
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the role of microwave thermal and non-thermal effects on WO3-graphene oxide composite synthesis†
Bárbara S. Rodrigues,
Marcos R. S. Vicente and
Juliana S. Souza *
*
Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, Santo André, 09210-580, SP, Brazil. E-mail: juliana.souza@ufabc.edu.br
First published on 6th September 2023
Abstract
The effects of microwave-assisted synthesis on the morphology and crystalline structure of WO3-graphene oxide (GO) composites have been investigated. Using two different microwave reactors, evidence supports that thermal and non-thermal effects significantly influence the properties of the synthesized materials. The findings reveal that the microwave cavity geometry affects how the microwaves are “delivered” to the reactional cavity as a function of time; it also orientates the growth process of the WO3 particles. Consequently, the crystalline structure and morphology are affected. As a result, the WO3-GO composites produced using a CEM reactor exhibit a rounded shape and hexagonal phase of WO3, besides enhanced reduction of GO. Whereas the composites made using an Anton-Paar reactor are composed of sheets and flowers of WO3 with hexagonal, triclinic and/or WO3 hydrate structures and cause a lower reduction on the GO.
1. Introduction
The efficiency of materials in various applications depends directly on their crystalline structure and shape, and the control of these properties, in turn, depends on the synthesis conditions. In this context, using microwaves as a heating source in liquid-phase inorganic synthesis becomes very attractive. This technique allows higher heating rates, reaction speed, yields, selectivity of the obtained product, and reproducibility. It also allows excellent control of the reaction parameters and automation.1–5 In general terms, microwave chemistry involves selective dielectric heating of ionic compounds and solvents based on their response to the oscillating electric field of microwaves. This way, microwave irradiation can heat the target material without heating the entire oven or oil bath, saving time and energy.1–5Some scientists believe microwaves do not influence the orientation of molecules during collisions nor the activation energy of the reaction but only act on the temperature parameter of the Arrhenius equation. In this sense, selective dielectric heating increases the reaction speed because it allows the activation energy of a chemical reaction to be reached more quickly, as the radiation can transfer much power to the response in a short time interval. From the point of view of obtaining inorganic particles, the thermal effects are traditionally associated with the nucleation and growth steps.2,4,6–8 Microwaves produce homogeneous heating of the reaction medium; thus, the nucleation step is improved, resulting in particles of controlled size. In addition, the nature of the reactants is a much more critical issue than in syntheses using traditional heating methods. Depending on the species employed, the dissolution rate of the reactants can change considerably, affecting the reaction kinetics.7 Also, many inorganic syntheses use organic species as capping agents or in producing reaction intermediates. These organic species generally interact very strongly with microwaves, creating a significant increase in the heating rate, solubility of reactants, and speed of reactions, which can promote very different results from those observed in syntheses using conventional heating.9,10
Other scientists believe microwaves affect molecular orientation during collisions, affecting the pre-exponential term of the Arrhenius equation. These effects are caused by the unique interaction of microwaves with the irradiated species and are called “microwave-specific effects” or “non-thermal effects”. The non-thermal effects include superheating, selective heating of polar species, formation of “molecular radiators” by directly coupling the microwave energy with specific reactant species, and a homogeneous solution. From the point of view of inorganic synthesis, these non-thermal effects directly affect the interface events.3,4,11 In the growth process of metal and metal oxide particles, for example, liquid phase growth occurs by the adsorption of different species on the surface of the growth nuclei formed in the initial steps, in addition to the oriented aggregation of nuclei.7,12–14 Thus, the unique interactions of microwaves with the different species in the reaction medium affect the adsorption capacity of the surfaces in the solid state and the reactivity of the species in solution with these surfaces.7 Another interesting non-thermal effect is related to the fact that the geometry of the microwave cavity can affect the morphology of the particles.15,16 Thus, different morphologies can be obtained simply by changing the equipment used in the procedure. This paper provides evidence of non-thermal effects through synthesizing WO3-graphene oxide composites using two additional microwave reactors.
WO3 was chosen due to its applicability to energy conversion devices. WO3 can be used to develop supercapacitors,17 lithium-ion batteries,18 electrochromic devices,19 electrocatalytic water splitting,20 photochromic devices,21 sensors22 and photocatalysts.23,24 This vast number of applications is correlated to its crystalline structure, morphology, bandgap energy and potential window.20 Few works explore microwave irradiation as a heating source in synthesizing WO3-based materials.25–37 Also, most of these studies only used the microwave to decrease the reaction time.25–28,31–33,36,37 A few studies suggested microwaves can affect the properties of the resultant materials.29,30,34,35 Also, most works used water as the solvent,25–30,32–37 a medium microwave absorber. Besides that, usually, the authors do not make clear necessary information, such as (i) the kind of oven used (domestic or dedicated);26–28,32–35,37 (ii) the microwave mode (monomode or multimode);26–28,32,34–37 (iii) kind of irradiation (constant power or constant heating);34 and (iv) irradiation power.25,33,34
A common strategy to improve the efficiency of WO3-based devices is to make a composite with graphene oxide.17,38–40 The study of the microwave-assisted synthesis of this kind of composite is also very restricted. The irradiation provided enough energy for this material to reduce GO to rGO. Also, it removes the oxygen functional groups. However, it has yet to be provided with any attempt mechanism.34
In this context, the present study shows a detailed development of the microwave-assisted synthesis of WO3-graphene oxide composites. The critical conditions that affect microwave chemistry are described and discussed. Also, we provide evidence on how the non-thermal effects of microwave-assisted synthesis can affect the morphology and interaction between both materials.
2. Material and methods
2.1. Synthesis of WO3, GO and WO3-GO materials
The synthesis procedure was based on a method previously published by the group41 with modifications. For the samples of WO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GO, 1:1 (molar ratio) 151.2 mg of graphene oxide was suspended in 81 mL of ethylene glycol and sonicated for 30 min. Then, 1.512 g of ammonium metatungstate and 32.24 g of oxalic acid were added to the solution previously prepared. The mixture was sonicated again for 15 min. 7 mL of the resultant solution was transferred to a 10 mL glass vial sealed and placed in the microwave cavity (Discovery CEM or Anton Paar). The heating mode was the constant temperature, where the irradiation power was kept at a maximum of 300 W. Table 1 summarizes the synthesis conditions.
:GO, 1:1 (molar ratio) 151.2 mg of graphene oxide was suspended in 81 mL of ethylene glycol and sonicated for 30 min. Then, 1.512 g of ammonium metatungstate and 32.24 g of oxalic acid were added to the solution previously prepared. The mixture was sonicated again for 15 min. 7 mL of the resultant solution was transferred to a 10 mL glass vial sealed and placed in the microwave cavity (Discovery CEM or Anton Paar). The heating mode was the constant temperature, where the irradiation power was kept at a maximum of 300 W. Table 1 summarizes the synthesis conditions.
| Reactional condition | WO3:GO |
Heating protocol | Step 1 | Step 2 |
|---|---|---|---|---|
| (A) | 1:1 |
Heat as fast as possible | 180 °C for 1 min | 200 °C for 1 min |
| (B) | 1:1 |
10 min ramp | — | 200 °C for 1 min |
The obtained suspensions were rinsed and centrifuged once with water and two times with isopropyl alcohol, drying for 24 h at 60 °C. After this, the materials were annealed for one hour at 380 °C.
For control, pure WO3 and GO materials were produced following the same procedures, without adding GO and W precursor, respectively. The materials were named WO3(X)–Y, GO(X)–Y and WO3-GO(X)–Y, where (X) are conditions A and B described in Table 1, and Y are AP (for the samples prepared using the Anton Paar reactor) and CEM (for the samples prepared using the CEM reactor).
2.2. Characterization
Scanning electron microscopy (SEM) images of the catalysts were collected on the SEM-FEG scanning electron microscope HR Inspect F50 (FEI). WO3-based materials were suspended in isopropanol, sonicated for 30 min, and deposited on thermic oxide 200 nm ultrathin of ø6, type 〈100〉 (Ted Pella).X-ray diffraction (XRD) patterns were obtained using a D8 (FOCUS) X-ray diffractometer with a Cu Kα radiation source operating at 40 kV and 40 mA. The XRD profiles were collected between 2θ: 10° and 60°. Raman spectra were obtained using an RFS100/S (Bruker) FT-Raman spectrometer with excitation at 1064 nm from an Nd:YAG laser. Diffuse reflectance spectra were measured on a Cary 50Scan (Varian) spectrometer using a reflectance accessory coupled optical fiber (Pike).
X-ray photoelectron spectra were obtained through a K-alpha XPS, Thermo Fisher Scientific, aiming to identify the composition of sample compounds. It used an Al Kα emission with applied vacuum <10−8 mbar. The experiments were performed with 1400 and 50 eV energy, with 1.0 and 0.1 eV resolutions, respectively.
3. Results and discussion
Traditionally, the properties of a material are strictly related to the synthesis conditions: the nature and proportion of reactants, solvent, temperature and reaction time. However, in this study, all the previously mentioned reaction conditions are the same for synthesizing all samples. In this sense, it is essential to highlight that the use of microwaves as a heating source adds other variables to be considered in the reaction conditions, being the volume of the reactional mixture and dimensions of the vial (since microwaves can penetrate about 2 cm in the reaction medium), irradiation mode (monomode or multimode), and operation mode (fixed power or fixed temperature).42In multimode microwaves, the electromagnetic field is distributed chaotically inside the cavity. These cavities are usually large, so the field density is low. Therefore, the equipment must operate at high irradiation powers to achieve high heating rates. On the other hand, this equipment allows the performance of several reactions simultaneously, generating a more significant amount of product.42 In the case of monomode microwaves, only a single mode of the electromagnetic wave interacts with the reaction mixture. These devices have a small cavity, which provides a high field density; as a result, low microwave power is required to achieve high heating rates.42,43
When fixed power operating mode is used, the equipment is set to maintain a single power during the entire reaction time. In this condition, there will invariably be a temperature rise.42 It is essential to point out that the heating rate is a crucial parameter in the synthesis result. Therefore, this is worth emphasizing since the magnetron used with a microwave source becomes less efficient with time. Thus, even if two devices of the same brand are used in the fixed power mode of operation, the heating rates can be different if the magnetrons are used for various lengths of time. Differences in the design of the cavity geometry can also lead to changes in the microwave power introduced into the reaction cavity.42 Thus, the use of two different brands of equipment can lead to other synthesis products when the fixed power mode is employed.
In this sense, theory indicates that the only way to ensure the reproducibility of a chemical reaction under microwave conditions in different equipment (of the same brand or not) is by controlling the temperature and reaction time.42 In this context, the fixed temperature mode is more interesting since, in this mode of operation, the temperature is kept constant for a determined time through microwave irradiation. Furthermore, the irradiation power is modulated to keep the temperature constant, and a power limit value is set.42
In this study, all reactions were performed using the same volume and type of reaction vial; the two devices employed the same irradiation mode (monomode); and the method of operation was fixed temperature, keeping the maximum power at 300 W. Theoretically, these conditions should guarantee the reproducibility of the reaction. Thus, we hypothesize that differences in the properties of the resultant materials may result in thermal and non-thermal effects associated with the microwave irradiation.
The first property evaluated was morphology. Fig. 1 shows the SEM images of the WO3, GO and WO3-GO samples. The images show changes in the morphologies of the WO3 obtained in the different reactors. WO3(A and B)-AP comprises sheets that aggregate into spherical architectures. EDS images of the WO3-GO samples (Fig. 2) shows clearly the spherical shape. WO3(A and B)-CEM SEM images (Fig. 1 and 2) also indicate spherical shapes but are smaller in this case. Also, the spheres are composed of smaller particles with irregular shapes (see Fig. S1†).
 | ||
| Fig. 1 Backscattered SEM images of WO3, GO and WO3-GO samples. | ||
 | ||
| Fig. 2 Secondary electron SEM images and EDS images of the WO3-GO samples. | ||
Thus, it is possible to observe a trend in the morphologies of the materials produced as a function of the reactor used for the synthesis. This fact should be related mainly to the geometry of the cavity, which affects how the electromagnetic radiation reaches the reaction medium, promoting unique orientations in crystal growth,15,16 and a non-thermal effect.
GO exhibits a distinctive aspect that does not vary significantly upon changing the microwave reactor (Fig. 1 and 2). Finally, the images of the WO3-GO composites show that the samples seem to be a mixture of the morphologies of the separated materials (Fig. 1 and 2). However, the size of the WO3 particles seems to change when the WO3(A and B)-CEM samples are compared to those observed on WO3(A and B)-GO-CEM samples.
Fig. 2 shows the secondary electron (SE) SEM images of the WO3-GO samples and the EDS images. The contrast observed in the SE images indicates the difference in the atomic mass of the species that compose the sample. Thus, it shows the regions rich in carbon (dark grey) and tungsten (light grey). Furthermore, EDS images show that element W is present in the same parts of bright areas in the SE images. In contrast, elements C and O are present in the darker areas of the SE images. Based on these images, it is possible to observe that the graphene and WO3 are mixed very inhomogeneously. This observation can be ascribed to the high molar ratio of GO used to prepare the material. However, it is impossible to assess whether there is a chemical interaction between the WO3 and GO particles at the interface between the two materials.
XPS spectra of the WO3-GO samples (Fig. 3) were collected to understand chemical changes promoted by the irradiation conditions. Table 2 summarizes the atomic percentage of the elements W, C and O calculated from the survey spectra (Fig. 3). It was observed that the samples produced using the microwave CEM (WO3-GO(A) and (B) CEM) exhibit a small percentage of nitrogen. We believe the nitrogen can result from adsorbed nitrogen species from the ammonium salt. On the other hand, the samples produced on the microwave Anton Paar (WO3-GO(A) and (B) AP) did not show the presence of nitrogen. Also, it was observed that the proportion of W produced is higher for the WO3-GO(A) and (B) CEM samples. Finally, when the same microwave reactor is compared, the heating mode “heat as fast as possible” produced samples with higher W content (WO3-GO(A)-CEM and WO3-GO(A)-AP samples). It is essential to highlight that XPS spectroscopy can detect species present in the 5 nm depth of the samples; thus, the atomic percentage values must be considered carefully.
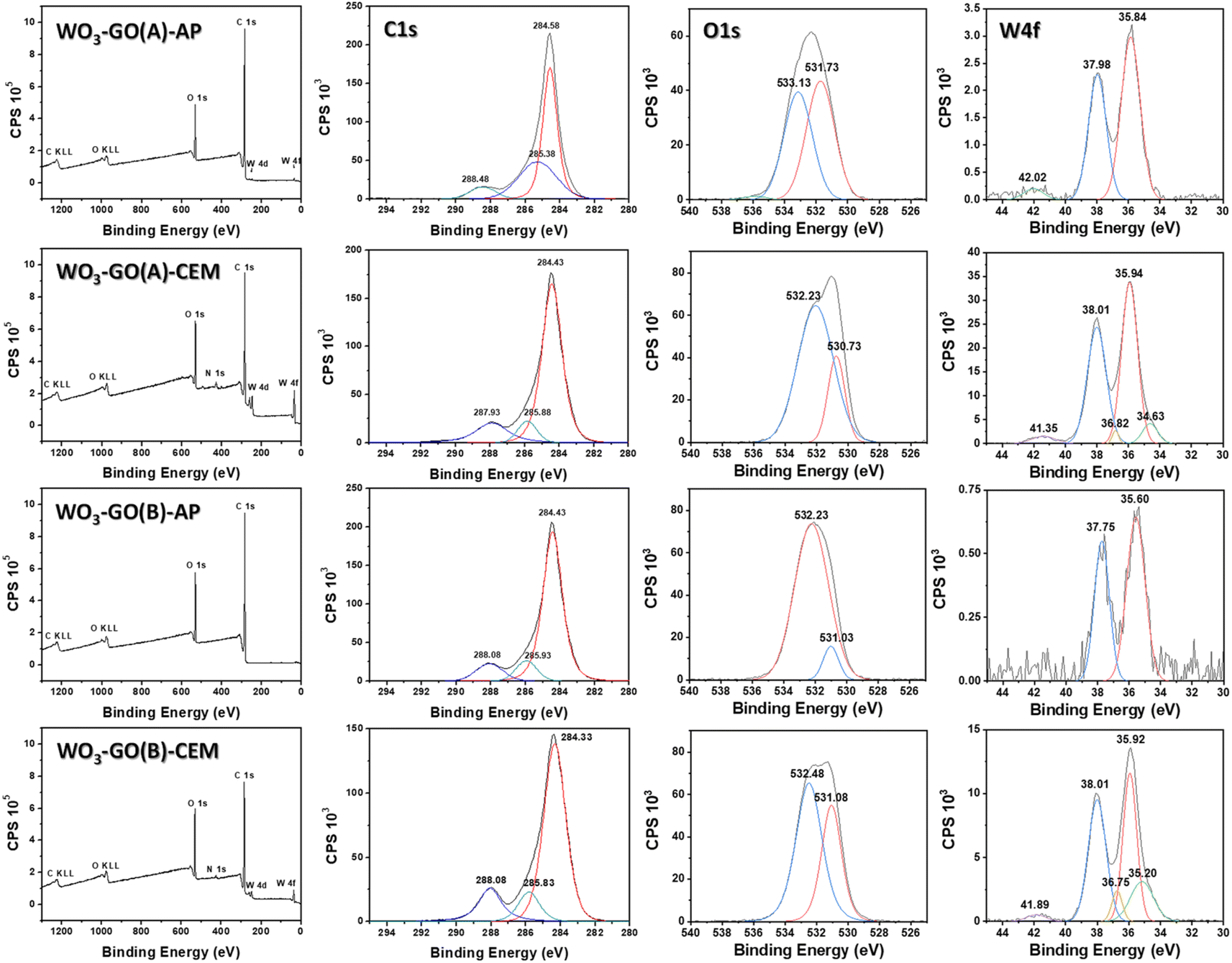 | ||
| Fig. 3 The survey, C 1s, O 1s and W 4f high-resolution spectra of the WO3-GO samples. | ||
| Material | Element (%) | |||
|---|---|---|---|---|
| W | C | O | N | |
| WO3-GO(A)-AP | 0.09 | 86.01 | 13.90 | — |
| WO3-GO(A)-CEM | 1.11 | 79.79 | 18.16 | 0.94 |
| WO3-GO(B)-AP | 0.02 | 84.58 | 15.40 | |
| WO3-GO(A)-CEM | 0.47 | 79.44 | 20.07 | 0.02 |
High-resolution XPS W spectra of core-level W 4f (Fig. 3) can be deconvoluted into two distinct peaks, W 4f7/2 and W 4f5/2, with binding energies of 35.60 to 35.95 eV and 38.01 to 37.75 eV, respectively.44–47 The presence of spin–orbit coupling of ∼2.1 suggests the existence of the WO6+ oxidation state.45,46 For the WO3-GO-CEM materials, these peaks can be deconvoluted in two pairs. For these samples, a distinct pair of XPS peaks are observed at lower binding energies of 36.82 to 36.75 eV and 35.20 to 34.63 eV, which can be correlated to the existence of W5+ oxidation states.46 This result suggests that for the samples produced using the CEM reactor, there is the formation of defective WO3−x. The materials also exhibit a weak emission at ∼42 eV, ascribed to W 5p3/2, which suggests the oxide formation of WO3.46 WO3-GO(B)-AP does not exhibit a peak around 42 eV, which can be ascribed to the low-intensity of the W emission.
Fig. 3 also shows the high-resolution XPS O 1s spectra. The spectra of all samples can be deconvoluted into two distinct peaks; however, the shape and binding energies are much different. The spectra of the samples WO3-GO(A)-CEM, WO3-GO(B)-AP and WO3-GO(B)-CEM exhibit a peak at ∼532 eV, which was previously ascribed to the bonds of the oxygen and tungsten in lower oxidation states, due to a small amount of water containing h-WO3.46 However, W at low oxidation states has been only observed on WO3-GO(A and B)-CEM samples. This peak has also been ascribed to the O–H in adsorbed H2O;48,49 to C–O–C bond;44 and to oxygen chemisorbed on the surface of prepared composites.50 The samples, WO3-GO(B)-AP and CEM, also show a peak at ∼531 eV, assigned to the chemisorbed oxygen of the surface hydroxyls.51 Also, 531 eV is the minimum energy required to disassemble W–O and the oxygen anion (O2−) on the lattice sites of Wolfram.45 Conversely, the sample WO3-GO(A)-CEM exhibits a peak at 530.73 eV, which may correspond to the O2− oxygen bond with W6+ atoms.46,51 Also, it could be ascribed to chemisorbed oxygen species present at the surface of the composite.52 Finally, the sample WO3-GO(A)-AP shows a peak at 533.13 eV, which can be ascribed to hydroxyl (–OH) groups on the surface of the composites or the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the composite.49 In summary, the high-resolution O 1s spectra show mainly ascribed to oxygen species non-chemically bonded to the W. This observation can be ascribed to the low atomic percentage of W compared to C and O in the samples. The only sample that shows binding energy that can be ascribed to the W–O bond exhibits a higher atomic percentage of W (Table 2).
O bond of the composite.49 In summary, the high-resolution O 1s spectra show mainly ascribed to oxygen species non-chemically bonded to the W. This observation can be ascribed to the low atomic percentage of W compared to C and O in the samples. The only sample that shows binding energy that can be ascribed to the W–O bond exhibits a higher atomic percentage of W (Table 2).
The deconvoluted C 1s core level of XPS high-resolution for the WO3-GO samples shows tree peaks in Fig. 3. The binding energies are around 284.38 ± 0.05, 285.91 ± 0.08 eV and 288.00 ± 0.08 eV for the samples WO3-GO(A)-CEM, WO3-GO(B)-AP and WO3-GO(B)-CEM. The binding energies for the sample WO3-GO(A) show small displacement; coincidently, the samples exhibit a higher atomic percentage of C (Table 2). The peak around 288 eV may correspond to carbonate species ascribed to OC–O bonds.44,49,51–54 The peak around 284 is associated with the CC–C bond, corresponding to sp2 bonding in graphene.48,50,52,54,55 The peak around 285 is associated with C–O bonds.45,49,50,52
Thus, different from the morphologies, the chemical composition varies significantly upon changing the reactor cavity and heating mode. These aspects can result from changes in the crystalline structure of the materials.
To understand the effect of microwave conditions on the crystalline structure, Raman spectra (Fig. 4) and XRD patterns (Fig. 5) of the samples were collected. Raman spectra of WO3-(GO) samples (Fig. 4) show the characteristics bands of WO3 around 806 cm−1 and 701 cm−1 ascribed to O–W–O antisymmetric stretching, and W2O6 and W3O8 stretching.56,57
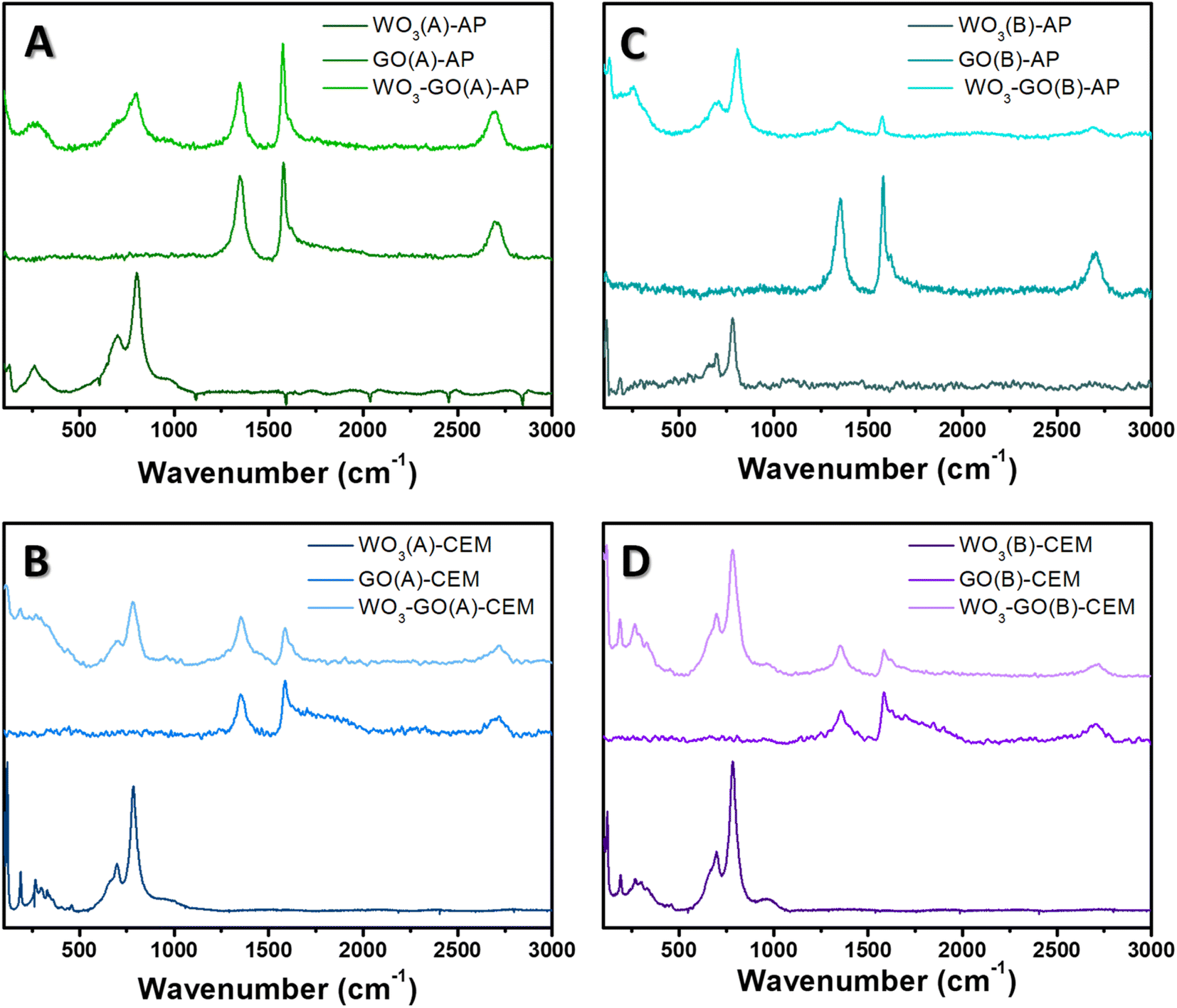 | ||
| Fig. 4 Raman spectra of (WO3)-(GO)-(A)-AP (a), (WO3)-(GO)-(A)-CEM (b), (WO3)-(GO)-(B)-AP (c) and (WO3)-(GO)-(B)-AP (d) samples. | ||
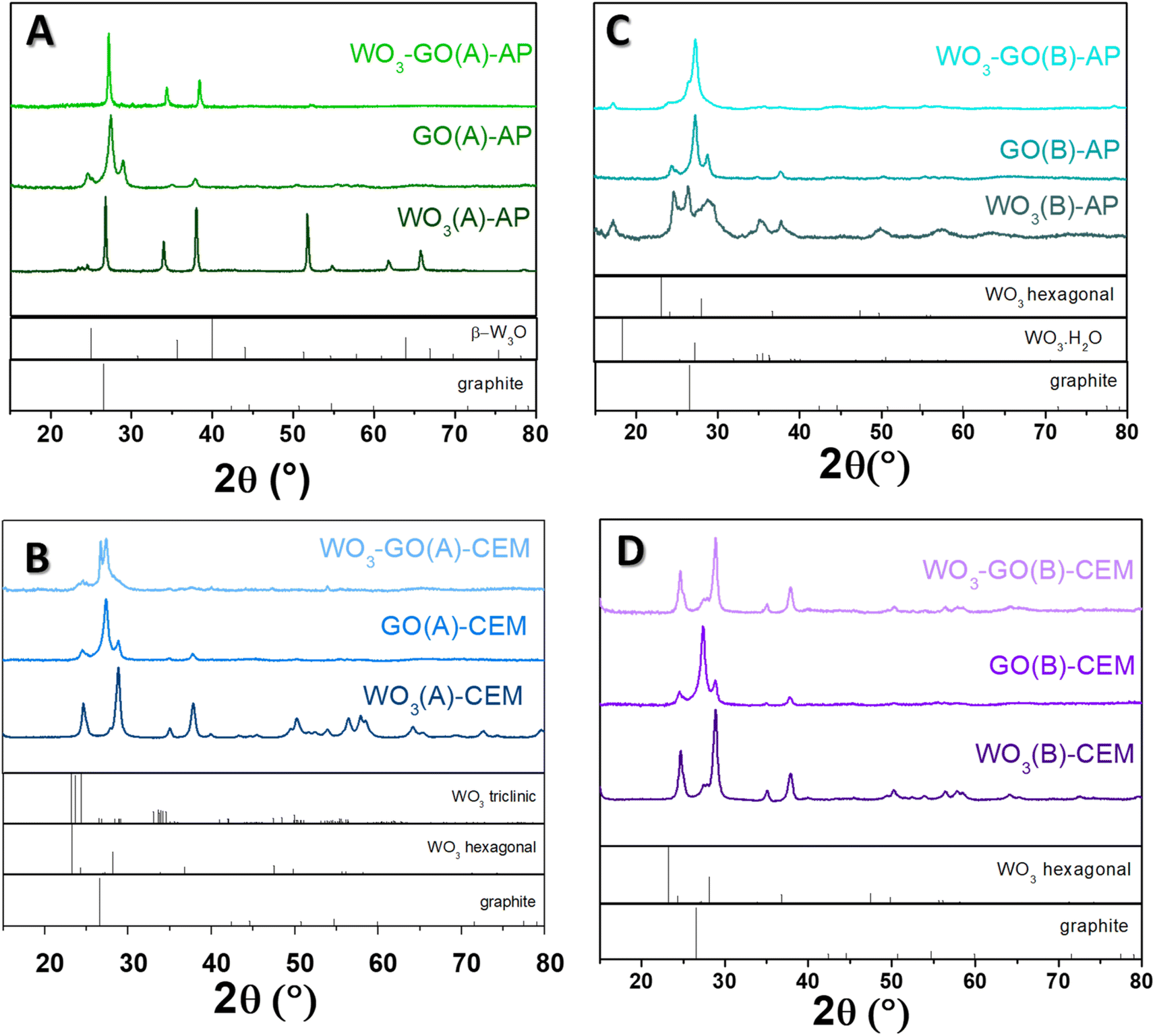 | ||
| Fig. 5 XRD diffractogram of (WO3)-(GO)-(A)-AP (a), (WO3)-(GO)-(A)-CEM (b), (WO3)-(GO)-(B)-AP (c) and (WO3)-(GO)-(B)-AP (d) samples. | ||
The Raman spectrum of GO and WO3-GO samples (Fig. 4) exhibits two bands at around 1350 and 1604 cm−1, corresponding to the D and G bands of graphitic structure. The G band is related to the C–C vibrations of the carbon with sp2 orbitals and is characteristic of graphitic sheets' distinct peaks. D band is associated with the vibrational defects of the C–C bond.58 The higher ratio between the ID/IG bands can be related to the reduction degree of GO.59 The ID/IG ratio calculated for GO(A)-AP (Fig. 4a) was 0.93; in comparison, WO3-GO(A)-AP (Fig. 4a) shows a smaller value (0.88), which is associated with partial oxidation of graphene. On the other hand, the ID/IG ratio for GO(A)-CEM (Fig. 4b) is smaller (0.79) than the value calculated for WO3-GO(A)-CEM (1.29). The ID/IG ratio is similar for the WO3-GO(B)-AP and GO(B)-AP samples (0.77) (Fig. 4c). Finally, the ID/IG ratio of WO3-GO(B)-CEM was calculated as (1.02), whereas the value calculated for GO (B)-CEM was 0.92.
These results show that for the samples prepared using the CEM reactor, the ID/IG ratio of GO-CEM was not affected by the heating mode. The formation of WO3 lead decreased the reduction degree of GO on the WO3-GO-CEM samples. Thus, that led us to believe that de GO takes part in the WO3 formation; this observation is consistent with the changes in the size of WO3 on the WO3(A and B)-CEM and WO3(A and B)-GO-CEM samples (Fig. 1). As a result, it may indicate the formation of a real heterojunction between both materials.
Among the GO-AP samples, a fast heating mode induced the reduction degree of GO(A)-AP compared to the GO(B)-AP. The ID/IG ratio value for the GO(B)-AP and WO3-GO(B)-AP may indicate that a mixture of the components forms these materials. Finally, the lower ID/IG ratio of WO3-GO(A)-AP compared to GO(B)-AP may also indicate that the GO takes part in the WO3 growth process. For this set of materials, no evident changes in morphology were observed (Fig. 1).
The crystalline phase of the samples was confirmed via XRD diffraction. All pristine GO samples (Fig. 5) have shown the same pattern, with an intense peak around 25° found in the GO structure. The peak around 24.5° was found in the few layers of rGO obtained from graphite flakes.60
The peaks observed for WO3(A)-AP (Fig. 5a) are associated with the crystalline phase of W3O (PDF number 01-071-3817).61,62 The same peaks are observed in WO3-GO(A)-AP; however, for this sample, no peaks can be attributed to the crystalline structure of graphene or graphite. On the other hand, WO3(A)-CEM (Fig. 5b) showed peaks associated with the hexagonal phase of WO3 (PDF 01-085-2459).63 In comparison, WO3-GO(A)-CEM exhibited some peaks of WO3 hexagonal in the mixture with the triclinic structure of WO3 (PDF 01-071-0305).64 Observing a hexagonal phase is consistent with the high-resolution W 4f XPS spectrum of this material (Fig. 3).
WO3(B)-AP (Fig. 5c) showed a mixed phase between WO3 hexagonal and the hydrated form of WO3 with a characteristic peak at 16.8°. WO3-GO(B)-AP, in turn, exhibited peaks related to the pure orthorhombic phase of WO3·H2O (PDF 01-084-0886).65 Finally, WO3(B)-CEM and WO3-GO(B)-CEM (Fig. 5d) showed both the pure hexagonal phase of WO3, which is consistent with the W 4f XPS results (Fig. 3).
Hexagonal WO3 can be obtained under conventional synthesis by heating the tungsten precursor hydrothermally at 180 °C for 12 to 24 h.66–68 However, the orthorhombic WO3 hydrate can also be prepared under milder conditions.69 In this sense, the microwave-assisted synthesis described here reduced the time required for the synthesis, which may be ascribed to the fast heating hates (a thermal effect). On the other hand, the triclinic structure is typically obtained at higher temperatures during an annealing process.70,71 Thus, the triclinic structure produced here may result from creating hot spots during the synthesis. These hot spots can make a higher temperature locally, which some scientists consider a non-thermal effect.
Interestingly, we are unaware of the use of hydrothermal treatment to obtain the W3O phase. Traditionally, this structure is produced by sputtering methods followed by calcination.72,73 Thus, the obtention of the W3O phase may also be ascribed to creating hot spots.
Understanding the reasons for such significant changes is a great challenge. The reactional condition (A) (Table 1) implies the fastest possible heating. In this sense, the magnetron lifespan and the design of the cavity geometry will impact the heating rate when AP and CEM equipment are considered. Therefore, changes in the characteristics of the GO, WO3, and WO3-GO(A)-AP materials when compared to the GO, WO3, and WO3-GO(A)-CEM materials are expected to be different. By observing the heating curves (ESI file†), it is possible to notice that both pieces of equipment take around 1 minute to reach 180 °C. However, when the CEM equipment is used, the irradiation power values are closer to 300 W during the heating process to 180 °C.
On the other hand, when the AP equipment is used, the irradiation power is more relative to the maximum value for less than 30 seconds. This observation leads us to believe that microwaves interact more intensely with the particles in the reaction media at the initial stages of the synthesis when the CEM equipment is used. Thus, the differences in microwave intensity at the beginning of the synthesis process could be a reason for the changes in the properties observed.
For reaction condition B (Table 1), both reactors must heat the reactional mixture to 200 °C for 10 minutes. The heating graphs (ESI-file†) show that the irradiation power delivered by the reactors as a function of time is very different. For the CEM instrument, the irradiation power is higher at the final stages of the process, whereas the AP instrument delivers higher power at the beginning of the process.
Considering all results, it is possible to observe that the irradiation power intensity as a function of time is more homogeneous when the CEM instrument is used. This characteristic may be responsible for producing crystalline structures, which are commonly reported under milder conditions through conventional synthesis. In this sense, microwaves show the advantage of reducing the reaction time due to a thermal effect (high heating rate).
On the other hand, the irradiation power is typically more intense at the beginning of the synthesis process when the Anton Paar instrument is used. This characteristic may create more hot spots (a non-thermal effect), leading to the formation of crystalline structures typically obtained under higher temperatures using conventional heating sources.
4. Conclusion
This work discusses the thermal and non-thermal effects associated with microwave irradiation as a heating source in the synthesis of WO3-GO composites. For this study, two reactors with different cavities operating in monomode and fixed temperature were used.It was observed that the microwave cavity of the CEM provides a more homogeneous irradiation intensity as a function of reaction time. On the other hand, the Anton Paar reactor concentrates the highest irradiation power intensity at the beginning of the synthesis process.
As a result, it is observed that the crystal structure of pure WO3 and WO3 in the composite with GO prepared in the microwave CEM is predominantly hexagonal. This structure is obtained when WO3 is synthesized under a conventional hydrothermal route and temperature similar to that employed in this work. However, under the conventional route, the synthesis time is much longer. Therefore, the main advantage of using microwaves is the reduction of time.
For pristine WO3 and the composite with GO prepared in the Anton Paar microwave, the crystalline structures observed are only produced at elevated temperatures when conventional heating methods are used. This result leads us to believe that hot spots create a significant local temperature increase (a non-thermal effect).
It was also observed that the geometry of the cavity guides the growth of the particles, generating distinct morphologies in the materials as a function of the reactor cavity; this is another evidence of a non-thermal effect. Finally, the data suggest that GO participates in the growth process of WO3, which evidences the formation of a heterojunction.
By focusing on the thermal and non-thermal effects of microwaves in WO3-graphene oxide synthesis, we demonstrate the potential for tunning the material properties, advancing material synthesis and microwave-assisted techniques in developing high-performance composites.
Ethical approval
The experiments reported in this paper do not involve tissue samples from human subjects (including blood or sweat) or animal experiments.Author contributions
Bárbara Scola Rodrigues was responsible for the synthesis and characterization of the materials; she also contributed to the writing of the manuscript. Marcos Roberto da Silva Vicente collected the SEM images and data for the analysis. Dr Juliana do Santos de Souza was responsible for the experimental conception and design, data analysis and manuscript writing.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by FAPESP (grants 2021/05958-4 and 2019/26010-9). This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. The authors also acknowledge Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). Finally, we thank LNNano-CNPEM for using the SEM facility and the Multi-users platform (CEM) at UFABC for instrumental facilities.References
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC
.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358–1374 RSC
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chem.–Eur. J., 2005, 11, 440–452 CrossRef CAS PubMed
- Y.-J. Zhu and F. Chen, Chem. Rev., 2014, 114, 6462–6555 CrossRef CAS PubMed
- B. L. Hayes, 2006.
- P. Chu, F. G. Dwyer and J. C. Vartuli, 1988.
- G. A. Tompsett, W. C. Conner and K. S. Yngvesson, ChemPhysChem, 2006, 7, 296–319 CrossRef CAS PubMed
- D. Serrano, M. Uguina, R. Sanz, E. Castillo, A. Rodrıguez and P. Sanchez, Microporous Mesoporous Mater., 2004, 69, 197–208 CrossRef CAS
- M. Niederberger and G. Garnweitner, Chem.–Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed
- G. Garnweitner and M. Niederberger, J. Mater. Chem., 2008, 18, 1171–1182 RSC
- W. C. Conner and G. A. Tompsett, J. Phys. Chem. B, 2008, 112, 2110–2118 CrossRef CAS PubMed
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef PubMed
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC
- Q. Zhang, S.-J. Liu and S.-H. Yu, J. Mater. Chem., 2009, 19, 191–207 RSC
- W. C. Conner, G. Tompsett, K.-H. Lee and K. S. Yngvesson, J. Phys. Chem. B, 2004, 108, 13913–13920 CrossRef CAS
- B. Panzarella, G. A. Tompsett, K. S. Yngvesson and W. C. Conner, J. Phys. Chem. B, 2007, 111, 12657–12667 CrossRef CAS PubMed
- Z. C. Yang, H. H. Zhang, B. Ma, L. Q. Xie, Y. T. Chen, Z. H. Yuan, K. L. Zhang and J. Wei, Appl. Surf. Sci., 2019, 463, 150–158 CrossRef CAS
- A. S. Lakhnot, T. Gupta, Y. Singh, P. Hundekar, R. Jain, F. D. Han and N. Koratkar, Energy Storage Mater., 2020, 27, 506–513 CrossRef
- X. L. Zhong, X. Q. Liu and X. G. Diao, J. Inorg. Mater., 2021, 36, 128–139 CrossRef
- T. H. Wondimu, A. W. Bayeh, D. M. Kabtamu, Q. Xu, P. Leung and A. A. Shah, Int. J. Hydrogen Energy, 2022, 47, 20378–20397 CrossRef CAS
- A. L. Popov, N. M. Zholobak, O. I. Balko, O. B. Balko, A. B. Shcherbakov, N. R. Popova, O. S. Ivanova, A. E. Baranchikov and V. K. Ivanov, J. Photochem. Photobiol., B, 2018, 178, 395–403 CrossRef CAS PubMed
- V. Ambardekar, T. Bhowmick and P. P. Bandyopadhyay, Int. J. Hydrogen Energy, 2022, 47, 15120–15131 CrossRef CAS
- B. Babu, R. Koutavarapu, J. Shim, J. Kim and K. Yoo, Chemosphere, 2021, 268, 129346 CrossRef CAS PubMed
- S. Kitano, M. Yamauchi, S. Hata, R. Watanabe and M. Sadakiyo, Green Chem., 2016, 18, 3700–3706 RSC
- M. Parthibavarman, M. Karthik and S. Prabhakaran, Vacuum, 2018, 155, 224–232 CrossRef CAS
- K. Santhi, C. Rani and S. Karuppuchamy, J. Mater. Sci.: Mater. Electron., 2016, 27, 5033–5038 CrossRef CAS
- S. M. Harshulkhan, K. Janaki, G. Velraj, R. S. Ganapthy and M. Nagarajan, J. Mater. Sci.: Mater. Electron., 2016, 27, 4744–4751 CrossRef
- K. Santhi, C. Rani, R. D. Kumar and S. Karuppuchamy, J. Mater. Sci.: Mater. Electron., 2015, 26, 10068–10074 CrossRef CAS
- D. B. Hernandez-Uresti, D. Sánchez-Martínez, A. Martínez-de La Cruz, S. Sepúlveda-Guzmán and L. M. Torres-Martínez, Ceram. Int., 2014, 40, 4767–4775 CrossRef CAS
- J. Sungpanich, T. Thongtem and S. Thongtem, J. Nanomater., 2014, 739251 Search PubMed
- C.-T. Lin and T.-H. Tsai, Asian J. Chem., 2013, 25, 7098–7102 CrossRef CAS
- Y.-C. Hsiao and C.-C. Hu, J. Electrochem. Soc., 2013, 160, H279 CrossRef CAS
- R. Adhikari, G. Gyawali, T. Sekino and S. W. Lee, J. Solid State Chem., 2013, 197, 560–565 CrossRef CAS
- S. Thangavel, M. Elayaperumal and G. Venugopal, Mater. Express, 2012, 2, 327–334 CrossRef CAS
- X. T. Yu, H. X. Liu, Y. Shen, J. Y. Xu, F. Y. Cai, T. Li, J. Lü and W. Cao, Inorg. Chem. Commun., 2020, 120, 108147 CrossRef CAS
- M. Parthibavarman, M. Karthik and S. Prabhakaran, J. Cluster Sci., 2019, 30, 495–506 CrossRef CAS
- M. Sumathi, A. Prakasam and P. Anbarasan, J. Mater. Sci.: Mater. Electron., 2019, 30, 3294–3304 CrossRef CAS
- X. T. Chang, S. B. Sun, L. H. Dong, X. Hu and Y. S. Yin, Electrochim. Acta, 2014, 129, 40–46 CrossRef CAS
- A. K. Gautam, M. Faraz and N. Khare, Ceram. Int., 2021, 47, 27885–27889 CrossRef CAS
- T. H. Wondimu, G. C. Chen, D. M. Kabtamu, H. Y. Chen, A. W. Bayeh, H. C. Huang and C. H. Wang, Int. J. Hydrogen Energy, 2018, 43, 6481–6490 CrossRef CAS
- M. Kuznetsova, S. A. Oliveira, B. S. Rodrigues and J. S. Souza, Top. Catal., 2021, 64, 748–757 CrossRef CAS
- J. Kremsner and A. Stadler, Basics, Equipment & Application, 2013, vol. 51 Search PubMed
- B. L. Hayes, Microwave synthesis: chemistry at the speed of light, Cem Corporation, 2002 Search PubMed
- J. Jayachandiran, M. Arivanandhan, O. Padmaraj, R. Jayavel and D. Nedumaran, Adv. Compos. Hybrid Mater.s, 2020, 3, 16–30 CrossRef CAS
- R. Samal, B. Chakraborty, M. Saxena, D. J. Late and C. S. Rout, ACS Sustain. Chem. Eng., 2018, 7, 2350–2359 CrossRef
- P. Bhojane and P. M. Shirage, J. Energy Storage, 2022, 55, 105649 CrossRef
- S. Zhang, H. Li and Z. Yang, J. Alloys Compd., 2017, 722, 555–563 CrossRef CAS
- Z. Yousaf, S. Sajjad, S. A. K. Leghari, S. Noor, A. Kanwal, S. H. Bhatti, K. H. Mahmoud and Z. M. El-Bahy, J. Environ. Chem. Eng., 2021, 9, 106746 CrossRef CAS
- A. Abd Elhakim, M. El-Kemary, M. M. Ibrahim, I. M. El-Mehasseb and H. S. El-Sheshtawy, Appl. Surf. Sci., 2021, 564, 150410 CrossRef
- Z. Jiang, W. Chen, L. Jin, F. Cui, Z. Song and C. Zhu, Nanomaterials, 2018, 8, 909 CrossRef PubMed
- R. Gayathri, P. Rajeswaran, G. Raja, S. Bavaji, N. Ameen and M. Shkir, Diamond Relat. Mater., 2021, 119, 108562 CrossRef CAS
- J. Kaur, K. Anand, N. Kohli, A. Kaur and R. C. Singh, Chem. Phys. Lett., 2018, 701, 115–125 CrossRef CAS
- X. Jie, D. Zeng, J. Zhang, K. Xu, J. Wu, B. Zhu and C. Xie, Sens. Actuators, B, 2015, 220, 201–209 CrossRef CAS
- T. Govindaraj, C. Mahendran, V. Manikandan, J. Archana, M. Shkir and J. Chandrasekaran, J. Alloys Compd., 2021, 868, 159091 CrossRef CAS
- S. Ansari, M. S. Ansari, S. Satsangee and R. Jain, Anal. Chim. Acta, 2019, 1046, 99–109 CrossRef CAS PubMed
- H. Yoon, M. G. Mali, M.-w. Kim, S. S. Al-Deyab and S. S. Yoon, Catal. Today, 2016, 260, 89–94 CrossRef CAS
- I. M. Szilágyi, B. Fórizs, O. Rosseler, Á. Szegedi, P. Németh, P. Király, G. Tárkányi, B. Vajna, K. Varga-Josepovits, K. László, A. L. Tóth, P. Baranyai and M. Leskelä, J. Catal., 2012, 294, 119–127 CrossRef
- J. Kaur, K. Anand, K. Anand and R. C. Singh, J. Mater. Sci., 2018, 53, 12894–12907 CrossRef CAS
- S. Muhammad Hafiz, R. Ritikos, T. J. Whitcher, N. Md. Razib, D. C. S. Bien, N. Chanlek, H. Nakajima, T. Saisopa, P. Songsiriritthigul, N. M. Huang and S. A. Rahman, Sens. Actuators, B, 2014, 193, 692–700 CrossRef
- G. Pavoski, T. Maraschin, F. d. C. Fim, N. M. Balzaretti, G. B. Galland, C. S. Moura and N. R. d. S. Basso, Mater. Res., 2016, 20, 53–61 CrossRef
- Y. S. Kwon, A. A. Gromov, A. P. Ilyin, A. A. Ditts, J. S. Kim, S. H. Park and M. H. Hong, Int. J. Refract. Met. Hard Mater., 2004, 22, 235–241 CrossRef CAS
- H. Hartman and F. Ebert, Z. Anorg. Allg. Chem., 1931, 198, 116 CrossRef
- J. Oi, A. Kishimoto, T. Kudo and M. Hiratani, J. Solid State Chem., 1992, 96, 13–19 CrossRef CAS
- R. Diehl, G. Brandt and E. Salje, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1105–1111 CrossRef
- J. T. Szymanski and A. C. Roberts, Can. Mineral., 1984, 22, 681–688 CAS
- L. Wang, J. Liu, Y. Wang, X. Zhang, D. Duan, C. Fan and Y. Wang, Colloids Surf., A, 2018, 541, 145–153 CrossRef CAS
- K. Huang, Q. Pan, F. Yang, S. Ni, X. Wei and D. He, J. Phys. D: Appl. Phys., 2008, 41, 155417 CrossRef
- J. Shi, Z. Cheng, L. Gao, Y. Zhang, J. Xu and H. Zhao, Sens. Actuators, B, 2016, 230, 736–745 CrossRef CAS
- S. Songara, V. Gupta, M. K. Patra, J. Singh, L. Saini, G. S. Gowd, S. R. Vadera and N. Kumar, J. Phys. Chem. Solids, 2012, 73, 851–857 CrossRef CAS
- A. Souza-Filho, V. Freire, J. Sasaki, J. Mendes Filho, J. Julião and U. Gomes, J. Raman Spectrosc., 2000, 31, 451–454 CrossRef CAS
- Q. Jia, H. Ji and X. Bai, J. Mater. Sci.: Mater. Electron., 2019, 30, 7824–7833 CrossRef CAS
- A. Y. Adesina, Q. A. Drmosh, A. M. Kumar and A. K. Mohamedkhair, Surf. Coat. Technol., 2021, 420, 127334 CrossRef CAS
- L. Maille, C. Sant, C. Le Paven-Thivet, C. Legrand-Buscema and P. Garnier, Thin Solid Films, 2003, 428, 237–241 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04113a |
| This journal is © The Royal Society of Chemistry 2023 |