
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of sulfamoyl benzamide derivatives as selective inhibitors for h-NTPDases†
Zahid Hussain Zaigham‡
a,
Saif Ullah‡b,
Julie Pelletierc,
Jean Sévigny cd,
Jamshed Iqbal*b and
Abbas Hassan*a
cd,
Jamshed Iqbal*b and
Abbas Hassan*a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan. E-mail: ahassan@qau.edu.pk
bCentre for Advanced Drug Research, COMSATS University Islamabad, Abbottabad Campus, Abbottabad, Pakistan. E-mail: drjamshed@cuiatd.edu.pk
cCentre de recherche du CHU de Québec-Université Laval, Québec City, QC, Canada
dDépartement de Microbiologie-Infectiologie et d'Immunologie, Faculté de Médecine, Université Laval, Québec City, QC, Canada
First published on 11th July 2023
Abstract
The aim of this research work is the synthesis of sulfamoyl-benzamides as a selective inhibitor for h-NTPDases. Sulfonamides are synthesized in aqueous medium from chlorosulfonylbenzoic acid while carboxamides are synthesized using carbodiimide coupling decorated with different biologically relevant substituents such as n-butyl, cyclopropyl, benzylamine, morpholine, and substituted anilines. In addition, sulfonamide-carboxamide derivatives were synthesized having the same substituents on either side. These compounds were screened against h-NTPDase activity, a main family of ectonucleotidases. Among the eight discovered isoforms of the h-NTPDases, four isoforms, h-NTPDase1, -2, -3, and -8, are involved in various physiological and pathological functions, for instance thrombosis, diabetes, inflammation, and cancer. The compound N-(4-bromophenyl)-4-chloro-3-(morpholine-4-carbonyl)benzenesulfonamide (3i) was found to be the most potent inhibitor of h-NTPDase1 with an IC50 value of 2.88 ± 0.13 μM. Similarly, the compounds N-(4-methoxyphenyl)-3-(morpholinosulfonyl)benzamide (3f), 5-(N-benzylsulfamoyl)-2-chloro-N-(4-methoxyphenyl)benzamide (3j) and 2-chloro-N-cyclopropyl-5-(N-cyclopropylsulfamoyl)benzamide (4d) reduced the activity of the h-NTPDases2 with IC50 in sub-micromolar concentrations. Against the h-NTPDase3, 3i was the potent compound with an IC50 concentration of 0.72 ± 0.11 μM. The h-NTPDase8 was selectively blocked by the most potent inhibitor 2-chloro-5-(N-cyclopropylsulfamoyl)benzoic acid (2d) with (IC50 = 0.28 ± 0.07 μM). Moreover, the molecular docking studies of the potent inhibitors showed significant interactions with the amino acids of the respective h-NTPDase homology model proteins.
Introduction
The ectonucleotidases are comprised of four families which include nucleoside triphosphate diphosphohydrolases (h-NTPDases), ecto-nucleotide pyrophosphatases/phosphodiesterases (NPPs), ecto-5′-nucleotidase and alkaline phosphatases (ALPs)1–6 The h-NTPDases belong to a diverse group of eight isozymes ranging from h-NTPDase1 to h-NTPDase8. Each isozyme has a specific capability to catalyse nucleotides. The h-NTPDase1, h-NTPDase3, and h-NTPDase8 have the ability of hydrolysing nucleoside triphosphates as well as nucleoside diphosphates with nearly similar preferences whereas, h-NTPDase2 preferentially catalyses the nucleoside triphosphate as substrate.7 The expression of the h-NTPDase isoforms was identified in almost every human body tissue i.e. h-NTPDase1 is expressed in the subsets of the activated T-cells, monocytes, blood vascular endothelium, and dendritic cells.4 h-NTPDase3 was found in the digestive system (epithelial cells), kidneys, reproductive system, respiratory tract, and islets of Langerhans, whereas, h-NTPDase8 was located in the kidney, liver, and intestine.8–11 Various physiological and pathological functions are associated with the h-NTPDases such as regulation of vascular fluidity, cardiac protection, inflammation of vascular vessels, maintenance of thrombosis, and relaxation of vascular vessels. Some unwanted effects due to the irregularity of the h-NTPDases include thrombosis, inflammation, diabetes, and cancer.8,12,13Sulfonamides are the most effective chemotherapeutic agents used as anti-hypertensive, anti-bacterial, antiprotozoal, anti-cancer, anti-inflammatory, and anti-diuretic agents.14 They are mostly considered bacteriostatic and exhibit exceptional anti-bacterial activity against both Gram-positive and Gram-negative bacteria.15,16 Hence, sulfonamide moieties are used to treat septicaemia, bacillary dysentery, tonsillitis, and urinary tract infections.17 They can also lower the blood glucose level and are employed as anti-diabetic agents.
Sulfamoyl-carboxamide is a continually growing class of derivatives possessing valuable and diverse biological activities. Some recent biological activities are reviewed to demonstrate their significance (Fig. 1). NLRP3 is an emerging target for inflammatory diseases such as neuroinflammation, immune inflammation, and metabolic inflammation. NLRP3 is a nucleotide oligomerization domain (NOD)-like receptor protein 3 which is activated by pathogens and damages the cell signal pathway by making NLRP3 inflammasome, a multimeric protein complex.18 Small molecule sulfonylurea urea such as glyburide (glibenclamide) have been identified as inhibitors for NLRP3 inflammasome.19 Further modification of the glyburide led to sulfamoyl-benzamide derivatives JC-171 with higher selectivity, increase solubility, and importantly, without affecting the glucose level (Fig. 1).20 Mincione et al. have reported sulfamoyl-benzamides as topically acting carbonic anhydrase inhibitors. These were tested against three isozymes of carbonic anhydrases and showed affinity close to clinical drugs both in vitro and in vivo as evidenced by compound I (Fig. 1).21 A series of sulfamoyl-4-oxoquinoline-3-carboxamides were screened for defective ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel. Several of the compounds showed correction for CFTR in sub-micromolar concentration, for instance compound II was more effective in comparison.22 A prominent example is the sulfonylurea-carboxamide based herbicide nicosulfuron, which works at low doses and high selectivity for corn.23 Zhao et al. have reported a series of indole sulfonamide-carboxamide as an excellent inhibitor for wild-type HIV reverse transcriptase through a systematic SAR. The optimization led to compound III which was found to have pyrrolidine sulfonamide at position 3 and bromo at position 5 of the indole structure.24 The 5-chlorosalicylamide scaffold holds biologically interesting properties and the core structure exists in glyburide and JC-171 (vide supra), however, Galal et al. have found their anti-cancer properties. The sulfamoyl-salicylamide derivatives were tested against five types of human cell lines, while compound IV was found to be the best against the breast cancer (MCF-7 and MDA-MB-231) cells (Fig. 1). Furthermore, the tubulin polymerization experiments and flow cytometric assays revealed that compound IV resulted in cell cycle arrest by preventing tubulin polymerization.25
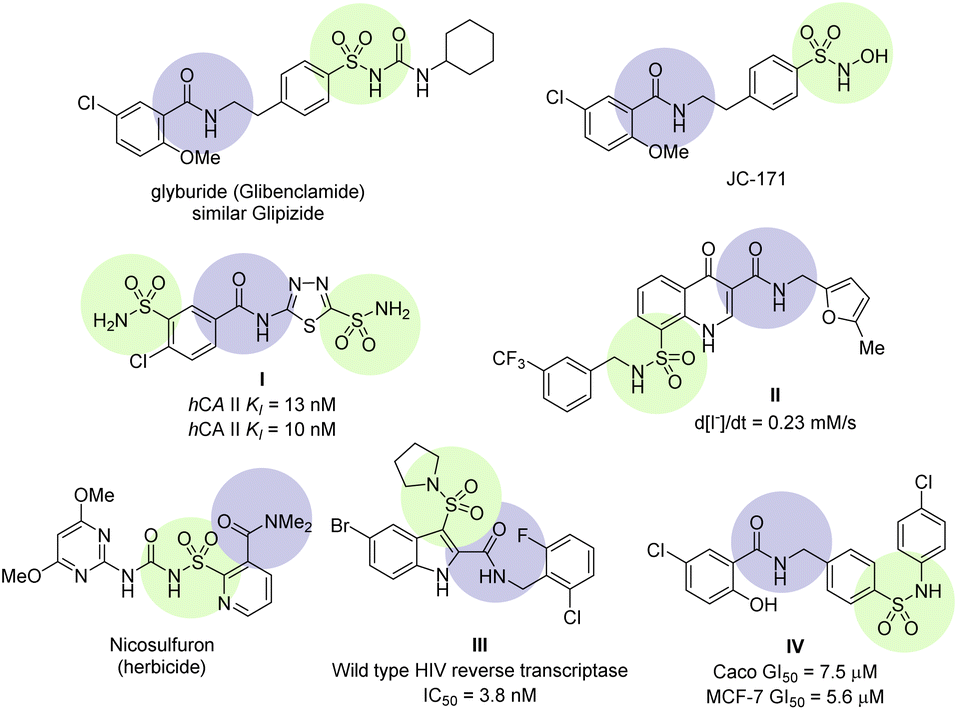 | ||
| Fig. 1 Selected examples of sulfamoyl-carboxamide containing biologically active compounds. | ||
Considering our interest in finding new inhibitors for h-NTPDases,26,27 and owing to the diverse biological profile of sulfamoyl-carboxamide, synthesis of planned derivatives of sulfamoyl-benzamides was carried out to identify their potential against the activity of the h-NTPDase-1, -2, -3, and -8 to develop more effective inhibitors of h-NTPDases for the potential treatment of the pathological conditions associated with the unwanted function of the h-NTPDases.
Results and discussion
Chemistry
The synthesis of the sulfamoyl-benzamide derivatives is outlined in Scheme 1. We synthesize the 2-substituted benzene sulfonamides-carboxamides through a linear approach, starting from chlorosulfonation of benzoic acids, followed by the sulfonamide and finally the carboxamide synthesis. The chlorosulfonation of the electron-deficient benzoic acid has required slight excess of chlorosulfonic acid and elevated temperature.28 The corresponding sulfonyl chloride was treated with different amines including cyclopropylamine, morpholine and p-bromoaniline for the synthesis of sulfamoylbenzoic acids (2a–2e).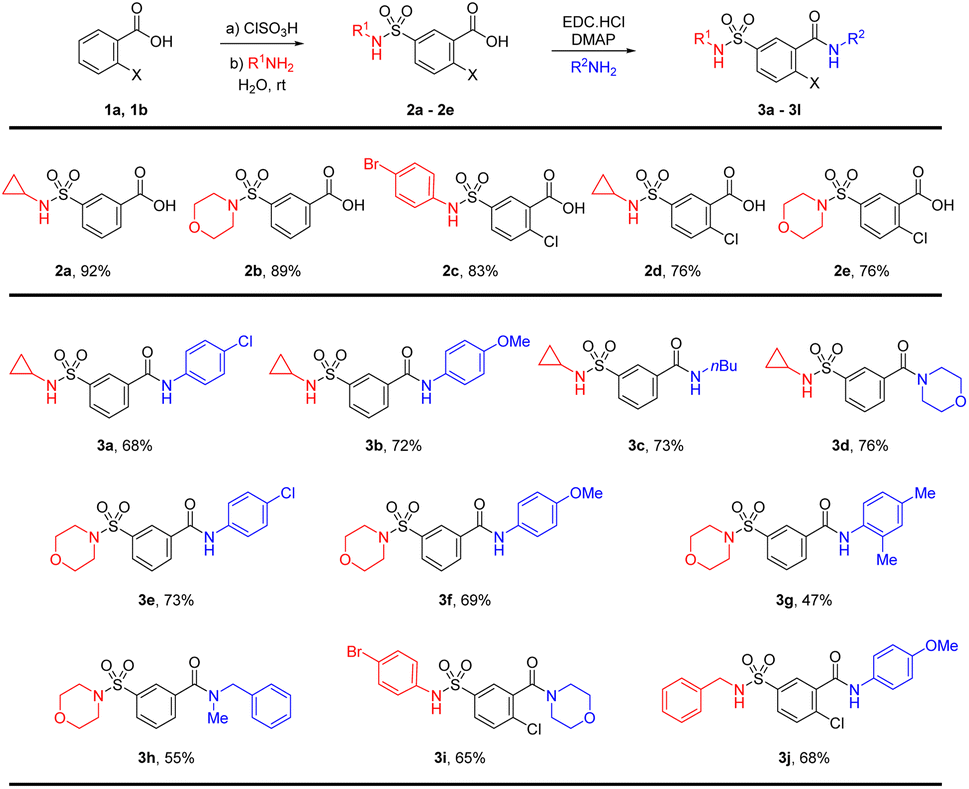 | ||
| Scheme 1 Linear synthesis of sulfamoyl-benzamide derivatives. | ||
The sulfamoyl benzoic acids were subjected to standard carbodiimide coupling conditions using EDC catalytic DMAP in DCM and DMF as co-solvents.29 The amide coupling generally worked well for anilines and primary and secondary aliphatic amines (Scheme 1). The cyclopropyl sulfonamide 2a was treated with p-chloroaniline and p-anisidine for the synthesis of the sulfamoyl-benzamides 3a and 3b in 68% and 72% isolated yields, respectively. There was little effect of the electronic nature of the aniline on the amide formation. Similar yields were found for aliphatic amines such as n-butylamine and morpholine resulting in product 3c and 3d. The morpholinosulfonamide 2b was also reacted with p-chloroaniline and p-anisidine for the synthesis of the benzamides 3e and 3f in 73% and 69% yields, respectively. In addition, 2,4-xylidine was able to furnish product 3g in 47% yield. Further reaction with N-methylbenzylamine resulted in the corresponding amide 3h in 55% yields. The morpholine substituted products 3e–3h have high polarity and if the pH is not properly maintained, the isolation of the product from the aqueous phase may pose a challenge. The p-bromoaniline and benzylamine substituted sulfonamides 2c and 2d were also converted to amides 3i and 3j in good yields (Scheme 1).
To introduce further diversity, a one-pot bis-sulfonamide-carboxamide synthesis resulted in parallel substitutions on either side of the aryl core. To this end, the chlorosulfonation product was telescoped and treated with excess amines for the synthesis of sulfonamide and carboxamide simultaneously for product 4a–4e in good yields. The bis-amidation was carried out with p-chloroaniline, morpholine, pyrrolidine, cyclopropylamine, and benzylamine (Scheme 2).
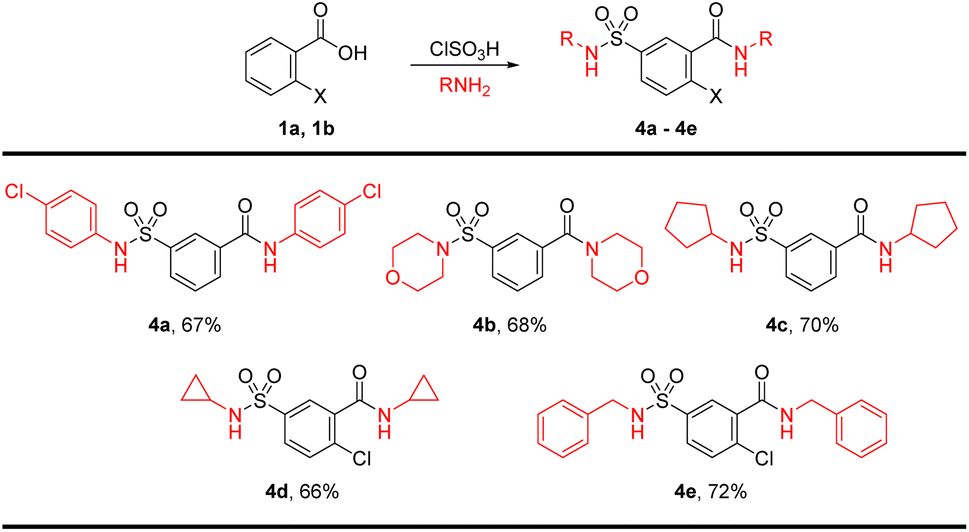 | ||
| Scheme 2 Direct synthesis of sulfamoyl-benzamide derivatives. | ||
Biological study
| Codes | h-NTPDase1 IC50 ± SEM (μM) or % inhibition ± S.Da | h-NTPDase2 IC50 ± SEM (μM) or % inhibition ± S.Da | h-NTPDase3 IC50 ± SEM (μM) or % inhibition ± S.Da | h-NTPDase8 IC50 ± SEM (μM) or % inhibition ± S.Da |
|---|---|---|---|---|
| a The results are presented as an average of the assays conducted in triplicate. IC50 values are determined for the compounds exhibiting over 50% inhibition of enzyme activity; SEM = standard error mean; SD = standard deviation. | ||||
| 2a | 25 ± 3.2% | 10 ± 1.2% | 1.32 ± 0.06 | 24 ± 2.5% |
| 2b | 15 ± 2.3% | 8 ± 0.7% | 35 ± 1.5% | 9 ± 3.3% |
| 2c | 3 ± 1.2% | 22 ± 1.2% | 37 ± 2.1% | 25 ± 2.4% |
| 2d | 3 ± 1.2% | 19 ± 0.9% | 41 ± 1.4% | 0.28 ± 0.07 |
| 2e | 12 ± 3.4% | 15 ± 1.4% | 46 ± 1.3% | 36 ± 2.3% |
| 3a | 5 ± 1.1% | 36 ± 1.7% | 1.33 ± 0.05 | 1.78 ± 0.08 |
| 3b | 34 ± 2.3% | 46.91 ± 2.31 | 46.16 ± 2.41 | 41.48 ± 2.7 |
| 3c | 42 ± 1.4% | 2.88 ± 0.08 | 1.49 ± 0.51 | 46 ± 2.1% |
| 3d | 28 ± 2.2% | 44 ± 2.3% | 41 ± 1.3% | 35 ± 2.4% |
| 3e | 17.49 ± 1.23 | 15 ± 1.2% | 9 ± 3.2% | 24 ± 3.1% |
| 3f | >100 | 0.27 ± 0.08 | 6.71 ± 0.04 | 29 ± 4.5% |
| 3g | 26 ± 3.1% | 31 ± 3.2% | 3.21 ± 0.09 | 39 ± 3.2% |
| 3h | 16 ± 2.3% | 32 ± 1.5% | 2.54 ± 0.05 | 38 ± 1.5% |
| 3i | 2.88 ± 0.13 | 43 ± 1.4% | 0.72 ± 0.11 | 32 ± 3.5% |
| 3j | 37 ± 1.3% | 0.29 ± 0.07 | 3.16 ± 0.12 | 36 ± 1.7% |
| 4a | 9 ± 2.3% | 35 ± 4.6% | 22 ± 2.1% | 9 ± 2.1% |
| 4b | 19 ± 2.2% | 25 ± 3.7% | 37 ± 3.4% | 31.79 ± 2.51 |
| 4c | 17 ± 4.1% | 46.91 ± 2.07 | 9 ± 1.2% | 35 ± 1.2% |
| 4d | 36 ± 2.4% | 0.13 ± 0.01 | 0.20 ± 0.07 | 2.46 ± 0.09 |
| 4e | 42 ± 1.3% | 2.24 ± 0.03 | 12 ± 2.4% | 3 ± 1.2% |
| Suramin | 16.1 ± 1.08 | 24.1 ± 0.15 | 4.30 ± 0.84 | 101.1 ± 2.34 |
The observation of the SAR of the compounds 3e–3h with morpholine ring attached to the sulfonyl group and variation with respect to the amide linkage, the compound 3e was found although least but selective inhibitor of the h-NTPDase1 which blocked the activity presenting the IC50 = 17.49 ± 1.23 μM, whereas the molecule 3f more specifically inhibited the activity of the isoform with IC50 values equals 0.27 ± 0.08 μM. The two compounds 3g and 3h selectively reduced the activity of the h-NTPDase3 to almost equal extent i.e., 3.21 ± 0.09 μM and 2.54 ± 0.05 μM. The effect of the compounds 3g and 3h may be attributed to the presence of the methyl and methylene groups which contributed to the sigma bond hyperconjugation bonding effect to interact with the amino acids of the h-NTPDase3 catalytic site. The presence of the two halogen atoms chlorine and bromine in the structure 3i (IC50 = 2.88 ± 0.13 μM) led the compounds towards the most potent and single active molecule against the isoform h-NTPDase1. Comparison of the SAR of the compounds 3e and 3i against the h-NTPDase1 revealed that the presence of the two halogen atoms on the chemically almost similar structures enhanced the activity of compound 3i to a greater extent. The compound 3j preferentially blocked the activity of the h-NTPDase2 demonstrating the IC50 concentration of 0.29 ± 0.07 μM in accordance with the 3f which reduced the h-NTPDase2 activity to the same level. The h-NTPDase2 inhibitory effect of the 3j might be attributed to the presence of the methoxy group and the additional potency of the 3j to inhibit the h-NTPDase3 could be due to the presence of the methylene group like the structures 3g and 3h.
Molecular docking study
For the purpose to predict the plausible mode of interaction of selected inhibitors of each isoform i.e., h-NTPDase1, h-NTPDase2, h-NTPDase3, and h-NTPDase8, the molecular docking study was carried out by using the homology models. Followed by the docking of the compounds with respective proteins, the most reliable pose which showed a significant binding affinity with the amino acid residues was selected to visualize.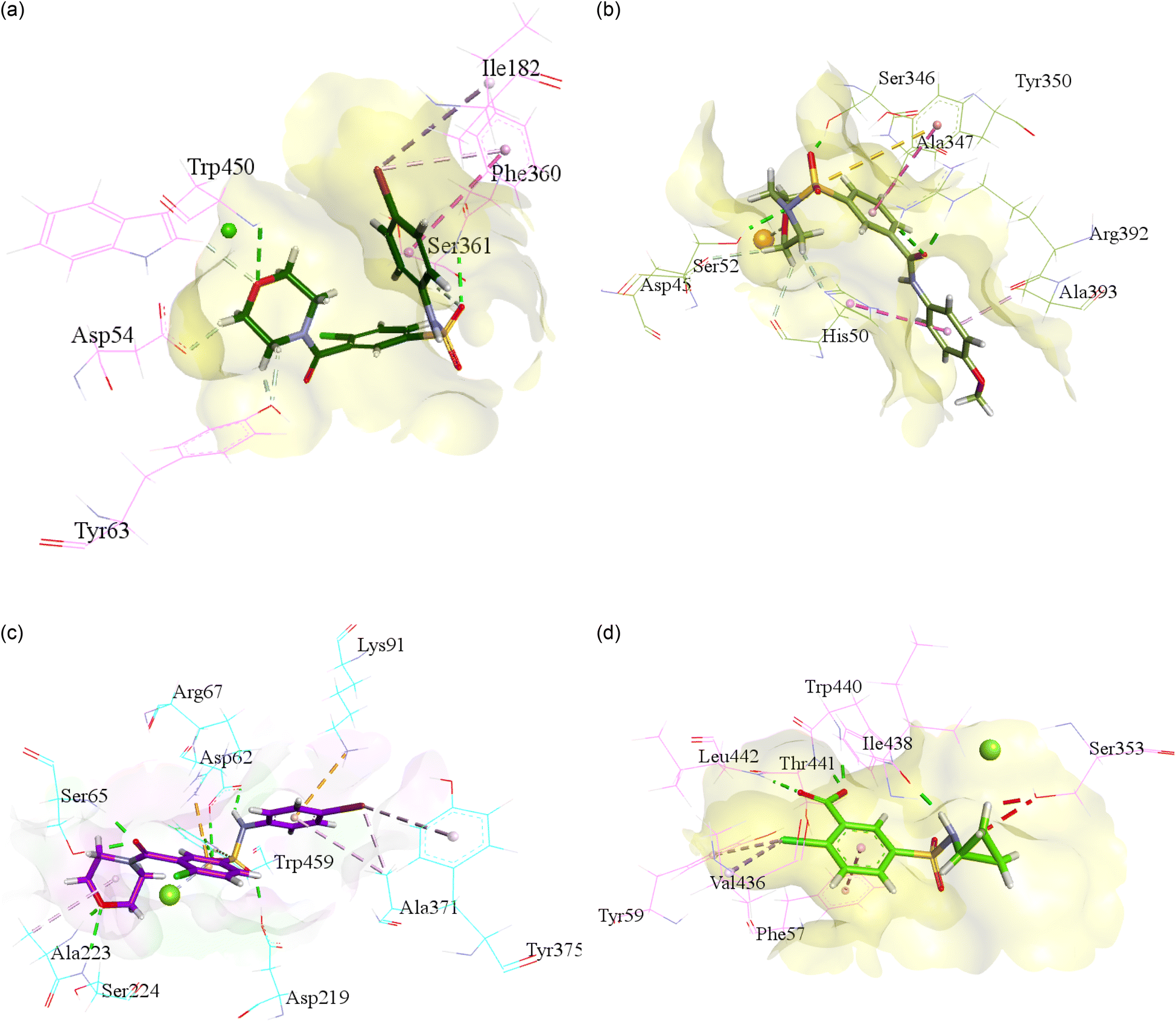 | ||
| Fig. 2 Illustration of molecular docking of the most potent NTPDases inhibitor against modelled h-NTPdase1, h-NTPdase2, h-NTPdase3, and h-NTPdase8. (a) Illustration of the plausible mode of interaction of compounds 3i within the h-NTPDase1 catalytic site. (b) Illustration of the plausible mode of interaction of compounds 3f within the h-NTPDase2 catalytic site. (c) Illustration of the plausible mode of interaction of compounds 3i within the h-NTPDase3 catalytic site. (d) Illustration of the plausible mode of interaction of compounds 3i within the h-NTPDase8 catalytic site. | ||
Conclusions
In conclusion, the sulfamoylbenzoic acids (2a–2e), sulfamoyl-benzamides (3a–3j), and bis-sulfonamide-carboxamide (4a–4e) were synthesized and tested for their effect on the activity of the h-NTPDase1, h-NTPDase2, h-NTPDase3, and h-NTPDase8. As a result, many potent and selective inhibitors of each isozyme were found which blocked the activity from micromolar to sub-micromolar concentrations. The compound 3i (IC50 = 2.88 ± 0.13) was obtained as most potent inhibitor of h-NTPDases1; compounds 3f (IC50 = 0.27 ± 0.08 μM), 3j (IC50 = 0.29 ± 0.07 μM) and 4d (IC50 = 0.13 ± 0.01 μM) preferably reduced the function of the h-NTPDase2. The potent inhibitor of h-NTPDase1 (3i) was also very active against the activity of the h-NTPDase3 with IC50 concentrations equal to 0.72 ± 0.11 μM and the compound 2d selectively blocked the activity of the h-NTPDase8 presenting the IC50 value of 0.28 ± 0.07 μM. Moreover, the molecular docking interactions of the most potent compounds with the amino acid residues of the h-NTPDases proteins justified the in vitro results. In short, the molecules screened as potent and selective inhibitors of the h-NTPDases isozymes may be further evaluated as drug candidates against the diseases associated with the aberrant activity of these enzymes.Experimental
Synthesis of the compounds
The detailed procedure for the synthesis of the sulfamoyl-benzamides and their analytical data is given in the ESI† file.Biological section
Conflicts of interest
We have no conflicts of interest to disclose.Acknowledgements
AH is grateful to the Higher Education Commission (HEC) Pakistan for NRPU research grant 20-3885/R&D/HEC/14. JI acknowledges the financial support provided by the HEC via NRPU project No. 20-15846/NRPU/R&D/HEC/2021.Notes and references
- I. Grković, D. Drakulić, J. Martinović and N. Mitrović, Curr. Neuropharmacol., 2019, 17, 84–98 CrossRef PubMed.
- S.-Y. Lee and C. E. Müller, MedChemComm, 2017, 8, 823–840 RSC.
- S.-Y. Lee, S. Sarkar, S. Bhattarai, V. Namasivayam, S. De Jonghe, H. Stephan, P. Herdewijn, A. El-Tayeb and C. E. Müller, Front. Pharmacol., 2017, 8, 54 Search PubMed.
- D. D. Nabinger, S. Altenhofen and C. D. Bonan, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2020, 98, 109770 CrossRef CAS PubMed.
- M. Tozzi and I. Novak, Front. Pharmacol., 2017, 8, 878 CrossRef PubMed.
- H. Ahmad, S. Ullah, F. Rahman, A. Saeed, J. Pelletier, J. Sévigny, A. Hassan and J. Iqbal, Eur. J. Med. Chem., 2020, 208, 112759 CrossRef CAS PubMed.
- K. Hayat, S. Afzal, A. Saeed, A. Murtaza, S. Ur Rahman, K. M. Khan, A. Saeed, S. Zaib, J. Lecka, J. Sévigny, J. Iqbal and A. Hassan, Bioorg. Chem., 2019, 87, 218–226 CrossRef CAS PubMed.
- A. J. Marcus, M. J. Broekman, J. H. F. Drosopoulos, K. E. Olson, N. Islam, D. J. Pinsky and R. Levi, Semin. Thromb. Hemostasis, 2005, 31, 234–246 CrossRef CAS PubMed.
- G. Kauffenstein, C. R. Fürstenau, P. D'Orléans-Juste and J. Sévigny, Br. J. Pharmacol., 2010, 159, 576–585 CrossRef CAS PubMed.
- S. C. Robson, J. Sévigny and H. Zimmermann, Purinergic Signalling, 2006, 2, 409–430 CrossRef CAS PubMed.
- F. Kukulski, S. A. Lévesque and J. Sévigny, in Advances in Pharmacology, ed. K. A. Jacobson and J. Linden, Academic Press, 2011, vol. 61, pp. 263–299 Search PubMed.
- C. E. Müller, J. Iqbal, Y. Baqi, H. Zimmermann, A. Röllich and H. Stephan, Bioorg. Med. Chem. Lett., 2006, 16, 5943–5947 CrossRef PubMed.
- S. Afzal, M. al-Rashida, A. Hameed, J. Pelletier, J. Sévigny and J. Iqbal, Front. Pharmacol., 2020, 11, 585876 CrossRef CAS PubMed.
- Y. Lavanya, Int. J. Pharm. Sci. Invent., 2017, 6, 1–3 Search PubMed.
- S. Seino, Diabetologia, 2012, 55, 2096–2108 CrossRef CAS PubMed.
- Y. Ö. Genç and Y. Reşit Bekdemir, Ann. Clin. Microbiol. Antimicrob., 2008, 7, 1–6 CrossRef PubMed.
- M. H. Mengelers and Janssen, J. Vet. Pharmacol. Ther., 1997, 20, 276–283 CrossRef CAS PubMed.
- X. Zhang, A. Xu, J. Lv, Q. Zhang, Y. Ran, C. Wei and J. Wu, Eur. J. Med. Chem., 2020, 185, 111822 CrossRef CAS PubMed.
- M. Lamkanfi, J. L. Mueller, A. C. Vitari, S. Misaghi, A. Fedorova, K. Deshayes, W. P. Lee, H. M. Hoffman and V. M. Dixit, J. Cell Biol., 2009, 187, 61–70 CrossRef CAS PubMed.
- C. Guo, J. W. Fulp, Y. Jiang, X. Li, J. E. Chojnacki, J. Wu, X.-Y. Wang and S. Zhang, ACS Chem. Neurosci., 2017, 8, 2194–2201 CrossRef CAS PubMed.
- F. Mincione, M. Starnotti, L. Menabuoni, A. Scozzafava, A. Casini and C. T. Supuran, Bioorg. Med. Chem. Lett., 2001, 11, 1787–1791 CrossRef CAS PubMed.
- Y. F. Suen, L. Robins, B. Yang, A. S. Verkman, M. H. Nantz and M. J. Kurth, Bioorg. Med. Chem. Lett., 2006, 16, 537–540 CrossRef CAS PubMed.
- K. R. Darren, W. M. David and R. S. Jonathan, Weed Technol., 1994, 8, 630–634 CrossRef.
- Z. Zhao, S. E. Wolkenberg, M. Lu, V. Munshi, G. Moyer, M. Feng, A. V. Carella, L. T. Ecto, L. J. Gabryelski, M.-T. Lai, S. G. Prasad, Y. Yan, G. B. McGaughey, M. D. Miller, C. W. Lindsley, G. D. Hartman, J. P. Vacca and T. M. Williams, Bioorg. Med. Chem. Lett., 2008, 18, 554–559 CrossRef CAS PubMed.
- A. M. F. Galal, M. M. Soltan, E. R. Ahmed and A. G. Hanna, MedChemComm, 2018, 9, 1511–1528 RSC.
- G. Zaman, S. Ullah, M. Uzair, S. Batool, H. Ahmad, F. Ullah, J. Pelletier, J. Sévigny, J. Iqbal and A. Hassan, ChemMedChem, 2023, e202300165 CrossRef PubMed.
- Z. Begum, S. Ullah, M. Akram, M. Uzair, F. Ullah, Ahsanullah, J. Pelletier, J. Sévigny, J. Iqbal and A. Hassan, Bioorg. Chem., 2022, 129, 106196 CrossRef CAS PubMed.
- J. P. Bassin, R. J. Cremlyn and F. J. Swinbourne, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 56, 245–275 CrossRef CAS.
- A. Murtaza, S. Afzal, G. Zaman, A. Saeed, J. Pelletier, J. Sévigny, J. Iqbal and A. Hassan, Bioorg. Chem., 2021, 115, 105240 CrossRef CAS PubMed.
- S. A. Lévesque, É. G. Lavoie, J. Lecka, F. Bigonnesse and J. Sévigny, Br. J. Pharmacol., 2007, 152, 141–150 CrossRef PubMed.
- M. Zebisch, M. Krauss, P. Schäfer and N. Sträter, J. Mol. Biol., 2012, 415, 288–306 CrossRef CAS PubMed.
- M. Zebisch and N. Sträter, Proc. Natl. Acad. Sci., 2008, 105, 6882–6887 CrossRef CAS PubMed.
- J. Iqbal and S. J. A. Shah, Sci. Rep., 2018, 8, 2581 CrossRef PubMed.
- MOE (Molecular Operating Environment) Version 2019.0201, Chemical Computing Group (CCG), 2019 Search PubMed.
- LeadIT version 2.3.2, BioSolveIT GmbH, Sankt Augustin, Germany, 2017 Search PubMed.
- Dassault Systemes BIOVIA, Discovery Studio Modeling Environment, Release 2017, Dassault Systemes, San Diego, 2016 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03874b |
| ‡ Both authors have equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |