
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe–Al binary composite filled dialysis membrane tubes (DMT-HFAO): a modified method for assessment of phosphate desorption from aqueous and soil solutions†
Ahmed Kassima,
Abi M. Taddesse *b,
Dechassa Nigussiec,
Isabel Diazd and
Nejat Redwan Habibe
*b,
Dechassa Nigussiec,
Isabel Diazd and
Nejat Redwan Habibe
aDepartment of Chemistry, Hawassa College of Teacher Education, Ethiopia
bDepartment of Chemistry, Haramaya University, P.O. Box 138, Dire Dawa, Ethiopia. E-mail: abi92003@yahoo.com; Tel: +251912018750
cSchool of Plant Sciences, Haramaya University, Ethiopia
dInstituto de Catálisis y Petroleoquímica, CSIC, C/Marie Curie 2, Madrid 28049, Spain
eDepartment of Applied Chemistry, Adama Science and Technology University, Ethiopia
First published on 25th July 2023
Abstract
Phosphorus (P) limits plant growth particularly in strongly acidic soils due to P fixation. P availability to a plant is a functional concept of time rather than a measurable quantity. Therefore, a method that can estimate P availability over time is required. This research work was intended to synthesize a nanocomposite material that can monitor soil P desorption kinetics. To this effect, a binary sorbent system filled in a dialysis membrane tube was developed. Accordingly, calcined and amorphous powder samples of Fe–Al binary mixed oxides were synthesized by a gel-evaporation method and characterized by XRD, FTIR, TGA-DTA, SEM-EDX and BET techniques. The performance, as a phosphate sink, of crystalline hydrous ferric aluminum oxide (HFAO) and hydrous amorphous ferric aluminum oxide (HAFAO) each filled in a dialysis membrane tube (DMT) was evaluated. A single hydrated ferric oxide (HFO) suspension filled in dialysis membrane tubes (DMT) designated DMT-HFO was used as a benchmark. For the aqueous system, the sorption capacity of the DMT-HFAO was found to be 260% (mg mg−1 phosphate) whereas the amorphous congener (DMT-HAFAO) was approximately 200% (mg mg−1 phosphate) times that of DMT-HFO during the 24 h equilibration. For the soil solution system, the phosphate desorbed by the DMT-HFAO was about 520% (mg mg−1 phosphate) compared with a single system, DMT-HFO, in 168 h. For the desorption experiment carried out with soil solution, the data fitted fairly well with first order kinetics for both sorbents (R2 = 0.946–0.998), the amount adsorbed by DMT-HFAO being greater than DMT-HFO. The soil data fitted an intra-particle diffusion model fairly well for both sorbents (R2 = 0.98–0.992) with rate constants, kp, following the order: DMT-HFAO > DMT-HAFAO > DMT-HFO. The DMT-HFAO approach also showed better fit to the two component first order model (R2 = 0.994 & 0.997) indicating that the modified method has promising potential for a long-term phosphate desorption kinetics study from soil, the implication of which is important both from agricultural and environmental perspectives. However, correlation of the P adsorbed by this sink method with actual plant P uptake in various soils should be carried out to validate the universality of this technique.
1. Introduction
Phosphorus (P) is a non-renewable resource, considered as one of the global environmental challenges in the decades to come.1 Phosphorus is inefficiently used in agriculture, the production of which is expected to peak by 2035.2 Hence, a more efficient use of P in agriculture is compulsory not only to overcome the consequences of progressive depletion of P rock reserves on agricultural production but also to circumvent the challenge of advanced deterioration of water quality. However, efficient use of P fertilizer should rely on more precise prediction of available P to plants in soil.3,4Phosphate availability is a function of chemical equilibrium-controlled solubility and rate limited processes and no simple direct measurements are available.5 Most methods for available P determination attempt to quantify P solubility using different extractants, but few relate this to P supply rates that are relevant to plant uptake. Plants obtain P from the soil solution plus P that enters the solution (via desorption), through its roots or root symbionts during the period used to define availability.6
Desorption can be studied by adding materials that bind phosphate strongly, keeping the solution concentration low so that desorption from the soil particles can continue.7 Van der Zee et al. (1987)8 proposed the use of Fe-oxide impregnated filter paper strips (Fe-oxide strips) as a promising method to study the short term (Myers et al., 2005)9 P release kinetics of soils. However, this method was found to be less applicable for long-term desorption studies.7,10 Use of dialysis membrane tube filled with hydrous ferric oxide (DMT-HFO) for studying long-term P dynamics has been proposed previously.11–15
The phosphate sink in the aforementioned cases is hydrated ferric oxide which is an example of single component system. However, considerable number of reports has shown that excellent and efficient phosphorus adsorbents are all characterized by high iron, aluminum, calcium and manganese contents.16–19 Thus, substrates with high contents of these materials can be efficient phosphate sinks in immobilizing phosphate from soils and water bodies. The surface characteristics studies of composited metal oxides are given importance because they imitate the natural systems like soils more closely than their individual congeners.20,21 Accordingly, various ions such as Al(III), Cr(III), Cu(II), La(IV),Mn(IV), Si(IV), Ti(IV), and Zr(IV)18,22–30 had been introduced into iron oxide to form bimetallic oxide adsorbents for phosphate sorption. However, the sorption studies in many of the studies mentioned above were carried out via batch or column study by placing the sorbent in aqueous system containing the target analyte. The batch or column approach has a limitation of using the sorbent repeatedly as recovering the sorbent is difficult when naked sorbent is in direct contact with the matrix. Furthermore, it would be more complicated when one attempts to perform similar study in soil solution. To circumvent these shortcomings, placing the sorbent in membrane bags that are selective to the target analyte (phosphate in our case) is critically important. To the best of our knowledge, no work has been reported on phosphate sorption or desorption study using amorphous/crystalline binary Fe–Al mixed oxide sorbent filled in dialysis membrane tubes placed in aqueous or soil solution system.
In view of the above facts, we hypothesized that the performance of single component Fe-hydr(oxide) sorbent filled in dialysis membrane tubes can be improve by using the mixed Fe–Al hydr(oxide) sorbent filled in the same dialysis bags. The objectives of this study were, therefore, to evaluate the phosphate sorption and desorption properties of the proposed method (DMT-HFAO) and compare the performance of the modified system with the single bench mark (DMT-HFO).
2. Materials and methods
2.1 Description of sampling sites and sampling procedure
Soil samples from two selected sites were collected randomly. These sites (Gununo and Bishoftu) were purposely selected based on their pH and exchangeable aluminum and hence different P-fixing capacity of the soils. Gununo is one of districts in Wolayita zone, Southern Nations, Nationalities and Peoples' Region (SNNPR), Ethiopia. It is located at 7° 58′′ N and 37° 35′′ E. Its annual average temperature is 21.86 °C and the mean annual rainfall is 1200 mm. The types of soils in the area according to FAO classification are dominantly Eutric Nitisols or according to USDA classification Alfisols.31,32 Bishoftu town is located on the escarpment of the Great Rift Valley, 47 km south of Addis Ababa, in Oromia National Regional State. Topographically, the town is located in tepid to cool sub-moist mid highland at an altitude of about 1920 masl with moderate weather condition. The absolute location of Bishoftu is 8° 44′ 40′′ N and 38° 59′ 9′′ E. The average temperature is 20 °C and the annual mean rainfall is 850 mm. The soil types are Haplic Andosol, Vitric Andosol and Eutric Vertisol.33 There are also black land clay soils which are medium to slightly acidic properties with majority of them are slightly acidic whereas gray soils are neutral.34 Top soil samples (0–20 cm) were collected randomly using Food and Agriculture Organization. (FAO) guidelines for soil sampling procedure (FAO, 2008b). From each site, twenty topsoil samples were taken from which composite samples were made up. The samples were then air dried and ground to pass through a 2 mm sieve. A composite sample of each site was used to determine the physicochemical properties of the soils and conduct P desorption experiment.2.2 Selected physical and chemical properties of the soils
The physical and chemical properties of the soils were determined before sorption–desorption procedure and tabulated in Table S1.† The pH (KCl) of the soil samples was determined in the soil suspension by dispersing 20 g of dried soil in 50 mL of 1 M KCl and shaking end-over-end at 20 rpm for 2 h.7 Available phosphorus was determined using Bray and Kurtz (Bray-1P) method (0.03 M NH4F + 0.025 M HCl).35 Total soil P (PT) was determined on sub-samples of 0.3 g soil with the addition of 5 mL concentrated H2SO4 and heating to 360 °C on a digestion block with subsequent stepwise (0.5 mL) additions of H2O2 until the solution was clear.36 Dithionite citrate bicarbonate (DCB)-extractable crystalline Fe and Al (FeDCB and AlDCB) were determined by the method of Mehra and Jackson.37 Ammonium oxalate dark (AOD) extractable Feox and Alox were determined as described by Shang and Zelazny (2008).38 Organic C was determined by dichromate oxidation method using GENESUS-20 spectrophotometer as described in Motsara and Roy (2008).39 Particle size distribution of the soils was determined using hydrometer method after dispersion of the soil with sodium hexametaphosphate, Na6(PO3)6.402.3 Synthesis of crystalline and amorphous Fe–Al binary mixed oxides powders
Using gel evaporation method,41 powder sample of Fe–Al (10% mol) mixed oxide was synthesized from Fe(NO3)3·9H2O (Sigma-Aldrich, Germany) and Al(NO3)3·9H2O (Uni-chem chemical reagents,AR) dissolved in ethylene glicol (Baker Analytical reagent,USA) at molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 of total metal ion to ethylene with addition of deionized water just enough to dissolve the starting materials. The solution was then refluxed in a 250 mL flask at 65 °C for 24 h to get hydrosol. The hydrosol was dried at 100 °C for 24 h to obtain xerogel. Finally, the xerogel was grounded and calcined at 300 °C for 2 h (crystalline phase). The calcined crystal sample is referred as calcined powder sample (CPS). Another sample (for amorphous phase) was also prepared by the same procedure without calcinations and referred as amorphous powder sample (APS) the hydrated suspensión of which is labeled as HFAO and HAHFAO respectively.
:3 of total metal ion to ethylene with addition of deionized water just enough to dissolve the starting materials. The solution was then refluxed in a 250 mL flask at 65 °C for 24 h to get hydrosol. The hydrosol was dried at 100 °C for 24 h to obtain xerogel. Finally, the xerogel was grounded and calcined at 300 °C for 2 h (crystalline phase). The calcined crystal sample is referred as calcined powder sample (CPS). Another sample (for amorphous phase) was also prepared by the same procedure without calcinations and referred as amorphous powder sample (APS) the hydrated suspensión of which is labeled as HFAO and HAHFAO respectively.
2.4 Characterization
The structure of the as-synthesized powder was identified by XRD equipped with CuKα radiation (λ = 1.5405 Å) operated at 40 kV tube voltage and 40 mA tube current with a scanning rate of 4 min−1 at a step scan of 0.02. The crystallite size was determined from the XRD peaks using Scherer formula.42 The surface functional groups of the as-synthesized samples were determined using Fourier transform infrared (FTIR) spectrometer (Spectrum 65, PerkinElmer) in the range 4000–400 cm−1 using KBr pelletizer. The morphology and particle size distribution of the solids were determined by scanning electron microscopy (SEM) using a Hitachi TM1000 with backscattered electrons detector and EDX detector. Sorption properties were measured under N2 atmosphere at −196 °C in a Micromeritics instrument ASAP 2420 device. The isotherms were registered, approximately 200 mg of each sample was outgassed at 150 °C for 16 h under high vacuum. Thus, the surface areas of the materials were estimated using the Brunauer–Emmet–Teller (BET) method and their micropore and external surface areas were determined by the t-plot method. Pore size distributions were predicted by applying the BJH method. The total pore volume was taken from the relative pressure close to unity (P/Po = 0.98). Thermo gravimetric analysis (TGA) was performed using a PerkinElmer TGA7 instrument in the temperature range of 30 °C to 900 °C under air flow at a heating rate of 20° min−1.2.5 Preparation of the sorbents suspensions
Suspensions of as-synthesized calcined samples (HFAO) and noncalcined amorphous powder samples (HAFAO) were prepared separately by adding 5 and 10 g powders respectively to 1 L volumetric flask and filling deionizer water to the mark. The masses of the calcined and amorphous powders are different considering the mass loss due to removal of surface adsorbed water and residual organic material during calcination at 300 °C. Equivalent amount of hydrous ferric oxide (HFO) to be used as a reference was also prepared following the procedure described by Freese et al. (1995).7 The pH of the suspensions were adjusted to pH = 5.6 for solution containing only deionized water and to the pH of soil for bottles containing soil solution. Twenty-centimeter-length DMT strips (Medicell International Ltd, London; dialysis tubing: visiting, size 320/32 inches, approximate pore size 2.5–5.0 nm; membrane thickness 3 μm) were boiled two times in deionized water for 5 min and thoroughly rinsed. Twenty milliliter of each suspension was filled in dialysis tube each in three replicates. During the filling of the DMTs, each type of suspension was stirred vigorously to obtain uniform quantities of HFAO, HAFAO and HFO for each DMT strips.2.6 Phosphate sorption and desorption study
This experiment consists of three stages. The first stage was to undertake a preliminary experiment to investigate possible leakage of the phosphate binding materials into surrounding solution. The second stage was carried out to determine the phosphate sorption capacity of the sink materials employed in the dialysis membrane tubes in phosphate contaminated aqueous systems. The last experiment was executed to employ the DMT-adsorbent system into soil solution to study the long-term P desorption kinetics.A preliminary experiment was conducted by placing DMTs filled with HFAO, HAFAOand HFO in contact with 80 mL de-ionized water in 500 mL capped polyethylene bottles and shaking gently for a week. This was performed to investigate possible leakage of the sink materials (HFAO and HAFAO) through the dialysis membrane tubes when in contact with deionized water. Any leakage of the sorbent materials in particular the iron component through the dialysis membrane tubes was checked both qualitatively and quantitatively taking 5 mL of deionized water sample for analysis at the interval of 24 h for a week. For the qualitative test, NH4SCN and [K4Fe(CN)6] were added to the acidified deionized water sample where as AAS was used for the quantitative determination of iron. The result of this experiment confirmed trace level leakage of iron for amorphous sorbent (HAFAO) (Table S5†).
In the second stage of this experiment, phosphate sorption characteristics of the as-synthesized sorbents were undertaken by placing appropriate amount of the respective sinks filled in the dialysis membrane tubes in 80 mL of 10, 30, 50 and 100 ppm KH2PO4 solution. In the third stage of the experiment, P desorption characteristics was studied by taking 1 g air dried soils from Bishoftu and Gununo in 80 mL 2 mM CaCl2 and 0.3 mM KCl solution taken in separate 500 mL capped plastic bottles. Two drops of chloroform were added to the soil suspension to inhibit microbial activity.7 Both pure P solutions and soil suspensions were kept in contact with dialysis membrane tube (DMT) filled with as-synthesized HFAO, HAFAO and the reference HFO as P sorbents. The aqueous solutions and soil suspensions were shaken at 120 rpm on rotary shaker for the intervals of 24, 48, 72, 96 and 168 h at 25 °C. At each interval of time, 2 mL samples of soil suspensions was taken and 2 mL deionized water was added to keep volume of solution constant (assuming negligible change in the concentration of P). All experiments were carried out in triplicates. At the mentioned time interval, the DMT-HFAO, DMT-HAFAO and DMT-HFO were changed for fresh one. Prior to changing the DMT-sorbent system, any soil material adhering to the DMT was rubbed off with a glass rod, to minimize loss of soil material from the soil suspension. The sorbents were then transferred to a separate clean polyethylene beakers. HFO and HAFAO were dissolved by adding 1 mL conc. H2SO4 while HFAO was dissolved by adding 7 mL. The blank was prepared by adding the indicated volume of conc. H2SO4 to 20 mL of each sorbent suspension. Finally, samples of the dissolved HFO, HAFAO and HFAO were taken for analysis and adjusted to final volume and analyzed spectrophotometrically according to Murphy and Riley (1962).43 Sorption efficiency of the sorbents at each interval of time was calculated as mg P determined per gram of the sorbent phosphate remaining in solution at time t. The amount of P sorbed, concentration of phosphate remaining in solution and initial P concentrations were employed in the data analysis using isotherm and kinetics models. The adsorption capacity of the phosphate ion is the concentration of the phosphate ion on the adsorbent mass and was calculated based on the mass balance principle,
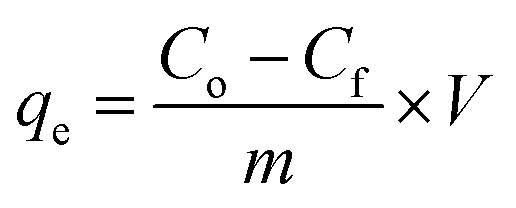
2.7 Data analysis
The sorption/desorption data obtained were statistically analyzed by using Statistical Analysis System (SAS Institute 2006, versión 9.1.3). Analysis of variance was done using the General Linear Model (GLM) procedure. The Tukey test was used to determine significant differences at α = 0.05.3. Results and discussion
3.1 Physicochemical properties of soils
Table S1† depicts physicochemical properties of the soils studied. The large difference between their pH (Gununo 3.5 and Bishoftu 6.8), exchangeable aluminium, DCB extractable Al and clay content could be attributed to the different physicochemical properties of the two soils. The organic carbon was low for Bishoftu and moderate for Gununo soil. Bray I indicated highly labile P in Bishoftu than Gununo soil. In light of these properties, Gununo Nitisol has high P fixing capacity where as Bishoftu Andisols exhibited low P fixing capacity.3.2 Characterization of the as-synthesized sorbents
X-ray diffraction patterns of the calcined and uncalcined Fe–Al binary oxide systems are shown in Fig. 1. The binary system exposed to a calcinations temperature of 300 °C (CPS) showed better crystallinity as compared to the uncalcined (APS) congener, which is amorphous. Peaks shown by CPS correspond to the primitive cubic system of maghemite (γ-Fe2O3) (JCPDS no. 39-1346). The weak diffraction peak observed at 35.47 can also be ascribed to the maghemite phase of the amorphous APS. Our findings agree with results from previous reports.18,44,45 No peak attributable to alumina was observed in CPS due to perhaps the amorphous nature of alumina under the calcinations temperature employed in this experiment (300 °C), as crystallized alumina such as γ-Al2O3 and α-Al2O3 could be expected under thermal treatment with temperatures at 800 °C and 1000 °C respectively.16,18,46 | ||
| Fig. 1 XRD patterns of uncalcined (APS) and calcined (CPS) Fe–Al binary oxide powder samples. | ||
The average crystallite size of the crystalline Fe–Al binary oxide (CPS) is found to be 21.40 nm. Our result concurs with the report made by Tofik et al. (2016)18 the size of which was 20.94 nm for the sample of the same composition sintered at 300 °C. We also estimated the specific surface area of the as-prepared samples using BET methods (Table S2†). The estimated specific surface áreas (m2 g−1) of HFAO (CPS), HAFAO (APS) and HFO were found to be 48.7, 11.4, and 17.1 respectively. The smallest specific surface área registered by of the amorphous binary system could be possibly due to residual solvents (ethylene glicol) present in the uncalcined product as evidenced in the FTIR result. In fact, the TGA/DTA curves of these samples also supported the above assertion. The termal stability of amorphus phase of the adsorbent (APS) was determined as shown in Fig. 2. It exhibited three distinct weight losses. The initial loss (≈23%) is between 100 and 150 °C as the result of external wáter adsorbed on the surface of the sorbent whereas pronounced loss (≈30%) occurred in the temperature range between 150 and 250 °C due to removal of trapped solvent such as ethylene glycol from the internal pore of the adsorbent. The third weight loss which accounts for (≈5%) was observed in the temperatura range from 250 to 350 °C possibly be due to phase change from maghemite to haematite form of the iron oxide. Above 350 °C, no change in weight was recorded evidencing the conversión of the adsorbent to its corresponding oxide forms. Contrary to the above observation, ≈2% weight loss was registered up to 200 °C accounting for the removal of physisorbed wáter and the remaining 2% loss exhibited between 200 and 550 °C signifies phase changes among the different iron oxide forms. The significantly larger loss from the amorphus sample justifies its smaller specific surface área compared with the crystalline counterpart.
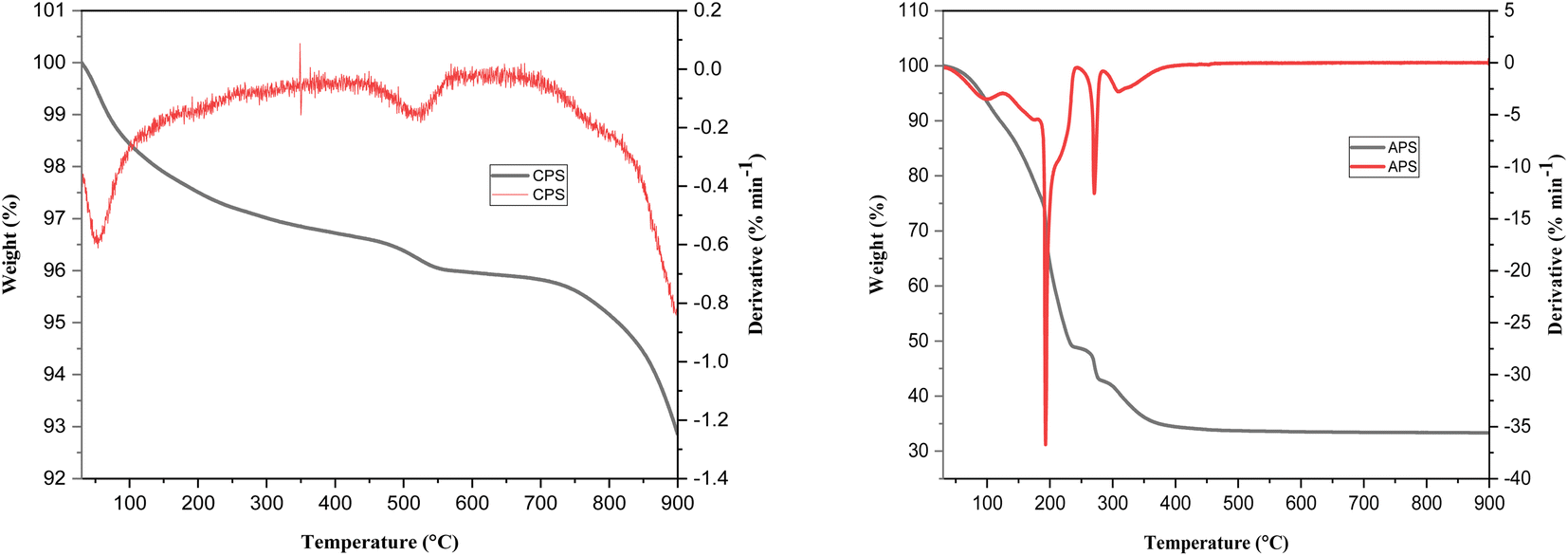 | ||
| Fig. 2 TGA/DTG plots of CPS and APS samples. | ||
The SEM micrographs of amorphous (APS) and crystalline (CPS) as-synthesized binary Fe–Al mixed oxides are displayed in Fig. S1.† In both cases, no distinct morphology was observed. EDX (data not shown) analyses confirmed the presence of both metals considered in the relatively wider range: 98.0–100% (Fe), 0–2% (Al) revealing the heterogeneous nature of the sorbent.
Fig. 3 shows FTIR spectra of CPS before and after sorption of phosphate. The peaks observed in both cases are similar. The broad bands at 3422 and 3430 cm−1 correspond to the stretching mode of O–H group of adsorbed water molecule. The peak observed around 1636 cm−1 can be associated with bending mode of physically adsorbed water molecule. The peak shown at 2338 cm−1 can be assigned to carbonate due to CO2 adsorbed from air during preparation.18 The peak at 1059 cm−1 observed on CPS is a characteristic of M–OH bending vibrations.47,48 The peaks observed in the range from 550 to 640 cm−1 could be ascribed to M–O stretching frequencies.
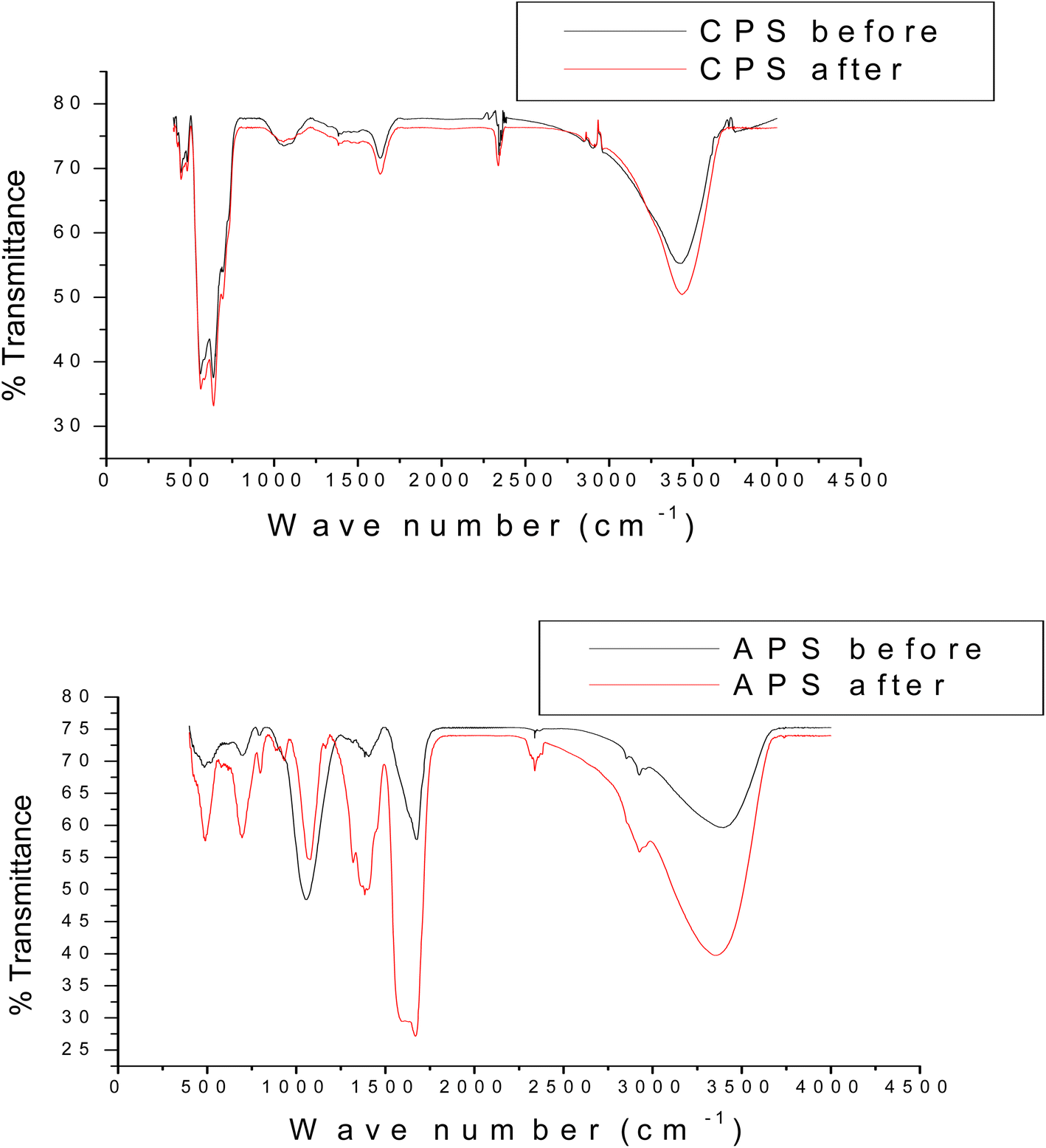 | ||
| Fig. 3 FTIR spectra of CPS and APS before and after phosphate sorption. | ||
Fig. 3 exhibits the FTIR peaks of APS before and after sorption. The major peaks seemed to be similar in both cases. The peaks around 3360–3382 cm−1 represent the νOH stretching vibration whereas the peaks observed in the range from 1655–1670 cm−1 could be attributed to δH–OH bending vibration of adsorbed water molecules. The peaks around 2925, 2338, 1385 and 1052 cm−1 represent the C–H, C–O symmetric stretching vibrations, w(OC2H4) of the ethylene glycol used in the synthesis and ν3 asymmetric vibrations of adsorbed carbonate anions respectively. The peak shown at 1052 cm−1 indicates M–OH bending vibrations whereas peaks shown at 700 and 484 cm−1 could be ascribed to M–O stretching vibrations.49 We noticed the presence of additional peak at 933 cm−1 and peaks with enhanced intensity in the FTIR spectrum carried out after the virgin sorbent was exposed to phosphate sorption. The additional peak exhibited could be due to asymmetric vibration of P–O26 or P–OM modes of vibration50 while the increased peak intensity could be attributed to overlap of P–O with those of the COO− and –OH groups.51–53 Our findings indicate the sorption of P onto the as-synthesized phosphate binding materials.
| DMT-sorbent | mg P g−1 sorbent |
|---|---|
| Means with different letters along the column are significantly different. | |
| DMTHFAO | 3.00a |
| DMTHAFAO | 2.53b |
| DMT-HFO | 0.83c |
| LSD = 0.296, CV = 5.57 | |
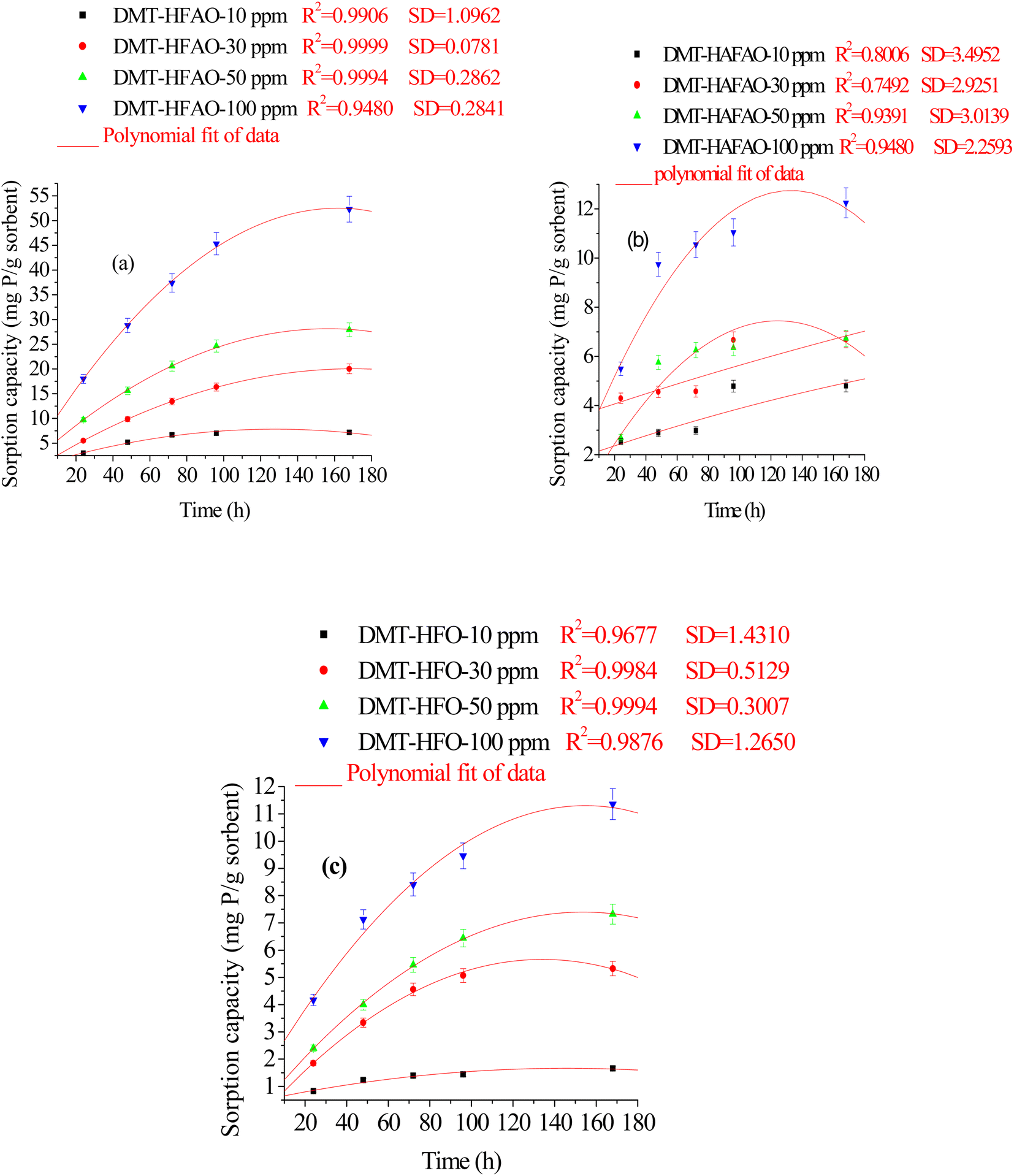 | ||
| Fig. 4 Cumulative P sorbed by DMT-sorbent (a-HFAO, b-HAFAO and c-HFO) at different initial concentrations of standard KH2PO4 solution in the intervals from 24–168 h. Vertical bars represent error. | ||
Fig. S2† illustrates the total amount of P sorbed by the three sorbent systems at 10 ppm phosphate concentration. At the plateau, the initial concentration of 10 ppm (0.32 mmol P/L) dropped to 4.6 × 10−6 and 1.5 × 10−5 mol P/L as the result of sorption by DMT-HFAO and DMT-HFO respectively. This indicated that for a unit mass of the sorbents, DMT-HFAO has more sorption capacity in lowering solution P than DMT-HFO. Freese et al. (1995)7 reported that HFO dropped 0.8 mmol P/L to 2 × 10−6 mol L−1 in 68 h. In this study, HFO also dropped 0.32 mmol P/L to 1.5 × 10−5 mol L−1 after 72 h. The sorption efficiency of the HFO in the present work was lower than the single system previously reported. Despite this difference, our sorbent is found to be efficient in adsorbing phosphate from aqueous system. Moreover, the binary system prepared under the same experimental condition showed better sorption efficiency than the single system. For DMT-HAFAO sorbent system, phosphate sorption rate was almost constant for the intervals of 24, 48 and 72 h. The loss seems relatively high for the intervals of 24–72 h and equilibrium was reached after 96 h.
Fig. 4a–c and S2† show cumulative phosphate extracted by the three sorbents from phosphate concentration in the range from 10–100 ppm in the intervals of 24–168 h. Phosphate extracted was significantly (p < 0.05) influenced by sorbent type, initial phosphate concentration and extraction time. The cumulative P extracted by DMT-HFAO was 52.3 mg P g−1 (sorbent) while HAFAO and HFO extracted 12.5 and 11.3 mg P g−1 (sorbent) respectively. The remarkable sorption efficiency of DMT-HFAO as compared to DMT-HAFAO and DMT-HFO is evidenced in this work.
The sorption of phosphate was fast at the initial stage tending to equilibrate after 48 and 72 h for DMT-HFO and DMT-HFAO respectively. The initially fast adsorption stage was due to ion-exchange with surface hydroxyl ions of the sorbents. The slow adsorption rate in the later stage represents a gradual uptake of phosphate as the result of diffusion in the internal matrix of the sorbents.54,55 The nano-sized DMT-HFAO extracted more phosphate than HFO in the equilibration time between 72 and 96 h. It appeared that as concentration of phosphate in solution in equilibrium with DMT-HFAO becomes low, DMT-HFAO extracts even more phosphate than HFO implying its potential to desorb phosphate from soil with low P concentration. Harvey and Rhue (2008)20 reported that Fe–Al hydr(oxide) were effective for P removal. According to Biswas et al. (2007),56 the sorption ability of Fe–Al mixed oxide was much higher than either of pure oxide. They also reported that the specific surface area of mixed oxide was greater than that of the pure oxide.
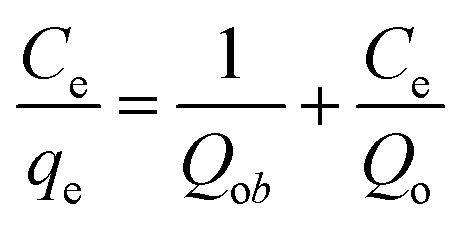 | (1) |
Freundlich model,
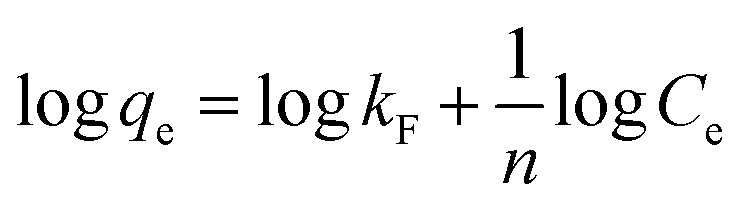 | (2) |
All the three sorbent systems viz., DMT-HFAO, DMT-HAFAO and DMT-HFO fitted better to both Freundlich and Langmuir models with the former showing consistently better fit to the data (Table 2, Fig. S3†) evidencing the heterogeneity in the sorption process. The lower n values in the Freundlich model are indicators of more heterogeneous sorption. Tofik et al. (2016)18 reported higher n value for sorption of phosphate by direct contact of nano-sized Fe–Al mixed oxide with phosphate solution for 24 h equilibration time.
| Isotherm models | ||||||
|---|---|---|---|---|---|---|
| Parameters | ||||||
| Langmuir | Freundlich | |||||
| R2 | Qo (mg g−1) | b | R2 | KF | n | |
| DMT-HFAO | 0.980–0.993 | 15.9 | 0.72 | 0.986–0.994 | 3.6 | 1.6 |
| DMT-HAFAO | 0.886–0.980 | 7.9 | 0.79 | 0.975–0.994 | 1.6 | 1.7 |
| DMT-HFO | 0.964–0.986 | 4.2 | 0.77 | 0.969–0.993 | 0.44 | 1.7 |
The observed difference could be attributed to longer equilibration and diffusion controlled P sorption in our case as direct contact of phosphate with the sink is restrained due to the membrane tubes. This allowed phosphate to diffuse into micropores that are not energetically equal because of heterogeneity.54 The higher Freundlich adsorption constant kF for DMT-HFAO also indicated higher sorption capacity of the sorbent as compared to DMT-HAFAO and DMT-HFO (Table 2).
3.3 Soil phosphate desorption by DMT-sorbents
Fig. 5a illustrates phosphate desorbed by the three sorbents from Bishoftu (low-P fixing capacity) and Gununo (high P fixing capacity) soils fitted with intra-particle diffusion model. The graph clearly shows two linear sections for DMT-HFAO and DMT-HFO which indicate series of sorption processes. The first linear stage was a fast stage. The second linear portion was a moderate adsorption process, where the rate of adsorption was governed by the intra-particle diffusion in the pore structure. Some literature54,60,61 indicated a third section due to steric hindrance from adsorbed phosphates, but this is unlikely to happen in our case since at each interval of time, the DMT-sorbent was replaced by new one. Phosphate sorption ability of the three DMT-sorbent systems of soil solution extract revealed that they have potentials in desorbing phosphate from soils with high and low P fixing capacities. In both soils, DMT-HFAO and DMT-HFO data fitted the second linear portion very well compared with DMT-HAFAO.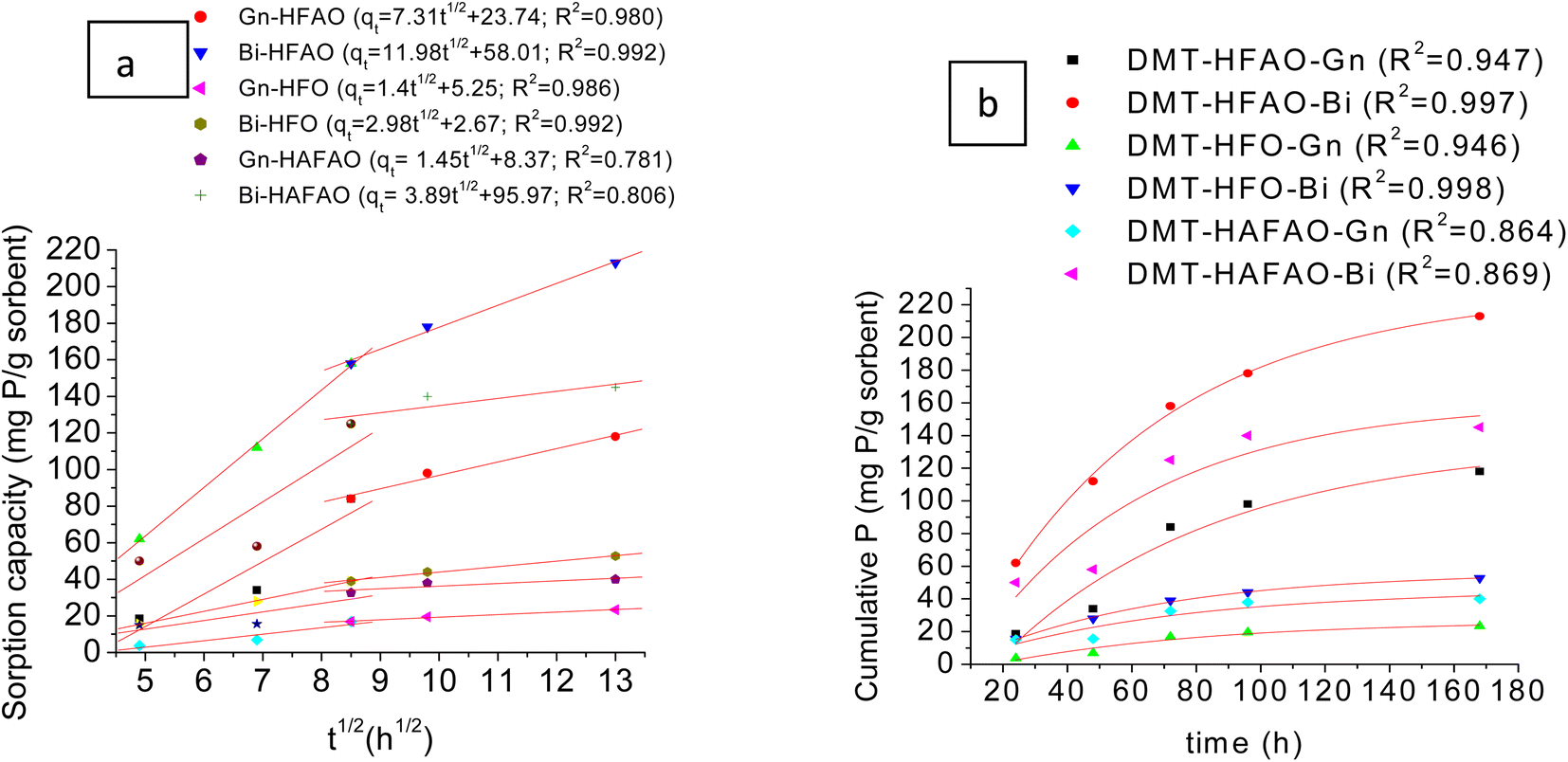 | ||
| Fig. 5 (a) P desorbed by DMT-sorbent-systems for Gununo and Bishoftu soils using intra-particle diffusion model; (b) Bishoftu and Gununo soils data fitted to two component first order model. | ||
The relative amounts of P desorbed by DMT-HFAO were 52 and 46% (mg mg−1, phosphate) of the total P for Gununo and Bishoftu soils respectively (Fig. S6†). In contrast to this, DMT-HFO desorbed 10 and 11% where as HAFAO desorbed 18 and 30% of the total P from Gununo and Bishoftu soils respectively. This indicated that among three sorbents, DMT-HFAO can be a potential candidate for desorbing more P from soils with both high and low P fixing capacity. From the positive intercepts indicated in Fig. 5a, one could discern the rapid desorption in shorter time. Most of the intercepts in literature were also positive.62–65 If the rate determining step was only intra-particle diffusion, the line would pass through the origin.66 But this was not observed in this study indicating that the initial stage attributed to the boundary effect. Under such case, soil P release is rate determining step. The larger intercept for DMT-HAFAO is an indication of greater boundary effect for HAFAO.67 The second portion of DMT-HFAO for both soils seemed to attain plateau.
Fig. 5b illustrates the cumulative P desorbed from the two soils fitted to two component first order model. In general, this model provided a better fit to all the sorbent systems in both soils despite the relatively lower R2 values exhibited by DMT-HAFAO (R2 = 0.869 and 0.864 for Bishoftu and Gununo soils respectively). The model assumes two different phosphate pools from soils distinguished by different release kinetics indicated by the change of the slope in the curve. The rate constants kA and kB of the soils P desorption were obtained from the slope of the plot of lnP against time for each pool (Fig. S6a–d†). For Gununo soil, rate constants for the labile pools, kA, were found to be 0.03146 h−1 and 0.03125 h−1 where as the rate constants for the less labile pool kB were estimated to be 0.00606 h−1 and 0.00588 h−1 for the sorbents DMT-HFAO and DMT-HFO respectively. In analogous way, the rate constants for the labile pools, kA, were found to be 0.01937 h−1 and 0.01917 h−1 where as the rate constants for the less labile pool, kB, were estimated to be 0.00537 h−1 and 0.00537 h−1 for the sorbents DMT-HFAO and DMT-HFO respectively in the case of Bishoftu soils. For both sorbents, a more labile phosphate pool (pool A) characterized by relatively higher phosphate release rate (kA > kB in each case) compared to the slow labile pool (pool B) was evidenced. The higher kA for Gununo soil is an indication of desorption of adsorbed phosphate directly in contact with soil solution.40,68 The higher and positive intercept for Bishoftu soil indicated short sorption of available phosphate in this soil.67
In all cases, the rate constant values for soils P desorption in this study were less than rate constant of phosphate transport, km through DMT (0.09 h−1) that was reported by Freese et al. (1995).7 This means, it is P desorption from soils that determine rate, not diffusion through membrane. The main difference in P sorption capacity is the ability of DMT-HFAO to extract more P than the other two sorbents even under very acidic soil condition.
Phosphate desorbed from soil solutions by DMT-HFAO was higher than the corresponding DMT-HFO and DMT-HAFAO for both sites (Fig. 5b). The difference between the two sites may be attributed to the difference in clay and organic matter content.13,69 The highest phosphate sorption from Bishoftu soil by DMT-HFAO was due to low fixing capacity of the soil. The amount of P extracted by DMT-HFAO, DMT-HFAO and DMT-HFO comprised 52, 18 and 10% (mg mg−1) of the total phosphate in 168 h from Gununo soil. For soil from Bishoftu site, DMT-HFAO extracted 46% of the total while DMT-HAFAO and DMT-HFO extracted 11 and 31% respectively in 168 h. This indicated that DMT-HFAO has remarkable phosphate desorption capacity from soils with high P fixing capacity than DMT-HFO and DMT-HAFAO. Freese et al. (1995)7 achieved 110% Pi extraction by single component system, HFO, for the duration of 500 h. But our binary system DMT-HFAO extracted about 520% (mg mg−1) Pi for 168 h and hence preferred to DMT-HFO for long-term phosphate desorption kinetics from acid soils. The DMT-HFAO system has also an advantage of shortening extraction time reasonably, provided that P desorption from soil by the system correlates with P uptake by a plant that in turn helps to recommend fertilizer with potentially less eutrophication to which this research work is targeted.
4. Conclusion
The synthesized Fe–Al mixed oxide binary sorbent, taking the advantages of individual oxide, has more sorption capacity than single component, HFO. The sorption data in all the three sorbent systems fitted to different isotherm and kinetic models. The relatively higher Qo = 15.9 mg g−1 and kF = 3.6 indicated that the sorption capacity of DMT-HFAO (crystalline form) is greater than the other two sorbents in the concentration range studied. The new binary system, DMT-HFAO extracted about 520% (mg mg−1, phosphate) of DMT-HFO in 168 h and hence preferred to study phosphate desorption kinetics from acid soils. It has also the advantage of shortening extraction time reasonably, provided that P desorption from soil by the system correlates with P uptake by a plant. This study evidenced the better performance of the two component sorbent than the single counterpart. The modified method we have designed (DMT-HFAO) keeps phosphate in aqueous soil very low due to its high sorption capacity as compared to the reference DMT-HFO and its binary amorphus congener DMT-HAFAO. This makes phosphate from less stable pools to replenish phosphate in aqueous solution. Therefore, the modified method is useful in predicting the time for depletion of P from slow labile P pools of soils. This in turn helps to recommend the appropriate time for fertilizer application, an advantage both from economic and environmental perspectives. However, more study is required relating the desorption parameters of this system with plant parameters. Moreover, soils of various pH should be entertained to check the universality of this system.Abbreviations
| APS | Amorphous Powder Sample |
| CPS | Calcined Powder Sample |
| DMT | Dialysis Membrane Tube |
| HFAO | Hydrous Ferric Aluminum Oxide |
| HAFAO | Hydrous Amorphous Ferric Aluminum Oxide |
| HFO | Hydrous Ferric Oxide |
| JCPDS | Joint Committee on Powder Diffraction Standards |
| SNNRG | Southern Nations Nationalities Region |
| TSP | Triple Super Phosphate |
| UV | Ultra Violet |
| XRD | X-ray Diffraction |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank SIDA and the Research and Extension Office of Haramaya University for providing the financial grant for the research (HURG-2014-03-03, HURG-2016-03-02 and HURG-2020-03-02-75). I. D. acknowledges funding from PID2022-136321OB-C21, MCIN/AEI/10.13039/501100011033 and European Union “NextGenerationEU”/PRTR through the project TED2021-131143B-I00.References
- D. Cordell and T.-S. S. Neset, Glob. Environ. Change, 2014, 24, 108–122 CrossRef.
- M. Keyzer, Economist, 2010, 158, 411–425 CrossRef.
- N. W. Menzies, B. Kusumo and P. W. Moody, Plant Soil, 2005, 269, 1–9 CrossRef CAS.
- R. Recena, I. Diaz, M. C. del Campillo, J. Torrent and D. Antonio, Sustainable Dev., 2016, 36, 54 Search PubMed.
- X. Y. W. M. Post, Biogeoscince, 2011, 8, 5907–5934 Search PubMed.
- H. Tiessen and J. O. Moir, in Soil Sampling and Methods of Analysis, ed. M. R. Carter, Lewis Publishers, Boca Raton, 1993, pp. 75–86 Search PubMed.
- D. Freese, R. Lookman, R. Merckx and W. H. Riemsdijk, Soil Sci. Soc. Am. J., 1995, 59, 1295–1300 CrossRef CAS.
- S. E. A. T. M. Van der Zee, L. G. J. Fokkink and W. H. Van Reimsdijk, Soil Sci. Soc. Am. J., 1987, 51, 599–604 CrossRef CAS.
- R. G. Myers, G. M. Pierzynski, S. J. Thien and A. N. Sharpley, Soil Sci. Soc. Am. J., 2005, 69, 511–521 CrossRef CAS.
- R. Lookman, D. Freese, R. Marks, K. Vlassak and W. H. Van Riemskdijk, Environ. Sci. Technol., 1995, 29, 1569–1575 CrossRef CAS PubMed.
- P. C. De Jager and A. S. Claassens, Commun. Soil Sci. Plant Anal., 2005, 36, 309–319 CrossRef CAS.
- S. Heidari, A. Reyhanitabar and S. Oustan, Geoderma, 2017, 305, 275–280 CrossRef CAS.
- V. A. Ochwoh, A. S. Claassenseand and P. C. De Jager, Commun. Soil Sci. Plant Anal., 2005, 36, 535–556 CrossRef CAS.
- A. Reyhanitabara, S. Heidar, S. Oustan and R. Gilkes, Commun. Soil Sci. Plant Anal., 2018, 49, 1281–1288 CrossRef.
- A. M. Taddesse, A. S. Claassens and P. C. De Jager, J. Plant Nutr., 2008, 31, 1507–1522 CrossRef CAS.
- B. Abebe, A. M. Taddesse, I. Diaz, T. Kebede and E. Teju, J. Environ. Chem. Eng., 2017, 5, 1330–1340 CrossRef CAS.
- J. Lu, H. Liu, R. Liu, X. Zhao, L. Sun and J. Qu, Powder Technol., 2013, 233, 146–154 CrossRef CAS.
- A. S. Tofik, A. M. Taddessea, K. T. Tesfahun and G. G. Girma, J. Environ. Chem. Eng., 2016, 4, 2458–2468 CrossRef CAS.
- Y. B. Dibabe, A. M. Taddesse, E. Teju and Y. Bogale, Environ. Nanotechnol., Monit. Manage., 2022, 18, 100723 CAS.
- O. R. Harvey and R. D. Rhue, J. Colloid Interface Sci., 2008, 322, 384–393 CrossRef CAS PubMed.
- M. G. Sujana and S. Anand, Appl. Surf. Sci., 2010, 256, 6956–6992 CrossRef CAS.
- A. F. De Sousa, T. P. Braga, Chagas, E. C. Gomes, A. Valentini and E. Longhinotti, Chem. Eng. J., 2012, 210, 143–149 CrossRef.
- L. Lai, Q. Xie, L. Chi, W. Gu and D. Wu, J. Colloid Interface Sci., 2016, 465, 76–82 CrossRef CAS PubMed.
- G. Li, S. Gao, G. Zhang and X. Zhang, Chem. Eng. J., 2014, 235, 124–131 CrossRef CAS.
- J. Lu, H. Liu, X. Zhao, W. Jefferson, F. Cheng and J. Qu, Colloids Surf., A, 2014, 455, 11–18 CrossRef CAS.
- C. Namasivayam and K. Prathap, J. Hazard. Mater., 2005, 123, 127–134 CrossRef CAS PubMed.
- A. Sarkar, S. K. Biswas and P. Pramanik, J. Mater. Chem., 2010, 20, 4417–4424 RSC.
- H. Duan, L. Zhang, Y. Wang, Y. Liu and Y. Wang, Environ. Sci. Pollut. Res., 2021, 28, 62662–62676 CrossRef CAS.
- Z. Wang, M. Xing, W. Fang and D. Wu, Appl. Surf. Sci., 2016, 366, 67–77 CrossRef CAS.
- G. Zhang, H. Liu, R. Liu and J. Qu, J. Colloid Interface Sci., 2009, 335, 168–174 CrossRef CAS PubMed.
- F. Laekemariam, K. Kibret, T. Mamo, E. Karltun and H. Gebrekidan, Environ. Syst. Res., 2016, 5, 24 CrossRef.
- T. Beshah, Understanding farmers: Explaining soil and water conservation inKonso, Wolaita, and Wollo, Ethiopia, PhD thesis, Wageningen University and Research Center, The Netherlands, 2003 Search PubMed.
- M. Abebe, Nature and Management of Ethiopian Soil, 1998 Search PubMed.
- H. F. Murphy, A report on the fertility status and other data on some soils of Ethiopia, Experimental Bulletin No. 44, An Oklahoma State University-USAID contract Publication, 1968 Search PubMed.
- R. H. Brayand and L. T. Kurtz, Soil Sci., 1945, 59, 39–45 CrossRef.
- R. L. Thomas, R. W. Sheard and J. R. Moyer, Agron. J., 1967, 59, 240–243 CrossRef CAS.
- O. P. Mehra and M. L. Jackson, Clays Clay Miner., 1958, 7, 317–327 CrossRef.
- C. Shang and L. W. Zelazny, in: Methods of Soil Analysis Part 5-Mineralogical Methods, ed. A. L. Ulery and L. R. Drees, Soil Science Society of America, Inc., Wisconsin, 2008, pp. 33–80 Search PubMed.
- M. R. Motsara and R. N. Roy, Guide to laboratory establishment for plant nutrient analysis, FAO fertilizer and plant nutrition bulletin, Rome, 2008 Search PubMed.
- S. Sertsu and T. Bekele, Procedure for Soil Analysis, National Soil Research Center, Ethiopian Agricultural Research Organization, 2000 Search PubMed.
- F. Gulshan, Y. Kameshima, A. Nakajima and K. Okada, J. Hazard. Mater., 2009, 169, 697–702 CrossRef CAS PubMed.
- P. Persson, N. Nilsson and S. Staffan, J. Colloid Interface Sci., 1996, 177, 263–275 CrossRef CAS PubMed.
- J. Murphy and J. P. Riley, Anal. Chim. Acta, 1962, 27, 31–36 CrossRef CAS.
- M. AliAhmed and N. N. Moghaddam, Mater. Sci. Polym., 2013, 31, 264–268 CrossRef.
- M. Nazari, N. Ghasemi, M. Heydar and M. M. Motlagh, J. Nanostruct. Chem., 2014, 4, 99 CrossRef.
- N. R. Habib, A. M. Taddesse and A. Temesgen, Bull. Chem. Soc. Ethiop., 2018, 32, 101–109 CrossRef CAS.
- C. Gagan, W. Dashan and A. Howard, Chem. Commun., 2007, 4809–4811 Search PubMed.
- N. Masami, Soil Sci. Plant Nutr., 1986, 32, 51–58 CrossRef.
- J. Lu, D. Liu, J. Hao and G. Zhang, Chem. Eng. Res. Des., 2015, 93, 652–661 CrossRef CAS.
- C.-G. Lee, J.-A. Lee, J.-A. Park, J.-H. Kim, S.-B. Kim, S. H. Lee and J.-W. Choi, Chem. Eng. J., 2014, 236, 341–347 CrossRef.
- W. Xie and D. Zhao, Sci. Total Environ., 2016, 542, 1020–1029 CrossRef CAS PubMed.
- Z. Ren, X. Xu, X. Wang, B. Gao, Q. Yue, W. Song, L. Zhang and H. Wang, J. Colloid Interface Sci., 2016, 468, 313–323 CrossRef CAS PubMed.
- S. Gypser, F. Hirsch, A. M. Schleicher and D. Freese, J. Environ. Sci., 2018, 70, 175–189 CrossRef CAS PubMed.
- T. Liu, K. Wu and L. Zeng, J. Hazard. Mater., 2012, 217–218, 29–35 CrossRef CAS PubMed.
- W. Rudzinski, W. A. Steele and G. Zgrablich, Equilibria and dynamics of gas adsorption on heterogeneous solid surfaces, Elsevier, Amsterdam, 1996, p. 202 Search PubMed.
- K. Biswas, S. K. Saha and U. C. Ghosh, Ind. Eng. Chem. Res., 2007, 46, 5346–5356 CrossRef CAS.
- I. Langmuir, Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- H. M. F. Freundlich, Z. Phys. Chem., 1906, 57, 385–470 CAS.
- D. Freese, R. Lookman, R. Merckx and W. H. Riemsdijk, Soil Sci. Soc. Am. J., 1995, 59, 1295–1300 CrossRef CAS.
- M. D'Arcy, D. Weiss, M. Bluck and R. Vilar, J. Colloid Interface Sci., 2011, 364, 205–212 CrossRef PubMed.
- Z. Ren, L. Shao and G. Zhang, Water, Air, Soil Pollut., 2012, 223, 4221–4231 CrossRef CAS.
- D. Ozer, G. Dursun and A. Ozer, J. Hazard. Mater., 2007, 144, 171–179 CrossRef.
- F.-C. Wu, R.-L. Tseng and R.-S. Juang, J. Colloid Interface Sci., 2005, 283, 49–56 CrossRef CAS PubMed.
- F.-C. Wu, R.-L. Tseng and R.-S. Juang, Water Res., 2001, 35, 613–618 CrossRef CAS PubMed.
- W. Zhang, Z. Xu, B. Pan, L. Lv, Q. Zhang, Q. Zhang, W. Du and B. P. Q. Zhang, Colloid Interface Sci., 2007, 311, 382–390 CrossRef CAS.
- L. Zhang, A. L. Hugo, M. Xu, C. Du and Y. Du, Int. J. Environ. Res. Public Health, 2015, 12, 11 Search PubMed.
- F.-C. Wu, R.-L. Tseng and R.-S. Juang, Chem. Eng. J., 2009, 153, 1–8 CrossRef CAS.
- d P. C. De Jager and A. S. Claassens, Commun. Soil Sci. Plant Anal., 2005, 36, 309–319 CrossRef.
- P. A. Sanchez, C. A. Palm and L. T. Szoth, Phosphorus dynamics in shifting cultivation systems in the Amazon, in SCOPE-UNEP Regional Workshop 3: South and Central America, ed. H. Tissen, D. Lopez-Hernandez, and I. H. Salcedo, Institute of Pedology, Saskatoon, Canada, 1991, p. 142 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04000c |
| This journal is © The Royal Society of Chemistry 2023 |