
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress in photocatalytic CO2 reduction based on single-atom catalysts
Wanyu Hua,
Haiyue Yang *ab and
Chengyu Wang*ab
*ab and
Chengyu Wang*ab
aCollege of Materials Science and Engineering Northeast Forestry University, Harbin 150040, China. E-mail: haiyueyang@nefu.edu.cn; wangcy@nefu.edu.cn
bKey Laboratory of Bio-based Material Science and Technology, Ministry of Education Northeast Forestry University, Harbin 150040, China
First published on 11th July 2023
Abstract
Reduced CO2 emissions, conversion, and reuse are critical steps toward carbon peaking and carbon neutrality. Converting CO2 into high-value carbon-containing compounds or fuels may effectively address the energy shortage and environmental issues, which is consistent with the notion of sustainable development. Photocatalytic CO2 reduction processes have become one of the research focuses, where single-atom catalysts have demonstrated significant benefits owing to their excellent percentage of atom utilization. However, among the crucial challenges confronting contemporary research is the production of efficient, low-cost, and durable photocatalysts. In this paper, we offer a comprehensive overview of the study growth on single-atom catalysts for photocatalytic CO2 reduction reactions, describe several techniques for preparing single-atom catalysts, and discuss the advantages and disadvantages of single-atom catalysts and present the study findings of three single-atom photocatalysts with TiO2, g-C3N4 and MOFs materials as carriers based on the interaction between single atoms and carriers, and finally provide an outlook on the innovation of photocatalytic CO2 reduction reactions.
Introduction
As mankind enters the era of the industrial revolution, raw fossil materials are widely used in production and life as the main energy materials, causing a sharp rise in emissions of CO2.1 CO2 as a typical greenhouse gas, will increase surface temperature by 1.5 to 4.5 °C for every 1-fold increase in concentration.2 As a result of modernization, significant CO2 emissions have brought severe ecological and environmental problems, for instance, global warming, the greenhouse effect, glacier and permafrost melting, sea level rising, etc.,2 which are threatening the survival and living environment of human beings at all times and posing significant obstacles to the future sustainable growth of human civilization. Countries are always paying attention to the global climate problem, and China has shown great concern and determination in this regard. China suggested during the 75th UN General Assembly in September 2020 that “China should strive to reach a peak in carbon dioxide emissions by 2030 and work towards achieving carbon neutrality by 2060”.4On the other hand, reliable energy and chemical sources are crucial to our contemporary lifestyles, which are necessary for mobility, wealth, and daily comfort.3–5 We now need to look for sustainable and environmentally acceptable alternative resources along with the depletion of resources derived from fossil fuels as well as their significant influence on environmental damage.6 There is now widespread agreement that CO2 emissions should be successfully decreased7 via renewable energy conversion techniques and possibly provide high-value carbon-containing compounds or fuels.8 Therefore, scientists put forward the concept of artificial catalytic light energy conversion. Through direct solar energy collecting, artificial converting photocatalytic energy is a practical solution to the problems of environmental protection and energy security9 because of its cleanliness, inexhaustibility, efficiency, and cost-effectiveness.10 To make use of unlimited and freely available solar energy, artificial photocatalytic energy conversion delivers a further deployable and encouraging strategy that can combat the global energy crisis and cope with the extremely volatile climate by converting greenhouse gas CO2 into high-value carbon-containing compounds and finally ending the carbon cycle.11,12 As a result, CO2 reduction reactions have attracted extensive attention from researchers across the country.13 Solar-powered CO2 conversion to fuel is an appealing approach for meeting rising energy needs.14 CO2 reduction reactions may be conducted in this technique by electrons/holes following photo-excitation of semiconductor photocatalysts by sun irradiation, which has major benefits since photocatalytic processes can be performed under moderate circumstances.15,16 The unique feature of photocatalytic CO2 reduction reactions is the wide variety of CO2 reduction products, including HCOOH,17–20 CO,21–23 CH4,24–28 CH3OH,29–32 etc., which is mostly determined by the kind of active sites on the photocatalyst's surface. Therefore, along with efforts to produce superior photocatalysts, the exact building of reaction sites that enable adequate selectivity for the desired products has been widely researched for effective photocatalytic CO2 reduction processes.33 Because the function of a conventional photocatalytic system is heavily dependent on the energy band structure and surface structure of the catalyst, sluggish separation of electron–hole pairs and a restricted number of surface active sites are still far from satisfactory.34 The lack of activity and poor output selection severely limit its practical application. In order to enhance the production of the required components as well as better understand the CO2 reduction reaction process, output selectivity must be tuned.35 Despite significant efforts, heterogeneous photocatalysts have so far suffered from many shortcomings, such as the inadequate photon absorption efficiency, fast charge carrier complexation, ineffective molecular activation36 and delayed charge transfer of carriers, which significantly inhibits the transfer of charges from the catalyst surface to reactant molecules and limits the performance of various systems.37 Therefore, single-atom catalysts have gained attention as prospective photocatalyst candidates due to their distinctive characteristics of very with excellent atomic efficiency and exceptional catalytic performance.11 In addition to boosting the amount of active sites, for photocatalytic reactions, the separated reaction centres in the single-atom photocatalytic systems are possible to broaden the scope of light collecting as well as enhance the effectiveness of charge segregation and transference. Moreover, with enough light traps and precise surface modification, the created single-atom photocatalyst is highly deployable and can adsorb and activate molecules.3 In addition, we can draw more precise connections between structure and performance thanks to the structural simplicity of single-atom photocatalysts, which improves our knowledge of the underlying principles of photocatalysis.
It is vital to outline the most recent research developments in single-atom photocatalytic CO2 reduction so as to encourage the quick growth of this new subject. This may both expose the principle operating mechanism and provide inspiration for future research directions. Although some excellent reviews have summarized the impact of single-atom reaction sites in photocatalysis3,11,38 in detail and emphasized the approaches to single-atom catalysts manufacturing,37,39,40 the underlying concept of single-atom photocatalytic reactions and the application of novel CO2 reduction photocatalysts were not discussed in the reviewed literature, which is crucial to comprehend single-atom photocatalytic CO2 reduction as well as its prospective large-scale use. In the overview paper, the key principles of single-atom photocatalytic CO2 reduction are firstly highlighted, so as to fully understand its mechanism of operation and therefore facilitate more efficient framework and preparation of single-atom photocatalysts. Secondly, the production techniques, advantages and disadvantages of single-atom photocatalysts are discussed. Then, the improvement and achievements of three popular photocatalysts for CO2 reduction processes with TiO2, g-C3N4 and MOFs (metal–organic frameworks) materials as carriers are presented based on the interactions between single atoms and carriers. Finally, we discuss some problems and potential opportunities for single-atom catalyst development in photocatalytic energy conversion.
Achievements and characteristics
In this paper, through the screening and statistics of single-atom photocatalytic CO2 publications gathered in the CNKI, SCIE, and Springer databases, we find that the number of published papers and related citations and downloads increased exponentially from 2015 to 2022, as shown in Fig. 1 and 2, and this topic was still in a highly discussed stage at the beginning of 2023. In the last decade, the research on single-atom photocatalytic CO2 shows the characteristics of the rapid growth of talents, extensive application fields, and continuous emergence of results. As one of the typical photocatalytic oxidation–reduction reactions, extensive research on photocatalytic reactions (e.g. water splitting) has established a technological basis towards photocatalytic CO2 reduction reactions.41 In 2014, Yang et al. were the first to disclose the production of hydrogen via water using a single-atom catalyst (0.2Pt/TiO2) by photocatalysis, which outperformed metal nanoparticles.42 Two years later, Zhang et al. exhibited that single Co atoms anchored in the metal–organic framework (MOF) significantly promoted the segregation of photoinduced carriers in the MOF-525-Co photocatalyst and augmented the directional mobility of photoinduced electrons from the carriers to the metal center.43 In 2017, Wei et al. further found that the intermediate energy gap state generated in the isolated Co1–P4 site anchored g-C3N4 photocatalyst may boost light trapping capacity and offer segregation centres to inhibit charge complexation at the same time prolong carrier life.44 In 2019, Wang et al. simulated the structure of chlorophyll and prepared a series of hollow Zr-porphyrin-MOF(HNTM-Au/Cu/CO-SA) photocoupled electrocatalysts supported by Au, Cu and Co atoms. When combined with light, HNTM-Au-SA and HNTM-Co-SA demonstrated excellent CO Faraday efficiencies of 95.2% and 92.2%, respectively.45 These ground-breaking efforts have significantly aided and sparked the explosive growth of single-atom photocatalytic CO2.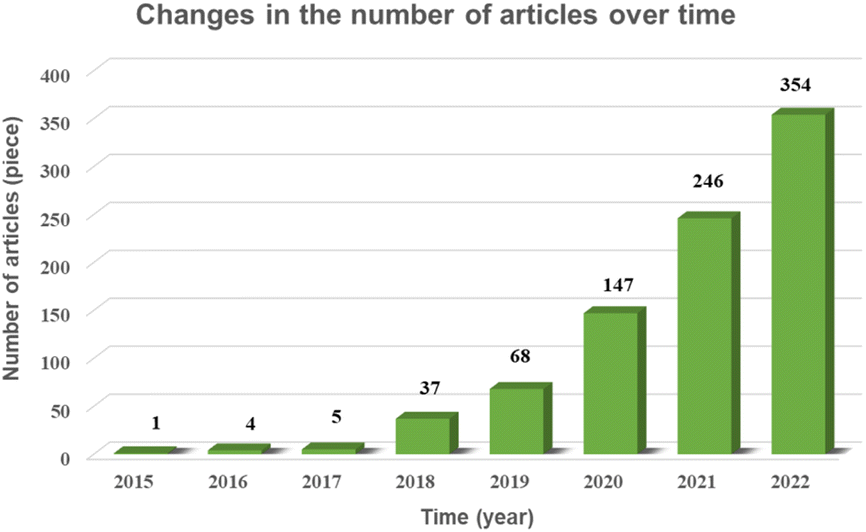 | ||
| Fig. 1 Graph of number of articles over time. | ||
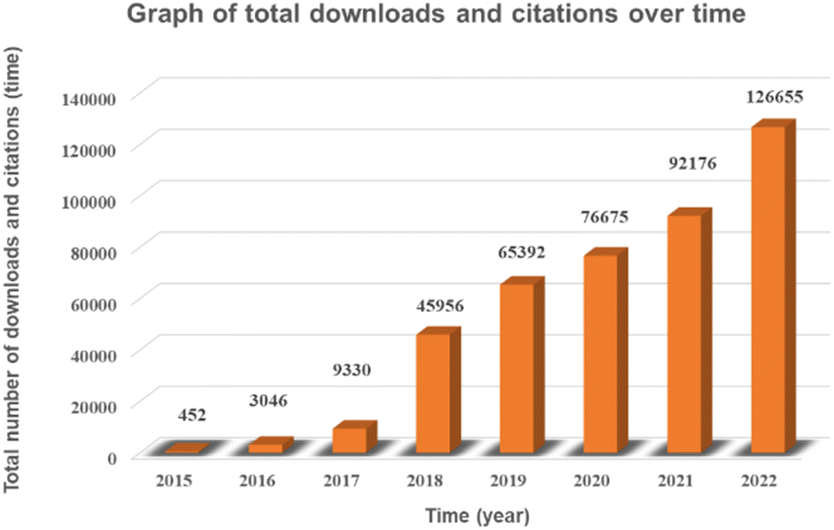 | ||
| Fig. 2 Graph of total downloads and citations over time. | ||
Photocatalysis is a chemical reaction where catalysts absorb photons to generate energizing electrons and holes, that are then used to initiate reduction and oxidation events, respectively.3,46–48 The photocatalytic water splitting process and single-atom photocatalytic CO2 reduction process have a similar reaction mechanism. Six stages are involved in the photocatalytic CO2 reduction process: light absorption, photoexcitation, segregation and transference of photo-generated electron–hole pairs, CO2 adsorption, interfacial CO2 reduction reaction, and product desorption.49
As depicted in Fig. 3, photocatalysts generate photo-generated electron–hole pairs underneath the influence of incident light, and the excitation wavelength of the incident light is controlled by the energy band structure of semiconductors. Once that energy produced by the incident light exceeds the semiconductor band gap, the excited semiconductor's electrons move from the valence to the conduction band, producing photo-generated electron–hole pairs.50 The conduction band and valence band of photocatalysts act as reduction/oxidation centres, which will supply electrons to reduce CO2 and oxidize the water vapor in the reactant mixture, respectively.51 Therefore, the modification of semiconductor photocatalysts is particularly important for photocatalytic reactions. Doping or solid solution structure can be used to enhance the density of electrons and holes in a photocatalyst, resulting in a broad spectrum of light response and an excellent light absorptivity.52 The thermodynamic properties of CO2 molecules are stable, which makes the photocatalytic CO2 reduction process more complicated. It is challenging to attain an acceptable selectivity since most photocatalytic CO2 reduction processes have near reduction potentials and there are competitive hydrogen desorption reactions.53 Furthermore, the bond energy of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is 799 kJ mol−1,54 making the activation of CO2 extremely difficult. The selectivity of products is intimately connected to the diffusion of reactants and intermediaries. Adsorbing CO2 molecules, decreasing the overpotential of CO2 reduction, capturing photo-generated electrons to facilitate charge separation, and enhancing selectivity by modifying the desorption energy of intermediaries are all functions of the active centre in a photocatalytic CO2 process.55 Furthermore, the efficiency of the photocatalytic CO2 reduction process is greatly reliant upon that quantity and complete exploitation of reaction sites.15 Therefore, it is essential to study the mechanism of reaction sites and control the types of reaction sites to trigger photocatalytic CO2 reduction reactions with good target product selectivity.
O is 799 kJ mol−1,54 making the activation of CO2 extremely difficult. The selectivity of products is intimately connected to the diffusion of reactants and intermediaries. Adsorbing CO2 molecules, decreasing the overpotential of CO2 reduction, capturing photo-generated electrons to facilitate charge separation, and enhancing selectivity by modifying the desorption energy of intermediaries are all functions of the active centre in a photocatalytic CO2 process.55 Furthermore, the efficiency of the photocatalytic CO2 reduction process is greatly reliant upon that quantity and complete exploitation of reaction sites.15 Therefore, it is essential to study the mechanism of reaction sites and control the types of reaction sites to trigger photocatalytic CO2 reduction reactions with good target product selectivity.
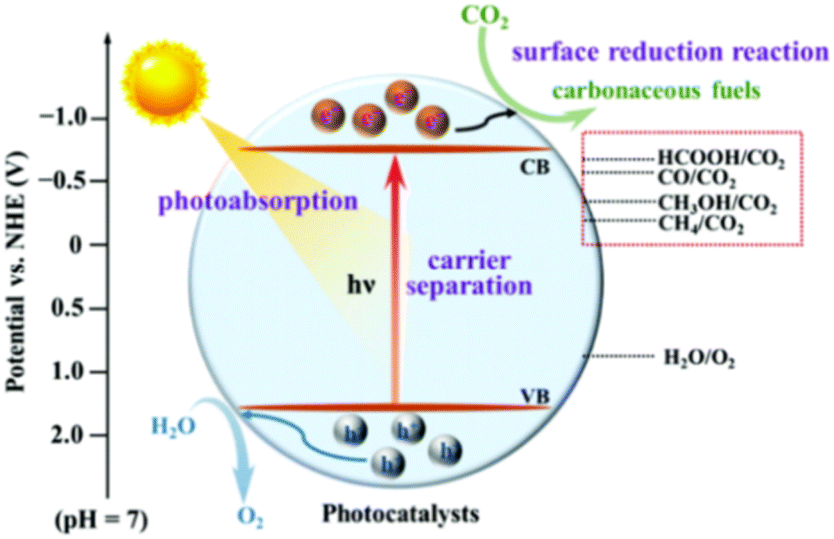 | ||
| Fig. 3 CO2 photocatalytic reduction process.56 Reproduced with permission. Copyright 2020, Royal Society of Chemistry. | ||
Preparation of single-atom catalysts
Because of its substantial advantages in terms of improved light trapping, accelerated charge transport kinetics, and interfacial reactions in photocatalytic systems,57 single-atom catalysts were proven to be extremely effective in many typical photocatalytic processes, particularly CO2 reduction. Scientists have made tremendous progress in the investigation of catalysts in synthetic chemistry in recent years, which has tremendously aided in the growth of single-atom catalysts.58 Due to the high interfacial energy of individual atoms in single-atom catalysts, active sites are easily relocated and aggregated into the clusters and nanoparticles.59 The coexistence of metal nanoparticles and metal monatomic sites in supported catalysts has been widely accepted by scientists,60 which provides extra challenges in high-loading active sites. In order to achieve sufficient distance and good distribution between each substance of the metal precursor, prevent metal site agglomeration after reprocessing, and provide enough anchoring positions for stabilizing a single atom, scientists have carried out a variety of synthetic studies.11 As depicted in Fig. 4A, especially for photocatalytic CO2 reduction processes, photocatalysts with single-atom reaction sites provide considerable advantages over traditional metal or metal oxide co-catalysts in the type of nanoparticles or clusters because of their distinctive structure.15 In this paper, we summarized some particular methods for making single-atom catalysts, mainly wet synthesis, atomic layer deposition, co-precipitation method, high-temperature calcination, chemical etching, freeze-drying, microwave-assisted and ball milling, and focused on wet synthesis method, atomic layer deposition method, co-precipitation method and dipping method.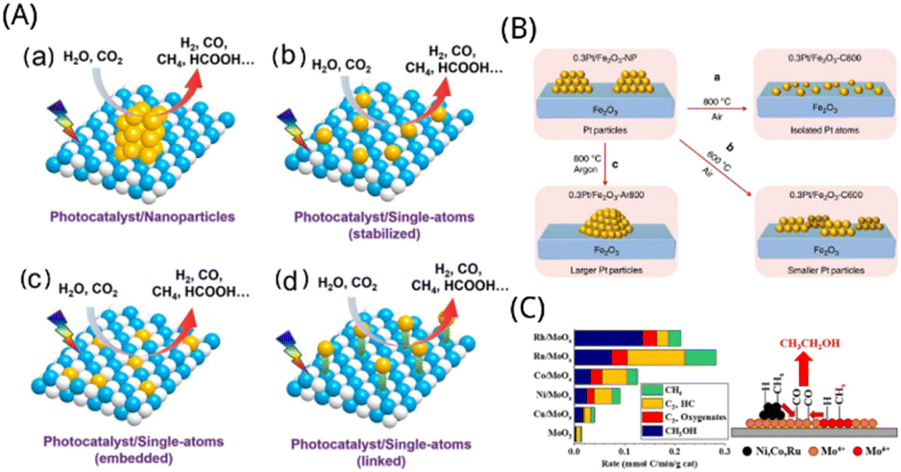 | ||
| Fig. 4 (A) Water splitting and CO2 reduction reactions on a photocatalyst containing (a) reactive sites for nanoparticles, (b)–(d) reactive sites for single atoms.15 Reproduced with permission. Copyright 2021, Wiley-VCH GmbH. (B) Restructuring of Pt nanoparticles caused by heat. (a and b) Calcination in an oxygen or inert environment (c), producing dispersion as single atoms or particle sintering, respectively.69 Reproduced with permission. Copyright 2019, Nature Communications. (C) Product formation rate and reaction mechanism of Mo-containing catalysts.77 Reproduced with permission. Copyright 2019, American Chemical Society. | ||
Wet synthesis method
Since it has the advantage of being simple to operate and requiring no additional equipment, wet synthesis is frequently utilized to produce stable single-atom sites.61 At the same time, wet synthesis offers good dispersibility and deployment.62 In a conventional wet synthesis procedure, metal antecedents in solution (including metal compounds and mononuclear metal complexes) are often supplied to semiconductor substrates by electrostatic adsorption or organic complexation, and then reduced or activated to obtain isolated single-atom reaction sites on the semiconductor.63Biasi et al. modified the catalysts with a catalyst wet pretreatment method, which was used for catalysts prepared after modification with different concentrations of aqueous NaBr solutions, and found that bromides have an active role in the reconstitution of the metal phase. They performed wet pretreatment of catalysts with 1% Pd/C catalyst sample (1PdC) using aqueous sodium bromide solution. Two dilute solutions (1.22 and 12.5 mM) were first prepared. Typically, 1.0 g of the original catalyst 1PdC was suspended in 15 cm3 of water. The modifier was then introduced, an exact aliquot of one of the two starting aqueous solutions (1.22 or 12.5 mM) was added, and the mixture was stirred for 2 h. The final volume of the pretreatment was always adjusted to 30 cm3. The solids were recovered by vacuum filtration and carefully washed with water (5 × 5 cm3) on a Buchner filter. The pretreated mother liquor was concentrated and analysed by ion chromatography to estimate the amount of bromides. Thus, the solid was dried overnight in an oven at 383 K. Approximately 50 mg of all catalysts obtained by the above method were mineralized and analysed for palladium content by ICP-OES.64 Chen et al. used vacancy defects on the layered nitride Ti3−xC2Ty to load Pt single atoms (Pt loading (mass fraction) was only 0.2%), which showed near 100% conversion and selectivity in the catalytic CO2 amination reaction at 140 °C. They dispersed 8 mL of Ti3−xC2Ty MXene suspension in 20 mL of distilled water under magnetic stirring to form clear solution A. Solution B was prepared by adding 10 μL of H2PtCl6·6H2O (0.1 g mL−1) solution to 50 mL of distilled water. Solutions A and B were then were sonicated for 1 h and 10 min. Solution B was slowly added dropwise to solution A under stirring. After the reaction for 8 h, Pt1/Ti3−xC2Ty was precipitated with acetone for half an hour and collected by centrifugation with acetone to obtain Pt1/Ti3−xC2Ty.65 Xu et al. compounded a single cobalt atom catalyst (Co–N–C) by an improved template etching method and applied it to the degradation of pollutants. The experimental test found that Co–N–C has more catalytic oxidation activity than Co3O4, and the peroxymonosulfate (PMS)/Co–N–C oxidation process with mixed reaction pathway has a broad prospect in removing complex water pollutants. First, they added 0.6 mmol of Co(CH3COO)2·4H2O and 1.8 mmol of C12H8N2·H2O to 60 mL of ethanol and sonicated for 10 min. Second, 3 g of MgO was further added and sonicated for another 15 min. The mixture was then sealed and magnetically stirred at 60 °C (water bath) overnight. After complete evaporation of ethanol, the dried solid was ground into a powder and heated in a tube oven. The oven was fluxed with N2 at room temperature for 30 min, after which it was ramped up to 700 °C at a rate of 2 °C min−1 and maintained for 2 h. The product was stirred in 1 M H2SO4 for 4 h to leach out the template MgO and any possible metals or oxides. The acid leaching was repeated 3 times. After washing 5 times with ultrapure water, the solid was dried at 60 °C overnight.66
Co-precipitation method
Co-precipitation is a method of adding a precipitant to a mixed metal salt solution so that two or more cations contained in the solution are precipitated together to produce a precipitation mixture or solid solution precursors, which are filtered, washed, and thermally decomposed to obtain a composite oxide.67 Currently, the main carriers that can be used for the preparation of single-atom catalysts are oxides and carbon nanotubes.68Lang et al. prepared 1.8 wt% Pt/FeOx catalysts, denoted as Pt1/FeOx, by co-precipitation method of an aqueous mixture of chloroplatinic acid (H2PtCl6·6H2O, 37 mgPt mL−1, 2.6 mL, 99.9%, Sigma-Aldrich) and Fe(NO3)3·9H2O (1 mol L−1, 40 mL) with Na2CO3 solution (11 g Na2CO3 in 100 mL H2O) at 50 °C under stirring for 3 h, and ageing static for a further 2 h. The obtained solid was recovered by filtration, washed with deionized water and dried at 60 °C overnight. Then a part of Pt1/FeOx was calcined at 800 °C according to the above method, and expressed as Pt1/FeOx-C800, as depicted in Fig. 4B. The specific activity is significantly higher than that of traditional nanoparticle catalysts because of the resultant large concentration of single atoms. This non-defective stabilizing method may be expanded to non-reducible carriers by easily blended with iron oxide, laying the foundation for development of highly loaded single-atom catalysts for various industrially important catalytic reactions.69 Extensive studies revealed that the single atoms served as the most important active sites. Lin et al. synthesized a series of Ir/FeOx catalysts with different Ir loadings through a co-precipitation method and HAADF-STEM revealed that the individual Ir atoms occupied exactly the positions of the Fe atoms.70 To solve the problem of low loading of single-atom catalysts, Ding et al. used MnO2 with special pore structure as a carrier to increase the loading of noble metals. They prepared Ag/Hollandite-type MnO2 single-atom catalysts using MnSO4 and KMnO4 as raw materials and AgNO3 as catalyst precursor by co-precipitation method. Hollandite-type MnO2 was first prepared by hydrothermal method, and then the prepared [Ag(NH3)2]OH solution and H2O2 solution was added to the MnO2 suspension.71 After drying, a loading of about 28.8 wt% of Ag/Hollandite type MnO2 single-atom catalyst.
Atomic layer deposition method
Because standard techniques of creating catalysts make it difficult to regulate the development and locations of metal particles,72 atomic layer deposition can control the metal particles as nanoparticles, clusters and single atoms by regulating the number of cycles or deposition sites.73 A technique called atomic layer deposition (ALD) may deposit materials as single atomic films on the surface of the substrate layer by layer.74Ye et al. fabricated the single atom of Pt1 loaded up to 4wt% on cerium oxide nanorods by atomic layer deposition technology. Ceria defect sites, metal loadings, and high-temperature calcination were discovered to be efficient techniques of tuning the stability of Pt1 single atoms in the hydrogen atmosphere. It revealed that at the defect and step-edge sites, a single atom of Pt1 on cerium oxide was largely in the +2 valence state, but the single atom at the step site was in the +4 valence state.75 Gorey et al. prepared Pt–Sn bimetallic catalyst by ALD technology combined with temperature programmed method, in which clusters of Pt4, Pt7 and Pt24 of sizes ranging were deposited on SiO2, then hydrogenated, and the seeds of Sn were selectively added by self-limiting reaction with SnCl4. Sn deposition was found to be self-limiting at fairly modest SnCl4 exposures, revealing that the reaction was self-contained, and non-selective Sn deposition efficiency on SiO2 carriers was 40-fold lower than that on hydrogenated Pt cluster sites. The activity test showed that the catalyst with an alloy structure can obtain higher catalytic performance. They used Pt4, Pt7, and Pt24 clusters. Mass-selected Ptn+ were introduced into the UHV system, where they were deposited on the SiO2 substrate by means of a 2 mm diameter exposure mask at a distance of 1 mm from the surface. The deposition energy of the Pt clusters was calibrated by delayed potential analysis of the beam on the sample and set to ∼1 eV per atom. Deposition was monitored by integrating the neutralization currents, and here all samples contained 1.5 × 1014 Pt atoms per cm2 (∼10% of a dense row of Pt monolayers) deposited as Pt4, Pt7, or Pt24.76 Zhang et al. investigated syngas reforming catalysts made from transition metal elements (Ni, Co, Cu, and Ru) loaded on molybdenum oxide which was produced by deposition of atomic layers on silica. Transition metals were added to molybdenum oxide, which significantly improved the catalytic performance. Furthermore, the formation of the CHx group may be accelerated further by ALD placing a sufficient quantity of transition metal on the Mo active site, thereby facilitating the formation of C2+ oxide species, as depicted in Fig. 4C. Bis(tert-butylimino)bis(dimethylamino) molybdenum (Strem Chemicals), bis(ethylcyclopentendien) manganese, titanium tetrachloride, and trimethyl aluminum were used as precursors for MoOx, MnOx, TiOx, and AlOx depositions. Water was used as a coreactant. ALD was done at 623 K with an alternative deposition of MoOx and TiOx (AlOx) with five cycles of deposition of each element for MoTiOx/SiO2 and MoAlOx/SiO2. For the MoMnOx/SiO2, five cycles of MoOx was deposited at 623 K, after which the reactor was cooled to 523 K to deposit five cycles of MnOx by ALD.77
Dipping method
The dipping method as one of the common methods for the preparation of single-atom catalysts is one of the most common methods for the preparation of single-atom catalysts. The method is to immerse the carrier into the catalyst precursor solution, load the metal ions on the carrier surface by electrostatic adsorption on the carrier surface, and prepare the single-atom catalyst with good catalytic performance by rotary evaporation, drying and reduction processes.78 Currently, metal oxides,79 1D nanowires80 and 2D nanosheets81 are often used as carriers for the preparation of single-atom catalysts by the impregnation method.Kim et al. prepared Pt1/ATO single-atom catalysts using H2PtCl6·6H2O as the catalyst precursor and antimony-doped oxidized (ATO) powder as the carrier by the dipping method. They first mixed Pt (1 wt%, 4 w% and 8 wt%) precursor solutions with ATO at different contents, dried and then prepared Pt1/ATO single-atom catalysts by reduction at 100 °C and 400 °C under H2 atmosphere, respectively. HAADF-STEM images confirm the presence of Pt single atoms and that Pt single atoms are located on the surface of Sn Sb or SnO2. Density flooding theory (DFT) calculations show that: the Sb sites in Sn Sb reach their most thermodynamically stable state when they are replaced by Pt. The catalyst showed high catalytic activity, selectivity and durability, and after 1800 cycles, Pt/ATO still maintained high activity in catalytic formic acid oxidation reaction.82 Zeng et al. prepared Pt1@Fe–N–C single-atom catalysts by impregnation with 2-methylimidazole (C4H6N2), ZnO and iron acetate, using H2PtCl6 as the catalyst precursor. The Pt1@Fe–N–C single-atom catalysts with a negative loading of about 2.1 wt% were prepared by adding H2PtCl6 solution to the Fe–N–C solution at 70 °C to fully adsorb Pt4+, and then stirred, filtered and dried in Ar atmosphere. After extensive testing, the catalyst showed good durability.83 Yang et al. prepared CoN4/nitrogen-doped graphene (NG) single-atom catalysts by impregnation method using g-C3N4 as the raw material, polyoxyethylene-polyoxypropylene-polyoxyethylene (F127 complex) as the surface active agent and Co(NO3)2·6H2O as the catalyst precursor. They first ultrasonically dispersed the g-C3N4, Co(NO3)2 and F127 complex precursors to make the F127-protected Co ions penetrate into the layer of g-C3N4, then pyrolyzed the precursors under N2 atmosphere and finally etched with hydrochloric acid at room temperature to obtain CoN4/NG catalysts with a loading of about 9 wt% and they exhibited excellent electrocatalytic activity.81 We list a variety of loaded single-atom catalysts with single atoms, carriers, fabrication methods and their applications, as shown in Table 1.
| Single-atoms | Support | Methods | Applications | Ref. |
|---|---|---|---|---|
| Pt1 | Co3O4 | Atomic layer deposition method | Ammonia borane dehydrogenation for room-temperature hydrogen production | 84 |
| Pt1 | Ceria nanorods | Atomic layer deposition method | Hydrogen reducing | 75 |
| PtnSnx | SiO2 | Atomic layer deposition method | Ethylene binding and dehydrogenation | 76 |
| Ni, Co, Cu, Ru | MoOx/SiO2 | Atomic layer deposition method | Synthesis gas conversion | 77 |
| Pt | MOF-NC | Atomic layer deposition method | Oxygen reduction reactions | 85 |
| Rh | Phosphotungstic acid | Co-precipitation method | CO oxidation reactions | 86 |
| Pt | CeO2 | Co-precipitation method | At 200 °C, the full conversion for the CO shift reaction | 87 |
| Pt1 | Co3O4 | Co-precipitation method | The total oxidation of methanol | 88 |
| Pt | g-C3N4 | Dipping method | Photocatalytic H2 evolution | 89 |
| Pd1Ag3 | Al2O3 | Dipping method | Liquid-phase hydrogenation of diphenylacetylene (DPA) | 90 |
| Pt–Co | HZSM-5 | Dipping method | Dichloromethane catalytic oxidation (DCM) | 91 |
| Rh | ZnO | Dipping method | Hydroformylation of olefins | 92 |
| CoN | Graphene | Dipping method | Cathode catalyst of Zn air battery | 81 |
| Ni | Graphene | Dipping method | Electrocatalytic hydrogen evolution | 93 |
| Ni | Graphene | Dipping method | Electroreduction of CO2 | 94 |
| Co, Ni, Zn, Pd, Pt | Metal oxides, nitrogen-doped carbon, polymeric carbon nitride | Dipping method and two-step annealing | Sustainable chemical and energy transformations | 95 |
| Pt1 | Co nanocrystals | Ball-milling method | 5-Hydroxymethylfurfural (HMF) hydrodeoxygenation to 2,5-dimethylfuran (DMF) | 96 |
| Pt1 | ZnO | Ball-milling method | Semi-hydrogenation and carbon monoxide oxidation | 97 |
| Pt | Ceramic MOF (Ce-MOF) | Low-temperature photoreduction method | CO oxidation | 98 |
| Ag | Antimony-doped tin oxide (ATO) | High-temperature cracking method | CO oxidation | 99 |
| Ni | Ultrathin 2D graphitized carbon nanosheets | High-temperature cracking method | The conversion of CO2 to CO | 100 |
| Ni | TiO2 nanoparticles | Molten salt method | Photocatalytic H2 evolution | 101 |
| Fe | Nitrogen-doped porous carbon | Method for thermal emission and trapping with the aid of molten salt | Column 4 cathode oxygen reduction reactions | 102 |
| NiCu | SiO2 | Successive reduction method | Ethanol dehydrogenation | 103 |
Advantages of single-atom catalysts in photocatalysis
It is crucial to investigate advantageous heterogeneous catalysts if alternative sources of energy like biomass, solar, or nuclear are to be used in the future.104,105 Single-atom catalysts offer enormous possible application in metal resource rationalization and the creation of an atomic economy.106 Moreover, loaded single-atoms have demonstrated significant promise in heterogeneous catalysis because of their improved activity, stability, and selectivity, as well as their peculiar electronic structure and unsaturated coordination centres.107 Single-atom catalysts that integrate separated single metal atoms on suitable supports, therefore open up new possibilities for heterogeneous catalysts in terms of the material fabrication and possible applications for various catalytic processes.108 We briefly outline the substantial benefits of single-atom catalysts for photocatalytic reactions in the following four categories in this paper.Improved metal utilization
Nanostructured materials have been extensively researched as heterogeneous catalysts for various catalytic processes.109 Despite the significant accomplishments of traditional nano-catalysts, they still continue to be numerous obstacles to overcome.110,111 Hardly no metal atoms accessible to the reaction mixture may function as catalytically active sites upon a cost standpoint, which results in the unsatisfactory usage efficiency of metal atoms (particularly for nobel metals).108 A single-atom catalyst is a revolutionary form of catalyst that has reactive metal atoms scattered on the substrate separately.112 High dispersion of single atoms with good stability allows each atom to be exposed as a catalytically active site.113 Constructing single-atom reaction sites is beneficial to maximize the usage of metal active centres for catalytic processes.114 The homogenous and well-dispersed active centres give an approximately 100% usage of metal atoms,115 increase the usage of precious metals, and accomplish the cost-effectiveness goal, which is also one of the initial goals of preparing single-atom catalysts.116Carbon-based ones offer outstanding qualities like as cheap cost, various structures, excellent stability, and strong conductance, and are thus commonly employed as SACs carriers.117 In the carbon skeleton, there exists a tight contact between metal atoms and neighboring carbon atoms, which changes the carrier concentration and electronic structure of metal atoms and promotes the development of new active sites within nearby carbon atoms.118 As a result, the carbon-based equipped metal monoatomic catalyst's atom usage rate is nearly 100%,119 resulting in high energy conversion efficiency while lowering expenses. Economic gains and energy sustainability can be realized via constructing a distinctive structure of the catalyst.120 Wang et al. demonstrated an effective strategy for the simultaneous design of Pd SAs, clusters and VOs on TiO2, resulting in enhanced photocatalytic activity for H2 production and selective oxidation of benzylamine at a very low Pd loading cost with greatly improved atom utilization. The optimized PdSA+C/TiO2-VO photocatalysts showed higher yields of H2 and N-benzylidene benzylamine than the PdSA/TiO2-VO containing only Pd SAs and other related photocatalysts.121 Chen et al. investigated Pt/TiO2 (anatase) sac for the reverse water–gas shift (rWGS) reaction. The potential of Pt single atoms (Pt1) for catalytic conversion is initially masked by their saturated coordination with TiO2 and can be released by reduction-oxidation treatment. We show that a controlled reducing atmosphere moves Pt1 to form small amorphous aggregates that can be redispersed into Pt1 by mild oxidation. Redispersed Pt1 shows less coordination to surface O than Pt1 of fresh catalysts, hence better accessibility and consequently higher activity in the rWGS reaction.122
Enhanced reaction activity
From the viewpoint of photocatalysis, light harvesting, charge creation and segregation, and catalytic reaction are the three fundamental reaction stages in a photocatalytic process. The total efficiency of a photocatalytic system is governed in this way by the equilibrium of kinetics and thermodynamics of these three major reaction stages.3 However, photocatalysis is hampered by fast coupling of photoexcited charge carriers and a scarcity of active sites for catalytic processes, culminating in disappointing results that disconnected from practical use. Along with their special geometric and electrical properties, single metal atom catalysts not only boost the amount of active sites owing to maximal atom-utilization efficiency, and moreover improve the fundamental activity of the each active site.123 What is more, for photocatalytic reactions with single atom reaction sites, semiconductors are the carriers of metal active centres and primary catalysts to provide photo-generated charge carriers to promote photocatalytic reactions,124 and single atoms attached to the surface of the photocatalyst serve as the reaction sites for oxidation and reduction. Charge separation is facilitated by photocatalysts with single atomic reaction sites,44 and it is helpful to change the electronic band structure of photocatalysts,125 thus Increasing activity through boosting light reception and photocarrier transmission. Furthermore, the unsaturated single atom contributes to the adsorption, activation together with selective conversion of CO2 molecules. Moreover, the surface structure of single-atom photocatalysts can enhance photocatalyst adsorption, activity and chemical stability through different metal-carrier interactions. Zeng et al. pretreated graphite carbon nitride (GCN) with H2O2 to introduce active C–OH groups capable of reacting with HAuCl4 to fix monatomic Au(I) in the GCN matrix via solid Au(I)–O coordination bonds. In the photocatalytic hydrogen evolution process, this mutual interaction can effectively limit the migration of intermediate Au(0) towards the production of Au particles. As a result, the single atom Au(I) integrated GCN demonstrated higher activity and stability for photocatalytic hydrogen production.126 Wang et al. prepared porous titanium dioxide loaded copper single-atom (Cu–SAs/TiO2) photocatalysts and found that the total yield of CH4 and CO from CO2 photoreduction on Cu–SAs/TiO2 reached 34.64 μmol gcat h−1 with good activity and stability of the photocatalyst when a small amount of oxygen (117.6 ppm) was added to the reaction system. This is because the competing electron transfer to O2 on Cu–SAs/TiO2 maintains the dynamic stability of the Cuδ+ active centre, the O2-derived *OOH species reduces the formation energy barrier of the key intermediate *COOH, and the transfer of *H species accelerates the intermediate protonation process.127Improved photocatalytic stability and selectivity
The interfacial free energy of metals grows significantly with decreasing particle size, from nanoparticles and subnanoclusters to individual metal atoms. Metals' interfacial free energy rises dramatically as particle size decreases. As a result, the mutual interaction between active sites and metal atoms becomes stronger.118 Thus individual metal atoms have stronger metal–atom–carrier interactions and are more stable. As a single atom is chemically anchored on the supporting photocatalyst, a single metal atom is firmly bonded with the adjacent anchoring sites, which raises the specific surface area, modifies the bonding and coordination environment of atoms, and forms a unique low coordination active centre structure with strong site activity and high uniformity.128 Therefore, single-atom catalysts demonstrate greater specific catalytic activity, stability, selectivity, as well as catalytic efficiency over typical nanoparticle-based multiphase catalysts For some photocatalytic processes.129 Nie et al. discovered a novel sort of active site on CeO2 near the ionic platinum Pt2+ that exhibited increased activity. Such active sites may be stabilized in an oxidative atmosphere when the temperature reaches 800 °C.130Single atoms have an evenly distributed, firmly attached, and often electron-deficient character. By varying the manner and intensity of the reaction mixture, intermediate, and/or output adsorption, or by altering the reaction route, one may control the selectivity of the reaction.104 By using a straightforward deposition technique, Huang et al. created photocatalysts with single Co2+ sites on C3N4 and showed high activity and product selectivity for CO production. Under visible-light irradiation, a turnover number of over 200 was achieved for the generation of CO employing the synthesized photocatalyst. The existence of single Co2+ sites on C3N4 and their critical role in accomplishing selective CO2 reduction were verified by further X-ray absorption spectroscopy experiments.131 Wang et al. demonstrated a voltage-measured electro filtering approach for the ambient temperature production of single-atom catalysts. Following the anchoring of the iron single atom, the free energy of the attracted CO decreases from +0.61e to 0.27e, so that the electrons can be transferred out of the 3d orbital of iron into the orbital of CO. Fe–SAs/N–C were shown to show outstanding photocatalytic performance for the conversion of aqueous CO2 to syngas with a controllable CO/H2 ratio when exposed to visible light. CO and H2 have gas evolution rates of 4500 and 4950 μmol g−1 h−1 for CO and H2, respectively.132 Additionally, the capacity of the metal atoms to attract other substances may be diminished and selectivity can be increased by alloying the metal atoms in single-atom photocatalysts133 and altering the electronic structure of the active core atom by the ligand atoms.134 Wang et al. designed and constructed a Cuδ+/CeO2–TiO2 photocatalyst through the pyrolytic conversion of Cu2+–Ce3+/MIL-125-NH2 precursor. In the designed photocatalyst, TiO2 acts as a light-trapping material for the generation of electron–hole pairs effectively separated by CeO2–TiO2 interface, and the Cu–Ce dual active sites synergistically promote the generation and dimerization of *CO intermediates, thus reducing the energy barrier of C–C coupling. The Cuδ+/CeO2–TiO2 photocatalyst showed a productivity of 4.51 μmol−1 gcat−1 h−1 and a selectivity of 73.9% for the conversion of CO2 to C2H4 under simulated sunlight with H2O as the hydrogen source and hole scavenger.135
Pointing the way to photocatalysis research at the atomic level
The simple geometry and electronic structure of single-atom photocatalysts,136 the variety of preparation methods,137 and the ease of identifying and understanding the reaction mechanism of single atoms as active sites138 facilitate the atomic-scale research of structure–activity connections and point the way to other photocatalytic studies at the atomic level. Wang et al. designed a Z-Scheme photocatalyst with N N–Cu1–S single-atom electron bridge (denoted as Cu-SAEB) for mediating CO2RR. In the absence of sacrificial agent, the yields of CO and O2 on Cu-SAEB were as high as 236.0 and 120.1 μmol g−1 h−1, respectively, which exceeded most of the previously reported photocatalysts. The designed Cu-SAEB was highly stable over a total of 30 reaction cycles of 300 h due to the enhanced contact interface of Cu-SAEB and modulated by the atomic structure of N–Cu1–S. Moreover, N–Cu1–S acts as SAEB to achieve an effective Z-Scheme charge transfer mode at the contact interface of Cu-SAEB, thus maintaining the photogenerated carriers with strong redox potential and ultimately enhancing the photocatalytic CO2 performance. This study provides a new direction for the design of highly active photocatalysts with atomic precision and demonstrates the role of interfacial structure in the photocatalytic process.139Disadvantages and improvements of single-atom catalysts
Although single-atom catalysts have many advantages mentioned above, they also have some limitations. When the metal particles are reduced to the single-atom level, the specific surface area increases dramatically, leading to a sharp increase in the free energy of the metal surface,140 which can easily cause agglomerative coupling to form large clusters during preparation and reaction, thus resulting in catalyst deactivation and affecting the activity and stability of the catalyst.141 In addition, single-atom catalysts require high structural characterization instruments and complex analysis of catalytic reaction mechanisms, which are engineering technical problems.142 In some reactions, strong interactions between intermediates and transition metal single atoms may deactivate the reaction sites.143 During the application of defective engineering to synthesize single-atom catalysts, the conjugation can be weakened, leading to electron–hole complexation, which can also disrupt charge transport and damage the carrier structure,144 affecting its performance. High stability, high activity and high loading are the great challenges for single-atom catalysts.The synergistic effect of single metal atoms and carriers can prevent the atomic diffusion of individual metal atoms from aggregating into particles, and the metal oxide carriers can directly participate in the activation of catalytic substrates, making the single-atom catalyst carriers promote the proximity effect of catalytic reactions and substantially enhance the activity of single-atom catalysts.145 Therefore, choosing a suitable carrier is an effective way to alleviate the above-mentioned defects of monatomic catalysts. Therefore, we found that to improve the performance of single-atom catalysts it is not enough to study just the metal single atoms. The choice of the carrier plays a crucial role in improving single-atom catalysts.146 It has been shown that the interaction between the metal and the carrier is often used to stabilize the metal particles with the aim of obtaining highly stable and long-lived catalysts. The carriers in loaded metal catalysts not only play the role of dispersing and stabilizing metal nanoparticles, but also interact with metal particles,147 which often leads to interfacial charge transfer, metal structure change, molecular adsorption modulation and other phenomena, and in turn affects the activity, selectivity and stability of catalysts.148
Commonly used carriers for single-atom catalysts are also abundant, mainly metals and metal oxides, such as Fe2O3, TiO2 and Al2O3.149 In addition, carbon-based carriers are also commonly used as catalyst carriers, such as carbon nanospheres, carbon nanofibers, graphene, metal–organic framework-derived carbon, covalent triazine framework, carbon nitride and phthalocyanine derived carbon, etc.150 TiO2 is an inexpensive, non-toxic and chemically stable metal oxide with three crystalline phases, rutile, anatase and plagioclase, which are commonly found in nature. Among them, rutile and anatase TiO2 have been widely used as catalyst carrier materials.151 Among the many carriers for SACs, carbon-based has excellent properties, such as low price, diverse structure, excellent stability and good electrical conductivity, and is widely used as a carrier material for SACs.117 Carbon-based carriers are able to introduce heteroatom anchor sites and immobilize metal single atoms; heteroatom-doped carbon can also play a positive role in photocatalytic processes.152 Carbon materials are preferred as carriers for catalysts, including carbon nanospheres, carbon nanofibers, graphene, metal–organic framework-derived carbon, covalent triazine framework, carbon nitride and phthalocyanine derived carbon.153 In this paper, we focus on the important roles of three widely used carriers, TiO2, g-C3N4, and MOFs, in improving single-atom catalysts.
TiO2
Apart from the role of the metal oxide carrier of single-atom catalysts as a ligand to stabilize the catalytic metal center, the metal atoms on the carrier adjacent to the single-atom dispersed metal center may also directly participate in the activation of the reactants and catalytic reaction, forming a significant proximity effect that enhances the catalytic performance, making the real active sites of the atom-dispersed metal catalysts may go far beyond the isolated metal atoms themselves.154In TiO2-loaded single-atom palladium catalysts, the Pd–O–Ti(III) atomic-level interface formed around the palladium can effectively activate oxygen to form superoxide ions at room temperature, giving the catalysts superior low-temperature catalytic carbon monoxide oxidation activity and excellent performance in the oxidative elimination of greenhouse gases (e.g., methane) and volatile organic pollutants (e.g., toluene).155
Liu et al. found that the Ti(III)–O–Pd interface in the atomically dispersed Pd1/TiO2 catalyst activated O2 to superoxide, thereby promoting catalytic oxidation, as depicted in Fig. 5A.155 Due to the unique O2 activation mechanism, the Pd1/TiO2 catalyst reported in this work exhibits the highest CO conversion frequency among the previously reported Pd-based catalysts and enhances the catalytic effect on the combustion of hazardous volatile organic compounds and greenhouse gases. The direct engagement of metal atoms on oxide carriers suggests that the actual active sites of the atomically dispersed metal catalysts may extend well beyond the isolated metal atoms themselves.156
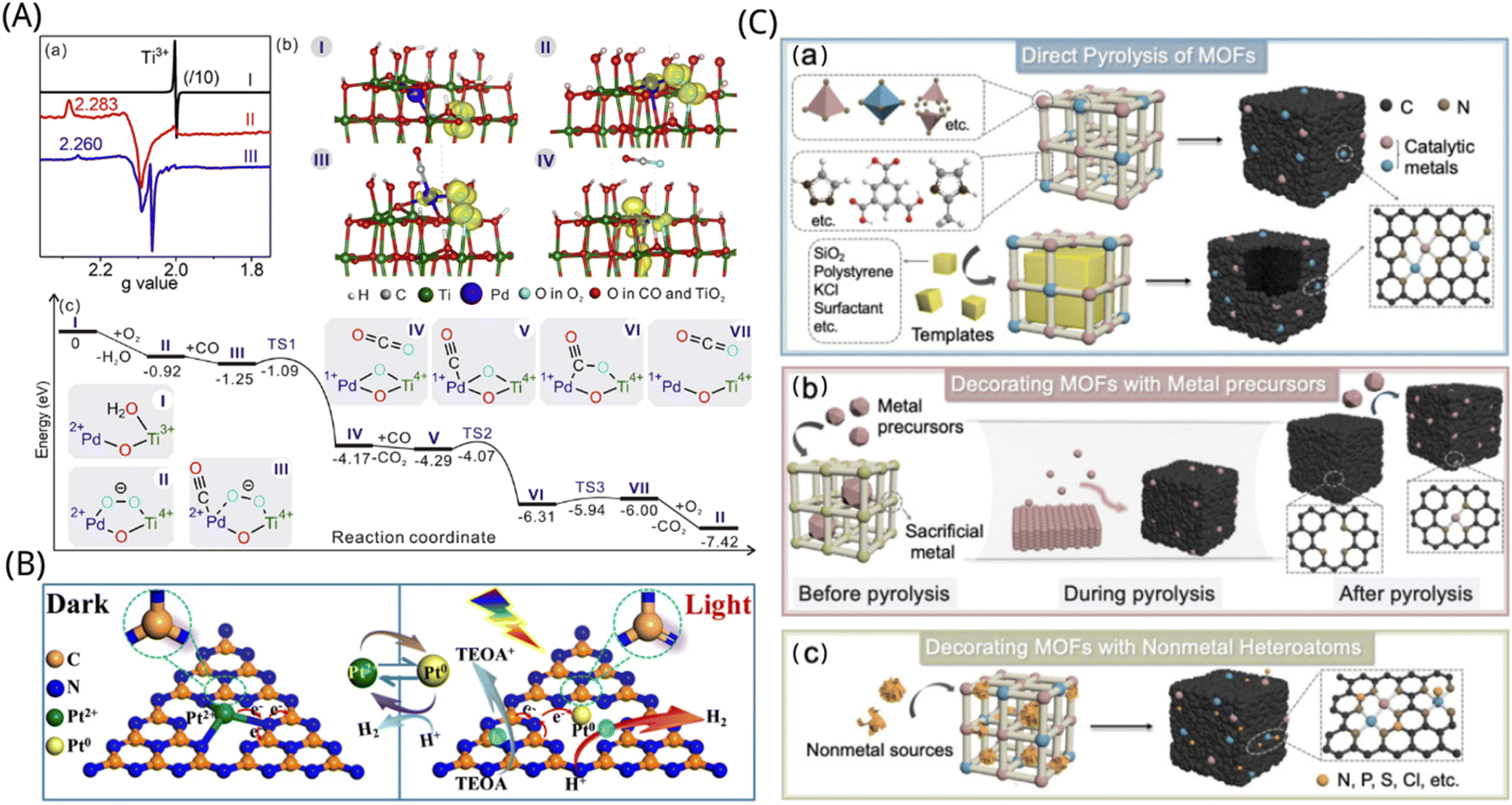 | ||
| Fig. 5 (A) Mechanism of Ti(III)–O–Pd(II) interface to promote CO oxidation. (a) EPR signal for monitoring the catalytic reaction and (b) structural model showing the spin state of the stable intermediate. (c) Energy of intermediates and transition states in the CO oxidation mechanism at the Ti(III)–O–Pd(II) interface calculated by DFT.155 Reproduced with permission. Copyright 2018, Elsevier. (B) Illustration of charge transfer and bond variation on S–Pt–C3N4 catalysts for photocatalytic hydrogen evolution. TEOA = triethanolamine.162 Reproduced with permission. Copyright 2020, Wiley-VCH GmbH (C) schematic diagram of the conversion of MOFs to sac by pyrolysis in ORR applications. (a) Direct pyrolysis of MOFs. (b) Decoration of MOFs with metallic precursors. (c) Modification of MOFs with non-metallic heteroatoms.167 Reproduced with permission. Copyright 2021, Elsevier. | ||
g-C3N4
Non-precious metal single-atom materials have received much attention in photocatalysis because of their low cost, high reactivity, high selectivity and high atomic utilization.157 However, the high surface energy of individual atoms causes agglomeration during preparation and catalytic measurements, leading to damage of the catalytic sites.141 Strong interactions between substrates and single atoms are a key factor in preventing the agglomeration of individual metal atoms and can modulate the geometry and electronic structure of the catalyst to optimize catalytic activity. Nitrogen-doped porous carbon has been widely investigated as an ideal non-precious metal single-atom carrier due to its multi-level pore structure, high specific surface area and defect effect,158 which synergistically enhances the catalytic performance for oxygen reduction reactions, oxygen precipitation reactions, hydrogen precipitation reactions, and carbon dioxide reduction reactions.159In general, more charge transfer between the low-coordinated non-precious metal single atoms and the strongly electronegative N atoms changes the electron distribution around the metal single atoms, and stronger chemical bonds are formed between the metal single atoms and the carriers, enabling a stronger and more stable dispersion. On the other hand, the defect effect caused by N doping and the intrinsic defects of the carbon material provide additional anchor sites for the metal single atoms.160 In addition, N doping sites provide confined space for metal single atoms by modulating the porous channels of carbon materials.161
It was demonstrated through the experiments of Zhang et al. that the Pt2+ atom can effectively attract electrons from the C3N4 planar layer and convert to Pt0 atoms under light illumination, accompanied by Pt–N bond breakage, as shown in Fig. 5B. At the same time, the reverse conversion of the –C–N to –CN bond and the reconstruction of the C3N4 molecular structure occurred, which has been confirmed by the similarity of the C 1s and N 1s spectra of S–Pt–C3N4 in the excited state to the C 1s and N 1s spectra of primitive g-C3N4 in the ground state. Consequently, in the excited state, a single Pt0 atom with abundant electrons and a C3N4 layer with abundant holes in the isolated state can participate in the reduction and oxidation of water, respectively, and the photocatalytic H2 precipitation performance of single-atom Pt/C3N4 is significantly higher than that of Pt-particle-C3N4 and C3N4.162
MOFs
For MOF-immobilized single-atom catalysts based on MOF, the atomic metal sites can be stabilized in the metal nodes, pore spaces or organic ligands of MOF.163 Compared with other carriers, MOF-immobilized single-atom catalysts have the following advantages. Firstly, three-dimensional ordered porosity is the main feature of MOF-immobilized single-atom catalysts, which can greatly promote the mass transfer process during the reaction and thus improve the catalytic efficiency.164 Moreover, MOF has rich chemical tunability, which can easily and precisely control the structure of the immobilized single-atom catalytic sites, thus making the prepared atomic-level metal catalysts rich in structural diversity.165 In addition, embedding atomic-level catalytic active sites in MOFs can achieve a significant increase in the activity of the non-homogeneous catalysts without compromising their long-lasting stability and reproducibility. What's more, by introducing more than one type of metal-based sites at the same time, synergistic interactions between different chemical components in the MOF can be effectively achieved, thus improving the catalytic performance. Last but not least, in general, catalysts prepared using MOFs can usually achieve a high loading of metal content.166 Hu et al. found that the strategy of converting MOFs into sacs by pyrolysis can be divided into three stages. Firstly, MOFs, including pristine MOFs and MOFs with hard/soft templates, are directly converted to sac by high temperature treatment. In the second stage, MOFs are modified with additional metal precursors by various methods. In the final stage, MOFs are combined with components containing nonmetallic heteroatoms to provide more coordination atoms and modulate the electronic structure of the active metal centre, as depicted in Fig. 5C.167Novel single-atom photocatalysts for CO2 reduction
With to the growth of constant technology and the enhancement of people's environmental awareness, new single-atom photocatalysts for CO2 reduction are emerging. In this paper, we mainly focus on three popular single-atom photocatalysts of TiO2, g-C3N4 and MOFs materials based on the interaction between single atoms and carriers.TiO2
The benefits of the photocatalytic reduction technique are its lack of toxicity, low energy need, and absence of secondary emissions. Widespread interest has emerged in CO2 photocatalytic reduction processes over TiO2-based catalysts.168 In 1979, Inoue et al. found that photocatalytic reduction of CO2 into CH3OH, HCOOH, CH4, etc. could be done by utilizing TiO2, CdS, SiC and other photosensitive semiconductor powders suspended in water as catalysts, and explained the kinetics of the photocatalytic reaction based on the charge transfer theory of photoexcited semiconductors,169 which was the first proposal of the reaction principle of photocatalytic reduction of CO2. The reaction equation is shown below. After continuous research, it was found that TiO2 as the most favoured photocatalyst170 can not only catalyse the conversion of CO2 into commercially viable organic compounds, but also remove pollutants in the atmosphere such as mercury by photocatalytic oxidation,171 which has a good application value and prospect compared with traditional emission reduction technologies.| CO2 + e− → CO2−, E = −1.9 eV | (1) |
| CO2 + H+ + 2e− → HCO2−, E = −0.49 eV | (2) |
| CO2 + 2H+ + 2e− → CO + H2O, E = −0.53 eV | (3) |
| CO2 + 4H+ + 4e− → HCHO + H2O, E = −0.48 eV | (4) |
| CO2 + 6H+ + 6e− → CH3OH + H2O, E = −0.38 eV | (5) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O, E = −0.24 eV | (6) |
| 2H+ + 2e− → H2, E = −0.41 eV | (7) |
| H2O → ½O2 + 2H+ + 2e−, E = 0.82 eV | (8) |
Due to the TiO2's high band gap, the photocatalytic reaction's solar light usage efficiency is poor. In order to optimize visible light consumption and TiO2 activity, it is necessary to dope and modify TiO2. The methods mainly include non-metal doping, metal cocatalyst deposition, heterostructure construction via coupling with other semiconductors, and organic photosensitizer modification. When metal is used to modify TiO2, charge carriers are moved from semiconductor to metal because of the different Fermi energy levels, enhancing the effectiveness of photo-generated transfer of electrons. Simultaneously, the Schottky energy barrier forms on the surface of the metal–TiO2, inhibiting the coupling of photo-generated electrons and holes and therefore enhancing TiO2's photocatalytic efficiency. At present, the metals that are widely used mainly include Pt, Ag, Au, Pd, Ru, Rh, etc.
TiO2 is a suitable supporting material for constructing single-atom photocatalysts based on metal–oxygen bonding configuration. Scientists synthesized single-atom photocatalysts based on TiO2 materials usually through the sol–gel technique, hydrothermal synthesis, and chemical vapor deposition.172 Pan et al. developed a binary component catalyst made up of single atoms (SAs–Pt and Au) anchored on self-doped TiO2 nanotubes (TNTs) to promote the effective transition of photogenerated electrons from defect sites to SAs via covalent interactions, which improved electron–hole pair separation and charge carrier transfer. The photocatalytic CO2 reduction efficiency of the Pt–Au/R-TNTs with 0.33 weight percent of SA metals was up to 149-fold more than that of unmodified R-TNT, and the overall apparent quantum yield (AQY) was 17.9%, with yields of CH4 and C2H6 reaching 360.0 and 28.8 μmol g−1 h−1, respectively.173 Compared with other metals, the benefits of the Cu element include affordability and low environmental impact, that may effectively increase photo-generated electron and hole segregation in the catalyst and broaden the spectrum absorption range. Therefore, scientists have undertaken substantial study on the metal Cu–TiO2 combination. Yu et al. suggested that the interaction of metal alloy nanoparticles (NPs) with single atoms might enhance catalytic performance. Therefore, they co-loaded the Cu SAs and Au–Cu alloy NPs on TiO2 to obtain the excellent performance photocatalyst by photo-deposition, as shown in Fig. 6A. The combinatorial action of Cu SAs and Au–Cu alloy NPs may improve the activation of CO2 and H2O adsorption and decrease the overall intrinsic limitations for CH4 and C2H4 production. The yields of 3578.9 mol g−1 h−1 for CH4 and 369.8 μmol g−1 h−1 for C2H4 make the production of sunlight-driven high-value solar fuels more feasible.174 Yin et al. further developed Cu single atoms anchored on nitrogen-doped carbon on TiO2, which showed 100% CO selectivity and high epitaxial quantum efficiency up to 2.0% for photocatalytic CO2 reduction with H2O vapor at 420 nm.175 SAs synthesis and characterization are shown in Fig. 6B. Lee et al. demonstrated the atomic-level CO2 photoreduction reaction on TiO2 photocatalysts with uniform and stable transition metal single atoms and found that the interaction of electrons with individual Cu atoms and neighboring TiO2 affects the reduction on the TiO2 surface, resulting in the automatic establishment of O-vacancies in the vicinity of copper atoms. Therefore, they controlled the space allocation of homogeneous individual copper atoms on TiO2 as to allow adjacent copper atoms to participate in the mutual effect with CO2 intermediates through controlled charge positioning. In comparison to the original TiO2, the improved Cu1/TiO2 photocatalyst exhibits a 66 times better CO2 photoreduction capability.176 Through the fixing of a single tungsten (W) atomic site with oxygen coordination on the intrinsic step of classical TiO2 nanoparticles, Feng et al. created a unique “single atomic site on an atomic step” approach. The composition of the active site for CO2 reduction may be adjusted by modifying the extra W5+ to produce a W5+–O–Ti3+ site, resulting in a CO2 reduction efficiency of 60.6 μmol g−1 h−1 and a CH4 selectivity for CO that exceeds that of pristine TiO2 by an order of magnitude.177 The fundamental principle of CO2 photocatalytic reduction in different reaction systems is a direction well worth discussing. Chen et al. used a template-assisted in situ pyrolysis approach to synthesize a Cu single-atom-incorporated three-dimensional-ordered macro-porous TiO2 (Cu0.01/3DOM-TiO2) photocatalyst. The 3DOM TiO2 framework that the Cu single atoms are evenly embedded in, not just to widens the spectrum of light absorption, and moreover offers particular active sites for the adsorption and conversion of CO2 molecules. In the gas–solid system, the Cu0.01/3DOM-TiO2 photocatalyst showed excellent selectivity and activity yet it tended to produce C2H4 in the liquid–solid system. With a selectivity of 83.3% and a generation rate of 43.15 μmol g−1 h−1, the photocatalytic CO2 reduction process in the gas–solid system primarily produced CH4, whereas the reaction in the liquid–solid system produced C2H4 as the major output with a selectivity of 58.4% and a formation rate of 6.99 μmol g−1 h−1.178 By rationally designing photocatalysts and fine-tuning the reaction conditions, this study contributes a few new knowledge on boosting photocatalytic CO2 reduction to desired products. Except for the application of such photocatalysts to reduce CO2 to hydrocarbons, the organic fertilizer urea can also be made catalytically with inorganic substances such as N2. Li et al. used a photoinduction approach based on TiO2 photocatalyst coupled with reversible monatomic copper (expressed as Cu SA–TiO2) to produce a moderate photocatalytic production of urea utilizing N2 and CO2 molecules in the environment of pure H2O, as shown in Fig. 6C. The urea yield was as high as 432.12 μg gcat−1 and the speed of photogenerated electron extraction was over 30-fold higher than that of the reference photocatalyst.179
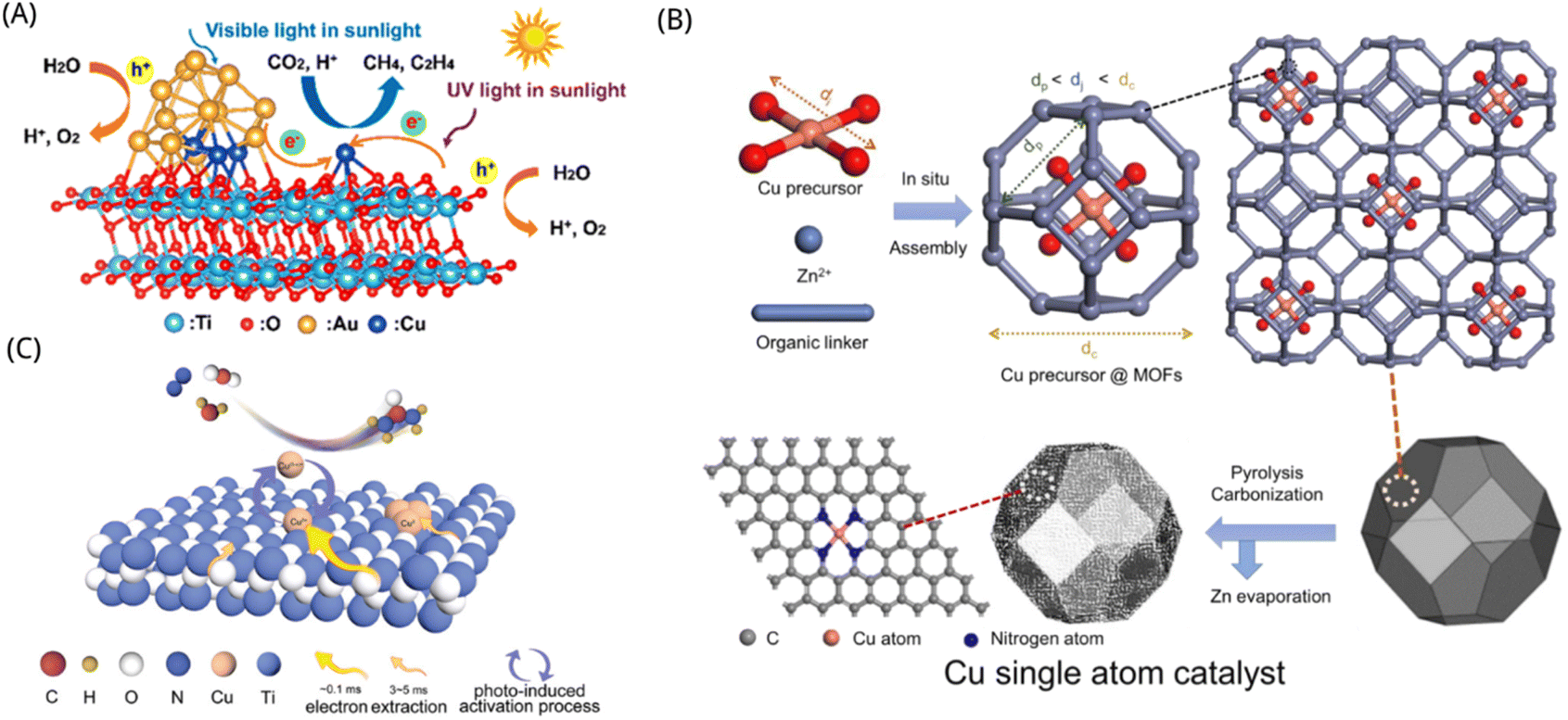 | ||
| Fig. 6 (A) Diagrammatic representation of the photocatalytic CO2 reduction process by Cu0.8Au0.2/TiO2.174 Reproduced with permission. Copyright 2021, American Chemical Society. (B) The scheme for fabricating Cu single atoms site catalysts.175 Reproduced with permission. Copyright 2022, American Chemical Society. (C) Diagram of urea photosynthesis on the surface with a variety of photocatalysts (pure TiO2, Cu SA–TiO2, and Cu0–TiO2).179 Reproduced with permission. Copyright 2022, Wiley-VCH GmbH. | ||
Photocatalytic CO2 reduction by TiO2-based photocatalysts has a promising future in emission reduction and energy redevelopment. However, owing to the defects of high recombination probability of photo-generated electron–hole pairs, poor CO2 adsorption performance of catalyst and difficulty in CO2 activation, and the poor rate of solar energy usage (especially in visible light), its catalytic efficiency is limited.180 Construction of TiO2-based single-atom photocatalysts is definitely a good way to realize efficient photocatalytic CO2 reduction performances. At present, the research has achieved good results and the catalytic efficiency has been significantly improved. Nevertheless, the main research area is dominated by noble single-atom metals and the fabrication method is relatively complicated. Further research is still needed to produce inexpensive, environmentally friendly and universally applicable photocatalysts based on TiO2 materials.
g-C3N4
Graphite nitrogen carbide (g-C3N4) has a band gap of 2.7 eV, which is a semiconductor with photocatalytic performance beneath visible light. It has a wider wavelength utilization range of sunlight the TiO2.181 Moreover, the catalyst is composed of two common nonmetallic elements, C and N, and has the characteristics of non-toxicity, no pollution, low preparation cost, etc.182 Its physical and chemical properties are stable at normal temperatures and pressure. Therefore, g-C3N4 has been extensively studied as a possible nonmetallic semiconductor photocatalyst. However, its high carrier recombination efficiency and low specific surface area reduce the photocatalytic activity of g-C3N4. Photocatalysts based on g-C3N4 often have poor charge separation and CO2 adsorption capabilities, which have a negative impact on their capacity to reduce CO2. Therefore, we can modify the original catalyst in different aspects by preparing composite materials or chemical doping to obtain high photocatalytic performance.Nano-materials can greatly increase the contact area of catalytic reaction, so most of the g-C3N4 on the modified photocatalyst is found as nanotubes. Qin et al. synthesized ultra-thin nanosheet g-C3N4 (NS-g-C3N4) by calcination method, as shown in Fig. 7A. Their extremely thin nanostructures and plentiful surface defect sites significantly increase visible light adsorption efficiency along with the segregation and transmission of photo-generated electrons, and provide powerful chemisorption sites for CO2. Moreover, the surface defects of the nanosheets can contribute to the selective photodegradation from CO2 to CO, and thus provide significant activity, selectivity and stability for the photocatalytic CO2 reduction process. They developed NS-g-C3N4 with a layer thickness of 10 nm that was more efficient than its bulk equivalent (B-g-C3N4) for the photocatalytic reduction of CO2 beneath the illumination of solar light, with CO being the sole product found in the system and yielding 5.8-fold more than B-g-C3N4.183 To modify the g-C3N4 photocatalyst's capacity for light absorption, redox potential, and electron removal efficiency, Liu et al. fabricated pore-like carbon nitride (g-C3N4) nanotubes. Moreover, they also added the appropriate quantities of translucent zeolite imidazolium framework-8 (ZIF-8) nanoclusters to the equipped tubular g-C3N4 to improve its capacity for capturing CO2. Through the collaborative impact of semiconductor nanostructure and surface metal–organic framework grafting agent, the improved ZIF-8 modified tubular g-C3N4 photocatalysts shown a remarkable increase in photocatalytic CH3OH process performance, which is three-fold more than the traditional block g-C3N4 (BCN) photocatalyst made via melamine pyrolysis.184
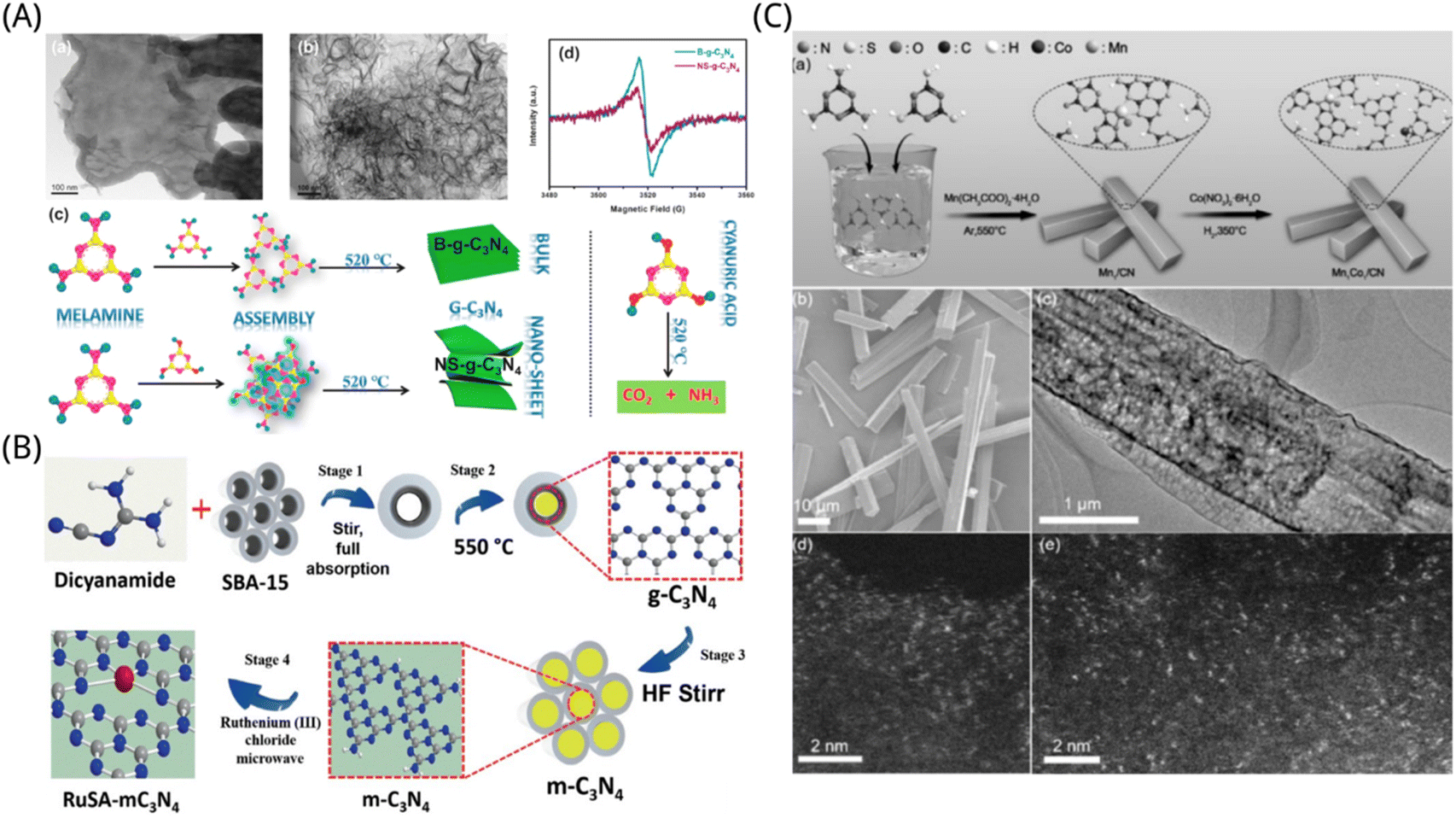 | ||
| Fig. 7 (A) TEM pictures of (a) B-g-C3N4 and (b) NS-g-C3N4. (c) The steps involved in synthesizing B-g-C3N4 and NS-g-C3N4. (d) The EPR spectrogram of B-g-C3N4 and NS-g-C3N4.183 Reproduced with permission. Copyright 2021, Elsevier Inc. (B) Schematic diagram of the synthetic procedure for a ruthenium single atom on mesoporous C3N4. (RuSA-mC3N4: Ruthenium single atom; HF: Hydrofluoric acid; mC3N4: mesoporous carbon nitride; SBA-15: template.)188 Reproduced with permission. Copyright 2021, Wiley-VCH GmbH. (C) (a) Preparation of the Mn1Co1/CN catalyst. (b) SEM and (c) TEM diagrams of Mn1Co1/CN. AC HAADF-STEM diagram of (d) Mn1/CN and (e) Mn1Co1/CN.192 Reproduced with permission. Copyright 2022, Wiley-VCH GmbH. | ||
For the purpose of increasing catalytic activity, stability and selectivity of g-C3N4 photocatalysts, scientists have often used single atoms decorations for refinement. Utilizing space-constrained single atom Fe and K ions, the graphitic carbon nitride (FeN4/K-g-C3N4) fabricated by Cheng et al. demonstrated remarkable performance and selectivity for the photocatalytic CO2 reduction process. The g-C3N4 layer's single atom of Fe forms a connection with its four neighboring N atoms, acting as the centre of activity for the capture and revitalization of CO2 molecules. In addition, the alkali metal K ions may efficiently enhance segregation and transference, which together with the spatial confinement of the single atom Fe and K ions in g-C3N4, promotes photocatalytic performance and selectivity of CO2 conversion to CO. The conversion rate of CO2 to CO can reach 20.00 μmol g−1 h−1 with a selectivity close to 100%, which outperforms pure g-C3N4 by a factor of more than ten.185 Zhao et al. further demonstrated the contribution of single-atom Fe to catalytic activity. They used density flooding theory (DFT) and time-dependent DFT (TDDFT) approaches, combined with experimental and computer-based mechanisms, to find out more about the photoreduction of CO2 with H2O that is facilitated by single-atom Fe-supported graphitic nitrides (g-C3N4) and to learn more about the function of single-atom Fe in g-C3N4. It was found that in the absence of the Fe atom, the speed-limiting step of the hydrogen bonding compound is the cleavage of the C–O bond in the COOH radical throughout the CO2RR process, involving both photophysical and photochemical reactions. In addition to activating CO2 in its ground state and increasing the rate constant of the restricting step in the photophysical reaction, the existence of the Fe atom also serves as a catalytically active centre, decreasing the restriction of the reaction for the cleavage of the C–O bond in COOH* in the photochemical process, thus improving photocatalytic activity.186 Cheng et al. ligated single-atom site Ni on porous multilayers of g-C3N4 (i.e. Ni5–CN) synthesized from the bottom up by a self-limiting approach and defined an unsaturated edge-limiting strategy. The few isolated Ni clusters in this Ni5–CN system are dispersed on the edges of g-C3N4, allowing non-edge single-atom site Ni species to be immobilized and obtaining a substantial density of single-atom active sites. The advantages in N–Ni–N combinatorial link and surface carrier transference are promoted by the cationic linkage environment of the monoatomic Ni centre generated by doping Ni–N into the primary bonding shell. The Ni5–CN system had significant photocatalytic performance for CO2 reduction, with a CO production rate of 8.6 μmol g−1 h−1 beneath visual light, which was 7.8-fold more than the pure porous several-layer g-C3N4 system (i.e. CN, 1.1 μmol g−1 h−1).187 In order to achieve photocatalytic CO2 reduction to methanol fuel utilizing water as an electron carrier and reaching an output of cat. 1500 μmol g−1 in 6 hours of response, Sharma et al. produced RuSA-mC3N4 photoactive catalysts by an innovative single-atom synthesis approach with single ruthenium atoms dispersed on a granular carbon nitride surface, as shown in Fig. 7B. The analysis by EXAFS absorption spectroscopy revealed that the Ru–N/C intercalation in the initial ligand shell layer formed a cationic ligand environment with a single atomic ruthenium centre, realizing a synergistic effect of N–Ru–N linkage and charge transfer on interface. The coupling of Ru with NC sites boosts the amount of electrons transferred and charge distribution on Ru, lowering the photo-carrier transference obstacle and improving the photocatalytic performance of RuSA-mC3N4, which makes the average carrier life of RuSA-mC3N4 system longer than that of m-C3N4.188 Zhang et al. improved the catalytic performance for CO2 in photochemistry by embedding single cobalt(II) sites on g-C3N4, resulting in CO yields of up to 464.1 μmol g−1 h−1, which are 3 and 222-fold higher than when employing Co-MOF and CoCl2 as the cobalt source, correspondingly. This was accomplished by pyrolyzing ultrathin cobalt metal organic framework (MOF) nanosheets (also known as metal organic layers; MOLs) in the process of forming g-C3N4. When g-C3N4 is being formed, Co(II) sites may be evenly and atomic scale disseminated over its surface because of the confinement impact of the MOF matrix and the tight engagement of the metal organic layer with the g-C3N4 predecessor.189
It has been demonstrated that a novel method for promoting multiphase processes is single-atom photocatalysis. Numerous single-atom metal species exist, each with unique functions. However, a significant obstacle still exists in the integration of representational benefits into dual single-atom photocatalysis. The catalytic performance of dual single-atom catalysts can be improved by combining their complementary functionalities and synergistic effects. Chen et al. synthesized a composite of rare earth single atom La onto carbon nitride. The La–N charge-transfer bridge was used as the active centre of the photocatalytic CO2 reduction reaction, thus promoting the activation of CO2, rapid generation of COOH* and desorption of CO, achieving a remarkable CO generation rate of 92 μmol g−1 h−1 and a CO selectivity of 80.3%, which is superior to the majority of g-C3N4-based photocatalytic CO2 reduction approaches. Moreover, the rate of CO generation remained nearly constant under five cycles of 20 hours of light, showing a more robust stability.190 Cheng et al. developed diatomic catalysts loaded on conjugated porous carbon nitride polymers featuring cobalt (Co) and ruthenium (Ru) functionalities for efficient photocatalytic CO2 reduction. During CO2 photoreduction, the active Co site promotes dynamic charge transfer and the Ru site promotes selective CO2 surface binding interactions. The association of particular atomic properties and the synergy between Co and Ru leads to a excellent photocatalytic CO2 reduction with a respective apparent quantum efficiency (AQE) of 2.8% at 385 nm, as well as a high turnover number (TON) of over 200 in the absence of any added sacrificial agent.191 Inspired by natural photosynthetic systems, Ou et al. created a bimetallic single-atom active site photocatalyst with Mn and Co single atoms, as shown in Fig. 7C, with two compatible Mn and Co active centres (Mn1Co1/CN) on carbon nitride (CN). The placement of Mn and Co bimetallic single atoms on CN as redox sites successfully improves hole–electron segregation and energy transfer by taking advantage of the synergistic impact of the atomic active centres. The Mn1Co1/CN photocatalyst showed excellent catalytic performance in the conversion of CO2 to CO with a CO output of 47 μmol g−1 h−1, significantly higher than that of the corresponding monometallic active centre photocatalyst.192
Although the catalytic performance and selectivity of the g-C3N4-based single-atom photocatalysts have been improved, their photocatalytic efficiency for CO2 reduction still falls short of expectations. Moreover, the modified materials mostly contain precious metals, so the high cost and complicated fabrication processes are major obstacles to practical applications. Therefore, the in-depth design and fabrication of high-efficiency and inexpensive single-atom photocatalysts based on g-C3N4 for CO2 conversion is still to be explored.
MOFs
Because MOFs are porous, there is a large specific surface area available for reactions. As catalytic active sites, the metal centres are evenly dispersed across the materials' interior and exterior surfaces, giving them numerous metal active sites and strong CO2 adsorption. The metal centres and ligands create a distinctive energy band structure through electron structure matching, which is suitable for light energy absorption and transfer. Furthermore, pore channel binding results in effects which including space-limited and charge-limited domains, optimization of process intermediates, and selectivity.193,194 Coupled with the high affinity of MOFs for CO2 facilitates the interaction of reactants and catalysts, advancing the reaction to proceed efficiently. In addition, MOFs materials can regulate of the electronic structure of the active centre by coupling with other materials to extend the carrier lifetime and improve the utilization efficiency, giving them a unique competitive advantage in the photocatalytic CO2 reduction process.By including coordinated unsaturated single atoms into the MOFs matrix, the modular optimization of MOFs is accomplished. This allows the newly created MOFs to effectively and selectively absorb and photolyze CO2 when exposed to visible light. Zhang et al. found that the inclusion of a single Co atom in MOFs can significantly enhance the efficiency of the porphyrin unit's ability to separate electrons from holes and can also encourage the directional migration of photogenerated excitons from the porphyrin to the catalytic Co centre, thereby providing long-lasting electrons for the conversion of CO2 molecules immobilized in the Co centre. The porphyrin MOFs consisting of atomic scale scattered catalytic centres demonstrated dramatically improved photocatalytic CO2 reduction that is equivalent to 3.13 times of CO precipitation rate (200.6 μmol g−1 h−1) and 5.93 times of CH4 production rate (36.67 μmol g−1 h−1) in comparison to the parent MOFs, as shown in Fig. 8A.43 The synthesis of photocatalysts from dilute metal elements and the efficient reduction of CO2 is a difficult yet extremely appealing technique for scientists. Wang et al. created an indium-porphyrin structure, In(H2TCPP)(1−n)[Fe(TCPP)(H2O)](1−n)[DEA](1−n) (In-FenTCPP-MOF; H2TCPP = tetrakis (4-benzoic acid) porphyrin; DEA = diethylamine),195 as shown in Fig. 8B, for high-performance conversion of CO2 to CO powered by visible light. Its porphyrin ring-supported iron centre is the effective active site to absorb electrons from photoexcited MOFs so as to facilitate CO2 reduction. A 24 hour photocatalytic process with excellent CO selectivity (approximately 99.5%) may obtain a high CO output of 3469 μmol g−1. Compared to its cobalt counterpart or indium-based MOFs without iron, its activity is significantly greater.
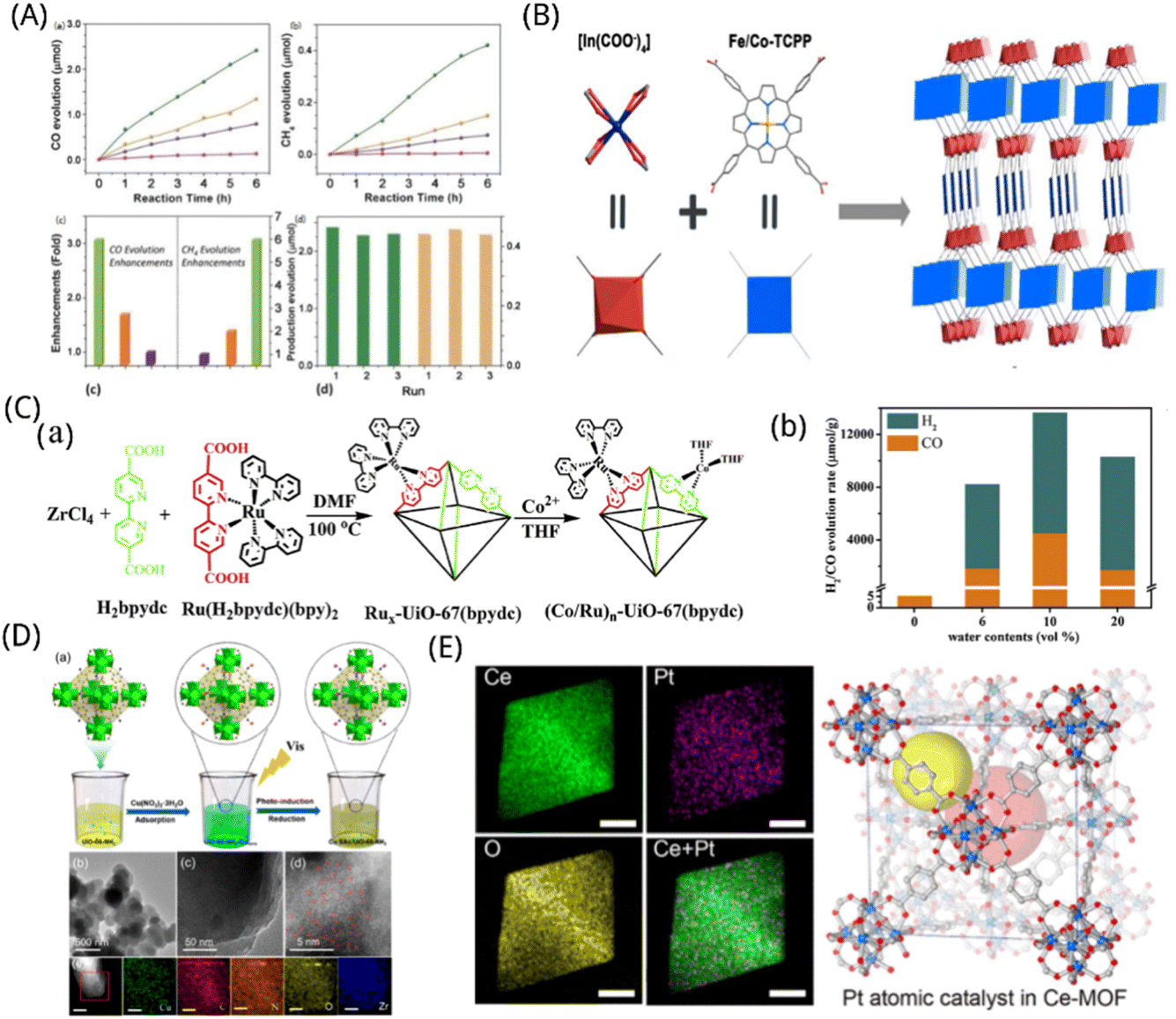 | ||
| Fig. 8 (A) Time-dependent evolution of (a) CO and (b) CH4 on MOF-525-Co (green), MOF-525-Zn (orange), MOF-525 (purple) photocatalysts and H6TCPP ligands (pink). (c) Enhanced production evolution on MOF-525-Co (green), MOF-525-Zn (orange) and MOF-525-Zn (purple). (d) Yields of CO (green) and CH4 (orange) production on MOF-525-Co photocatalyst as a measure of cycle reproducibility.43 Reproduced with permission. Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (B) Synthesis and structure of the In–Fe/CoTCPP-MOFs.195 Reproduced with permission. Copyright 2020, American Chemical. Society. (C) (a) Schematic diagram of the synthesis of (Co/Ru)n-UiO-67(bpydc); (b) comparison of H2/CO evolution rate with different water contents.198 Reproduced with permission. Copyright 2019, Elsevier B.V. (D) (a) The synthesis procedure of Cu SAs/UiO-66-NH2 photocatalyst. C (gray), O (red), Zr–O clusters (green), N (blue), Cu ions (orange), and Cu SAs (purple). (b) TEM and (c) HRTEM of Cu SAs/UiO-66-NH2. (d) Corrected STEM images of Cu SAs/UiO-66-NH2. Cu SAs were marked with red circles. (e) EDS profiles of Cu SAs/UiO-66-NH2.200 Reproduced with permission. Copyright 2020, American Chemical Society. (E) Elemental mappings of Ce, Pt, O, and overlay of Ce and Pt in the Pt–SA–Ce–MOF catalyst and two types of pores in the C-MOF structure (indicated by two large yellow and red spheres).98 Reproduced with permission. Copyright 2020, American Chemical Society. | ||
It is well documented that establishing functional groupings into the skeleton of UiO series MOFs is simple and easy, and can be achieved either directly by using functionalized ligands as starting ligands, or by post-synthetic modification and replacement, with the topology of the final product remaining unchanged. These features make the UiO series MOFs have a lot of promise for use in gas separation, CO2 capture and catalysis.196,197 One of the environmentally friendly methods for turning CO2 recycling into products with additional value is the photocatalytic reduction of CO2 to syngas (CO and H2). Utilizing a single central catalyst, Liu et al. created a straightforward and efficient two-step self-assembly procedure to functionalize phosphorescent metal–organic frameworks. The generated (Co/Ru)n-UiO-67(bpydc) provided the molecular platform for the rapid injection of multiple electrons from photosensitizers (PSs) into the Co-catalyst, resulting in the efficient production of syngas from the MOF-based compound photocatalyst in 16 hours with a production of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 μmol g−1 (H2:CO = 2:1), which is 29.2 times higher than its homogeneous counterpart. Moreover, through carefully controlling the PS/catalyst molar ratio in the MOFs platform as well as the content of H2O in the photocatalytic system, the H2/CO ratio could be successfully regulated, as shown in Fig. 8C.198 UiO-66 is a class of MOFs material with the chemical formula Zr6O4(OH)4(CO2)12, which was discovered and prepared by Cavka et al. in 2008. Due to its good hydrothermal stability, UiO-66 offers a variety of uses for gas adsorption and separation, especially in CO2 capture, which has been studied in a variety of investigations by scholars.197 Cmarik et al. explored the study of different functional groups on gas adsorption properties and found that UiO-66-NH2 has great potential for CO2 adsorption and separation because of the powerful interaction between polar functional group aminos and CO2 as well as the properties of its own structural dimensions.199 Wang et al. achieved the establishment of Cu single atoms on UiO-66-NH2 carriers (Cu SAs/UiO-66-NH2) by a photoinduced method, as shown in Fig. 8D, which greatly facilitated the photoreduction of CO2 to liquid fuels with conversion rates of 5.33 and 4.22 μmol g−1 h−1 for CH3OH and CH3CH2OH, respectively, with the superior performance than the pristine UiO-66-NH2 and Cu nanoparticles/UiO-66-NH2 composites. This was owing to the addition of Cu SAs to UiO-66-NH2, which considerably aided CO2 conversion to CHO* and CO* intermediates, leading to the high selectivity for CH3OH and CH3CH2OH.200 Chu et al. presented a photo-induced reduction method for producing catalysts by attaching single atom Zn on UiO-66-NH2. Moreover, the low-liganded Zn–N2 site can greatly facilitate the synthesis of COOH*, which is the restrictive step in the production of CO2, making UiO-66-NH2-0.7Zn SAs have exceptional capacity in converting CO2 into CO. Compared with the original UiO-66-NH2 in the lack of the photosensitizer or hole sacrificial agent, its performance under ultraviolet-visible light is improved by about 5 times.201 The outcomes of this study open up new possibilities for the production of highly effective photocatalysts with single-atom sites for photocatalytic CO2 conversion.
600 μmol g−1 (H2:CO = 2:1), which is 29.2 times higher than its homogeneous counterpart. Moreover, through carefully controlling the PS/catalyst molar ratio in the MOFs platform as well as the content of H2O in the photocatalytic system, the H2/CO ratio could be successfully regulated, as shown in Fig. 8C.198 UiO-66 is a class of MOFs material with the chemical formula Zr6O4(OH)4(CO2)12, which was discovered and prepared by Cavka et al. in 2008. Due to its good hydrothermal stability, UiO-66 offers a variety of uses for gas adsorption and separation, especially in CO2 capture, which has been studied in a variety of investigations by scholars.197 Cmarik et al. explored the study of different functional groups on gas adsorption properties and found that UiO-66-NH2 has great potential for CO2 adsorption and separation because of the powerful interaction between polar functional group aminos and CO2 as well as the properties of its own structural dimensions.199 Wang et al. achieved the establishment of Cu single atoms on UiO-66-NH2 carriers (Cu SAs/UiO-66-NH2) by a photoinduced method, as shown in Fig. 8D, which greatly facilitated the photoreduction of CO2 to liquid fuels with conversion rates of 5.33 and 4.22 μmol g−1 h−1 for CH3OH and CH3CH2OH, respectively, with the superior performance than the pristine UiO-66-NH2 and Cu nanoparticles/UiO-66-NH2 composites. This was owing to the addition of Cu SAs to UiO-66-NH2, which considerably aided CO2 conversion to CHO* and CO* intermediates, leading to the high selectivity for CH3OH and CH3CH2OH.200 Chu et al. presented a photo-induced reduction method for producing catalysts by attaching single atom Zn on UiO-66-NH2. Moreover, the low-liganded Zn–N2 site can greatly facilitate the synthesis of COOH*, which is the restrictive step in the production of CO2, making UiO-66-NH2-0.7Zn SAs have exceptional capacity in converting CO2 into CO. Compared with the original UiO-66-NH2 in the lack of the photosensitizer or hole sacrificial agent, its performance under ultraviolet-visible light is improved by about 5 times.201 The outcomes of this study open up new possibilities for the production of highly effective photocatalysts with single-atom sites for photocatalytic CO2 conversion.
However, the poor porosity of the support structure, the poor affinity of SAs to the support, and the high-temperature synthesis led to bottlenecks in the practical utilization of SAs, such as high manufacturing expense, poor catalytic performance, and low metal atom usage. To achieve higher catalytic efficiency and atom utilization efficiency, it is necessary to synthesize SAs in a scalable, low-energy manner that closely matches the atomic scale planned 3D nanostructures. Guo et al. used a low-expense ceramic MOF (Ce-MOF) with tailored flaws spanning porous and crystalline structures to produce a simple synthesis approach. The SA(Pt) produced by low-temperature photoreduction could be encased in the Ce-MOF flaws. The conjugated catalyst weighing 0.12 wt% showed 100% CO2 conversion at a low temperature of 150 °C because of the uniform dispersion and the distinctive electronic hybridization with Ce-MOF. The amount of platinum consumed is only 10% of that consumed by the most advanced catalysts under the same conditions and has high stability, as shown in Fig. 8E, making it the current catalyst with the highest recorded efficiency.98 Gas permeable metal–organic framework membranes can alter the electronic structure and catalytic characteristics of metal single atoms to promote diffusion, activation, and reduction of gas molecules (e.g., carbon dioxide) and generate liquid fuels beneath visible illumination in the mild circumstances. Hao et al. found that porous metal–organic framework membranes embellished with metal single atoms could facilitate the photoreduction of CO2 and O2 at the high flux gas–solid interface. Using Ir SAs as active centres, defective engineered MOFs (e.g., activated NH2-UiO-66) particles may convert CO2 to HCOOH with an apparent quantum efficiency (AQE) of 2.51% at the gas–liquid–solid interface of the reaction at 420 nm. The gas-permeable SA/MOFs membrane could directly convert moist CO2 gas to HCOOH with high HCOOH selectivity due to the promotion of gas diffusion at the porous gas–solid interface, with a significant increase in AQE of 15.76% at 420 nm.202 By adding structural flaws to the MOFs framework, the interatomic distance among metal sites can be extended band the aggregation of metals can be inhibited, thus increasing the yield of SACs.
By modulating the MOFs structure and defects, scientists have made photocatalysts based on MOFs materials more qualified contenders for the efficient fabrication of single-atom photocatalysts. However, the stability, cost and many uncertainties of MOFs-based materials are still a concern when put into industrial production.
Conclusions
In the paper, we succinctly make a summary about the research and promotion of new photocatalysts for CO2 reduction from four aspects: the mechanism of the photocatalytic CO2 reduction process, the preparation of single-atom catalysts, advantages and disadvantages of single-atom catalysts and their applications which show good promise in photocatalysis. At this stage, research on photocatalytic CO2 conversion is still in its infancy, and its solar energy conversion efficiency has certain limitations, and still faces many challenges in research and application.(1) Since single-atom catalysts have a low density of unsaturated active centres, researchers have mostly used highly loaded metal single-atom catalysts to raise the density of photocatalytic electron pumps and photocatalytic active centres. However, most metal single-atom catalysts have a large surface free energy, such that they are prone to agglomeration thus affecting catalytic activity and efficiency. Therefore, researchers have further modified the supporting semiconductor materials and anchored the isolated sites to reduce the agglomeration phenomenon to a certain extent, but it still needs to find more efficient, economical and workable methods to improve the stability of single-atom photocatalysts.
(2) The selectivity and activity of CO2 reduction remain to be enhanced. Single-atom photocatalysts for CO2 reduction remain not widely used due to their low stability and catalytic activity.
(3) The lack of in-depth comprehension of the synthesis and catalytic mechanism of single-atom catalysts, and lacking an intuitive way to characterize them, has hindered the study of the synthesis process-structure–property relationship of single atom-catalysts.
Given the aforementioned difficulties, in purpose to promote the maturation of single atom photocatalytic CO2 reduction technology and its widespread use in production, we must methodically analyse the findings of previous studies and investigate the principle of photocatalytic processes to fill theoretical gaps and promote the creation of effective and affordable novel photocatalytic materials, which can actively promote the cross-fertilization of photocatalysis with other fields and explore the application and development of efficient and stable photocatalyst in more fields. As technology and science advance and the deep understanding of the “carbon peaking and carbon neutrality” theory grows, photocatalytic CO2 reduction technology will move towards a more mature and broader field.
Author contributions
Miss Wanyu Hu: writing – original draft and conceptualization. Prof. Haiyue Yang: supervision and writing – review & editing. Prof. Chengyu Wang: resources and supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This project was supported by China Postdoctoral Science Foundation (grant no. 2021M700736) and Postdoctoral Foundation of Heilongjiang (grant no. LBH-Z21089).Notes and references
- S. Hernandez-Aldave and E. Andreoli, Catal. Sci. Technol., 2022, 12, 3412–3420 RSC.
- M. I. Alam, R. Cheula, G. Moroni, L. Nardi and M. Maestri, Catal. Sci. Technol., 2021, 11, 6601–6629 RSC.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proceedings of the National Academy of Sciences, 2006, 103, 15729–15735 CrossRef CAS PubMed.
- W. Dessie, X. Luo, M. Wang, L. Feng, Y. Liao, Z. Wang, Z. Yong and Z. Qin, Appl. Microbiol. Biotechnol., 2020, 104, 4757–4770 CrossRef CAS PubMed.
- L. Jiang, Q. Yang, Z. Xia, X. Yu, M. Zhao, Q. Shi and Q. Yu, RSC Adv., 2023, 13, 5833–5850 RSC.
- H. Li, P. H. Opgenorth, D. G. Wernick, S. Rogers, T.-Y. Wu, W. Higashide, P. Malati, Y.-X. Huo, K. M. Cho and J. C. Liao, Science, 2012, 335, 1596 CrossRef CAS PubMed.
- S. Chen, K. Li, H. Liu, J. Zhang and T. Peng, Catal. Sci. Technol., 2022, 12, 1637–1650 RSC.
- M. Irshad, Q. tul Ain, M. Zaman, M. Z. Aslam, N. Kousar, M. Asim, M. Rafique, K. Siraj, A. N. Tabish and M. Usman, RSC Adv., 2022, 12, 7009–7039 RSC.
- Z.-H. Xue, D. Luan, H. Zhang and X. W. D. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- Y. Lu, Z. Zhang, H. Wang and Y. Wang, Appl. Catal., B, 2021, 292, 120162 CrossRef CAS.
- S. Wang, G. Li and C. Fang, Renewable Sustainable Energy Rev., 2018, 81, 2144–2159 CrossRef.
- S. Davis, D. Nugegoda, J. Tropp, J. D. Azoulay and J. H. Delcamp, MRS Commun., 2020, 10, 252–258 CrossRef CAS.
- X. Guan, S. S. Mao and S. Shen, ChemNanoMat, 2021, 7, 873–880 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- H. Pan and M. D. Heagy, Nanomaterials, 2020, 10, 2422 CrossRef CAS PubMed.
- M. Zhang, X. Wang, X. Qi, H. Guo, L. Liu, Q. Zhao and W. Cui, J. Catal., 2022, 413, 31–47 CrossRef CAS.
- S. Wang and X. Wang, Appl. Catal., B, 2015, 162, 494–500 CrossRef CAS.
- K. Niu, Y. Xu, H. Wang, R. Ye, H. L. Xin, F. Lin, C. Tian, Y. Lum, K. C. Bustillo, M. M. Doeff, M. T. M. Koper, J. Ager, R. Xu and H. Zheng, Sci. Adv., 2017, 3, e1700921 CrossRef PubMed.
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- R. Wang, J. Shen, K. Sun, H. Tang and Q. Liu, Appl. Surf. Sci., 2019, 493, 1142–1149 CrossRef CAS.
- K. Jiang, L. Zhu, Z. Wang, K. Liu, H. Li, J. Hu, H. Pan, J. Fu, N. Zhang, X. Qiu and M. Liu, Appl. Surf. Sci., 2020, 508, 145173 CrossRef CAS.
- Y. Wang, H. Huang, Z. Zhang, C. Wang, Y. Yang, Q. Li and D. Xu, Appl. Catal., B, 2021, 282, 119570 CrossRef CAS.
- W. Li, D.-K. Ma, X. Hu, F. Gou, X. Yang, W. MacSwain, C. Qi and W. Zheng, J. Catal., 2022, 415, 77–86 CrossRef CAS.
- M. R. U. D. Biswas, A. Ali, K. Y. Cho and W.-C. Oh, Ultrason. Sonochem., 2018, 42, 738–746 CrossRef PubMed.
- A. Lais, M. A. Gondal, M. A. Dastageer and F. F. Al-Adel, Int. J. Energy Res., 2018, 42, 2031–2049 CrossRef CAS.
- S. Navarro-Jaen, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef CAS PubMed.
- Y. Zhang, L. Zheng, J. Jia, K. Li, T. Zhang and H. Yu, Colloids Surf., A, 2022, 639 Search PubMed.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- Q. Zhang and J. Guan, Sol. RRL, 2020, 4, 2000283 CrossRef CAS.
- Q. Wang, D. Zhang, Y. Chen, W.-F. Fu and X.-J. Lv, ACS Sustainable Chem. Eng., 2019, 7, 6430–6443 CrossRef CAS.
- S. Liang, C. Hao and Y. Shi, Chemcatchem, 2015, 7, 2559–2567 CrossRef CAS.
- Y. Xia, M. Sayed, L. Zhang, B. Cheng and J. Yu, Chem Catal., 2021, 1, 1173–1214 CrossRef CAS.
- S. Liang, C. Hao and Y. Shi, ChemCatChem, 2015, 7, 2559–2567 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang, H. J. Zhao, Y. Wang and H. G. Yang, Chem. - Eur. J., 2014, 20, 2138–2144 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14308–14312 Search PubMed.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312 CrossRef CAS PubMed.
- D. Yang, H. Yu, T. He, S. Zuo, X. Liu, H. Yang, B. Ni, H. Li, L. Gu, D. Wang and X. Wang, Nat. Commun., 2019, 10, 3844 CrossRef PubMed.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- C. Gao, J. Wang, H. Xu and Y. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- C. Peng, G. Reid, H. Wang and P. Hu, J. Chem. Phys., 2017, 147, 030901 CrossRef PubMed.
- A. Folli, J. Z. Bloh and D. E. Macphee, J. Electroanal. Chem., 2016, 780, 367–372 CrossRef CAS.
- S. S. Tan, L. Zou and E. Hu, Catal. Today, 2006, 115, 269–273 CrossRef CAS.
- M.-Q. Yang, M. Gao, M. Hong and G. W. Ho, Adv. Mater., 2018, 30, 1802894 CrossRef PubMed.
- Y. Chen, Y. Liu, F. Wang, X. Guan and L. Guo, J. Energy Chem., 2021, 61, 469–488 CrossRef CAS.
- Y. Zhang, B. Xia, J. Ran, K. Davey and S. Z. Qiao, Adv. Energy Mater., 2020, 10, 1903879 CrossRef CAS.
- Y. Xia and J. Yu, Chem, 2020, 6, 1039–1040 CAS.
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- C. B. Hiragond, N. S. Powar, J. Lee and S.-I. In, Small, 2022, 18, 2201428 CrossRef CAS PubMed.
- N. Zheng and T. Zhang, Natl. Sci. Rev., 2018, 5, 625 CrossRef.
- K. Qi, M. Chhowalla and D. Voiry, Mater. Today, 2020, 40, 173–192 CrossRef CAS.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- Q. Xu, J. Zhang, D. Wang and Y. Li, Chin. Chem. Lett., 2021, 32, 3771–3781 CrossRef CAS.
- D. V. R. Kumar, K. Woo and J. Moon, Nanoscale, 2015, 7, 17195–17210 RSC.
- P. Chen, W. Zhang, Y. Sun and F. Dong, Environ. Funct. Mater., 2022, 1, 127–138 Search PubMed.
- P. Biasi, S. Sterchele, F. Bizzotto, M. Manzoli, S. Lindholm, P. Ek, J. Bobacka, J.-P. Mikkola and T. Salmi, Catal. Today, 2015, 246, 207–215 CrossRef CAS.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.-C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- H. Xu, N. Jiang, D. Wang, L. Wang, Y. Song, Z. Chen, J. Ma and T. Zhang, Appl. Catal., B, 2020, 263, 118350 CrossRef CAS.
- J. Xing, J. F. Chen, Y. H. Li, W. T. Yuan, Y. Zhou, L. R. Zheng, H. F. Wang, P. Hu, Y. Wang and H. J. Zhao, Chem. – Eur. J., 2014, 20, 2138–2144 CrossRef CAS PubMed.
- Y. Lu, C. Zhang, X. Li, A. R. Frojd, W. Xing, A. Z. Clayborne and W. Chen, Nano Energy, 2018, 50, 316–322 CrossRef CAS.
- R. Lang, W. Xi, J.-C. Liu, Y.-T. Cui, T. Li, A. F. Lee, F. Chen, Y. Chen, L. Li and L. Li, Nat. Commun., 2019, 10, 234 CrossRef PubMed.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- J. Ding, M. Fan, Q. Zhong and A. G. Russell, Appl. Catal., B, 2018, 232, 348–354 CrossRef CAS.
- J. A. Schwarz, C. Contescu and A. Contescu, Chem. Rev., 1995, 95, 477–510 CrossRef CAS.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- M. Leskela and M. Ritala, Angew. Chem., Int. Ed., 2003, 42, 5548–5554 CrossRef CAS PubMed.
- X. Ye, H. Wang, Y. Lin, X. Liu, L. Cao, J. Gu and J. Lu, Nano Res., 2019, 12, 1401–1409 CrossRef CAS.
- T. J. Gorey, B. Zandkarimi, G. Li, E. T. Baxter, A. N. Alexandrova and S. L. Anderson, J. Phys. Chem. C, 2019, 123, 16194–16209 CrossRef CAS.
- L. Zhang, M. R. Ball, K. R. Rivera-Dones, S.-c. Wang, T. F. Kuech, G. W. Huber, I. Hermans and J. A. Dumesic, ACS Catal., 2020, 10, 365–374 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- J. D. Kistler, N. Chotigkrai, P. Xu, B. Enderle, P. Praserthdam, C. Y. Chen, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed., 2014, 53, 8904–8907 CrossRef CAS PubMed.
- X.-K. Gu, B. Qiao, C.-Q. Huang, W.-C. Ding, K. Sun, E. Zhan, T. Zhang, J. Liu and W.-X. Li, ACS Catal., 2014, 4, 3886–3890 CrossRef CAS.
- L. Yang, L. Shi, D. Wang, Y. Lv and D. Cao, Nano Energy, 2018, 50, 691–698 CrossRef CAS.
- J. Kim, C. W. Roh, S. K. Sahoo, S. Yang, J. Bae, J. W. Han and H. Lee, Adv. Energy Mater., 2018, 8, 1701476 CrossRef.
- X. Zeng, J. Shui, X. Liu, Q. Liu, Y. Li, J. Shang, L. Zheng and R. Yu, Adv. Energy Mater., 2018, 8, 1701345 CrossRef.
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan, J. Zhu, S. Chen, W. Zhang, S. Wei and J. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed.
- Z. Song, Y.-N. Zhu, H. Liu, M. N. Banis, L. Zhang, J. Li, K. Doyle-Davis, R. Li, T.-K. Sham, L. Yang, A. Young, G. A. Botton, L.-M. Liu and X. Sun, Small, 2020, 16, 2003096 CrossRef CAS PubMed.
- M. J. Huelsey, B. Zhang, Z. Ma, H. Asakura, D. A. Do, W. Chen, T. Tanaka, P. Zhang, Z. Wu and N. Yan, Nat. Commun., 2019, 10, 1330 CrossRef PubMed.
- A. Jan, J. Shin, J. Ahn, S. Yang, K. J. Yoon, J.-W. Son, H. Kim, J.-H. Lee and H.-I. Ji, RSC Adv., 2019, 9, 27002–27012 RSC.
- Z. Jiang, X. Feng, J. Deng, C. He, M. Douthwaite, Y. Yu, J. Liu, Z. Hao and Z. Zhao, Adv. Funct. Mater., 2019, 29, 1902041 CrossRef.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- A. V. Rassolov, I. S. Mashkovsky, G. O. Bragina, G. N. Baeva, P. V. Markov, N. S. Smirnova, J. Wärnå, A. Y. Stakheev and D. Y. Murzin, Mol. Catal., 2021, 506, 111550 CrossRef CAS.
- Y. Su, K. Fu, Y. Zheng, N. Ji, C. Song, D. Ma, X. Lu, R. Han and Q. Liu, Appl. Catal., B, 2021, 288, 119980 CrossRef CAS.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. T. Cui, Y. Tan, B. Qiao, L. Li and A. Wang, Angew. Chem., Int. Ed., 2016, 55, 16054–16058 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty and D. Su, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- Y. Cheng, S. Zhao, H. Li, S. He, J.-P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan and M. F. Chisholm, Appl. Catal., B, 2019, 243, 294–303 CrossRef CAS.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li and T. Sun, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- T. Gan, Y. Liu, Q. He, H. Zhang, X. He and H. Ji, ACS Sustainable Chem. Eng., 2020, 8, 8692–8699 CrossRef CAS.
- J. Hu, W. Ma, Y. Pan, Z. Cheng, S. Yu, J. Gao, Z. Zhang, C. Wan and C. Qiu, Chemosphere, 2021, 276, 130170 CrossRef CAS PubMed.
- S. Guo, Y. Zhao, C. Wang, H. Jiang and G. J. Cheng, ACS Appl. Mater. Interfaces, 2020, 12, 26068–26075 CrossRef CAS PubMed.
- Z. Huang, T. Ban, Y. Zhang, L. Wang, S. Guo, C.-R. Chang and G. Jing, Appl. Catal., B, 2021, 283, 119625 CrossRef CAS.
- S.-G. Han, D.-D. Ma, S.-H. Zhou, K. Zhang, W.-B. Wei, Y. Du, X.-T. Wu, Q. Xu, R. Zou and Q.-L. Zhu, Appl. Catal., B, 2021, 283, 119591 CrossRef CAS.
- M. Xiao, L. Zhang, B. Luo, M. Lyu, Z. Wang, H. Huang, S. Wang, A. Du and L. Wang, Angew. Chem., 2020, 132, 7297–7301 CrossRef.
- Y. Cao, H. Peng, S. Chu, Y. Tang, C. Huang, Z. Wang, F. Liu, J. Wu, B. Shan and R. Chen, Chem. Eng. J., 2021, 420, 129713 CrossRef CAS.
- J. Shan, J. Liu, M. Li, S. Lustig, S. Lee and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2018, 226, 534–543 CrossRef CAS.
- W.-H. Lai, Z. Miao, Y.-X. Wang, J.-Z. Wang and S.-L. Chou, Adv. Energy Mater., 2019, 9, 2002473 Search PubMed.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- E. J. Jang, J. Lee, D. G. Oh and J. H. Kwak, ACS Catal., 2021, 11, 5894–5905 CrossRef CAS.
- U. Kerketta, A. B. Tesler and P. Schmuki, Catalysts, 2022, 12, 1223 CrossRef CAS.
- D. Liu, Q. He, S. Ding and L. Song, Adv. Energy Mater., 2020, 10, 2001482 CrossRef CAS.
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746–2762 RSC.
- L. Lin, T. Hisatomi, S. Chen, T. Takata and K. Domen, Trends Chem., 2020, 2, 813–824 CrossRef CAS.
- Y. Liu, Z. Liu, D. Huang, M. Cheng, G. Zeng, C. Lai, C. Zhang, C. Zhou, W. Wang, D. Jiang, H. Wang and B. Shao, Coord. Chem. Rev., 2019, 388, 63–78 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- J. Liu, H. Wu, F. Li, X. Feng, P. Zhang and L. Gao, Adv. Sustainable Syst., 2020, 4, 2000151 CrossRef CAS.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- T. Cui, L. Li, C. Ye, X. Li, C. Liu, S. Zhu, W. Chen and D. Wang, Adv. Funct. Mater., 2022, 32, 2108381 CrossRef CAS.
- S. G. Han, D. D. Ma and Q. L. Zhu, Small Methods, 2021, 5, 2100102 CrossRef CAS PubMed.
- H. He, H. H. Wang, J. Liu, X. Liu, W. Li and Y. Wang, Molecules, 2021, 26, 6501 CrossRef CAS PubMed.
- Y. Zeng, E. Almatrafi, W. Xia, B. Song, W. Xiong, M. Cheng, Z. Wang, Y. Liang, G. Zeng and C. Zhou, Coord. Chem. Rev., 2023, 475, 214874 CrossRef CAS.
- R. Zheng, Z. Liu, Y. Wang, Z. Xie and M. He, Joule, 2022, 6, 1148–1159 CrossRef CAS.
- T. Wang, X. Tao, X. Li, K. Zhang, S. Liu and B. Li, Small, 2021, 17, 2006255 CrossRef CAS PubMed.
- L. Chen, R. R. Unocic, A. S. Hoffman, J. Hong, A. H. Braga, Z. Bao, S. R. Bare and J. Szanyi, JACS Au, 2021, 1, 977–986 CrossRef CAS PubMed.
- B. Wang, H. Cai and S. Shen, Small Methods, 2019, 3, 1800447 CrossRef.
- S. Tu, Y. Guo, Y. Zhang, C. Hu, T. Zhang, T. Ma and H. Huang, Adv. Funct. Mater., 2020, 30, 2005158 CrossRef CAS.
- Q. Tay, P. Kanhere, C. F. Ng, S. Chen, S. Chakraborty, A. C. H. Huan, T. C. Sum, R. Ahuja and Z. Chen, Chem. Mater., 2015, 27, 4930–4933 CrossRef CAS.
- L. Zeng, C. Dai, B. Liu and C. Xue, J. Mater. Chem. A, 2019, 7, 24217–24221 RSC.
- T. Wang, F. Sun, S. Liu, G. Zhuang and B. Li, Appl. Catal., B, 2023, 325, 122339 CrossRef CAS.
- N. Fu, X. Liang, Z. Li and Y. Li, Phys. Chem. Chem. Phys., 2022, 24, 17417–17438 RSC.
- W. Qu, C. Chen, Z. Tang, H. Wen, L. Hu, D. Xia, S. Tian, H. Zhao, C. He and D. Shu, Coord. Chem. Rev., 2023, 474, 214855 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2019, 363, 1419–1423 Search PubMed.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS PubMed.
- Z. Wang, J. Yang, J. Cao, W. Chen, G. Wang, F. Liao, X. Zhou, F. Zhou, R. Li and Z.-Q. Yu, ACS Nano, 2020, 14, 6164–6172 CrossRef CAS PubMed.
- Y. Pan, Y. Qian, X. Zheng, S.-Q. Chu, Y. Yang, C. Ding, X. Wang, S.-H. Yu and H.-L. Jiang, Natl. Sci. Rev., 2021, 8, nwaa224 CrossRef CAS PubMed.
- J. Liu, ACS Catal., 2017, 7, 34–59 CrossRef CAS.
- T. Wang, L. Chen, C. Chen, M. Huang, Y. Huang, S. Liu and B. Li, ACS Nano, 2022, 16, 2306–2318 CrossRef CAS PubMed.
- D. Zhang, Y. Li, Y. Li and S. Zhan, SmartMat, 2022, 3, 417–446 CrossRef CAS.
- L. Wang, L. Huang, F. Liang, S. Liu, Y. Wang and H. Zhang, Chin. J. Catal., 2017, 38, 1528–1539 CrossRef CAS.
- H. Liu, M. Cheng, Y. Liu, J. Wang, G. Zhang, L. Li, L. Du, G. Wang, S. Yang and X. Wang, Energy Environ. Sci., 2022, 15, 3722–3749 RSC.
- G. Wang, Y. Wu, Z. Li, Z. Lou, Q. Chen, Y. Li, D. Wang and J. Mao, Angew. Chem., Int. Ed., 2023, 62, e202218460 CAS.
- F. Doherty, H. Wang, M. Yang and B. R. Goldsmith, Catal. Sci. Technol., 2020, 10, 5772–5791 RSC.
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef.
- Y. Chen, Z. Huang, Z. Ma, J. Chen and X. Tang, Catal. Sci. Technol., 2017, 7, 4250–4258 RSC.
- Z. Gao, Y. Meng, A. Koso, J. Mishima, B. Xie, Z. Ni and S. Xia, Colloids Surf., A, 2022, 648, 129365 CrossRef CAS.
- Q. Wang, X. Y. Kong, Y. Wang, L. Wang, Y. Huang, H. Li, T. Ma and L. Ye, ChemSusChem, 2022, e202201514 CAS.
- P. Liu, Y. Zhao, R. Qin, L. Gu, P. Zhang, G. Fu and N. Zheng, Sci. Bull., 2018, 63, 675–682 CrossRef CAS PubMed.
- H. Li, H.-x. Zhang, X.-l. Yan, B.-s. Xu and J.-j. Guo, New Carbon Mater., 2018, 33, 1–11 CrossRef CAS.
- J. Liu, ChemCatChem, 2011, 3, 934–948 CrossRef CAS.
- T. A. Shifa and A. Vomiero, Adv. Energy Mater., 2019, 9, 1970158 CrossRef CAS.
- L. Zhang, L. Xue, B. Lin, Q. Zhao, S. Wan, Y. Wang, H. Jia and H. Xiong, ChemSusChem, 2022, 15, e202102494 CAS.
- Y. Yao, Y. Hu, H. Hu, L. Chen, M. Yu, M. Gao and S. Wang, J. Colloid Interface Sci., 2019, 554, 376–387 CrossRef CAS PubMed.
- X. Liu, W. Chen and W. Wang, ACS Omega, 2021, 6, 35799–35809 CrossRef CAS PubMed.
- J. Su, R. Ge, Y. Dong, F. Hao and L. Chen, J. Mater. Chem. A, 2018, 6, 14025–14042 RSC.
- J. Gu, Y. Peng, T. Zhou, J. Ma, H. Pang and Y. Yamauchi, Nano Res. Energy, 2022, 1, e9120009 CrossRef.
- X. Cui, W. Li, P. Ryabchuk, K. Junge and M. Beller, Nat. Catal., 2018, 1, 385–397 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, L. Gu, P. Zhang, G. Fu and N. Zheng, Sci. Bull., 2018, 63, 675–682 CrossRef CAS PubMed.
- Y. Z. Wang, M. Yang, Y.-M. Ding, N.-W. Li and L. Yu, Adv. Funct. Mater., 2022, 32, 2108681 CrossRef CAS.
- J. Kim, H. E. Kim and H. Lee, ChemSusChem, 2018, 11, 104–113 CrossRef CAS PubMed.
- Q. Li, Q. Shao, Q. Wu, Q. Duan, Y. Li and H.-g. Wang, Catal. Sci. Technol., 2018, 8, 3572–3579 RSC.
- P. Li, H. Wang, X. Tan, W. Hu, M. Huang, J. Shi, J. Chen, S. Liu, Z. Shi and Z. Li, Appl. Catal., B, 2022, 316, 121674 CrossRef CAS.
- J. Zhang, B. Guan, X. Wu, Y. Chen, J. Guo, Z. Ma, S. Bao, X. Jiang, L. Chen and K. Shu, Catal. Sci. Technol., 2023, 13, 1932–1975 RSC.
- Z. Shi, W. Yang, Y. Gu, T. Liao and Z. Sun, Adv. Sci., 2020, 7, 2001069 CrossRef CAS.
- L. Zhang, R. Long, Y. Zhang, D. Duan, Y. Xiong, Y. Zhang and Y. Bi, Angew. Chem., Int. Ed., 2020, 59, 6224–6229 CrossRef CAS PubMed.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- M.-L. Hu, V. Safarifard, E. Doustkhah, S. Rostamnia, A. Morsali, N. Nouruzi, S. Beheshti and K. Akhbari, Microporous Mesoporous Mater., 2018, 256, 111–127 CrossRef CAS.
- L. Hu, W. Li, L. Wang and B. Wang, EnergyChem, 2021, 3, 100056 CrossRef CAS.
- Z. Xiong, Z. Lei, B. Gong, X. Chen, Y. Zhao, J. Zhang, C. Zheng and J. C. Wu, Catal. Commun., 2017, 89, 4–8 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- H. S. Zakria, M. H. D. Othman, R. Kamaludin, S. H. S. A. Kadir, T. A. Kurniawan and A. Jilani, RSC Adv., 2021, 11, 6985–7014 RSC.
- Y. Li and C.-Y. Wu, Environ. Sci. Technol., 2006, 40, 6444–6448 CrossRef CAS PubMed.
- M. A. Hossen, H. Solayman, K. H. Leong, L. C. Sim, Y. Nurashikin, A. Abd Aziz, L. Wu and M. U. Monir, Results Eng., 2022, 100795 CrossRef CAS.
- H. Pan, X. Wang, Z. Xiong, M. Sun, M. Murugananthan and Y. Zhang, Environ. Res., 2021, 198, 111176 CrossRef CAS PubMed.
- Y. Yu, X. a. Dong, P. Chen, Q. Geng, H. Wang, J. Li, Y. Zhou and F. Dong, ACS Nano, 2021, 15, 14453–14464 CrossRef CAS PubMed.
- H. Yin, F. Dong, D. Wang and J. Li, ACS Catal., 2022, 12, 14096–14105 CrossRef CAS.
- B.-H. Lee, E. Gong, M. Kim, S. Park, H. R. Kim, J. Lee, E. Jung, C. W. Lee, J. Bok and Y. Jung, Energy Environ. Sci., 2022, 15, 601–609 RSC.
- Y. Feng, C. Wang, P. Cui, C. Li, B. Zhang, L. Gan, S. Zhang, X. Zhang, X. Zhou and Z. Sun, Adv. Mater., 2022, 34, 2109074 CrossRef CAS PubMed.
- C. Chen, T. Wang, K. Yan, S. Liu, Y. Zhao and B. Li, Inorg. Chem. Front., 2022, 9, 4753–4767 RSC.
- D. Li, Y. Zhao, Y. Miao, C. Zhou, L.-P. Zhang, L.-Z. Wu and T. Zhang, Adv. Mater., 2022, 34, 2207793 CrossRef CAS PubMed.
- Q. Liu, Z.-X. Low, L. Li, A. Razmjou, K. Wang, J. Yao and H. Wang, J. Mater. Chem. A, 2013, 1, 11563–11569 RSC.
- S. Yan, Z. Li and Z. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- P. P. Singh and V. Srivastava, RSC Adv., 2022, 12, 18245–18265 RSC.
- Y. Qin, G. Dong, L. Zhang, G. Li and T. An, Environ. Res., 2021, 195, 110880 CrossRef CAS PubMed.
- S. Liu, F. Chen, S. Li, X. Peng and Y. Xiong, Appl. Catal., B, 2017, 211, 1–10 CrossRef CAS.
- X. Cheng, J. Wang, K. Zhao and Y. Bi, Appl. Catal., B, 2022, 316, 121643 CrossRef CAS.
- Z. Zhao, W. Liu, Y. Shi, H. Zhang, X. Song, W. Shang and C. Hao, Phys. Chem. Chem. Phys., 2021, 23, 4690–4699 RSC.
- L. Cheng, H. Yin, C. Cai, J. Fan and Q. Xiang, Small, 2020, 16, 2002411 CrossRef CAS PubMed.
- P. Sharma, S. Kumar, O. Tomanec, M. Petr, J. Zhu Chen, J. T. Miller, R. S. Varma, M. B. Gawande and R. Zbořil, Small, 2021, 17, 2006478 CrossRef CAS PubMed.
- J.-H. Zhang, W. Yang, M. Zhang, H.-J. Wang, R. Si, D.-C. Zhong and T.-B. Lu, Nano Energy, 2021, 80, 105542 CrossRef CAS.
- P. Chen, B. Lei, X. a. Dong, H. Wang, J. Sheng, W. Cui, J. Li, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 15841–15852 CrossRef CAS PubMed.
- L. Cheng, X. Yue, L. Wang, D. Zhang, P. Zhang, J. Fan and Q. Xiang, Adv. Mater., 2021, 33, 2105135 CrossRef CAS PubMed.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., 2022, 134, e202206579 CrossRef.
- X. Zeng, C. Xiao, L. Liao, Z. Tu, Z. Lai, K. Xiong and Y. Wen, Nanomaterials, 2022, 12, 4049 CrossRef CAS PubMed.
- B. Smit and T. L. Maesen, Nature, 2008, 451, 671–678 CrossRef CAS PubMed.
- S.-S. Wang, H.-H. Huang, M. Liu, S. Yao, S. Guo, J.-W. Wang, Z.-M. Zhang and T.-B. Lu, Inorg. Chem., 2020, 59, 6301–6307 CrossRef CAS PubMed.
- X. Song, Q. Guan, Z. Cheng and W. Li, Appl. Catal., B, 2018, 227, 13–23 CrossRef CAS.
- M. Usman, A. Helal, M. M. Abdelnaby, A. M. Alloush, M. Zeama and Z. H. Yamani, Chem. Rec., 2021, 21, 1771–1791 CrossRef CAS PubMed.
- M. Liu, Y.-F. Mu, S. Yao, S. Guo, X.-W. Guo, Z.-M. Zhang and T.-B. Lu, Appl. Catal., B, 2019, 245, 496–501 CrossRef CAS.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed.
- G. Wang, C.-T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- M. Chu, Y. Li, X. Chen, G. Hou, Y. Zhou, H. Kang, W. Qin and X. Wu, J. Mater. Chem. A, 2022, 10, 23666–23674 RSC.
- Y.-C. Hao, L.-W. Chen, J. Li, Y. Guo, X. Su, M. Shu, Q. Zhang, W.-Y. Gao, S. Li, Z.-L. Yu, L. Gu, X. Feng, A.-X. Yin, R. Si, Y.-W. Zhang, B. Wang and C.-H. Yan, Nat. Commun., 2021, 12, 2682 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |