
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence[Cs©C18]+ and [Na©C14]+: perfect planar alkaline-metal-centered polyynic cyclo[n]carbon complexes with record coordination numbers†
Min Zhang,
Rui-Nan Yuan,
Yan-Bo Wu ,
Qiang Chen,
Zhihong Wei and
Si-Dian Li*
,
Qiang Chen,
Zhihong Wei and
Si-Dian Li*
Shanxi University, China. E-mail: lisidian@sxu.edu.cn; weizhihong@sxu.edu.cn; chenqiang@sxu.edu.cn
First published on 10th August 2023
Abstract
Searching for the maximum coordination number (CN) in planar species with novel bonding patterns has fascinated chemists for many years. Using the experimentally observed polyynic cyclo[18]carbon D9h C18 and theoretically predicted polyynic cyclo[14]carbon D7h C14 as effective ligands and based on extensive first-principles theory calculations, we predict herein their perfect planar alkaline-metal-centered complexes D9h Cs©C18+ (1) and D7h Na©C14+ (4) which as the global minima of the systems possess the record coordination numbers of CN = 18 and 14 in planar polyynic species, respectively. More interestingly, detailed energy decomposition and adaptive natural density partitioning bonding analyses indicate that the hypercoordinate alkaline-metal centers in these complexes exhibit obvious transition metal behaviors, with effective in-plane (π-6s)σ, (π-7p)σ, and (π-5d)σ coordination bonds formed in Cs©C18+ (1) and (π-3s)σ, (π-3p)σ, and (π-3d)σ coordination interactions fabricated in Na©C14+ (4) to dominate the overall attractive interactions between the metal center and its cyclo[n]carbon ligand. Similarly, alkaline-metal-centered planar Cs Cs©C17B (2), C2v Cs©C17− (3), C2v Na©C13B (5), and C2v Na©C13− (6) have also been obtained with CN = 18, 17, 14, and 13, respectively.
1 Introduction
The successive discoveries of fullerenes in 1985,1 carbon nanotubes in 1991,2 and graphene in 2004![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 which all consist exclusively of 3-coordinate carbon atoms have sparked a new field of synthetic carbon allotropes in chemistry. The recent characterization of polyynic cyclo[18]carbon D9h C18 with bond length alternation (BLA) in 2019 using high-resolution atomic force microscopy marked the onset of an alternative family of molecular carbon allotropes consisting solely of 2-coordinate carbon atoms in the C4N+2 cyclo[n]carbon ring series.4 Previous gas-phase experiments indicated that cyclo[n]carbon rings as primary precursors may coalescence to form fullerenes and carbon nanotubes.5,6 Electronic spectroscopic measurements showed that both C18 and C14 possess monocyclic geometries, though these studies did not reveal whether they have cumulantic or polyynic structures.7,8 High level quantum Monte Carlo (QMC) simulation and coupled cluster methods with single and double excitations (CCSD) investigations indicated that both polyynic D9h C18 and D7h C14 are the ground states of the systems due to second-order Jahn-Taller effects, with their cumulantic counterparts with no BLA always behaving as transition states.9,10 Such perfect monocyclic D(2N+1)h C4N+2 polyynic species have aroused considerable interests among chemists and presented viable possibilities to form planar metal-doped cyclo[n]carbon complexes with super-high coordination numbers (CN). A recent theoretical investigation11 suggested that the Li-doped C18 complex may serve as a potential optical switch which transforms between two stable Cs configurations with Li inside (Li@C18in) and outside the carbon ring (Li@C18out). However, in the ground state (Li@C18in) of such an alkaline-metal-doped cyclo[18]carbon complex, the Li atom with the coordination number of CN = 5 appears to be severely off-centered due to the size mismatch between Li and its monocyclic C18 ligand. Similar situation happens in the recently proposed metal-doped M@C16 complexes (M = Ca, Sc, Ti, V, Ce, U) in which the off-centered alkaline-earth, lanthanide, or actinide metal atoms have the coordination numbers between CN = 4 ∼ 6,12 again due to size effect. A recent first-principles theory investigation by our group indicated that, in the experimentally observed La©C13+, the La center with the large atomic radius of rLa = 1.83 Å13 matches the C13 ligand perfectly both geometrically and electronically to form the perfect planar La-centered D13h La©C13+ which has the highest coordination number of CN = 13 in planar species reported to date, demonstrating the unique coordinating capability of cyclo[n]carbon rings as effective ligands to metal centers in chemistry.14 However, it still remains unknown to date in both experiments and theory whether or not metal-centered hypercoordinate planar cyclo[n]carbon complexes with CN > 13 can be achieved in chemistry. To achieve higher CNs (CN = n ≥ 14) in metal-centered cyclo[n]carbon complexes, it requires in chemical intuition that the metal centers have atomic radii greater than that of La.
3 which all consist exclusively of 3-coordinate carbon atoms have sparked a new field of synthetic carbon allotropes in chemistry. The recent characterization of polyynic cyclo[18]carbon D9h C18 with bond length alternation (BLA) in 2019 using high-resolution atomic force microscopy marked the onset of an alternative family of molecular carbon allotropes consisting solely of 2-coordinate carbon atoms in the C4N+2 cyclo[n]carbon ring series.4 Previous gas-phase experiments indicated that cyclo[n]carbon rings as primary precursors may coalescence to form fullerenes and carbon nanotubes.5,6 Electronic spectroscopic measurements showed that both C18 and C14 possess monocyclic geometries, though these studies did not reveal whether they have cumulantic or polyynic structures.7,8 High level quantum Monte Carlo (QMC) simulation and coupled cluster methods with single and double excitations (CCSD) investigations indicated that both polyynic D9h C18 and D7h C14 are the ground states of the systems due to second-order Jahn-Taller effects, with their cumulantic counterparts with no BLA always behaving as transition states.9,10 Such perfect monocyclic D(2N+1)h C4N+2 polyynic species have aroused considerable interests among chemists and presented viable possibilities to form planar metal-doped cyclo[n]carbon complexes with super-high coordination numbers (CN). A recent theoretical investigation11 suggested that the Li-doped C18 complex may serve as a potential optical switch which transforms between two stable Cs configurations with Li inside (Li@C18in) and outside the carbon ring (Li@C18out). However, in the ground state (Li@C18in) of such an alkaline-metal-doped cyclo[18]carbon complex, the Li atom with the coordination number of CN = 5 appears to be severely off-centered due to the size mismatch between Li and its monocyclic C18 ligand. Similar situation happens in the recently proposed metal-doped M@C16 complexes (M = Ca, Sc, Ti, V, Ce, U) in which the off-centered alkaline-earth, lanthanide, or actinide metal atoms have the coordination numbers between CN = 4 ∼ 6,12 again due to size effect. A recent first-principles theory investigation by our group indicated that, in the experimentally observed La©C13+, the La center with the large atomic radius of rLa = 1.83 Å13 matches the C13 ligand perfectly both geometrically and electronically to form the perfect planar La-centered D13h La©C13+ which has the highest coordination number of CN = 13 in planar species reported to date, demonstrating the unique coordinating capability of cyclo[n]carbon rings as effective ligands to metal centers in chemistry.14 However, it still remains unknown to date in both experiments and theory whether or not metal-centered hypercoordinate planar cyclo[n]carbon complexes with CN > 13 can be achieved in chemistry. To achieve higher CNs (CN = n ≥ 14) in metal-centered cyclo[n]carbon complexes, it requires in chemical intuition that the metal centers have atomic radii greater than that of La.
Searching for the maximum coordination number in planar species is more than a curiosity, it is to push the limits and ultimately to understand the essential concepts in chemistry.14,15 To successfully design a metal-centered hyper-coordinate planar complex, the metal center and its ligand must match both geometrically and electronically, i.e., they must have the right geometrical sizes and electronic configurations. The prototypical electron-deficient planar cyclo[n]boron rings have proven to be effective ligands to coordinate transition metal centers. Perfect σ + π dually aromatic wheel-like D8h Co©B8−, D9h Ru©B9−, D9h Rh©B9−, D9h Ir©B9−, D10h Ta©B10−, and D10h Nb©B10− with CN = 8, 9, 9, 9, 10, and 10 have been observed in gas phases in recent joint photoelectron spectroscopy and first-principles theory investigations.15–20 These results present the possibility to form metal-centered hyper-coordinate planar complexes using CnBm binary monocyclic rings as effective ligands, as indicated in the cases of the previously reported C2v Y©B6C6+ and C2v Sc©B5C6.14
Alkaline-earth metal centers in their perfect body-centered cubic carbonyl complexes Oh M(CO)8+ (M = Ca, Sr, or Ba) in low-temperature neon matrixes have been confirmed to be honorary transition metals with effective M–(CO)8 (π) coordination interactions.21 Similar M(dπ)–(CO)8 (π) coordination bonds were predicted to exist in Oh M(CO)8− complexes (M = K, Rb) in which the alkaline metal centers K and Rb exhibit transition metal behaviours.22 Given the fact that alkaline metals possess the largest atomic radii in the periodical table23 and have the potential to form complexes with transition metal behaviors, it is possible to form alkaline-metal-doped cyclo[n]carbon complexes (n ≥ 14) or their boron-substituted derivatives with CN ≥ 14 if the alkaline metal center and its ligand are chosen properly to match both geometrically and electronically.
Keeping the inspirations in mind, using the experimentally observed perfect planar ring-like D9h C18 and theoretically predicted D7h C14 as ligands and based on extensive global minimum searches augmented with first-principles theory calculations, we predict in this work the perfect planar alkaline-metal-centered D9h Cs©C18+ (1) and D7h Na©C14+ (4) which have the record coordination numbers of CN = 18 and 14 in planar species, respectively. Cs and Na with the atomic radii of rCs = 2.65 Å and rNa = 1.86 Å13 prove to match the D9h C18 and D7h C14 ligands perfectly both geometrically and electronically, respectively. Effective in-plane (π-s)σ, (π-p)σ, and (π-d)σ coordination bonds are formed to dominate the attractive interactions in these novel complexes in which the alkaline-metal centers exhibit transition metal behaviors. The iso-chemical shielding surfaces and out-of-plane π and in-plane σ ring current maps of the concerned species are computationally simulated to evidence their σ + π dual aromaticity.
2 Computational details
Extensive global-minimum (GM) searches were performed on Cs©C18+, Na©C14+, Cs©C17B, Cs©C17−, Na©C13B, and Na©C13− using the TGmin2 code24 at DFT level based on the basin-hopping algorithm.25 Over 1000 stationary points were explored for each species at PBE/DZVP level employing the CP2K program.26,27 The low-lying isomers were then fully optimized at both M06-2X and ωB97XD levels28,29 with vibrational frequencies checked, with the aug-cc-pvtz basis set for C, B, Na, and K and Stuttgart relativistic small-core pseudopotentials30,31 for Rb, Cs, and Fr, using the Gaussian16 program.32 The fact that M06-2X produces essentially the same polyynic D9h C18 and D7h C14 structures (Fig. S1†) as that obtained at the more accurate QMC and CCSD levels9,10 evidences the reliability of the optimized geometries depicted in Fig. 1. Natural bond orbital (NBO) analyses were performed using NBO 7.0 program.33 The energy decomposition analyses (EDA) together with the natural orbitals for chemical valence (NOCV) calculations, denoted as EDA-NOCV,34–36 were carried out with the ADF program package37 at M06-2X/TZ2P38 level where scalar relativistic effects were considered for Cs using the zero order regular approximation (ZORA).39 The frozen core approximation was not employed in EDA-NOCV computations. The overall interaction energy (ΔEint) between two fragments is divided into three main terms: the electrostatic interaction energy (ΔEelstat), Pauli repulsion (ΔEPauli), and orbital interaction energy (ΔEorb) in eqn (1):| ΔEint = ΔEelstat + ΔEPauli + ΔEorb. | (1) |
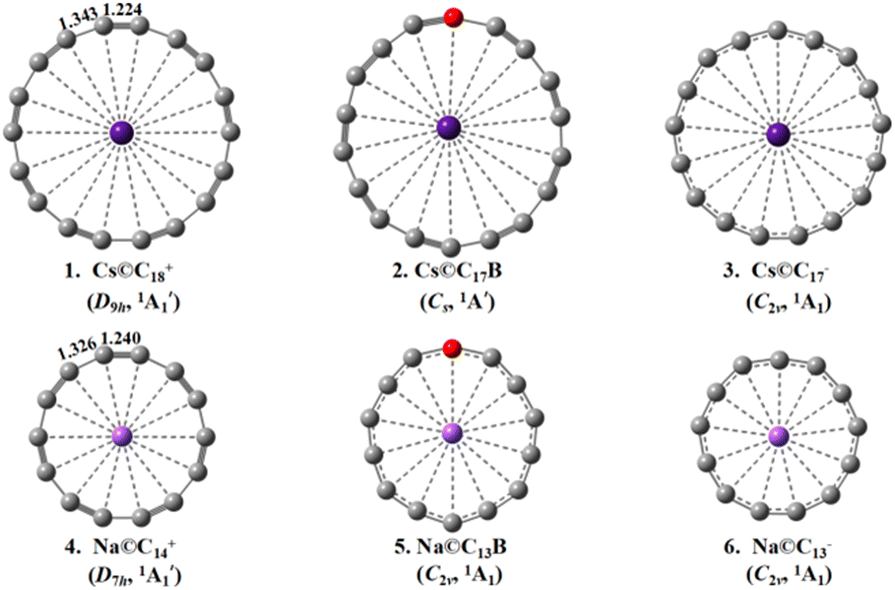 | ||
| Fig. 1 Optimized structures of D9h Cs©C18+ (1), Cs Cs©C17B (2), C2v Cs©C17− (3), D7h Na©C14+ (4), C2v Na©C13B (5), and C2v Na©C13− (6) at M06-2X level. | ||
Detailed bonding analyses on D9h Cs©C18+ (1), D7h Na©C14+ (4), and Cs Cs©C17B (2) were implemented using the adaptive natural density partitioning (AdNDP 2.0) approach40,41 at the M06-2X/6-31G level, with the isosurface maps of the orbitals visualized using the Visual Molecular Dynamics (VMD) software.42 The iso-chemical shielding surfaces (ICSSs)43,44 and isosurfaces of localized orbital locators (LOL)45 were obtained with Multiwfn 3.8 code.46 The anisotropy of the current-induced density (ACID) analyses were realized by the ACID code,47 with the maps finally generated by POV-Ray render.48
3 Results and discussions
3.1 Structures and stabilities
The optimized GM structures of D9h Cs©C18+ (1), Cs Cs©C17B (2), C2v Cs©C17− (3), D7h Na©C14+ (4), C2v Na©C13B (5), and C2v Na©C13− (6) are collectively plotted in Fig. 1, with more alternative isomers summarized in Fig. S3–S8.† Fig. S2† depicts the optimized GM structures of (a) the alkaline-metal-centered cyclo[18]carbon complexes M©C18+ with M = Li, Na, K, Rb, Cs, and Fr and (b) alkaline-metal-centered cyclo[14]carbon complexes M©C14+ with M = Li, Na, and K at M06-2X. It is noticed that the alkaline metal atoms in the GMs are all located inside the cyclo[n]carbon rings, with the alkaline metal atoms severely off-centered in C2v Li©C18+, C2v Na©C18+, C2v K©C18+, and C2v Li©C14+ and slightly off-centered in Cs Rb©C18+ and Cs Fr©C18+. The K atom in C7v K©C14+ lies about 1.14 Å above the ligand plane along the C7 molecular axis due to its large atomic radius (rK = 2.32 Å) which appears to be too big to be hosted inside the C14 ring.Encouragingly, the Cs center with the NBO net atomic charge of qCs = +0.99 |e| proves to have the right atomic radius of rCs = 2.65 Å to be coordinated exactly at the center of the D9h C18 ligand in D9h Cs©C18+ (1) to achieve the highest coordination number of CN = 18 reported to date. As the well-defined GM of the complex (Fig. S3†), Cs©C18+ (1) exhibits the alternating bond lengths of rC–C = 1.343 Å and rC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C = 1.224 Å at M06-2X which are well inherited from its parent ligand D9h C18 ligand with rC–C = 1.343 Å and rCC = 1.223 Å at the same theoretical level (Fig. S1 and Table S1†).
C = 1.224 Å at M06-2X which are well inherited from its parent ligand D9h C18 ligand with rC–C = 1.343 Å and rCC = 1.223 Å at the same theoretical level (Fig. S1 and Table S1†).
The large calculated HOMO–LUMO gap of ΔEgap = 5.38 eV at M06-2X well supports its high chemical stability. The second isomer C2v Cs©C18+ with a Cs+ located outside the C18 ring and the seventh isomer C2v Cs©C18+ with a Cs+ inserted into the C18 ring appear to be 0.38 eV and 4.79 eV less stable than D9h GM at M06-2X, respectively (Fig. S3†). The slightly off-centered planar Cs Rb©C18+ and Cs Fr©C18+ also possess the coordination numbers of CN = 18 (Fig. S2†). Both the planar neutral Cs Cs©C17B (2) which is isoelectronic with Cs©C18+ (1) with obviously bond-length alternations and C2v Cs©C17− (3) with roughly the same averaged bond lengths are the well-defined GMs of the systems with CN = 18 and 17, respectively (Fig. S4 and S5†). However, the severely off-centered C2v Li©C18+, C2v Na©C18+, and C2v K©C18+ with obvious smaller alkaline metal centers Li, Na, and K appear to have much smaller coordination numbers with CN = 4 ∼ 6 (Fig. S2†).
Similarly, the Na center with qNa = +0.95 |e| appears to have the right atomic radius (rNa = 1.86 Å) to be hosted exactly at the center of the D7h C14 ligand to form the perfect planar polyynic D7h Na©C14+ (4) (Fig. S6†) with CN = 14. The second lowest-lying isomer Cs Na©C14+ with the Na+ center located outside the C14 ring lies only 0.23 eV higher than Na©C14+ (4) (Fig. S6†). The two close-lying lowest-lying isomers of Cs©C18+ and Na©C14+ discussed above (Fig. S3 and S6†) may transform between each other with low energy barriers under certain conditions. Na©C14+ (4) as the GM of the system has the alternating bond lengths of rC–C = 1.326 Å and rCC = 1.240 Å at M06-2X well comparable with the corresponding values of rC–C = 1.324 Å and rCC = 1.237 Å calculated for D7h C14 at the same theoretical level (Fig. S1 and Table S1†), while Li with the atomic radius of rLi = 1.52 Å proves to be too small and K with rK = 2.32 Å appears to be too big to be hosted at the ring center of the C14 ligand, they form severely off-centered and off-planed structures, respectively (Fig. S2†). With the HOMO–LUMO gap of ΔEgap = 5.87 eV, Na©C14+ (4) is expected to have a high chemical stability. The slightly off-centered planar C2v Na©C13B (5) with CN = 14 and vibrationally averaged C2v Na©C13− (6) with CN = 13 with roughly the averaged bond lengths also appear to be the well-defined GMs of the systems (Fig. S7 and S8†).
As expected, the high-symmetry Cs©C18+ (1) and Na©C14+ (4) exhibit highly characteristic calculated vibrational spectroscopic features as shown in their simulated IR spectra in Fig. S9,† with the former possessing well characterized IR peaks at 513 and 2202 cm−1 and Raman active vibrations at 1792 and 2293 cm−1, respectively, while the latter having two well separated IR peaks at 545 and 2160 cm−1 and one dominant Raman feature at 1252 cm−1. Such well-defined spectral features can help facilitate future experimental characterizations of these species. Their simulated UV-vis spectra are also shown in Fig. S9† with 100 excited states included to better understand their electronic structures.
3.2 EDA-NOCV bonding scheme analyses
To shed insights into the bonding nature of D9h Cs©C18+ (1) and D7h Na©C14+ (4), detailed EDA-NOCV analyses were carried out at M06-2X/TZ2P. The D3h subgroup was applied to D9h Cs©C18+ (1) because the highest point group supported by ADF program is D8h. It was found that Cs+ and C18 as the most possible reacting fragments give the most favourite orbital interaction energy of ΔEorb = −13.95 kcal mol−1 for Cs©C18+ (1) in different fragmental schemes (Table S2†). They are thus chosen as interacting species to demonstrate the bonding scheme of Cs©C18+ (1) in Fig. 2(a). Similarly, Na+ and C14 as reacting fragments with ΔEorb = −22.41 kcal mol−1 are chosen for Na©C14+ (4) in Fig. 2(b). | ||
| Fig. 2 (a) MO bonding scheme of D3h Cs©C18+ with the fragments of C18 and Cs+ as interacting species and (b) MO bonding scheme of D7h Na©C14+ with C14 and Na+ as interacting species at M06-2X/TZ2P-ZORA level. | ||
The bonding molecular orbitals (MOs) 15a1′, 19e1′ and 20e1′ of D3h Cs©C18+ representing covalent bonding MOs between Cs+ and C18 are connected with the corresponding fragmental orbitals by bold dashed lines in Fig. 2(a), with the orbital compositions tabulated in Table S3.† The non-degenerate 15a1′ mainly originates from the occupied 8a1′ of C18 with in-plane π characteristics (abbreviated as πin) and vacant 6s of Cs+ via (π-6s)σ coordination interaction, the doubly degenerate 19e1′ is composed of the occupied in-plane 13e1′ (πin) of C18 with one nodal plane and vacant 7px and 7py of Cs+ via (π-7p)σ coordination, while the doubly degenerate 20e1′ is composed of the occupied 14e1′ of C18 with πin characteristics with two nodal planes and vacant 5dxy and 5dx2–y2 of Cs+ via (π-5d)σ coordination. As detailed in Table 1, EDA analyses demonstrate that the overall interaction energy of ΔEint = −15.22 kcal mol−1 between the Cs+ and C18 in Cs©C18+ consists of the Pauli repulsion ΔEPauli = 1.89 kcal mol−1, coulombic attraction ΔEelstat = −3.16 kcal mol−1, and orbital interaction ΔEorb = −13.95 kcal mol−1, with covalent orbital interaction making a dominating contribution of 81.5% to the overall attraction interaction (−17.11 kcal mol−1), while electrostatic attraction contributing only 18.5%. The decompositions of the orbital interactions ΔEorb into pairwise contributions between occupied and vacant MOs of the fragments provide quantitative insight into the charge flow. The strongest orbital interaction ΔEorb(1) (20e1′, 27.5%) arises mainly from [C18 (πin)] → [Cs+ (5d)] where C18 serves as a πin-donor to coordinate the 5dxy and 5dx2–y2 orbitals of the Cs+ as σ-acceptors. The orbital interaction ΔEorb(2) (19e1′, 16.8%) originates from [C18 (πin)] → [Cs+ (7p)] where the 7px and 7py orbitals of the Cs+ serve as σ-acceptors. The orbital interaction ΔEorb(3) (15a1′, 14.1%) originates from [C18 (πin)] → [Cs+ (6s)] where the 6s orbital of the Cs+ is a σ-acceptor. Fig. S10† shows the corresponding deformation densities Δρ associated with the pairwise interactions ΔEorb(1), ΔEorb(2) and ΔEorb(3) in Cs©C18+, further indicating that C18 serves as a πin-donor while Cs+ is a σ-acceptor in the complex.
| Energy terms | Interaction | Cs+ + C18 | Interaction | Na+ + C14 |
|---|---|---|---|---|
| a The value in parentheses gives the percentage contribution to the total attractive interactions (ΔEelstat + ΔEorb).b The value in parentheses gives the percentage contribution to the total orbital interaction (ΔEorb). | ||||
| ΔEint | −15.22 | −24.44 | ||
| ΔEelstata | −3.16 (18.5%) | −1.42 (6.0%) | ||
| ΔEPauli | 1.89 | 3.45 | ||
| ΔEorba | −13.95 (81.5%) | −22.41 (94.0%) | ||
| ΔEorb(1)b | C18 (πin) donation → [Cs+ (5d)] | −3.84 (27.5%) | C14 (πin) donation → [Na+ (3p)] | −5.58 (24.9%) |
| ΔEorb(2)b | C18 (πin) donation → [Cs+ (7p)] | −2.34 (16.8%) | C14 (πin) donation → [Na+ (3s)] | −4.30 (19.2%) |
| ΔEorb(3)b | C18 (πin) donation → [Cs+ (6s)] | −1.96 (14.1%) | C14 (πin) donation → [Na+ (3d)] | −4.06 (18.1%) |
| ΔEorb(rest)b | −5.81 (41.6%) | −8.47 (37.8%) | ||
Detailed EDA-NOCV calculations for D7h Na©C14+ (4) gives a similar trend as shown in Fig. 2(b) and Table 1. The bonding MOs 6a1′, 6e1′ and 5e2′ representing covalent bonding interactions between the Na+ and C14 fragmental orbitals are connected by bold dashed lines, with the orbital compositions listed in Table S4.† The 6a1′ mainly originates from the occupied 4a1′ of C14 with πin characteristics and vacant 3s of Na+ via (π-3s)σ coordination interaction, the doubly degenerate 6e1′ is composed of occupied 5e1′ of C14 with πin characteristics and vacant 3px and 3py of Na+ via (π-3p)σ coordination, while the 5e2′ is composed of C14 with πin characteristics and vacant 3dxy and 3dx2–y2 of Na+ via (π-3d)σ coordination.
EDA analyses (Table 1) indicate that overall attraction interaction is overwhelmingly dominated by covalent orbital contribution (94.0%), while electrostatic attraction makes only a marginal contribution (6.0%). The decompositions of ΔEorb into pairwise contributions between occupied and vacant MOs of the fragments reveals that the strongest orbital interaction ΔEorb(1) (24.9%) originates mainly from [C14 (πin)] → [Na+ (3p)], the orbital interaction ΔEorb(2) (19.2%) arises mainly from [C14 (πin)] → [Na+ (3s)], while the orbital interaction ΔEorb(3) (18.1%) originates from [C14 (πin)] → [Na+ (3d)]. The corresponding deformation densities Δρ associated with the pairwise interactions ΔEorb(1), ΔEorb(2) and ΔEorb(3) in Na©C14+ in Fig. S11† clearly indicate that C14 serves as a πin-donor while Na+ is a σ-acceptor.
The EDA-NOCV results detailed above quantitatively indicate that the cyclo[4N + 2]carbon ligands (N = 4, 3) serve as good πin-donors to stabilize alkaline metal centers in both Cs©C18+ (1) and Na©C14+ (4) by donating their πin valence electrons partially to the vacant s, p, and d orbitals of Cs+ and Na+ through effective in-plane (π-s)σ, (π-p)σ, and (π-d)σ coordination interactions.
Localized orbital locator (LOL) is an effective space function in revealing the distributions of delocalized electrons on conjugated rings in molecules. We calculated in-plane LOL-σ, in-plane LOL-πin, and out-of-plane LOL-πout separately based on the corresponding in-plane σ MOs, in-plane π MOs, and out-of-plane π MOs of the systems, respectively. To better reflect spatial distributions of LOL-σ, LOL-πin, and LOL-πout in Cs©C18+ (1) and Na©C14+ (4), the color-filled maps of LOL-σ on the ring plane, LOL-πin on the ring plane, and LOL-πout 1 Å above the ring plane are plotted in Fig. 3(a) and (b) comparatively. By comparing the area colors on the maps, it can be clearly seen that both LOL-πin and LOL-πout exhibit heavy density distributions over the short CC bonds and light density distributions over the long C–C bonds, well supporting the alternating of triple and single bonds in different bond lengths in both polyynic Cs©C18+ (1) and Na©C14+ (4).
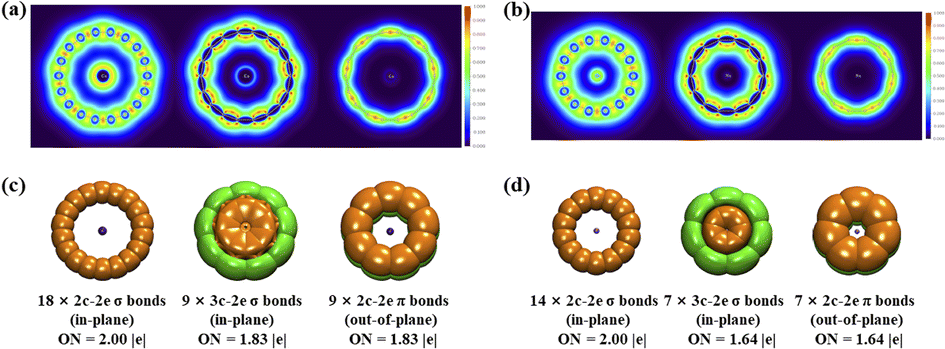 | ||
| Fig. 3 Color-filled maps of the localized orbital locator isosurfaces of (a) D9h Cs©C18+ (1) and (b) D7h Na©C14+ (4) and AdNDP bonding patterns of (c) D9h Cs©C18+ (1) and (d) D7h Na©C14+ (4) with the occupation numbers (ON) indicated. | ||
3.3 AdNDP bonding pattern analyses
Detailed AdNDP analyses in Fig. 3(c) and (d) unveil both the localized and delocalized bonds in D9h Cs©C18+ (1) and D7h Na©C14+ (4) more vividly. As expected, out of the 72 valence electrons in Cs©C18+ (1), 36 electrons form 18 equivalent 2c–2e C–C peripheral in-plane σ bonds with the occupation numbers of ON = 2.00 |e|. The remaining 36 valence electrons are distributed in two types of chemical bonds, including 9 equivalent in-plane 3c–2e σ bonds on nine CsC2 triangles with ON = 1.83 |e| and 9 equivalent out-of-plane 2c–2e C–C π bonds with ON = 1.83 |e|, respectively. Such a bonding pattern follows the 4N + 2 aromatic rule for σ aromaticity with Nσ = 4 and π aromaticity with Nπ = 4, respectively, making the planar complex σ + π dually aromatic in nature and rendering extra stability to the system, similar to the situation in the previously reported D9h C18.11Similarly, as shown in Fig. 3(d), D7h Na©C14+ (4) possesses 7 equivalent 2c–2e C–C periphery in-plane σ bonds, 7 equivalent 3c–2e in-plane σ bonds on seven NaC2 triangles, and 7 equivalent 2c–2e out-of-plane C–C π electrons, again following the 4N + 2 aromatic rule with Nσ = Nπ = 3 for σ + π dual aromaticity. Similar bonding patterns exist in Cs Cs©C17B (2) (Fig. S12†). The dual aromaticities of both Cs©C18+ (1) and Na©C14+ (4) are also well supported by numbers of their delocalized in-plane σ MOs and delocalized out-of-plane π MOs shown in Fig. S13.†
The simulated ICSS isosurfaces of D9h Cs©C18+ (1) and D7h Na©C14+ (4) based on the ZZ components of the calculated nuclear-independent chemical shifts (NICS-ZZ) are presented as Fig. 4(a), in comparison with that of the previously reported σ + π dually aromatic D9h C18 and D7h C14. It can be clearly seen that, similar to D9h C18 and D7h C14, both D9h Cs©C18+ (1) and D7h Na©C14+ (4) are aromatic in nature, with the spaces inside the cyclo[n]carbon rings and within ∼1.0 Å above the ring planes belonging to chemical shielding areas with negative NICS-ZZ values (highlighted in yellow) and the blet-like regions around the cyclo[n]carbon rings in horizontal direction belonging to chemical deshielding areas with positive NICS-ZZ values (highlighted in green).
 | ||
| Fig. 4 (a) Calculated iso-chemical shielding surfaces (ICSSs) of D9h Cs©C18+ (1), D9h C18, D7h Na©C14+ (4), and D7h C14. Yellow and green regions stand for chemical shielding and deshielding areas, respectively. (b) Calculated out-of-plane-π and in-plane-σ ring current maps of D9h Cs©C18+ (1) and D7h Na©C14+ (4), compared with the corresponding ring current maps of D9h C18 and D7h C14, respectively. The external magnetic field is perpendicular to the ring plane. The red arrows indicate the directions of the ring currents on the ACID iso-surfaces. | ||
The widely used ACID method can be employed to display graphically the ring currents induced by an external magnetic field in vertical directions perpendicular to the cyclo[n]carbon ring. Fig. 4(b) presents the calculated out-of-plane π and in-plane σ ring currents maps for both D9h Cs©C18+ (1) and D7h Na©C14+ (4), in comparison with the corresponding ring currents obtained for D9h C18 and D7h C14 at the same theoretical level, respectively. As clearly indicated in Fig. 4(b), these alkaline-metal-centered polyynic complex monocations do possess intrinsic σ aromaticity and π aromaticity simultaneously, similar to their neutral parent ligands D9h C18 and D7h C14 in ring current distributions.
4 Conclusions
In summary, based on extensive first-principles theory calculations, we have predicted in this work a series of alkaline-metal-centered perfect planar complexes Cs©C18+ (1), Cs©C17B (2), Cs©C17− (3), Na©C14+ (4), Na©C13B (5), and Na©C13− (6) which turn out to be GMs of the systems with the record coordination numbers of CN = 18 ∼ 13 in planar species. These hyper-coordinate planar complexes possess effective in-plane (π-s)σ, (π-p)σ, and (π-d)σ coordination interactions which dominate the attractive interaction between the alkaline metal center as σ-acceptor and its cyclo[n]carbon ligand as in-plane π-donor, evidencing the transition metal behaviors of the alkaline metal centers in them. Similar to the situation in the recently observed alkaline-earth metal carbonyl species,21 the proposed perfect planar alkaline-metal-centered polyynic D(2N+1)h cyclo[4N + 2]carbon complexes with relatively low coordination energies may be produced in gas phases by laser ablation of alkaline-metal-carbon mixed binary targets and characterized by spectroscopic measurements at low temperatures to further push the boundary of coordination chemistry.Author contributions
S. D. Li, Z. H. Wei, and Q. Chen conceived the project and finalized the manuscript. M. Z. and R. N. Y. did the calculations and prepared the first draft. Y. B. Wu helped analyze the data. All authors approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (21973057, 21720102006 and 22003034) and Natural Science Foundation of Shanxi Province of China (20210302124002).Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS
.
- S. Iijima and T. Ichihashi, Nature, 1993, 363, 603–605 CrossRef CAS
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- K. Kaiser, L. M. Scriven, F. Schulz, P. Gawel, L. Gross and H. L. Anderson, Science, 2019, 365, 1299–1301 CrossRef CAS PubMed
- G. von Helden, N. G. Gotts and M. T. Bowers, Nature, 1993, 363, 60–63 CrossRef CAS
- Y. Rubin, M. Kahr, C. B. Knobler, F. Diederich and C. L. Wilkins, J. Am. Chem. Soc., 1991, 113, 495–500 CrossRef CAS
- A. E. Boguslavskiy, H. Ding and J. P. Maier, J. Chem. Phys., 2005, 123, 034305 CrossRef CAS PubMed
- A. E. Boguslavskiy and J. P. Maier, Phys. Chem. Chem. Phys., 2007, 9, 127–130 RSC
- T. Torelli and L. Mitas, Phys. Rev. Lett., 2000, 85, 1702–1705 CrossRef CAS PubMed
- S. Arulmozhiraja and T. Ohno, J. Chem. Phys., 2008, 128, 114301 CrossRef PubMed
- Z. y. Liu, X. Wang, T. Lu, A. h. Yuan and X. f. Yan, Carbon, 2022, 187, 78–85 CrossRef CAS
- Y. h. Jiang, Y. b. Wu, J. j. Deng and Z. y Wang, Phys. Chem. Chem. Phys., 2021, 23, 8817–8824 RSC
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill Book Co., 15th edn, 1999 Search PubMed
- X. Q. Lu, H. G. Lu and S. D. Li, RSC Adv., 2021, 11, 27193–27198 RSC
- T. Heine and G. Merino, Angew. Chem., Int. Ed., 2012, 51, 4275–4276 CrossRef CAS PubMed
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2012, 51, 2101–2105 CrossRef CAS PubMed
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2011, 50, 9334–9337 CrossRef CAS PubMed
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Chem. Phys., 2013, 138, 134315 CrossRef PubMed
- W. L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 165–168 CrossRef CAS PubMed
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Acc. Chem. Res., 2013, 46, 350–358 CrossRef CAS PubMed
- X. Wu, L. L. Zhao, J. Y. Jin, S. D. Pan, W. Li, X. Y. Jin, G. J. Wang, M. F. Zhou and G. Frenking, Science, 2018, 361, 912–916 CrossRef CAS PubMed
- H.-R. Li, X.-Q. Lu, Y.-Y. Ma, Y.-W. Mu, H.-G. Lu, Y.-B. Wu and S.-D. Li, J. Clust. Sci., 2019, 30, 621–626 CrossRef CAS
- J. A. Dean, Lange's handbook of Chemistry, McGraw-Hill Book Co, 15th edn, 1999 Search PubMed
- X. Chen, Y.-F. Zhao, Y.-Y. Zhang and J. Li, J. Comput. Chem., 2019, 40, 1105–1112 CAS
- D. J. Wales and H. A. Scheraga, Science, 1999, 285, 1368–1372 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC
- D. Feller, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS
- L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef PubMed
- M. J. Frisch, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS
- M. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576–6580 CrossRef CAS
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597 CrossRef CAS
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC
- N. V. Tkachenko and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2019, 21, 9590–9596 RSC
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans., 2001, 2, 1893–1898 Search PubMed
- E. Kleinpeter, S. Klod and A. Koch, J. Mol. Struct., 2007, 811, 45–60 CrossRef CAS
- H. L. Schmider and A. D. Becke, J. Mol. Struct. (THEOCHEM), 2000, 527, 51–61 CrossRef CAS
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed
- Povray, Persistence of vision raytracer, POV-Ray 3.7, https://www.povray.org/ Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03930g |
| This journal is © The Royal Society of Chemistry 2023 |