated angiogenesis effect.
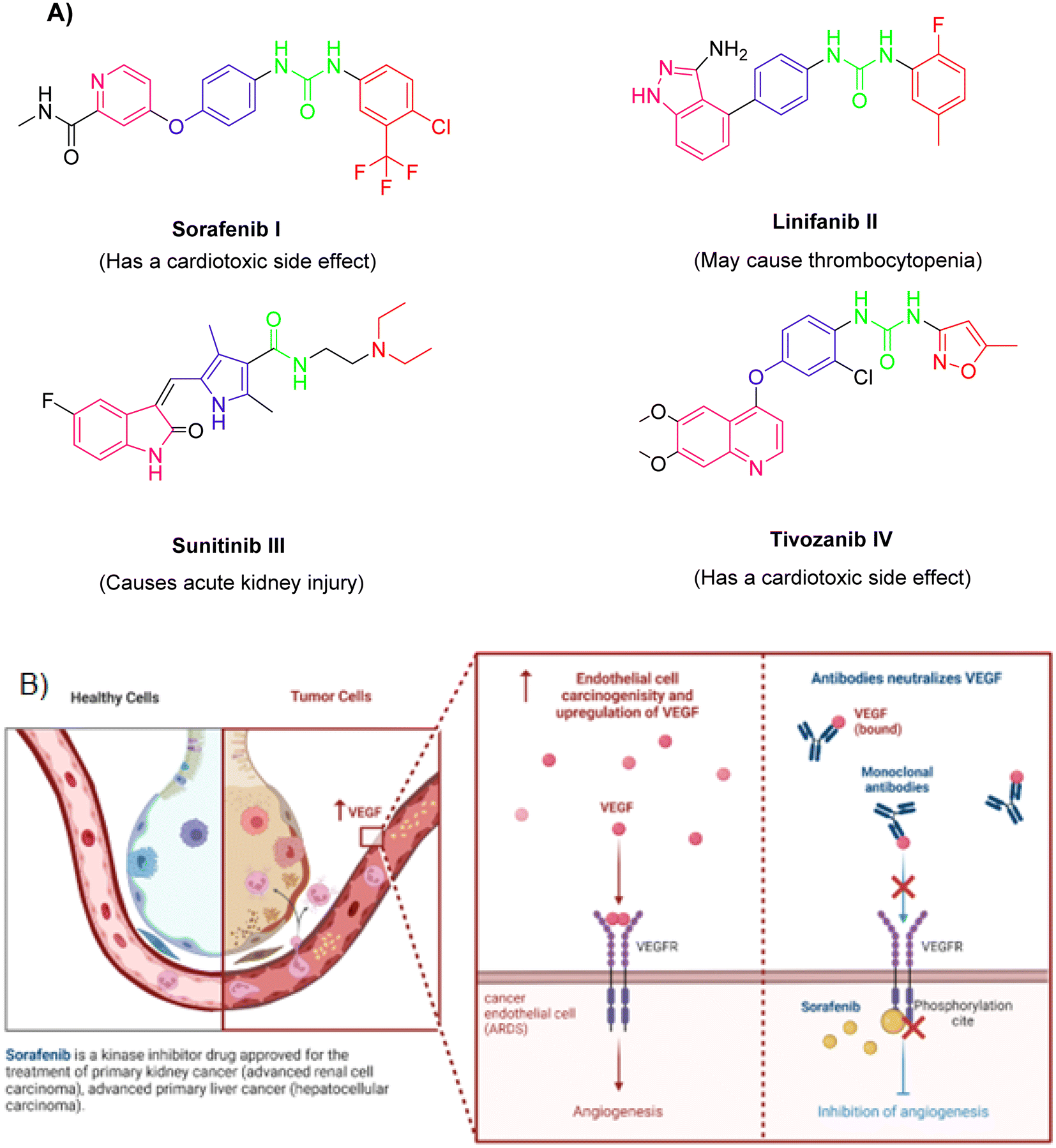 | ||
| Fig. 1 (A) Some reported VEGFR-2 inhibitors and their necessary four parts. (B) Function of VEGFR-2 and effects of sorafenib. | ||
In light of the abovementioned facts, we were encouraged to build a new scaffold constructed from the four key reported VEGFR-2 inhibitors features. The rationale of our molecular design depended basically on the bioisosteric modification of the well-defined VEGFR-2 inhibitors at their four main parts. However, the 6-aryl-5-cyano-thiouracil moiety was used in the current as a heterocyclic head. The choice of such a moiety was suitable as it contains a pyrimidine core that can fill the large size space of the ATP binding region. Besides, the pyrimidine's nitrogen atoms may serve as H-bond acceptors that may potentiate binding of the designed compounds with the VEGFR-2 receptor. Supporting with the structure of sorafenib I, the potent VEGFR-2 inhibitor, we have designed the spacer part of the target members to share the same length of that of sorafenib. The spacer part, hence, consisted of a four-carbon bridge attached to an NH group which are directly linked to heterocyclic head. The length of the designed linker was then convenient to occupy the gate keeper region in VEGFR-2 pocket (Fig. 2).
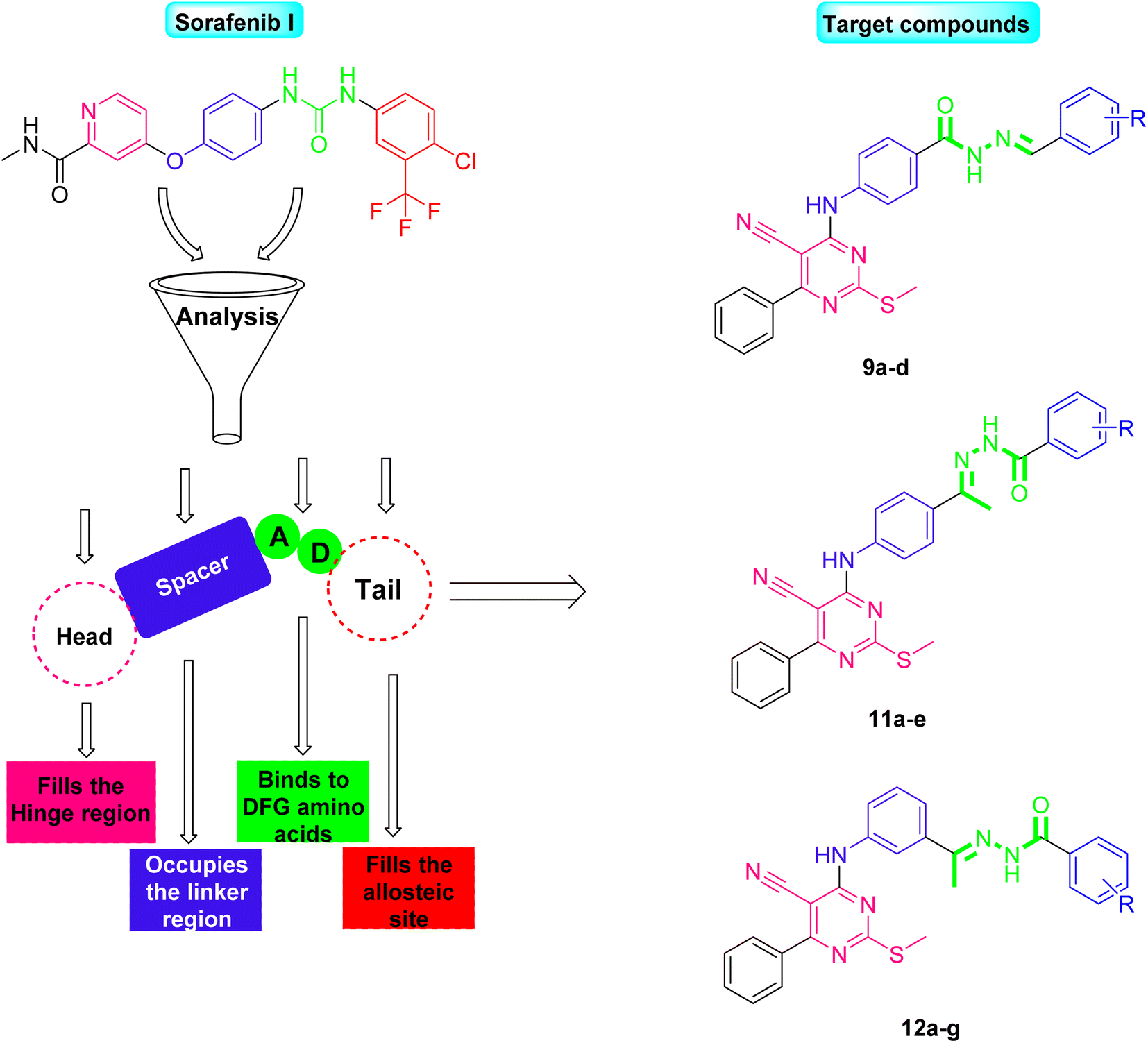 | ||
| Fig. 2 Rationale of molecular design for synthesis of novel VEGFR-2 inhibitors. | ||
Two different hydrophilic moieties were selected in the current study to play the role of the compounds' pharmacophore part. The choice of the moieties was based on their ability to act either as H-bond donor or acceptor. Thus, they could bind to the two essential receptor's amino acids, Glu883 and Asp1044. The final position to be modified was the terminal hydrophobic tail. Herein, we fixed the sorafenib phenyl hydrophobic tail with a change in its substitutions as we selected a wide variety of substitutions that enabled us to study the SAR of the designed compounds against VEGFR-2 tyrosine kinase receptor (Fig. 2).
2. Results and discussion
2.1. Chemistry
Chemical reactions for the synthesis of the target derivatives were depicted in Schemes 1–3. First, the starting material 2-mercapto-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitril 1 was obtained in a good yield by the cyclocondensation of benzaldehyde with ethyl cyanoacetate and thiourea in alkaline media according to the reported method.21–23 The derivative 1 was subjected to the alkylation with an equivalent amount of methyl iodide in 10% alcoholic KOH to afforded 2-(methylthio)-6-oxo-4-phenyl-1,6-dihydropyrimidine-5-carbonitrile 2. Thus, refluxing of compound 2 with phosphorus oxychloride afforded the corresponding the key intermediate 3. The latter 3 was refluxed in n-butanol with the commercially available aromatic amines namely, 4-aminobenzoic acid, 4-aminoacetophenone, and 3-aminoacetophenone to yield the corresponding target intermediates 4, 5, and 6, respectively (Scheme 1).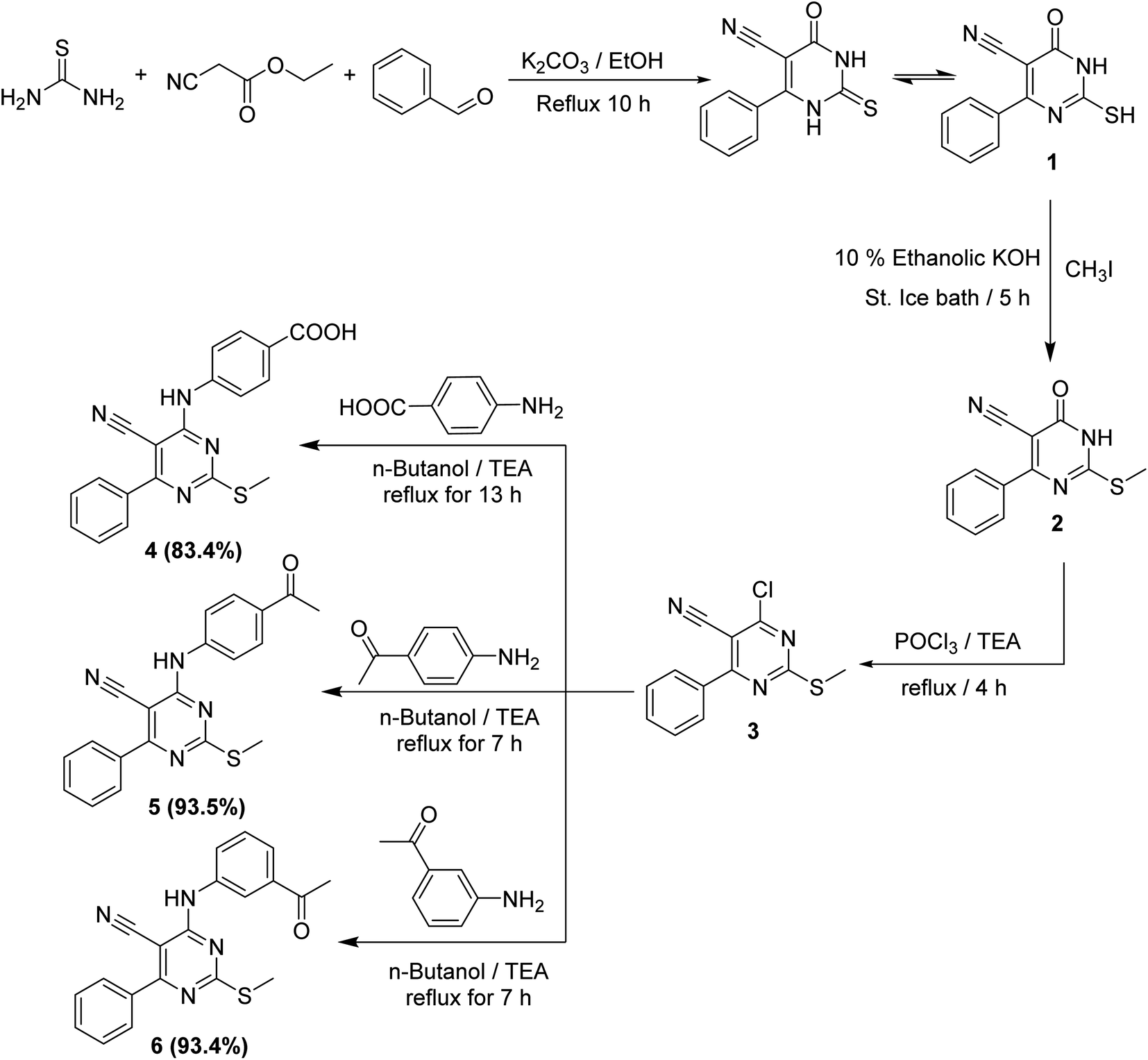 | ||
| Scheme 1 General procedure for synthesis of intermediate compounds 4, 5, and 6. | ||
The 1H NMR analyses of such intermediates (4, 5, and 6) showed three exchangeable signals at δ 10.12, 10.15, and 10.10 ppm corresponding to the NH protons, respectively. Besides, the 1H NMR spectra of 5 and 6 exhibited characteristic singlet signal for COCH3 group resonating at rang of δ 2.53–2.58 ppm.
In order to accomplish the target final compounds, the intermediate 4 was firstly activated by refluxing with thionyl chloride according to the reported method16 to achieve the acid chloride derivative 7 which was then utilized as freshly prepared without further purification to react with hydrazine hydrate in absolute ethanol to afford acid hydrazide analog 8. Such reaction was performed using an ice bath to avoid desulfurization which may take place through the attack hydrazine hydrate on the SCH3 group. The evidences for the formation of the acid hydrazide was verified by spectral data. The IR spectrum displayed strong bands at 3296, 3202 and 1658 cm−1 corresponding to NH, NH2 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, respectively. 1H NMR spectrum revealed appearance of a broad singlet signal at δ 4.72 ppm that refers to NH2 protons and another two singlet signals of two NH groups at δ 9.97 and 9.74 ppm. Then, the hydrazide 8 was allowed to react with appropriate aldehydes at refluxing ethanol temperature in the presence of a catalytic amount of glacial acetic acid to obtain the respective imine compounds or Schiff's bases 9a–d (Scheme 2).
O groups, respectively. 1H NMR spectrum revealed appearance of a broad singlet signal at δ 4.72 ppm that refers to NH2 protons and another two singlet signals of two NH groups at δ 9.97 and 9.74 ppm. Then, the hydrazide 8 was allowed to react with appropriate aldehydes at refluxing ethanol temperature in the presence of a catalytic amount of glacial acetic acid to obtain the respective imine compounds or Schiff's bases 9a–d (Scheme 2).
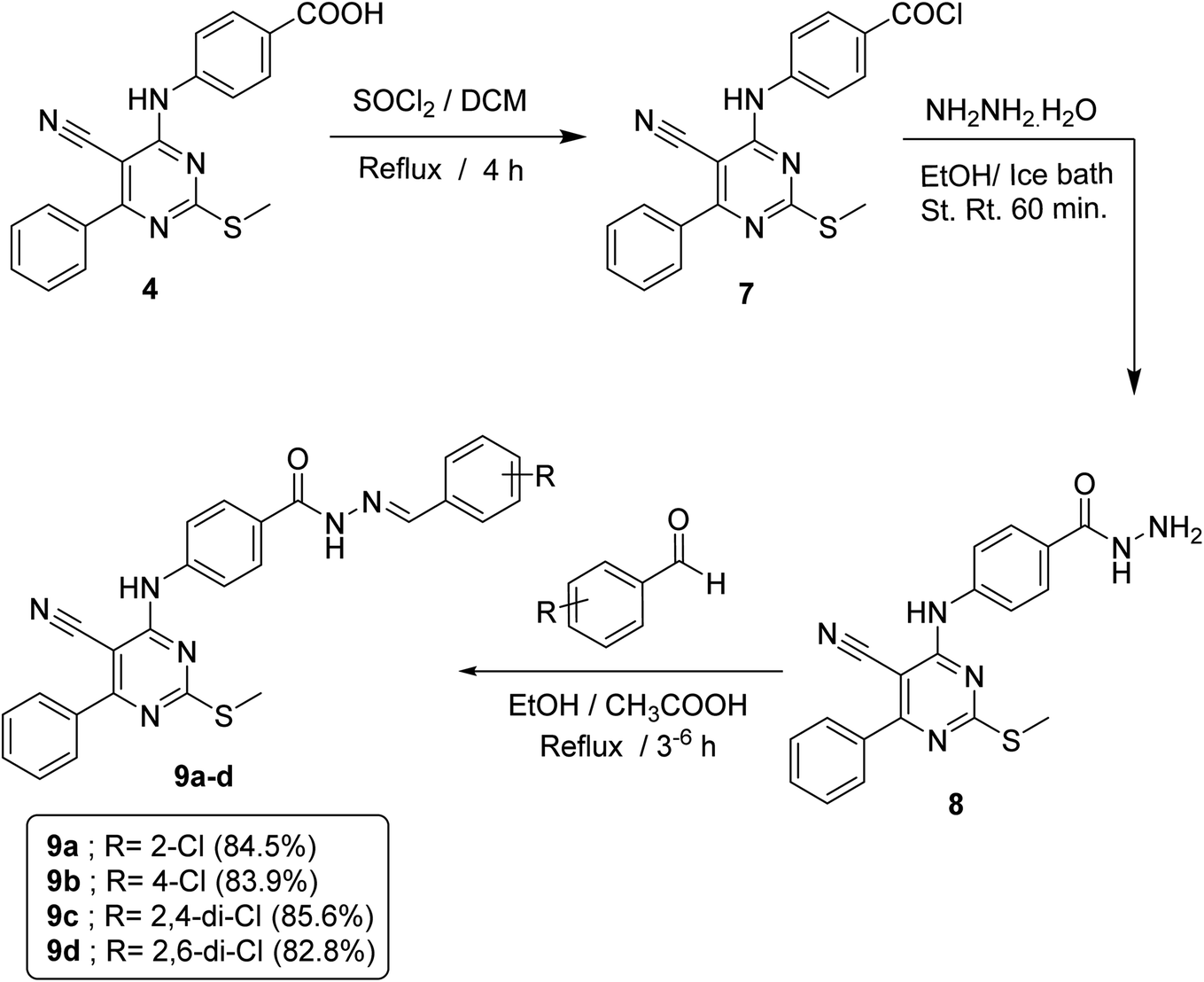 | ||
| Scheme 2 General procedure for synthesis of target compounds 9a–d. | ||
IR spectra of these compounds showed the disappearance of NH2 absorption band of the hydrazide compound 8. On the other hand, 1H NMR spectra of such compounds displayed a down-field singlet signal at a range of δ 8.84–8.00 ppm that refers to the new benzylidene proton, in addition to an increase in the aromatic protons by five, four or three protons according to the aldehyde used.
Condensation of the keton derivatives 5 and 6 with the appropriate acid hydrazides 10a–g (obtained according to the reported procedures)16,23,24 at heating and refluxing temperature in absolute ethanol containing catalytic amount of glacial acetic acid afforded the respective hydrazono compounds 11a–e and 12a–g, respectively (Scheme 3).
 | ||
| Scheme 3 General procedure for synthesis of target compounds 11a–e and 12a–g. | ||
IR spectra of 11a–e and 12a–g series confirmed presence of the carbonyl absorption band at a range of 1666–1637 cm−1 and NH bands at a range of 3341–3196 cm−1. On the other hand, the 1H NMR spectra showed up-field singlet signals of the aliphatic SCH3 and CH3 protons at δ range of 2.51–2.42 ppm and 2.44–2.20 ppm as well as the presence of NH protons at a δ range of 11.13–9.90 ppm. An important point was noticed regarding a couple of compounds, 11b and 12b, as the 1H NMR analyses revealed that they existed as E/Z isomers. This conclusion was reached through the investigation of signaling patterns of two methyl groups and two NH groups. For compound 11b, the two isomers appeared as about 60%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40%. While, for compound 12b, the two isomers appeared as about 65%:35%. Complete details for signaling ratio of each signal group were illustrated in ESI (Table S-1 and 1H NMR charts).† Moreover, the 13C NMR spectra of such series showed signals of the two methyl carbons at δ range of 19.20–13.77 ppm.
:40%. While, for compound 12b, the two isomers appeared as about 65%:35%. Complete details for signaling ratio of each signal group were illustrated in ESI (Table S-1 and 1H NMR charts).† Moreover, the 13C NMR spectra of such series showed signals of the two methyl carbons at δ range of 19.20–13.77 ppm.
2.2. Biological evaluation
| Comp. no. | In vitro cytotoxicity IC50a (μM) | VEGFR-2a (μM) | ||
|---|---|---|---|---|
| HCT 116 | MCF-7 | WI-38 | ||
| a Data are presented as mean ± S. D of the IC50 values from two different experiments.b NT: not tested. | ||||
| 9a | 30.15 ± 0.25 | 28.66 ± 0.2 | NTb | NTb |
| 9b | 12.34 ± 0.09 | 15.26 ± 0.12 | NTb | NTb |
| 9c | 20.62 ± 0.15 | 26.75 ± 0.24 | NTb | NTb |
| 9d | 7.14 ± 0.025 | 9.85 ± 0.035 | NTb | 2.41 ± 0.16 |
| 11a | 23.58 ± 0.18 | 32.16 ± 0.28 | NTb | NTb |
| 11b | 10.7 ± 0.05 | 13.48 ± 0.1 | NTb | 1.55 ± 0.25 |
| 11c | 10.45 ± 0.05 | 10.92 ± 0.05 | NTb | 1.38 ± 0.03 |
| 11d | 13.78 ± 0.11 | 14.31 ± 0.15 | NTb | 2.32 ± 0.21 |
| 11e | 1.14 ± 0.01 | 1.54 ± 0.01 | 63.41 ± 0.015 | 0.61 ± 0.01 |
| 12a | 17.98 ± 0.15 | 25.18 ± 0.19 | NTb | NTb |
| 12b | 8.65 ± 0.05 | 9.77 ± 0.06 | NTb | 0.53 ± 0.07 |
| 12c | 9.25 ± 0.07 | 10.17 ± 0.05 | NTb | 0.74 ± 0.15 |
| 12d | 7.15 ± 0.06 | 10.33 ± 0.05 | NTb | 1.61 ± 0.18 |
| 12e | 19.44 ± 0.14 | 30.14 ± 0.26 | NTb | NTb |
| 12f | 21.36 ± 0.15 | 26.11 ± 0.18 | NTb | NTb |
| 12g | 16.39 ± 0.12 | 21.72 ± 0.15 | NTb | NTb |
| Sorafenib | 8.96 ± 0.05 | 11.83 ± 0.07 | NTb | 0.19 ± 0.15 |
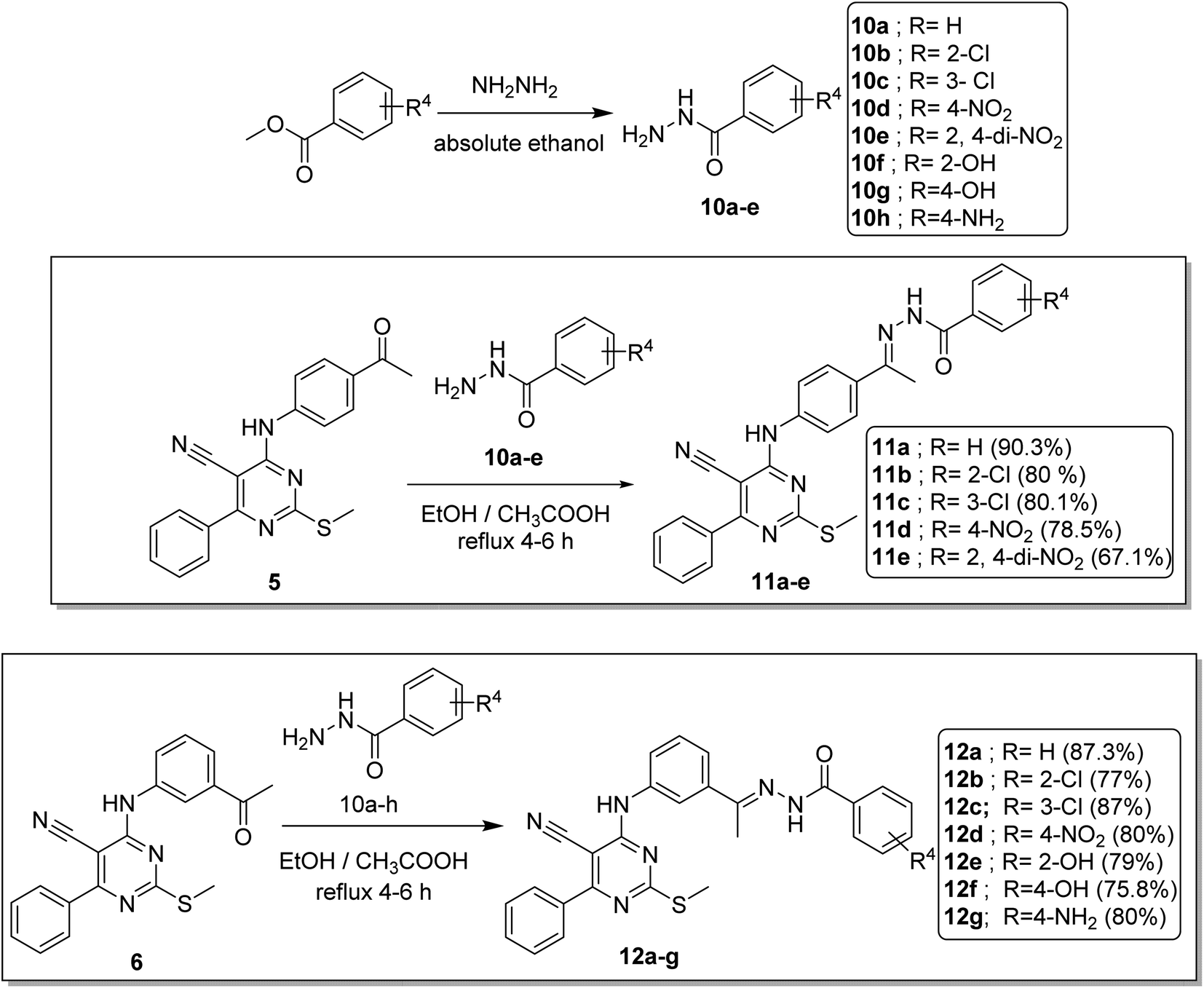
• Comparing the cytotoxicity of compounds containing m-disubstituted linkers (12d, 12b, 12a, and 12g) with their corresponding compounds of the p-disubstituted linkers (11d, 11b, and 11a), respectively, indicated that, the m-disubstituted derivatives are more active than the p-disubstituted ones (Fig. 3).
 | ||
| Fig. 3 SAR studies of 12a, 12d, and 12b compared to 11a, 11d, and 11b, respectively. | ||
With regard to the benzylidene derivatives (containing hydrazone moiety) 9a–d:
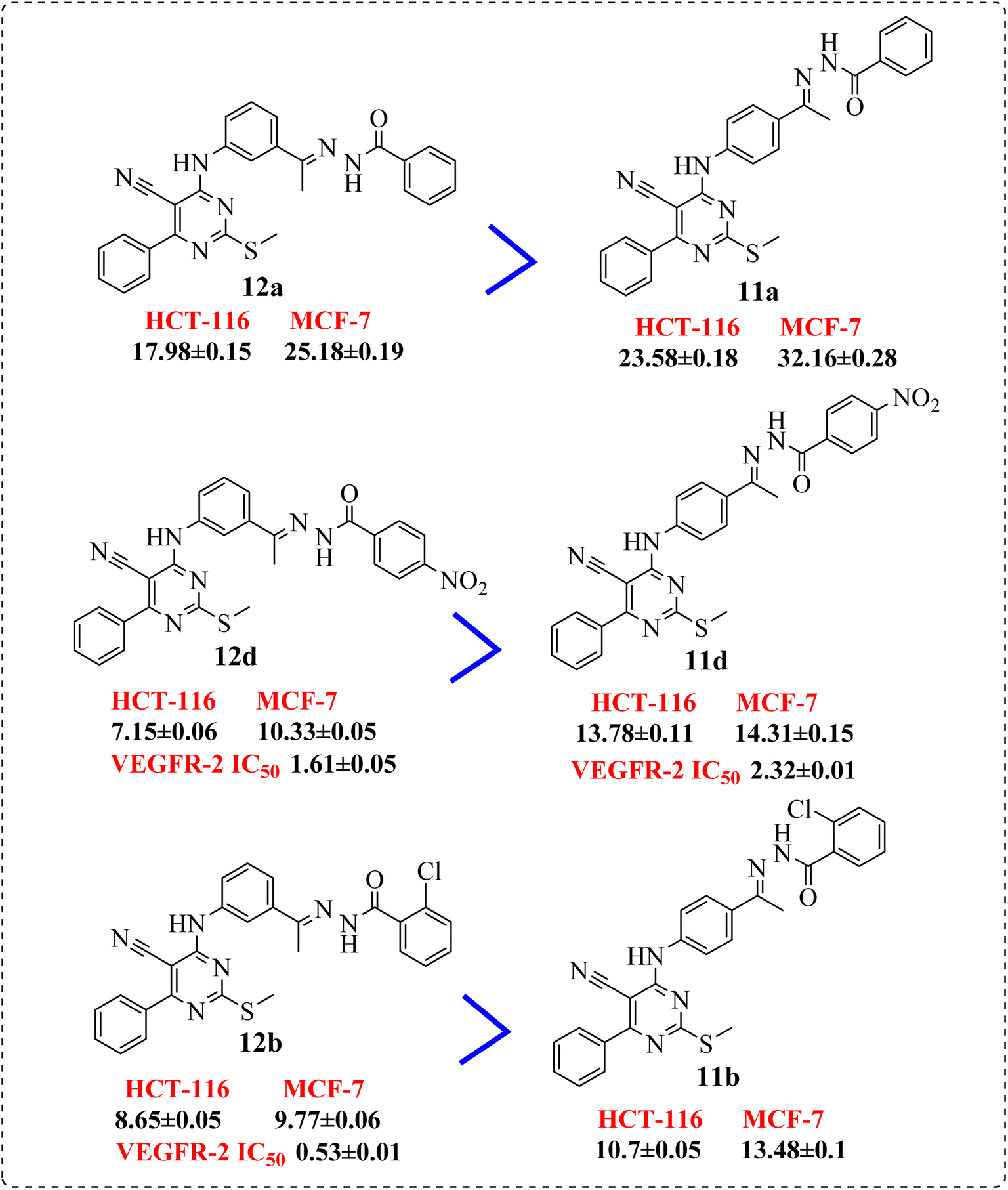
• The p-electron withdrawing substitution derivatives (9b) were more active than these with o-electron withdrawing ones (9a) (Fig. 4).
 | ||
| Fig. 4 SAR studies of 9b compared to 9a. | ||
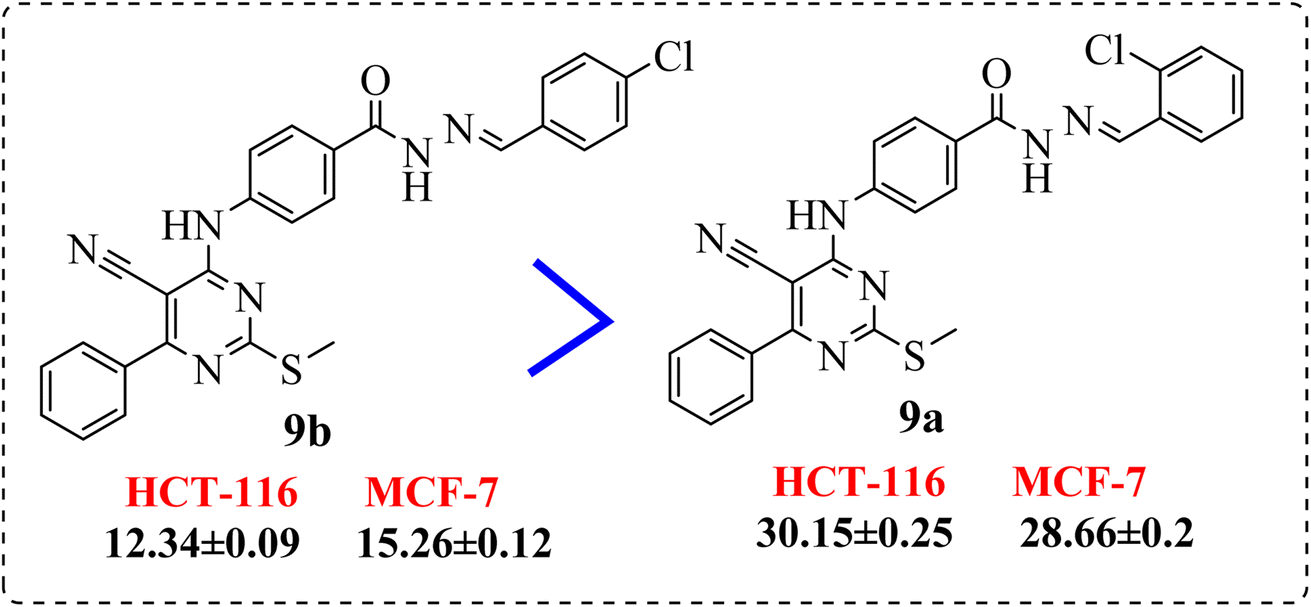
• Introduction of a chlorine atom at p-position exhibited preferable activity than o-position. Meanwhile, di-chloro substitution at 2,6-positions (9d) showed an activity stronger than the p-substituted one, but 2,4-dichloro derivative (9c), elicited lower activity than both (Fig. 5).
 | ||
| Fig. 5 SAR studies of 9d and 9c compared to 9b and 9a, respectively. | ||
• Compound 11e containing di-nitro substitution at 2,4-positions showed the highest cytotoxic activity on the both tested cell lines, moreover, shifting the nitro group from o-position showed a sever decreasing in cytotoxicity and VEGFR-2 TK inhibitory activity (Fig. 6).
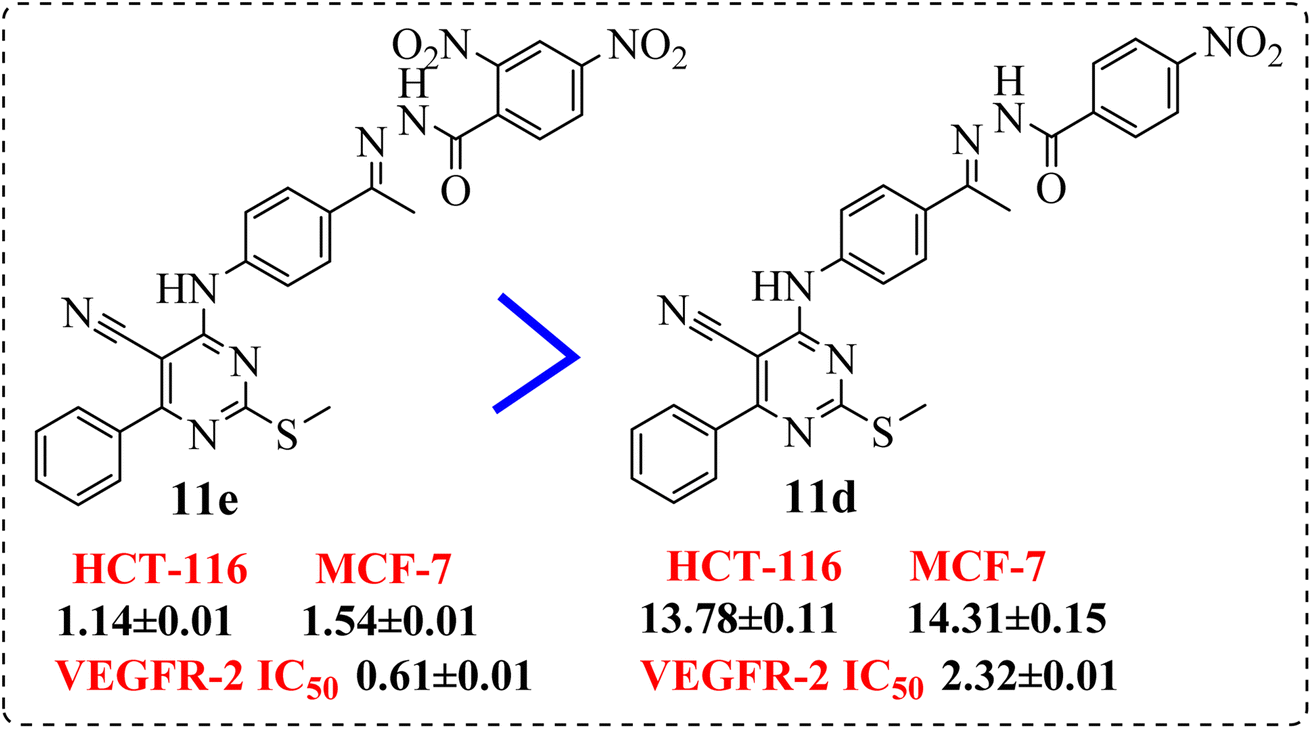 | ||
| Fig. 6 SAR studies of 11e compared to 11d. | ||
• Shifting the chloro group from m-position (11c) to o-position (11b) showed notable decrease in the cytotoxic activity (Fig. 7).
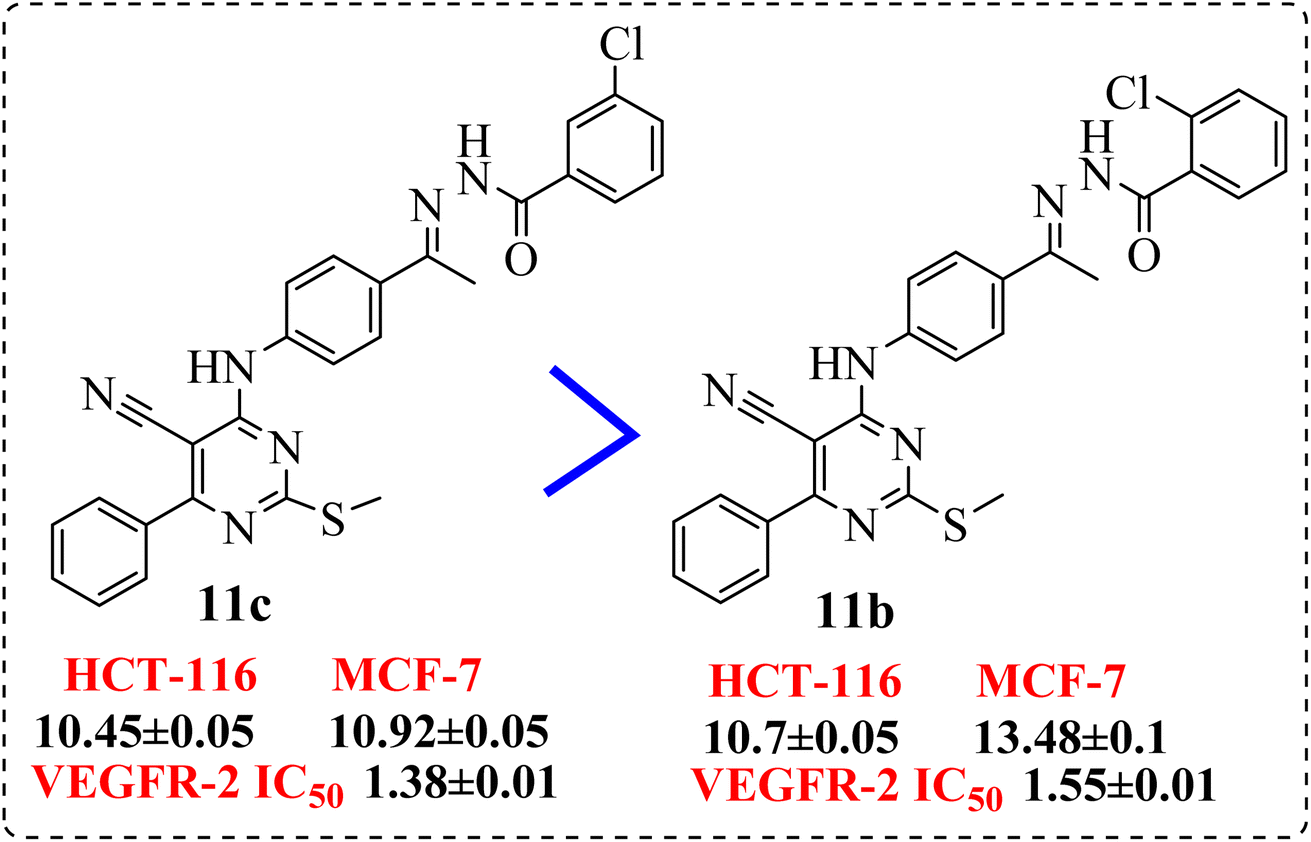 | ||
| Fig. 7 SAR studies of 11c compared to 11b. | ||
Regarding to the derivatives 12a–g containing m-disubstituted linkers, observation of the cytotoxic activity revealed that:
• Compounds 12b and 12d, containing terminal hydrophobic tail substituted with electron withdrawing groups with their corresponding derivatives with electron donating ones 12e and 12g, indicated the derivatives of electron withdrawing groups were more active than those donating one (Fig. 8).
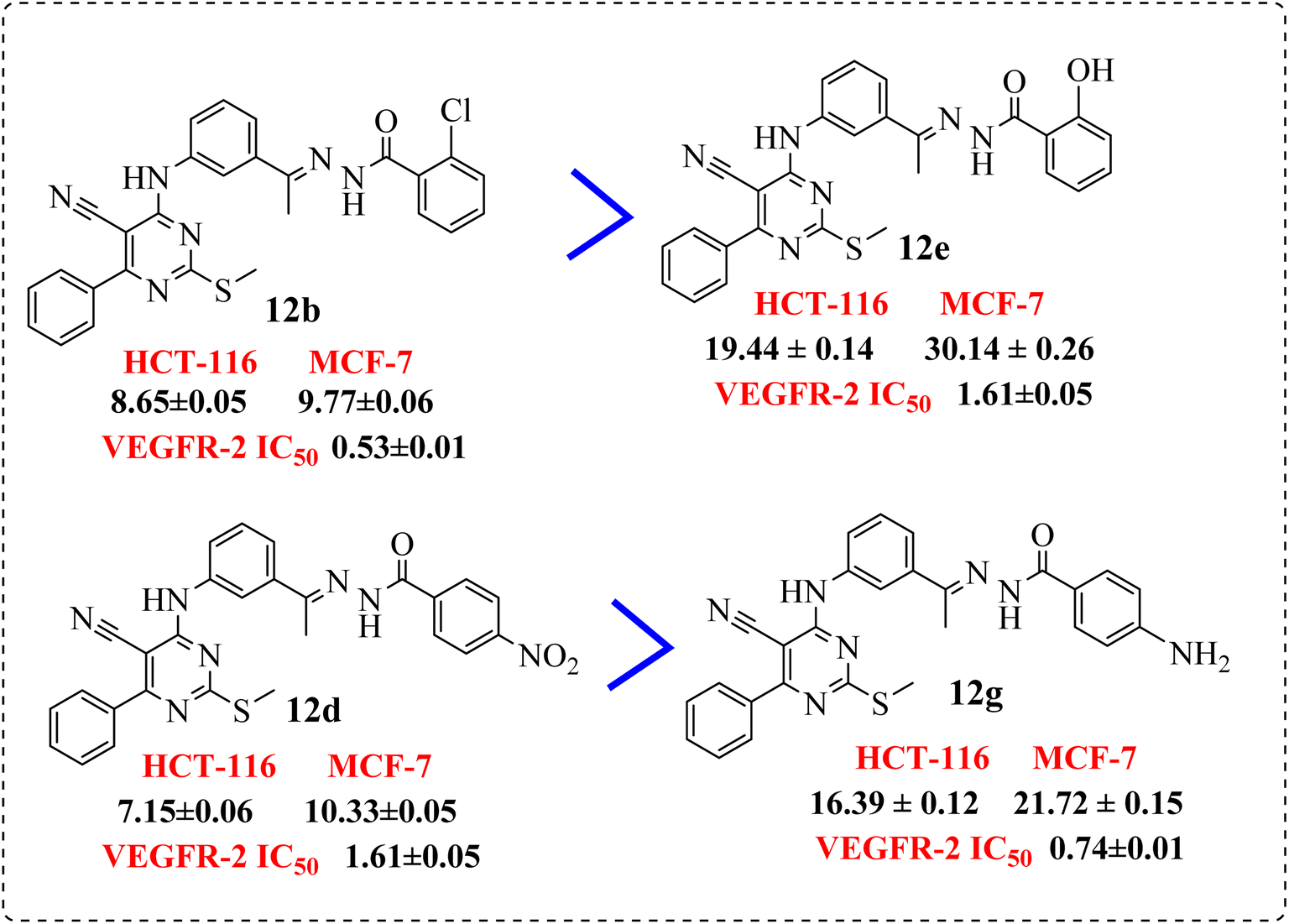 | ||
| Fig. 8 SAR studies of 12b and 12d compared to 12e and 12g, respectively. | ||
• Introduction of electron withdrawing group at o-position and m-position exhibited preferable activity than p-position (Fig. 9).
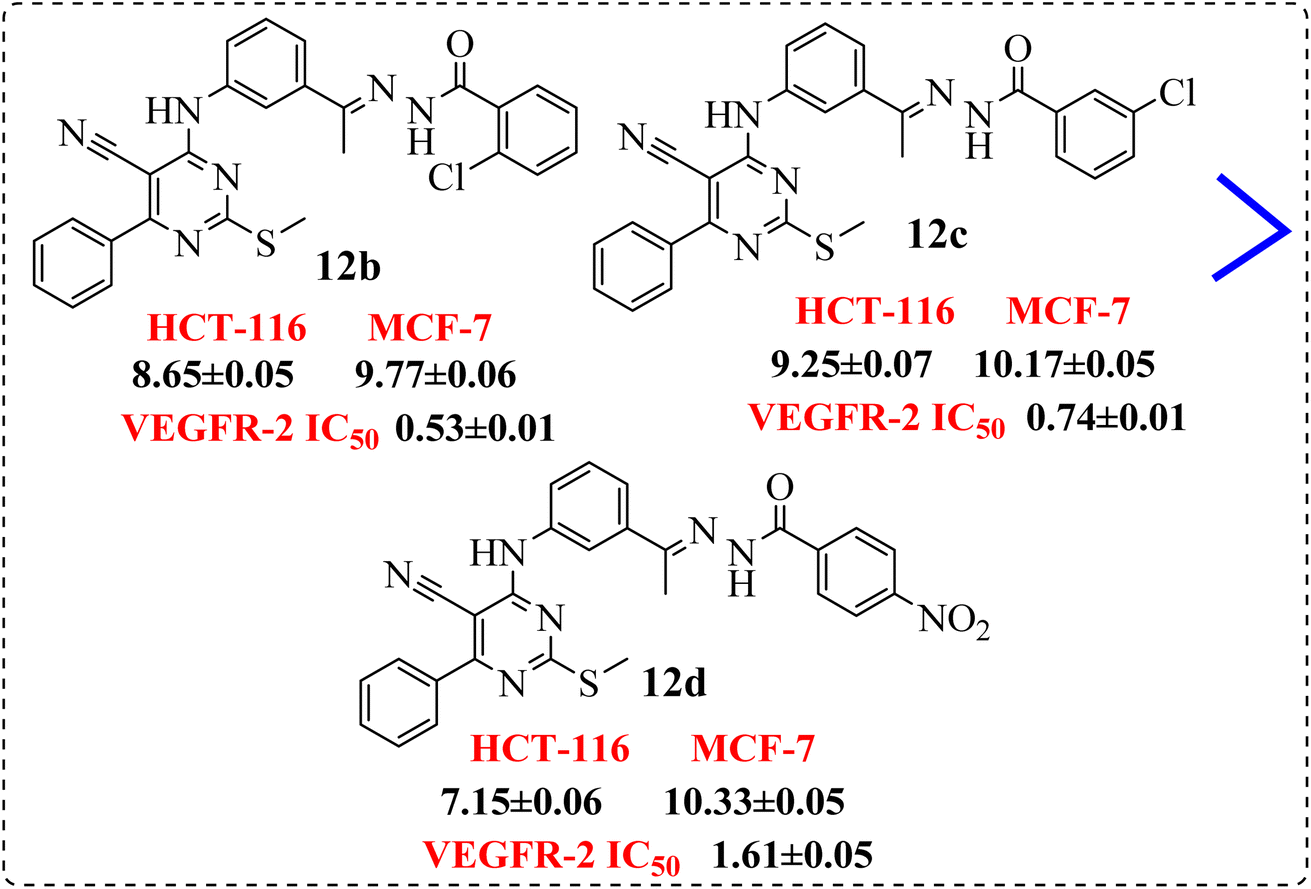 | ||
| Fig. 9 SAR studies of 12b and 12c compared to 12d. | ||
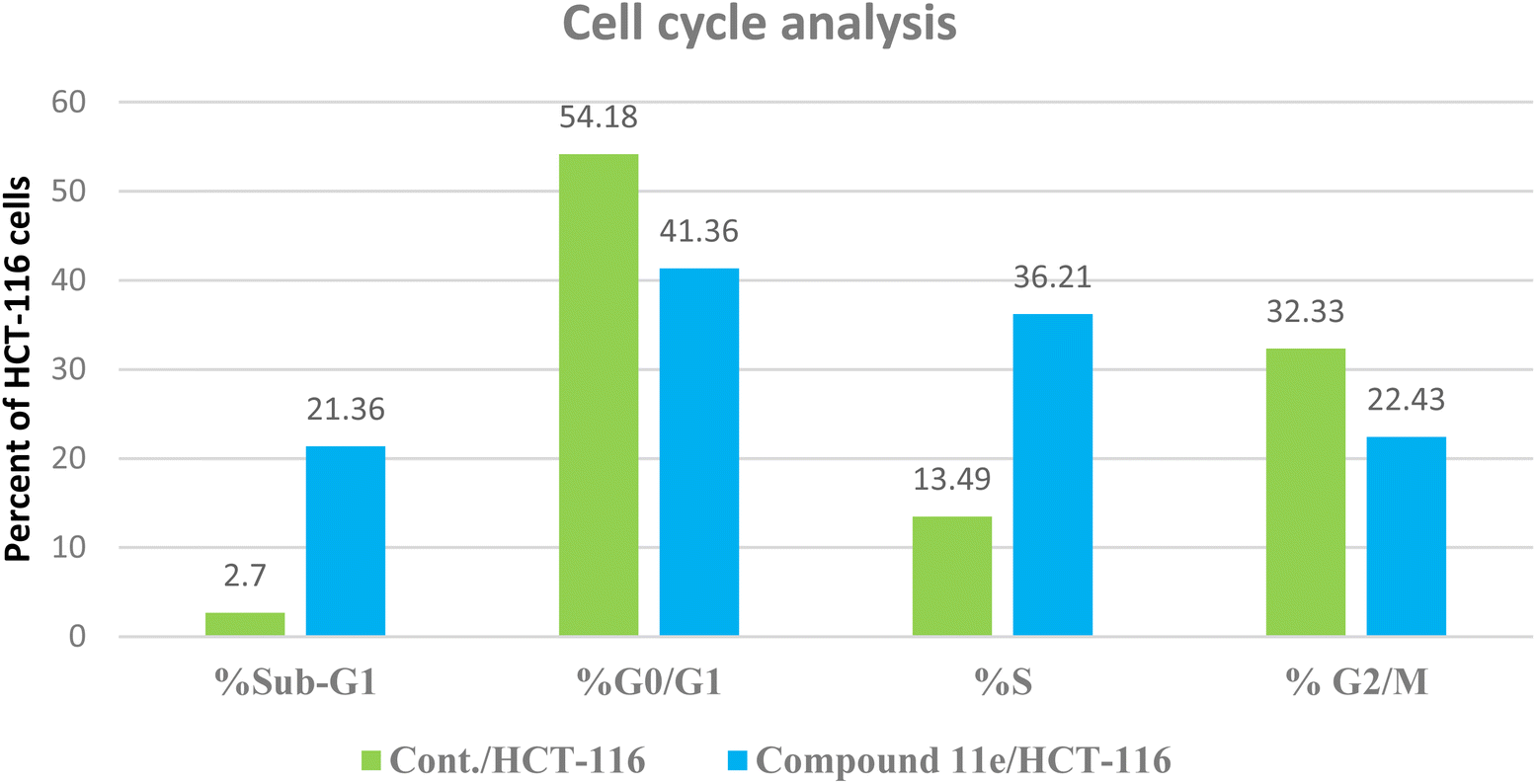 | ||
| Fig. 10 The representative histograms show the cell cycle distribution of control (HCT-116), and cells treated with compound 11e. | ||
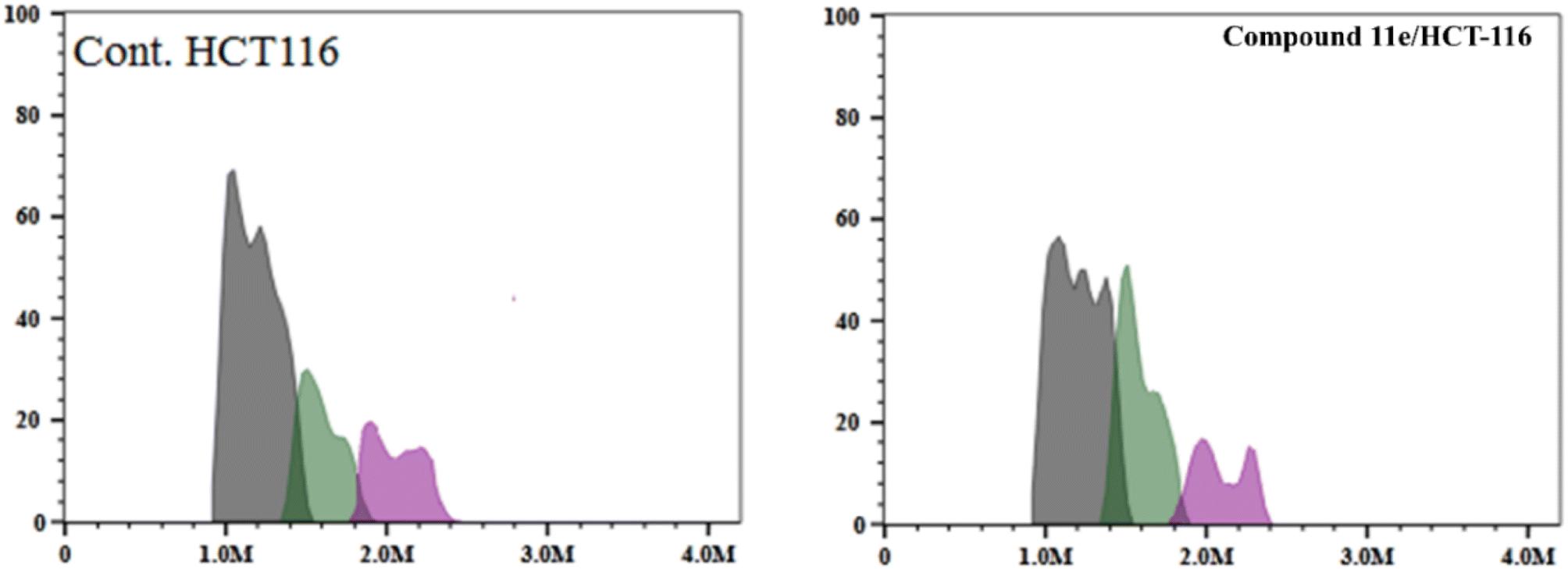 | ||
| Fig. 11 Flow cytometric analysis of cell cycle phases post the compound 11e treatment. | ||
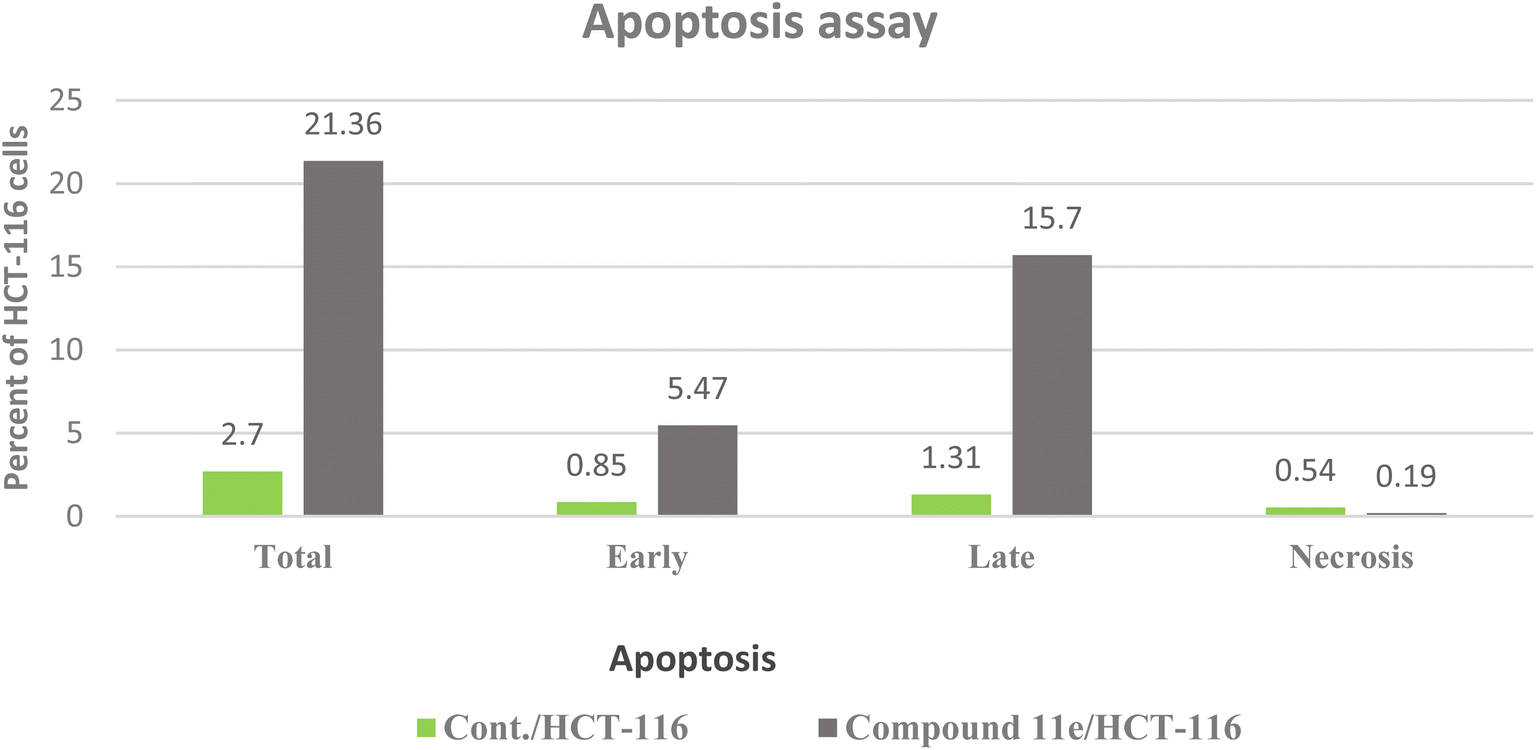 | ||
| Fig. 12 The representative flow cytometric charts for control (HCT-116) and the cells treated with compound 11e. | ||
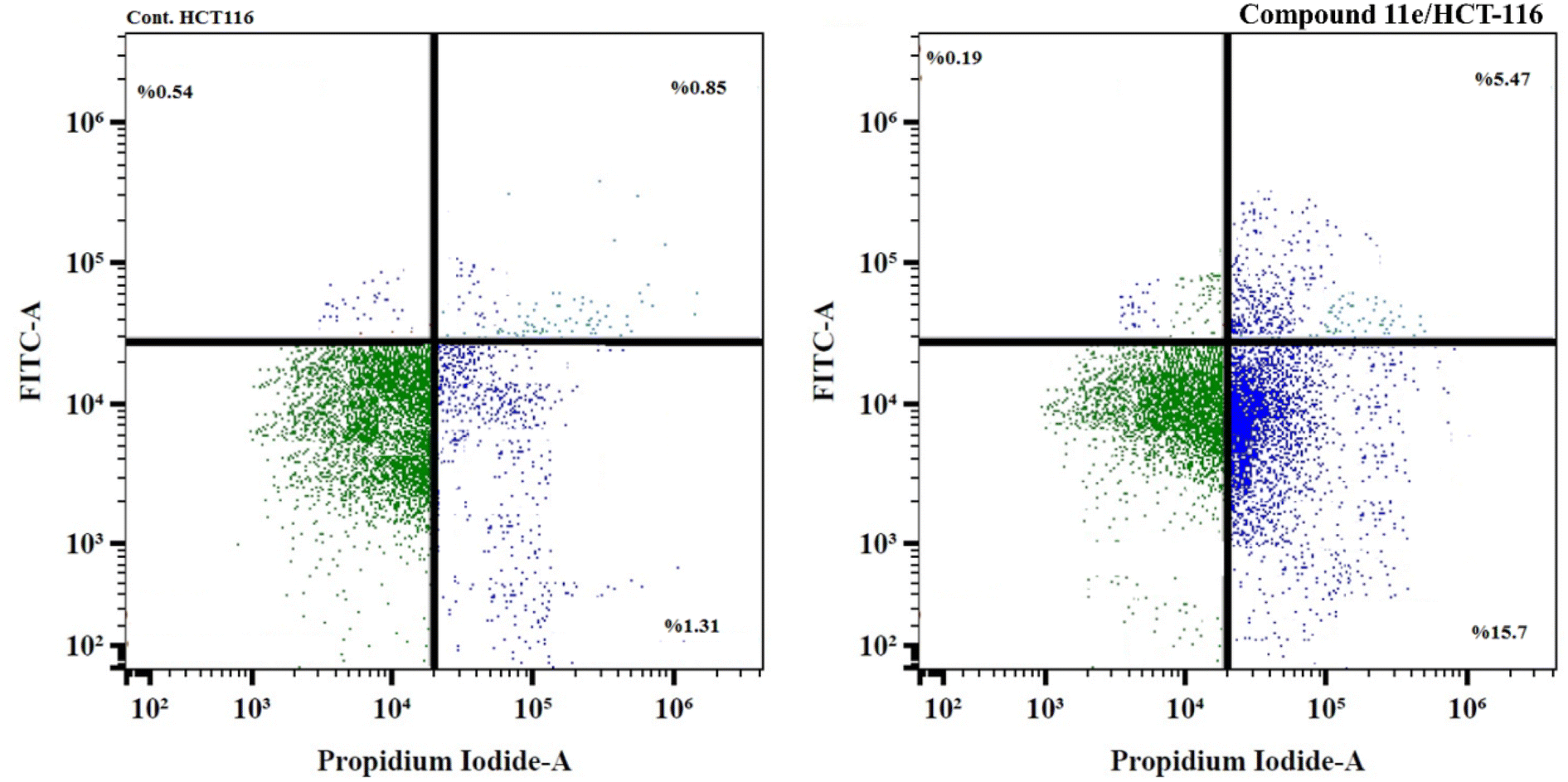 | ||
| Fig. 13 Flow cytometric analysis of apoptosis in HCT-116 cells exposed to compound 11e. For each dot plot chart, the quadrant regions represent the cells in each sub-population; lower left quadrant (viable cells), lower right quadrant (late apoptosis), upper right quadrant (early apoptosis), and upper left quadrant (necrosis). | ||
In these experiments, we calculated the changes in the expression levels of such proteins that resulted from the treatment of HCT-116 cells for 24 h with the selected molecule at their IC50. The data outlined in (Table 4) showed the significance of compound 11e as potent inhibitor for the pro-inflammatory factors TNF-α and IL-6, which played main role in immunomodulatory activity (Fig. 14). Compound 11e caused remarkable decrease in the levels of TNF-α and IL-6, with inhibition percent 70.22 pg per mL % for TNF-α & 81.39, 79.52 and 84.46 pg per mL % for IL-6, respectively.
| Sample | Pro-inflammatory factors inhibition (pg per mL %) | |
|---|---|---|
| TNF-α | IL-6 | |
| Dexamethasone | 82.47 | 93.15 |
| Compound 11e/HCT-116 | 70.22 | 84.46 |
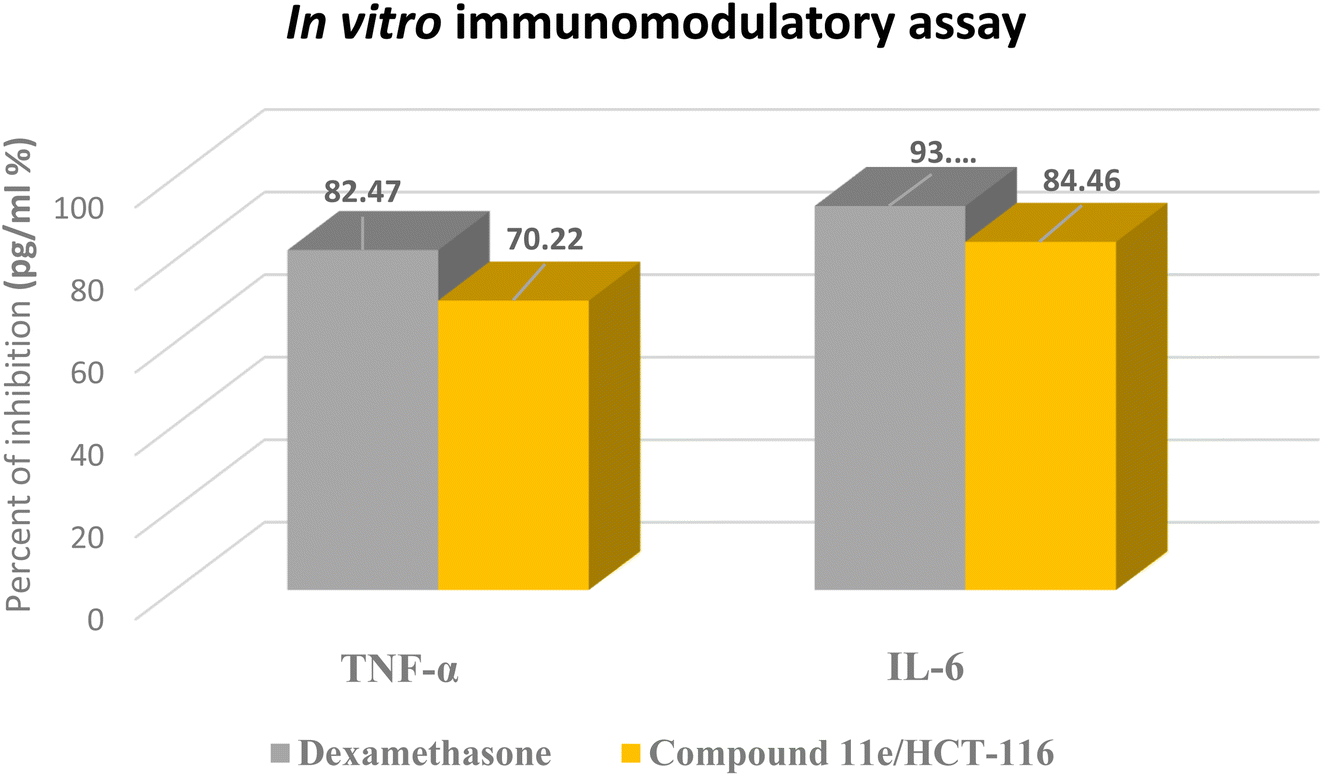 | ||
| Fig. 14 The representative in vitro immunomodulatory assay charts for dexamethasone as positive control and the tested compound 11e. | ||
| Sample | Apoptosis markers assay (pg mL−1) | |
|---|---|---|
| Caspase-3 | Fold | |
| Cont./HCT-116 | 56.48 | 1 |
| Compound 11e/HCT-116 | 304.67 | 5.4 |
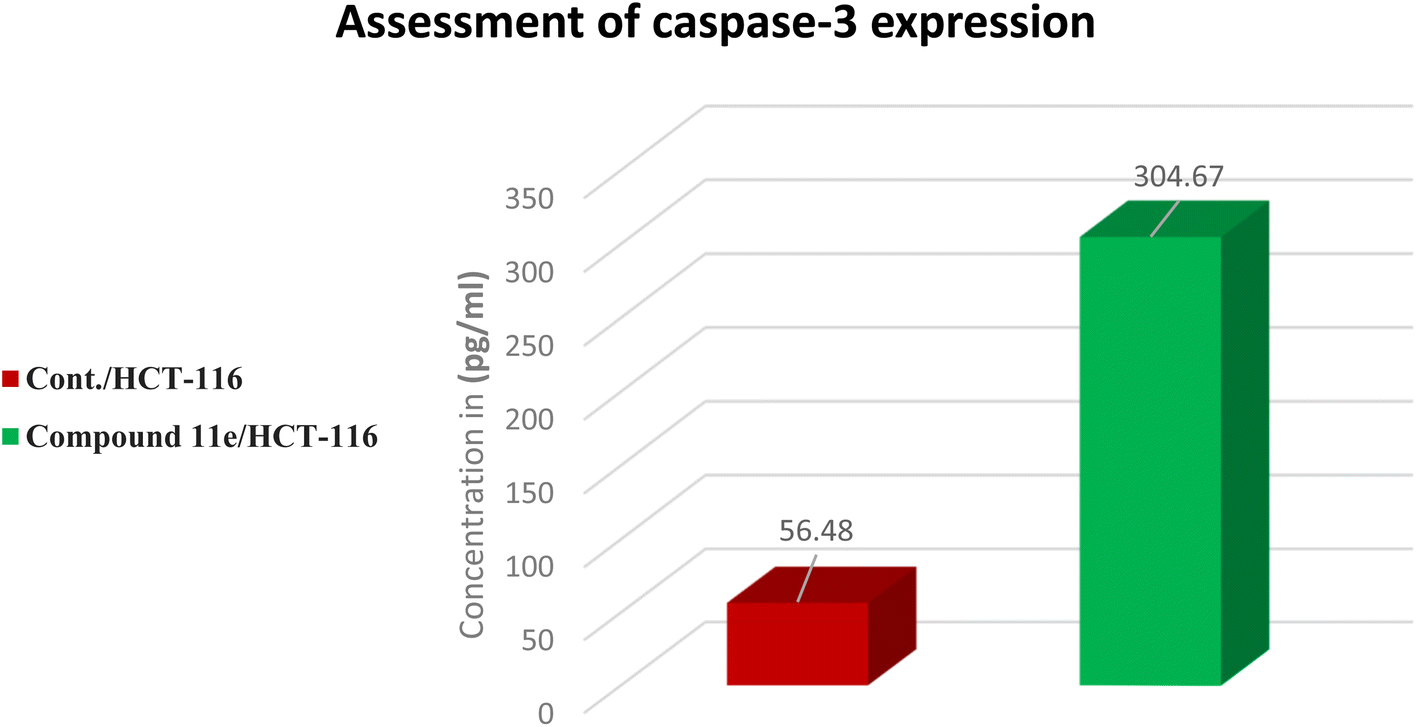 | ||
| Fig. 15 The representative in vitro caspase-3 expression chart for the tested compound 11e. | ||
2.3. In silico studies
| Comp. | ΔG [kcal mol−1] | Comp. | ΔG [kcal mol−1] |
|---|---|---|---|
| 9a | −8.70 | 12a | −8.95 |
| 9b | −8.23 | 12b | −9.17 |
| 9c | −8.95 | 12c | −8.99 |
| 9d | −8.85 | 12d | −9.01 |
| 11a | −9.32 | 12e | −9.34 |
| 11b | −8.82 | 12f | −8.60 |
| 11c | −9.06 | 12g | −8.90 |
| 11d | −9.30 | Sorafenib | −8.70 |
| 11e | −9.36 |
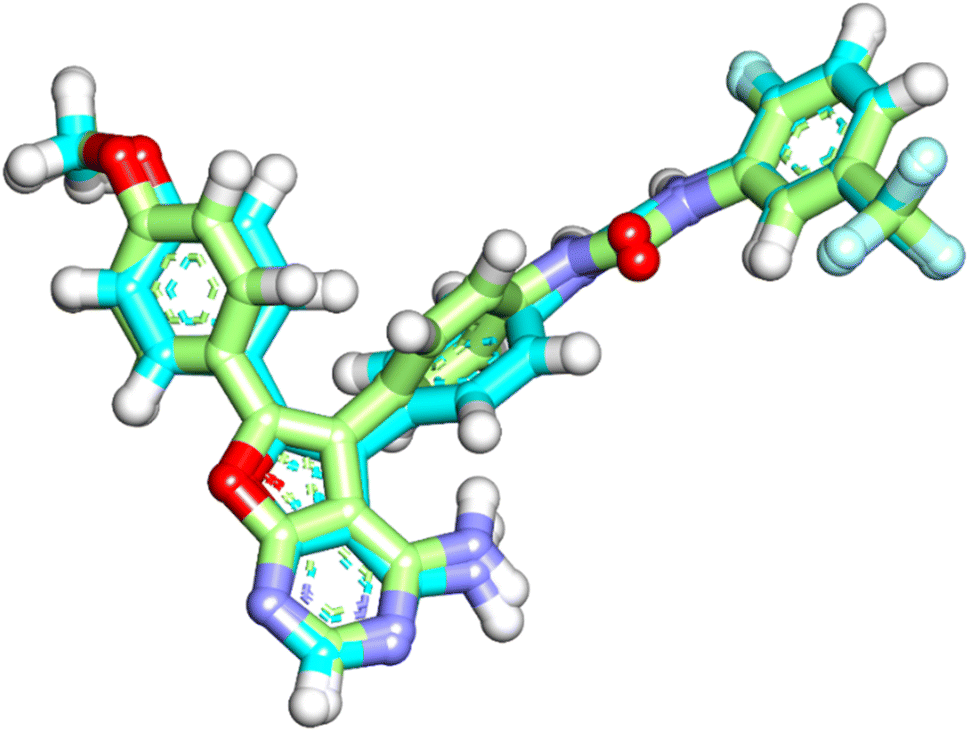 | ||
| Fig. 16 Superimposition of the native ligand (turquoise) and the re-docked one (green) in the VEGFR-2 TK active site (RMSD = 0.49 Å). | ||
The native ligand was exhibited an affinity score of −10.90 kcal mol−1 with RMSD value equal 0.49 Å (Fig. 17). It showed many important interactions with the residues at the active site of VEGFR-2 TK. The urea side chain possessed both hydrogen bond acceptor and donor, which bound with two crucial residues (Glu883 and Asp1044) of the receptor, where the NH motifs of the urea moiety usually formed two hydrogen bonds with the carboxylate of Glu883 with distance of 2.01 and 1.94 Å. Whereas the CO motif formed another hydrogen bond with the backbone NH of with Asp1044 with a distance of 2.05 Å at DFG region. The heterocyclic moiety showed two extra hydrogen bonds with Glu915 and Cys917. Besides, the phenyl group (spacer) interacted with the hydrophobic site (Val914, Ala864, Cys1043 and Val846) in the linker region by pi interactions. These results were found to be identical to the reported data.35
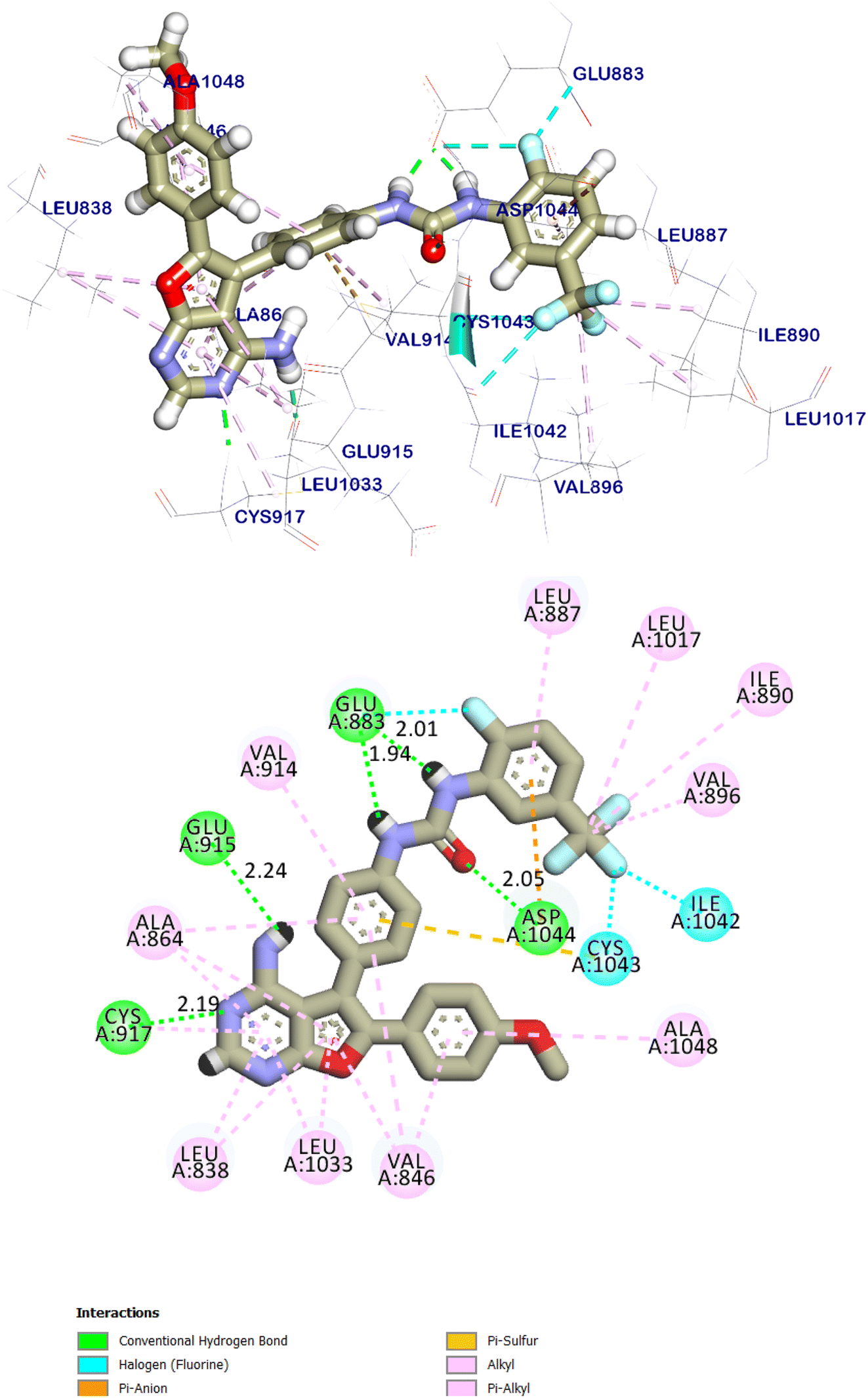 | ||
| Fig. 17 The predicted binding mode for the co-crystalized ligand with the active site of VEGFR-2 TK. | ||
Sorafenib interactions with the amino acids of the pocket have been studied and displayed in 2D and 3D style in (Fig. 18). The proposed binding mode of sorafenib, revealed an affinity value of −8.70 kcal mol−1. It demonstrated the important interactions with the residues at the active site of VEGFR-2 TK. The urea moiety possessed both hydrogen bond acceptor and donor that bound with two crucial residues (Glu883 and Asp1044) of the receptor, where the NH motifs of the urea moiety formed two hydrogen bonds with the carboxylate of Glu883 with distances of 2.02 and 1.98 Å. Whereas the CO motif formed another hydrogen bond with the backbone NH of with Asp1044 with a distance of 2.03 Å at DFG region. Besides, the phenyl group (spacer) interacted with the hydrophobic site (Cys917, Cys1043 and Phe916) in the linker region. Moreover, the distal hydrophobic moiety attached to the urea linker occupied the hydrophobic pocket formed by Cys1043, Val896, Asp1044, Ile886, Ile890, Ile1042, and Leu887. Furthermore, the N-methylpicolinamide moiety occupied the hinge region, it was formed one hydrogen bond with Cys917 with a distance of 2.79 Å and pi interactions with Ala864, Phe916, Leu838, Leu1033 and Cys917 (Fig. 18). The urea linker played an important role in the binding affinity towards VEGFR-2 TK, which was responsible for the higher binding affinity of sorafenib. These results were found to be identical to the reported data.36
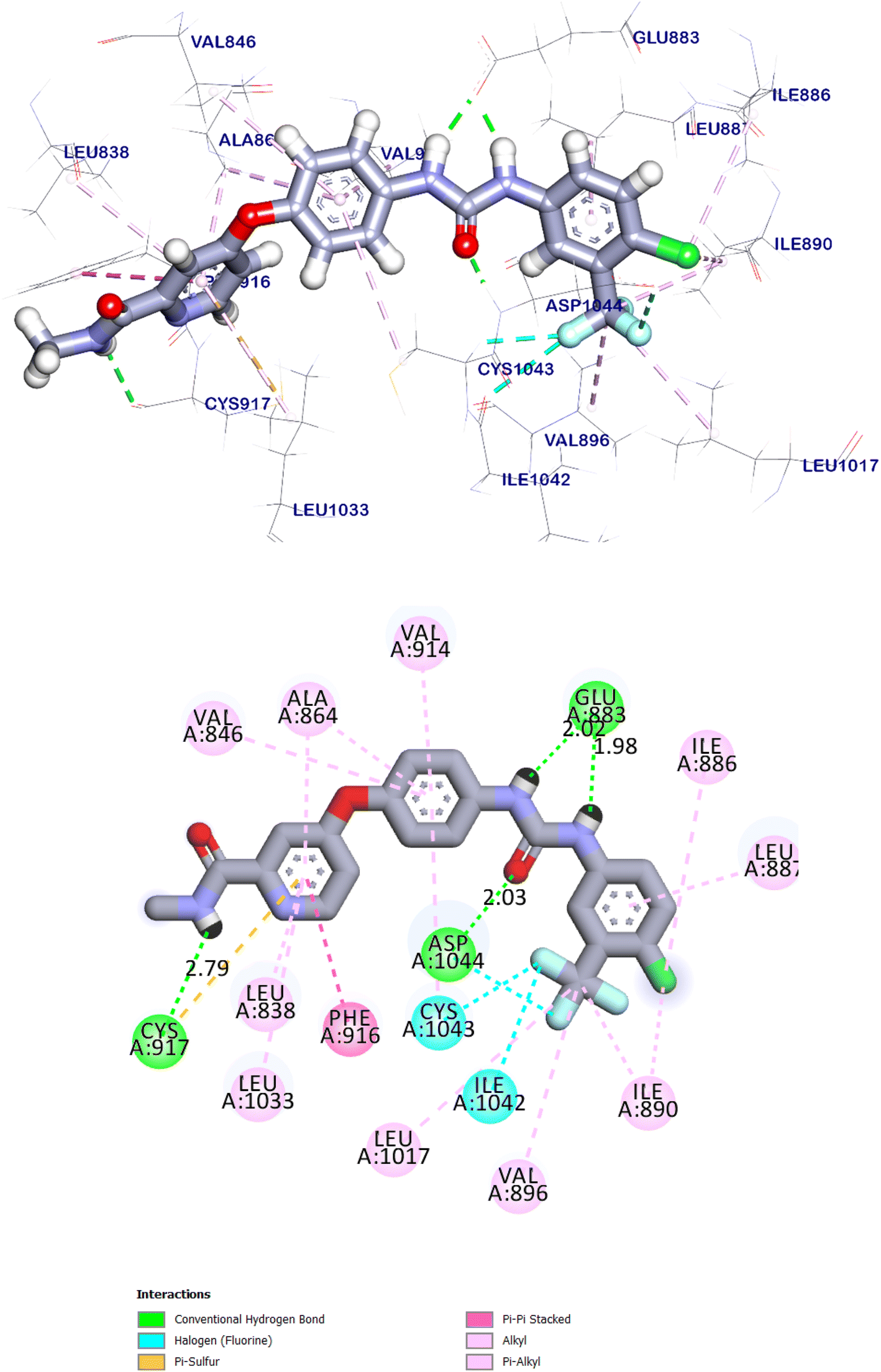 | ||
| Fig. 18 The predicted binding mode for sorafenib with the active site of VEGFR-2 TK. | ||
The docking score for compound 9d was −8.85 kcal mol−1. Through its pharmacophoric group, it formed two hydrogen bonds with Glu883 and Asp1044. The spacer moiety (4-aminophenyl ethylidene) interacted hydrophobically with Val914, and Cys1043. Many hydrophobic interactions in the hinge region occurred between the 5-cyano-2-(methylthio)-6-phenyl-pyrimidin moiety and different amino acid residues including Leu838, Val846, Gly1046, Phe843, and Ala1048. Furthermore, in the allosteric pocket, the 2,6-dichloro phenyl group formed numerous hydrophobic interactions with Val896, Val897, Ile890, Ile886, Phe1045 and Leu887. It also had a pi–anion interaction with Asp1044 (Fig. 19).
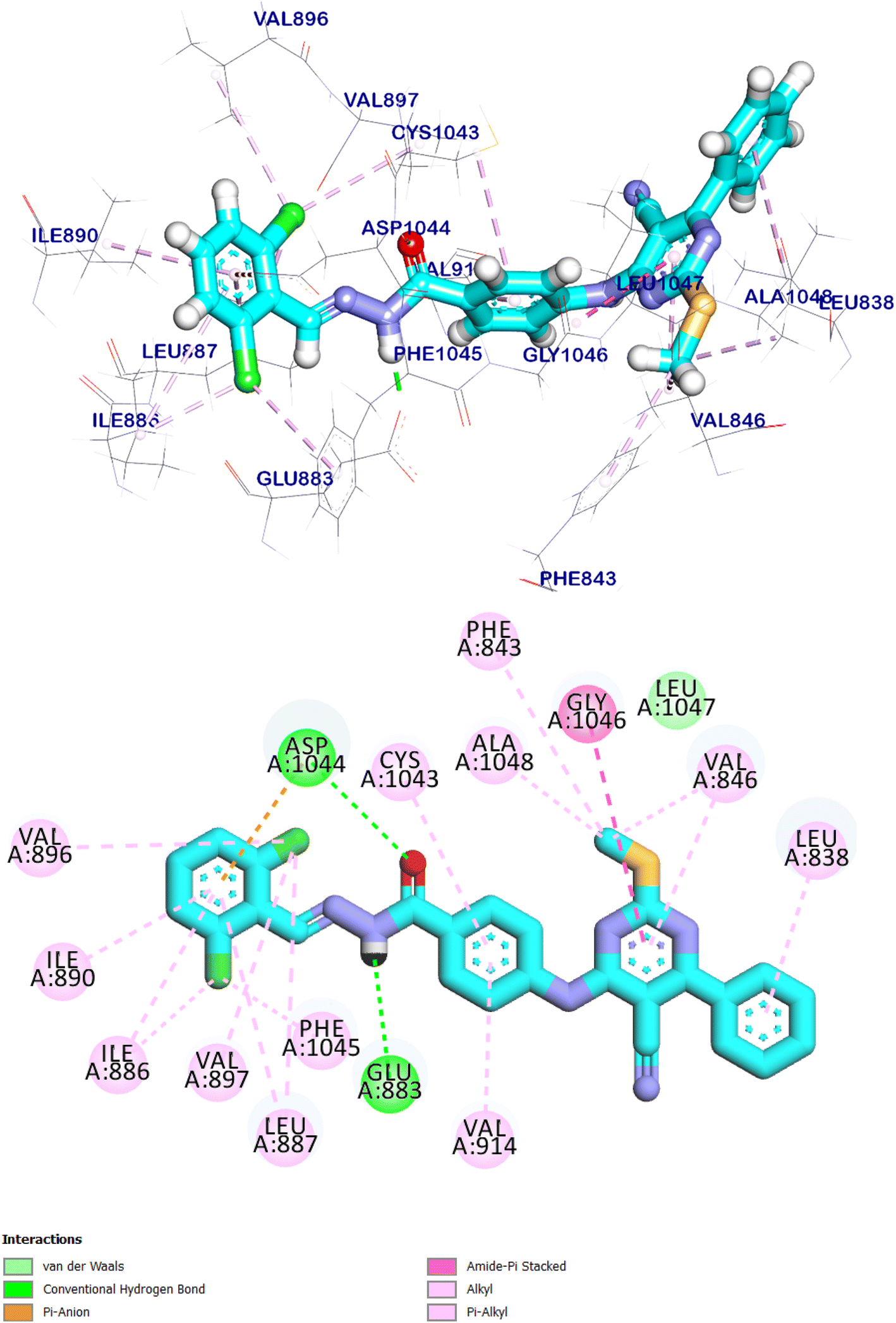 | ||
| Fig. 19 The predicted binding mode of compound 9d with the active site of VEGFR-2 TK. | ||
The docking findings of compound 11e revealed that it can interact with the essential amino acids in the VEGFR-2 TK active site. As displayed in (Fig. 20), it was showed docking energy of −9.36 kcal mol−1. Both 4-amino phenyl and 5-cyano-2-(methylthio)-6-phenylpyrimidin moieties were oriented towards the gate keeper area and the hinge region, respectively, which bound with Ala1048 and Arg1049 by two hydrogen bonds with distances 2.75 and 2.80 Å, additionally, it was interacted with Val846, Ala864, Lys866, Val914, Cys1043, Ala1048 and Arg1049 by seven hydrophobic interactions. Also, compound 11e incorporated in two hydrogen bonds with the key amino acids; Glu883 (2.18 Å) and Asp1044 (2.52 Å) in the DFG motif. Additionally, the of the later moieties' orientation allowed the hydrophobic substituents in the docked compound to fit inside the hydrophobic allosteric pocket. Such a binding pattern of compound 11e encouraged us to study its MD simulation.
 | ||
| Fig. 20 The predicted binding mode of compound 11e with the active site of VEGFR-2 TK. | ||
Compound 12a showed docking energy of −8.95 kcal mol−1. The 5-cyano-2-(methylthio)-6-phenylpyrimidin moiety formed seven hydrophobic interactions in the hinge region with Arg1049, Ala1048, Leu1047, Leu1033, and Cys1043. Additionally, it was formed two hydrogen bonds with Arg1049 and Arg1030. The 4-amino phenyl (linker) moiety formed an extra hydrogen bond with Gly1046 in addition to two hydrophobic interactions with Val846, and Ala864. The pharmacophore moiety occupied the DFG region forming two hydrogen bonds with Glu883 Asp1044. The terminal hydrophobic moiety occupied the allosteric pocket forming four hydrophobic interactions with Leu887, Lys866, Asp1044, and Val917 (Fig. 21).
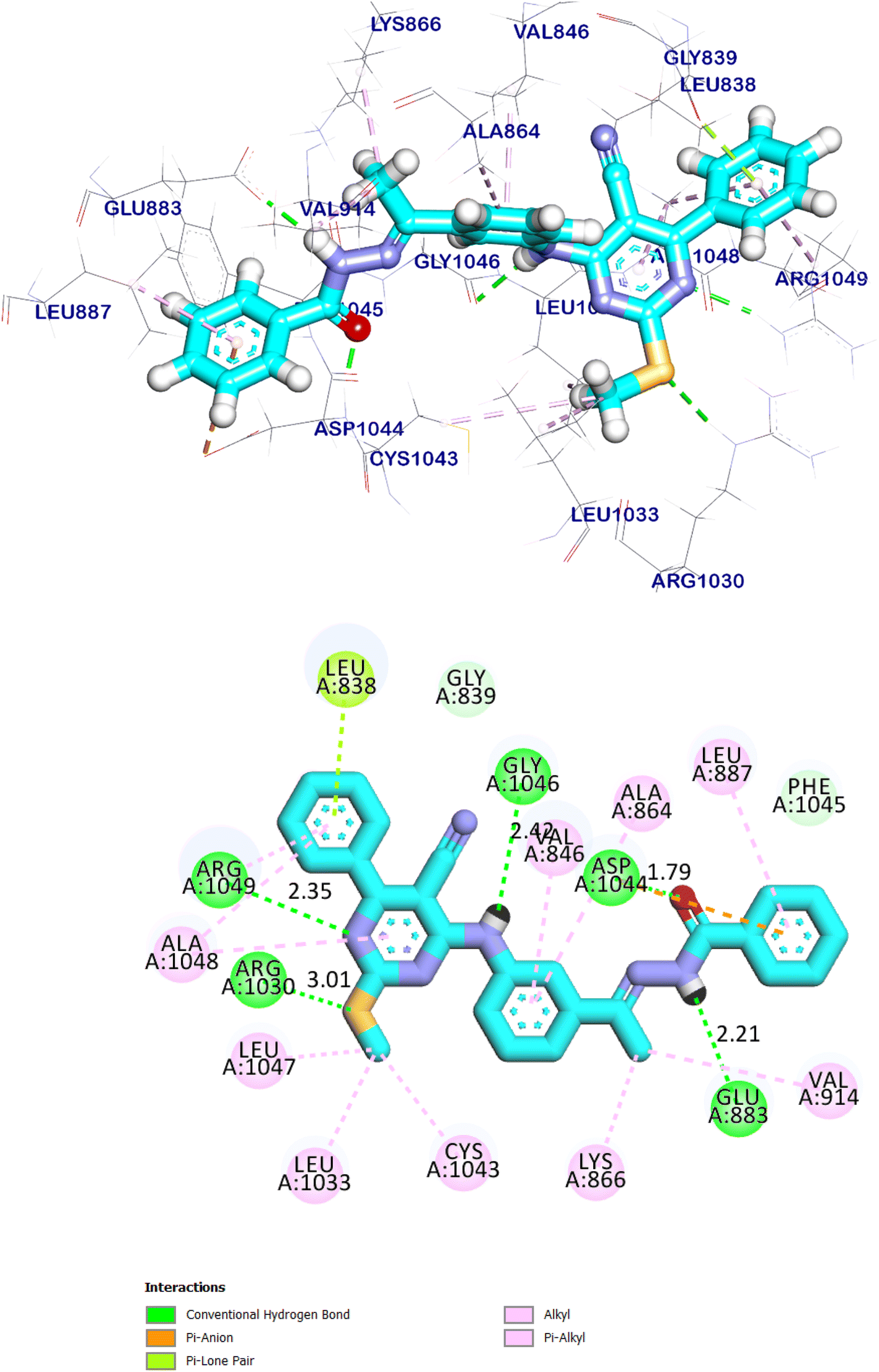 | ||
| Fig. 21 The predicted binding mode of compound 12a with the active site of VEGFR-2 TK. | ||
The net result of computational studies of the designed molecules indicates that most of the synthesized compounds showed significant VEGFR-2 TK inhibition affinities.
Comparing to the binding modes of the reference drugs, we found a lot of compounds have excellent binding modes with ΔG values higher than or at least similar to that of the reference drugs (sorafenib), against VEGFR-2 TK with ΔG values ranging from −8.23 to −9.36 compared with sorafenib ΔG value −8.70. Other compounds showed binding affinities lower than that of the reference drugs. The results may give guidance for further structural modifications.
The p-di-substituted and m-di-substituted benzohydrazide derivatives 11a–e and 12a–g exhibited a higher ability to form hydrogen bonds interaction than other groups, which explains their higher cytotoxic activity. Specifically, compounds with benzohydrazide side chains, such as 11e, 11b, 11d, 11c, 12b, 12d, and 12c, showed high affinity toward VEGFR-2 TK with (ΔG −9.36, −8.82, −9.30, −9.06, −9.17, −9.01, and −8.99), respectively, and these results were consistent with the cytotoxicity MTT assay. Furthermore, the series with a benzylidene side chain exhibited a remarkable affinity score against VEGFR-2 TK, such as compounds 9c which had affinity score (−8.95 kcal mol−1) higher than sorafenib (−8.70 kcal mol−1).
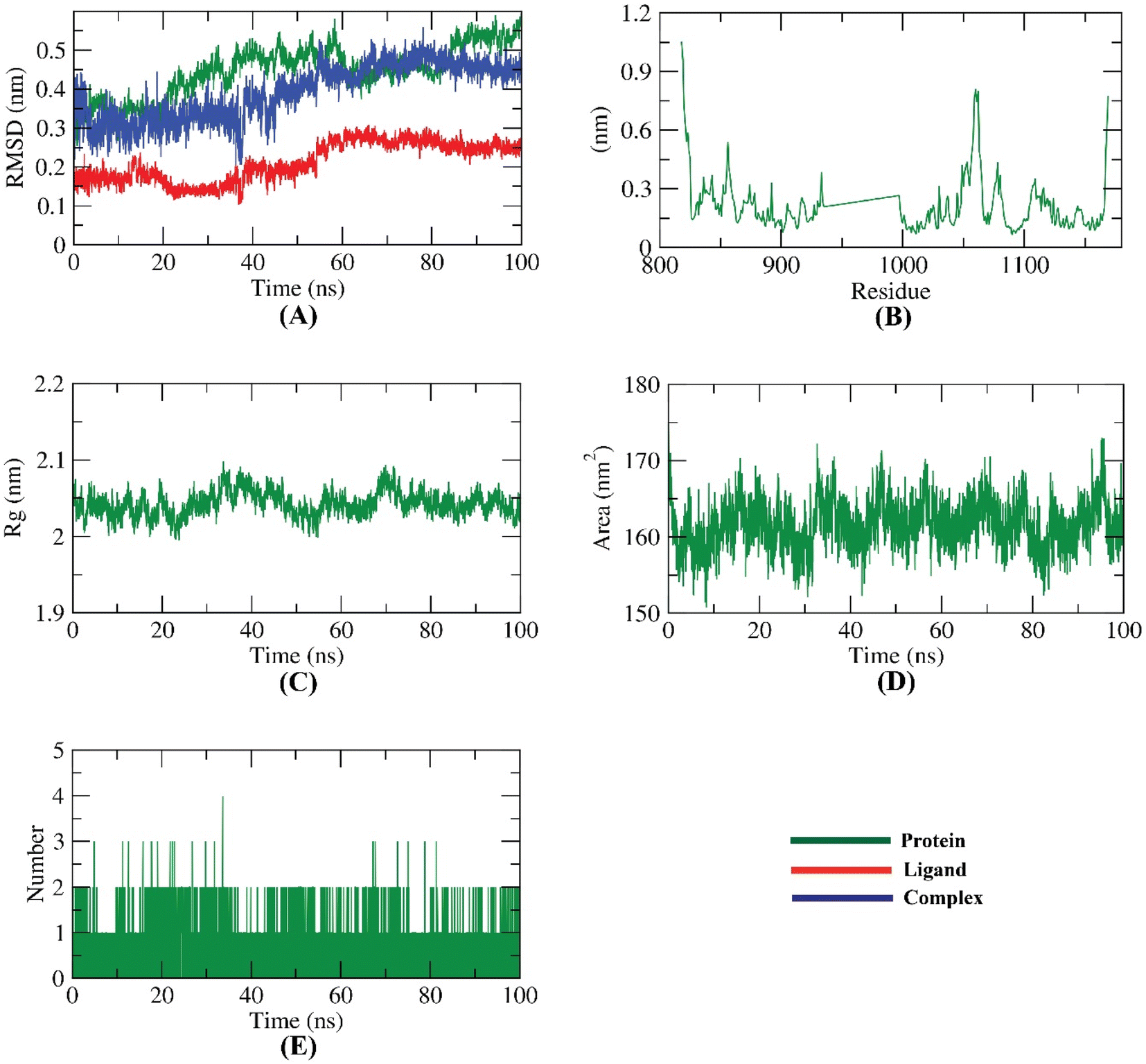 | ||
| Fig. 22 MD simulations; (A) RMSD, (B) RMSF (C) Rg (D) SASA, and (E) hydrogen bonding for compound 11e-VEGFR-2 TK complex over the MD run (100 ns). | ||
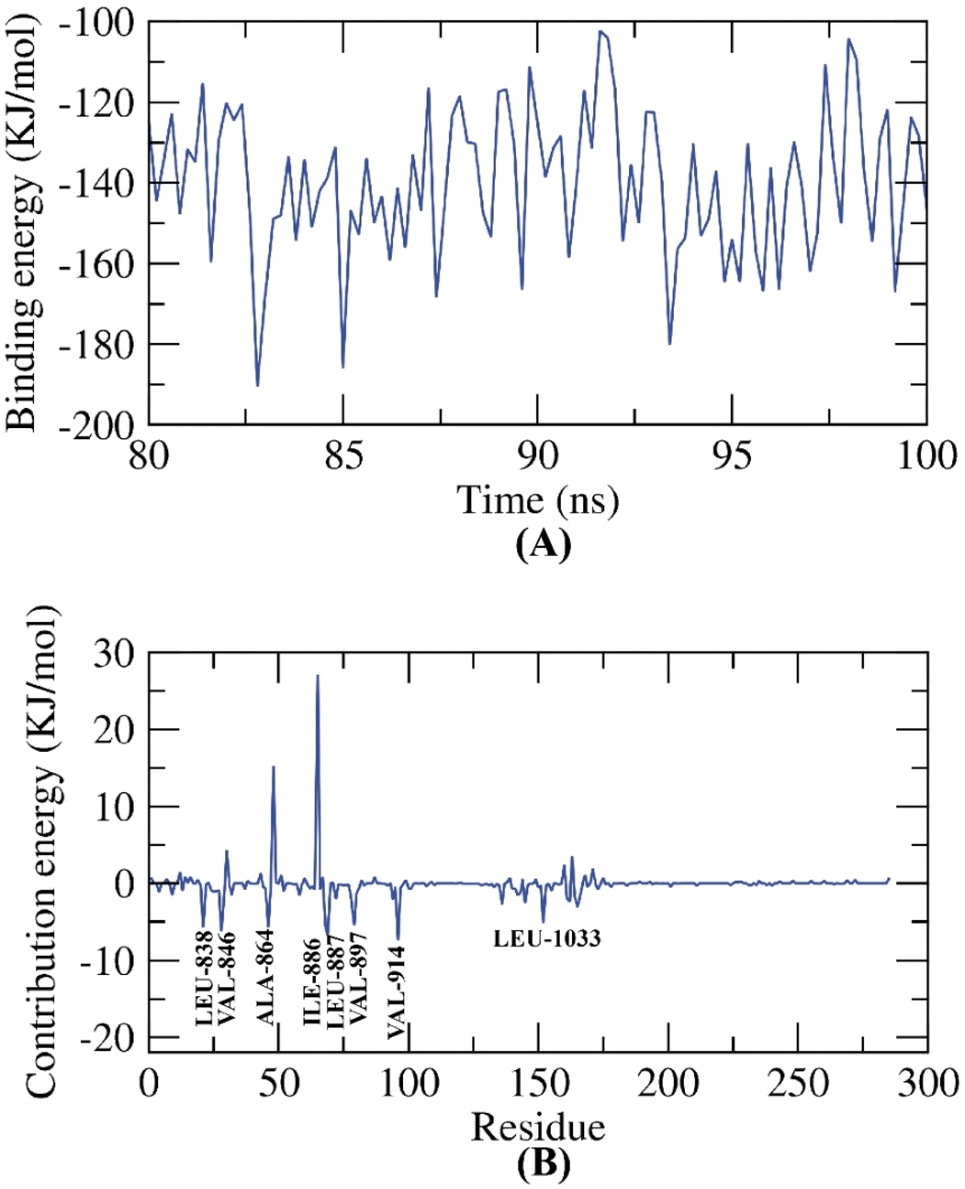 | ||
| Fig. 23 MM-PBSA outputs of the compound 11e-VEGFR-2 TK complex. | ||
3. Conclusion
A new sixteen pyrimidine-5-carbonitrile derivatives were designed and synthesized as VEGFR-2 TK inhibitors. The synthesized compounds were evaluated in vitro for their anti-proliferative activities against colon cancer (HCT-116), and breast cancer (MCF-7) cell lines. Compounds 9b, 9d, 11b, 11c, 11e, 12b, 12c, and 12d exhibited the highest activities towards both cell lines. In particular, compound 11e exhibited the best cytotoxic activity against HCT-116 and MCF-7 with IC50 values of 1.14 ± 0.01 and 1.54 ± 0.01 μM, respectively, compared to sorafenib as a reference drug with IC50 values of 8.96 ± 0.05 & 11.83 ± 0.07 μM, respectively. In addition, the most active cytotoxic agents 11e was further assessed for their inhibitory action on VEGFR-2 TK activity to confirm the mechanism responsible for their induced cytotoxic activities. The in vitro analysis of VEGFR-2 TK inhibition revealed that, compounds 11e and 12b were the most potent VEGFR-2 TK inhibitors with an IC50 values of 0.61 ± 0.01 and 0.53 ± 0.07 μM, respectively, in comparing with that of sorafenib (IC50 = 0.19 ± 0.15 μM). Moreover, compound 11e arrested the cell growth in HCT-116 cells at S and sub-G1 phase and induced a significant increase in the apoptotic cells. Finally, the binding patterns of the target derivatives were investigated through the docking study against the proposed molecular target (VEGFR-2 TK, PDB ID 1YWN). The results of molecular docking studies showed similar binding modes and interactions to sorafenib against VEGFR-2 TK active site.4. Experimental
4.1. Chemistry
Compounds, 4-oxo-6-phenyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile 1,21,22 2-(methylthio)-6-oxo-4-phenyl-1,6-dihydro-pyrimidine-5-carbonitrile 2,37 4-chloro-2-(methylthio)-6-phenylpyrimidine-5-carbonitrile 3,37 substitutedbenzohydrazide 10a–e24,38–40 were prepared according to the reported procedures.4.1.1.1. 4-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]benzoic acid 4. Off-white powder (yield 83.4%, 3.1 g); mp = 273–274 °C; IR (KBr) ν cm−1: 3451 (OH), 3319 (NH), 2975 (CH aromatic), 2928 (CH aliphatic), 2210 (C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1696 (CO), 1614 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm: 12.85 (s, 1H, OH), 10.12 (s, 1H, NH), 7.96 (d, J = 8.5 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.90 (d, J = 6.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.79 (d, J = 8.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.65–7.57 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 2.51 (s, 3H, SCH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.72, 168.71, 167.38, 160.39, 142.53, 136.24, 131.78, 130.24 (2C), 129.25 (2C), 129.06 (2C), 126.81, 122.93 (2C), 116.40, 85.84, 14.30; molecular formula C19H14N4O2S (362.41).
N), 1696 (CO), 1614 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm: 12.85 (s, 1H, OH), 10.12 (s, 1H, NH), 7.96 (d, J = 8.5 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.90 (d, J = 6.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.79 (d, J = 8.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.65–7.57 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 2.51 (s, 3H, SCH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.72, 168.71, 167.38, 160.39, 142.53, 136.24, 131.78, 130.24 (2C), 129.25 (2C), 129.06 (2C), 126.81, 122.93 (2C), 116.40, 85.84, 14.30; molecular formula C19H14N4O2S (362.41).
4.1.1.2. 4-[(4-Acetylphenyl)amino]-2-(methylthio)-6-phenylpyrimidine-5-carbonitrile 5. Beige fluffy powder (yield 93.5%, 3.36 g); mp = 236–235 °C; 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.15 (s, 1H, NH), 7.99 (d, J = 8.5 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.90 (d, J = 6.2 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.83 (d, J = 8.4 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.66–7.58 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 2.58 (s, 3H, COCH3), 2.52 (s, 3H, SCH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 197.21, 174.73, 168.73, 160.34, 142.88, 136.23, 133.11, 131.80, 129.30 (2C), 129.26 (2C), 129.07 (2C), 122.73 (2C), 116.40, 85.99, 27.05, 14.34; molecular formula C20H16N4OS (360.44).
4.1.1.3. 4-[(3-Acetylphenyl)amino]-2-(methylthio)-6-phenylpyrimidine-5-carbonitrile 6. Faint brown fluffy powder (yield 93.4%, 3.36 g); mp = 226–225 °C; 1H NMR (500 MHz, DMSO-d6) δ ppm: 10.10 (s, 1H, NH), 7.93 (d, J = 8.8 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.85 (s, 1H, Ar–H, H-2 of –C6H4), 7.83 (d, J = 2.3 Hz, 1H, Ar–H, H-4 of –C6H4), 7.76–7.73 (m, 2H, Ar–H, H-5 & H-6 of –C6H4), 7.57–7.53 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 2.53 (s, 3H, COCH3), 2.45 (s, 3H, SCH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 197.26, 174.64, 168.73, 160.40, 142.76, 136.38, 132.96, 131.80, 129.36 (2C), 129.30 (2C), 129.12 (2C), 122.83 (2C), 116.65, 86.16, 27.11, 14.37; molecular formula C20H16N4OS (360.44).
White crystals (yield 90%, 3.4 g); mp = 294–295 °C; IR (KBr) ν cm−1: 3296 (NH), 3202 (NH2), 3035 (CH aromatic), 2922 (CH aliphatic), 2213 (CN), 1658 (CO), 1602 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 9.97 (s, 1H, CONH), 9.74 (s, 1H, NH), 7.84 (d, J = 6.6 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.79 (d, J = 8.7 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.67 (d, J = 8.8 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.58–7.53 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 4.72 (s, 2H, NH2), 2.44 (s, 3H, SCH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.71, 168.71, 165.98, 160.46, 140.92, 136.35, 131.81, 129.62, 129.29 (2C), 129.12 (2C), 127.81 (2C), 123.17 (2C), 116.51, 85.61, 14.32; molecular formula C19H16N6OS (376.44).
4.1.4.1. N-(2-Chlorobenzylidene)-4-[(5-cyano-2-(methylthio)-6-phenyl pyrimidin-4-yl)-amino]benzohydrazide 9a. White fluffy powder (yield 84.5%, 0.27 g); mp = 300–301 °C; IR (KBr) ν cm−1: 3276, 3236 (NH), 3062 (CH aromatic), 2931 (CH aliphatic), 2222 (C
N), 1649 (CO), 1599 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 12.05 (s, 1H, CONH), 10.08 (s, 1H, NH), 8.84 (s, 1H, NCH), 8.00 (d, J = 5.6 Hz, 1H, Ar–H, H-6 of 2-ClC6H4), 7.93 (d, J = 8.4 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.85 (d, J = 7.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.79 (d, J = 8.2 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.60–7.53 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 7.50 (d, J = 7.5 Hz, 1H, Ar–H, H-3 of 2-Cl–C6H4), 7.43–7.39 (m, 2H, Ar–H, H-4 & H-5 of 2-Cl–C6H4), 2.48 (s, 3H, SCH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.78, 168.76, 163.05, 160.43, 143.89, 141.79, 136.32, 133.71, 132.17, 132.02, 131.85, 130.50, 129.32 (3C), 129.14 (3C), 128.64, 128.20, 127.39, 123.06 (2C), 116.50, 85.80, 14.40; molecular formula C26H19ClN6OS (498.99).
4.1.4.2. N-(4-Chlorobenzylidene)-4-[(5-cyano-2-(methylthio)-6-phen-ylpyrimidin-4-yl)-amino]benzohydrazide 9b. Off-white fluffy powder (yield 83.9%, 0.275 g); mp = 309–310 °C; HPLC purity 99.53%; IR (KBr) ν cm−1: 3300, 3227 (NH), 3062 (CH aromatic), 2924 (CH aliphatic), 2214 (C
N), 1650 (CO), 1607 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 11.89 (s, 1H, CONH), 10.07 (s, 1H, NH), 8.41 (s, 1H, NCH), 7.91 (d, J = 8.4 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.85 (d, 2H, J = 6.8 Hz, Ar–H, H-2 & H-6 of –C6H5), 7.77 (d, J = 8.4 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.72 (d, J = 8.3 Hz, 2H, Ar–H, H-2 & H-6 of 4-ClC6H4), 7.57–7.49 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 7.50 (d, J = 8.1 Hz, 2H, Ar–H, H-3 & H-5 of 4-ClC6H4), 2.48 (s, 3H, SCH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.77, 168.76, 163.06, 160.44, 146.65, 141.70, 136.33, 134.99, 133.88, 131.85, 129.51 (2C), 129.31 (3C), 129.25, 129.14 (3C), 128.59 (2C), 123.09 (2C), 116.52, 85.79, 14.39; MS (m/z): 497.99 (M+, 31.63%), 500.61 (M2+, 28.55%), 462.04 (100%, base peak); molecular formula C26H19ClN6OS (498.99).
4.1.4.3. 4-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]-N-(2,4-dichloro-benzylidene)benzohydrazide 9c. Off-white powder (yield 85.6%, 0.30 g); mp = 290–291 °C; HPLC purity 97.46%; IR (KBr) ν cm−1: 3303, 3201 (NH), 3043 (CH aromatic), 2925 (CH aliphatic), 2215 (C
N), 1650 (CO), 1612 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 12.09 (s, 1H, CONH), 10.07 (s, 1H, NH), 8.78 (s, 1H, NCH), 7.99 (d, J = 8.5 Hz, 1H, Ar–H, H-6 of –C6H3), 7.93 (d, J = 8.5 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.85 (d, J = 7.4 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.79 (d, J = 8.4 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.70 (s, 1H, Ar–H, H-3 of –C6H3), 7.60–7.49 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 7.50 (d, J = 8.6 Hz, 1H, Ar–H, H-5 of –C6H3), 2.48 (s, 3H, SCH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.78, 168.77, 163.05, 160.43, 142.81, 141.89, 136.32, 135.57, 134.37, 131.86, 131.31, 129.96, 129.32 (3C), 129.14 (3C), 129.03, 128.66, 128.58, 123.07 (2C), 116.51, 85.82, 14.40; molecular formula C26H18Cl2N6OS (533.43).
4.1.4.4. 4-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]-N-(2,6-dichloro-benzylidene)benzohydrazide 9d. White fluffy powder (yield 82.8%, 0.29 g); mp = 298–299 °C; IR (KBr) ν cm−1: 3311, 3202 (NH), 3062 (CH aromatic), 2926 (CH aliphatic), 2218 (C
N), 1645 (CO), 1610 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 12.11 (s, 1H, CONH), 10.07 (s, 1H, NH), 8.62 (s, 1H, NCH), 7.93 (d, J = 8.5 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.85 (d, J = 7.2 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.78 (d, J = 8.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.59–7.54 (m, 5H, Ar–H, H-3 & H-5 of –C6H3 and H-3, H4 & H5 of –C6H5), 7.42 (t, J = 8.1 Hz, 1H, Ar–H, H-4 of –C6H3), 2.50 (s, 3H, SCH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 177.55, 171.12, 165.64, 160.46, 147.49, 136.34, 131.85, 129.67 (4C), 129.31 (5C), 129.14 (4C), 123.09, 115.53, 113.17, 112.36, 81.83, 14.38; molecular formula C26H18C12N6OS (533.43).
4.1.5.1. N-(1-(4-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)ethylidene)benzohydrazide 11a. Off-white powder (yield 90.3%, 0.30 g); mp = 251–252 °C; IR (KBr) ν cm−1: 3294, 3229 (NH), 3055 (CH aromatic), 2921 (CH aliphatic), 2216 (C
N), 1650 (CO), 1612 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 10.74 (s, 1H, CONH), 9.97 (s, 1H, NH), 7.85–7.82 (m, 4H, Ar–H, H-2 & H-6 of –C6H5 and H-2 & H-6 of –COC6H5), 7.69 (d, J = 8.1 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.61–7.51 (m, 6H, Ar–H, H-3 & H-5 of –C6H4, H-3 & H-5 of –C6H5 and H-3 & H-5 of –COC6H5), 7.50–7.47 (m, 2H, Ar–H, H-4 of –COC6H5 and H-4 of –C6H5), 2.46 (s, 3H, SCH3), 2.34 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.71, 168.69, 160.40, 155.46, 139.55, 136.40, 134.65, 131.79, 129.29 (4C), 129.12 (4C), 128.91, 128.43, 127.10, 123.41 (2C), 122.81, 116.58, 111.45, 85.45, 14.94, 14.37; molecular formula C27H22N6OS (478.57).
4.1.5.2. 2-Chloro-N-(1-(4-[(5-cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)benzohydrazide 11b. Off-white fluffy powder (yield 80%, 0.28 g); mp = 232–233 °C; HPLC purity 99.01%; IR (KBr) ν cm−1: 3300, 3224 (NH), 3057 (CH aromatic), 2924 (CH aliphatic), 2214 (C
N), 1658 (CO), 1612 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 11.09 (d, J = 125.9 Hz, 1H, CONH), 9.90 (d, J = 67.3 Hz, 1H, NH), 7.85–7.81 (m, 3H, Ar–H, H-2 & H-6 of –C6H5 and H-6 of 2-ClC6H4), 7.69 (d, J = 8.4 Hz, 1H, Ar–H, H-5 of 2-ClC6H4), 7.58–7.51 (m, 4H, Ar–H, H-2, H-3, H-5 & H-6 of –C6H4), 7.50–7.45 (m, 2H, Ar–H, H-3 & H-5 of –C6H5), 7.43–7.35 (m, 3H, Ar–H, H-4 of –C6H4 and H-3 & H-4 of 2-ClC6H4), 2.42 (d, J = 32.2 Hz, 3H, SCH3), 2.26 (d, J = 16.6 Hz, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.73–174.61, 170.13, 168.69–168.62, 163.53, 160.40–160.33, 154.52, 148.58, 139.59, 139.03, 137.29, 136.38, 136.31, 134.69, 134.50, 131.77, 131.67, 131.01, 130.81, 130.24, 130.11, 129.35, 129.28, 129.10 (2C), 127.68, 127.38, 127.12, 126.40, 123.37, 116.54, 85.46, 85.37, 14.96, 14.39, 14.2, 14.15; molecular formula C27H21ClN6OS (513.02).
4.1.5.3. 3-Chloro-N-(1-(4-[(5-cyano-2-(methylthio)-6-phenylpyramid-in-4-yl)amino]-phenyl)ethylidene)benzohydrazide 11c. Yellow powder (yield 80.1%, 0.285 g); mp = 262–263 °C; HPLC purity 96.25%; IR (KBr) ν cm−1: 3306, 3194 (NH), 3060 (CH aromatic), 2926 (CH aliphatic), 2222 (C
N), 1666 (CO), 1620 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.88 (s, 1H, CONH), 9.98 (s, 1H, NH), 7.96–7.86 (m, 5H, Ar–H, H-2 & H-6 of –C6H5 and H-2, H-4 & H-6 of 3-ClC6H4), 7.74 (d, J = 9.1 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.67 (d, J = 9.9 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.63–7.57 (m, 4H, Ar–H, H-3, H-4 & H-5 of –C6H5 and H-5 of 3-ClC6H4), 2.51 (s, 3H, SCH3), 2.40 (s, 3H, –CH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.66, 168.61, 163.03, 160.36, 136.35, 134.48, 131.71, 131.07, 129.21 (4C), 129.04 (4C), 128.08, 127.12, 125.52, 123.31 (3C), 116.46, 85.45, 56.51, 19.02, 14.30; MS (m/z): 513.59 (M+, 14.25%), 515.73 (M+ + 2, 10.12%), 172.61 (100%, base peak); molecular formula C27H21ClN6OS (513.02).
4.1.5.4. N-(1-(4-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)-4-nitrobenzohydrazide 11d. Yellow fluffy powder (yield 78.5%, 0.285 g); mp = 268–269 °C; HPLC purity 93.41%; IR (KBr) ν cm−1: 3307, 3210 (NH), 3007 (CH aromatic), 2924 (CH aliphatic), 2213 (C
N), 1657 (CO), 1611 (CN), 1528, 1343 (NO2); 1H NMR (500 MHz, DMSO-d6) δ ppm: 11.06 (s, 1H, CONH), 9.98 (s, 1H, NH), 8.32 (d, J = 8.4 Hz, 2H, Ar–H, H-3 & H-5 of 4-NO2C6H4), 8.09 (d, J = 8.7 Hz, 2H, Ar–H, H-2 & H-6 of 4-NO2C6H4), 7.87 (d, J = 8.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.84 (d, J = 7.1 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.70 (d, J = 8.2 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.58–7.54 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 2.46 (s, 3H, SCH3), 2.36 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.72, 168.68, 162.95, 160.39, 156.81, 149.62, 140.43, 139.82, 136.38, 134.33, 131.79, 131.13, 129.99, 129.36, 129.29 (2C), 129.11 (2C), 127.23, 126.73, 124.02, 123.37, 122.80, 116.56, 85.49, 15.19, 14.38; molecular formula C27H21N7O3S (523.57).
4.1.5.5. N-(1-(4-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)-2,4-dinitrobenzohydrazide 11e. Reddish-orange fine powder (yield 67.1%, 0.265 g); mp = 298–299 °C; IR (KBr) ν cm−1: 3297, 3204 (NH), 3113 (CH aromatic), 2925 (CH aliphatic), 2209 (C
N), 1614 (CN), 1550, 1362 (NO2); 1H NMR (500 MHz, DMSO-d6) δ ppm: 11.11 (s, 1H, CONH), 10.01 (s, 1H, NH), 8.87 (d, J = 3.1 Hz, 1H, Ar–H, H-3 of –C6H3), 8.38 (dd, J = 9.6, 2.5 Hz, 1H, Ar–H, H-5 of –C6H3), 8.11 (d, J = 9.6 Hz, 1H, Ar–H, H-6 of –C6H3), 7.96 (d, J = 8.8 Hz, 2H, Ar–H, H-2 & H-6 of –C6H4), 7.85 (d, J = 6.9 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.74 (d, J = 8.7 Hz, 2H, Ar–H, H-3 & H-5 of –C6H4), 7.60–7.54 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 2.48 (s, 3H, SCH3), 2.44 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.74, 168.71, 166.67, 160.35, 146.91, 144.96, 142.13, 140.07, 136.38, 133.43, 131.53, 130.72, 130.16, 129.31 (2C), 129.14 (2C), 127.28 (2C), 124.09, 123.40 (2C), 117.17, 116.13, 86.39, 14.41, 13.77; molecular formula C27H20N8O5S (568.57).
4.1.6.1. N-(1-(3-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)benzohydrazide 12a. Beige fluffy powder (yield 87.3%, 0.29 g); mp = 242–225 °C; HPLC purity 99.38%; IR (KBr) ν cm−1: 3341, 3254 (NH), 3056 (CH aromatic), 2926 (CH aliphatic), 2206 (C
N), 1649 (CO), 1615 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.80 (s, 1H, CONH), 9.98 (s, 1H, NH), 7.92–7.87 (m, 4H, Ar–H, H-2, H-6 of –C6H5 and Ar–H, H-2 & H-6 of –COC6H5), 7.70–7.67 (m, 2H, Ar–H, H-4 of –C6H4 and H-4 –COC6H5), 7.62–7.59 (m, 5H, Ar–H, H-3, H-4, H-5 of –C6H5 and H-3 & H-5 of –COC6H4), 7.54 (d, J = 7.3 Hz, 2H, Ar–H, H-2 & H-5 of –C6H4), 7.47 (d, J = 8.00 Hz, 1H, Ar–H, H-6 of –C6H4), 2.47 (s, 3H, SCH3), 2.40 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.63, 168.58, 161.83, 160.58, 155.46, 138.85, 138.33, 136.40, 134.55, 132.02, 131.65, 129.19 (3C), 129.03 (3C), 128.83 (2C), 128.42, 125.10, 123.44, 122.10, 116.49, 85.10, 15.03, 14.22; molecular formula C27H22N6OS (478.57).
4.1.6.2. 2-Chloro-N-(1-(3-[(5-cyano-2-(methylthio)-6-phenyl pyri-midin-4-yl)amino]-phenyl)ethylidene)benzohydrazide 12b. White fluffy powder (yield 77%, 0.27 g); mp = 221–222 °C; IR (KBr) ν cm−1: 3396 (NH), 3147, 3026 (CH aromatic), 2993 (CH aliphatic), 2202 (C
N), 1664 (CO), 1613 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 11.13 (d, J = 136.2 Hz, 1H, CONH), 9.96 (d, J = 72.0 Hz, 1H, NH), 8.17 (d, J = 34.5 Hz, 1H, Ar–H, H-2 of –C6H4), 7.84 (d, J = 7.2 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.69–7.64 (m, 1H, Ar–H, H-6 of –C6H4), 7.60 (dd, J = 15.0, 7.2 Hz, 1H, Ar–H, H-6 of –C6H4), 7.56–7.47 (m, J = 12.8, 5.9 Hz, 3H, Ar–H, H-3 of 3-ClC6H4 and Ar–H, H-3 & H-5 of –C6H5), 7.46–7.37 (m, 3H, Ar–H, H-4 of –C6H4, 4 H-4 of 3-ClC6H4 & Ar–H, H-4 of –C6H5), 7.35–7.27 (m, 1H, Ar–H, H-5 of –C6H4), 7.25–7.15 (m, 1H, Ar–H, H-5 of 3-ClC6H4), 2.45 (s, 3H, SCH3), 2.27 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.70, 170.21, 168.66, 163.60, 160.61, 154.48, 148.32, 138.76, 138.59, 137.14, 136.44, 131.73, 130.99, 130.19, 129.27, 129.10, 128.90, 127.69, 125.22, 24.86, 123.58, 122.53, 116.58, 85.16, 84.92, 15.38, 14.30; molecular formula C27H21ClN6OS (513.02).
4.1.6.3. 3-Chloro-N-(1-(3-[(5-cyano-2-(methylthio)-6-phenylpyramidin-4-yl)amino]phenyl)-ethylidene)benzohydrazide 12c. Beige crystals (yield 87%, 0.31 g); mp = 230–231 °C; HPLC purity 100%; IR (KBr) ν cm−1: 3308, 3178 (NH), 3067 (CH aromatic), 2923 (CH aliphatic), 2216 (C
N), 1669 (CO), 1628 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.91 (s, 1H, CONH), 10.00 (s, 1H, NH), 8.18 (s, 1H, Ar–H, H-2 of 3-ClC6H4), 7.96 (s, 1H, Ar–H, H-6 of 3-ClC6H4), 7.90–7.88 (m, 3H, Ar–H, H-4 of –C6H4 and H-2 & H-6 of –C6H5), 7.63–7.53 (m, 6H, Ar–H, H-2 of –C6H4, H-4, H-5 of 3-ClC6H4 and Ar–H, H-3, H-4 & H-5 of –C6H5), 7.57–7.53 (m, 1H, Ar–H, H-5 of –C6H4), 7.46 (d, J = 4.00 Hz, 1H, Ar–H, H-6 of –C6H4), 2.47 (s, 3H, SCH3), 2.41 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.62, 168.56, 162.95, 160.60, 150.58, 138.71, 138.37, 137.74, 136.42, 136.35, 133.22, 131.65, 131.07, 129.17 (2C), 129.02 (2C), 128.86, 128.47, 126.99, 125.04, 123.39, 122.13, 116.47, 85.11, 27.23, 14.20; molecular formula C27H21ClN6OS (513.02).
4.1.6.4. N-(1-(3-[(5-cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)-4-nitrobenzohydrazide 12d. Off-white crystals (yield 80%, 0.29 g); mp = 265–266 °C; IR (KBr) ν cm−1: 3305, 3196 (NH), 3076 (CH aromatic), 2927 (CH aliphatic), 2213 (C
N), 1670 (CO), 1597 (CN), 1543, 1345 (NO2); 1H NMR (400 MHz, DMSO-d6) δ ppm: 11.10 (s, 1H, CONH), 9.98 (s, 1H, NH), 8.36 (d, J = 8.0 Hz, 2H, Ar–H, H-3 & H-5 of 4-NO2C6H4), 8.16 (d, 2H, Ar–H, H-2 & H-6 of 4-NO2C6H4), 8.06 (s, 1H, Ar–H, H-2 of –C6H4), 7.89 (d, 2H, J = 8.0 Hz, Ar–H, H-2 & H-6 of –C6H5), 7.71–7.68 (m, 1H, Ar–H, H-5 of –C6H4), 7.61–7.59 (m, 4H, Ar–H, H-3, H-4 & H-5 of –C6H5 and H-4 of –C6H4), 7.47 (d, 1H, Ar–H, H-6 of –C6H4), 2.51 (s, 3H, SCH3), 2.20 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.61, 168.58, 164.39, 163.07, 160.56, 138.15, 137.19, 136.39, 131.65, 129.95 (3C), 129.19 (2C), 129.05 (2C), 129.01, 128.92, 125.36, 124.02, 123.93, 123.70, 122.14, 116.50, 85.11, 15.04, 14.21; molecular formula C27H21N7O3S (523.57).
4.1.6.5. N-(1-(3-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)-2-hydroxybenzohydrazide 12e. Beige crystals (yield 79%, 0.27 g); mp = 204–205 °C; IR (KBr) ν cm−1: 3392 (OH), 3297 (NH), 3060 (CH aromatic), 2925 (CH aliphatic), 2213 (C
N), 1649 (CO), 1602 (CN); 1H NMR (500 MHz, DMSO-d6) δ ppm: 11.39 (s, 1H, OH), 9.97 (s, 1H, CONH), 8.14 (s, 1H, NH), 7.96 (d, J = 5.00 Hz, 1H, Ar–H, H-6 of 2-OHC6H4), 7.84 (d, J = 7.2 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.66–7.61 (m, 2H, Ar–H, H-5 of –C6H4 & H-4 of 2-OHC6H4), 7.60–7.51 (m, 4H, Ar–H, H-3, H-4 & H-5 of –C6H5 and H-4 of –C6H4), 7.43–7.37 (m, 2H, Ar–H, H-2 & H-6 of –C6H4), 6.99 (d, J = 8.2 Hz, 1H, Ar–H, H-3 of 2-OHC6H4), 6.94 (t, J = 7.6 Hz, 1H, Ar–H, H-5 of 2-OHC6H4), 2.43 (s, 3H, SCH3), 2.32 (s, 3H, CH3); 13C NMR (126 MHz, DMSO-d6) δ ppm: 174.71, 168.65, 162.55, 160.62, 157.21, 152.12, 138.69, 138.39, 136.45, 133.94, 131.73, 131.21, 129.27 (2C), 129.10 (2C), 128.93, 125.11, 123.56, 122.04, 120.13, 118.44, 117.47, 116.58, 85.13, 14.42, 14.30; molecular formula C27H22N6O2S (494.57).
4.1.6.6. N-(1-(3-[(5-Cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)-4-hydroxybenzohydrazide 12f. Beige fine powder (yield 75.8%, 0.26 g); mp = 231–232 °C; HPLC purity 94.88%; IR (KBr) ν cm−1: 3316 (OH), 3270, 3196 (NH), 3011 (CH aromatic), 2806 (CH aliphatic), 2204 (C
N), 1659 (CO), 1614 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.53 (s, 1H, CONH), 9.99 (s, 1H, OH), 9.49 (s, 1H, NH), 7.89 (d, J = 7.5 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.81 (d, J = 8.3 Hz, 1H, Ar–H, H-4 of –C6H4), 7.71–7.67 (m, 3H, Ar–H, H-2 & H-6 of 4-OHC6H4 and H-2 of –C6H4), 7.64–7.59 (m, 3H, Ar–H, H-3, H-4 & H-5 of –C6H5), 7.45 (t, J = 8.0 Hz, 1H, Ar–H, H-5 of –C6H4), 6.87 (d, J = 8.4 Hz, 1H, Ar–H, H-6 of –C6H4), 6.79 (d, J = 8.2 Hz, 2H, Ar–H, H-3 & H-5 of 4-OHC6H4), 2.47 (s, 3H, SCH3), 2.38 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.61, 168.57, 166.40, 161.01, 160.57, 160.47, 139.00, 136.42, 131.64, 129.28 (3C), 129.19 (2C), 129.02 (2C), 128.80, 124.90, 124.44, 123.34, 122.03, 115.29 (3C), 85.10, 14.80, 14.21; MS (m/z): 494.37 (M+, 54.40%), 419.31 (100%, base peak); molecular formula C27H22N6O2S (494.57).
4.1.6.7. 4-Amino-N-(1-(3-[(5-cyano-2-(methylthio)-6-phenylpyrimidin-4-yl)amino]phenyl)-ethylidene)benzohydrazide 12g. White fluffy powder (yield 80%, 0.27 g); mp = 227–228 °C; IR (KBr) ν cm−1: 3478, 3363, 3293 (2NH, NH2), 3071 (CH aromatic), 2924, (CH aliphatic), 2215 (C
N), 1637 (CO), 1606 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm: 10.29 (s, 1H, CONH), 9.97 (s, 1H, NH), 8.12 (s, 1H, Ar–H, H-4 of –C6H4), 7.91 (d, J = 8.00 Hz, 2H, Ar–H, H-2 & H-6 of –C6H5), 7.68 (d, J = 8.4 Hz, 2H, Ar–H, H-2 & H-6 of 4-NH2C6H4), 7.65 (d, J = 3.9 Hz, 1H, Ar–H, H-6 of –C6H4), 7.64–7.56 (m, 4H, Ar–H, H-2 of –C6H4, and H-3, H-4 & H-5 of –C6H5), 7.45 (t, J = 7.9 Hz, 1H, Ar–H, H-5 of –C6H4), 6.61 (d, J = 8.3 Hz, 2H, Ar–H, H-3 & H-5 of 4-NH2C6H4), 5.75 (s, 2H, NH2), 2.47 (s, 3H, SCH3), 2.37 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ ppm: 174.61, 168.57, 164.59, 160.57, 152.66, 139.18, 138.35, 136.43, 131.63, 130.31, 129.19 (3C), 129.02 (3C), 128.76, 124.79, 123.26, 121.97, 120.53, 116.52, 113.00 (2C), 85.09, 14.60, 14.22; molecular formula C27H23N7OS (493.59).
4.2. Biological evaluation
The cell lines were obtained from the Holding company for biological products and vaccines (VACSERA), Cairo, Egypt.
4.3. In silico studies
Conflicts of interest
This work was funded by the authors and there is no any conflict of interest.Acknowledgements
This paper is based upon work supported by Science, Technology & Innovation Funding Authority (STIFA) under grant number 44434.References
- V. Ganapathy, M. Thangaraju and P. D. Prasad, Nutrient transporters in cancer: relevance to Warburg hypothesis and beyond, Pharmacol. Therapeut., 2009, 121(1), 29–40 CrossRef CAS PubMed.
- H. Varmus, The new era in cancer research, Science, 2006, 312(5777), 1162–1165 CrossRef CAS PubMed.
- Z. Wang, X.-H. Shi, J. Wang, T. Zhou, Y.-Z. Xu and T.-T. Huang, et al., Synthesis, structure–activity relationships and preliminary antitumor evaluation of benzothiazole-2-thiol derivatives as novel apoptosis inducers, Bioorg. Med. Chem. Lett., 2011, 21(4), 1097–1101 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, Ca-Cancer J. Clin., 2018, 68(1), 7–30 CrossRef PubMed.
- N. M. Saleh, A. A. H. Abdel-Rahman, A. M. Omar, M. M. Khalifa and K. El-Adl, Pyridine-derived VEGFR-2 inhibitors: rational design, synthesis, anticancer evaluations, in silico ADMET profile, and molecular docking, Arch. Pharm., 2021, 354(8), 2100085 CrossRef CAS PubMed.
- R. G. Yousef, W. M. Eldehna, A. Elwan, A. S. Abdelaziz, A. B. Mehany and I. M. Gobaara, et al., Design, Synthesis, In Silico and In Vitro Studies of New Immunomodulatory Anticancer Nicotinamide Derivatives Targeting VEGFR-2, Molecules, 2022, 27(13), 4079 CrossRef CAS PubMed.
- F. Ran, W. Li, Y. Qin, T. Yu, Z. Liu and M. Zhou, et al., Inhibition of vascular smooth muscle and cancer cell proliferation by new VEGFR inhibitors and their immunomodulator effect: design, synthesis, and biological evaluation, Oxid. Med. Cell. Longev., 2021, 2021, 1–21 Search PubMed.
- R. G. Yousef, A. Ibrahim, M. M. Khalifa, W. M. Eldehna, I. M. Gobaara and A. B. Mehany, et al., Discovery of new nicotinamides as apoptotic VEGFR-2 inhibitors: virtual screening, synthesis, anti-proliferative, immunomodulatory, ADMET, toxicity, and molecular dynamic simulation studies, J. Enzym. Inhib. Med. Chem., 2022, 37(1), 1389–1403 CrossRef CAS PubMed.
- M. S. Taghour, H. A. Mahdy, M. H. Gomaa, A. Aglan, M. G. Eldeib and A. Elwan, et al., Benzoxazole derivatives as new VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, in silico studies, and antiproliferative evaluation, J. Enzym. Inhib. Med. Chem., 2022, 37(1), 2063–2077 CrossRef CAS.
- E. B. Elkaeed, R. G. Yousef, M. M. Khalifa, A. Ibrahim, A. B. Mehany and I. M. Gobaara, et al., Discovery of New VEGFR-2 Inhibitors: Design, Synthesis, Anti-Proliferative Evaluation, Docking, and MD Simulation Studies, Molecules, 2022, 27(19), 6203 CrossRef CAS PubMed.
- M. S. Taghour, H. Elkady, W. M. Eldehna, N. M. El-Deeb, A. M. Kenawy and E. B. Elkaeed, et al., Design and synthesis of thiazolidine-2, 4-diones hybrids with 1, 2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: in vitro anticancer evaluation and in silico studies, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 1903–1917 CrossRef CAS PubMed.
- M. M. Alanazi, H. Elkady, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani and M. M. Alanazi, et al., New quinoxaline-based VEGFR-2 inhibitors: design, synthesis, and antiproliferative evaluation with in silico docking, ADMET, toxicity, and DFT studies, RSC Adv., 2021, 11(48), 30315–30328 RSC.
- A. R. Kotb, D. A. Bakhotmah, A. E. Abdallah, H. Elkady, M. S. Taghour and I. H. Eissa, et al., Design, synthesis, and biological evaluation of novel bioactive thalidomide analogs as anticancer immunomodulatory agents, RSC Adv., 2022, 12(52), 33525–33539 RSC.
- I. H. Eissa, M. A. Dahab, M. K. Ibrahim, N. A. Alsaif, A. Alanazi and S. I. Eissa, et al., Design and discovery of new antiproliferative 1, 2, 4-triazin-3 (2H)-ones as tubulin polymerization inhibitors targeting colchicine binding site, Bioorg. Chem., 2021, 112, 104965 CrossRef CAS PubMed.
- M. A. Abdelgawad, K. El-Adl, S. S. El-Hddad, M. M. Elhady, N. M. Saleh and M. M. Khalifa, et al., Design, molecular docking, synthesis, anticancer and anti-hyperglycemic assessments of thiazolidine-2, 4-diones bearing sulfonylthiourea moieties as potent VEGFR-2 inhibitors and PPARγ agonists, Pharmaceuticals, 2022, 15(2), 226 CrossRef CAS PubMed.
- I. H. Eissa, R. El-Haggar, M. A. Dahab, M. F. Ahmed, H. A. Mahdy and R. I. Alsantali, et al., Design, synthesis, molecular modeling and biological evaluation of novel benzoxazole-benzamide conjugates via a 2-thioacetamido linker as potential anti-proliferative agents, VEGFR-2 inhibitors and apoptotic inducers, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 1587–1599 CrossRef CAS PubMed.
- M. A. Dahab, H. A. Mahdy, H. Elkady, M. S. Taghour, A. Elwan and M. A. Elkady, et al., Semi-synthesized anticancer theobromine derivatives targeting VEGFR-2: in silico and in vitro evaluations, J. Biomol. Struct. Dyn., 2023, 1–20 CrossRef PubMed.
- N. A. Alsaif, H. A. Mahdy, M. M. Alanazi, A. J. Obaidullah, H. M. Alkahtani and A. M. Al-Hossaini, et al., Targeting VEGFR-2 by new quinoxaline derivatives: design, synthesis, antiproliferative assay, apoptosis induction, and in silico studies, Arch. Pharm., 2022, 355(2), 2100359 CrossRef CAS.
- N. A. Alsaif, A. Elwan, M. M. Alanazi, A. J. Obaidullah, W. A. Alanazi and A. F. Alasmari, et al., Design, synthesis and molecular docking of new [1, 2, 4] triazolo [4, 3-a] quinoxaline derivatives as anticancer agents targeting VEGFR-2 kinase, Mol. Diversity, 2022, 26(4), 1915–1932 CrossRef CAS.
- H. A. Mahdy, M. K. Ibrahim, A. M. Metwaly, A. Belal, A. B. Mehany and K. M. El-Gamal, et al., Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4 (3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers, Bioorg. Chem., 2020, 94, 103422 CrossRef CAS PubMed.
- S. Kambe, A one step synthesis of 4-oxo-2-thioxopyrimidine derivatives by the ternary condensation of ethyl cyanoacetate, aldehydes, and thiourea, Synthesis, 1979, 4, 287–289 CrossRef.
- A. A. Nasser, I. H. Eissa, M. R. Oun, M. A. El-Zahabi, M. S. Taghour and A. Belal, et al., Discovery of new pyrimidine-5-carbonitrile derivatives as anticancer agents targeting EGFR WT and EGFR T790M, Org. Biomol. Chem., 2020, 18(38), 7608–7634 RSC.
- I. A. Osman, R. R. Ayyad and H. A. Mahdy, New pyrimidine-5-carbonitrile derivatives as EGFR inhibitors with anticancer and apoptotic activities: design, molecular modeling and synthesis, New J. Chem., 2022, 46(24), 11812–11827 RSC.
- M. A. El-Zahabi, H. Sakr, K. El-Adl, M. Zayed, A. S. Abdelraheem and S. I. Eissa, et al., Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents, Bioorg. Chem., 2020, 104, 104218 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS PubMed.
- F. Denizot and R. Lang, Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability, J. Immunol. Methods, 1986, 89(2), 271–277 CrossRef CAS PubMed.
- M. Thabrew, R. D. Hughes and I. G. McFarlane, Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay, J. Pharm. Pharmacol., 1997, 49(11), 1132–1135 CrossRef CAS.
- T. Aiebchun, P. Mahalapbutr, A. Auepattanapong, O. Khaikate, S. Seetaha and L. Tabtimmai, et al., Identification of vinyl sulfone derivatives as egfr tyrosine kinase inhibitor: in vitro and in silico studies, Molecules, 2021, 26(8), 2211 CrossRef CAS.
- E. M. Mohi El-Deen, M. M. Anwar, A. A. A. El-Gwaad, E. A. Karam, M. K. El-Ashrey and R. R. Kassab, Novel Pyridothienopyrimidine Derivatives: Design, Synthesis and Biological Evaluation as Antimicrobial and Anticancer Agents, Molecules, 2022, 27(3), 803 CrossRef CAS PubMed.
- N. A. Alsaif, A. Elwan, M. M. Alanazi, A. J. Obaidullah, W. A. Alanazi and A. F. Alasmari, et al., Design, synthesis and molecular docking of new [1, 2, 4] triazolo [4, 3-a] quinoxaline derivatives as anticancer agents targeting VEGFR-2 kinase, Mol. Diversity, 2021, 1–18 Search PubMed.
- H. Van De Waterbeemd and E. Gifford, ADMET in silico modelling: towards prediction paradise?, Nat. Rev. Drug Discov., 2003, 2(3), 192–204 CrossRef CAS PubMed.
- A. Elwan, A. E. Abdallah, H. A. Mahdy, M. A. Dahab, M. S. Taghour and E. B. Elkaeed, et al., Modified benzoxazole-based VEGFR-2 inhibitors and apoptosis inducers: design, synthesis, and anti-proliferative evaluation, Molecules, 2022, 27(15), 5047 CrossRef CAS PubMed.
- T. Al-Warhi, M. F. Abo-Ashour, H. Almahli, O. J. Alotaibi, M. M. Al-Sanea and G. H. Al-Ansary, et al., Novel [(N-alkyl-3-indolylmethylene) hydrazono] oxindoles arrest cell cycle and induce cell apoptosis by inhibiting CDK2 and Bcl-2: synthesis, biological evaluation and in silico studies, J. Enzym. Inhib. Med. Chem., 2020, 35(1), 1300–1309 CrossRef CAS.
- A. E. Abdallah, I. H. Eissa, A. B. Mehany, H. Sakr, A. Atwa and K. El-Adl, et al., Immunomodulatory quinazoline-based thalidomide analogs: design, synthesis, apoptosis and anticancer evaluations, J. Mol. Struct., 2023, 1281, 135164 CrossRef CAS.
- Y. Miyazaki, S. Matsunaga, J. Tang, Y. Maeda, M. Nakano and R. J. Philippe, et al., Novel 4-amino-furo [2, 3-d] pyrimidines as Tie-2 and VEGFR2 dual inhibitors, Bioorg. Med. Chem. Lett., 2005, 15(9), 2203–2207 CrossRef CAS PubMed.
- M. Hagras, M. A. Saleh, R. R. Ezz Eldin, A. A. Abuelkhir, E. G. Khidr and A. A. El-Husseiny, et al., 1, 3, 4-Oxadiazole-naphthalene hybrids as potential VEGFR-2 inhibitors: design, synthesis, antiproliferative activity, apoptotic effect, and in silico studies, J. Enzym. Inhib. Med. Chem., 2022, 37(1), 386–402 CrossRef CAS.
- A. M. Farghaly, O. M. AboulWafa, Y. A. Elshaier, W. A. Badawi, H. H. Haridy and H. A. Mubarak, Design, synthesis, and antihypertensive activity of new pyrimidine derivatives endowing new pharmacophores, Med. Chem. Res., 2019, 28, 360–379 CrossRef CAS.
- N. A. Alsaif, M. A. Dahab, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia and M. M. Alanazi, et al., New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: Design, molecular modeling, and synthesis, Bioorg. Chem., 2021, 110, 104807 CrossRef CAS.
- I. H. Eissa, A.-G. A. El-Helby, H. A. Mahdy, M. M. Khalifa, H. A. Elnagar and A. B. Mehany, et al., Discovery of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation, Bioorg. Chem., 2020, 105, 104380 CrossRef CAS PubMed.
- M. M. Alanazi, H. A. Mahdy, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani and A. A. Al-Mehizia, et al., New bis ([1, 2, 4] triazolo)[4, 3-a: 3′, 4′-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation, Bioorg. Chem., 2021, 112, 104949 CrossRef CAS PubMed.
- N. A. Alsaif, M. S. Taghour, M. M. Alanazi, A. J. Obaidullah, W. A. Alanazi and A. Alasmari, et al., Identification of new [1, 2, 4] triazolo [4, 3-a] quinoxalines as potent VEGFR-2 tyrosine kinase inhibitors: Design, synthesis, anticancer evaluation, and in silico studies, Bioorg. Med. Chem., 2021, 46, 116384 CrossRef CAS PubMed.
- R. Talaat, W. El-Sayed, H. Agwa, A. Gamal-Eldeen, S. Moawia and M. Zahran, Novel thalidomide analogs: Anti-angiogenic and apoptotic effects on Hep-G2 and MCF-7 cancer cell lines, Biomed. Aging Pathol., 2014, 4(3), 179–189 CrossRef CAS.
- T. Inoue, K. Kibata, M. Suzuki, S. Nakamura, R. Motoda and K. Orita, Identification of a vascular endothelial growth factor (VEGF) antagonist, sFlt-1, from a human hematopoietic cell line NALM-16, FEBS Lett., 2000, 469(1), 14–18 CrossRef CAS PubMed.
- R. M. Talaat, Soluble angiogenesis factors in sera of Egyptian patients with hepatitis C virus infection: correlation with disease severity, Viral Immunol., 2010, 23(2), 151–157 CrossRef CAS.
- E. B. Elkaeed, I. H. Eissa, H. Elkady, A. Abdelalim, A. M. Alqaisi and A. A. Alsfouk, et al., A Multistage In Silico Study of Natural Potential Inhibitors Targeting SARS-CoV-2 Main Protease, Int. J. Mol. Sci., 2022, 23(15), 8407 CrossRef CAS.
- E. B. Elkaeed, F. S. Youssef, I. H. Eissa, H. Elkady, A. A. Alsfouk and M. L. Ashour, et al., Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease, Int. J. Mol. Sci., 2022, 23(13), 6912 CrossRef CAS PubMed.
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. M. Gobaara, B. A. Alsfouk and D. Z. Husein, et al., Design, Synthesis, Docking, DFT, MD Simulation Studies of a New Nicotinamide-Based Derivative: In Vitro Anticancer and VEGFR-2 Inhibitory Effects, Molecules, 2022, 27(14), 4606 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04182d |
| This journal is © The Royal Society of Chemistry 2023 |