
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel series of dipeptide derivatives containing indole-3-carboxylic acid conjugates as potential antimicrobial agents: the design, solid phase peptide synthesis, in vitro biological evaluation, and molecular docking study†
Sunil R. Tivari‡
a,
Siddhant V. Kokate‡b,
Enrique Delgado-Alvaradocd,
Manoj S. Gaykee,
Amol Kotmalef,
Harun Patelg,
Iqrar Ahmadg,
Elizabeth M. Sobhiah,
Siva G. Kumari,
Bianey García Larab,
Vicky D. Jaina and
Yashwantsinh Jadeja *a
*a
aDepartment of Chemistry, Marwadi University, Rajkot-360003, Gujarat, India. E-mail: drysjadeja@gmail.com
bDepartamento de Química, Universidad de Guanajuato, Noria Alta S/N, Guanajuato-36050, Guanajuato, Mexico
cMicro and Nanotechnology Research Center, Universidad Veracruzana, Blvd. Av. Ruiz Cortines No. 455 Fracc. Costa Verde, Boca del Río 94294, Mexico
dFacultad de Ciencias Quimicas, Universidad Veracruzana, Blvd. Av. Ruiz Cortines No. 455 Fracc. Costa Verde, Boca del Río 94294, Mexico
eIndrashil University, Rajpur, Kadi, Mehsana 382740, Gujarat, India
fDepartment of Chemistry, Savitribai Phule Pune University, Pune 411007, Maharashtra, India
gDepartment of Pharmaceutical Chemistry, R C. Patel Institute of Pharmaceutical Education and Research, Shirpur District, Dhule 425405, Maharashtra, India
hDepartment of Pharmacoinformatics, NIPER, Mohali 160062, Punjab, India
iDepartment of Medicinal Chemistry, Sri Venkateswara College of Pharmacy, Chittoor 517127, Andhra Pradesh, India
First published on 14th August 2023
Abstract
A new library of peptide-heterocycle hybrids consisting of an indole-3-carboxylic acid constituent conjugated with short dipeptide motifs was designed and synthesized by using the solid phase peptide synthesis methodology. All the synthesized compounds were characterized by spectroscopic techniques. Additionally, the synthesized compounds were subjected to in vitro antimicrobial activities. Two Gram-negative bacteria (Escherichia coli and Pseudomonas aeruginosa) and two Gram-positive (Streptococcus pyogenes and Staphylococcus aureus) were used for the evaluation of the antibacterial activity of the targeted dipeptide derivatives. Good antibacterial activity was observed for the screened analogues by comparing their activities with that of ciprofloxacin, the standard drug. Also, two fungi (Aspergillus niger and Candida albicans) were employed for the evaluation of the antifungal activity of the synthesized compounds. When compared to the standard drug Fluconazole, it was observed that the screened analogues exhibited good antifungal activity. In continuation, all the synthesized derivatives were subjected to integrated molecular docking studies and molecular dynamics simulations to investigate binding affinities, intermolecular interaction networks, and conformational flexibilities with deoxyribonucleic acid (DNA) gyrase and lanosterol-14-alpha demethylase. The molecular docking studies revealed that indole-3-carboxylic acid conjugates exhibited encouraging binding interaction networks and binding affinity with DNA gyrase and lanosterol-14 alpha demethylase to show antibacterial and antifungal activity, respectively. Such synthesis, biological activity, molecular dynamics simulations, and molecular docking studies of short peptides with an indole conjugate unlock the door for the near future advancement of novel medicines containing peptide-heterocycle hybrids with the ability to be effective as antimicrobial agents.
1 Introduction
As the 21st century unfolds, antimicrobial resistance (AMR) poses a grave threat to the health of mankind. The term antimicrobial therapeutics refers to therapeutics that fight infections caused by fungi, bacteria, viruses, etc.1,2 The development of AMR to antibiotics occurs whenever a pathogen develops resistance to them and certain developments in pathogens make medicines less potent at curing diseases. The AMR can happen naturally throughout the course of evolution. However, the misuse or overuse of medicines can hasten the process of AMR. Whenever an ill individual consumes the wrong medication or takes it incorrectly, the pathogen that leads to the illness withstands the exposure to the medicine that normally kills it or hinders its growth. Then that survived pathogen can propagate causing an upsurge in the number of pathogens, which are resistant to medication. This in turn leads to an increase in the number of diseases that are resistant to that particular medication. Therefore, if the rise in resistance to antimicrobial therapeutics is not controlled, many pathogens could become extremely life threatening. The resistance to antimicrobial therapeutics may cause 10 million deaths annually by the year 2050, according to a report released by the government of the United Kingdom.3 World health organization and experts around the world concurred that the surge in resistance to antimicrobial therapeutics is a critical issue requiring an immediate and concerted global action plan.4,5 Therefore, efforts to discover novel therapeutics that can be utilized as potent antimicrobial medicines are crucial for the future to evade this AMR.6,7Antimicrobial peptides (AMPs) have demonstrated good capability to prevent AMR, which has rendered many therapeutics ineffective.8 AMPs have shown a broad spectrum of antimicrobial characteristics when tested against a diverse range of pathogens.9–11 Lots of peptides with antimicrobial properties are effective against pathogens that are resistant towards many medications and have a very low tendency to achieve resistance. Peptides with antimicrobial properties are seen as promising and viable therapeutic prospects for the coming future because of their lesser toxicity, a wide range of activities, and depleted resistance. Numerous different types of organisms are capable of producing AMPs, commonly referred to as host defence peptides, which are extremely vital parts of their inherent immune system. Even though predominantly AMPs with cationic motifs are seen,12–14 the anionic motif of AMPs has also been reported.15,16 The three most universal structural characteristics of AMPs consist of their relatively smaller peptide sequence (often 12–50 amino acids), cationic character (owing to the inclusion of one or more arginine, lysin, and/or histidine), and amphipathic motif (both hydrophobic and hydrophilic characteristics are observed in it). Numerous studies have demonstrated the importance of the net separation of hydrophobic and hydrophilic sections as well as the peptide's net charge.17,18 A sudden rise in bacterial resistance is observed when traditional antibiotics mark specific receptors. Peptides with antimicrobial properties have different modes of action than this. The mechanism of action involves either disruption of the stability of bacterial membranes or the important internal cell units of bacteria.19,20 The membrane lytic action is one of the main advantages of AMPs, which has long been believed that is unaffected by the mechanism of action underlying resistance to antibiotics.21 Despite the fact that AMPs have many benefits, there are several obstacles to their profitable commercial and therapeutic expansion as medicines. The most significant downside in the use of AMPs as therapeutics lies in their inability to be administered orally owing to the ease with which gastrointestinal enzymes could inactive AMPs.22,23 AMPs are susceptible to protease and their synthesis cost is huge, these two additional drawbacks make the application of AMPs as therapeutics challenging. With the goal to boost stability against protease and oral bioavailability, many ground-breaking studies have been conducted that consists of the design and synthesis of short peptides from long series of natural AMPs while maintaining integral properties of natural AMPs, such as their net cationic peptide charge and the amphipathic motif.24–27
Worldwide, the synthetic route of peptides using solid support (solid phase peptide synthesis or SPPS) is usually favoured by researchers for the synthesis of AMPs over the synthetic route of peptides using liquid phase (liquid phase peptide synthesis or LPPS), particularly for peptides with long sequences. The SPPS route is much more effective than the conventional LPPS route because it is exceptionally efficient, utilizes straightforward synthetic processes and simple purification methods, rapidly synthesizes linear peptides, and produces products with high purity. Additionally, unlike LPPS, in SPPS there is no need for isolation of the prepared intermediates or high-speed procedures. SPPS route can be used for the synthesis of any sort of peptide sequence, such as short–long chain, complex, or cyclic. Some of the most compelling features of the SPPS protocol are that the peptide synthesis can be fully automated, and its scalability can be drastically improved by using an automated peptide synthesizer.28
Scientists all over the globe are highly interested in the research and development of novel therapeutics containing heterocyclic scaffolds due to the fact that, in nature, heterocyclics are omnipresent and have a wide range of synthetic applications and biological activities.29–33 Among all the available heterocyclic analogues nitrogen-containing heterocycles are the amplest and have great biological applicability than those containing oxygen and sulphur.34–36 For medicinal chemists to obtain novel potent therapeutics, indole which is also known as benzopyrrole, an aromatic heterocyclic compound containing a nitrogen atom, is of great significance. Derivatives of indole have plenty of notable biological activities like antiviral,37 anticancer,38 anti-inflammatory and analgesic,39anti-HIV,40 antioxidant,41 antimicrobial,42 antituberculosis,43 etc. Indole is one of the most important and widely used conjugate of the heterocyclic family. In the pharmaceutical market, plenty of drugs are available with indole moieties, like, Ondansetron,44 Alosetron,45 Indomethacin,46 Yohimbine,47 etc.
Considering the aforementioned facts, we designed and synthesized a novel library of hybrids of peptide-heterocycle (short dipeptides) consisting of indole-3-carboxylic acid constituent at the N-terminus and tyrosine at the C-terminus by employing the protocol of SPPS (Fig. 1). Additionally, we evaluated the antimicrobial activities of the obtained hybrids. The observed activities were compared to Ciprofloxacin, a standard drug, (for antibacterial activities) and Fluconazole, a standard drug, (for antifungal activities). Moreover, we have performed molecular docking studies on the short peptide derivatives that we have synthesized. The obtained dipeptide derivatives using this methodology involved effortless purification stages and the obtained intermediates during the synthesis of the required dipeptides do not require isolation. The combination of experiential and in silico studies of prepared derivatives 6a–jwould aid to establish the harmless and effective therapeutics to address the AMR.
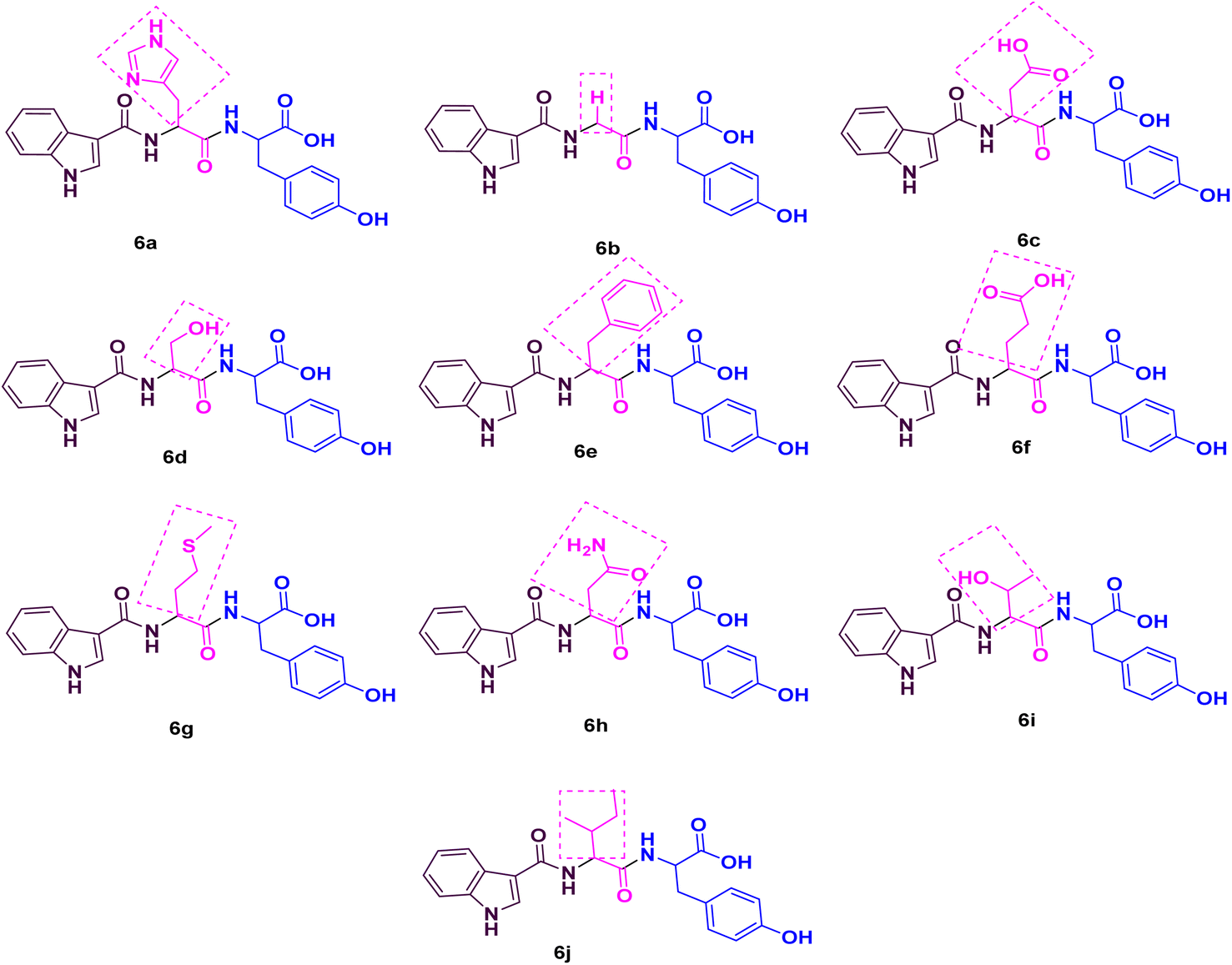 | ||
| Fig. 1 Peptide library of title compounds 6a–j. | ||
2 Material and methods
The 2-chlorotrityl chloride (2-CTC) resin was utilized for the synthesis of a series of short peptide derivatives with indole-3-carboxylic acid constituent. Highly efficient loading estimation is obtained from using such resin. This resin is mainly used for the synthesis of short-chain peptides.48 It was purchased from Merck. Fmoc (9-fluorenylmethoxycarbonyl)-protected L-amino acids were utilized and acquired from Sichuan, China. HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium) and triisopropylsilane (TIS) were acquired from survival chemical. HOBt·H2O (N-hydroxybenztriazole monohydrate), diisopropylethylamine (DIPEA) and trifluoroacetic acid (TFA) acquired from Spectrochem. Phenol acquired from SD fine chemicals. A fully automated CSBio peptide synthesizer (CS136X) was utilized for the synthesis of derivatives.An open capillary method was utilized to determine the melting points which were uncorrected. The Kaiser test was used for monitoring the deprotection and coupling reactions. A mixture of ethyl acetate and n-hexanes was used for the purification of the derivatives. Nicolet impact 400 Fourier-transform infrared (FTIR) spectrometer was employed to determine the IR spectra and potassium bromide (KBr) pressed pellet technique was used for that purpose. Shimadzu liquid chromatography mass spectrometry (LC-MS) (at 70 eV) Mass Spectrometer (ESI) was used to determine the mass spectra. Bruker Avance 400 MHz NMR (nuclear magnetic resonance) spectrometer was used to determine the 1H and 13C NMR spectra using DMSO-d6 (deuterated dimethyl sulfoxide) solvent.
3 Experimental section
3.1 Synthetic route for the formation of intermediate 1
To the peptide synthesizer, the 2-CTC resin was added. The substitution of the resin was 1.0 mmol g−1. Then the resin was washed with dichloromethane (DCM; 10 volumes (v)) and drained. Then, DCM (10 volumes) was added, stirred the reaction mass for 60 minutes (swelling), and drained. The Fmoc-Tyr(tBu)-OH (3.0 equiv.) was dissolved in DCM and transferred into the reaction vessel. Further, added DIPEA (6 equiv.) into the reaction vessel and stirred at 25 °C for 2 h. The peptidyl resin was filtered after 2 h and washed two times with DCM and one time with dimethylformamide (DMF).49 A solution of DIPEA, methanol (MeOH), and DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:8) was utilized to cap the unreacted functional groups of resin. The ultraviolet (UV) spectrophotometer was used for monitoring the loading percentage.
:2:8) was utilized to cap the unreacted functional groups of resin. The ultraviolet (UV) spectrophotometer was used for monitoring the loading percentage.
3.2 Synthetic route for the formation of intermediates 5a–j
Standard Fmoc/t-Bu(t-butyl)/Boc(t-butyloxycarbonyl) protocol was utilized to obtain the desired intermediates 5a–j using the SPPS method. In the SPPS reaction vessel, swelling of Fmoc-Tyr(Ot-Bu)-2-CTC resin was carried out in 20 volumes of DMF (with respect to the initial weight of the resin) for half an hour. CSBio peptide synthesizer was used for the synthesis of the desired intermediates. After swelling, desired intermediates were synthesized by repeating the subsequent steps: (a) deprotection of the Fmoc group was performed by utilizing 20% of piperidine/DMF (v/v) (10 volumes). The resin was washed two times (5 min and 10 min washings). Kaiser test was employed to check the complete deprotection. The blue colour of the test solution confirmed the complete deprotection. After this step, the reaction mass (peptidyl resin) was filtered and washed two times with DMF, one time with isopropyl alcohol (IPA), and three times with DMF. (b) Fmoc amino acid (3.0 equiv., 3 mmol with respect to the initial resin loading), HBTU (3.0 equiv., 3 mmol), HOBt·H2O (3.0 equiv., 3 mmol), and DIPEA (6.0 equiv., 6 mmol) in 6 volumes of DMF was used for all the coupling reactions. One hour of stirring time was required for all the coupling reactions. Kaiser test was employed to check the completion of the reaction. Colourless test solution and beads were detected in the test, which verified the completion of the coupling step.50 After this step, the reaction mass (peptidyl resin) was filtered and washed five times with DMF.3.3 Synthetic route for the synthesis of desired derivatives 6a–j
A mixture of TFA:TIS:H2O (80:10:10) (10 mL g−1) was used as a cleavage cocktail for the side chain and CTC resin cleavage. The protected peptidyl resin was stirred in the cleavage cocktail for 3 h at 20–25 °C.51 After 3 h, the reaction mass was filtered, and the obtained filtrate was precipitated using diisopropyl ether (DIPE) (50 v). Then the reaction mass was filtered and washed three times with DIPE (10 v). Dried the wet cake at 30 °C under a vacuum to get the desired products.
4 Results and discussion
4.1 Chemistry
As noted from Scheme 1, a new library of hybrids of peptide-heterocycle consisting of indole-3-carboxylic acid constituent conjugated with short dipeptide motifs 6a–j were designed and synthesized by using the methodology of SPPS via HBTU/HOBt coupling protocol. Yields of novel derivatives were in the range of 89–92%. All synthesized peptide derivatives were obtained in the form of solid powder and showed excellent physical stability.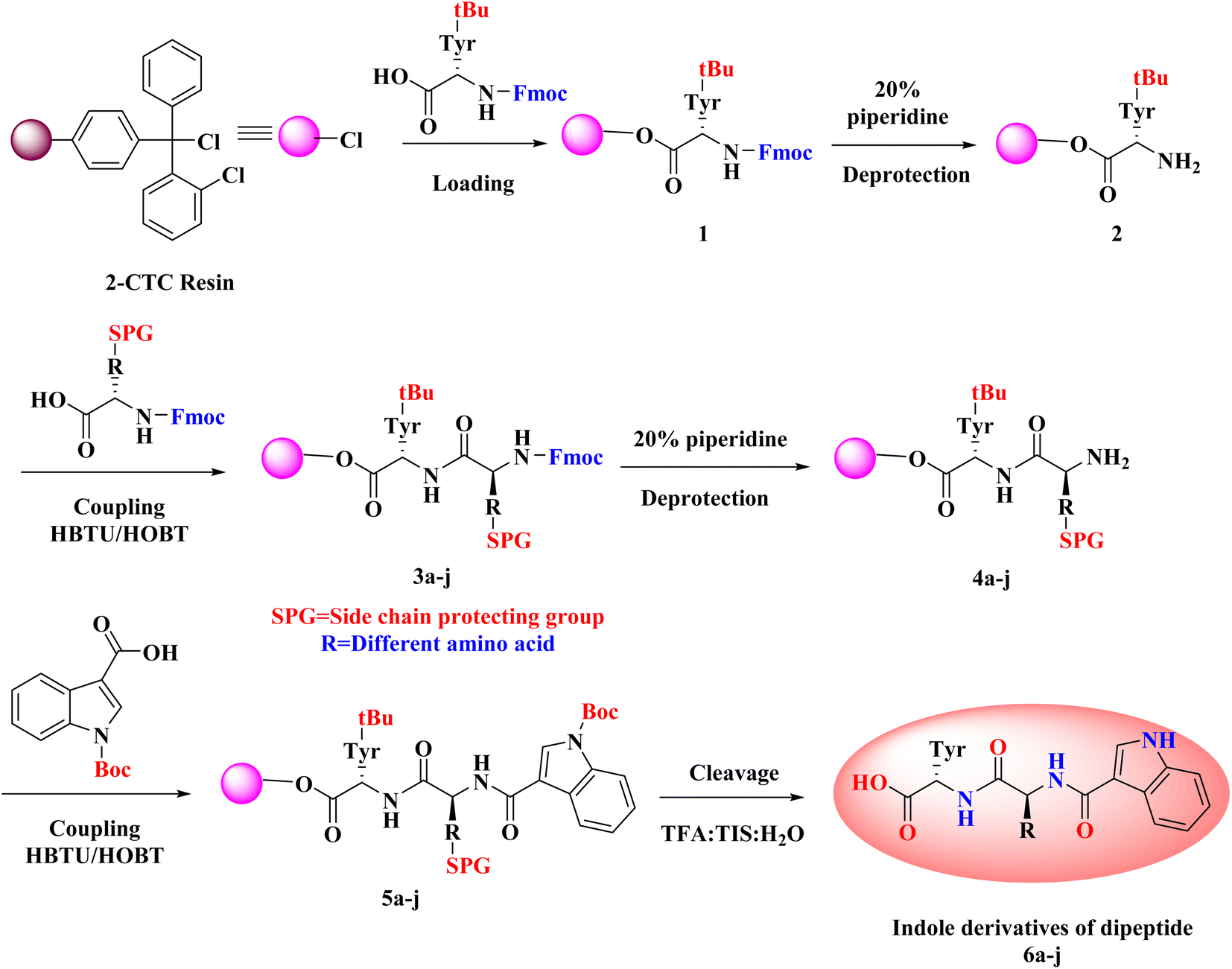 | ||
| Scheme 1 Route of synthesis of indole dipeptide derivatives 6a–j. | ||
The synthesis of dipeptide derivatives consisting of indole moiety 6a–j utilizing the 2-CTC resin is illustrated in Scheme 1. It involves repetitive cycles of coupling and deprotection. The 1st step is the formation of Fmoc-Tyr-2-CTC resin 1. It was obtained by loading Fmoc-Tyr-OH on 2-CTC resin by using DIPEA in DCM. 20% piperidine in DMF was utilized to deprotect the Fmoc group from 1 to give H2N-Tyr-2-CTC resin 2, having the free –NH2 (amino) functionality at N-terminal. The presence of free –NH2 functionality and thus complete deprotection were confirmed by carrying out the Kaiser test. The complete deprotection was confirmed when a blue coloured test solution was observed. Further, coupling of different Fmoc protected amino acids, Fmoc-R(SPG)-OH, with 2 was carried out employing coupling reaction conditions involving HBTU/HOBt. This step yielded Fmoc-R(SPG)-Tyr-2-CTC resin 3a–j, here, ‘SPG’ is a side chain protecting group and ‘R’ is a different amino acid. Completion of the coupling reaction is monitored by the Kaiser test. The test solution and beads were colourless, which concluded the success of the coupling reaction. Further, the deprotection of the Fmoc group of 3a–j yielded H2N–R(SPG)-Tyr-2-CTC resin 4a–j. Here also, the Kaiser test was used to validate the complete deprotection of the Fmoc group. Coupling of 4a–j with indole (N-Boc)3-carboxylic acid by employing above mentioned HBTU/HOBt coupling reaction conditions yielded indole (N-Boc)3-R(SPG)-Tyr-2-CTC resin moieties 5a–j. The global cleavage of 5a–j for three hours at room temperature is the final step in the Scheme 1. It uses the cleavage cocktail of TIS:H2O:TFA in the ratio 10:10:80. DIPE was used for precipitation to give the required dipeptides with indole conjugates. CTC resin and SPGs were cleaved from the desired peptide sequence by this final step – “acidolysis”. Ethyl acetate and n-hexanes were used for further purification of crude to obtain the pure form of desired compounds 6a–j in 89–92% yields.
The IR analysis of 6a was carried out to determine its structure, which exhibited the typical absorption bands at 3500–3200, 1700–1500, and 3600–2500 cm−1 for –NH, –CONH–, and –COOH stretching, respectively. In general, the 1H NMR spectrum of compound 6a showed 13 aromatic protons between 6.5 and 9 δ ppm, whereas 6 aliphatic protons were distributed between 3 and 5 δ ppm. Mass spectrometry data of 6a was found to be 462.28 [M + 1]+, which also confirmed the formation of the peptide conjugate 6a. Similarly, the rest of the derivatives have been characterized, and their characterization data is given in the ESI file.†
4.2 Biological evaluation
The organisms, Escherichia coli MTCC 443 and Pseudomonas aeruginosa MTCC 1688, Streptococcus pyogenes MTCC 442 and Staphylococcus aureus MTCC 96, Aspergillus niger MTCC 282 and Candida albicans MTCC 227 were collected from King Abdullah University Hospital (KAUH), Irbid, Jordan and were stored at −70 °C in trypticase-soy broth with 20% glycerol form BBL Microbiology Systems (Cockeysville, Md, USA) until ready for batch susceptibility testing. They were thawed and passed 3 times to assure purity and viability. The conventional Mueller–Hinton Broth-microdilution method was utilized to study the in vitro antimicrobial activities of the synthesized compounds 6a–j.52 It is a quantitative antimicrobial susceptibility testing method and is used to determine the minimum inhibitory concentration (MIC) of antimicrobial agents. In this test, under in vitro conditions, microorganisms are screened for their capability to produce visible growth in broth (broth dilution) consisting different dilutions of the antimicrobial agents. MIC value is the lowest concentration of an antimicrobial agent at which the visible growth of a microorganism is prohibited within a definite period of time. When compared to macrobroth dilution method, microbroth dilution method shows many advantages, like miniaturization and mechanization due to utilization of disposable small plastic “microdilution” trays. Additionally, advantages like reproducibility, prepared panels, cheap reagents and space that resulted owing to its miniaturization makes Mueller–Hinton Broth-microdilution method practical and popular among the research across the world.53,54The procedures used for the antimicrobial assay were:
Source of drugs obtained from Marwadi University, Gujarat. The findings of the biological evaluation of peptide derivatives 6a–j versus bacterial and fungal strains show that compared to other strains the synthesized novel derivatives are most active against the bacterial strain E. coli (Table 1).
| Entry | Name of derivative | MIC (antibacterial activity) in μg mL−1 | MIC (antifungal activity) in μg mL−1 | ||||
|---|---|---|---|---|---|---|---|
| E. C. | P. A. | S. A. | S. P. | C. A. | A. N. | ||
| a E. C.: Escherichia coli, P. A.: Pseudomonas aeruginosa, S. A.: Staphylococcus aureus, S. P.: Streptococcus pyogenes, C. A.: Candida albicans, A. N.: Aspergillus niger. | |||||||
| 6a | Indole-His-Tyr-OH | 25 | 125 | 150 | 90 | 80 | 70 |
| 6b | Indole-Gly-Tyr-OH | 250 | 250 | 500 | 500 | 500 | >1000 |
| 6c | Indole-Asp-Tyr-OH | 125 | 200 | 250 | 250 | 500 | 500 |
| 6d | Indole-Ser-Tyr-OH | 25 | 125 | 200 | 250 | 100 | 50 |
| 6e | Indole-Phe-Tyr-OH | 200 | 200 | 500 | 500 | 500 | 500 |
| 6f | Indole-Glu-Tyr-OH | 125 | 200 | 250 | 250 | 500 | 500 |
| 6g | Indole-Met-Tyr-OH | 12.5 | 80 | 200 | 125 | 50 | 80 |
| 6h | Indole-Asn-Tyr-OH | 125 | 200 | 250 | 250 | 500 | 500 |
| 6i | Indole-Thr-Tyr-OH | 25 | 125 | 200 | 250 | 50 | 80 |
| 6j | Indole-Ile-Tyr-OH | 500 | 500 | 500 | 500 | >1000 | >1000 |
| Std-1 | Ciprofloxacin | 15 | 50 | 25 | 50 | — | — |
| Std-2 | Fluconazole | — | — | — | — | 50 | 50 |
The peptide conjugate 6g was found to be 8 times more potent against E. coli with a MIC value of 12.5 μg mL−1 as compared with standard Ciprofloxacin which had a MIC of 15 μg mL−1. Indole dipeptide conjugates 6a, 6d, and 6i had MIC values of 25 μg mL−1 and were 4 times more potent than the control drug. Other conjugates 6c, 6f, and 6h were slightly less active than the standard Ciprofloxacin with a MIC value of 25 μg mL−1 against the bacterial strain E. coli. Against P. aeruginosa, apart from 6g, the activity of indole dipeptide conjugates was found to be less as compared to the control drug. Conjugate 6g exhibited a MIC value of 80 μg mL−1, whereas 6a, 6d, and 6i were active at a MIC value of 125 μg mL−1 against the bacterial strain P. aeruginosa. Similarly, peptide conjugate 6a, 6d, 6g, and 6i were more active than the standard drug, as 6a inhibited the Staphylococcus aureus at a MIC value of 150 μg mL−1 and 6d, 6g, and 6i exhibited inhibition at 200 μg mL−1, whereas the control drug inhibited the bacterial strain at 25 μg mL−1. For the activity against S. pyrogens, the derivative 6a was active with a MIC value of 90 μg mL−1, while the rest of the conjugates did not show significant activity as compared with standard Ciprofloxacin which had a MIC of 50 μg mL−1 (Fig. 2).
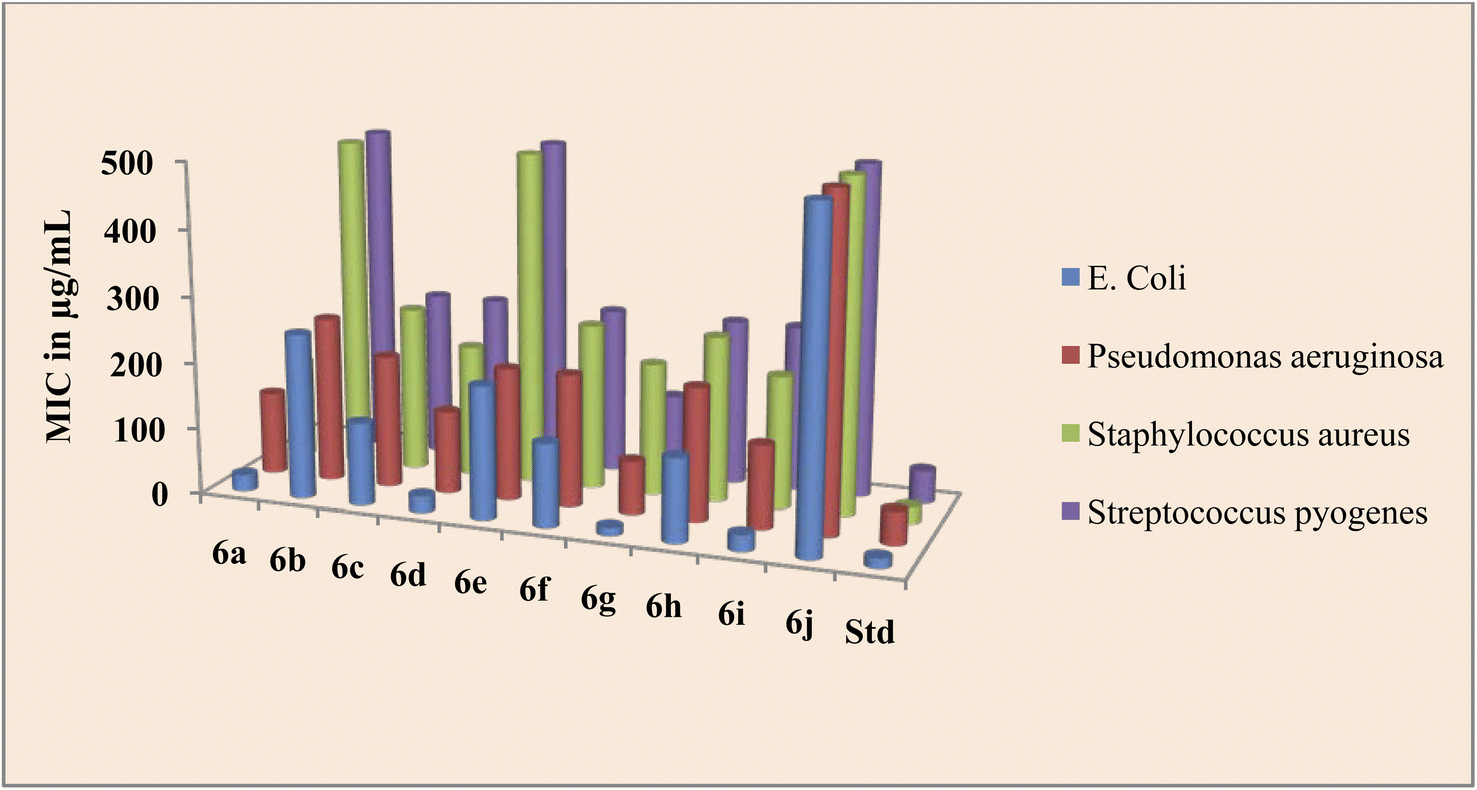 | ||
| Fig. 2 Antibacterial activity of the title compounds 6a–j. | ||
The novel indole dipeptide conjugate 6a–j also displayed very good activity against invasive fungal strains. Conjugates 6g and 6i were most active against C. albicans at a MIC value of 50 μg mL−1 compared to Fluconazole which was active at 50 μg mL−1 (Table 1). Therefore, they were 2 times more effective than the standard drug against the C. albicans. Conjugates 6a and 6d also displayed good activity against C. albicans at a MIC value of 80 μg mL−1 and 100 μg mL−1, respectively. Other conjugates were found to be less active against C. albicans with MIC values ≥500 μg mL−1. When screened against the fungal strain A. niger, the conjugate 6d was found to be most active at a MIC value of 50 μg mL−1 as compared to Fluconazole which was active against A. niger at 50 μg mL−1 (Table 1). It was 2 times more effective than the standard drug against A. niger. Conjugates 6a (70 μg mL−1), 6g (80 μg mL−1), and 6i (80 μg mL−1) also displayed good inhibition against A. niger as compared to the control drug which had a MIC of 100 μg mL−1. However, other conjugates were found to be less active against A. niger with MIC values in a range of ≥500 μg mL−1 (Fig. 3).
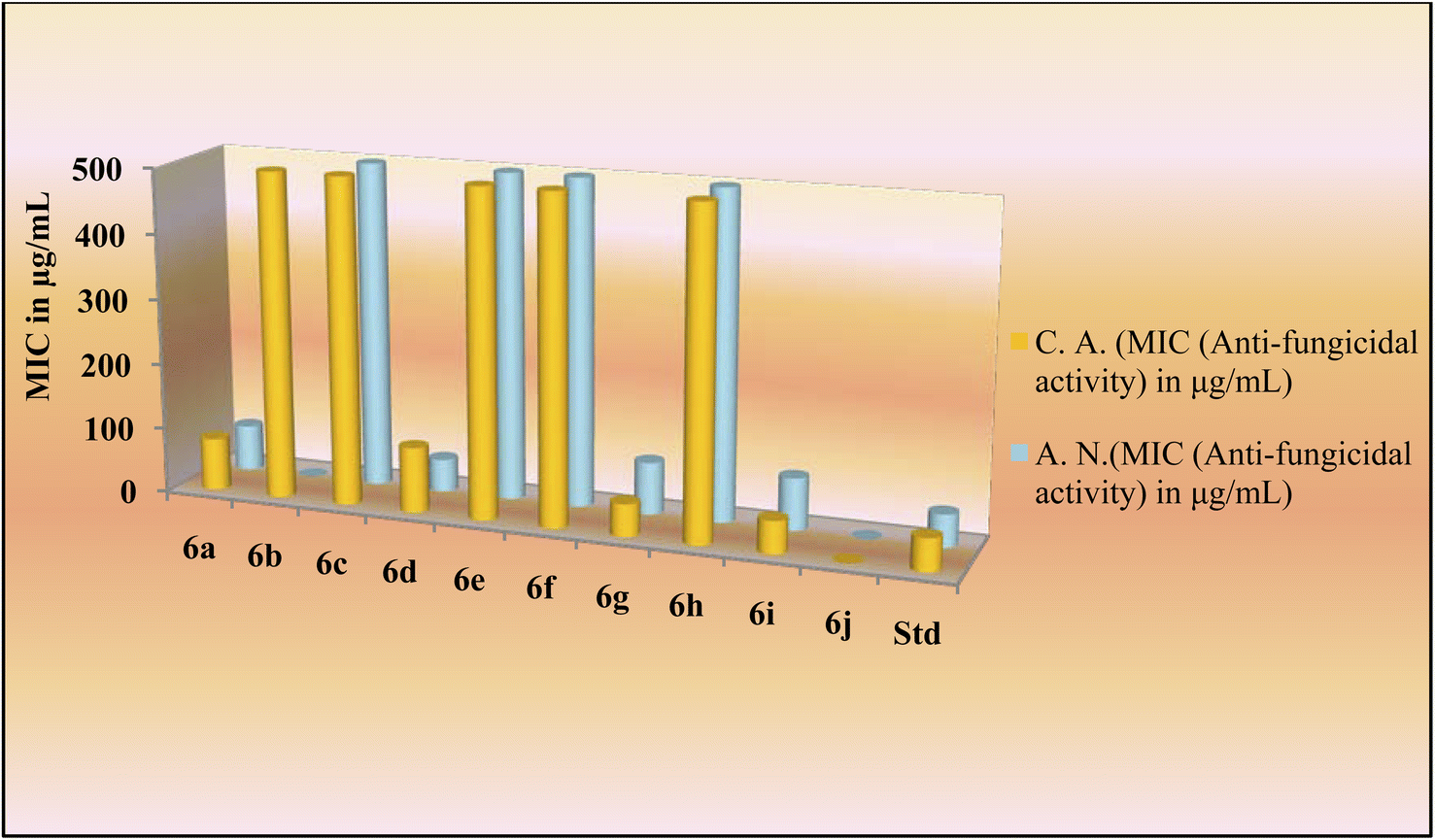 | ||
| Fig. 3 Antifungal activity of the title compounds 6a–j. | ||
4.3 Molecular docking
Crystal structure of DNA gyrase (PDB ID: 5BS8) and lanosterol 14-alpha demethylase (PDB ID: 4LXJ) are chosen from Protein Data Bank (PDB) based on the resolution quality parameter to perform binding affinity studies of indole derivatives.55,56 Many research groups have reported the application of indole-containing peptides as DNA gyrase inhibitors, along with the information on their synthesis, characterization, and biological evaluation. They described the design of new compounds, their inhibitory activity against DNA gyrase, and their potential as antimicrobial agents.57–60 Similarly, the design, synthesis, and biological evaluation of indole derivatives as inhibitors of lanosterol demethylase, a key enzyme in the biosynthesis of sterols in fungi, is also reported by many research groups. The studies showed that indole derivatives can inhibit the activity of this enzyme and have potential as antifungal agents.61–63 A protein preparation wizard was employed to repair both crystal structures by adding missing information such as adding hydrogens, assigning the right bond order, converting selenomethionine to methionine, and optimizing the orientation of hydroxyl and amine groups in serine (Ser), threonine (Thr), tyrosine (Tyr), and asparagine (Asn), glutamine (Gln), respectively.64 Restrained minimization was performed using OPLS-3e forcefield.65 Receptor grid generation was employed to generate the grid based on the centroid of the bound ligand in both enzyme crystal structures. The grid box was extended up to 15 Å as the inner box and 20 Å as the outer box covering the entire binding site. The 3D (three dimensional) builder module was employed to sketch the 10 indole derivative molecules followed by energy minimized to obtain the lowest energy state of ligands. Further, indole ligands were prepared using the LigPrep module to generate the proper ionization state of the ligand molecules at the default pH. Finally, prepared ligand molecules were docked in the generated grids in DNA gyrase and lanosterol 14-alpha demethylase using the extra precision (XP) docking protocol. Each of the poses was analysed for intermolecular interactions, Glide docking, and Glide emodel score.4.4 Molecular dynamics (MD) simulation
MD simulation was used to assess the conformational changes and stability of the target proteins (DNA gyrase and lanosterol 14-alpha demethylase) in the presence of the test compound 6c. An MD simulation study was carried out using the Desmond module of the Schrödinger 2021-1 suite, which has been run on an Ubuntu 18.04 HP Z2 G2 TOWER workstation integrated with an NVIDIA Quadro 6000 graphics processing unit (GPU).66,67 The docked ligand–protein complex of compound 6c was chosen, and the system was modelled using a preset solvent system of SPC water molecules with orthorhombic boundary conditions. Counter-ions (Na+ and Cl−) were added to neutralize the simulation box, the constructed system was submitted to energy minimization until a gradient threshold of 1 kcal mol−1 was obtained at 300 K and 1 bar of pressure using the NPT (N = number of particles, P = pressure, and T = temperature) (isothermal–isobaric) ensemble class.68–71 Lastly, both systems of compound 6c with DNA gyrase and lanosterol 14-alpha demethylase were subjected to a production run for 100 ns with a time step of 100 ps under NPT ensemble conditions.4.5 DNA gyrase binding study
Molecular docking studies of the indole derivatives 6a–j were performed on DNA gyrase and lanosterol 14-alpha demethylase to study their binding poses, intermolecular interactions, and binding strength of indole derivatives (Fig. 4–8 and S1–S15†). The indole derivative 6c (ID-6c) displayed a reasonable number of interactions with the DNA gyrase enzyme. The benzene moiety of ID-6c showed π–π stacking interaction with DNA nucleotide ‘adenine’ however, Lys441, Asp461, and Ptr129 residues displayed hydrogen bond interaction networks with ligand molecules as shown in Fig. 4. The computed binding affinity of ID-6c was found to be −7.8 kcal mol−1. The Glide emodel was calculated to be −101.1 for the ID-6c molecule. Whereas ID-6h ligand molecule also projected a good amount of intermolecular interaction networks with DNA gyrase as shown in Fig. 5. The benzene moiety of ID-6h ligand displayed π–π stacking interaction with the pyrimidine moiety of DT 10 and DA base of DNA. It also displayed π–cation interaction with Arg482. The other important residues such as Ptr129 and DA displayed hydrogen bond interaction with a ligand molecule. The binding affinity of ID-6h was computed to be 7.7 kcal mol−1. The calculated Glide emodel score of ID-6h was found to be −106.9 which is slightly better than the ID-6c molecule.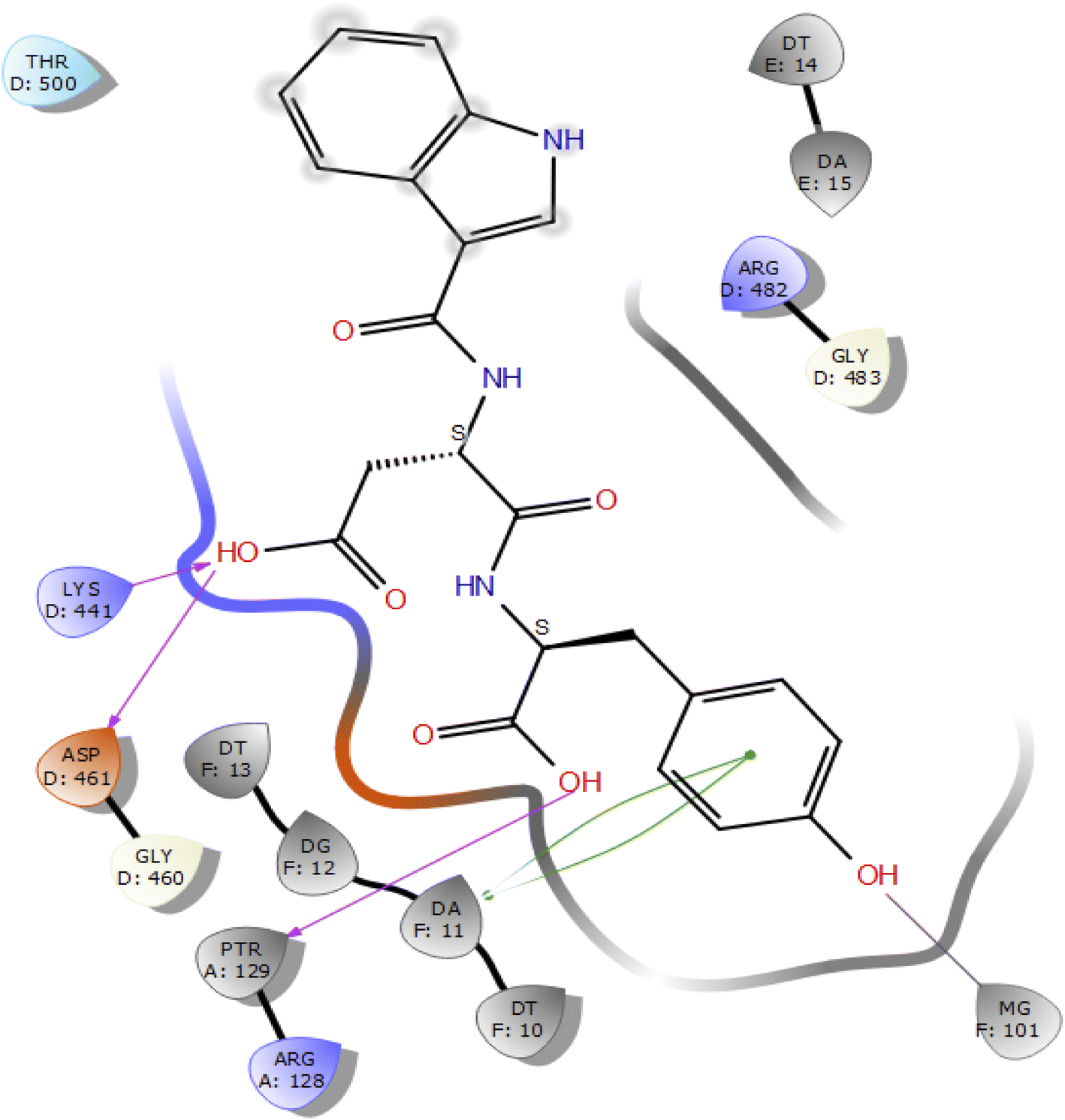 | ||
| Fig. 4 2D interaction diagram representation of ID-6c with DNA gyrase showing intermolecular interactions. | ||
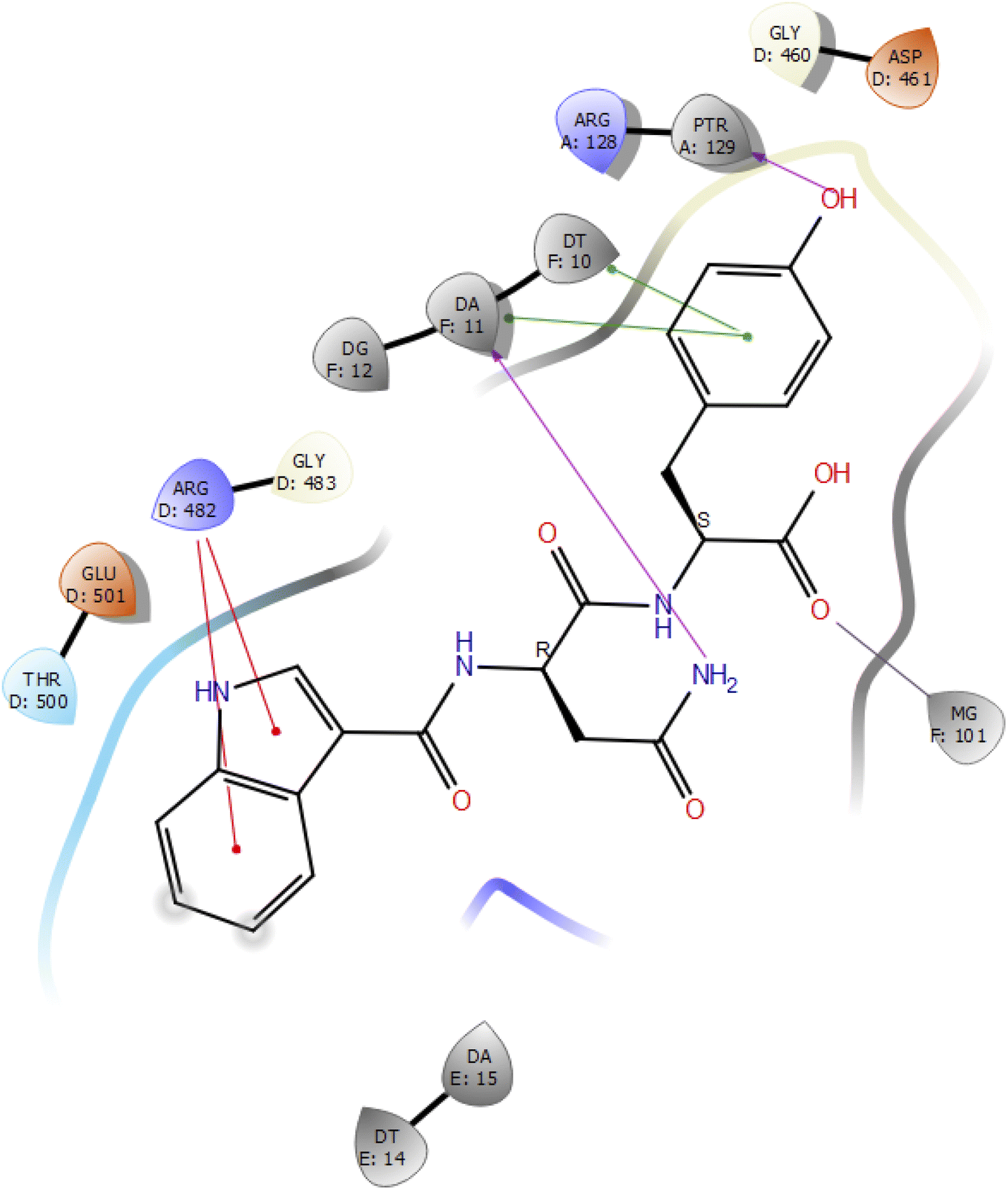 | ||
| Fig. 5 2D interaction diagram representation of ID-6h with DNA gyrase showing intermolecular interactions. | ||
Further, we have compared the results with the standard drug ciprofloxacin. Ciprofloxacin (quinoline ring) showed π–π stacking interaction with DNA nucleotide adenine-15 and guanine-11, whereas the carboxylic acid group established the hydrogen bond and salt bridge interaction with the Arg-128 as shown in Fig. 6. The overall docking study highlighted that indole derivatives are comfortably binding in the quinolone binding pocket (QBP) by forming interaction networks with Arg482, Tyr129, DT10, and DA11 majorly. All the calculated docking energy parameters of indole derivatives 6a–j are listed in Table 2.
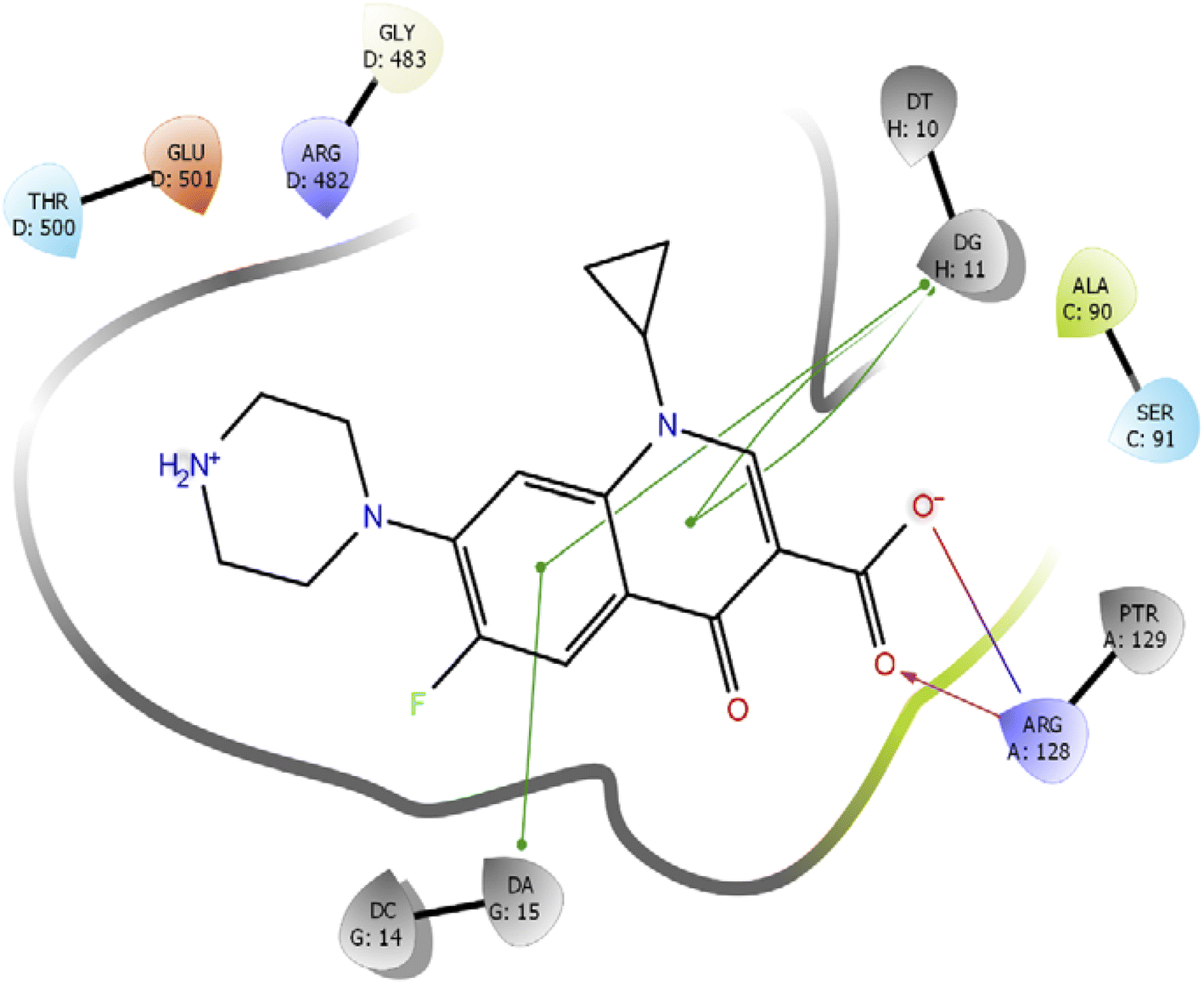 | ||
| Fig. 6 2D interaction diagram representation of Ciprofloxacin with DNA gyrase showing intermolecular interactions. | ||
| Molecule ID | Docking score (kcal mol−1) | Glide emodel |
|---|---|---|
| ID-6c | −7.8 | −101.1 |
| ID-6f | −7.8 | −96.9 |
| ID-6h | −7.7 | −106.9 |
| ID-6g | −7.7 | −100.2 |
| ID-6b | −7.5 | −88.1 |
| ID-6a | −6.9 | −103.8 |
| ID-6d | −6.8 | −94.3 |
| ID-6j | −6.8 | −85 |
| ID-6i | −6.5 | −86.2 |
| Ciprofloxacin | −9.475 | −89.761 |
4.6 Lanosterol 14-alpha demethylase binding study
In the case of lanosterol 14-alpha demethylase (CYP51), the ID-6c molecule displayed very good binding strength due to a greater number of intermolecular interactions. The binding poses of ID-6c projected hydrogen bond interaction networks with Arg98, Phe241, His381, Ser382, and Ser504 residues. The benzene moieties of the ligand molecule also displayed π–π stacking interaction networks with Tyr126 and Phe241 as shown in Fig. 7. The computed binding affinity of ID-6c was found to be −12.8 kcal mol−1 and the Glide emodel score was calculated to be −130.5. From the docking study, it is understood that indole derivatives are comfortably binding to the enzyme by making hydrogen bonds and π–π stacking interactions with active site residues. These results were compared with the standard drug fluconazole, which showed the π–π stacking interactions with the Phe-236 with docking score of the −6.913 kcal mol−1 as shown in Fig. 8. Overall, binding poses and interaction networks of indole molecules revealed that Tyr126, Tyr129, Phe241, His381, Ser382, and Met509 residues are vital for binding. The calculated various docking energy parameters of indole derivatives with lanosterol 14-alpha demethylase are listed in Table 3.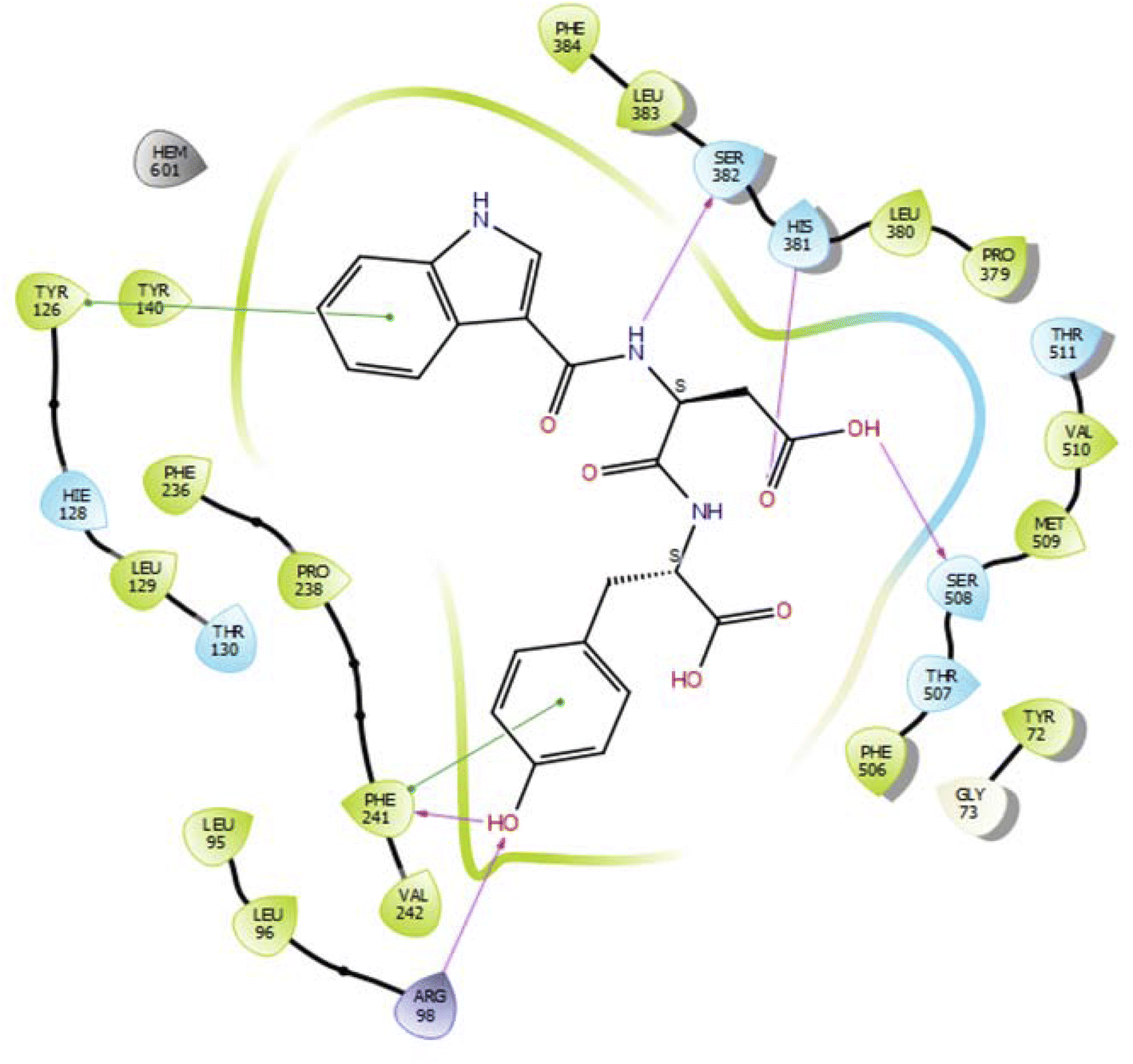 | ||
| Fig. 7 2D interaction diagram representation of ID-6c with lanosterol 14-alpha demethylase showing intermolecular interactions. | ||
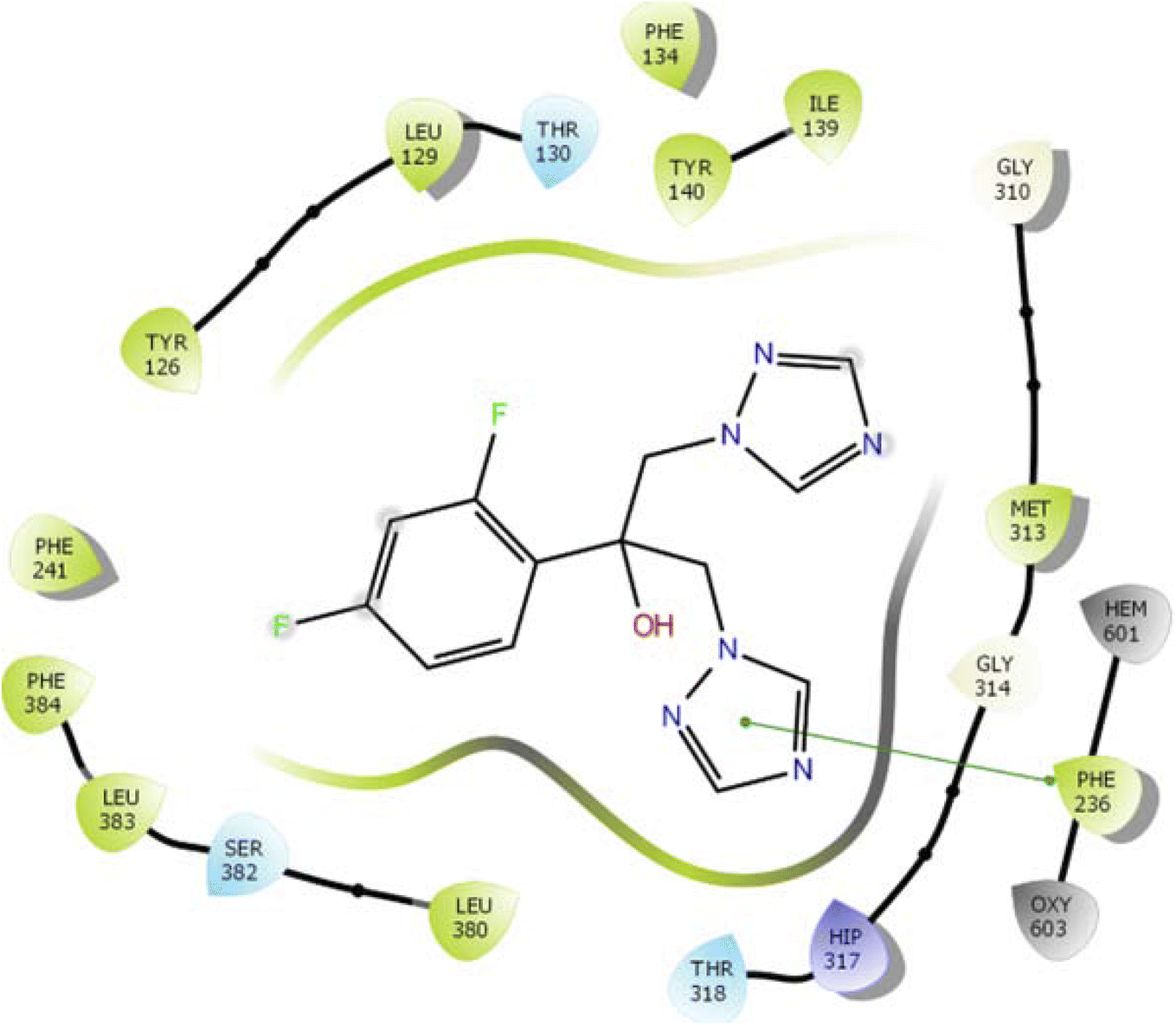 | ||
| Fig. 8 2D interaction diagram representation of Fluconazole with lanosterol 14-alpha demethylase showing intermolecular interactions. | ||
| Molecule ID | Docking score (kcal mol−1) | Glide emodel |
|---|---|---|
| ID-6c | −12.8 | −130.5 |
| ID-6d | −12.4 | −115.3 |
| ID-6e | −12.1 | −129.3 |
| ID-6i | −12 | −124.5 |
| ID-6f | −10.9 | −112 |
| ID-6j | −10.8 | −116.3 |
| ID-6a | −10.1 | −136.2 |
| ID-6h | −9.7 | −121.7 |
| ID-6b | −9.6 | −109.2 |
| ID-6g | −9.6 | −123 |
| Fluconazole | −6.913 | −62.202 |
4.7 Molecular insights from MD simulations
In molecular docking study of all the compounds it was seen that compound 6c exhibited the highest docking score towards the DNA gyrase and lanosterol 14-alpha demethylase, while compound 6g demonstrated significant antibacterial activity. Therefore, considering the docking score as a determining factor, compound 6c was subsequently chosen for further simulation. The Desmond Simulation Package was used to run MD simulations for 100 ns following the docking calculations to assess the steady nature and conformational stability of compound 6c in complex with DNA gyrase and lanosterol 14-alpha demethylase. The MDS trajectories have been used to calculate the root-mean square deviation (RMSD) of the ligand, the RMSD of protein Cα atoms, the root-mean square fluctuation (RMSF) of protein amino acids, and a histogram indicating ligand–protein interactions of the complexes in order to explore their structural stabilities, and binding modes. Total structural changes of protein–ligand complexes were initially investigated in terms of RMSD in the coordinates of protein Cα atoms and ligand from the original docked conformation. It was observed that when compound 6c is in the system, the maximum RMSD values of protein and ligand were ∼3.6 Å and ∼4.9 Å, respectively. The Cα atoms of GyrB were found to fluctuate slightly, while in the ligand a mean RMSD of ∼2.2 Å was observed for the duration between 0 and 72 ns, after which drift and fluctuation could be observed from 72 to 76 ns and remained stable till 100 ns. Fig. 9A demonstrated clearly that the compound 6c-5BS8 systems were consistent in sustaining the molecular interaction profile throughout the simulation run and, more importantly, did not exhibit significant fluctuation, which is an absolutely acceptable range for judging the protein–ligand complex interaction stability. Similarly, the RMSD plot of the 6c-4LXJ complex shows initial fluctuation during ∼0–29 ns with an average RMSD of ∼3.8 Å. With slight drift, RMSD increased to ∼4.9 Å at ∼32 ns, after minor fluctuation was observed, and a stable equilibrium was continued for the remaining period of simulation. However, the ligand plot shows an initial fluctuation due to equilibration and remains consistent around the RMSD value of ∼3.6 Å till 100 ns (Fig. 10A). From the above data, it is clear that the Cα atoms of DNA gyrase and lanosterol 14-alpha demethylase bound with compound 6c remained stable during the simulation.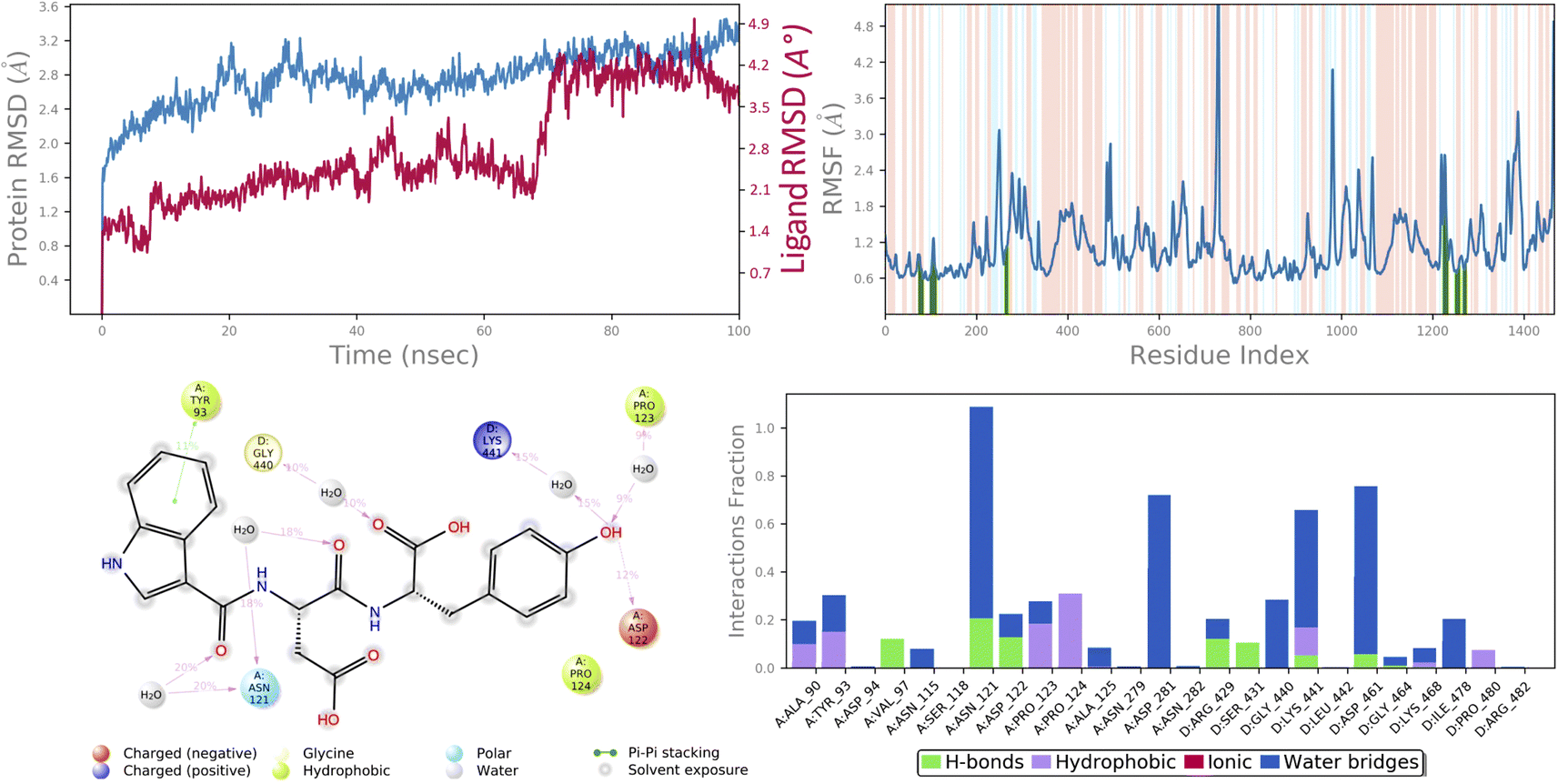 | ||
| Fig. 9 MD simulation analysis of 6c in complex with DNA gyrase (PDB ID: 5BS8) (A) time dependent RMSD of 6c-5BS8 complex (protein Cα-atoms RMSD is shown in grey while RMSD of compound 6c with respect to protein are shown in red) (B) protein amino acids RMSF (C) ligand 2d interaction diagram and (D) protein–ligand contact histogram obtained from MD trajectory. | ||
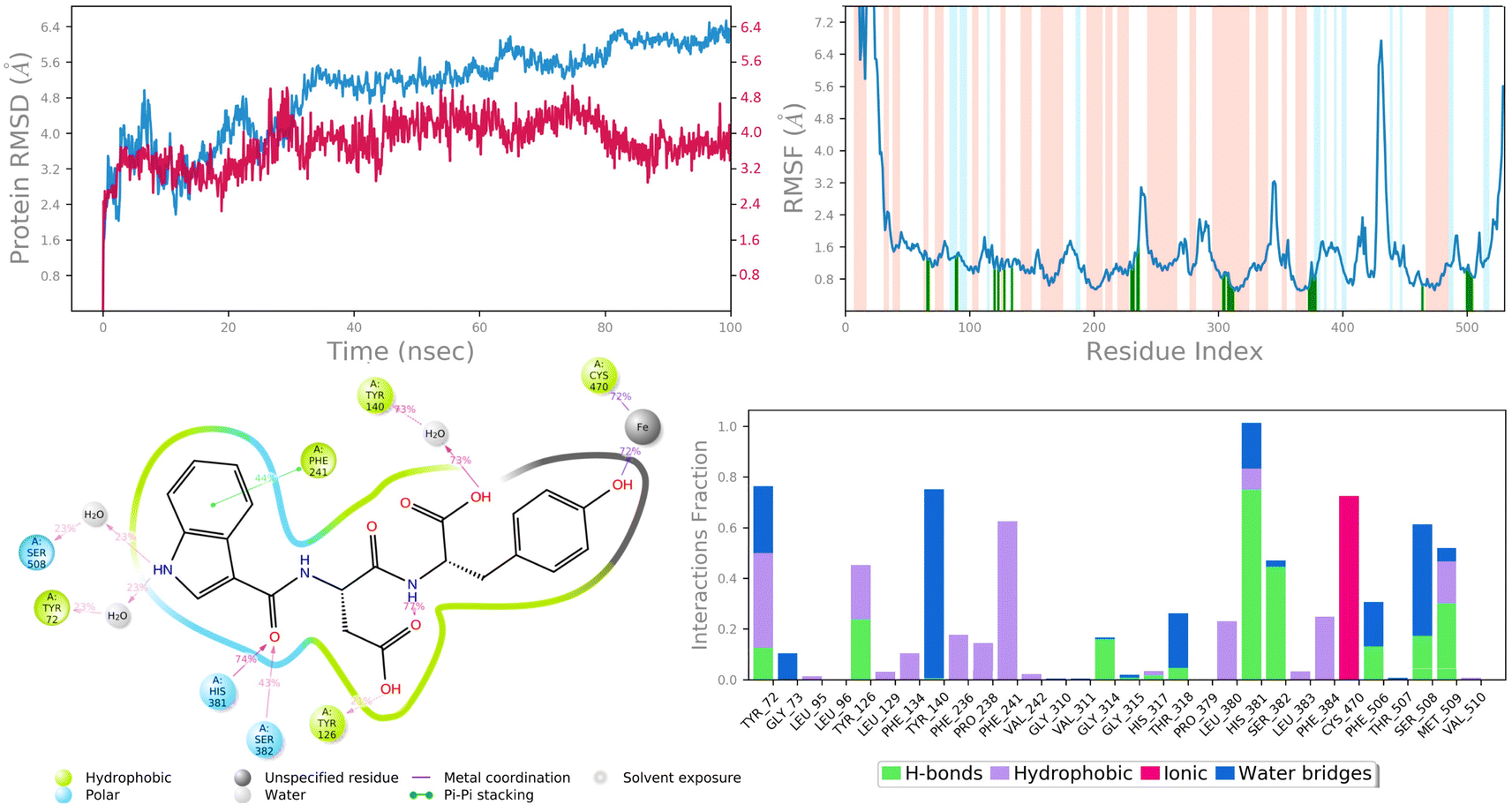 | ||
| Fig. 10 MD simulation analysis of 6c in complex with lanosterol 14-alpha demethylase (PDB ID: 4LXJ) (A) time dependent RMSD of 6c-LXJ complex (protein Cα-atoms RMSD is shown in grey while RMSD of compound 6c with respect to protein are shown in red) (B) protein amino acids RMSF (C) ligand 2d interaction diagram and (D) protein–ligand contact histogram obtained from MD trajectory. | ||
To assess the flexibility of the residues, the individual amino acid based RMSF in the trajectory was computed. More flexibility during the MD simulation is shown by higher RMSF values. The findings reveal that the RMSF of the protein molecule varies in different areas of the protein. Protein secondary structural elements, such as α-helical and β-strand, are displayed in red and blue backgrounds, respectively, in the RMSF plot, whereas the loop region is portrayed in white shades. Excluding the loop areas and the N/C terminus, the values of most residues are less than 2 Å, indicating that the residue conformation is relatively consistent during the simulation illustrated in Fig. 9B and 10B. The RMSF plot also showed that in the 6c-5BS8 complex, 25 amino acids interacted with the ligand, while in the 6c-4LXJ complex, 30 amino acids interacted with the ligand. All of these interacting residues are marked by a green vertical line. The average RMSF was found to be 1.14 Å and 1.7 Å in the case compound 6c was bound with DNA gyrase and lanosterol 14-alpha demethylase protein, respectively. The low values observed undoubtedly revealed that there was an insignificant fluctuation in any interacted amino acid residues during the simulation. In the 6c-5BS8 complex, ligand 2d interaction showed that indole moiety interacted with Tyr93 through π–π stacking at 11% of simulation time.
Through amino acid mediated hydrogen bonding and direct hydrogen bonding, compound 6c's terminal hydroxyl group formed H-bonds with lys441, pro123, and asp122 amino acid residues (Fig. 9C). The phenyl ring of the indole scaffold showed 44% simulation trajectories of parallel π–π stacking with the hydrophobic amino acid Phe241, while the NH group interacted with Ser308 and Tyr72 via amino acid mediated hydrogen bonding in the case of the 6c-4LXJ complex. In this complex, one ionic interaction is observed between the hydroxyl group and Cys470 through the protein Fe ion. In particular, polar amino acids His381, and Ser382 are involved in notable hydrogen bonding with the compound 6c at 74% and 43% of the simulation time, respectively (Fig. 10C).
The simulation interaction diagram can be used to study the four types of protein–ligand interactions (hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridge interactions) that occurred throughout the 100 ns MD simulation study. The interactions in 6c-5BS8 and 6c-4LXJ complexes are given in Fig. 9D and 10D. It can be observed that compound 6c formed major amino acid mediated hydrogen bonding, followed by hydrophobic interactions with several amino acids of the catalytic site cavity of DNA gyrase. While in lanosterol 14-alpha demethylase protein, compound 6c is stabilized by crucial hydrogen bonding and hydrophobic interaction. In particular, the significant polar contacts and particular hydrogen bond interactions (>30% of occupancy) were higher in 6c-4LXJ than in 6c-5BS8.
5 Conclusion
In this research article, a new library of hybrids of peptide-heterocycle consisting of indole-3-carboxylic acid constituent conjugated with short dipeptide motifs was outlined and prepared by using the methodology of solid phase peptide synthesis. All the synthesized compounds were characterized by elemental analysis and techniques of spectroscopy. Also, in vitro antimicrobial activities of the synthesized peptides were studied with the anticipation of finding out new therapeutics with potent antimicrobial properties. On analysing against Ciprofloxacin, the standard drug, derivatives 6a, 6d, 6g, and 6i demonstrated better activities versus bacterial strains. Compound 6g demonstrated excellent activity versus both bacterial and fungal species. It was evident from the findings that on incorporating methionine into the peptide chain, the microbial activities versus both bacterial and fungal strains have remarkably surged. Molecular docking studies of synthesized compounds against DNA GyrB and lanosterol 14-alpha demethylase exhibited a correlation between the docking score and biological activities. The 100 ns MD study investigations on compound 6c complex with DNA GyrB and lanosterol 14-alpha demethylase displayed that both complexes attain stability of interaction networks and conformational flexibilities. The obtained results from the current research aid to elucidate further advancement of antimicrobial agents consisting of hybrids of peptide-heterocycle.Conflicts of interest
The authors declare no conflicts of interest regarding the publication of this paper.Acknowledgements
The authors are thankful to the University Grants Commission, New Delhi, and Marwadi University, for technical and academic support. The authors are also thankful to the Department of Pharmacoinformatics, NIPER, Mohali, for providing Molecular docking studies report and Indrashil University, Kadi, Gujarat, for providing spectral data, also, Dr Ramesh Chandra Gupta (Banaras Hindu University) for his guidance throughout this project.References
- A. C. Singer, H. Shaw, V. Rhodes and A. Hart, Front. Microbiol., 2016, 7, 1728 CrossRef PubMed.
- B. M. Marshall and S. B. Levy, Clin. Microbiol. Rev., 2011, 24, 718–733 CrossRef CAS PubMed.
- J. O'Neill, Tackling drug-resistant infections globally: final report and recommendations, Review on Antimicrobial Resistance, London, 2016 Search PubMed.
- WHO, Antimicrobial resistance, accessed Jan 08, 2023, 2021, https://www.who.int/newsroom/factsheets/detail/antimicrobial-resistance Search PubMed.
- F. Prestinaci, P. Pezzotti and A. Pantosti, Pathog. Global Health, 2015, 109, 309–318 CrossRef.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam, M. U. Qamar, M. K. F. Salamat and Z. Baloch, Infect. Drug Resist., 2018, 11, 1645–1658 CrossRef CAS.
- S. B. Zaman, M. A. Hussain, R. Nye, V. Mehta, K. T. Mamun and N. Hossain, Cureus, 2017, 9, e1403 Search PubMed.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- K. K. Sharma, R. Ravi, I. K. Maurya, A. Kapadia, S. I. Khan, V. Kumar, K. Tikoo and R. Jain, Eur. J. Med. Chem., 2021, 223, 113635–113650 CrossRef CAS PubMed.
- K. K. Sharma, I. K. Maurya, S. I. Khan, M. R. Jacob, V. Kumar, K. Tikoo and R. Jain, J. Med. Chem., 2017, 60, 6607–6621 CrossRef CAS PubMed.
- S. R. Tivari, S. V. Kokate, E. M. Sobhia, S. G. Kumar, U. B. Shelar and Y. S. Jadeja, ChemistrySelect, 2022, 7(27), e202201481 CrossRef CAS.
- C. T. Mant, Z. Jiang, L. Gera, T. Davis, K. L. Nelson, S. Bevers and R. S. Hodges, J. Med. Chem., 2019, 67(7), 3354–3366 CrossRef.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- B. Mishra, S. Reiling, D. Zarena and G. Wang, Curr. Opin. Chem. Biol., 2017, 38, 87–96 CrossRef CAS PubMed.
- S. R. Dennison, M. Mura, F. Harris, L. H. G. Morton, A. Zvelindovsky and D. A. Phoenix, Biochim. Biophys. Acta, Biomembr., 2015, 1848, 1111–1118 CrossRef CAS PubMed.
- M. Zheng, M. Pan, W. Zhang, H. Lin, S. Wu, C. Lu, S. Tang, D. Liu and J. Cai, Bioact. Mater., 2021, 6(7), 1878–1909 CrossRef CAS PubMed.
- Yolandani, H. Ma, Y. Li, D. Liu, H. Zhou, X. Liu, Y. Wan and X. Zhao, Ultrason. Sonochem., 2023, 95, 106414 CrossRef CAS PubMed.
- Y. Xie, T. L. Lopez-Silva and J. P. Schneider, Org. Lett., 2022, 24(40), 7378–7382 CrossRef CAS PubMed.
- I. Lace, E. R. Cotroneo, N. Hesselbarth and N. A. Simeth, J. Pept. Sci., 2023, 29, e3466 CrossRef CAS.
- C. An, S. Wei, Y. Dao, X. Wang, W. Dong, X. You, C. Tian, Z. Zhang and S. Dong, Bioorg. Chem., 2023, 134, 106424 CrossRef CAS PubMed.
- S. Reardon, Nature, 2015, 521, 402–403 CrossRef CAS PubMed.
- X. Bao, K. Qian, M. Xu, Y. Chen, H. Wang, T. Pan, Z. Wang, P. Yao and L. Lin, J. Nanobiotechnol., 2023, 21, 16 CrossRef CAS PubMed.
- S. Maher, R. J. Mrsny and D. J. Brayden, Adv. Drug Delivery Rev., 2016, 106, 277–319 CrossRef CAS.
- J. D. Steckbeck, B. Deslouches and R. C. Montelaro, Expert Opin. Biol. Ther., 2014, 14, 11–14 CrossRef CAS PubMed.
- W. M. Hewitt, S. S. Leung, C. R. Pye, A. R. Ponkey, M. Bednarek, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2015, 137, 715–721 CrossRef CAS PubMed.
- S. R. Tivari, S. V. Kokate, U. B. Shelar and Y. S. Jadeja, Rasayan J. Chem., 2022, 15(2), 875–884 CrossRef CAS.
- S. R. Tivari, S. V. Kokate, M. S. Gayke, I. Ahmad, H. Patel, S. G. Kumar and Y. S. Jadeja, ChemistrySelect, 2022, 7(48), e202203462 CrossRef CAS.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- Y. N. Mabkhot, F. Alatibi, N. N. E. El-Sayed, S. Al-Showiman, N. A. Kheder, A. Wadood, A. Rauf, S. Bawazeer and T. B. Hadda, Molecules, 2016, 21(2), 222 CrossRef PubMed.
- N. B. Perry, J. W. Blunt and M. H. Munro, Tetrahedron, 1988, 44, 1727–1734 CrossRef CAS.
- J. I. Kobayashi, J. F. Cheng, M. Ishibashi, H. Nakamura, Y. Ohizumi, Y. Hirata, T. Sasaki, H. Lu and J. Clardy, Tetrahedron Lett., 1987, 28, 4939–4942 CrossRef CAS.
- M. Al-Smadi and F. Al-Momani, Molecules, 2008, 13, 2740–2749 CrossRef CAS PubMed.
- S. Tivari, S. V. Kokate, U. B. Shelar and Y. Jadeja, Rasayan J. Chem., 2022, 15(2), 875–884 CrossRef CAS.
- M. V. Raimondi, A. Presentato, G. L. Petri, M. Buttacavoli, A. Ribaudo, V. de Caro, R. Alduina and P. Cancemi, Antibiotics, 2020, 9, 292 CrossRef CAS PubMed.
- M. V. Raimondi, R. Listro, M. G. Cusimano, M. la Franca, T. Faddetta, G. Gallo, D. Schillaci, S. Collina, A. Leonchiks and G. Barone, Bioorg. Med. Chem., 2019, 27, 721–728 CrossRef CAS PubMed.
- V. Spanò, R. Rocca, M. Barreca, D. Giallombardo, A. Montalbano, A. Carbone, M. V. Raimondi, E. Gaudio, R. Bortolozzi, R. Bai, P. Tassone, S. Alcaro, E. Hamel, G. Viola, F. Bertoni and P. Barraja, J. Med. Chem., 2020, 63, 12023–12042 CrossRef.
- S. Xue, L. Ma, R. Gao, Y. Lin and Z. Linn, Acta Pharm. Sin. B, 2014, 4, 313–321 CrossRef PubMed.
- K. S. Prasad, R. R. Pillai, M. P. Ghimire, R. Ray, M. Richter, C. Shivamallu, A. S. Jain, S. K. Prasad, P. Sushma, S. Armaković, S. J. Armaković and R. G. Amachawadi, J. Mol. Struct., 2020, 1217, 128445 CrossRef CAS.
- A. Ozdemir, M. D. Altıntop, G. T. Zitouni, G. A. Çiftçi, I. Ertorun, O. Alatas and Z. A. Kaplancıklı, Eur. J. Med. Chem., 2015, 89, 304–309 CrossRef CAS PubMed.
- H. M. Kasralikar, S. C. Jadhavar and S. R. Bhusare, Bioorg. Med. Chem. Lett., 2015, 25, 3882–3886 CrossRef CAS PubMed.
- H. Gurer-Orhan, C. Karaaslan, S. Ozcan, O. Firuzi, M. Tavakkoli, L. Saso and S. Suzen, Bioorg. Med. Chem., 2016, 24, 1658–1664 CrossRef CAS PubMed.
- W. Hong, J. Li, Z. Chang, X. Tan, H. Yang, Y. Ouyang, Y. Yang, S. Kaur, I. C. Paterson, Y. F. Ngeow and H. Wang, J. Antibiot., 2017, 70, 832–844 CrossRef CAS PubMed.
- V. Velezheva, P. Brennan, P. Ivanov, A. Kornienko, S. Lyubimov, K. Kazarian, B. Nikonenko, K. Majorov and A. Apt, Bioorg. Med. Chem. Lett., 2016, 26, 978–985 CrossRef CAS PubMed.
- J. A. Generali and D. J. Cada, Hosp. Pharm., 2009, 44, 670–671 CrossRef.
- R. Horton, Lancet, 2001, 357, 1544–1545 CrossRef CAS PubMed.
- S. H. Ferreira, S. Moncada and J. R. Vane, Nature (London), New Biol., 1971, 231, 237–239 CrossRef CAS PubMed.
- K. E. Andersson, Pharmacol. Rev., 2001, 53, 417–450 CAS.
- D. M. M. Jaradat, Amino Acids, 2017, 50, 39–68 CrossRef PubMed.
- V. Made, S. Els-Heindl and A. G. Beck-Sickinger, Beilstein J. Org. Chem., 2014, 10, 1197–1212 CrossRef PubMed.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34(2), 595–598 CrossRef CAS PubMed.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
- N. C. Desai, K. A. Jadeja, D. J. Jadeja, V. M. Khedkar and P. C. Jha, Synth. Commun., 2021, 51(6), 952–963 CAS.
- B. Kowalska-Krochmal and R. Dudek-Wicher, Pathogens, 2021, 10(2), 165 CrossRef CAS PubMed.
- J. H. Jorgensen and M. J. Ferraro, Clin. Infect. Dis., 2009, 49, 1749–1755 CrossRef CAS PubMed.
- T. R. Blower, B. H. Williamson, R. J. Kerns and J. M. Berger, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(7), 1706–1713 CrossRef CAS PubMed.
- B. C. Monk, T. M. Tomasiak, M. V. Keniya, F. U. Huschmann, J. D. Tyndall, J. D. O'Connell, R. D. Cannon, J. G. McDonald, A. Rodriguez, J. S. Finer-Moore and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(10), 3865–3870 CrossRef CAS PubMed.
- M. A. Salem, A. Ragab, A. El-Khalafawy, A. H. Makhlouf, A. A. Askar and Y. A. Ammar, Bioorg. Chem., 2020, 96, 103619 CrossRef PubMed.
- H. Liu, Z.-W. Chu, D.-G. Xia, H.-Q. Cao and X.-H. Lv, Bioorg. Chem., 2020, 99, 103807 CrossRef CAS PubMed.
- Y. Hu, H. Shi, M. Zhou, Q. Ren, W. Zhu, W. Zhang, Z. Zhang, C. Zhou, Y. Liu, X. Ding, H. C. Shen, S. F. Yan, F. Dey, W. Wu, G. Zhai, Z. Zhou, Z. Xu, Y. Ji, H. Lv, T. Jiang, W. Wang, Y. Xu, M. Vercruysse, X. Yao, Y. Mao, X. Yu, K. Bradley and X. Tan, J. Med. Chem., 2020, 63(17), 9623–9649 CrossRef CAS PubMed.
- M. A. Salem, A. Ragab, A. A. Askar, A. El-Khalafawy and A. H. Makhlouf, Eur. J. Med. Chem., 2020, 188, 111977 CrossRef CAS PubMed.
- Pooja, P. Prasher, P. Singh, K. Pawar, K. S. Vikramdeo, N. Mondal and S. S. Komath, Eur. J. Med. Chem., 2014, 80, 325–339 CrossRef CAS PubMed.
- A. Angarita-Rodríguez, D. Quiroga and E. Coy-Barrera, Molecules, 2019, 25(1), 45 CrossRef PubMed.
- P. Singh, P. Verma, B. Yadav and S. S. Komath, Bioorg. Med. Chem. Lett., 2011, 21(11), 3367–3372 CrossRef CAS PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27(3), 221–234 CrossRef PubMed.
- K. Roos, C. Wu, W. Damm, M. Reboul, J. M. Stevenson, C. Lu, M. K. Dahlgren, S. Mondal, W. Chen, L. Wang and R. Abel, J. Chem. Theory Comput., 2019, 15(3), 1863–1874 CrossRef CAS PubMed.
- R. Pawara, I. Ahmad, S. Surana and H. Patel, In Silico Pharmacol., 2021, 6(1), 54 CrossRef PubMed.
- K. J. Bowers, D. E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, Proceedings of the 2006 ACM/IEEE Conference on Super-computing, IEEE, 2006, p. 43 Search PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118(45), 11225–11236 CrossRef CAS.
- R. Girase, I. Ahmad, R. Pawara and H. Patel, SAR QSAR Environ. Res., 2022, 33(3), 1–21 Search PubMed.
- G. Kalibaeva, M. Ferrario and G. Ciccotti, Mol. Phys., 2003, 101(6), 765–778 CrossRef CAS.
- G. J. Martyna, Phys. Rev. E, 1994, 50(4), 3234–3236 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectral data of all newly synthesized compounds along with methods for their biological activities. See DOI: https://doi.org/10.1039/d3ra04100j |
| ‡ Both authors have equal contributions. |
| This journal is © The Royal Society of Chemistry 2023 |