DOI:
10.1039/D3RA03862A
(Review Article)
RSC Adv., 2023,
13, 21890-21925
Recent advances in the synthesis of new benzothiazole based anti-tubercular compounds
Received
9th June 2023
, Accepted 7th July 2023
First published on 21st July 2023
Abstract
This review highlights the recent synthetic developments of benzothiazole based anti-tubercular compounds and their in vitro and in vivo activity. The inhibitory concentrations of the newly synthesized molecules were compared with the standard reference drugs. The better inhibition potency was found in new benzothiazole derivatives against M. tuberculosis. Synthesis of benzothiazole derivatives was achieved through various synthetic pathways including diazo-coupling, Knoevenagel condensation, Biginelli reaction, molecular hybridization techniques, microwave irradiation, one-pot multicomponent reactions etc. Other than recent synthetic developments, mechanism of resistance of anti-TB drugs is also incorporated in this review. Structure activity relationships of the new benzothiazole derivatives along with the molecular docking studies of selected compounds have been discussed against the target DprE1 in search of a potent inhibitor with enhanced anti-tubercular activity.
 Rakhi Yadav | Rakhi Yadav completed her MSc in 2020 from Kurukshetra University, Kurukshetra, Haryana, India. She joined Glycochemistry Laboratory of School of Physical Sciences, Jawaharlal Nehru University, New Delhi, as a research scholar in 2022. She is currently pursuing her PhD degree under the supervision of Prof. Ram Sagar. Her expertise lies in heterocyclic molecules, medicinal chemistry, organic synthesis, and the development of new methods for the natural product inspired bioactive glycohybrids. |
 Dilkhush Meena | Dilkhush Meena completed his BSc in 2021 from Kirorimal college, University of Delhi, New Delhi-110007, India. Since then, he has been pursuing his MSc from School of Physical Sciences, Jawaharlal Nehru University, New Delhi. He is working under the supervision of Prof. Ram Sagar for his MSc Research project. He is interested in Organic Synthesis, the development of carbohydrate derived materials, natural product-inspired hybrid analogues and molecular modeling, especially in protein-ligand interaction via in silico docking tools. |
 Kavita Singh | Kavita Singh has been completed her MSc from Deen Dayal Upadhyaya University, Gorakhpur, UP, India in 2019. She qualified CSIR-JRF then joined Glycochemistry Laboratory of School of Physical Sciences, Jawaharlal Nehru University, New Delhi, as a junior research fellow in 2021. She is currently persuing her PhD degree under the supervision of Prof. Ram Sagar. Her work is mainly focused on development of new methods for the synthesis of carbohydrate fused heterocyclic molecules as bioactive glycohybrids. She is also interested in medicinal chemistry and synthesis of natural product inspired bioactive scaffolds. |
 Rajdeep Tyagi | Rajdeep Tyagi has completed his MSc in 2018 from Kirorimal college, University of Delhi, New Delhi-110007, India. He joined Glycochemistry laboratory of School of Physical Sciences, Jawaharlal Nehru University, New Delhi, as a UGC junior research fellow in 2020. He is currently pursuing his PhD degree under the supervision of Prof. Ram Sagar. His expertise lies in heterocyclic molecules, medicinal chemistry, organic synthesis, and the synthesis of indole based bioactive glycohybrids. He is also interested in developing new methods for glycoconjugate synthesis and their bioapplications. |
 Yogesh Yadav | Yogesh Yadav has completed his MSc in 2021 from Kurukshetra University, Kurukshetra, Haryana, India. He joined Glycochemistry Laboratory of School of Physical Sciences, Jawaharlal Nehru University, New Delhi, as a CSIR Junior Research Fellow in 2022. He is currently pursuing his PhD degree under the supervision of Prof. Ram Sagar. His work is mainly focused on development of new methodology in organic synthesis, the synthesis of carbohydrate fused-linked heterocyclic molecules as bioactive molecules. He is also interested in synthesis of natural product inspired bioactive scaffolds as antiviral agents. |
 Ram Sagar | Prof. Ram Sagar received his MSc degree from University of Lucknow, Lucknow, UP, India. Prof. Sagar has completed his PhD degree in Organic Chemistry from Central Drug Research Institute (CDRI) Lucknow and University of Agra in 2006. After his PhD, he pursued his Research Associate with Prof. Y. D. Vankar at IIT Kanpur during 2006–2007. Then he pursued his first post-doctoral research at Seoul National University South Korea with Prof. Seung Bum Park during 2007–2008. He moved to University of Oxford in 2008 and worked with Prof. Benjamin G. Davis as BBSRC postdoctoral fellow until August 2012. He returned to India in August 2012 and held a faculty position at Shiv Nadar University (SNU), Greater Noida. He moved to Department of Chemistry, Banaras Hindu University (BHU) as Associate Professor in February 2018 and worked there till December 2020. He subsequently got full professor at Jawaharlal Nehru University (JNU), New Delhi in December 2020 and presently working as Professor of Chemistry in School of Physical Sciences. His current research interests include devising newer methods for the efficient synthesis of natural product inspired small molecules, glycohybrids and glycopeptides implicated in various diseases including tuberculosis and cancer. His interested also lies the preparation of carbohydrate based materials. |
Introduction
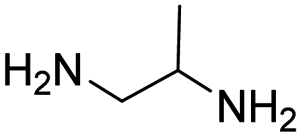
Tuberculosis (TB) is one of the most precarious and contagious infectious illnesses in the world caused by Mycobaterium tuberculosis, Mtb.1,2 Moreover, the rapid growth of drug resistant bacteria has contributed to a rise in incidence of both extensively drug resistant (XDR) and multidrug resistant (MDR) tuberculosis.3 Under this situation, only the recently developed Delamanid, Pretomanid,4 Bedaquiline5 and Fluoroquinolone antibiotics5 have proven to be effective novel pharmaceuticals with distinct modes of action to treat TB infection. This highlights the inherent challenges of creating and evaluating novel chemical agents by medicinal chemists, as well as the constraints brought on by a deficit of drug discovery research in the pharmaceutical sector.6 New drug development is the main objective of medicinal chemistry, which operates at the interface between synthetic organic chemistry and molecular biology. One of the most common, yet equally significant, sections of organic chemistry is the synthesis and study of heterocyclic chemistry, which has been the subject of extensive research for more than a century.7 Benzothiazole is a heterocyclic compound with benzene nucleus attached to a five membered ring having nitrogen and sulphur atoms placed at 1 and 3 positions.7 Benzothiazole analogues are most ubiquitous and an integral part of many pharmaceutical agents.8,9 Benzothiazole is considered as a fundamental building block in the search of a novel class of drug molecules with diverse pharmacological activities like anti-tubercular,5,10–14 anti-convulsant,15,16 anti-HIV,17 anti-mosquito,13 anti-microbial,16 anti-tumor,18,19 analgesic,20 anti-leishmanial,21 and anti-inflammatory.22,23 Additionally, the logical design and development of novel anti-TB agents incorporating a benzothiazole nucleus can assist in addressing the need for an effective anti-microbial therapy for the treatment tuberculosis.10 Anti-TB drugs are basically divided into two categories, (i) first line drugs, (ii) second line drugs.
First-line medications (FLD), such as Isoniazid (INH), Rifampicin (RIF) and its derivatives, Pyrazinamide (PZA), Ethambutol (EMB), Streptomycin (STM) (Fig. 1) can be used to treat TB infection.24
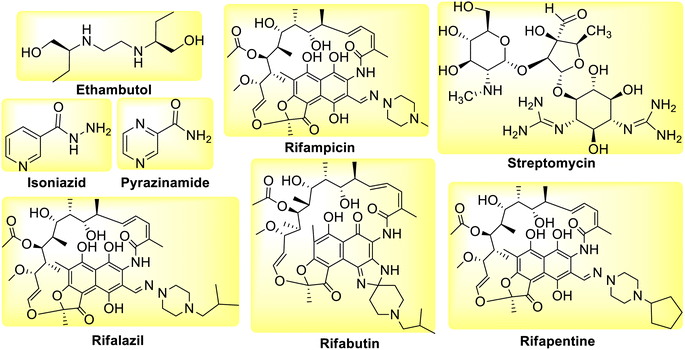 |
| | Fig. 1 Molecular structure of first line anti-tubercular drugs. | |
However, as drug-resistant bacteria proliferate, causing relapse and disease progression, there are numerous cases and fatalities reported due to a decline in the effectiveness of these first-line medications. The combination of these drugs is used to increase patient adherence to the treatment and avoid the emergence of new resistant strains of bacteria that utilize different mechanisms of action. The rise of multidrug resistant tuberculosis (MDR-TB), which is resistant to at least Isoniazid (INH) and Rifampicin (RIF) is extremely concerning since it necessitates the use of second-line medications that are more toxic and expensive as compared to first line anti-tuberculosis drugs (Fig. 2). Whereas XDR-TB refers to resistance to three or more of the six classes of second-line medications.25 Active TB patient shows symptoms and can spread the disease while latent TB patient has no symptoms and cannot spread the disease.26
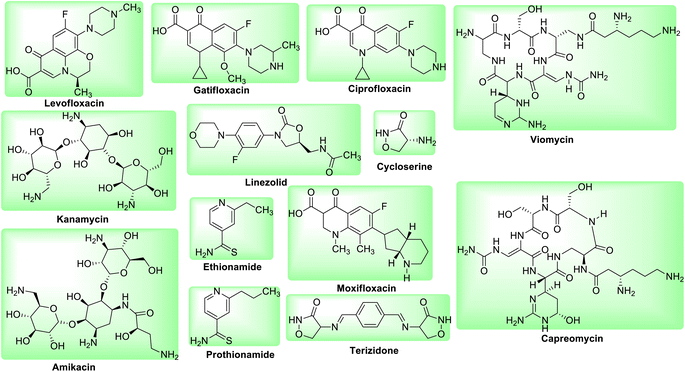 |
| | Fig. 2 Molecular structures of second line anti-tubercular drugs. | |
Among the new class of drugs Delamanid and Pretomanid belongs to nitroimidazole class of antibiotics while Bedaquiline belongs to diarylquinoline class of antibiotics (Fig. 3). These drugs are crucial for the treatment of MDR-TB. Bedaquiline blocks the proton pump for ATP synthase while Delamanid and Pretomanid prevents the production of mycolic acid in cell walls.27,28
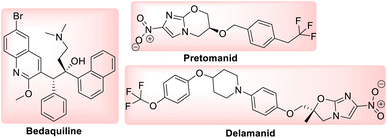 |
| | Fig. 3 Molecular structure of newly approved anti-tubercular drugs against MDR and XDR TB. | |
Drug resistance for TB and mechanism of resistance
Spontaneous change in Mtb strains make them resistant to at least one anti-TB medication. Basically, drug resistance develops due to gene mutations. Thus, exposure to a single anti-TB medicine could slow the expansion of the Mtb population but not totally eradicate it. Like first-line medications, second-line medications are also linked to genetic alterations. Resistance to Rifampicin and its derivatives (rifabutin, rifapentine, and rifalazil) is linked to genetic changes in the rpoB gene, genetic alterations involving the embCAB operon cause Ethambutol resistance, mutations in the rpsl are linked to Streptomycin resistance, mutations in gyrA are linked to resistance to the drugs belonging to group quinolones, while mutations in rrs are linked to Kanamycin and Amikacin resistance (Table 1).26
Table 1 Classification of anti-TB drugs according to their mechanism of resistance and route of intake
| Drugs lines |
Groups |
Drugs |
Mechanism of resistance |
References |
| First line anti-TB drugs |
Group 1 (oral) |
Isoniazid |
Mutations in katG and inhA |
29 |
| Rifampicin/Rifampin |
Mutations in rpoB gene |
30 |
| Pyrazinamide |
Mutations in RpsA, pncA |
31 and 32 |
| Rifapentine |
Mutations in rpoB gene |
33 |
| Rifabutin |
Mutations in rpoB gene |
34 |
| Ethambutol |
embCAB operon |
24 |
| Injectable |
Streptomycin |
Mutations in rpsL |
35 |
| Second-line anti-TB drugs |
Group 2 (injectable) |
Kanamycin |
Mutations in rrs |
36 |
| Amikacin |
Mutations in rrs |
|
| Viomycin |
Mutations in rrs |
|
| Capreomycin |
Mutations in thyA |
|
| Group 3 (oral and injectable) |
Moxifloxacin |
Mutations in gyrA |
37 |
| Levofloxacin |
Mutations in gyrA |
|
| Group 4 (oral) |
Linezolid |
Mutations in G2576T (23S) |
38 |
| Prothionamide |
Mutations in etha |
39 |
| Ethionamide |
Mutations in etha and inhA |
40 |
| Terizidone |
Non |
|
| Cycloserine |
Mutation in alrA |
41 |
| Para-aminosalicylic acid (PAS) |
Mutations in thyA |
41 |
Because of development of MDR and XDR-TB the medicinal chemists are in continuous search of new molecules which can combat drug resistance tuberculosis. Several research groups throughout the globe are working towards this objective utilizing various natural product inspired molecular scaffolds. The benzothiazole is one of such privileged drugs like scaffold. There are several compilations of reports on benzothiazole nucleus and associated various biological activities. But detailed review on the recent synthetic developments of benzothiazole derivatives and their anti-TB activity was of absolute necessity. Keeping this in mind the current review focused on the recent developments towards synthesis of new benzothiazole derivatives and associated anti-TB activity.
Recent synthesis of benzothiazole based anti-tubercular molecules
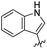
R. Chikhale and co-workers took decaprenylphosphoryl-β-D-ribose 20-epimerase (DprE1) as a possible therapeutic target for the creation of anti-tubercular drugs and synthesized novel derivatives of benzothiazolylpyrimidine-5-carboxamides 7a–g from three component one-pot classical Biginelli reaction between benzothiazolyloxobutanamide 4, substituted aromatic benzaldehydes 5 and thiourea 6 (Scheme 1, Table 2).42 Benzothiazolyloxobutanamide 4 was prepared from 2-aminobenzothiazole 3a in presence of sodium hydroxide and ethylacetate. Compound 3a in turn was prepared from aniline 1 via a two-step reaction involving an intermediate 2.
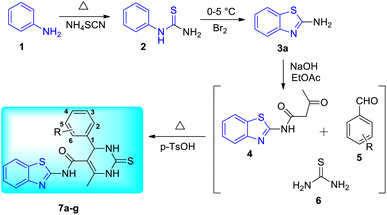 |
| | Scheme 1 Synthesis of benzothiazolylpyrimidine-5-carboxamide analogues. | |
Table 2 Anti-tubercular activity of benzothiazolylpyrimidine-5-carboxamide analoguesa
| Compounds |
R |
IC50 (μM) |
MIC (μM) |
| NT: not tested. |
| 7a |
H |
7.7 ± 0.8 |
0.08 |
| 7b |
2-Cl |
NT |
0.32 |
| 7c |
4-Cl |
NT |
0.32 |
| 7d |
2,4-Di Cl |
NT |
0.25 |
| 7e |
4-F |
9.2 ± 1.5 |
0.09 |
| 7f |
CF3 |
11.1 ± 1.8 |
0.09 |
| 7g |
4-N(Me)2 |
10.3 ± 2.6 |
0.08 |
| INH |
— |
0.2 |
— |
All synthesized compounds were evaluated for their anti-tubercular activity against the pathogenic strain of Mtb H37Rv ATCC 27294. MIC and IC50 values revealed that compounds 7a and 7g had comparative better activity than INH (Table 2). DprE1 selectivity and pharmacokinetics studies of these derivatives were carried out which showed compounds 7a and 7g were highly selective with better bioavailability (>52%) by oral dose. A pharmacophore model of these compounds suggested that, presence of aromatic, aliphatic carbon center and hydrogen bond donor is essential for better anti-tubercular activity and DprE1 inhibition.
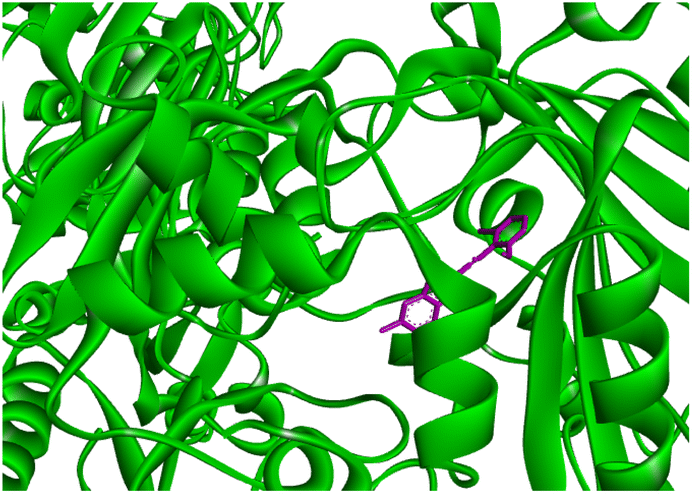
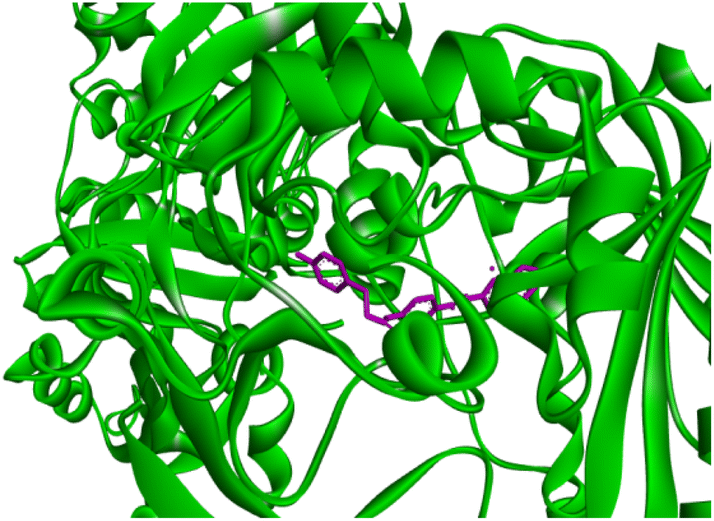
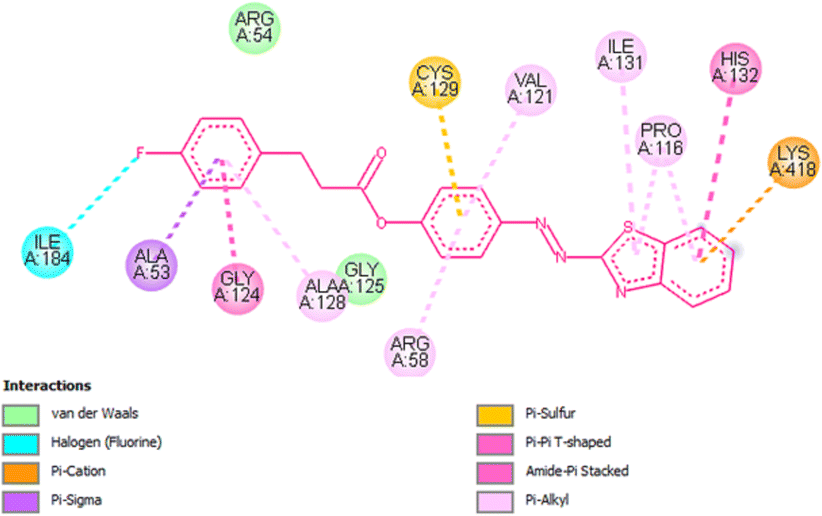
Docking studies of compound 7a against 4FDN protein of potential therapeutic site DprE1 revealed that it displays better binding affinity of −8.4 kcal mol−1 with several amino acids at active site of the protein chain. This finding suggests that, this could be a potential target of 7a and responsible for its anti-tubercular activity (Fig. 4 and 5).
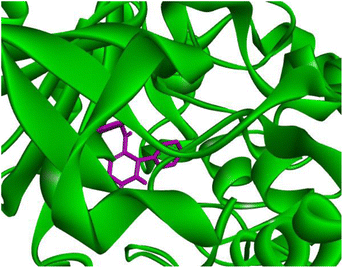 |
| | Fig. 4 3D representation of ligand 7a and its interactions at the active site of 4FDN protein. | |
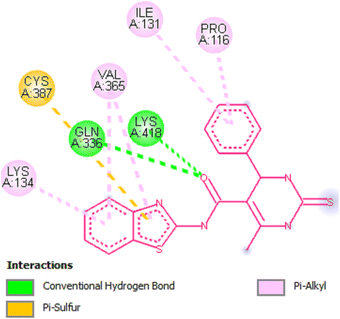 |
| | Fig. 5 2D representation of docking results showing interactions of ligand 7a with 4FDN protein. | |
Shaikh and co-workers synthesized some acetamide linked benzothiazole derivatives through various intermediates. Initial step involved the synthesis of (E)-5-arylidenethiazolidine-2,4-diones 9a–n (Scheme 2) from the Knoevenagel condensation reaction of 1,3-thiazolidine-2,4-dione 8 with various aromatic aldehydes in ethanol solvent in the presence of a piperidine catalyst. Next to this the reaction of aniline 1 with acetic acid in presence of bromine and ammonium thiocyanate lead to the formation of 2-amino-6-thiocyanato benzothiazole 10. The later 10 on further reaction with chloroacetyl chloride produced 2-chloro-N-(6-thiocyanatobenzo[d]thiazol-2-yl)acetamide 11 (Scheme 3). Finally the reaction of (E)-5-arylidenethiazolidine-2,4-diones 9a–n and 2-chloro-N-(6-thiocyanatobenzo[d]thiazol-2-yl)acetamide 11 in presence of anhydrous K2CO3 in DMF (Scheme 4) furnished the desired compounds 12a–n (Scheme 4, Table 3).43
 |
| | Scheme 2 Synthesis of (E)-5-arylidenethiazolidine-2,4-diones. | |
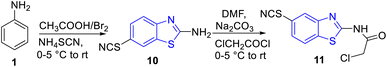 |
| | Scheme 3 Synthesis of 6-thiocyanatobenzo[d]thiazol-2-amine 10 and 2-chloro-N-(6-thiocyantobenzo[d]thiazol-2-yl)acetamide 11. | |
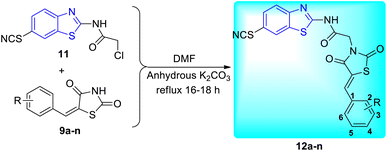 |
| | Scheme 4 Synthesis of 2,4-thiazolidinediones incorporated 2-amino-6-thiocyanato benzothiazole derivatives. | |
Table 3 Anti-tubercular activity of the synthesized compounds
| Compounds |
R |
MIC (μg mL−1) |
Inhibition (%) |
Compounds |
R |
MIC (μg mL−1) |
Inhibition (%) |
| 9a |
H |
250 |
98 |
12a |
H |
100 |
99 |
| 9b |
2-Cl |
500 |
97 |
12b |
2-Cl |
500 |
98 |
| 9c |
4-Cl |
250 |
99 |
12c |
4-Cl |
50 |
99 |
| 9d |
4-F |
100 |
98 |
12d |
4-F |
100 |
99 |
| 9e |
3-Br |
250 |
99 |
12e |
3-Br |
25 |
99 |
| 9f |
4-Me |
200 |
99 |
12f |
4-Me |
1000 |
98 |
| 9g |
4-OMe |
250 |
98 |
12g |
4-OMe |
100 |
99 |
| 9h |
4-N(Me)2 |
1000 |
98 |
12h |
4-N(Me)2 |
62.5 |
99 |
| 9i |
4-OH |
250 |
99 |
12i |
4-OH |
500 |
98 |
| 9j |
3-OMe-4-OH |
100 |
99 |
12j |
3-OMe-4-OH |
500 |
99 |
| 9k |
2-C4H3O |
1000 |
98 |
12k |
2-C4H3O |
500 |
99 |
| 9l |
3-OC6H5 |
200 |
98 |
12l |
3-OC6H5 |
250 |
99 |
| 9m |
3,4,5-Tri-OMe |
50 |
99 |
12m |
3,4,5-Tri-OMe |
1000 |
98 |
| 9n |
4-N[(CH2)5CH3]2 |
500 |
98 |
12n |
4-N[(CH2)5CH3]2 |
250 |
97 |
| 10 |
— |
500 |
99 |
RIF |
— |
40 |
99 |
| 11 |
— |
62.5 |
99 |
|
|
|
|
Biological evaluation of the synthesized compounds showed moderate to good anti-tubercular activity against M. tuberculosis H37RV with reference drug Rifampicin. The L–J agar (MIC) method was used to assess drug susceptibility and the MIC of the test compounds against M. tuberculosis H37Rv (Table 3). The compounds 10, 11 and 12g showed better activity (MIC = 25–50 μg mL−1). All other compounds showed moderate to modest anti-tubercular activity against M. tuberculosis H37RV. MIC values of 62.5–100 μg mL−1 were similar for compounds 9f, 9l, 12b, 12c, 12f, 12i and 12j while the remaining compounds showed minimal to moderate activity (MIC = 200–1000 μg mL−1).
Abdel-Aziz and co-workers synthesized few benzothiazole based halophenyl bis-hydrazones and their sulfone derivatives 15, 16a–b. Bis-hydrazone derivative of benzothiazole 15 was produced by the reaction of benzo[d]thiazole-2-carbohydrazide 13 with 2-oxo-N′-(4-substituted phenyl)propane hydrazonoyl chloride 14 in tetrahydrofuran (THF) under reflux conditions (Scheme 5). Resulting bis-hydrazones 15 on further reaction with sodium benzenesulfinate or sodium 4-methylbenzenesulfinate furnished the corresponding sulfones 16a and 16b respectively (Scheme 6, Table 4).44
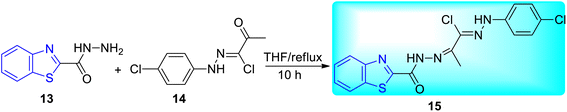 |
| | Scheme 5 Synthesis of benzothiazole based halophenyl bis-hydrazone. | |
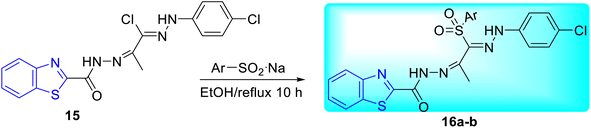 |
| | Scheme 6 Synthesis of sulfone derivative of benzothiazole based halophenyl bis-hydrazone compounds. | |
Table 4 Anti-tubercular activity of halophenyl bis-hydrazone and its sulfone derivativesa
| Compounds |
Ar |
MIC (μg mL−1) |
| NA: not active. |
| 15 |
— |
NA |
| 16a |
Ph |
NA |
| 16b |
4-Me-C6H4 |
125 |
| INH |
— |
0.40 |
| PZA |
— |
3.21 |
These benzothiazole based halophenyl bis-hydrazones derivatives when tested against mycobacterial strain were found to be less active against M. tuberculosis as compared to standard reference drugs Isoniazid and Pyrazinamide (Table 4).
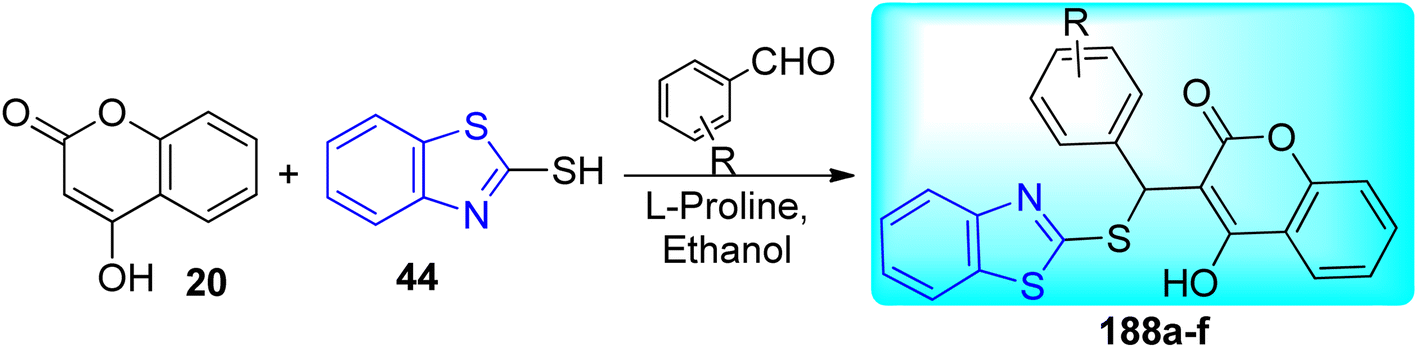
A. B. Patel and co-workers synthesized benzothiazole based derivatives of coumarin substituted quinazolines 22a–j (Scheme 7, Table 5). 2-Aminobenzoic acid 17 was used to create the first analogue, 2,4-dihydroquinazoline 18 which on further reaction with POCl3 in DMA (dimethylacetamide) gave 2,4-dichloroquinazoline 19. The intermediate analogue 21 was formed by the condensation of 4-hydroxycoumarin 20 with 2, 4-dichloroquinazoline 19 in the presence of potassium carbonate base. Intermediate 21 on reaction with various 2-aminobenzothiazole derivatives 3a–f furnished the desired compounds 22a–j in good yields.45
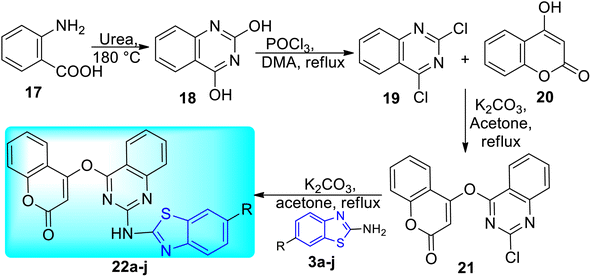 |
| | Scheme 7 Synthesis of benzothiazole based derivatives of coumarin substituted quinazolines. | |
Table 5 Anti-tubercular activity of benzothiazole based derivatives of coumarin substituted quinazolines
| Compounds |
R |
BACTEC MGIT method MIC (μg mL−1) |
L. J. MIC method MIC (μg mL−1) |
| 22a |
H |
>6.25 |
250 |
| 22b |
Cl |
>6.25 |
25 |
| 22c |
Br |
>6.25 |
12.5 |
| 22d |
F |
>6.25 |
3.12 |
| 22e |
NO2 |
>6.25 |
100 |
| 22f |
CN |
>6.25 |
200 |
| 22g |
Me |
>6.25 |
250 |
| 22h |
OMe |
>6.25 |
100 |
| 22i |
OEt |
>6.25 |
50 |
| 22j |
OH |
6.25 |
6.25 |
| EMB |
— |
3.12 |
— |
| PZA |
— |
6.25 |
— |
| RIF |
— |
0.25 |
— |
| INH |
— |
0.20 |
— |
According to the results of in vitro screening against H37Rv strain of M. tuberculosis, all newly synthesized compounds demonstrated moderate to good suppression of M. tuberculosis H37Rv at 3.12–25 μg mL−1 (Table 5). For the first selection of active compounds, the primary screening was carried out using the BACTEC MGIT technique45 at a concentration of 6.25 μg mL−1. Using primary screening 22d and 22j showed the maximum inhibition (99%) of all the investigated drugs. However, analogue 22d with a fluoro group attached to the benzothiazole ring demonstrated the best inhibition against M. tuberculosis H37Rv with MIC value of 3.12 μg mL−1, according to the results of secondary biological screening using the Lowenstein–Jensen MIC method.45
K. Chakraborti and co-workers designed and synthesized some new anti-mycobacterial chemotypes as benzo[d]thiazol-2-yl(piperazin-1-yl)methanones 24a–l, 25a–l and 26a–l (Scheme 8, Table 6) from the molecular hybridization of N-benzyl benzo[d]thiazole-2-carboxamides and alicyclic piperazines (Fig. 6) in solvent free conditions from good to moderate yields. Intermediates 5-substituted benzo[d]thiazole-2-carboxylate 23a–c were formed from the reaction of 2-aminothiophenol 17a–c with ethyl glyoxylate in presence of micellar solution of SDOSS (sodium dioctyl sulfosuccinate). The intermediates 23a–c on further coupling with alicyclic amines produced the diverse library of compounds 24a–l, 25a–l and 26a–l.46
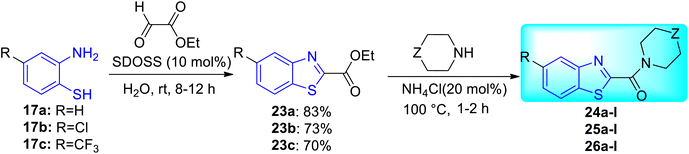 |
| | Scheme 8 Synthesis of benzo[d]thiazole-2-carboxamide analogues. | |
Table 6 Anti-tubercular activity of 5-substituted benzo[d]thiazole-2-carboxylates 23a–c and carboxamides 24/25/26a–l
| Compounds |
R |
Z |
R1 |
Yields (%) |
MIC (μg mL−1) |
Compounds |
R |
Z |
R1 |
Yields (%) |
MIC (μg mL−1) |
| 23a |
H |
— |
— |
83 |
25 |
25h |
Cl |
NR1 |
4-OMe-C6H4 |
54 |
3.125 |
| 23b |
Cl |
— |
— |
73 |
3.125 |
25i |
Cl |
NR1 |
4-COMe-C6H4 |
54 |
1.56 |
| 23c |
CF3 |
— |
— |
70 |
6.25 |
25j |
Cl |
NR1 |
4-Pyridyl |
58 |
12.5 |
| 24a |
H |
O |
— |
88 |
1.56 |
25k |
Cl |
NR1 |
2-Pyrazinyl |
47 |
6.25 |
| 24b |
H |
S |
— |
83 |
25 |
25l |
Cl |
NR1 |
CH(C6H5)2 |
77 |
0.78 |
| 24c |
H |
CH2 |
— |
67 |
25 |
26a |
CF3 |
O |
— |
88 |
0.78 |
| 24d |
H |
NR1 |
Me |
56 |
3.125 |
26b |
CF3 |
S |
— |
92 |
0.78 |
| 24e |
H |
NR1 |
COMe |
55 |
1.56 |
26c |
CF3 |
CH2 |
— |
74 |
12.5 |
| 24f |
H |
NR1 |
C6H5 |
76 |
1.56 |
26d |
CF3 |
NR1 |
Me |
77 |
6.25 |
| 24g |
H |
NR1 |
2-OMe-C6H4 |
55 |
3.125 |
26e |
CF3 |
NR1 |
COMe |
88 |
1.56 |
| 24h |
H |
NR1 |
4-OMe-C6H4 |
53 |
12.5 |
26f |
CF3 |
NR1 |
C6H4 |
72 |
12 |
| 24i |
H |
NR1 |
4-COMe-C6H4 |
51 |
1.56 |
26g |
CF3 |
NR1 |
2-OMe-C6H4 |
77 |
25 |
| 24j |
H |
NR1 |
4-Pyridyl |
54 |
3.125 |
26h |
CF3 |
NR1 |
4-OMe-C6H4 |
88 |
3.125 |
| 24k |
H |
NR1 |
2-Pyrazinyl |
77 |
6.25 |
26i |
CF3 |
NR1 |
4-COMe-C6H4 |
77 |
6.25 |
| 24l |
H |
NR1 |
CH(C6H5)2 |
63 |
0.78 |
26j |
CF3 |
NR1 |
4-Pyridyl |
74 |
3.125 |
| 25a |
Cl |
O |
— |
77 |
6.25 |
26k |
CF3 |
NR1 |
2-Pyrazinyl |
75 |
3.125 |
| 25b |
Cl |
S |
— |
67 |
6.25 |
26l |
CF3 |
NR1 |
CH(C6H5)2 |
74 |
25 |
| 25c |
Cl |
CH2 |
— |
77 |
12.5 |
INH |
— |
— |
— |
— |
0.098 |
| 25d |
Cl |
NR1 |
Me |
60 |
25 |
RIF |
— |
— |
— |
— |
0.19 |
| 25e |
Cl |
NR1 |
COMe |
82 |
12.5 |
EMB |
— |
— |
— |
— |
1.56 |
| 25f |
Cl |
NR1 |
C6H5 |
78 |
6.25 |
PYZ |
— |
— |
— |
— |
6.25 |
| 25g |
Cl |
NR1 |
2-OMe-C6H4 |
57 |
3.125 |
CIP |
— |
— |
— |
— |
1.56 |
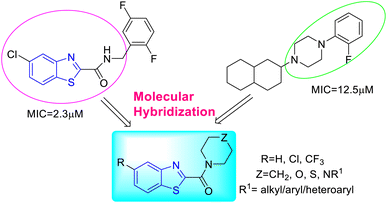 |
| | Fig. 6 Synthesis of benzo[d]thiazol-2-yl (piperazin-1-yl) methanones by the molecular hybridization method. | |
Synthesized benzo[d]thiazole-2-carboxamide derivatives were tested in vitro for their anti-tubercular activity against H37Rv strain of M. tuberculosis (Table 6). From this structurally diverse library, eighteen compounds 24a, 24d–f, 24g, 24i–j, 24l, 25g–i, 25l, 26a–b, 26e, 26h, 26j–k showed MICs value in the range of 0.78–3.125 μg mL−1. The compounds 24l, 25l, 26a, and 26b with MIC value of 0.78 μg mL−1 were found to be more powerful than the standard medicines Ethambutol (1.56 μg mL−1), Ciprofloxacin (1.56 μg mL−1), and Pyrazinamide (6.25 μg mL−1). The compounds 26a and 26b were found to be less cytotoxic against RAW 264.7 cell lines (mouse macrophage cell line) with inhibition of 24.56% and 18.12% having therapeutic index >60. As Mtb grow inside macrophages therefore any new molecule should remain nontoxic to these cells. SAR study revealed that, presence of –CF3 group on 26a and 26b improve their anti-tubercular activity.
Amongst the all-tested compounds the most active compound 26a was choosen for molecular docking studies to find its binding target. It shown better affinity towards 4P8N protein of DprE1 enzyme (Fig. 7 and 8) with a good binding affinity of −8.9 kcal mol−1 and MIC value of 0.78 μg mL−1. This compound 26a may be considered as lead compound in further search of a better ligand to fit within the target site of DprE1 of M. tuberculosis.
 |
| | Fig. 7 3D representation of ligand 26a and its interactions with 4P8N protein. | |
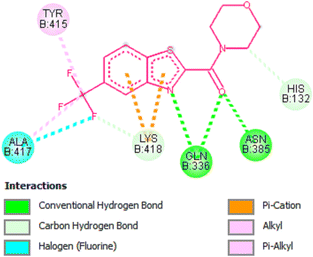 |
| | Fig. 8 2D representation of docking results showing interactions of compound 26a with 4P8N protein. | |
N. Bhoi and co-workers designed 4H-pyrimido [2,1-b] benzothiazole with an isoniazid nucleus 33a–n and its biological profile was investigated (Scheme 9, Table 7). The traditional approach was used to complete the synthesis in the hopes of finding novel analogue leads that could work as an anti-mycobacterial agent. Synthesis of adduct 31a–n involved the dropwise addition of hydrazine hydrate solution in presence of catalytic amount of H2SO4 to the previously synthesized derivatives 30a–n. Further reaction of adduct 31a–n with triethylamine and hydrochloride salt of isonicotinoyl chloride 32 produced the N-isonicotinoyl-2-methyl-4-(pyridin-2-yl)-4H-benzo[4,5]thiazolo[3,2-a]pyrimidine-3-carbohydrazide analogues 33a–n.47,48
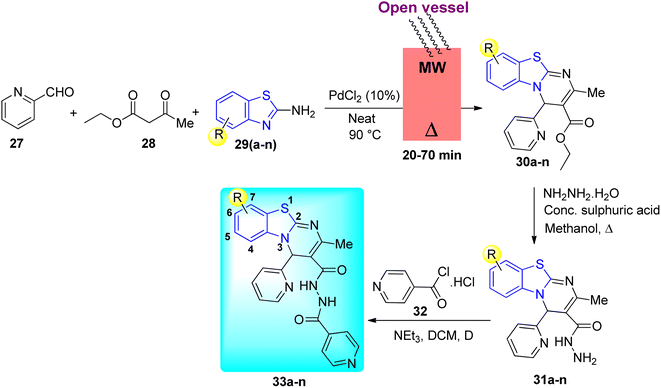 |
| | Scheme 9 Synthesis of isoniazid linked 4H-pyrimido [2,1-b] benzothiazole. | |
Table 7 Anti-mycobacterial activity of isoniazid linked 4H-pyrimido [2,1-b] benzothiazole analogues
| Compounds |
R |
Yields (%) |
Inhibition (%) |
MIC value (μg mL−1) |
| 33a |
H |
71.2 |
69.91 |
125 |
| 33b |
6-Br |
68.4 |
73.17 |
62.5 |
| 33c |
6-Me |
74.1 |
54.47 |
500 |
| 33d |
4-Me |
65.2 |
47.15 |
1000 |
| 33e |
6-NO2 |
69.2 |
56.91 |
250 |
| 33f |
6-Cl |
75.6 |
71.54 |
125 |
| 33g |
4-Cl |
66.1 |
81.30 |
50 |
| 33h |
6-F |
78.5 |
80.48 |
25 |
| 33i |
6-OMe |
79.6 |
49.59 |
500 |
| 33j |
6-OEt |
80.0 |
63.41 |
62.5 |
| 33k |
6-OCF3 |
67.6 |
79.67 |
6.25 |
| 33l |
6-OH |
71.2 |
68.29 |
100 |
| 33m |
4-OMe |
70.2 |
50.40 |
125 |
| 33n |
5,6 di Me |
69.5 |
82.11 |
12.5 |
| INH |
— |
— |
99.18 |
0.20 |
In a standard primary screen, all the newly synthesized compounds 33a–n were evaluated in vitro for their anti-mycobacterial activity against M. tuberculosis H37Rv using a well-known Lowenstein–Jensen (L–J) method. The results of anti-mycobacterial activity indicated that the synthesized compounds displayed diverse tuberculostatic activity (Table 7). Among them, compound 33k was found to be most potent compound with MIC value 6.25 mg mL−1, while compound 33n (MIC 12.5 mg mL−1) showed good anti-mycobacterial activity. Compounds 33b, 33g–h and 33j were found to display good to moderate anti-mycobacterial activity.
Samala and co-workers developed benzo[d]imidazo[2,1-b]thiazole derivatives from previously reported imidazo[1,2-a]pyridine-based pantothenate synthetase (PS) inhibitors for M. tuberculosis (Schemes 10 and 11, Table 8). Synthesis of final desired compounds involved three steps process. Step one was initiated from the reaction between 2-aminobenzothiazole 3a and 2-chloroethylacetoacetate 34 in 1,2-dimethoxyethane at 90 °C to give tricyclic compound 35. Step 2 involved two reaction pathways on ester group. Among these two pathways one was the conversion of ester group to acid hydrazide 36 and another one was the conversion of ester to acid 37. Compound 36 reacted with substituted aromatic carboxylic acids and substituted aldehydes to furnish desired compounds 38a–e and 39a–e respectively while compound 37 reacted with aromatic/aliphatic primary amines in order to furnish desired compounds 40a–e.49,50
 |
| | Scheme 10 Designing approach for the synthesis of new class of benzothiazoles. | |
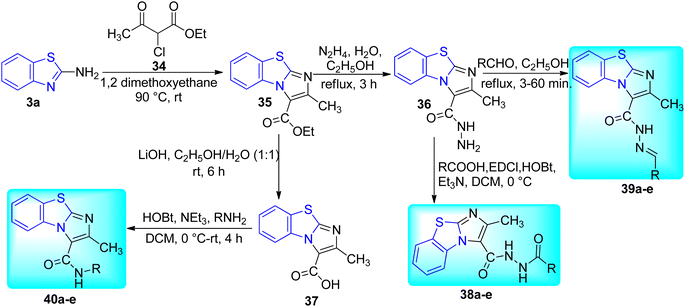 |
| | Scheme 11 Synthesis of benzo[d]imidazo[2,1-b]thiazole derivatives. | |
Table 8 Anti-tubercular activities of benzo[d]imidazo[2,1-b]thiazole derivatives
|
| Compounds |
R |
Yields (%) |
PanC IC50 (μM) |
MIC against Mtb (μM) |
Compounds |
R |
Yields (%) |
PanC IC50 (μM) |
MIC against Mtb (μM) |
| 38a |
Phenyl |
80 |
1.10 ± 0.4 |
35.67 |
39d |
3,4,5-Trimethoxyphenyl |
91 |
2.07 ± 0.20 |
29.45 |
| 38b |
4-Tolyl |
87 |
5.83 ± 0.24 |
17.15 |
39e |
4-N,N-Dimethylphenyl |
82 |
1.46 ± 0.12 |
4.13 |
| 38c |
4-Phenoxyphenyl |
74 |
0.53 ± 0.13 |
3.53 |
40a |
4-Bromophenyl |
63 |
0.52 ± 0.04 |
16.18 |
| 38d |
1-Naphthyl |
69 |
1.39 ± 0.08 |
15.60 |
40b |
Phenyl |
81 |
1.03 ± 0.11 |
40.67 |
| 38e |
Cyclohexyl |
89 |
2.91 ± 0.11 |
17.53 |
40c |
4-Ethoxyphenyl |
83 |
2.10 ± 0.09 |
41.95 |
| 39a |
4-Bromophenyl |
87 |
1.02 ± 0.13 |
15.12 |
40d |
Benzyl |
72 |
0.84 ± 0.1 |
19.44 |
| 39b |
4-Trifluoromethylphenyl |
93 |
5.31 ± 0.11 |
16.53 |
40e |
Cyclohexyl |
81 |
1.02 ± 0.11 |
19.94 |
| 39c |
Phenyl |
90 |
2.15 ± 0.8 |
9.35 |
INH |
— |
— |
>25 |
0.72 |
| |
|
|
|
|
EMB |
— |
— |
>25 |
7.64 |
Synthesized compounds were evaluated in vitro for their anti-TB activity against replicative and non-replicative Mtb (Table 8). All of the synthesized compounds were found to be active against Mtb with MICs ranging from 3.53 to 41.95 μM. Compound 38c with MIC of 3.53 μM emerged as a powerful molecule against Mtb (Table 8).50 The cytotoxicity study of the compound 38c was done against RAW 264.7 cell lines (mouse macrophage cell line) which showed better results with cytotoxicity of 10.4% at 50 μM.
A. Yardily and co-workers synthesized 2-(4-amino-2-aryl/alkyl aminothiazol-5 oyl)benzothiazole derivatives 43a–i from the reaction of amidinothioureas 41a–i and 2-(2-bromoacetyl)benzothiazole 42 in the presence of triethylamine at 35 °C (Scheme 12, Table 9).51
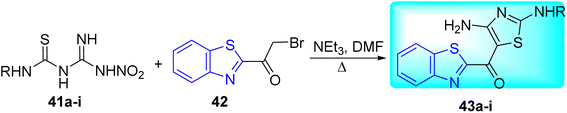 |
| | Scheme 12 Synthesis of 2-(4-amino-2-aryl/alkyl aminothiazol-5-oyl)benzothiazole derivatives. | |
Table 9 Anti-tubercular activity of 2-(4-amino-2-aryl/alkyl aminothiazol-5-oyl)benzothiazole derivativesa
| Compounds |
R |
Zone of inhibition (mm) |
| 0.5 mg |
1 mg |
1.5 mg |
2 mg |
Control |
| NA: not active. |
| 43a |
C6H5 |
NA |
NA |
2 |
3 |
3 |
| 43b |
4-ClC6H4 |
NA |
1 |
2 |
4 |
3 |
| 43c |
4-MeOC6H4 |
NA |
NA |
NA |
2 |
3 |
| 43d |
4-EtOC6H4 |
3 |
5 |
6 |
8 |
3 |
| 43e |
4-MeC6H4 |
NA |
3 |
4 |
4 |
2 |
| 43f |
C2H5 |
3 |
6 |
7 |
8 |
3 |
| 43g |
N–C3H7 |
NA |
NA |
NA |
NA |
NA |
| 43h |
N–C4H9 |
NA |
NA |
2 |
2 |
4 |
| 43i |
Allyl |
2 |
3 |
3 |
5 |
2 |
All the synthesized compounds were evaluated for their anti-tubercular activity. Compounds 43d, 43f, and 43i demonstrated the highest activity against M. tuberculosis when compared to control penicillin (Table 9).51
V. M. Patel and co-workers aimed to create powerful anti-mycobacterial molecules based on thiazolidine-4-one motif through Knoevenagel condensation via conventional heating as well as microwave irradiation as a green protocol (Scheme 13, Table 10). 4-(Benzo[d]thiazol-2-ylthio) aniline 45 was synthesized from the reaction of mercaptobenzothiazole 44 with 4-iodoaniline in the presence of CuI and TBAB (tetrabutylammonium bromide). Compound 45 on further reaction with pyridine-4-carbaldehyde in presence of glacial acetic acid formed (E)-N-(4-(benzo[d]thiazol-2-ylthio)phenyl)-1-(pyridin-4-yl)methanimine 46. Compound 46 on reaction with thioglycolic acid in presence of ZnCl2 gave 47, which on further reaction with substituted benzaldehydes in presence of piperidine and acetic acid furnished the target compounds 48a–j (Scheme 13).52
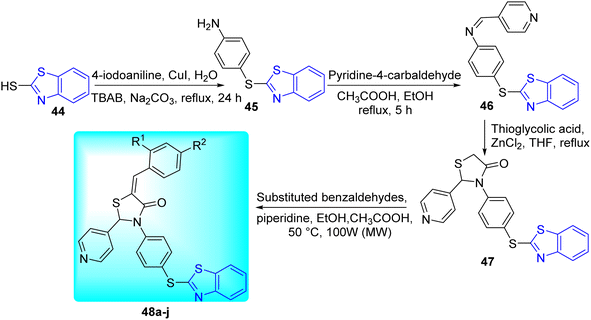 |
| | Scheme 13 Synthesis of series of (Z)-3-(4-(benzo[d]thiazol-2-ylthio) phenyl)-5-benzylidene-2-(pyridine-4-yl)thiazolidine-4-one. | |
Table 10 Anti-tubercular activity of thiazolidine-4-one substituted benzothiazoles
| Compounds |
R1 |
R2 |
% inhibition |
MIC values (μM) |
Compounds |
R1 |
R2 |
% inhibition |
MIC values (μM) |
| 47 |
— |
— |
74 |
>100 |
48g |
H |
Cl |
99 |
<50 |
| 48a |
H |
H |
69 |
>100 |
48h |
H |
Br |
10 |
>100 |
| 48b |
H |
OH |
71 |
>100 |
48i |
Pyridine-2-carbaldehyde |
100 |
<50 |
| 48c |
Me |
OMe |
13 |
>100 |
48j |
Pyridine-4-carbaldehyde |
99 |
<50 |
| 48d |
H |
NO2 |
95 |
>100 |
INH |
— |
99 |
0.25 |
| 48e |
H |
F |
98 |
<50 |
RIF |
— |
99 |
40 |
|
| 48f |
F |
H |
100 |
<50 |
|
|
|
|
|
In vitro anti-tubercular activity of the synthesized benzothiazole derivatives 47, 48a–j was assessed by using MABA approach against H37Rv strain of M. tuberculosis taking Isoniazid and Rifampicin as the standard reference drugs (Table 10).
S. S. Jawoor and co-workers created the ligand 52 by the dropwise addition of 2-hydrazinobenzothiazole 51a in ethanol to a solution of 8-formyl-7-hydroxy-4-methylcoumarin 50 in ethanol (Scheme 14, Table 11). Later on novel Co(II), Ni(II), and Cu(II) complexes of the Schiff base 53–55 (Fig. 9) were synthesized by the reaction of an ethanolic solution of the ligand 52 with CoCl2·6H2O/NiCl2·6H2O/CuCl2·2H2O under reflux conditions in search of potent anti-tubercular molecules.53
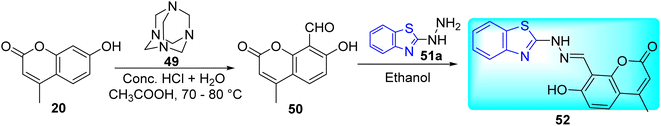 |
| | Scheme 14 Synthesis of benzothiazole based Schiff base. | |
Table 11 Anti-tubercular activity of Schiff base and the formed metal complexes
| Compounds |
MIC (μg mL−1) |
| 52 |
1.6 |
| 53 |
0.8 |
| 54 |
1.6 |
| 55 |
0.8 |
| STM |
6.25 |
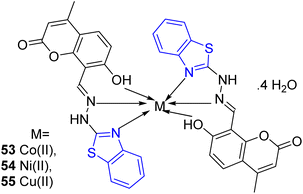 |
| | Fig. 9 Structure of metal complexes. | |
Synthesized metal complexes 53–55 along with ligand 52 were evaluated for their anti-tubercular activity against M. tuberculosis using Microplate Alamar Blue Assay (MABA) technique while taking Streptomycin (STM) as the reference drug. The MIC results showed that the metal complexes had higher anti-tubercular activity than that of the free ligand (Table 11).
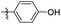
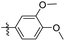
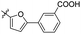
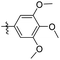
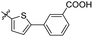
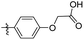
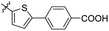
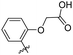
Reshma and co-workers synthesized some benzothiazole derivatives from a pre-existing lead to create a potent molecule against Mtb LAT, a critical enzyme for controlling the amino acid pool, which is essential for antibiotic resistance and persistence. It serves as potential target in management of latent tuberculosis. The initial step in the synthetic process involved the construction of the benzothiazole ring 57 by condensation of the 2-amino thiophenol 17a with malononitrile 56 in the presence of catalytic amounts of acetic acid in ethanol. Synthesis of final products 59a–v was achieved by Knoevenagel condensation reaction between 2-(benzo[d]thiazol-2-yl)acetonitrile 57 and aryl/heteroaryl aldehydes 58a–v (Scheme 15, Table 12).54
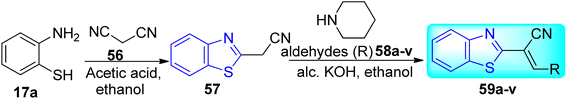 |
| | Scheme 15 Synthesis of acrylonitrile derivatives of benzothiazole. | |
Table 12 Anti-mycobacterial activity of acrylonitrile derivatives of benzothiazole
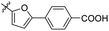
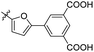
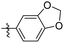
| Compounds |
R |
MIC (μM) |
LAT IC50 (μM) |
Compounds |
R |
MIC (μM) |
LAT IC50 (μM) |
| 59a |
 |
>89.93 |
10.38 ± 1.21 |
59m |
 |
>99.60 |
17.05 ± 1.21 |
| 59b |
 |
89.29 |
4.11 ± 0.78 |
59n |
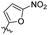 |
>84.18 |
19.59 ± 0.32 |
| 59c |
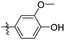 |
20.29 |
7.83 ± 0.31 |
59o |
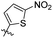 |
>79.87 |
54.76 ± 0.21 |
| 59d |
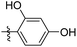 |
10.61 |
23.19 ± 0.89 |
59p |
 |
>83.06 |
37.91 ± 0.48 |
| 59e |
 |
>77.64 |
61.41 ± 1.56 |
59q |
 |
>67.20 |
3.74 ± 0.27 |
| 59f |
 |
>71.02 |
64.89 ± 2.31 |
59r |
 |
>67.20 |
14.06 ± 0.16 |
| 59g |
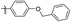 |
67.95 |
47.93 ± 1.82 |
59s |
 |
60.09 |
1.15 ± 0.27 |
| 59h |
 |
81.69 |
65.98 ± 0.63 |
59t |
 |
64.43 |
5.73 ± 0.79 |
| 59i |
 |
4.64 |
3.08 ± 0.37 |
59u |
 |
2.01 |
6.72 ± 0.27 |
| 59j |
 |
2.32 |
53.78 ± 0.96 |
59v |
 |
>57.60 |
2.62 ± 0.37 |
| 59k |
 |
49.60 |
16.23 ± 0.26 |
INH |
— |
0.4 |
— |
| 59l |
 |
>93.28 |
92.57 ± 1.94 |
RIF |
— |
0.5 |
— |
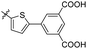
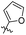
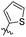
The MABA approach was used to screen all substances for their effectiveness against the replicative stage of Mtb. Compound 59u was found to be most potent with a MIC value of 2.01 μM. Compounds 59d, 59i, 59j also demonstrated good activity with MIC values of 10.61, 4.64 and 2.32 μM respectively (Table 12).54 Molecular docking of these active compounds with LAT from Mtb revealed that, these molecules binds to the hydrophobic pocket having Leu414, Val63 and Phe167.
A. C. Pinheiro and co-workers synthesized 2-arylidene-benzylidene hydrazinyl benzothiazole derivatives 61a–i from the reaction between 2-hydrazinobenzothiazole 51a, and substituted benzaldehydes 60 in refluxing methanol from moderate to good yields and investigated their anti-mycobacterial activity (Schemes 16 and 17, Table 13).55
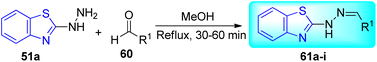 |
| | Scheme 16 Synthesis of 2-arylidene-benzylidene hydrazinyl benzothiazole derivatives. | |
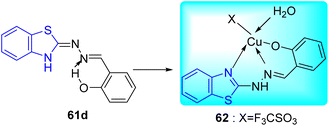 |
| | Scheme 17 Synthesis of metal complex. | |
Table 13 Anti-tubercular activity of 2-arylidene-benzylidene hydrazinyl benzothiazole derivatives and metal complex
| Compounds |
R1/X |
MIC (μM) |
| 61a |
Ph |
>100 |
| 61b |
2-ClC6H4 |
>100 |
| 61c |
2-NO2C6H4 |
10.5 |
| 61d |
2-OHC6H4 |
11.6 |
| 61e |
4-OMeC6H4 |
8.8 |
| 61f |
2-OH-4-OMeC6H3 |
167 |
| 61g |
2-OH-5-NO2C6H3 |
>100 |
| 61h |
Pyridin-2-yl |
4.9 |
| 62 |
2-OH-5-MeC6H3 |
12.4 |
| EMB |
— |
15.3 |
| INH |
— |
0.46 |
The most potent anti-mycobacterial compounds were 61c (aryl = 2-O2NC6H4), 61d (aryl = 2-HOC6H4), 61e and 61h and all these compounds showed greater anti-mycobacterial activities as compared to standard drug Ethambutol. Based on the MIC values of the ligand and its complex, which ranged from 4.9 to 12.4 μM for the M. tuberculosis H37Rv strain, complex 62 was found to be less active than that of ligand 61d. The diminished potency of the complex can be explained by the fact that less of the active ligand is available for activity against M. tuberculosis ATTC 27294 due to strong complexation by Cu(II) (Table 13).
T. M. Dhamelia and co-workers synthesized benzo[d]thiazole-2-carbanilides 66a–d, 67a–c, 68a–e (Scheme 18, Table 14) from CDI mediated direct reaction between benzo[d]thiazole-2-carboxylic acids 64a–c and aromatic amines 1a–l via three step synthetic pathway which involved green protocol for the synthesis of ethylbenzo[d]thiazole-2-carboxylates 63a–c, which were the precursors of desired carboxylic acids 64a–c.56
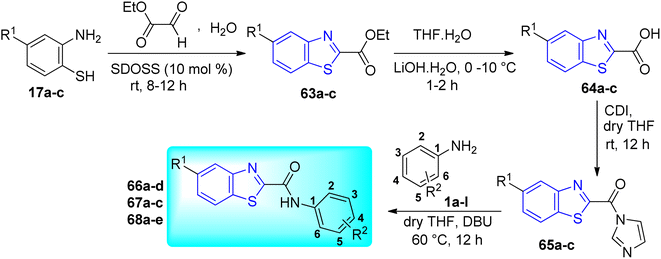 |
| | Scheme 18 Synthesis of N-arylbenzothiazole-2-carbanilides. | |
Table 14 Anti-mycobacterial activity of N-arylbenzothiazole-2-carbanilides
| Compounds |
R1 |
R2 |
MIC (μg mL−1) |
| 63a |
–H |
— |
25 |
| 63b |
–Cl |
— |
3.125 |
| 63c |
–CF3 |
— |
6.25 |
| 66a |
–H |
3-Cl |
0.78 |
| 66b |
–H |
4-CF3 |
0.78 |
| 66c |
–H |
3-NO2 |
0.78 |
| 66d |
–H |
3,4,5-Tri-OMe |
0.78 |
| 67a |
–Cl |
3-OMe |
0.78 |
| 67b |
–Cl |
4-Cl |
0.76 |
| 67c |
–Cl |
4-Morpholinyl |
0.78 |
| 68a |
–CF3 |
4-OMe |
0.78 |
| 68b |
–CF3 |
4-Cl |
0.78 |
| 68c |
–CF3 |
2-CF3 |
0.78 |
| 68d |
–CF3 |
4-NO2 |
0.78 |
| 68e |
–CF3 |
3,4,5-Tri-OMe |
0.78 |
| INH |
— |
— |
0.098 |
| RIF |
— |
— |
0.197 |
| EMB |
— |
— |
1.56 |
The anti-tubercular efficacy of the synthesized compounds was assessed in vitro against M. tuberculosis H37Rv (ATCC 27294 strain). With a therapeutic index of 64, the most potent molecules 66a–d, 67a–c, 68a–e were found to have MICs of 0.78 μg mL−1 (Table 14). Molecular docking of these active compounds suggested that, they bind to the catalytic site of enzyme ATP phosphoribosyl transferase and this binding might be responsible for their anti-tubercular activity.
Matada and co-workers synthesized new dispersion azo dye ligand and its bioactive Cu(II), Co(II), Ni(II), and Fe(III) complexes 72a–d (Fig. 10). Synthesis of azo dye ligand 71 was achieved via diazo-coupling reaction between 5,5,7-trimethyl-4,5,6,7-tetrahydro-1,3-benzothiazol-2-amine 69 and 2-thioxodihydropyrimidine-4,6(1H,5H)-dione 70 at 0–10 °C (Scheme 19, Table 15).57
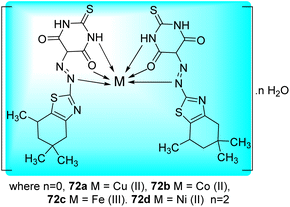 |
| | Fig. 10 Metal complexes of azo-dye ligand. | |
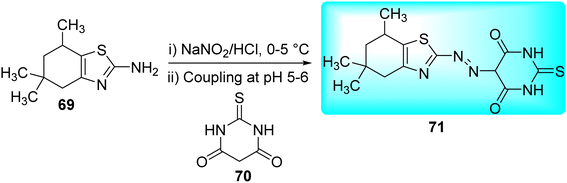 |
| | Scheme 19 Synthesis of Azo dye ligand. | |
Table 15 Anti-tubercular activity of azo-dye ligand and metal complexes at variable concentrationsa
| Ligands/complexes |
100 μg mL−1 |
50 μg mL−1 |
25 μg mL−1 |
12.5 μg mL−1 |
6.25 μg mL−1 |
3.12 μg mL−1 |
1.6 μg mL−1 |
0.8 μg mL−1 |
| S: sensitive, R: resistance. |
| 71 L |
S |
S |
S |
S |
S |
R |
R |
R |
| 72a [Cu(L)2] |
S |
S |
S |
S |
S |
S |
R |
R |
| 72b [Co(L)2] |
S |
S |
S |
S |
S |
S |
R |
R |
| 72c [Fe(L)2] |
S |
S |
S |
S |
S |
S |
S |
R |
| 72d [Ni(L)2(H2O)2] |
S |
S |
S |
S |
S |
R |
R |
R |
The Microplate Alamar Blue Assay (MABA) was used to investigate the anti-tubercular activity of the azo dye ligand (L) and its metal chelates against M. tuberculosis (H37Rv strain, ATCC 27294) (Table 15). The newly synthesized azo-dye showed tridentate behavior, and when it interacted with the different metal ions, it formed a six-membered chelate ring with octahedral geometry, apart from the Cu(II) complex, which had distorted octahedral geometrical environment (Fig. 10).
Bhat and co-workers synthesized 1-phenyl-2-(1-phenylethylidene) hydrazines 75a–i from the reaction of phenyl hydrazines 74 and different acetophenones 73a–i. Then 75a–i reacted with POCl3 under reflux conditions to give pyrazole-conjugated benzothiazoleanalogues 76a–i which further reacted with 2-hydrazinyl benzothiazole 51a and benzothiazole-2-carbohydrazide 77 to furnish the desired compounds 78a–i and 79a–i respectively (Scheme 20, Table 16).58
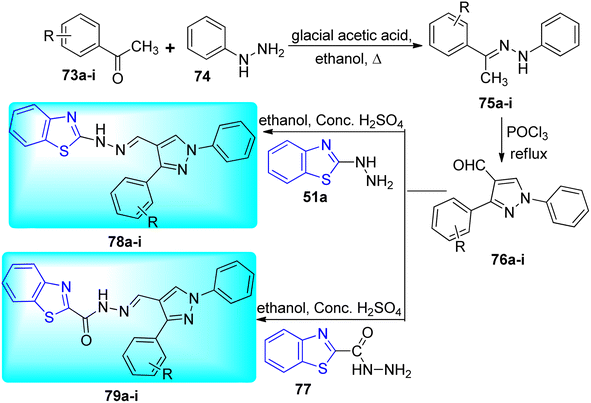 |
| | Scheme 20 Synthesis of pyrazole conjugated benzothiazole derivatives. | |
Table 16 Anti-tubercular activity of pyrazole conjugated benzothiazole derivatives
| Compounds |
R |
MIC (μg mL−1) |
Compounds |
R |
MIC (μg mL−1) |
| 78a |
H |
12.5 |
79a |
H |
25 |
| 78b |
p-OCH3 |
6.25 |
79b |
p-OCH3 |
25 |
| 78c |
p-OH |
6.25 |
79c |
p-OH |
25 |
| 78d |
p-CH3 |
1.6 |
79d |
p-CH3 |
25 |
| 78e |
p-Cl |
1.6 |
79e |
p-Cl |
100 |
| 78f |
p-Br |
6.25 |
79f |
p-Br |
50 |
| 78g |
p-NO2 |
6.25 |
79g |
p-NO2 |
100 |
| 78h |
m-NO2 |
50 |
79h |
m-NO2 |
50 |
| 78i |
p-N(CH3)2 |
25 |
79i |
p-N(CH3)2 |
50 |
| PYZ |
— |
3.125 |
CIP |
— |
3.125 |
In vitro screening was done for the anti-tubercular activity of the synthesized compounds 78a–i and 79a–i using Microplate Alamar Blue Assay (MABA) technique. Compared to benzothiazole carbohydrazide derivatives, which had MIC values of 100 to 25 μg mL−1, benzothiazole hydrazine compounds displayed greater activity (MIC values 25 to 1.6 μg mL−1) (Table 16). Molecular docking of most active compounds 78d and 78e were in accordance with anti-tubercular activity with docking score of −7.68 and −8.12 kcal mol−1 and these molecules were non-toxic in cytotoxicity assay.
Krause and co-workers synthesized some benzothiazole derivatives 82–84 (Scheme 21, Table 17) from the reaction of Methanesulfonic acid (MSA) and the appropriate carboxylic acid at 140 °C for 72 hours with 2-amino-4-chlorothiophenol or 2-amino-4-trifluoromethylthiophenol and the silica gels.59
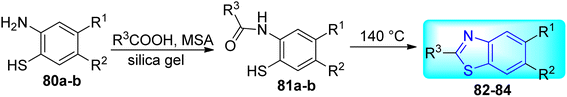 |
| | Scheme 21 Synthesis of substituted benzothiazoles. | |
Table 17 Anti-tubercular activity of substituted benzothiazoles
| Compounds |
R1 |
R2 |
R3 |
MIC (μg mL−1) H37Rv Spec. 210 |
| 82 |
Cl |
H |
–(CH2)3-Cy |
100 |
100 |
| 83 |
Cl |
H |
–(CH2)3-Cy |
100 |
100 |
| 84 |
CF3 |
H |
–(CH2)3-Cy |
100 |
100 |
| PZA |
— |
— |
— |
25 |
>400 |
| INH |
— |
— |
— |
0.125 |
12.5 |
| RIF |
— |
— |
— |
1.2 |
2.5 |
The synthesized compounds 82–84 were evaluated for their anti-tubercular activity against H37Rv strain of Mtb and a wild strain Spec. 210 extracted from tuberculosis patients. Rifampicin, Pyrazinamide and Isoniazid were used as standard reference drugs. All these benzothiazole analogues were found to possess moderate anti tubercular activity (Table 17).
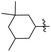
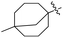
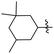
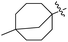
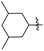
J. Graham and co-workers identified numerous hits with moderate activity from the screening of available libraries against M. tuberculosis and developed numerous benzothiazoleamide anti-tubercular agents 86a–j after extensive medicinal chemistry optimization. Under amide coupling conditions, utilizing 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) in the presence of N,N-diisopropylethylamine (DIEA) in dichloroethane (DCE), the synthesis began with substituted 2-amino-benzothiazole 3 intermediates and variously substituted cycloalkyl carboxylic acids 85a–j (Scheme 22, Table 18).60
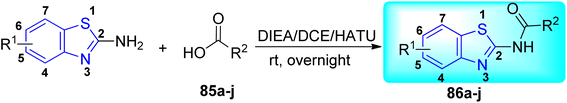 |
| | Scheme 22 Synthesis of benzothiazole amide derivatives. | |
Table 18 Structure activity relationship of cyclohexane derivatives towards M. tuberculosis H37Rv
| Compounds |
R1 |
R2 |
MIC (μg mL−1) |
Compounds |
R1 |
R2 |
MIC (μg mL−1) |
| 86a |
5-CF3 |
 |
≤0.12 |
86f |
5,7-Di-F |
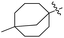 |
0.25 |
| 86b |
5,7-Di-Me |
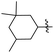 |
0.25 |
86g |
OCF3 |
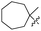 |
0.5 |
| 86c |
5,7-Di-F |
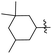 |
0.25 |
86h |
5-Br |
 |
≤0.12 |
| 86d |
CF3 |
 |
2 |
86i |
5,7-Di-Cl |
 |
≤0.12 |
| 86e |
CF3 |
 |
4 |
86j |
4,5,6-Tri-F |
 |
≤0.12 |
Anti-tubercular activity of the synthesized compounds 86a–j was evaluated by introducing differently substituted cyclohexane and bycyclo derivatives to the benzothiazole moiety. In order to predict the structure activity relationship with respect to cyclohexane derivatives, their MIC values were compared (Table 18). The preliminary mechanism of action studies revealed that these molecules targeting MmpL3, a mycobacterial mycolic acid transporter. These compounds were having better in vivo efficacy.
Deng and co-workers reported the novel selective triple-cleavage of bromodifluoroacetamides 87 by S8 for the first time. Using a cascade protocol, they synthesized 2-amido substituted benzothiazoles 88–89 in good to outstanding yields. In the absence of ligands, exogenous oxidants, or transition metal catalysts, this transformation simultaneously broke the three halogen–carbon bonds of the halogenated difluoro compounds 87 with a broad substrate range, to assemble the desired N-containing heterocycles 88–89 in good to exceptional yields (Scheme 23). Activity against M. tuberculosis was observed in some of the synthesized compounds.61
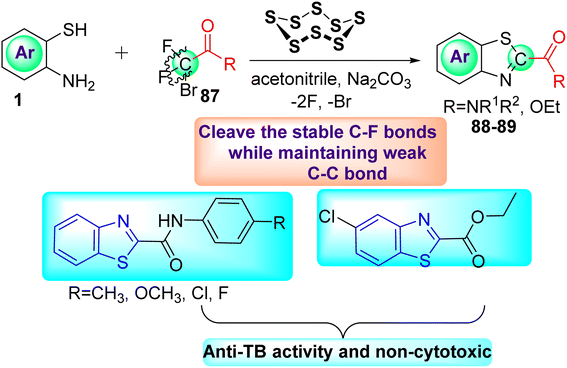 |
| | Scheme 23 Synthesis of amido substituted benzothiazole analogues. | |
A. P. Chavan and co-workers synthesized a new series of 4-(substituted benzylidene)-3-((benzo[d]thiazol-2-ylthio)methyl)isoxazol-5(4H)-one 92a–g by the reaction of mercapto benzothiazole 44 with 4-[(4-methoxyphenyl)-methylidene-]-3-chloro-methyl-5(4H)-isoxazolone 91a–g, prepared from 90, in the presence of NaHCO3 in ethanol in good yields (Scheme 24, Table 19).62
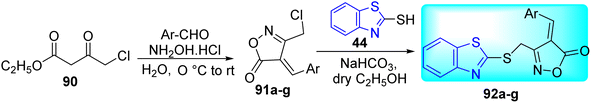 |
| | Scheme 24 Synthesis of oxazolone linked benzothiazole analogues. | |
Table 19 Anti-mycobacterial activity of oxazolone linked benzothiazole analogues (30 μg mL−1)a
| Compounds |
Ar |
Yields (%) |
Inhibition (%) |
Compounds |
Ar |
Yields (%) |
Inhibition (%) |
| % inhibition = (activity of mycobacteria without compounds − activity of mycobacteria in presence of compounds)/(activity of mycobacteria without compounds − blank) × 100. |
| 92a |
4-OH (C6H4) |
96 |
99.4 |
92e |
3,4-OCH2O-(C6H3) |
89 |
26.1 |
| 92b |
4-OCH3(C6H4) |
88 |
96.5 |
92f |
4-N(CH3)2C6H4 |
88 |
49.1 |
| 92c |
3-OCH3, 4-OH(C6H3) |
89 |
80 |
92g |
3-Indole |
85 |
16.1 |
| 92d |
4-CH3(C6H4) |
86 |
92.3 |
RIF |
— |
— |
99.5 |
The anti-tubercular activity of synthesized compounds 92a–g was carried out against M. tuberculosis H37Ra (ATCC 25177) using XTT reduction menadione assay (XRMA). Among the synthesized derivatives compound 92b was found to be most potent against M. tuberculosis and all compounds from 92a–g were found to be non-cytotoxic (Table 19).
Gawad and co-workers created a pharmacophore model by utilizing a ligand-based drug discovery method with a single ligand (Scheme 25, Table 20). The essential elements causing DprE1 inhibitory action were considered while creating the pharmacophore. The first step in the synthesis of 6-nitrobenzo[d]thiazol-2-amine 3 [27] involved simmering 4-nitroaniline 1a, potassium thiocyanate, and dropwise addition of bromine while using acetic acid as a diluent. A suitable aryl benzaldehyde and 6 nitrobenzo[d]thiazol-2-amine 3 were condensed in ethanol with a catalytic quantity of glacial acetic acid to create N-benzylidene-6-nitrobenzo[d]thiazol-2-amine 93. Finally 2-(6-nitrobenzo[d]thiazol-2-ylthio)-N-benzyl-N-(6-nitrobenzo[d]thiazol-2-yl)acetamide derivatives 96a–o were formed after a series of reduction, acetylation and nucleophilic substitution (SN2) reaction.63
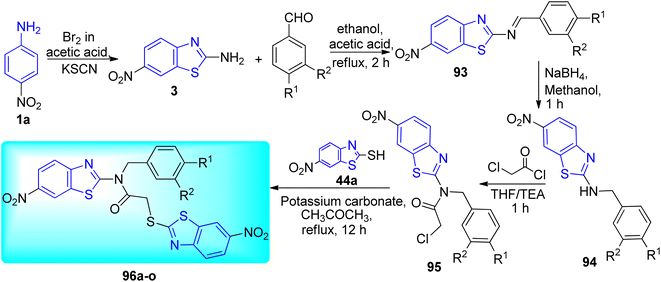 |
| | Scheme 25 Synthesis of acetamide derivatives of benzothiazole. | |
Table 20 Anti-tubercular activity of acetamide derivatives of benzothiazolea
| Compounds |
R1 |
R2 |
MIC (μM) H37Rv |
IC50 (μM) DprE1 |
| NT: not tested. |
| 96a |
Me |
Me |
2.41 |
NT |
| 96b |
OMe |
OMe |
3.74 |
NT |
| 96c |
F |
H |
3.23 |
NT |
| 96d |
SMe |
H |
2.48 |
NT |
| 96e |
OMe |
H |
2.81 |
NT |
| 96f |
H |
OH |
2.10 |
NT |
| 96g |
H |
OMe |
1.01 |
14.1 ± 1.7 |
| 96h |
OH |
OMe |
2.06 |
NT |
| 96i |
Cl |
H |
0.91 |
12.7 ± 0.9 |
| 96j |
H |
NO2 |
3.35 |
NT |
| 96k |
H |
H |
0.82 |
14.8 ± 2.4 |
| 96l |
COMe |
H |
2.79 |
NT |
| 96m |
F |
F |
3.04 |
NT |
| 96n |
NH2 |
H |
2.16 |
NT |
| 96o |
H |
Br |
1.04 |
11.2 ± 1.5 |
| INH |
— |
— |
0.31 |
— |
Using Isoniazid as a standard reference, anti-mycobacterial activity of the synthesized compounds was tested against M. tuberculosis H37Rv (ATCC 27294). Compounds 96g, 96i, 96k and 96o were found to have MIC values in between 0.82–1.04 μM, which was reported to be somewhat closer to the MIC of the standard reference Isoniazid, which is 0.31 μM. From this, authors concluded that by altering aliphatic and aromatic carbon centres more powerful DprE1 inhibitors can be synthesized (Table 20). Molecular docking of the synthesized compounds was done against BTZ043 to evaluate their DprE1 inhibition ability. Docking results suggested that di-halogen substituted compound was found to exhibit strong enzyme inhibition.
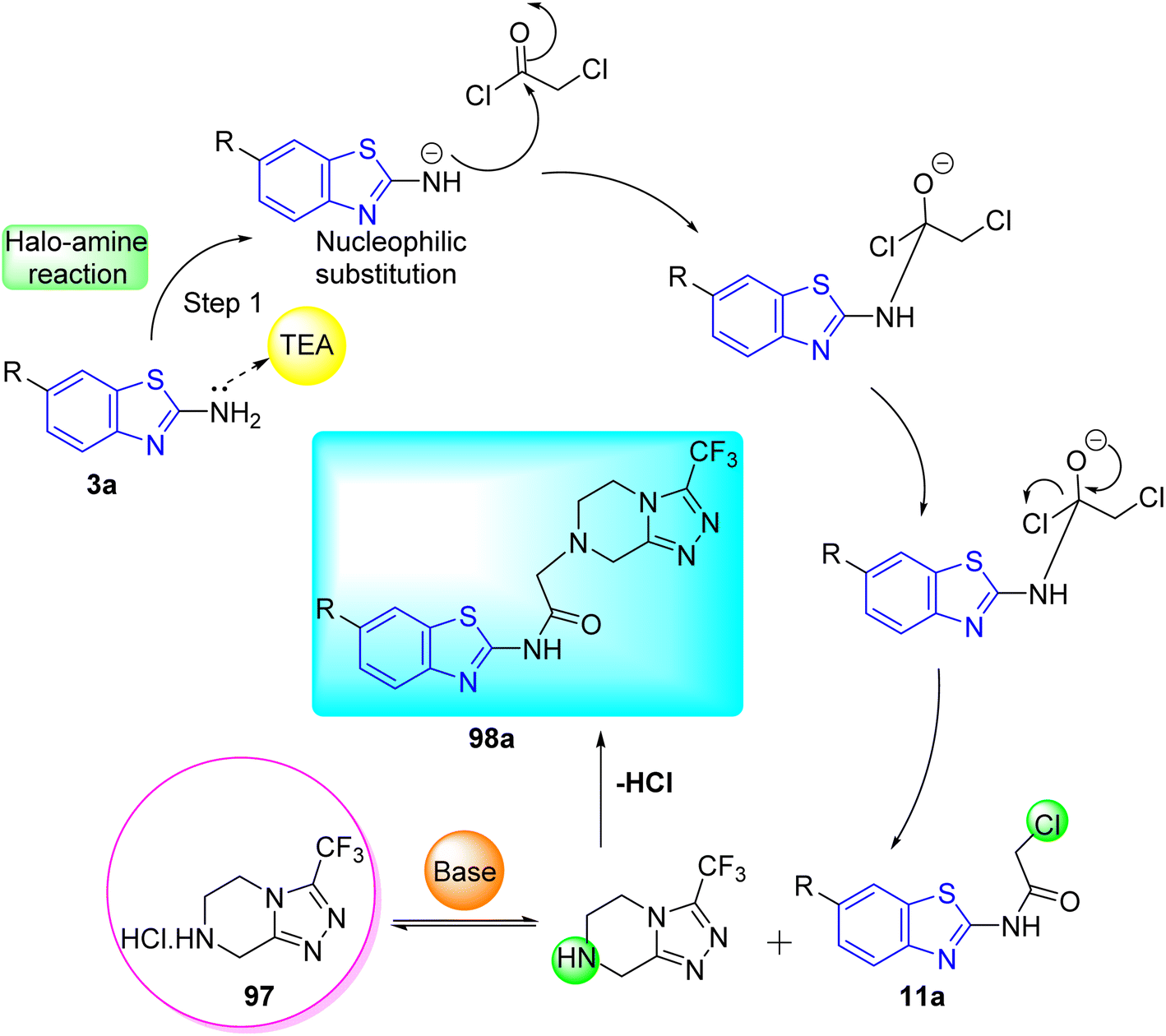
D. J. Jethava and co-workers synthesized N-(benzo[d]thiazol-2-yl)-2-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo [4,3-a]pyrazin-7 (8H)-yl) acetamide derivatives 98a–e after acetylation of benzothiazole 3a–e in presence of base NEt3 followed by nucleophilic substitution from triazolo-triazine 97 in presence of potassium carbonate in DMF solvent (Scheme 26, Table 21).64
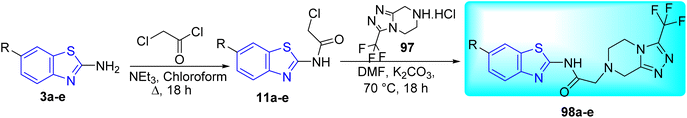 |
| | Scheme 26 Synthesis of triazolo-pyrazinyl linked benzothiazole analogues. | |
Table 21 Anti-tubercular activities of triazolo-pyrazinyl linked benzothiazole analogues
| Benzothiazole derivatives |
Final products |
Yields (%) |
MIC (μg mL−1) |
 3a 3a |
 98a 98a |
72 |
500 |
 3b 3b |
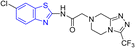 98b 98b |
75 |
250 |
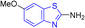 3c 3c |
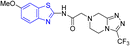 98c 98c |
70 |
250 |
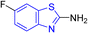 3d 3d |
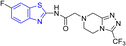 98d 98d |
81 |
500 |
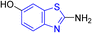 3e 3e |
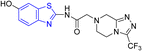 98e 98e |
69 |
500 |
Using the well-known Lowenstein–Jensen (L–J) technique, all novel compounds were tested against the M. tuberculosis H37Rv strain with Isoniazid as a positive control. A common MIC value of 500 mg mL−1 for the intended pathogenic strain of M. tuberculosis H37Rv was observed for compounds 98a, 98d and 98e (Fig. 11, Table 21).
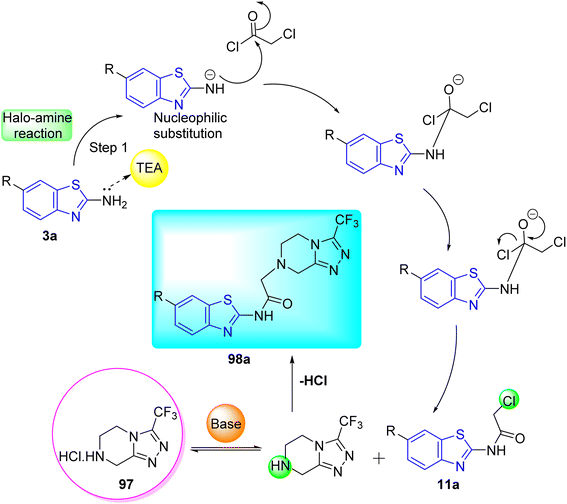 |
| | Fig. 11 Mechanistic pathway showing synthesis of compound 98a. | |
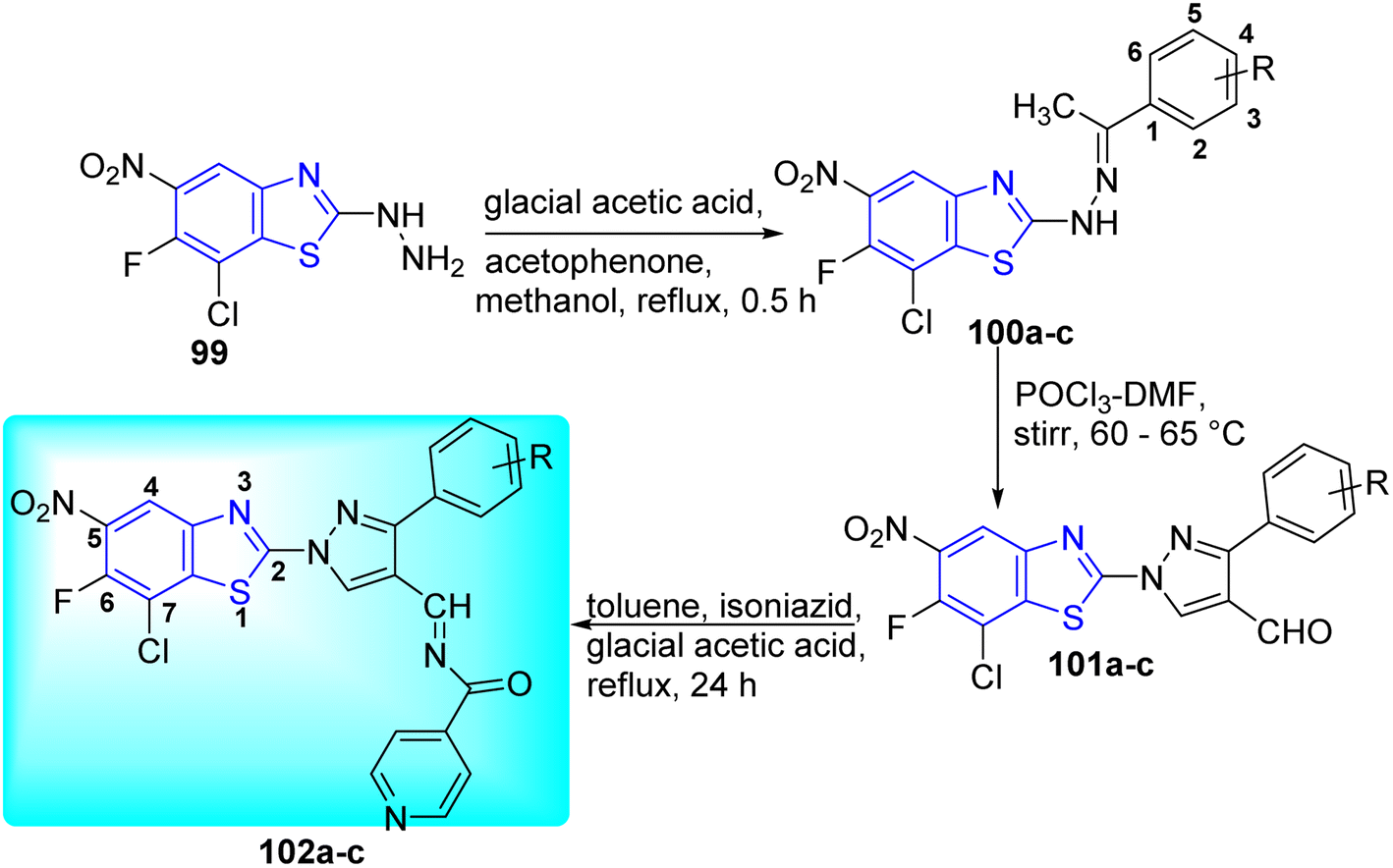
Hazra and co-workers synthesized N-((1-(7-chloro-6-fluoro-5-nitrobenzo[d] thiazol-2-yl) phenyl-1H-pyrazol-4-yl)methylene)-3-substituted isonicotino hydrazide 102a–c and N'-((1-(7-chloro-6-fluorobenzo[d]thiazol-2-yl)-3-phenyl-1H-pyrazol-4-yl)ethylene) isonicotinohydrazide 106a–c for improved anti-tubercular efficacy. Initial step involved the reaction of 7-chloro-6-fluoro-5-nitro-2-hydrazinylbenzo[d]thiazole 99 and 7-chloro-6-fluoro-4-nitro-2-hydrazinylbenzo[d]thiazole 103 with substituted acetophenones in presence of glacial acetic acid to produce 100a–c and 104a–c respectively. Compounds 102a–c and 106a–c underwent Vilsmeyer–Haack reaction in presence of POCl3 in DMF to produce 101a–c and 105a–c respectively. The later 101a–c and 105a–c after being treated with isoniazid in presence of glacial acetic acid furnished the desired compounds 102a–c and 106a–c respectively (Schemes 27 and 28, Table 22).65
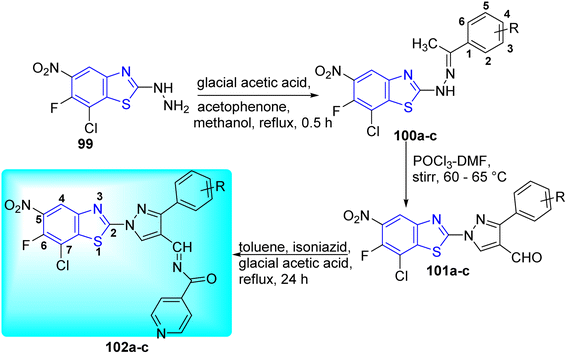 |
| | Scheme 27 Synthesis of new series of N-((1-(7-chloro-6-fluoro-5-nitrobenzo[d]thiazol-2-yl)phenyl-1H-pyrazol-4-yl)methylene)-3-substituted isonicotinohydrazide. | |
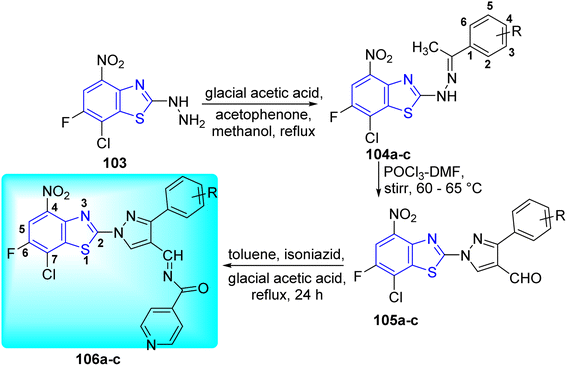 |
| | Scheme 28 Synthesis of new series of N-((1-(7-chloro-6-fluoro-4-nitrobenzo[d]thiazol-2-yl)phenyl-1H-pyrazol-4-yl)methylene)-3-substituted isonicotinohydrazide. | |
Table 22 Anti-tubercular activity of 5-nitro and 4-nitro substituted isonicotino-hydrazide analogues of benzothiazole
| Compounds |
R |
MIC (nM) |
| 102a |
H |
95.80 |
| 102b |
2,4 di Cl |
42.31 |
| 102c |
4-F |
46.30 |
| 106a |
H |
47.90 |
| 106b |
2,4 di Cl |
42.31 |
| 106c |
4-F |
46.30 |
| PYZ |
— |
60.095 |
| STM |
— |
14.387 |
The compounds 102a–c and 106a–c were found to be effective anti-tubercular agents (MIC = 40.19 to 64.96 nM) through in vitro anti-mycobacterial activity against M. tuberculosis H37Rv (ATCC 27294). All the substances examined had low cytotoxicity when evaluated on the THP-1 cell line. Even though this concentration is much higher than the concentration evaluated for the anti-tubercular action, the presence of a nitro group in the compound is demonstrated to increase the toxicity (Table 22).
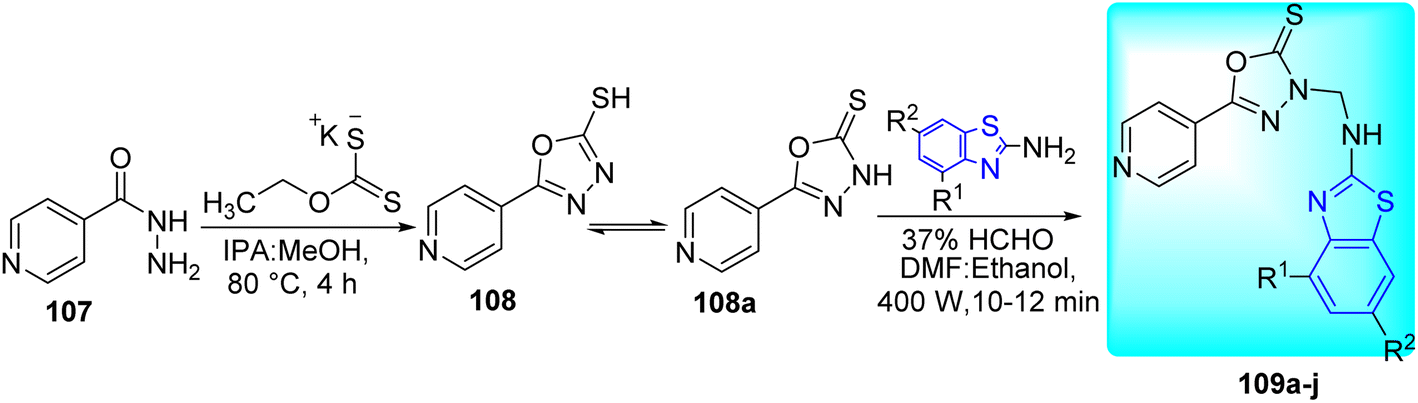
Sahoo and co-workers synthesized a variety of new analogues of 5-(pyridine-4-yl)-1,3,4-oxadiazole-2(3H)-thione 109a–j (Scheme 29, Table 23) by combining 1,3,4-oxadiazole 108a and benzo[d]thiazole via Mannich reaction under conventional heating and improved microwave irradiations.66
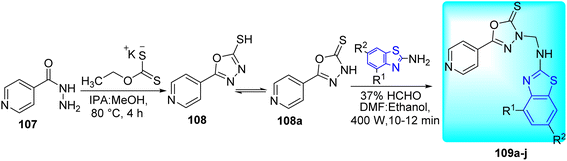 |
| | Scheme 29 Synthesis of 3-((substituted-benzo[d]thiazol-2-ylamino)methyl)-5-(pyridine-4-yl)-1,3,4-oxadiazole-2(3H)-thione. | |
Table 23 Anti-tubercular activity of 3-((substituted-benzo[d]thiazol-2-ylamino)methyl)-5-(pyridine-4-yl)-1,3,4-oxadiazole-2(3H)-thione analogues
| Compounds |
R1 |
R2 |
Conventional method yields (%) |
Microwave irradiation yields (%) |
MIC (μM) |
| 108a |
— |
— |
90 |
— |
>100 |
| 109a |
H |
H |
63 |
85 |
>50 |
| 109b |
H |
CH3 |
66 |
82 |
>100 |
| 109c |
CH3 |
H |
59 |
80 |
>50 |
| 109d |
H |
NO2 |
58 |
75 |
>50 |
| 109e |
NO2 |
H |
55 |
78 |
>100 |
| 109f |
H |
F |
63 |
80 |
>100 |
| 109g |
F |
H |
60 |
75 |
>100 |
| 109h |
H |
Br |
58 |
78 |
>100 |
| 109i |
H |
Cl |
54 |
75 |
>100 |
| 109j |
H |
OCH3 |
60 |
80 |
>100 |
| INH |
— |
— |
|
— |
0.25 |
| RIF |
— |
— |
|
— |
40 |
All the synthesized compounds were evaluated in vitro for their anti-tubercular activity against H37Ra strain of M. tuberculosis. Compound 109c, with a methyl group at the ortho position of an aromatic ring, displayed higher anti-tubercular activity. Change in the activity was also observed with the addition of various electron-releasing and electron-withdrawing substituents to the benzo[d]thiazole ring (Table 23). All the synthesized compounds were found to be non-cytotoxic (<50% inhibition at 50 μg mL−1) to HEK 293T cell lines with therapeutic index ranging from 8–64.
P. T. Acharya and co-workers synthesized a series of N-(1, 3-benzothiazole-2-yl)-2(pyridine-3-yl) formohydrazido acetamide derivatives 113a–i by using a simple and effective conventional technique (Scheme 30, Table 24). Initial step involved the synthesis of N-(1,3 benzothiazole-2-yl)-2chloroacetamide 111a–i from the acetylation of 2-amino benzothiazole derivatives 110a–i in presence of TEA in chloroform. Next step involved the reaction of nicotinohydrazide 112 with 111a–i in presence of base K2CO3 under reflux conditions to produce the desired compounds 113a–i.67
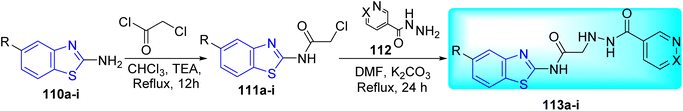 |
| | Scheme 30 Synthesis of acetamide derivatives of benzothiazole. | |
Table 24 Anti-tubercular activity of acetamide derivatives of benzothiazole
| Compounds |
R |
X |
MIC (mg mL−1) |
| 113a |
H |
H |
50 |
| 113b |
OCH3 |
H |
250 |
| 113c |
OC2H5 |
H |
100 |
| 113d |
OH |
H |
250 |
| 113e |
Cl |
H |
500 |
| 113f |
F |
H |
250 |
| 113g |
H |
N |
62.5 |
| 113h |
OCH3 |
N |
62.5 |
| 113i |
OC2H5 |
N |
100 |
| INH |
— |
— |
0.20 |
All synthesized compounds 113a–i were tested in vitro for their anti-tuberculosis activity against the H37Rv strain of M. tuberculosis using Lowenstein–Jensen media (conventional method). Compound 113a displayed promising activity against H37Rv strains with mean IC50 of 50 mg mL−1. Compounds 113g–h showed potent anti-tubercular action with mean IC50 of 62.5 mg mL−1 (Table 24). All the synthesized compounds were found to exhibit good pharmacokinetics properties (ADME) with good oral absorption percentage in the tolerable range of 65–100%. Docking of the synthesized compounds was done against PDB 1ENY of M. tuberculosis. Compound 113a was found to exhibit good binding affinity of −8.423 kcal mol−1 to the active site of 1ENY with reference to the standard drug Isoniazid (−6.33 kcal mol−1). Here PDB 1ENY was chosen in order to target enoyl-acyl-carrier protein reductase.
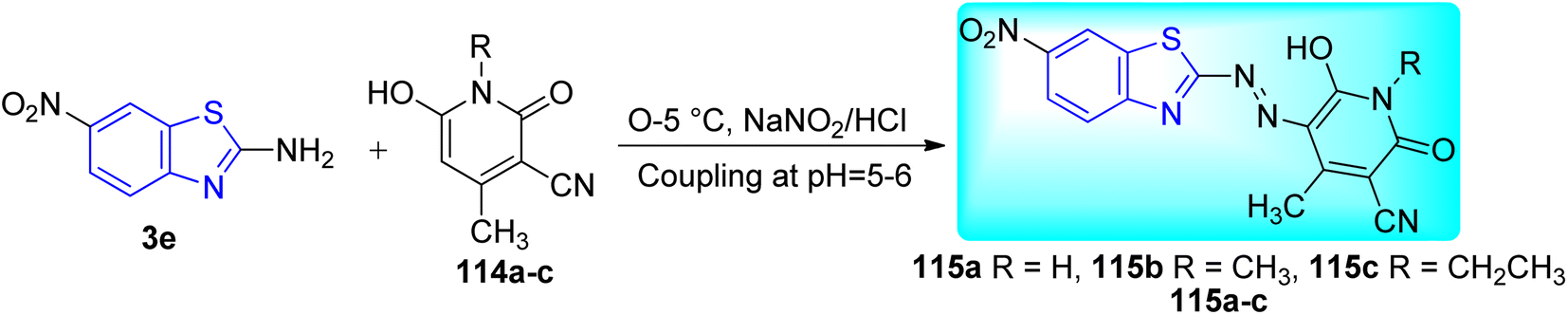
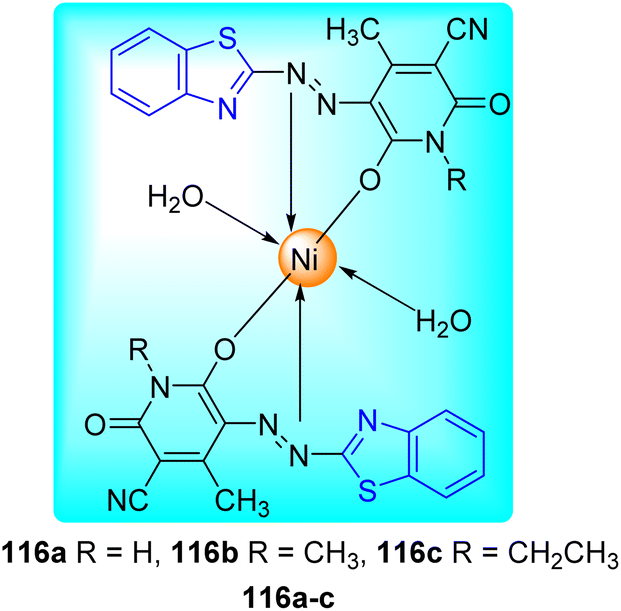
B. N. Ravi and co-workers described the synthesis of bioactive Ni(II) complexes 116a–c from azo dye ligands 115a–c. Azo dyes were formed from the diazo-coupling of 6-nitro-1,3-benzothiazole 3e with substituted pyridinone derivatives 114a–c in presence of NaNO2 in HCl at low temperature range (Scheme 31, Table 25). These Ni(II) complexes possess a structure of [Ni(L)2(H2O)2] with a metal–ligand ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (Fig. 12) where L is the deprotonated azo dye ligand which show bidentate behavior.68
:2 (Fig. 12) where L is the deprotonated azo dye ligand which show bidentate behavior.68
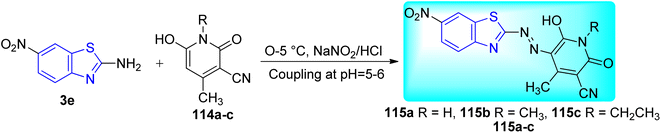 |
| | Scheme 31 Synthesis of azo-dye ligands. | |
Table 25 Anti-mycobacterial activity of the synthesized azo-dyes and their Ni(II) complexesa
| Compounds |
100 μg mL−1 |
50 μg mL−1 |
25 μg mL−1 |
12.5 μg mL−1 |
6.25 μg mL−1 |
3.12 μg mL−1 |
1.60 μg mL−1 |
0.80 μg mL−1 |
| S: sensitive, R: resistance. |
| 115a |
S |
S |
S |
S |
S |
S |
S |
R |
| 115b |
S |
S |
S |
S |
S |
S |
S |
R |
| 115c |
S |
S |
S |
S |
S |
R |
R |
R |
| 116a |
S |
S |
S |
S |
S |
S |
S |
R |
| 116b |
S |
S |
S |
S |
S |
R |
R |
R |
| 116c |
S |
S |
S |
S |
S |
S |
S |
R |
| STM |
S |
S |
S |
S |
S |
R |
R |
R |
| CIP |
S |
S |
S |
S |
S |
S |
R |
R |
| PYZ |
S |
S |
S |
S |
S |
S |
R |
R |
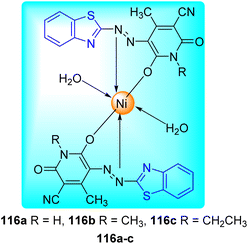 |
| | Fig. 12 Structure for Ni(II) complexes of azo dyes. | |
By using the Microplate Alamar Blue Assay (MABA), the anti-tubercular activity of the azo dye ligands and their Ni(II) complexes was assessed against M. tuberculosis (H37Rv strain, ATCC 27294). Some Ni(II) complexes of azo dyes showed good inhibitory activity with MIC value of 1.60 μg mL−1. Additionally, all other substances showed good to moderate activity, with MIC values in between 6.25–3.12 μg mL−1. The increased lipophilicity of the metal ion caused by the overlapping of the ligand's orbitals and partial sharing of the metal ion's positive charge with the donor atoms was responsible for the greater activity metal chelates than the ligand (Table 25).
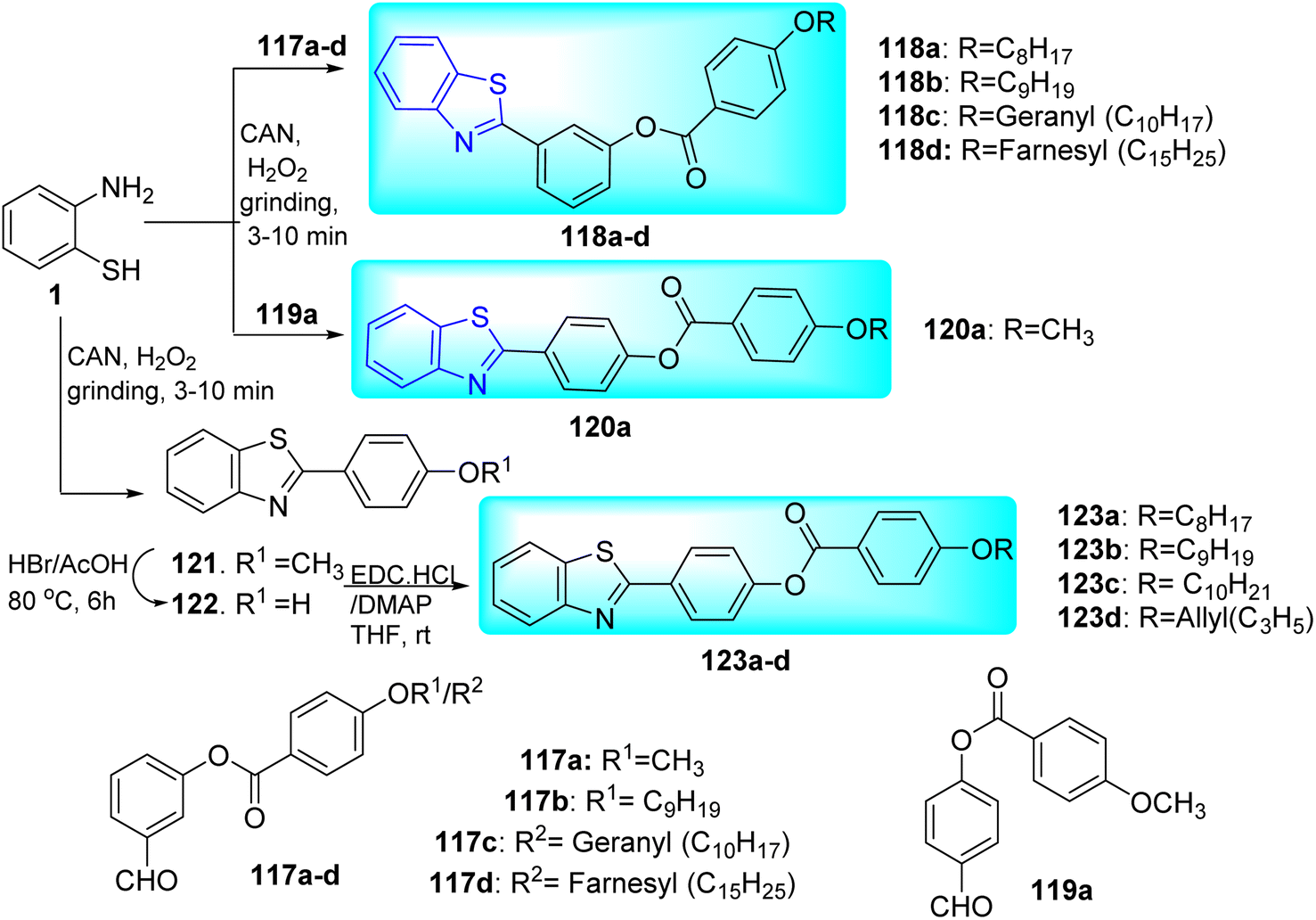
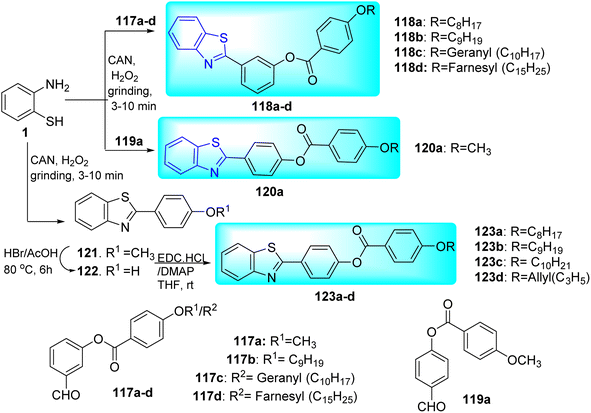
Velappan and co-workers synthesized 2-aryl benzothiazole based dual targeted compounds 118a–d, 120a, 123a–d through the reaction of 2-amino thio phenol 1 with various heterocyclic derivatives (Scheme 32, Table 26).69
 |
| | Scheme 32 Synthesis of 2-aryl substituted benzothiazole analogues. | |
Table 26 Anti-tubercular activity of 2-aryl substituted benzothiazole analoguesa
| Compounds |
MIC (μg mL−1) against H37Rv |
| MABA |
LORA |
| NT: not tested. |
| 118a |
30.12 |
47.31 |
| 118b |
39.52 |
40.63 |
| 118c |
56.13 |
40.09 |
| 118d |
31.49 |
27.81 |
| 120a |
29.51 |
70.42 |
| 123a |
>100 |
NT |
| 123b |
>100 |
NT |
| 123c |
>100 |
NT |
| 123d |
52.37 |
43.11 |
| INH |
0.40 |
>100 |
| RIF |
0.01 |
0.04 |
Their anti-tubercular activity was checked by using MABA for replicating form of Mtb and Low Oxygen Recovery Assay (LORA) for non-replicating form of Mtb. Compound 118a (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C8H17) showed MIC value of 30.12 μg mL−1 against replicating Mtb. Contrarily, compound 118b (RC9H19) was discovered to be the most effective against the non-replicating Mtb. The MIC values were determined in between 56–32 μg mL−1 against replicating Mtb and 40–28 μg mL−1 against non-replicating Mtb for molecules having geranyl 118c and farnesyl 118d chains. On the other hand, they discovered that the activity of the meta-isomers against replicating Mtb reduced as the length of the alkyl chain increased, with the best activity being observed for 120a with a methyl chain. The alkenyl chain once more exhibited better anti-tubercular action (<50 μg mL−1). 123a–c did not show any significant difference in activity against replicating and non-replicating Mtb. It was concluded that their effectiveness against replicating and non-replicating forms of Mtb is significantly influenced by their isomers (meta or para) and the presence of heteroatom's in the aromatic ring (Table 26).
C8H17) showed MIC value of 30.12 μg mL−1 against replicating Mtb. Contrarily, compound 118b (RC9H19) was discovered to be the most effective against the non-replicating Mtb. The MIC values were determined in between 56–32 μg mL−1 against replicating Mtb and 40–28 μg mL−1 against non-replicating Mtb for molecules having geranyl 118c and farnesyl 118d chains. On the other hand, they discovered that the activity of the meta-isomers against replicating Mtb reduced as the length of the alkyl chain increased, with the best activity being observed for 120a with a methyl chain. The alkenyl chain once more exhibited better anti-tubercular action (<50 μg mL−1). 123a–c did not show any significant difference in activity against replicating and non-replicating Mtb. It was concluded that their effectiveness against replicating and non-replicating forms of Mtb is significantly influenced by their isomers (meta or para) and the presence of heteroatom's in the aromatic ring (Table 26).
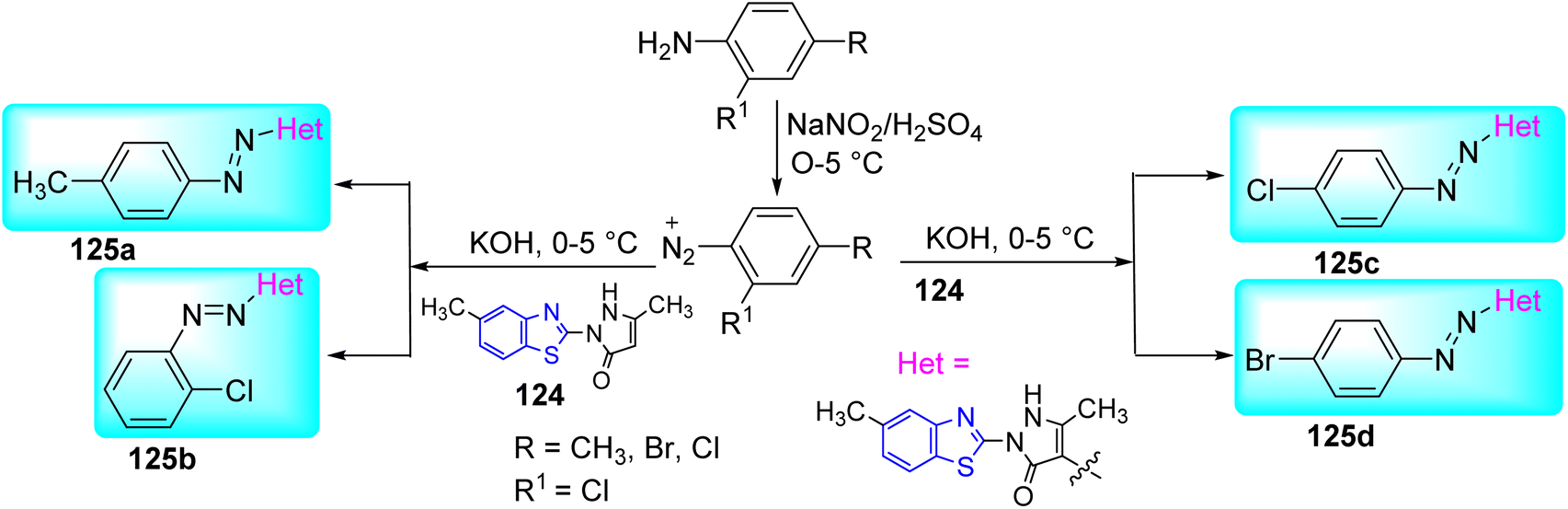
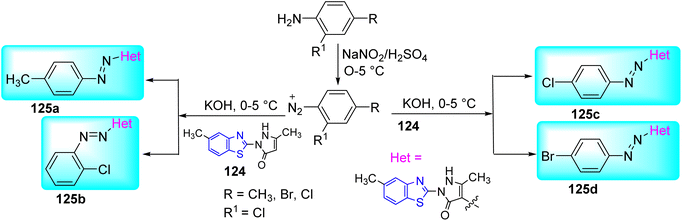
Maliyappa and co-workers created four heterocyclic azo dyes 125a–d using the standard diazo-coupling process between aniline derivatives and 5-methyl-2-(6-methyl-1,3-benzothiazol-2-yl)-2,4-dihydro-3H pyrazol-3-one 124 at lower temperature. Initial step involved the diazotization of substituted anilines in presence of NaNO2/H2SO4. Diazotized product on further coupling with benzothiazole derivatives in presence of base KOH at low temperature furnished the desired compounds 125a–d (Scheme 33, Table 27).70
 |
| | Scheme 33 Synthesis of azo dye ligands. | |
Table 27 Anti-mycobacterial activity of the synthesized azo dye ligandsa
| Compounds |
12.5 μg mL−1 |
6.25 μg mL−1 |
3.12 μg mL−1 |
1.6 μg mL−1 |
| S: sensitive, R: resistant. |
| 125a |
S |
S |
S |
S |
| 125b |
S |
S |
S |
S |
| 125c |
S |
S |
R |
R |
| 125d |
S |
S |
R |
R |
| PZA |
S |
S |
S |
R |
The synthesized compounds were screened for their anti-mycobacterial activity against Mtb by using MABA method. From the synthesized compounds 125a–b showed better activity than 125c–d (Table 27).
Abozeid and co-workers synthesized benzothiazole based naphthyl ketone 129 scaffold by refluxing formylchromone 126 and cyanoacetanilide 127 in ethanol in the presence of triethyl amine as catalyst (Scheme 34).71
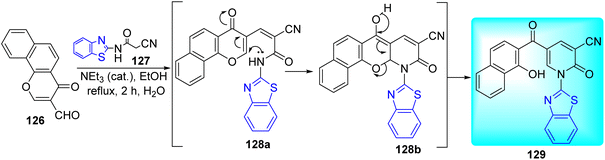 |
| | Scheme 34 Synthesis of benzothiazole based naphthyl ketone. | |
The synthesized compound was tested in vitro against Mtb using Isoniazid as positive control. Compound 129 was found to exhibit anti-tubercular activity against Mtb with a MIC value of 1.95 μg mL−1. Molecular docking of this active compound 129 against InhA enzyme showed better binding affinity of −9.3 kcal mol−1.
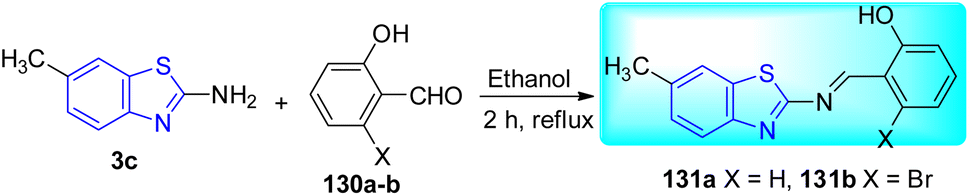
J. K. Suyambulingam and co-workers synthesized two Schiff bases, 2-[6-methylbenzothiazol-2-ylimino] methyl phenol 131a and 3-bromo-2-[6-methylbenzothiazol-2-ylimino] methyl phenol 131b utilizing a straightforward condensation reaction between amino benzothiazole derivative 3c and salicylaldehyde/bromosalicylaldehyde 130a–b (Scheme 35, Table 28).72
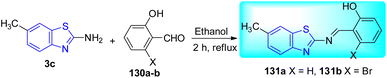 |
| | Scheme 35 Synthesis of benzothiazole based Schiff bases. | |
Table 28 Anti-tubercular activity of Schiff basesa
| Compounds |
100 μg mL−1 |
50 μg mL−1 |
25 μg mL−1 |
12.5 μg mL−1 |
6.25 μg mL−1 |
3.12 μg mL−1 |
1.6 μg mL−1 |
0.8 μg mL−1 |
| S: sensitive, R: resistant. |
| 131a |
S |
S |
S |
S |
S |
R |
R |
R |
| 131b |
S |
S |
S |
S |
S |
S |
S |
R |
Anti-tubercular activity of the synthesized compounds was evaluated against H37Rv strain of M. tuberculosis. Compound 131a showed moderate activity while compound 131b was found to exhibit better activity with a MIC value of 1.6 μg mL−1 which was lesser than standard drugs like Pyrazinamide, Streptomycin and Ciprofloxacin which have MIC values 3.125 μg mL−1, 6.25 μg mL−1, 3.125 μg mL−1 respectively (Table 28).
In order to find the inhibition potency of benzothiazole based Schiff bases the molecular docking of compound 131b was performed against 4P8N protein of M. tuberculosis DprE1. It was observed from the docking results that compound 131b interacts better with active site of 4P8N protein with a binding affinity of −9.2 kcal mol−1. The interactions involved were different types of pi–pi and hydrogen bond interactions (Fig. 13 and 14). The increase in protein–ligand interaction surface results in strong van der Waal's interactions and hence greater binding affinity.
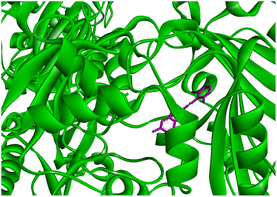 |
| | Fig. 13 3D representation of ligand 131b and its interactions with active site of 4P8N protein. | |
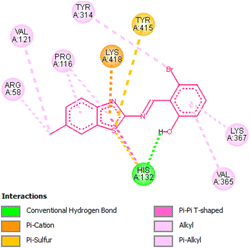 |
| | Fig. 14 2D representation of docking results showing interactions of compound 131b with 4P8N. | |
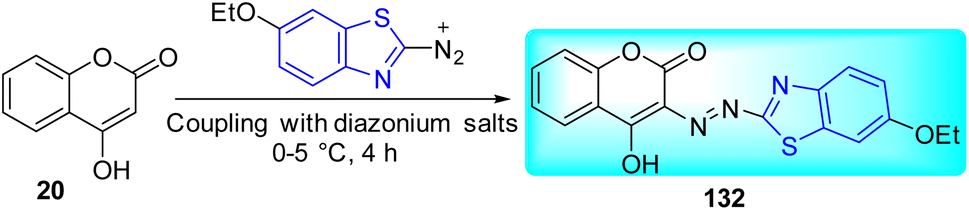
Nagaraja and co-workers synthesized 4-hydroxy coumarin containing benzothiazole based azo dye 132. Initial step involved the diazotization of 2-amino substituted benzothiazoles in presence of NaNO2/H2SO4. Final step involved diazo-coupling of 4-hydroxy coumarin 20 and diazotized benzothiazole analogue to furnish the desired compound 132 (Scheme 36).73
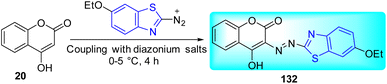 |
| | Scheme 36 Synthesis of coumarin based azo dye. | |
By using the MABA method, the compound 132 was tested for its anti-tubercular activity against M. tuberculosis. The outcome was compared to standard medications Pyrazinamide, Ciprofloxacin, and Streptomycin. Synthesized compound was found to be sensitive at a concentration range of 100–1.6 μg mL−1 and resistant at 0.8 μg mL−1.
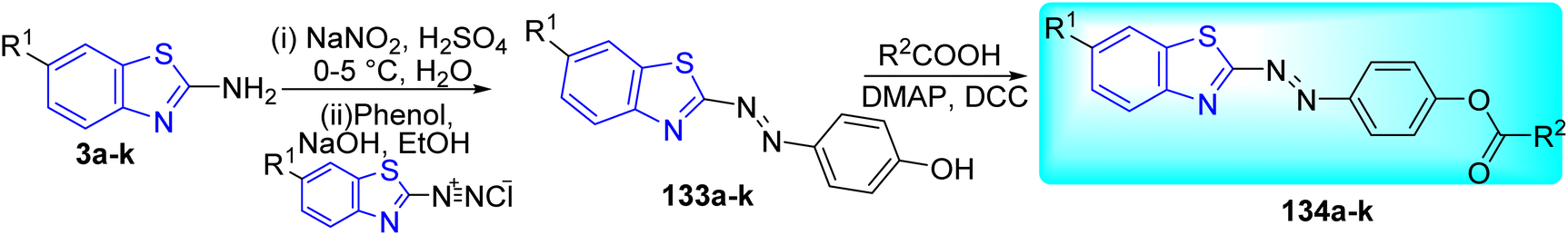
M. Bhat and co-workers synthesized a series of azo-ester derivatives of benzothiazole 134a–k via Steglich esterification reaction by using dicyclohexylcarbodiimide (DCC) as a coupling reagent and 4-(dimethylamino)pyridine (DMAP) as nucleophile. Initial step involved the formation of diazotized product 133a–k from the diazotization of 2-amino substituted benzothiazoles 3a–k. Compound 133a–k on further coupling with phenol in presence of base NaOH gave the azo-dye complex 134a–k. This complex on further reaction with suspension of substituted carboxylic acid in presence of DCC and DMAP furnished the desired compounds 134a–k (Scheme 37, Table 29).74
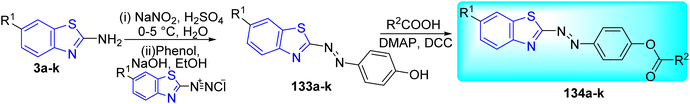 |
| | Scheme 37 Synthesis of benzothiazole azo-ester derivatives. | |
Table 29 Anti-tubercular activity of benzothiazole azo-ester derivatives
| Compounds |
R1 |
R2 |
Yields (%) |
MIC (μg mL−1) |
Compounds |
R1 |
R2 |
Yields (%) |
MIC (μg mL−1) |
| 134a |
OEt |
 |
55 |
2.5 ± 0.24 |
134h |
OEt |
 |
83 |
25 ± 0.25 |
| 134b |
OEt |
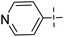 |
92 |
6.25 ± 0.18 |
134i |
H |
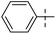 |
81 |
12.5 ± 0.13 |
| 134c |
OEt |
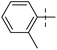 |
78 |
25 ± 0.39 |
134j |
H |
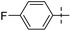 |
86 |
1.6 ± 0.08 |
| 134d |
OEt |
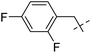 |
80 |
1.6 ± 0.15 |
134k |
H |
 |
67 |
25 ± 0.24 |
| 134e |
OEt |
 |
71 |
25 ± 0.43 |
STM |
— |
— |
— |
6.25 ± 0.16 |
| 134f |
OEt |
 |
92 |
50 ± 0.40 |
CIP |
— |
— |
— |
3.125 ± 0.22 |
| 134g |
OEt |
 |
88 |
50 ± 0.37 |
PZA |
— |
— |
— |
3.125 ± 0.35 |
By using the Microplate Alamar blue assay technique for M. tuberculosis, the produced compounds were tested for anti-tubercular activity. Among the synthesized compounds 134d and 134j showed better anti-tubercular activity with a MIC value of 1.6 μg mL−1 which was less than that of standard drugs like Streptomycin (MIC 6.25 μg mL−1) and Pyrazinamide (MIC 3.125 μg mL−1). Rest of the synthesized compounds displayed moderate activity (Table 29).
In order to predict the interaction of ligand 134j with M. tuberculosis DprE1 we performed docking against 4P8N protein. Along with different types of interactions with the protein chain compound 134j was found to exhibit best docking results with a binding affinity of −10.3 kcal mol−1 towards 4P8N (Fig. 15 and 16).
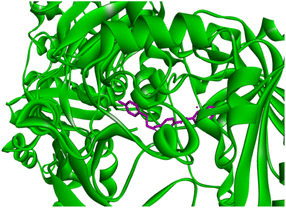 |
| | Fig. 15 3D representation of ligand 134j and its interactions with active site of 4P8N protein. | |
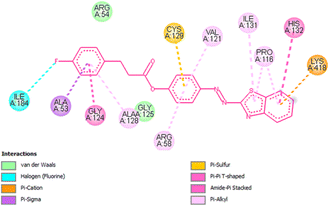 |
| | Fig. 16 2D representation of docking results showing interaction of compound 134j with 4P8N protein. | |
Chen and co-workers synthesized the benzothiazole based sulfonamide compounds 137a–d by treating different aryl amines 136a–d with 4-acetamido benzene sulfonyl chloride 135 followed by base catalyzed hydrolysis of the acetyl group (Scheme 38, Table 30).75
 |
| | Scheme 38 Synthesis of benzothiazole based sulfonamide compounds. | |
Table 30 Anti-tubercular activity of benzothiazole based sulfonamides against XDR-TB
| Compounds |
R |
MIC (μg mL−1) |
| 137a |
 |
14.26 |
| 137b |
 |
>32 |
| 137c |
 |
>32 |
| 137d |
 |
>32 |
| SPA |
 |
5.51 |
After screening of the synthesized compounds against M. tuberculosis H37Rv the selected compounds were tested against an isolated clinical strain of XDR-TB. Isoniazid (INH) and sulfaphenazole (SPA) were used as reference standards for anti-tubercular evaluation of the synthesized compounds. Among the synthesized compounds compound 137a displayed modest activity (MIC = 14.26 μg mL−1). Altering the position and introduction of phenyl group to benzothiazole moiety leads to decrease in anti-tubercular activity of the compounds 137b and 137d compounds (Table 30).
S. V. Mamatha and co-workers synthesized the target compounds 142a–d, 143a–d, 144a–d, 145a–d via several steps. Initial step involved the reaction of aniline derivatives 138a–d with bromine in acetic acid in presence of potassium thiocyanate to give 2-amino substituted benzothiazoles 139a–d. The later on reaction with hydrazine hydrate produced hydrazine benzothiazoles 140a–d. Compounds 140a–d underwent cyclization with carbon disulfide in presence of NaOH to produce triazol-2-thiol derivatives 141a–d which ultimately furnished the desired compounds 142a–d, 143a–d, 144a–d, 145a–d after being alkylated with several heterocyclic compounds (Scheme 39, Table 31).76
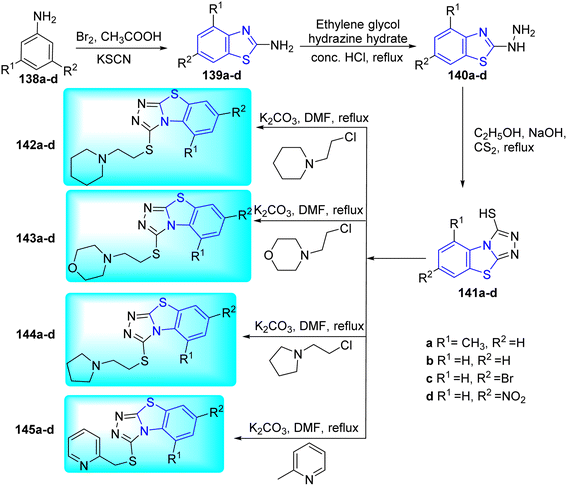 |
| | Scheme 39 Schematic pathway for the synthesis of triazole conjugated benzothiazole derivatives. | |
Table 31 Anti-tubercular activity of triazole conjugated benzothiazole derivatives
| Compounds |
MIC (μg mL−1) |
Docking score (kJ mol−1) |
Compounds |
MIC (μg mL−1) |
Docking score (kJ mol−1) |
| 142a |
25 |
−5.999 |
144c |
1.6 |
−5.568 |
| 142b |
1.6 |
−7.443 |
144d |
12.5 |
−5.698 |
| 142c |
1.6 |
−5.986 |
145a |
50 |
−6.186 |
| 142d |
1.6 |
−7.865 |
145b |
1.6 |
−6.176 |
| 143a |
50 |
−5.036 |
145c |
50 |
−6.392 |
| 143b |
50 |
−4.864 |
145d |
1.6 |
−6.338 |
| 143c |
50 |
−4.034 |
INH |
0.40 |
−6.617 |
| 143d |
6.25 |
−5.833 |
PZA |
3.125 |
— |
| 144a |
1.6 |
−6.424 |
CIP |
3.125 |
— |
| 144b |
3.12 |
−8.643 |
STM |
6.25 |
— |
Microplate Alamar Blue Assay (MABA) was used to test the anti-mycobacterial activity of synthesized compounds 142a–d, 143a–d, 144a–d and 145a–d against M. tuberculosis and MIC values are summarized in Table 31. The best action was demonstrated by the benzothiazolyltriazoles with piperidine (142b–d), pyrrolidine (144a–c) and pyrimidine (145b and 145d) moieties with MIC values ranging from 3.12 to 1.6 μg mL−1. A unique and promising hit molecule that shown good anti-TB properties as well as good docking score was compound 144b which possess benzothiazolyltriazole with a pyrrolidine moiety (Table 31). Molecular docking studies of these compounds against inhA of M. tuberculosis suggested that compound 144b is superior compound with a binding affinity of −8.654 kJ mol−1 as compared to the standard dug Isoniazid (−6.617 kJ mol−1).
B. Manjunatha and co-workers described the synthesis of various azo dyes 147a–e based on coumarin and benzothiazole in this study. Synthetic process involved the diazotization of 2-amino benzothiazole derivatives 146a–e in presence of NaNO2/HCl. Diazotized solution was then added to 4-hydroxycoumarin 20 in order to obtain azo dyes 147a–e while maintaining the pH of the reaction mixture (Scheme 40, Table 32).77
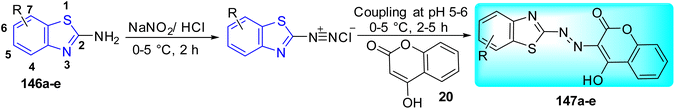 |
| | Scheme 40 Schematic pathway for the synthesis of coumarin azo dyes. | |
Table 32 Anti-tubercular activity of synthesized coumarin dyesa
| Compounds |
R |
100 μg mL−1 |
50 μg mL−1 |
25 μg mL−1 |
12.5 μg mL−1 |
6.25 μg mL−1 |
3.12 μg mL−1 |
1.6 μg mL−1 |
0.8 μg mL−1 |
| S: sensitive, R: resistant. |
| 147a |
H |
S |
S |
S |
S |
S |
S |
S |
R |
| 147b |
6-Cl |
S |
S |
S |
S |
S |
S |
S |
R |
| 147c |
6-NO2 |
S |
S |
S |
S |
S |
S |
S |
R |
| 147d |
6-OEt |
S |
S |
S |
S |
S |
R |
R |
R |
| 147e |
4-CH3 |
S |
S |
S |
S |
S |
S |
S |
R |
In vitro screening of the synthesized compounds was done against H37Rv strain of M. tuberculosis using MABA technique. Using Streptomycin as a reference point, the study's findings were interpreted in terms of minimum inhibitory concentration (MIC). The results of the anti-TB activity tests showed that compounds 147a–c and 147e had outstanding and comparable sensitivity (MIC = 1.6 μg mL−1). However, among the synthesized dyes, compound 147d with an ethoxy substitution at the benzothiazole's 6th position exhibited lower sensitivity (MIC = 3.2 μg mL−1) (Table 32).
Ethambutol, an anti-TB medicine, is known to target the arabinosyl transferases EmbA, EmbB, and EmbC, which are known to be involved in the manufacturing of the cell walls in M. tuberculosis. The donor and acceptor interactions as observed from docking predicts the mechanism of inhibition of arabinosyl transferases. Herein we observed the better interaction of ligand 147e to the active site of 7BVF protein (cryo-EM structure of M. tuberculosis in complex with Ethambutol) with a binding affinity of −9.4 kcal mol−1 (Fig. 17 and 18). These findings certainly will help in predicting the biochemical function and development of new anti-tubercular agents.
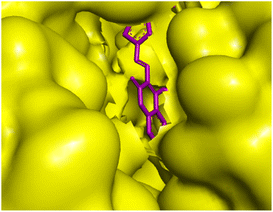 |
| | Fig. 17 Surface representation of docking between ligand 147e and 7BVF protein. | |
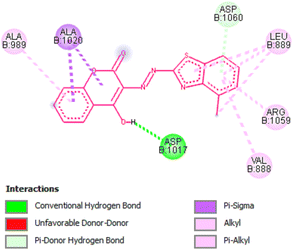 |
| | Fig. 18 2D view showing interaction between ligand 147e and various amino acids of 7BVF protein. | |
Satyadev and co-workers synthesized benzothiazole-linked-chalcones 151a–n from the reaction of 1-(2-aminobenzo[d]thiazol-5-yl)ethan-1-one 149 with aldehydes 150a–n in ethanol with pyridine as catalyst (Schemes 41 and 42, Table 33).78 The intermediate 149 in turn was synthesized from the reaction of 3-aminoacetophenone 148 with Br2 and potassium thiocyanate in glacial acetic acid.
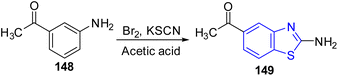 |
| | Scheme 41 Synthesis of 1-(2-aminobenzo[d]thiazol-5-yl) ethan-1-one. | |
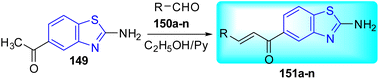 |
| | Scheme 42 Synthesis of benzothiazole linked substituted chalcones. | |
Table 33 Anti-tubercular activity of benzothiazole linked substituted chalcones
| Compounds |
R |
MIC (μg mL−1) |
| 151a |
C6H5 |
25 |
| 151b |
4-MeC6H4 |
25 |
| 151c |
4-OHC6H4 |
100 |
| 151d |
4-OMeC6H4 |
12.5 |
| 151e |
4-NMe2C6H4 |
12.5 |
| 151f |
4-NO2C6H4 |
50 |
| 151g |
4-ClC6H4 |
25 |
| 151h |
Furan-2-yl |
25 |
| 151i |
Furan-3-yl |
50 |
| 151j |
Thiophen-2-yl |
6.25 |
| 151k |
Thiophen-3-yl |
50 |
| 151l |
Pyridin-2-yl |
6.25 |
| 151m |
Pyridin-3-yl |
100 |
| 151n |
Pyridin-4-yl |
50 |
| PZA |
— |
3.125 |
In vitro screening of the synthesized compounds showed that 149, 151j and 151l were found to be most potent anti-tubercular compounds with a MIC value of 6.25 μg mL−1. Moreover, compounds 151d and 151e had notable inhibitory action with values of 12.5 and 12.5 μg mL−1 respectively. Rest other compounds showed moderate to less activity (Table 33).
Van Der Westhuyzen and co-workers discovered a powerful benzoheterocyclic oxime carbamate hit series 154–165 (Schemes 43 and 44, Tables 34 and 35) through the screening of a library of small polar compounds against M. tuberculosis.79 The reaction between 2-(benzo[d]thiazol-2-yl)acetonitrile 152 and sodium nitrite produced oxime 153. This oxime-based compound on further reaction with dimethyl carbamoyl chloride, mesyl chloride and alkyl chlorides in presence of base under reflux conditions produced the desired compounds 154, 155–156 and 157–165 respectively.
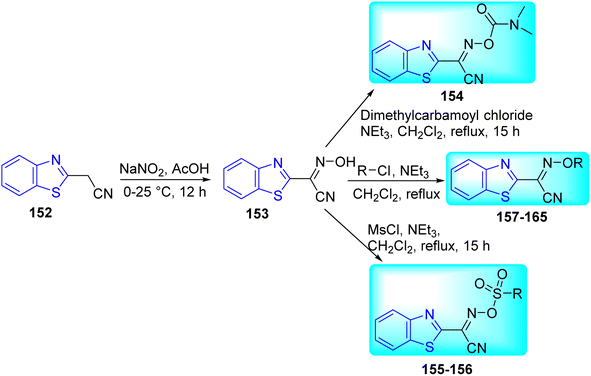 |
| | Scheme 43 Synthesis of benzothiazole oxime derivatives. | |
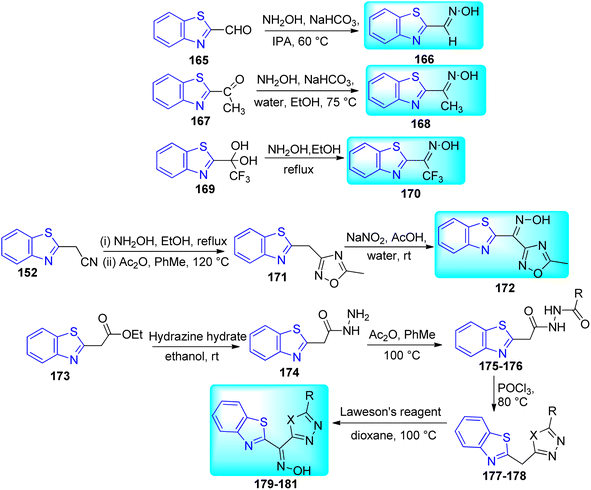 |
| | Scheme 44 Synthesis of oxadiazole and thiadiazole linked benzothiazole analogues. | |
Table 34 Anti-tubercular activity of benzoheterocyclic oxime carbamate analogues
| Compounds |
R |
MIC (μM) |
| 153 |
— |
>160 |
| 154 |
–CON(CH3)2 |
<0.16 |
| 155 |
–SO2N(CH3)2 |
0.30 |
| 156 |
–SO2CH3 |
5.0 |
| 157 |
–CH2COOCH2CH3 |
>160 |
| 158 |
–CH2CON(CH2CH3)2 |
>160 |
| 159 |
–CH3 |
160 |
| 160 |
–Propyl |
>160 |
| 161 |
–Bn |
20 |
| 162 |
2-Picolyl |
>160 |
| 163 |
3-Picolyl |
>160 |
| 164 |
4-Picolyl |
20 |
| RIF |
— |
0.009 |
| INH |
— |
0.14 |
Table 35 Anti-tubercular activity of oxadiazole and thiadiazole linked benzothiazole analogues
Biological activity results of these compounds 154–165 predicted that due to inability to penetrate the Mtb cell wall the free oxime 153 was very poor active whereas its carbamate derivative 154 shown great potency with MIC value lower than 0.16 μM. Whereas sulfamoyl masked derivatives 155 and 156 possess good anti-tubercular activity with MIC value of 0.30 μM and 5.0 μM respectively. When the oxime moiety was masked with alkyl ethers the anti-tubercular activity was decreased this may be due to these alkyl ethers groups are not falling inside the cell (intracellular) and releasing free oxime. These results indicated that the active anti-tubercular species is benzothiazole oxime 153. This study further suggested that there is need to work on these benzothiazole oxime derivatives 154–165 to optimize this chemical series and/or develop formulation strategies to improve permeation across the Mtb cell-wall (Tables 34 and 35).79
A commercially available aldehyde 165 or ketone 167 reacted with hydroxylamine under basic conditions to create free oximes 166, 168 with the nitrile group in 153 replaced by H and Me respectively. A CF3 substituted oxime 170 was prepared from hydrated compound 169 using hydroxyl amine under reflux condition. A reaction of 2-benzothiazoleacetonitrile 152 and hydroxylamine, followed by cyclization with acetic anhydride, was used to create 1, 2, 4-oxadiazole 171 which further get converted to respective oxime 172. Ester 173 and hydrazine were combined to create intermediate 174. After being acylated, the hydrazide was subsequently reacted with POCl3 and Lawesson's reagent to produce oxadiazole and thiadiazoles 179–181 (Scheme 44, Table 35).79
M. J. Zala and co-workers synthesized some novel pyrazolyl-pyrazoline derivatives 187a–d from green method of synthesis. The Vilsmeier–Hack reaction was used to create the starting material, 5-chloro-3-methyl-1-phenyl-1H-pyrazole-4-carbaldehyde 182. Further reaction of 182 with substituted thiophenols 183 and 184 in presence of K2CO3 and DMF produced substituted aromatic aldehydes 185a–b as key intermediates. Substituted aldehydes 185a–b underwent multicomponent one pot reaction with 2-acetyl pyrrole or 2-acetyl-1,3-thiazole 186a–b in presence of sodium hydroxide in ethanol at room temperature under sonification to furnish desired pyrazolylpyrazoline derivatives 187a–d after getting cyclized with 1,3-benzothiazol-2-ylhydrazine 51a (Scheme 45, Table 36).80
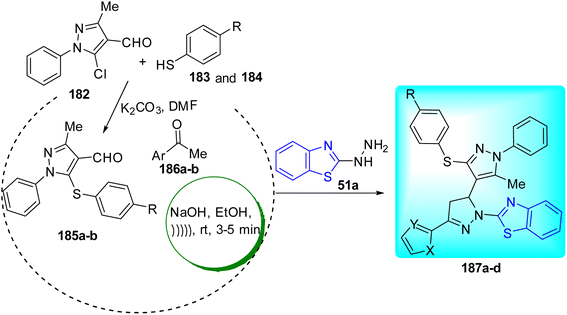 |
| | Scheme 45 Synthesis of pyrazolyl-pyrazoline derivatives of benzothiazole. | |
Table 36 Anti-tubercular evaluation of pyrazolyl-pyrazoline derivatives of benzothiazole
| Compounds |
R |
X |
Y |
% inhibition |
| 187a |
Me |
NH |
CH |
91 |
| 187b |
Me |
S |
N |
96 |
| 187c |
F |
NH |
CH |
75 |
| 187d |
F |
S |
N |
98 |
| RIF |
— |
— |
— |
98 |
| INH |
— |
— |
— |
99 |
Using a Lowenstein–Jensen medium, the synthesized compounds were assessed for their in vitro anti-tubercular activity against the H37Rv strain (Table 36). 187b and 187d exhibited excellent activity with inhibition of 96% and 98% respectively. It is quite interesting to note that the compound 187d can be introduced as new anti-tubercular compound in upcoming years.
Docking studies of ligand 187d revealed that it interacts in an efficient manner with the active site of 4DRE protein of inhA in M. tuberculosis. Basically, the enol-acyl carrier protein reductase inhA of M. tuberculosis is an attractive, validated target for anti-TB drug development. Moreover, direct inhibitors of inhA remain effective against inhA variants with mutations associated with Isoniazid resistance. With very good binding affinity of −10.5 kcal mol−1 ligand 187d can act as an alternative in case of Isoniazid resistance due to mutations in inhA gene (Fig. 19 and 20).
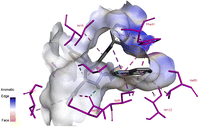 |
| | Fig. 19 3D view of interactions shown by ligand 187d and active site of 4DRE protein. | |
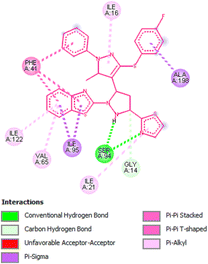 |
| | Fig. 20 2D view of interactions between ligand 187d and various amino acids of 4DRE protein chain. | |
P. R. Kadam and co-workers performed a one pot three component Knoevenagel condensation reaction between 4-hydroxycoumarin 20, substituted aldehydes, and 2-mercapto benzothiazole 44 in presence of L-proline as catalyst to synthesize 3-[(1,3-benzothiazol-2-ylsulfanyl) (phenyl)methyl]-2H-chromen-4-ol derivatives 188a–f (Scheme 46, Table 37).81
 |
| | Scheme 46 Synthesis of [(1,3-benzothiazol-2-ylsulfanyl) (phenyl)methyl]-2H-chromen-4-ol derivatives. | |
Table 37 Anti-tubercular activity of [(1,3-benzothiazol-2-ylsulfanyl) (phenyl)methyl]-2H-chromen-4-ol analoguesa
| Compounds |
R |
100 μg mL−1 |
50 μg mL−1 |
25 μg mL−1 |
12.5 μg mL−1 |
6.25 μg mL−1 |
3.12 μg mL−1 |
1.6 μg mL−1 |
0.8 μg mL−1 |
| S: sensitive, R: resistance. |
| 188a |
H |
S |
R |
R |
R |
R |
R |
R |
R |
| 188b |
Br |
S |
S |
S |
S |
R |
R |
R |
R |
| 188c |
Cl |
S |
S |
S |
S |
S |
R |
R |
R |
| 188d |
OCH3 |
S |
S |
S |
S |
S |
S |
S |
R |
| 188e |
OH |
S |
S |
S |
S |
R |
R |
R |
R |
| 188f |
CH3 |
S |
S |
S |
R |
R |
R |
R |
R |
In vitro anti-tubercular evaluation of all the synthesized compounds against H37Rv strain of M. tuberculosis was done using Microplate Alamar Blue Assay (MABA) technique. Due to the presence of the –OCH3 group, compound 188d demonstrated good activity with a MIC value of 1.6 μg mL−1 compared to the reference drug Streptomycin. Compound 188c and 188b demonstrated inhibition at 6.25 μg mL−1 and 12.5 μg mL−1 while compounds 188a and 188f demonstrated activity at 50 and 25 μg mL−1 (Table 37).
R. Moodley and co-workers synthesized a series of novel benzothiazole-urea-quinoline hybrid molecules via a three-step synthetic process that included an amidation coupling reaction as a crucial step. Initial step started from the reaction of 4,7-dichloroquinoline and various excess diamines to give the intermediate 4-aminoquinoline diamines 190a–e and 191 (Routes A and B respectively). Using the 1,1′carbonyldiimidazoles (CDIs), several 2-amino-6-substituted benzothiazoles 192a–g were converted to benzothiazole-1H-imidazole-1-carboxamide intermediates 193a–g also in excellent yields. The last step involved the synthesis of desired compounds 194a–y from the coupling of 4-aminoquinoline diamines with benzothiazole-1H-imidazole-1-carboxamide derivatives 193a–g (Scheme 47, Table 38).82
 |
| | Scheme 47 Synthesis of benzothiazole-urea-quinoline hybrid analogues. | |
Table 38 Anti-tubercular activity of benzothiazole-urea-quinoline hybrid analoguesa
| Compounds |
X |
Diamine linker |
b7H9/CAS/GLU/Tx 7 days (μM) |
c7H9/ADC/GLU/Tw 7 days (μM) |
Compounds |
X |
Diamine linker |
b7H9/CAS/GLU/Tx 7 days (μM) |
c7H9/ADC/GLU/Tw 7 days (μM) |
| NT: not tested. Protein-deficient Mtb media. Protein rich Mtb media. |
| 194a |
H |
H2N–NH2 |
21.001 |
>125 |
194o |
Cl |
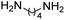 |
7.455 |
23.529 |
| 194b |
CF3 |
4.943 |
6.85 |
194p |
Br |
|
7.812 |
15.609 |
| 194c |
F |
>125 |
>125 |
194q |
F |
|
>125 |
>125 |
| 194d |
NO2 |
>125 |
>125 |
194r |
Cl |
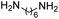 |
7.597 |
14.617 |
| 194e |
CF3 |
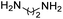 |
NT |
NT |
194s |
Br |
|
8.76 |
20.954 |
| 194f |
Cl |
125 |
>125 |
194t |
F |
|
6.974 |
31.25 |
| 194g |
Br |
8.89 |
14.898 |
194u |
CF3 |
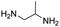 |
0.968 |
5.732 |
| 194h |
F |
62.5 |
62.5 |
194v |
F |
|
>125 |
>125 |
| 194i |
CF3 |
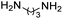 |
7.812 |
12.837 |
194w |
Br |
|
8.191 |
14.001 |
| 194j |
Cl |
4.389 |
11.748 |
194x |
CH3 |
|
7.219 |
10.35 |
| 194k |
CH3 |
31.25 |
62.33 |
194y |
Cl |
|
2.331 |
8.455 |
| 194l |
H |
9.628 |
9.447 |
RIF |
— |
|
0.03 |
0.001 |
| 194m |
Br |
15.924 |
16.863 |
|
| 194n |
F |
>125 |
125 |
|
All the synthesized compounds were evaluated in vitro against H37Rv strain of M. tuberculosis over seven days of incubation in two different media 7H9/CAS/GLU/Tx and 7H9/ADC/GLU/Tw. The main difference among the media was that the former contained tyloxapol (Tx) and casitone (CAS), whereas the later contained Tween-80 (Tw) and albumin-dextrose-catalase (ADC). Compound 194u was found to be most active against tuberculosis with MIC value of 0.968 μM with MIC90 values between 1–10 μM. Thirteen compounds 194b, 194g, 194i–j, 194l, 194o–p, 194r–t and 194x–y demonstrated potential anti-tubercular activity (Table 38). From cytotoxicity assay it was observed that compound 194t exhibited the highest cell viability at the MIC90 (92%) as compared to 194r (72%) and 194s (76%). In silico ADME and drug likeness properties suggested high percentage human oral absorption (>80%). Most of these compounds fulfilled Lipinski's rules for drug-like properties.
Conclusions and future perspectives
It became evidenced from above discussions that, benzothiazole nucleus is an important structural motif in medicinal chemistry for the search of new anti-tubercular compounds. Therefore, various analogues of benzothiazole nucleus have been synthesized and evaluated for their anti-tubercular activity by several research groups. There is much scope in benzothiazole derivatives as a source of molecular targets and research into this nucleus has recently received a lot of attention. Carbanilide derivatives of benzothiazole exhibited excellent anti-tubercular activity with MIC of 0.78 μg mL−1 as compared to Ethambutol (1.56 μg mL−1). Benzothiazole based Schiff bases were also potent against Mtb with MIC of 0.8–1.6 μg mL−1 which was better than standard drug Streptomycin (6.25 μg mL−1). Azo-ester complexes of benzothiazoles emerges as potential anti-TB molecules with good docking score and MIC value of 1.6 μg mL−1, this activity was much better than the standard drugs Streptomycin (MIC 6.25 μg mL−1) and Pyrazinamide (MIC 3.125 μg mL−1). Further coumarin based azo dye molecules were found as excellent anti-tubercular compounds along with good docking score and MIC value of 1.6 μg mL−1 as compared to the standard drug Streptomycin (6.25 μg mL−1). Pyrazole conjugates of benzothiazole derivatives were identified another potent molecules having better potency than standard drugs like Streptomycin and Ciprofloxacin with a MIC value of 1.6 μg mL−1. Among the hydrazine sub-series, compound containing CF3 was found to exhibit outstanding activity in both mediums. It is also evidenced from this discussion that, most of the synthesized compounds having C-6 substitution of benzothiazole ring is more potent than C-6 unsubstituted compounds. Benzothiazole based azo dyes and their metal complexes were also observed to inhibit the growth of M. tuberculosis. We performed docking of some selected most active compounds in order to find potent inhibitory action against DprE1, enol–acyl carrier protein reductase inhA and arabinosyl transferase. From the molecular docking studies it can be concluded that the selected compounds can be taken as lead to work and develop potent anti-tubercular molecules, which may works against drug resistance strains as well. As highlighted in current review that, recently benzothiazole derivatives are becoming molecules of interest for drug development against tuberculosis. However, further research is needed to completely understand the molecular mechanism of these active compounds to fully comprehend the molecular basis of the anti-tubercular activity in order to develop new anti-tubercular drugs that can obliterate mycobacterial infections.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are thankful to Jawaharlal Nehru University for providing facilities to write this detailed review on tuberculosis. RY, DM is grateful to JNU for institute fellowships. KS is thankful to CSIR and RT is thankful to UGC New Delhi for Research Fellowships. We are grateful to Ultra International and Sanganeria Foundation for supporting lab furniture to Glycochemistry Laboratory at JNU.
References
- S. H. E. Kaufmann and A. J. McMichael, Nat. Med., 2005, 11, S33 CrossRef CAS PubMed.
- A. Zumla, P. Nahid and S. T. Cole, Nat. Rev. Drug Discovery, 2013, 12, 388–404 CrossRef CAS PubMed.
- S. Landge, A. B. Mullick, K. Nagalapur, J. Neres, V. Subbulakshmi, K. Murugan, A. Ghosh, C. Sadler, M. D. Fellows, V. Humnabadkar, J. Mahadevaswamy, P. Vachaspati, S. Sharma, P. Kaur, M. Mallya, S. Rudrapatna, D. Awasthy, V. K. Sambandamurthy, F. Pojer, S. T. Cole, T. S. Balganesh, B. G. Ugarkar, V. Balasubramanian, B. S. Bandodkar, M. Panda and V. Ramachandran, Bioorg. Med. Chem., 2015, 23, 7694–7710 CrossRef CAS PubMed.
- L. D’Ambrosio, R. Centis, G. Sotgiu, E. Pontali, A. Spanevello and G. B. Migliori, ERJ Open Res., 2015, 1, 00010–02015 Search PubMed.
- K. N. Venugopala, M. A. Khedr, M. Pillay, S. K. Nayak, S. Chandrashekharappa, B. E. Aldhubiab, S. Harsha, M. Attimard and B. Odhav, J. Biomol. Struct. Dyn., 2019, 37, 1830–1842 CrossRef CAS PubMed.
- R. J. O’BRIEN and P. P. NUNN, Am. J. Respir. Crit. Care Med., 2001, 163, 1055–1058 CrossRef PubMed.
- K. A. Gaidhani, M. Harwalkar and P. S. Nirgude, World J. Pharm. Res., 2014, 3, 5041–5048 Search PubMed.
- B. Berk, Y. A. Tahirovic, E. F. Bülbül and S. N. Biltekin, Acta Pharm. Sci., 2017, 55, 17–28 CAS.
- X. Gao, J. Liu, X. Zuo, X. Feng and Y. Gao, Molecules, 2020, 25, 1674, DOI:10.3390/molecules25071675.
- R. S. Keri, S. A. Patil, S. Budagumpi and B. M. Nagaraja, Chem. Biol. Drug Des., 2015, 86, 410–423 CrossRef CAS PubMed.
- M. A. Khedr, M. Pillay, S. Chandrashekharappa, D. Chopra, B. E. Aldhubiab, M. Attimarad, O. I. Alwassil, K. Mlisana, B. Odhav and K. N. Venugopala, J. Biomol. Struct. Dyn., 2018, 36, 2163–2178 CrossRef CAS PubMed.
- S. K. Nayak, K. N. Venugopala, D. Chopra and T. N. G. Row, CrystEngComm, 2011, 13, 591–605 RSC.
- S. K. Nayak, K. N. Venugopala, T. Govender, H. G. Kruger and G. E. M. Maguire, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, o70 CrossRef CAS PubMed.
- V. N. Telvekar, V. K. Bairwa, K. Satardekar and A. Bellubi, Bioorg. Med. Chem. Lett., 2012, 22, 649–652 CrossRef CAS.
- M. Sharma, P. Chaudhary, P. K. Sharma, A. Sharma and J. Varshney, Int. J. Pharm. Res., 2010, 2, 5–11 Search PubMed.
- I. Yildiz-Oren, I. Yalcin, E. Aki-Sener and N. Ucarturk, Eur. J. Med. Chem., 2004, 39, 291–298 CrossRef CAS PubMed.
- L. Racanè, V. Tralić-Kulenović, D. W. Boykin and G. Karminski-Zamola, Molecules, 2003, 8, 342–349 CrossRef.
- R. S. Keri, B. S. Sasidhar, B. M. Nagaraja and M. A. Santos, Eur. J. Med. Chem., 2015, 100, 257–269 CrossRef CAS PubMed.
- A. El-Mekabaty, M. A. Sofan, A. M. Hasel and S. B. Said, ChemistrySelect, 2021, 6, 2569–2575 CrossRef CAS.
- J. B. Baell, A. J. Harvey and R. S. Norton, J. Comput.-Aided Mol. Des., 2002, 16, 245–262 CAS.
- F. Delmas, A. Avellaneda, C. Di Giorgio, M. Robin, E. De Clercq, P. Timon-David and J. P. Galy, Eur. J. Med. Chem., 2004, 39, 685–690 CrossRef CAS PubMed.
- K. Oketani, N. Nagakura, K. Harada and T. Inoue, Eur. J. Pharmacol., 2001, 422, 209–216 CrossRef CAS PubMed.
- R. Paramashivappa, P. Phani Kumar, P. V. Subba Rao and A. Srinivasa Rao, Bioorg. Med. Chem. Lett., 2003, 13, 657–660 CrossRef CAS PubMed.
- H. Nath and S. Ryoo, in Tuberculosis - Current Issues in Diagnosis and Management, ed. B. H. Mahboub and M. G. Vats, IntechOpen Limited, London, UK, 2013, pp. 163–180, DOI:10.5772/54960.
- M. A. Espinal, A. Laszlo, L. Simonsen, F. Boulahbal, S. J. Kim, A. Reniero, S. Hoffner, H. L. Rieder, N. Binkin, C. Dye, R. Williams and M. C. Raviglione, N. Engl. J. Med., 2001, 344, 1294–1303 CrossRef CAS.
- H. W. Al-Humadi, R. J. Al-Saigh and A. W. Al-Humadi, Front. Pharmacol., 2017, 8, 1–10 Search PubMed.
- R. V. Chikhale, M. A. Barmade, P. R. Murumkar and M. R. Yadav, J. Med. Chem., 2018, 61, 8563–8593 CrossRef CAS PubMed.
- H. B. Rode, D. M. Lade, R. Grée, P. S. Mainkar and S. Chandrasekhar, Org. Biomol. Chem., 2019, 17, 5428–5459 RSC.
- C. Vilchèze, Appl. Sci., 2020, 10, 2278 CrossRef.
- A. Telenti, P. Imboden, F. Marchesi, L. Matter, K. Schopfer, T. Bodmer, D. Lowrie, M. J. Colston and S. Cole, Lancet, 1993, 341, 647–651 CrossRef CAS.
- Y. Zhang, M. M. Wade, A. Scorpio, H. Zhang and Z. Sun, J. Antimicrob. Chemother., 2003, 52, 790–795 CrossRef CAS PubMed.
- W. Shi, X. Zhang, X. Jiang, H. Yuan, J. S. Lee, C. E. Barry, H. Wang, W. Zhang and Y. Zhang, Science, 2011, 333, 1630–1632 CrossRef CAS PubMed.
- J. G. Y. Chan, X. Bai and D. Traini, Expert Opin. Drug Delivery, 2014, 11, 421–431 CrossRef CAS.
- L. Yan, Med. Chem., 2015, 5, 412–414 Search PubMed.
- N. Honore and S. T. Cole, Antimicrob. Agents Chemother., 1994, 38, 238–242 CrossRef CAS PubMed.
- A. Sowajassatakul, T. Prammananan, A. Chaiprasert and S. Phunpruch, BMC Microbiol., 2014, 14, 1–7 CrossRef.
- A. D. Pranger, R. Van Altena, R. E. Aarnoutse, D. Van Soolingen, D. R. A. Uges, J. G. W. Kosterink, T. S. Van Der Werfe and J. W. C. Alffenaar, Eur. Respir. J., 2011, 38, 888–894 CrossRef CAS.
- M. H. Scheetz, S. A. Knechtel, M. Malczynski, M. J. Postelnick and C. Qi, Antimicrob. Agents Chemother., 2008, 52, 2256–2259 CrossRef CAS PubMed.
- F. Wang, R. Langley, G. Gulten, L. G. Dover, G. S. Besra, W. R. Jacobs and J. C. Sacchettini, J. Exp. Med., 2007, 204, 73–78 CrossRef CAS PubMed.
- K. A. Wolff and L. Nguyen, Expert Rev. Anti-Infect. Ther., 2012, 10, 971–981 CrossRef CAS.
- D. M. Patel, S. D. Patel, P. S. Jaiswal and K. J. Brahmbhatt, Int. J. Drug Dev. Res., 2012, 4, 76–91 Search PubMed.
- R. Chikhale, S. Menghani, R. Babu, R. Bansode, G. Bhargavi, N. Karodia, M. V. Rajasekharan, A. Paradkar and P. Khedekar, Eur. J. Med. Chem., 2015, 96, 30–46 CrossRef CAS PubMed.
- F. M. Shaikh, N. B. Patel, G. Sanna, B. Busonera, P. La Colla and D. P. Rajani, Med. Chem. Res., 2015, 24, 3129–3142 CrossRef CAS.
- H. A. Abdel-Aziz, W. M. Eldehna, M. Fares, S. T. A. Al-Rashood, K. A. Al-Rashood, M. M. Abdel-Aziz and D. H. Soliman, Int. J. Mol. Sci., 2015, 16, 8719–8743 CrossRef CAS.
- A. B. Patel and R. M. Raval, Res. Chem. Intermed., 2016, 42, 2163–2175 CrossRef CAS.
- S. Pancholia, T. M. Dhameliya, P. Shah, P. S. Jadhavar, J. P. Sridevi, P. Yogeshwari, D. Sriram and A. K. Chakraborti, Eur. J. Med. Chem., 2016, 116, 187–199 CrossRef CAS.
- M. N. Bhoi, M. A. Borad, E. A. Pithawala and H. D. Patel, Arabian J. Chem., 2019, 12, 3799–3813 CrossRef CAS.
- M. N. Bhoi, M. A. Borad, D. J. Jethava, P. T. Acharya, E. A. Pithawala, C. N. Patel, H. A. Pandya and H. D. Patel, Eur. J. Med. Chem., 2019, 177, 12–31 CrossRef CAS PubMed.
- G. Samala, P. B. Devi, S. Saxena, N. Meda, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2016, 24, 1298–1307 CrossRef CAS PubMed.
- G. Samala, R. Nallangi, P. B. Devi, S. Saxena, R. Yadav, J. P. Sridevi, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2014, 22, 4223–4232 CrossRef CAS PubMed.
- A. Yardily, V. Anu Danie, B. A. Rosy and T. F. Abbs Fen Reji, J. Saudi Chem. Soc., 2016, 20, 278–281 CrossRef CAS.
- V. M. Patel and N. B. Patel, Chem. Biol. Interface, 2017, 7, 145–153 Search PubMed.
- S. S. Jawoor, S. A. Patil and S. S. Toragalmath, J. Coord. Chem., 2018, 71, 271–283 CrossRef CAS.
- R. S. Reshma, V. U. Jeankumar, N. Kapoor, S. Saxena, K. A. Bobesh, A. R. Vachaspathy, P. E. Kolattukudy and D. Sriram, Bioorg. Med. Chem., 2017, 25, 2761–2771 CrossRef CAS.
- A. C. Pinheiro, M. V. N. de Souza, M. C. S. Lourenço, C. F. da Costa, T. C. Baddeley, J. N. Low, S. M. S. V. Wardell and J. L. Wardell, J. Mol. Struct., 2019, 1178, 655–668 CrossRef CAS.
- T. M. Dhameliya, R. Tiwari, A. Banerjee, S. Pancholia, D. Sriram, D. Panda and A. K. Chakraborti, Eur. J. Med. Chem., 2018, 155, 364–380 CrossRef CAS.
- M. N. Matada and K. Jathi, J. Mol. Struct., 2019, 1180, 196–208 CrossRef CAS.
- M. Bhat and S. L. Belagali, Future Med. Chem., 2018, 10, 71–87 CrossRef CAS PubMed.
- M. Krause, H. Foks, E. Augustynowicz-Kopeć, A. Napiórkowska, M. Szczesio and K. Gobis, Molecules, 2018, 23, 1–15 CrossRef PubMed.
- J. Graham, C. E. Wong, J. Day, E. McFaddin, U. Ochsner, T. Hoang, C. L. Young, W. Ribble, M. A. DeGroote, T. Jarvis and X. Sun, Bioorg. Med. Chem. Lett., 2018, 28, 3177–3181 CrossRef CAS.
- S. Deng, H. Chen, X. Ma, Y. Zhou, K. Yang, Y. Lan and Q. Song, Chem. Sci., 2019, 10, 6828–6833 RSC.
- A. P. Chavan, R. R. Deshpande, N. A. Borade, A. Shinde, P. C. Mhaske, D. Sarkar and V. D. Bobade, Med. Chem. Res., 2019, 28, 1873–1884 CrossRef CAS.
- J. Gawad and C. Bonde, Synth. Commun., 2019, 49, 2696–2708 CrossRef CAS.
- D. J. Jethava, P. T. Acharya, M. S. Vasava, M. N. Bhoi, Z. A. Bhavsar, S. K. Rathwa, D. P. Rajani and H. D. Patel, J. Mol. Struct., 2019, 1184, 168–192 CrossRef CAS.
- K. Hazra and B. Nandha, Int. J. Chem., 2020, 59, 1388–1399 Search PubMed.
- B. M. Sahoo, B. K. Banik, A. K. Mahato, C. N. Shanthi and B. C. Mohantad, in Green Approaches in Medicinal Chemistry for Sustainable Drug Design, Elsevier, 2020, pp. 779–818 Search PubMed.
- P. T. Acharya, D. J. Jethava, M. S. Vasava, Z. A. Bhavsar, M. N. Bhoi, D. P. Rajani and H. D. Patel, Int. J. Chem., 2020, 59, 1721–1737 Search PubMed.
- B. N. Ravi, J. Keshavayya and N. M. Mallikarjuna, J. Inorg. Organomet. Polym. Mater., 2020, 30, 3781–3796 CrossRef CAS.
- A. B. Velappan, D. Datta, R. Ma, S. Rana, K. S. Ghosh, N. Hari, S. G. Franzblau and J. Debnath, Bioorg. Chem., 2020, 103, 104170 CrossRef CAS PubMed.
- M. R. Maliyappa, J. Keshavayya, N. M. Mallikarjuna and I. Pushpavathi, J. Mol. Struct., 2020, 1205, 127576 CrossRef CAS.
- M. A. Abozeid, A. A. El-Sawi, M. Abdelmoteleb, H. Awad, M. M. Abdel-Aziz, A. R. Hassan Abdel-Rahman and E. S. Ibrahim El-Desoky, RSC Adv., 2020, 10, 42998–43009 RSC.
- J. K. Suyambulingam, R. Karvembu, N. S. P. Bhuvanesh, I. V. M. V. Enoch, P. M. Selvakumar, D. Premnath, C. Subramanian, P. Mayakrishnan, S. H. Kim and I. M. Chung, J. Adhes. Sci. Technol., 2020, 34, 2590–2612 CrossRef CAS.
- O. Nagaraja, Y. D. Bodke, I. Pushpavathi and S. Ravi Kumar, Heliyon, 2020, 6, e042456 CrossRef.
- M. Bhat and S. L. Belagali, Anti-Infect. Agents, 2019, 18, 15–23 CrossRef.
- H. Chen, B. Wang, P. Li, H. Yan, G. Li, H. Huang and Y. Lu, Bioorg. Med. Chem. Lett., 2021, 40, 1–6 Search PubMed.
- S. V. Mamatha, S. L. Belagali and M. Bhat, Anti-Infect. Agents, 2019, 18, 362–374 Search PubMed.
- B. Manjunatha, Y. D. Bodke, O. Nagaraja, T. N. Lohith, G. Nagaraju and M. A. Sridhar, J. Mol. Struct., 2021, 1246, 131170 CrossRef.
- S. A. Satyadev, Int. J. Pharm. Res., 2021, 12, 576–586 CrossRef CAS.
- R. Van Der Westhuyzen, A. Mabhula, P. M. Njaria, R. Müller, D. Ngumbu Muhunga, D. Taylor, N. Lawrence, M. Njoroge, C. Brunschwig, A. Moosa, V. Singh, S. P. S. Rao, U. H. Manjunatha, P. W. Smith, D. F. Warner, L. J. Street and K. Chibale, J. Med. Chem., 2021, 64, 9444–9457 CrossRef CAS PubMed.
- M. J. Zala and J. J. Vora, Russ. J. Org. Chem., 2021, 57, 1725–1732 CrossRef CAS.
- P. R. Kadam, Y. D. Bodke, M. D. Naik, O. Nagaraja and B. Manjunatha, Results Chem., 2022, 4, 100303 CrossRef CAS.
- R. Moodley, C. Mashaba, G. H. Rakodi, N. B. Ncube, M. V. Maphoru, M. O. Balogun, A. Jordan, D. F. Warner, R. Khan and M. Tukulula, Pharmaceuticals, 2022, 15, 576 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*






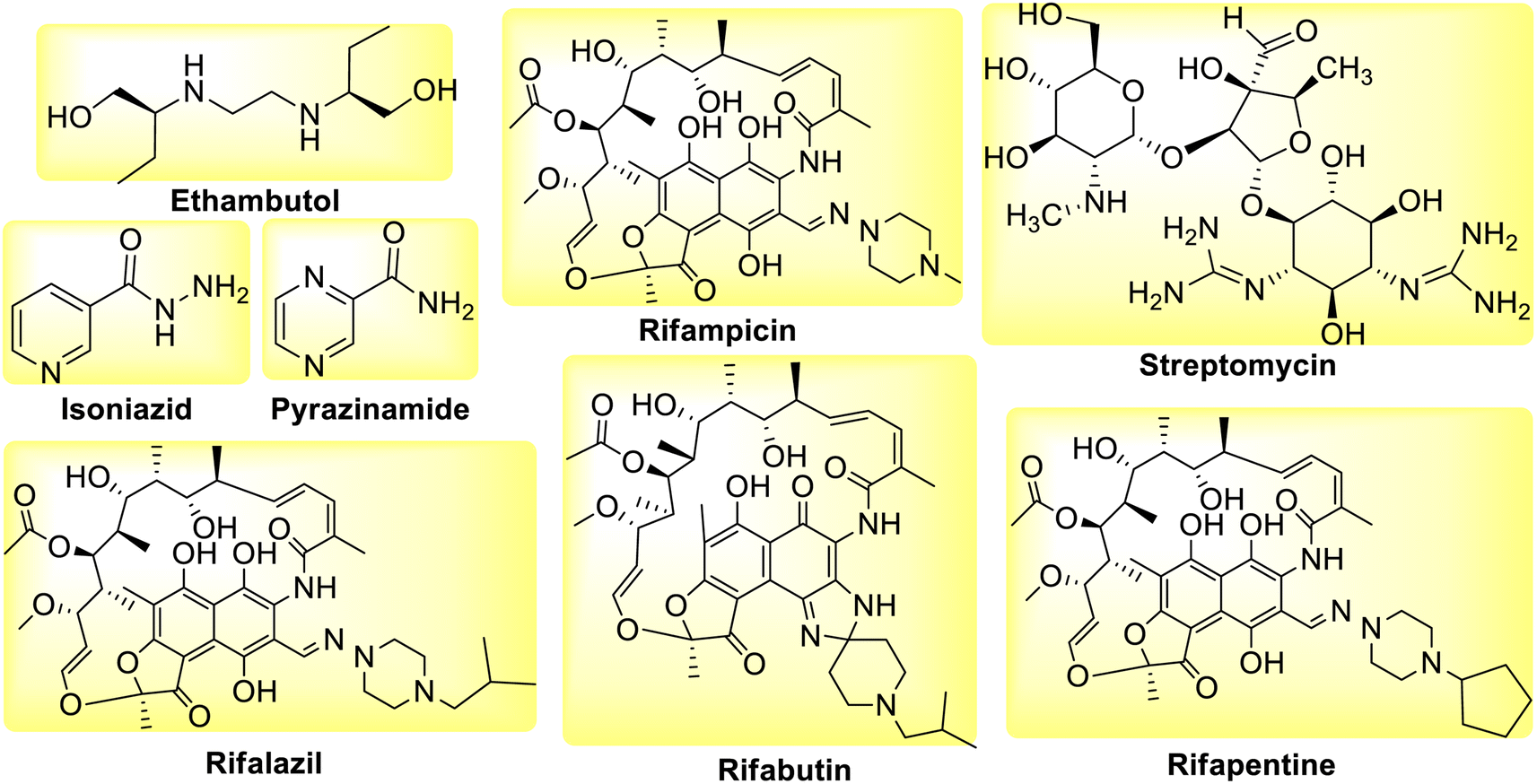
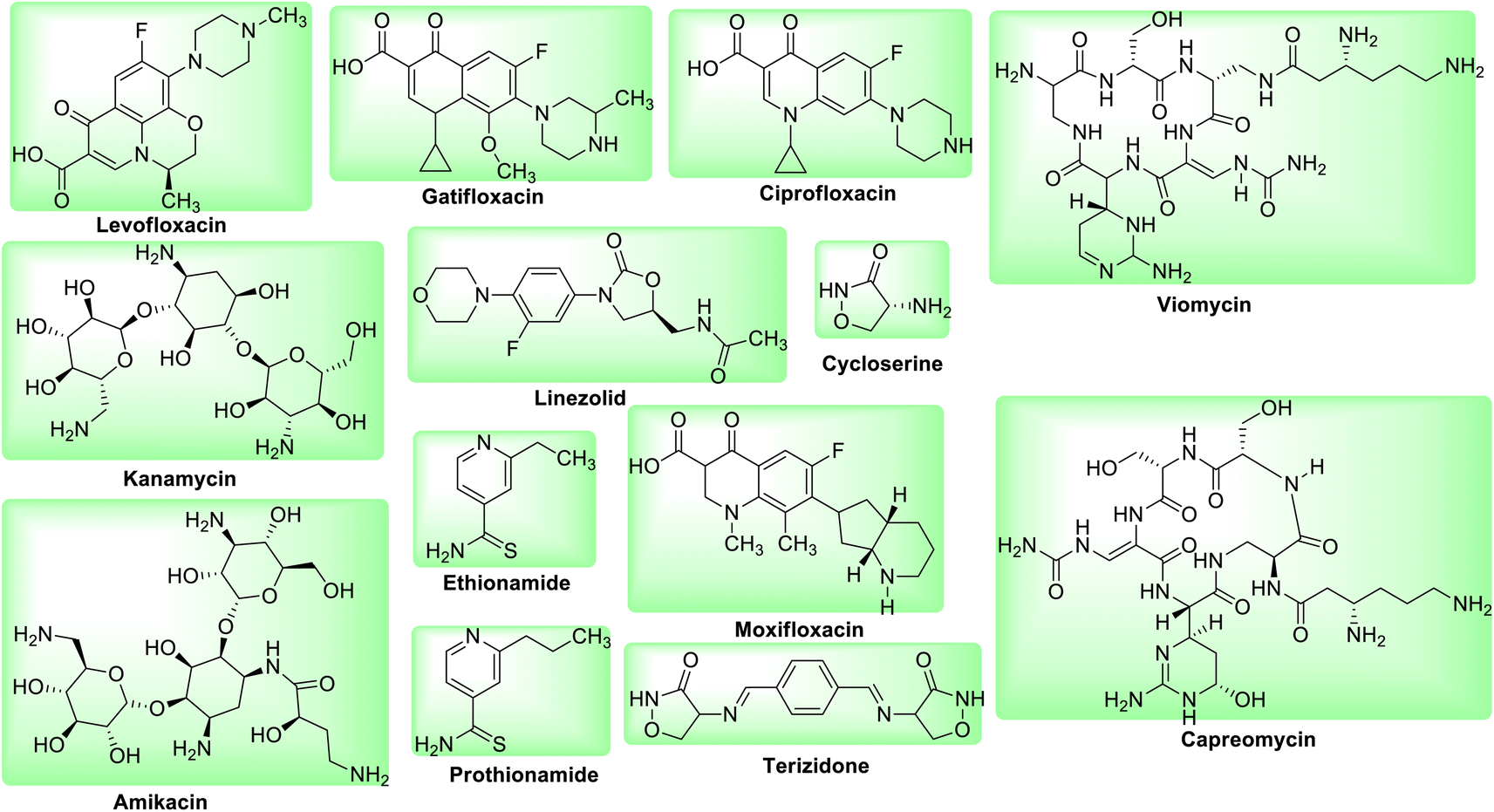
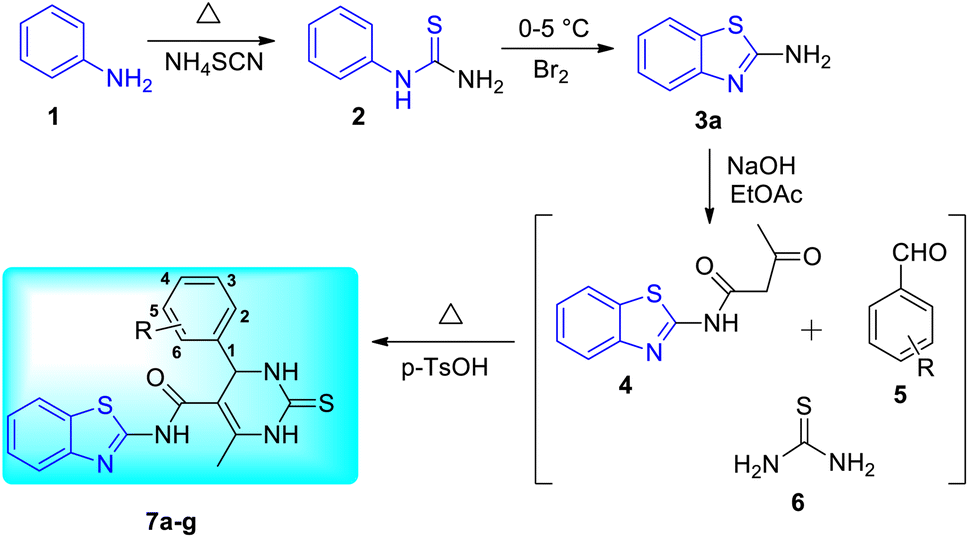
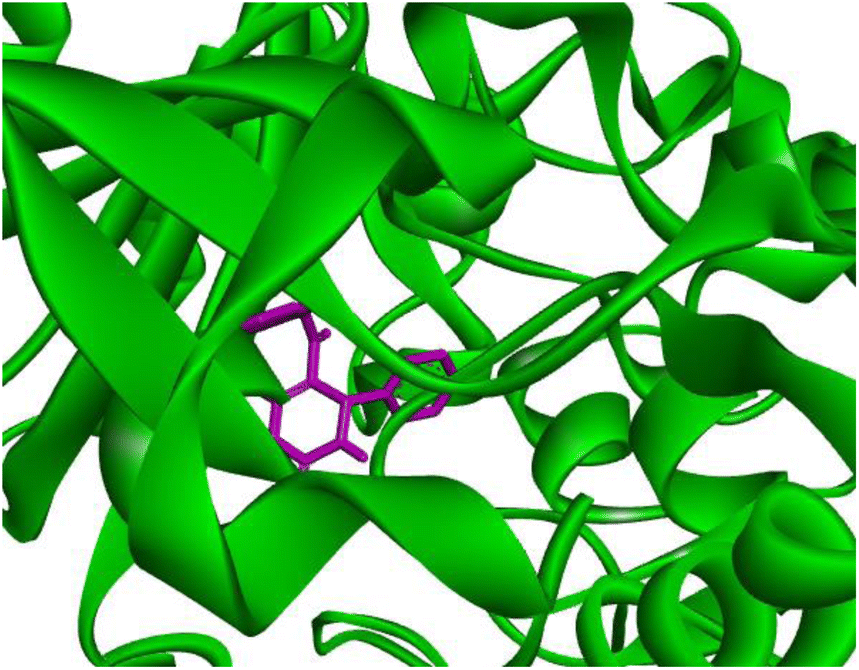
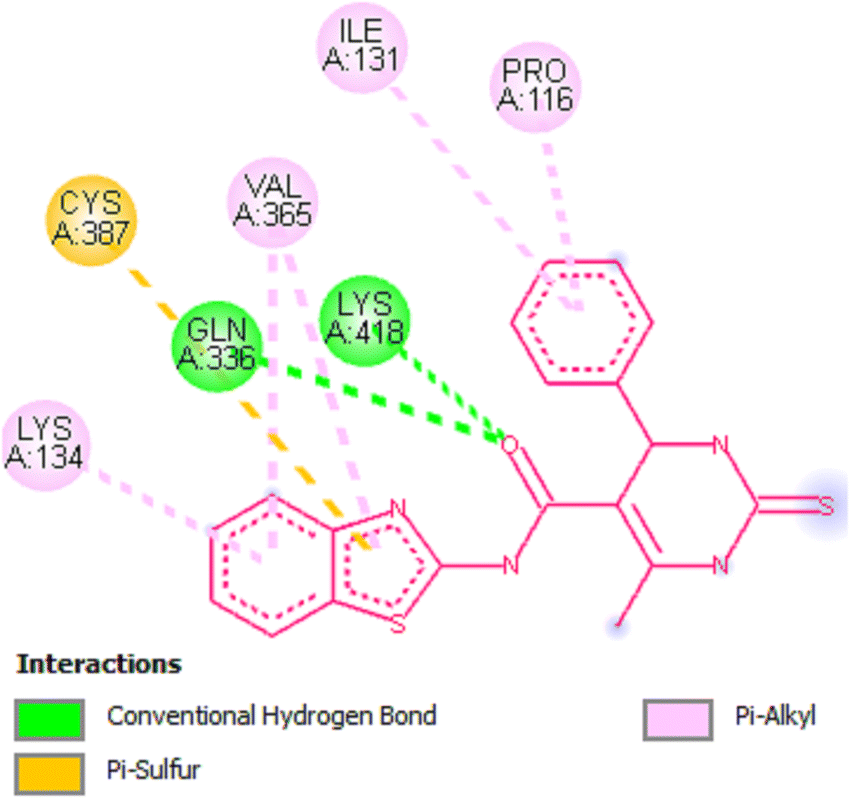
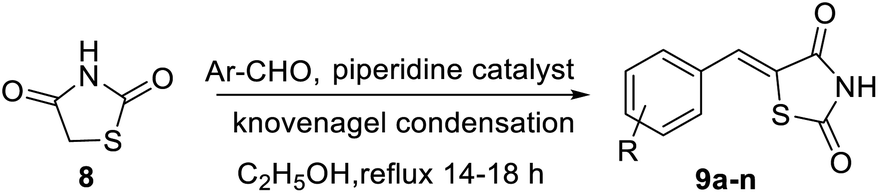
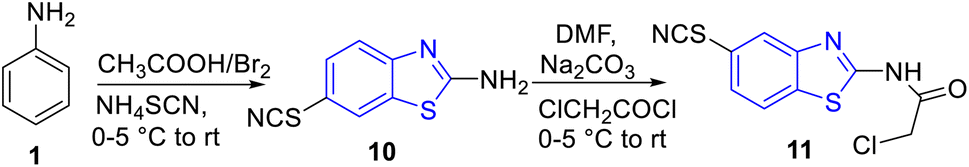
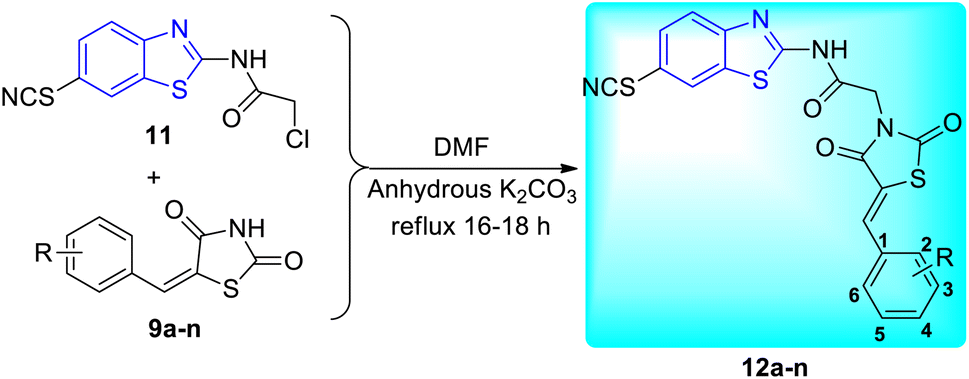
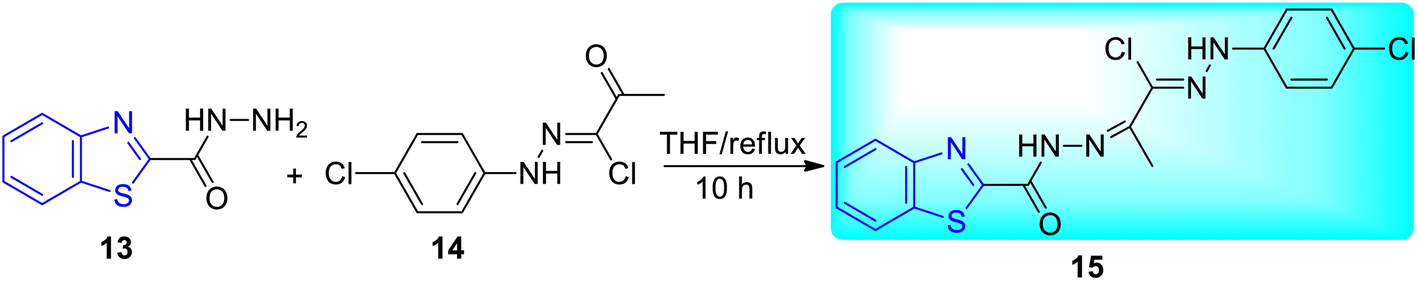
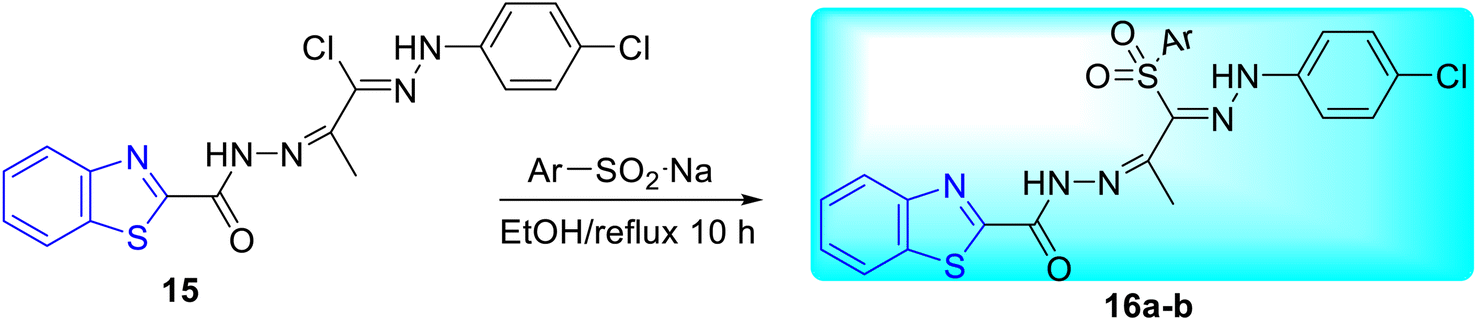
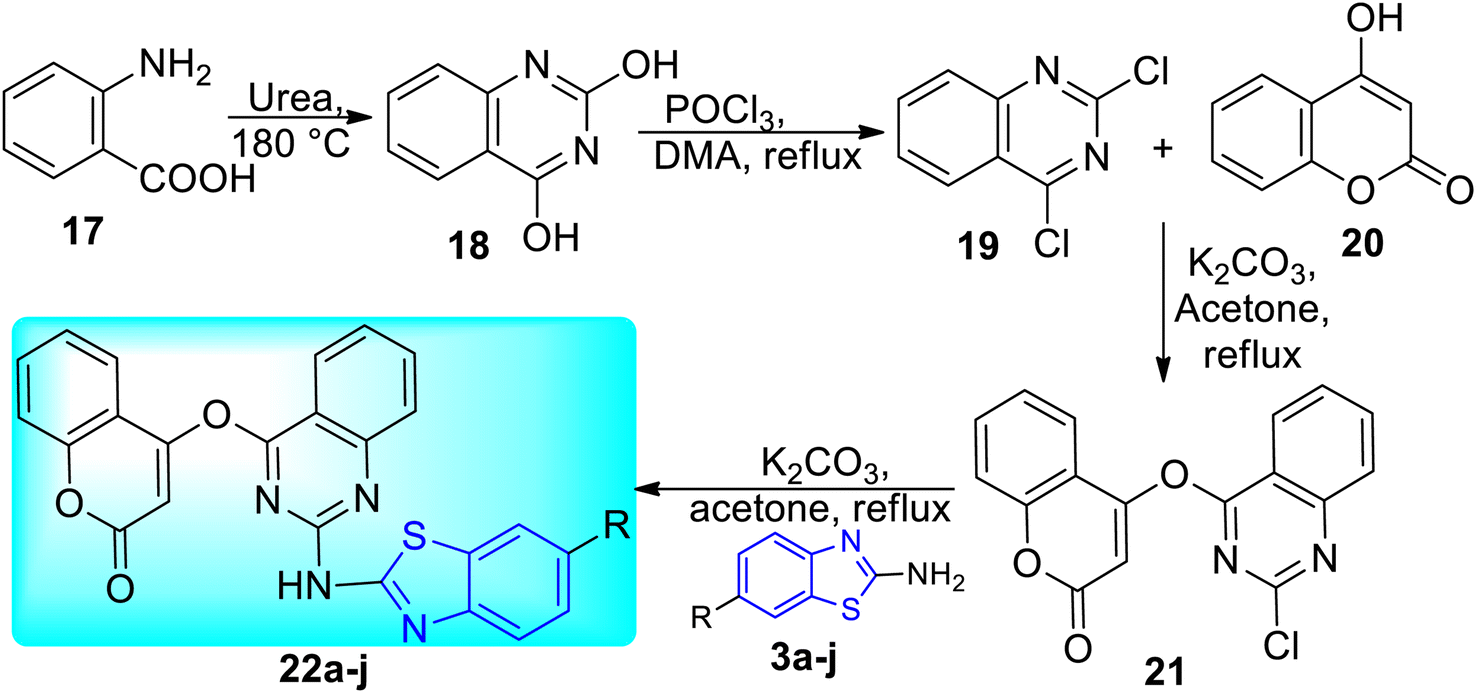
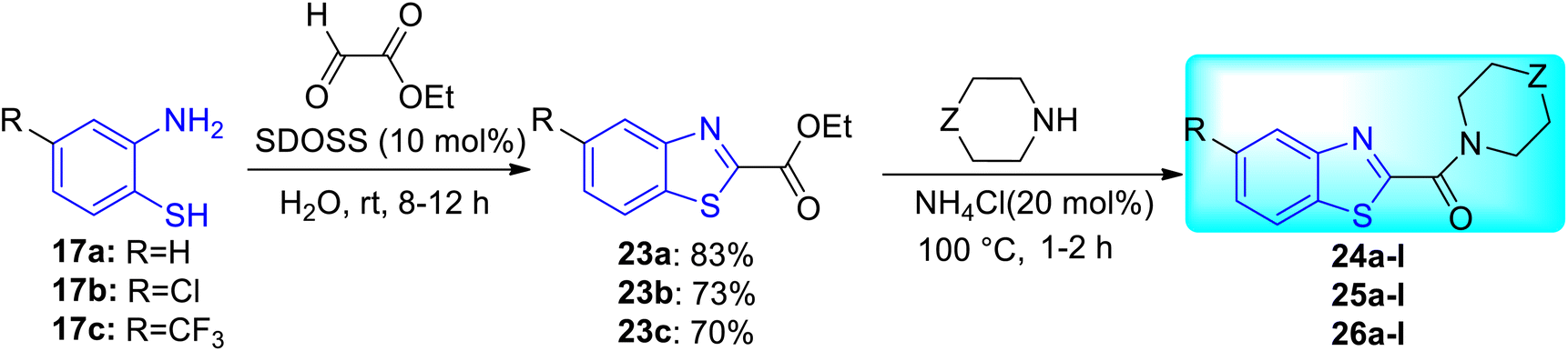
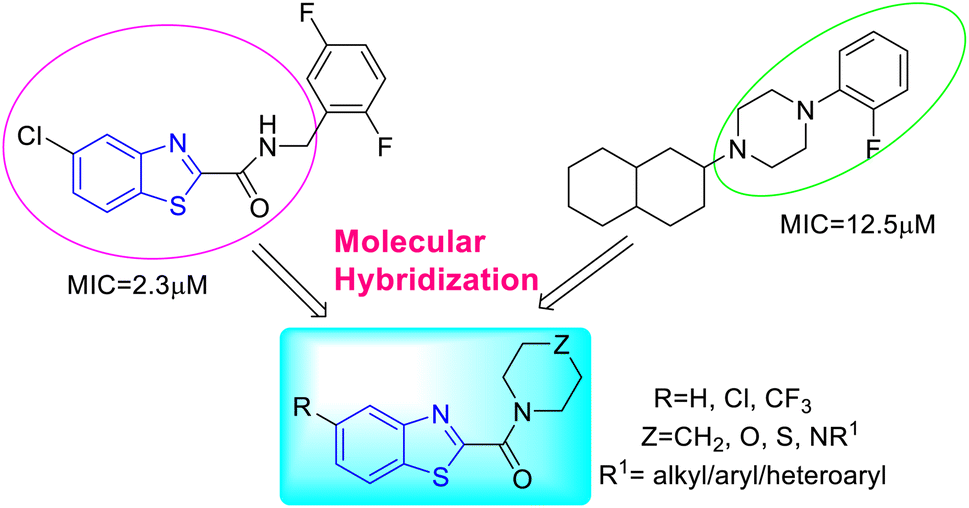

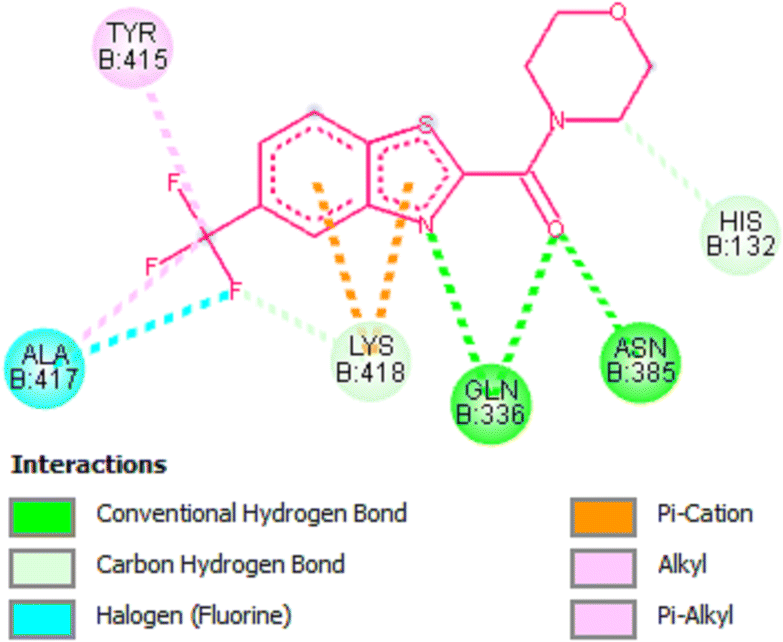
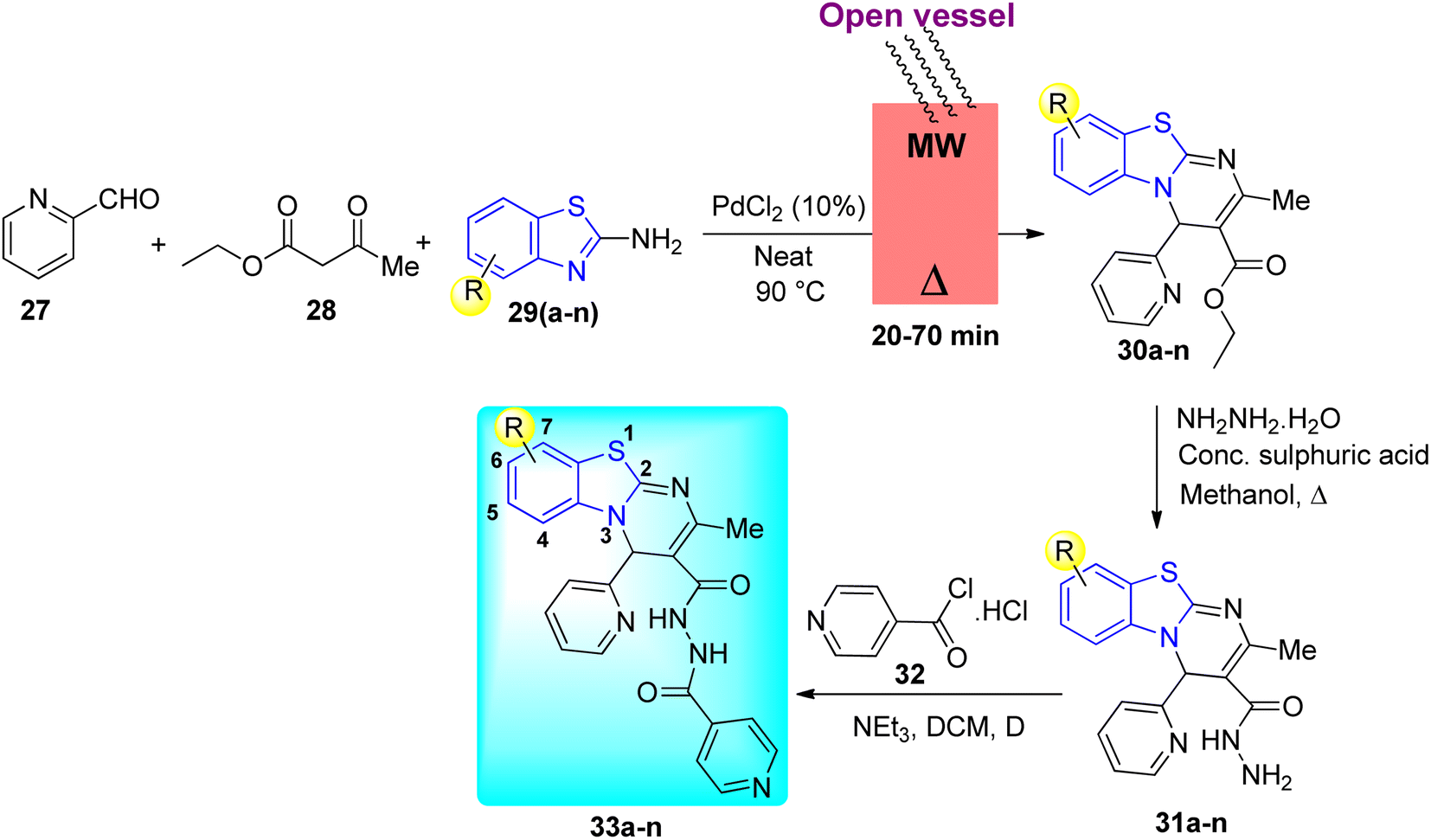
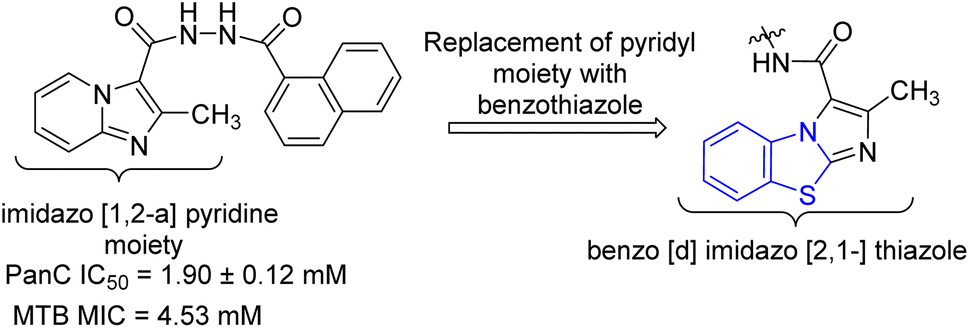
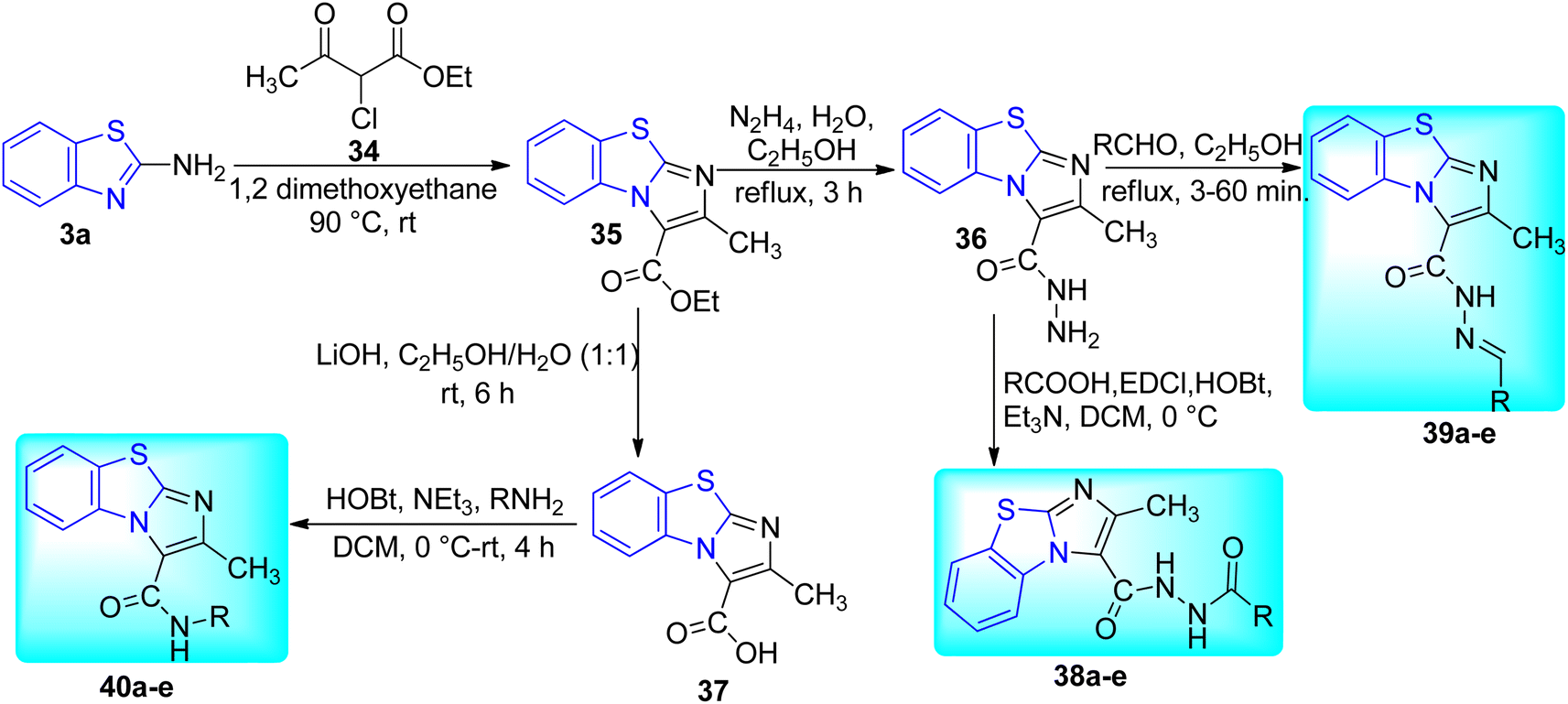

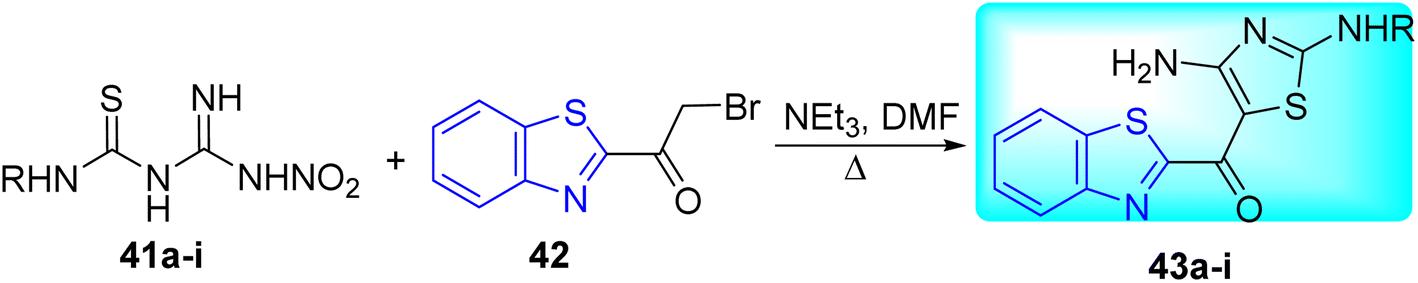
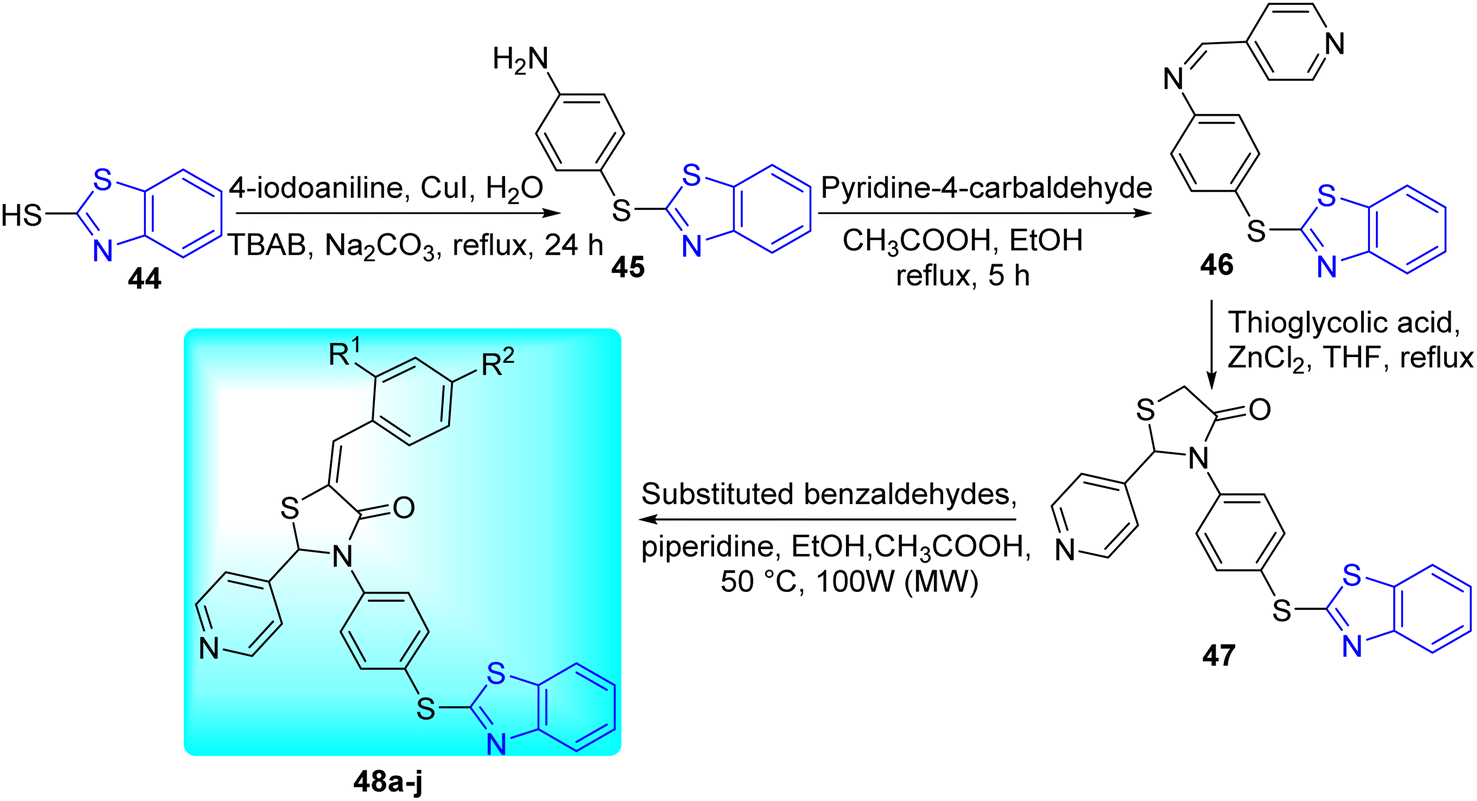
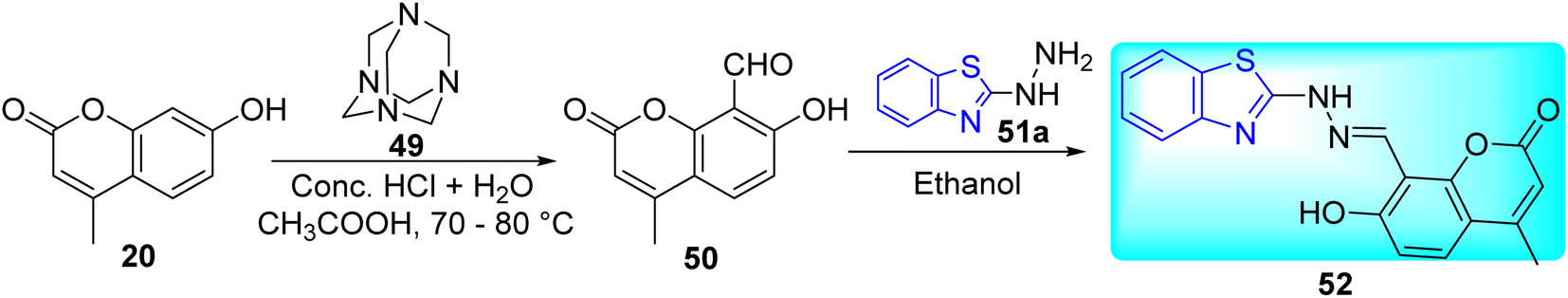
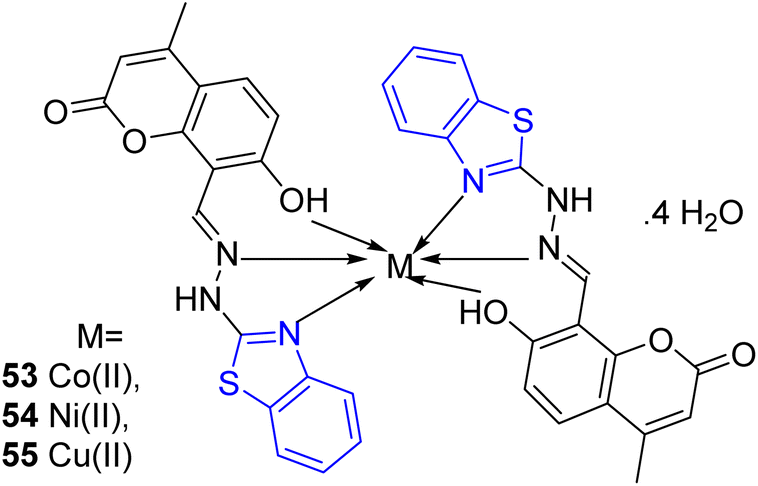
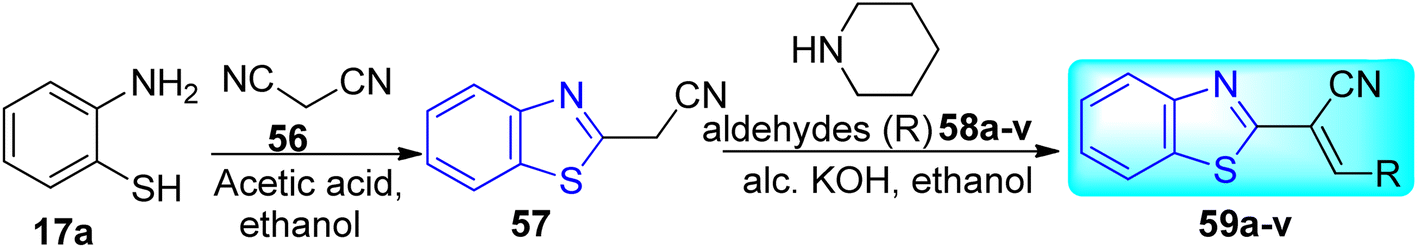


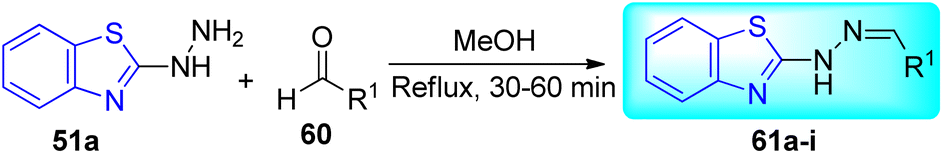
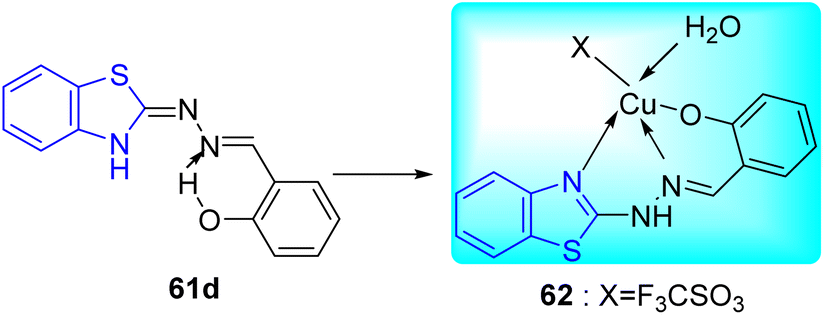
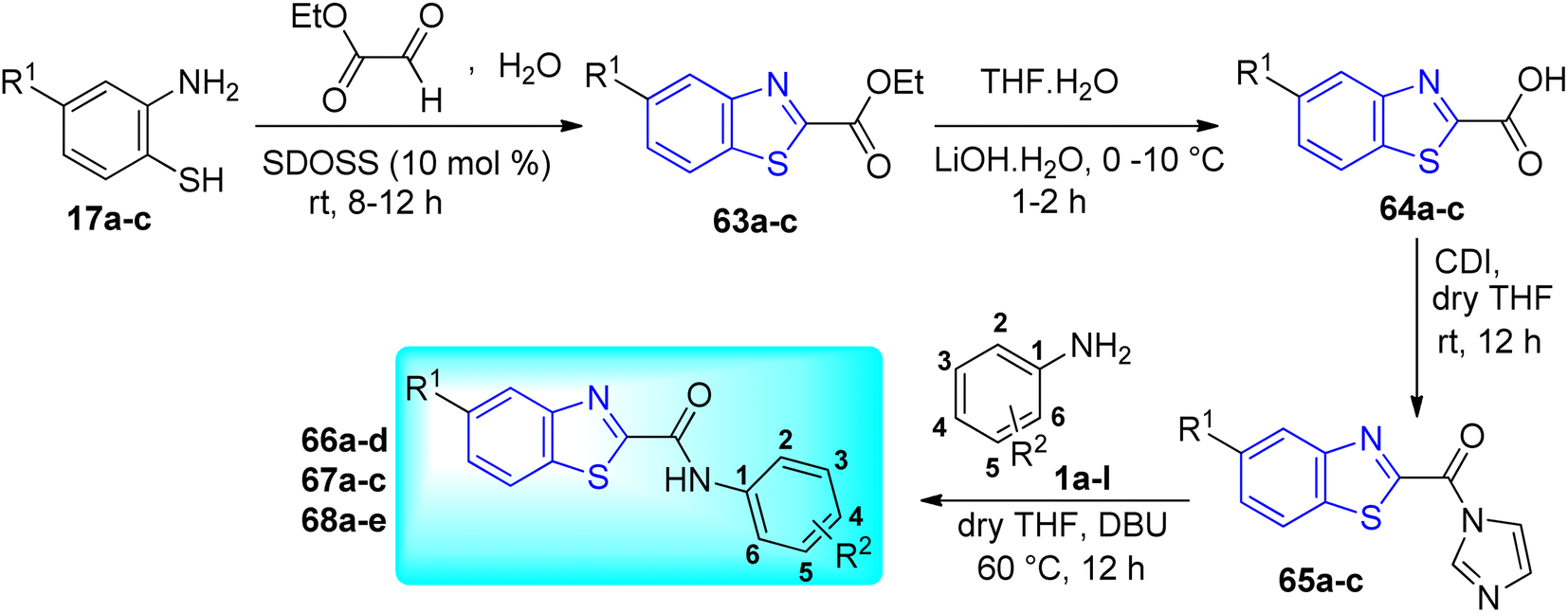
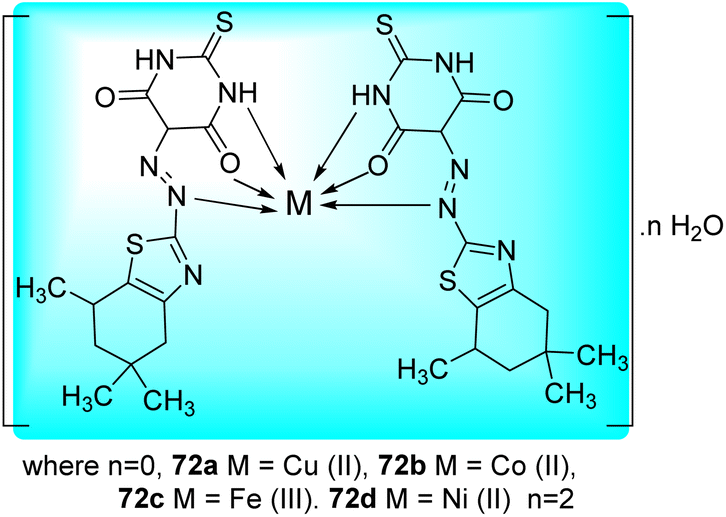
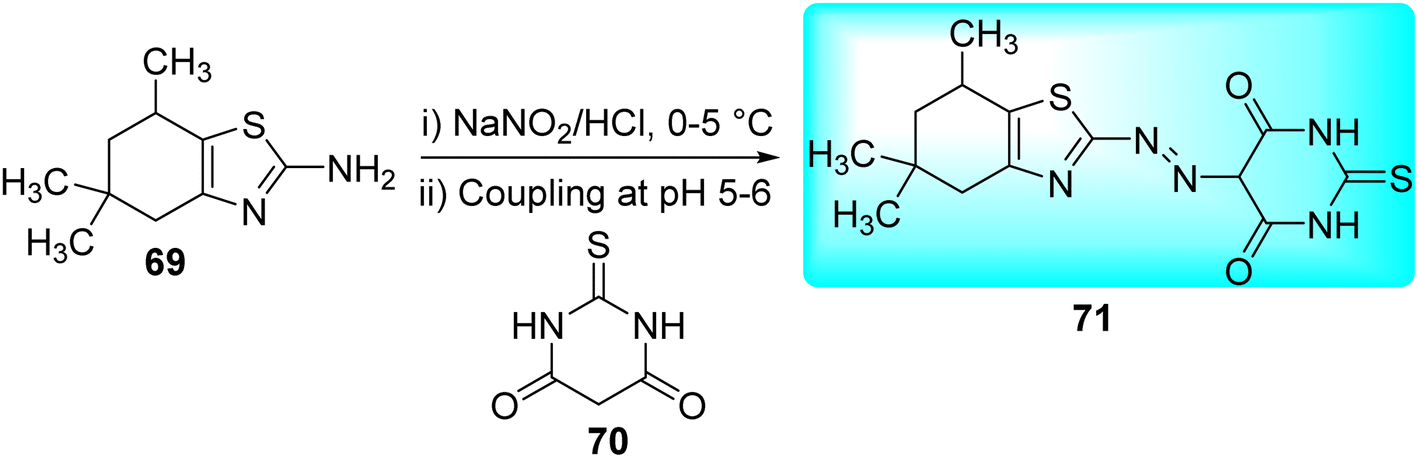
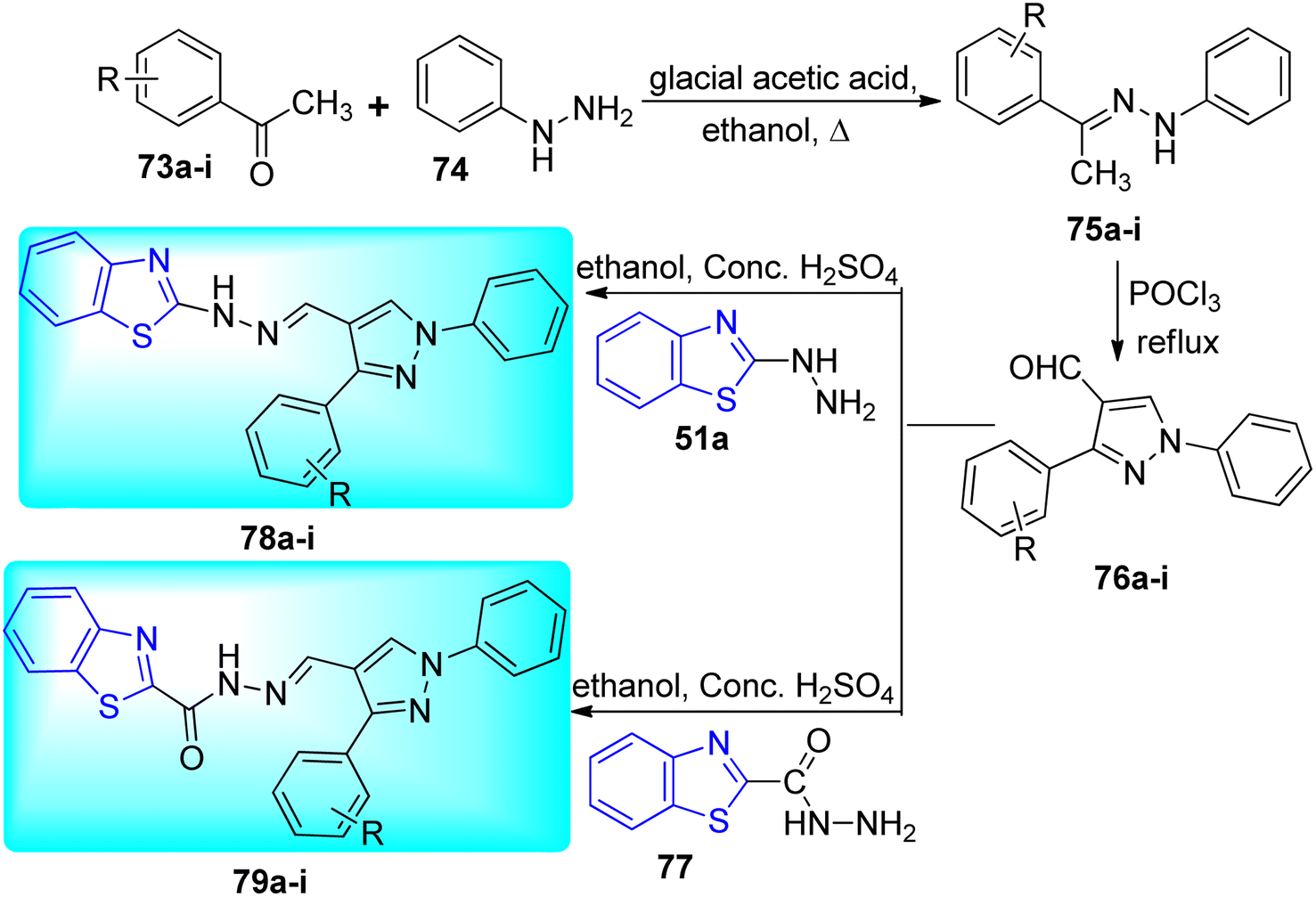
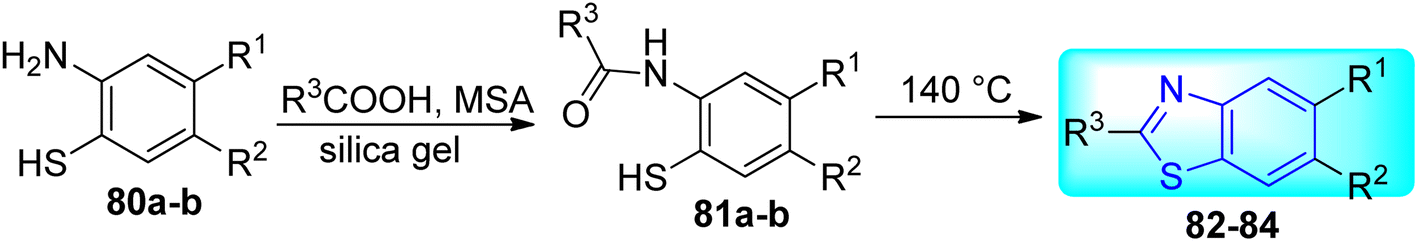
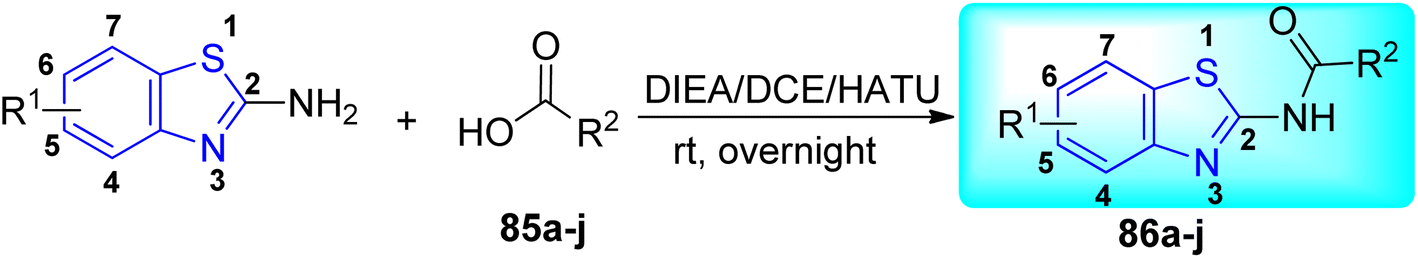
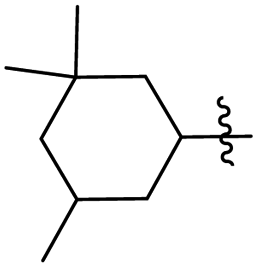
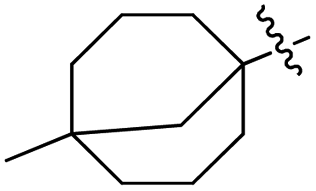

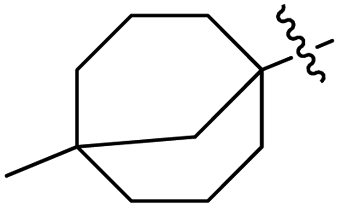
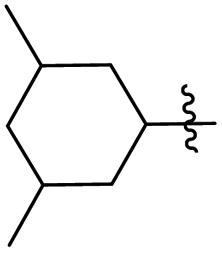
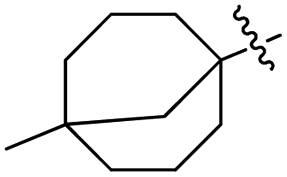
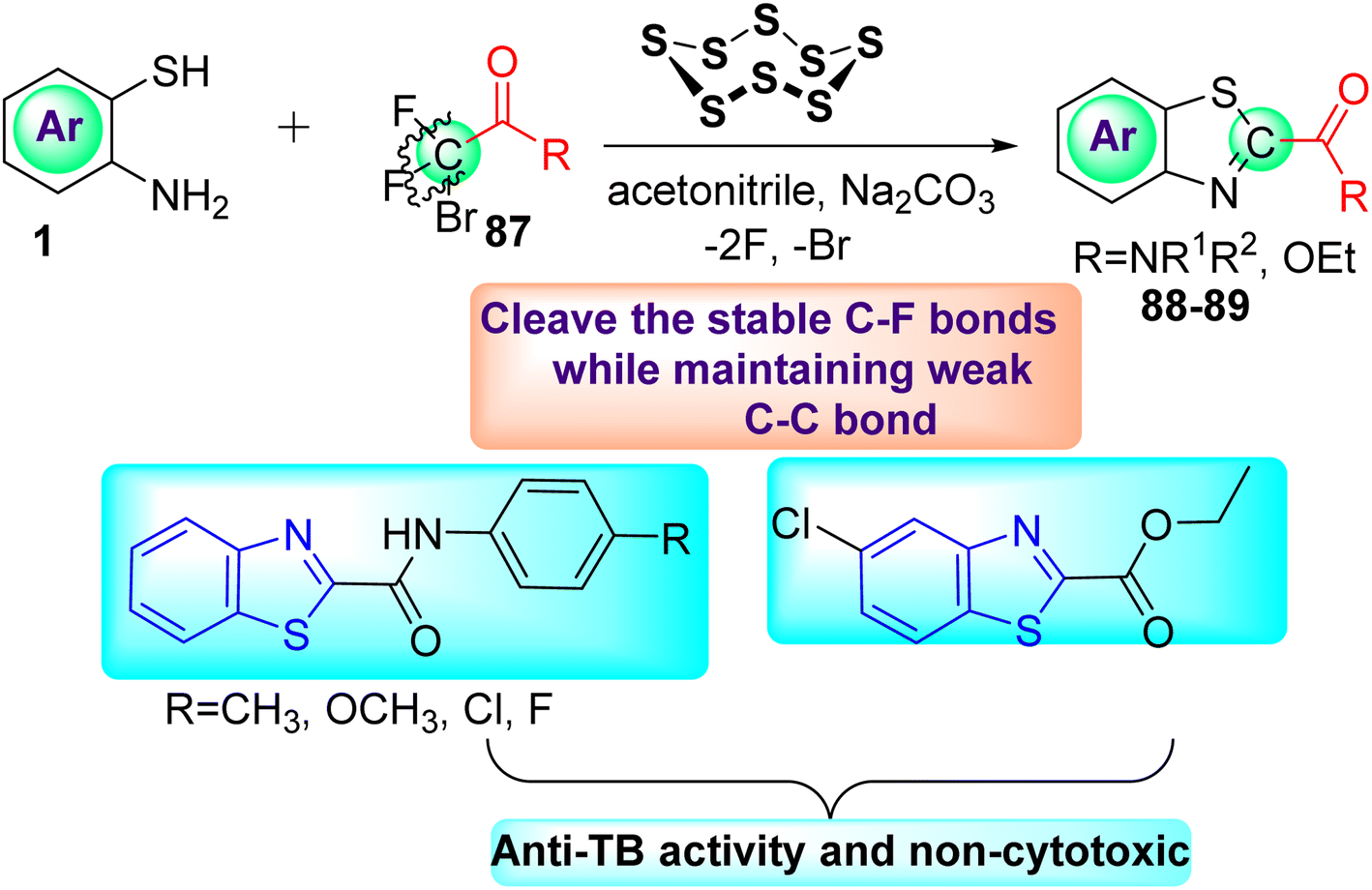
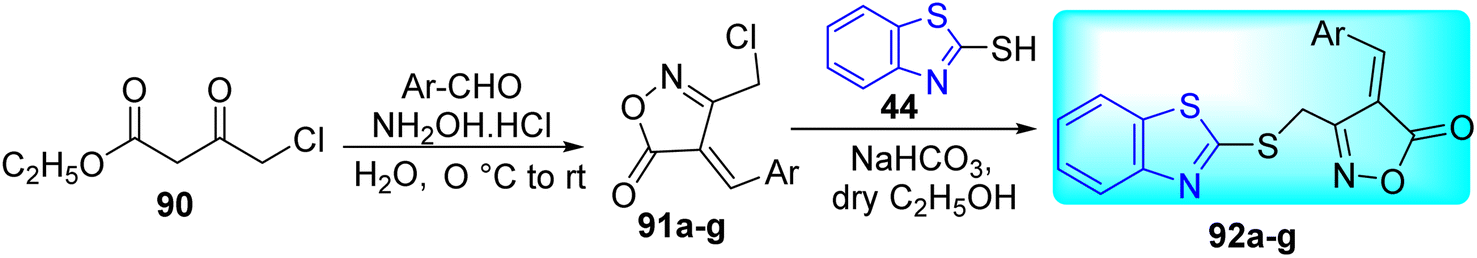
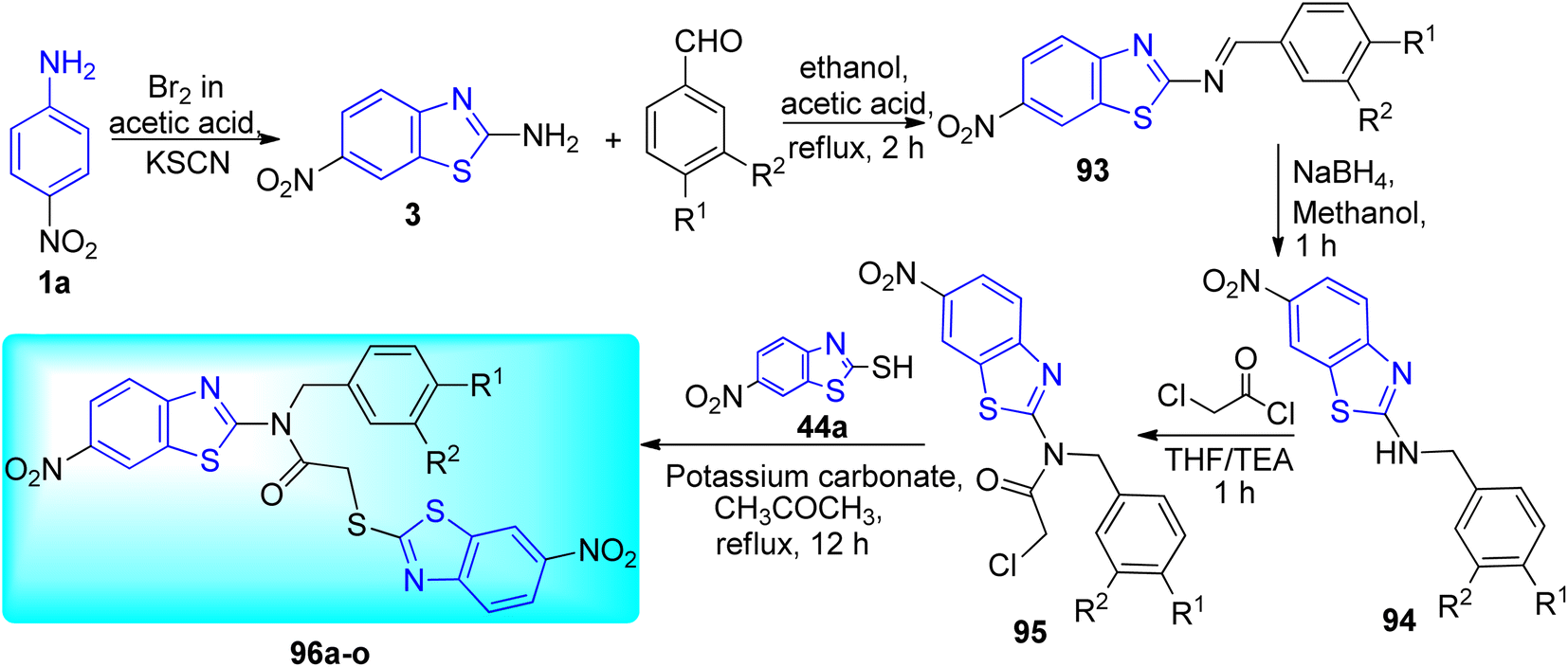
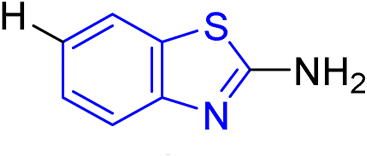 3a
3a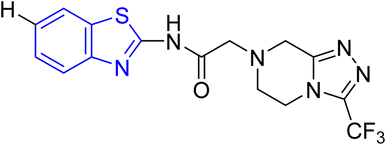 98a
98a 3b
3b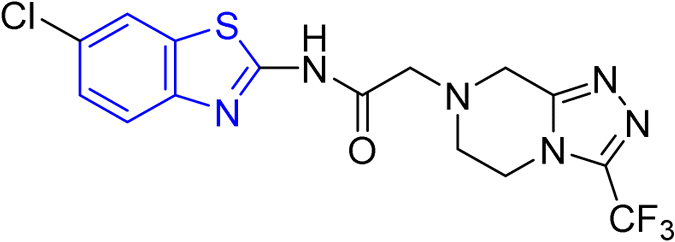 98b
98b 3c
3c 98c
98c 3d
3d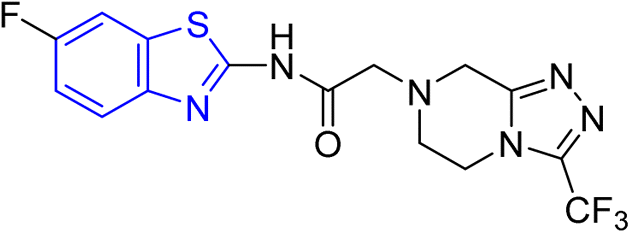 98d
98d 3e
3e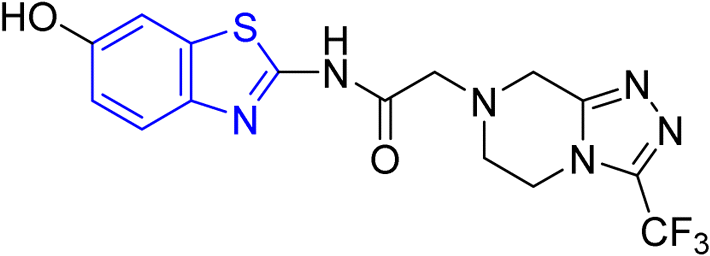 98e
98e