
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiodivergent synthesis of sulfone-tethered lactam–lactones bearing four contiguous stereocenters†
Timothy K. Beng *,
Jane Eichwald,
Jolyn Fessenden,
Kaiden Quigley,
Sapna Sharaf,
Nanju Jeon and
Minh Do
*,
Jane Eichwald,
Jolyn Fessenden,
Kaiden Quigley,
Sapna Sharaf,
Nanju Jeon and
Minh Do
Department of Chemistry, Central Washington University, Ellensburg, WA 98926, USA. E-mail: Timothy.beng@cwu.edu
First published on 13th July 2023
Abstract
Sulfone-tethered lactones/amides/amines display a diverse spectrum of biological activities, including anti-psychotic and anti-hypertensive. Sulfones are also widely present in functional materials and fragrances. We therefore reasoned that a regiodivergent and stereocontrolled strategy that merges the sulfone, lactone, and lactam motifs would likely lead to the discovery of new pharmacophores and functional materials. Here, we report mild conditions for the sulfonyllactonization of γ-lactam-tethered 5-aryl-4(E)-pentenoic acids. The annulation is highly modular, chemoselective, and diastereoselective. With respect to regioselectivity, trisubstituted alkenoic acids display a preference for 5-exo-trig cyclization whereas disubstituted alkenoic acids undergo exclusive 6-endo-trig cyclization. The lactam-fused sulfonyllactones bear angular quaternary as well as four contiguous stereocenters. The products are post-modifiable, especially through a newly developed Co-catalyzed reductive cross-coupling protocol.
Introduction
Biologically active and pharmaceutically-pertinent molecules are replete with the sulfone structural motif, presumably due to its overwhelmingly positive effects on metabolism, liposolubility, and stability.1,2 As shown in Fig. 1, ester/amide-tethered sulfones are marketed drugs for the treatment of human diseases, including anti-migraine Vioxx (A), anti-androgen Casodex (B), antipsychotic Amisulpride (C), CXCR2 antagonist Danirixin (D), r-secretase inhibitor E, and anti-hypertensive renin inhibitor Remikiren (F).3 Sulfone-bearing architectures are also widely present in functional materials where they exhibit remarkable activity.4 Importantly, the organic chemistry literature is saturated with examples of sulfones serving different roles in chemical synthesis.5 As a result, several methodologies for the construction of sulfonyl lactones have been developed.6 Among them, the vicinal sulfonyl functionalization of alkenes is heralded because it enables the simultaneous introduction of a sulfonyl group and other synthetically versatile functionalities such as lactones and lactams.6c,7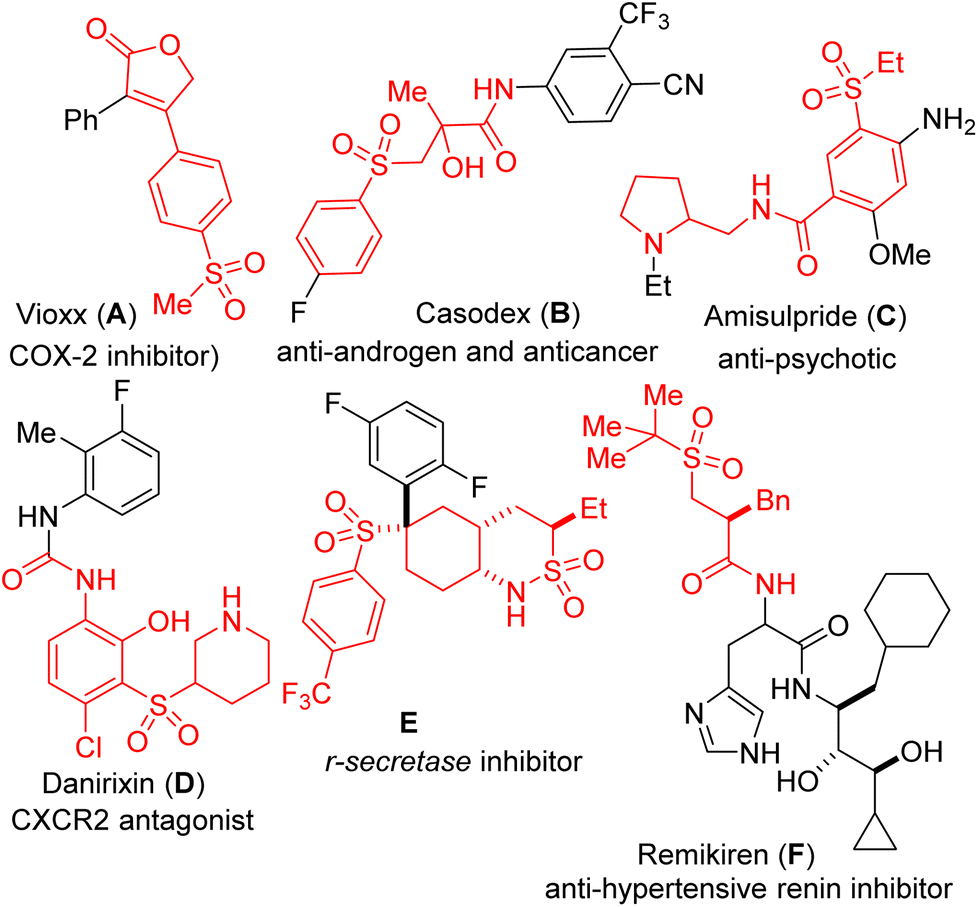 | ||
| Fig. 1 Examples of bioactive ester- or amide-tethered sulfones. | ||
Diversity-Oriented Synthesis (DOS) is gradually transitioning from structural diversity to biological as well as functional relevance.7 A complementary strategy for generating biologically-relevant chemical libraries is to employ complexity-generating transformations that produce compounds with conformationally rigid cyclic frameworks with a high ratio of sp3 carbon atoms.8 Increasing the three-dimensional character of a molecule has indeed been associated with a more successful outcome in drug discovery.9 We therefore surmised that a diastereoselective and modular strategy that merges the pharmaceutically pertinent lactam, lactone, and sulfone motifs would likely expand the 3D-structural space for the discovery of new small molecules with medicinal value.
In the last decade, we have sought to popularize the 1,3-azadiene-anhydride reaction for the stereocontrolled synthesis of lactam-bearing 5-aryl-4(E)-pentenoic acids of type 1 (ref. 10) (Fig. 2A) and subsequent post-diversification. Recently, we developed practical and flexible methodologies toward halogen-containing, trans-fused lactam–lactones bearing quaternary and contiguous stereocenters (see C and D, Fig. 2A).10f,g Seeking to further leverage the synthetic versatility of the aforementioned 1,3-azadiene-anhydride reaction, we decided to interrogate these sterically challenged and electronically diverse 5-aryl-4(E)-pentenoic acids (i.e., 1) in a sulfonyllactonization protocol (Fig. 2B). Although significant advances have been achieved in the arenas of hydrolactonization,11 halolactonization,12 aminolactonization,13 or trifluoromethyllactonization,14 of 4-pentenoic acids, sulfonyllactonization is still at the incipient stages. Recent advances are starting to bring these once elusive building blocks into the mainstream.5c,6 However, most of these advances hinge on the sulfonyllactonization of electronically suitable 4-aryl-4-pentenoic acids (Fig. 2C) where the regioselectivity of the transformation can be predicted a priori.
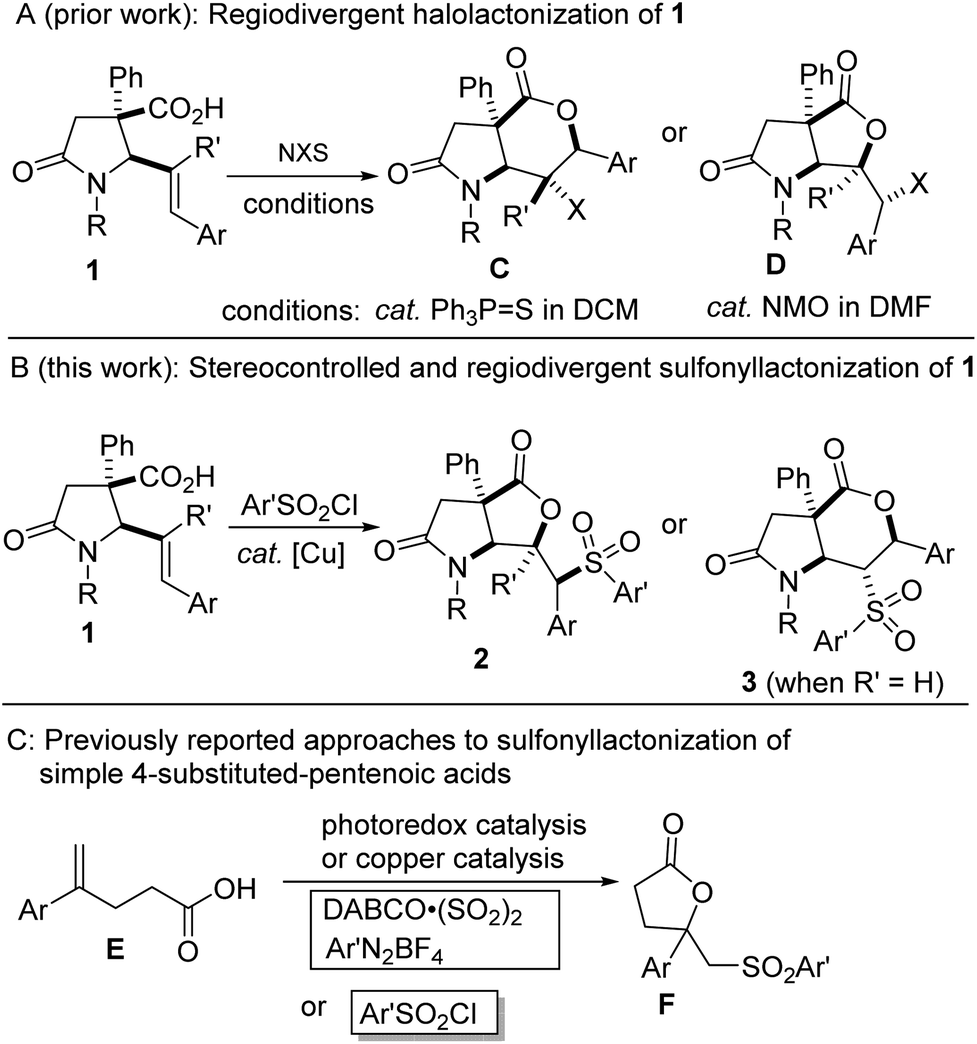 | ||
| Fig. 2 (A) Our prior synthesis of fused lactam-halolactones from 5-aryl-4-(E)-pentenoic acids 1, (B) proposed plan for stereocontrolled sulfonyllactonization of 1, (C) reported approaches for sulfonyllactonization of simple 4-substituted pentenoic acids. | ||
We were therefore motivated by current methodological limitations and the relevance of sulfonylated lactones to develop a regiodivergent, diastereoselective, flexible, and scalable method for the preparation of highly decorated sulfone-containing lactam–lactones bearing at least two tetrasubstituted- and four contiguous stereocenters. The successful sulfonyllactonization of trisubstituted alkenoic acids of type 1 (R′ ≠ H) would represent a significant advance given that such substrates are inherently problematic from the standpoints of regioselectivity and diastereoselectivity. Efforts towards the manifestation of our ideals are described herein. Regarding the regioselectivity of this intramolecular vicinal difunctionalization protocol, we find that trisubstituted alkenoic acids display a preference for 5-exo-trig cyclization whereas 1,2-disubstituted alkenoic acids undergo exclusive 6-endo-trig cyclization.
Results and discussion
We initiated studies toward the construction of sulfone-tethered fused lactam-γ-lactones by benchmarking our optimization efforts for sulfonyllactonization of alkenoic acid 1a (see the ESI† for the preparation of the lactam-tethered alkenoic acids) with the Buchwald-inspired5c reaction conditions described in Table 1. Trisubstituted alkenoic acid 1a was chosen as the model substrate with the view of testing the power/limits of the transformation since the sulfonyllactonization of trisubstituted alkenes is quite challenging.
|
||
|---|---|---|
| Entry | Deviation from conditions A | % Yield of 2a |
| a Relative configuration established by NOESY noe’s between a and b as well as between b and c. | ||
| 1 | EtOAc as solvent | 61 |
| 2 | THE as solvent | 69 |
| 3 | MTBE as solvent | 53 |
| 4 | Nitromethane as solvent | 68 |
| 5 | 1,4-Dioxane as solvent | 49 |
| 6 | Dichloroethane as solvent | 22 |
| 7 | DMSO as solvent | 48 |
| 8 | PhMe | 12 |
| 9 | DMF | 21 |
| 10 | MeOH | 0 |
| 11 | CF3CH2OH | 0 |
| 12 | TMO | 72 |
| 13 | Cu(MeCN)4PF6 omitted | 0 |
| 14 | Cul in place of Cu(MeCN)4PF6 | <5 |
| 15 | CuBr in place of Cu(MeCN)4PF6 | 22 |
| 16 | CuCI in place of Cu(MeCN)4PF6 | 48 |
| 17 | CuOTf in place of Cu(MeCN)4PF6 | <5 |
| 18 | Cu(OAc)2 in place of Cu(MeCN)4PF6 | 0 |
| 19 | Cu(NO3)2 3H2O in place of Cu(MeCN)4PF6 | 0 |
| 20 | DMAP omitted | 18 |
| 21 | DBU in place of DMAP | 73 |
| 22 | KF in place of DMAP | 51 |
| 23 | Et3N in place of DMAP | 38 |
| 24 | 2,2′-Bipyridine in place of DMAP | 15 |
| 25 | After 4 h at 60 °C | 53 |
| 26 | After 36 h at room temperature | 75 |
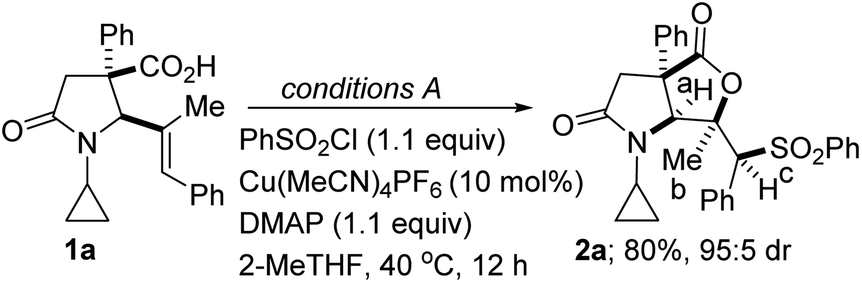
Reaction optimization was carried out and it was established that 2-methyltetrahydrofuran (2-MeTHF) out-performs other reaction media (e.g., ethyl acetate (EtOAc), tetrahydrofuran (THF), methyl tert-butyl ether (MTBE), nitromethane, 1,4-dioxane, dichloroethane, dimethyl sulfoxide (DMSO), toluene, N,N-dimethylformamide (DMF), methanol, trifluoromethyl ethanol, and 2,2,5,5-tetramethyloxolane (TMO); entries 1–12). This is noteworthy since 2-MeTHF offers several advantages,15 including that: (a) it readily phase-separates from aqueous layers (in contrast to THF); (b) it is obtained from furfural, which originates from a renewable feedstock; (c) it is not easily oxidized; and (d) it has minimal health risks. No background reaction occurs in the absence of the copper catalyst (entry 13). Other copper(I) precatalysts that were surveyed did not perform as well as Cu(MeCN)4PF6 (entries 14–19). The reaction could not be catalyzed by a cationic copper(II) salt (entries 18 and 19). Of the base additives that were surveyed, DMAP emerged as the optimal base (entries 20–24). As described in Table 1, the relative configuration of 5-exo cyclization product 2a was established by NOESY analysis. We are however not oblivious to the possibility of anti-stereoselective sulfonyllactonization followed by free rotation around the C–C sigma bond, which then places the hydrogen on the sulfone-bearing carbon (i.e., Hc) on the same side as the methyl group.
The scope of the transformation with respect to the lactam-tethered alkenoic acid has been explored. Knowing that the nature of the nitrogen substituent present on a nitrogen heterocycle can have a dramatic effect on its biological activity16 and reactivity, the effect of the N-substituent on the sulfenolactonization was first explored. Encouragingly, N-alkyl-substituted allylic lactam acids are competent substrates for the annulation (see 2a/b). N-arylated γ-lactam-tethered alkenoic acids underwent productive 6-exo-cyclization (see 2c/d), which is noteworthy since N-aryl γ-lactams are embedded in several pharmacologically pertinent targets.17 Lactam-tethered alkenoic acids harboring electronically diverse N-benzyl substituents undergo satisfactory cyclization (see 2e–g). The successful construction of sulfonylated lactam–lactones harboring the N-phenethyl group (see 2h–i) is noteworthy given that the latter is often employed as a precursor to the indolizidine/quinolizidine scaffold.18 The transformation displays excellent chemoselectivity given that a lactam-tethered alkenoic acid bearing an N-allyl substituent reacts with phenylsulfonyl chloride to afford bicycle 2k, without complications arising from sulfonyllactonization of the kinetically more accessible allyl group. We attribute this chemoselective sulfonyllactonization to conformational constraints and to the more activated nature of the styrenyl double bond. The addition process of the sulfonyl radical to the alkenoic acid is dependent on the steric hindrance of the olefin seeing as replacement of the internal methyl substituent with an unbranched hexyl group leads to a compromise in the efficiency of the annulation (2i vs. 2l/m). Indeed, our luck runs out when the methyl group is replaced by a sterically imposing tert-butyl substituent given that no cyclization takes place (2h vs. 2n). As expected, when the internal methyl group is replaced by a phenyl group, the lactam-tethered 4,5-diphenyl-4(E)-pentenoic acid undergoes exclusive 5-exo cyclization in modest efficiency (2h vs. 2o). The yield can be improved slightly when the reaction is performed at 60 °C. In these low-yielding cases, the remaining mass balance is mostly accounted for by recovered 1. Lactam-tethered 5-aryl-4(E)-pentenoic acids bearing electron-rich aryl groups are as competent as their electron-deficient congeners (2p vs. 2q).
In a mechanistically intuitive outcome, these studies have revealed that when disubstituted alkenoic acids are employed in place of the trisubstituted acids featured so far, a complete reversal in regioselectivity is observed and 6-endo cyclization predominates over 5-exo cyclization, leading to products of type 3 (see 3a/b). A direct comparison between 2b and 3a is illuminating and clearly highlights how the internal substituent on the alkene alters the regioselectivity.
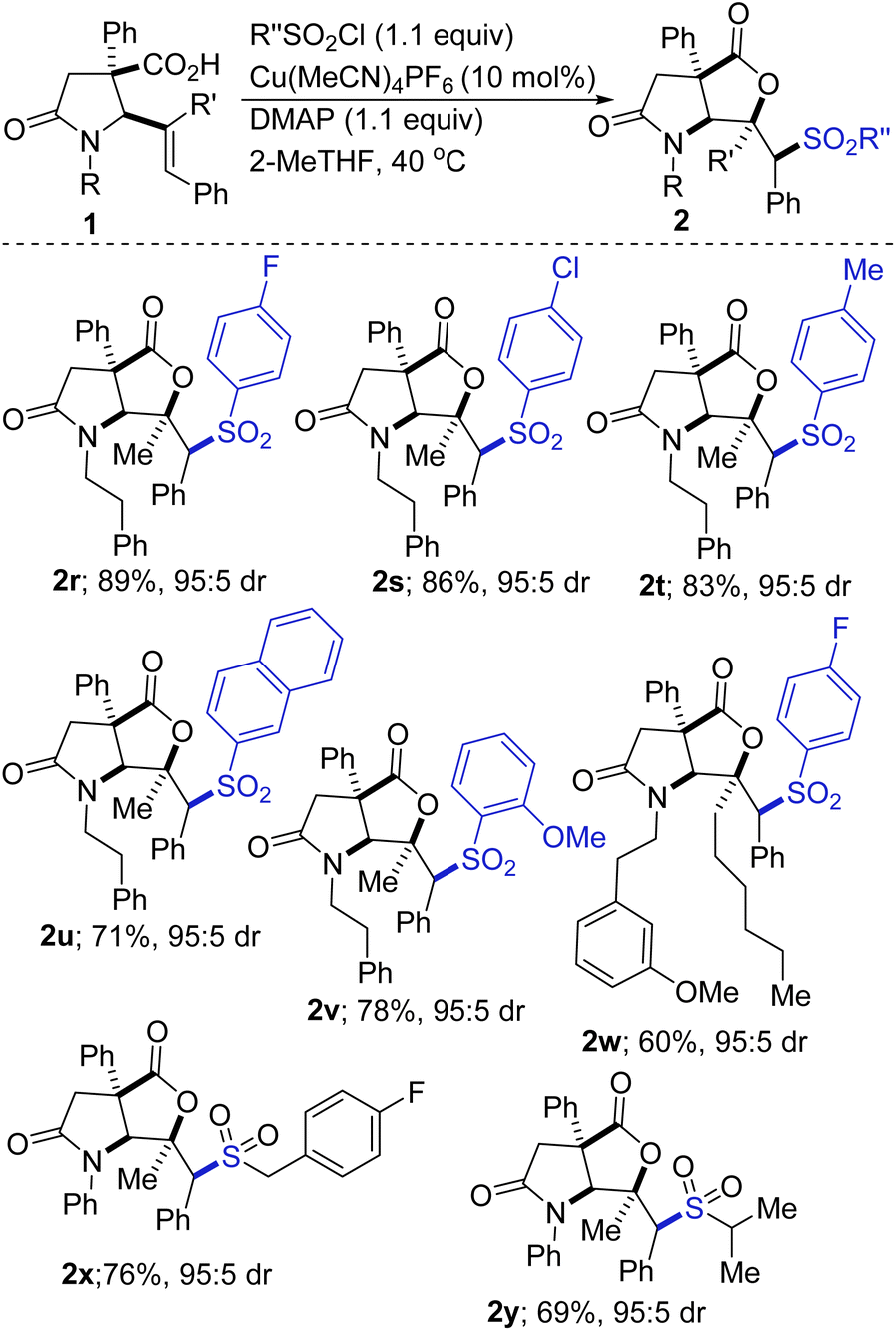
As shown in Scheme 2, the scope of the sulfonyllactonization with respect to the sulfonyl chloride has been explored, albeit briefly. The results indicate that both electron-deficient and electron-rich aryl sulfonyl chlorides are competent coupling partners (2r vs. 2t). A sterically imposing 2-naphthyl group can be installed, but the efficiency is unsurprisingly compromised (see 2t). Ortho-substituted aryl sulfonyl chlorides are tolerated as exemplified through the synthesis of sulfone 2v. The incorporation of a fluorinated moiety into organic molecules generally increases the solubility, lipophilicity, and metabolic stability of the parent molecules, thus, explaining why about 20% of prescribed and 30% of blockbuster drugs contain at least one fluorine atom. Fluorine-functionalized materials also have applications in optical, electronic, and agrochemical technologies.19 It is therefore noteworthy that fluorinated products 2r and 2w are obtainable in satisfactory yields. Other halogenated precursors are well tolerated (see 2s), which bodes well for late-stage diversification as the halogen group may be utilized as a functional handle for cross-coupling purposes. Our studies have revealed that alkyl sulfonyl chlorides undergo satisfactory vicinal sulfonyllactonization of 1, giving rise to the alkyl sulfones depicted in Scheme 2 (see 2x and 2y).
Based on prior mechanistic studies performed on the sulfonyllactonization of 4-aryl-4-pentenoic acids,5c and on the results obtained so far, a tentative mechanism is depicted in Fig. 3. An initial reaction between the Cu(I) catalyst and the radical source (i.e., the aryl sulfonyl chloride) generates a Cu(II) species and sulfonyl radical G. Regioselective addition of G to the alkene motif resident in lactam-tethered alkenoic acid 1 affords tertiary alkyl radical intermediate H, which is captured intramolecularly by the pendant acid to furnish sulfone-tethered bicyclic lactam–lactone 2, with regeneration of the Cu(I) species. As the results indicate, there is a reversal in the regioselectivity of addition of G to a disubstituted alkenoic acid, which instead leads to significantly more stable benzylic radical intermediate I (compared to the corresponding secondary nonbenzylic radical). Subsequent intramolecular capture of I by the tethered acid leads to 6-endo cyclization product 3. The formation of 3 is not surprising since the regioselectivity of radical addition to alkenes is generally dictated by the stability of the resulting radical and the addition onto aryl-alkenes results in a stabilized benzylic radical.5c The formation of 5-exo cyclization products of type 2 is quite intriguing since one would expect the tertiary nonbenzylic radical (i.e., H) to be less stable than the competing secondary benzylic radical. Although speculative, we surmise that the formation of 2 is mostly governed by sterics imposed by the bulky aryl substituents whereas the formation of 3 is almost entirely governed by the pronounced electronic effects.
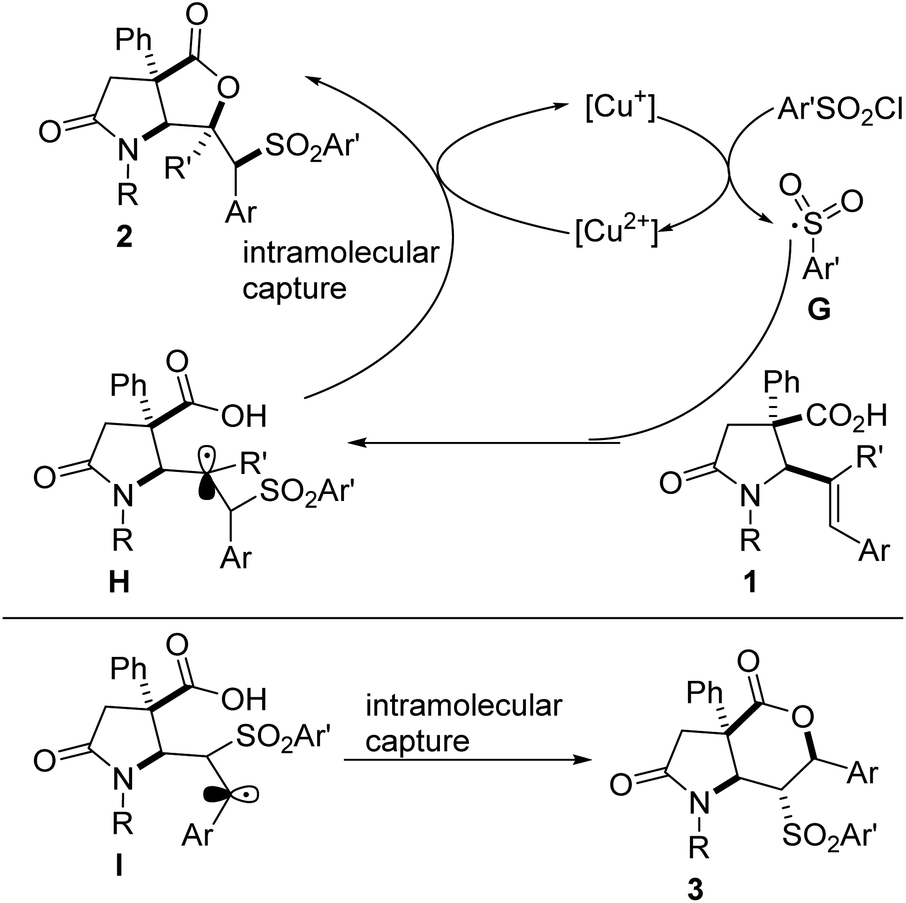 | ||
| Fig. 3 Plausible mechanism for sulfonyllactonization of 1 with aryl sulfonyl chlorides. | ||
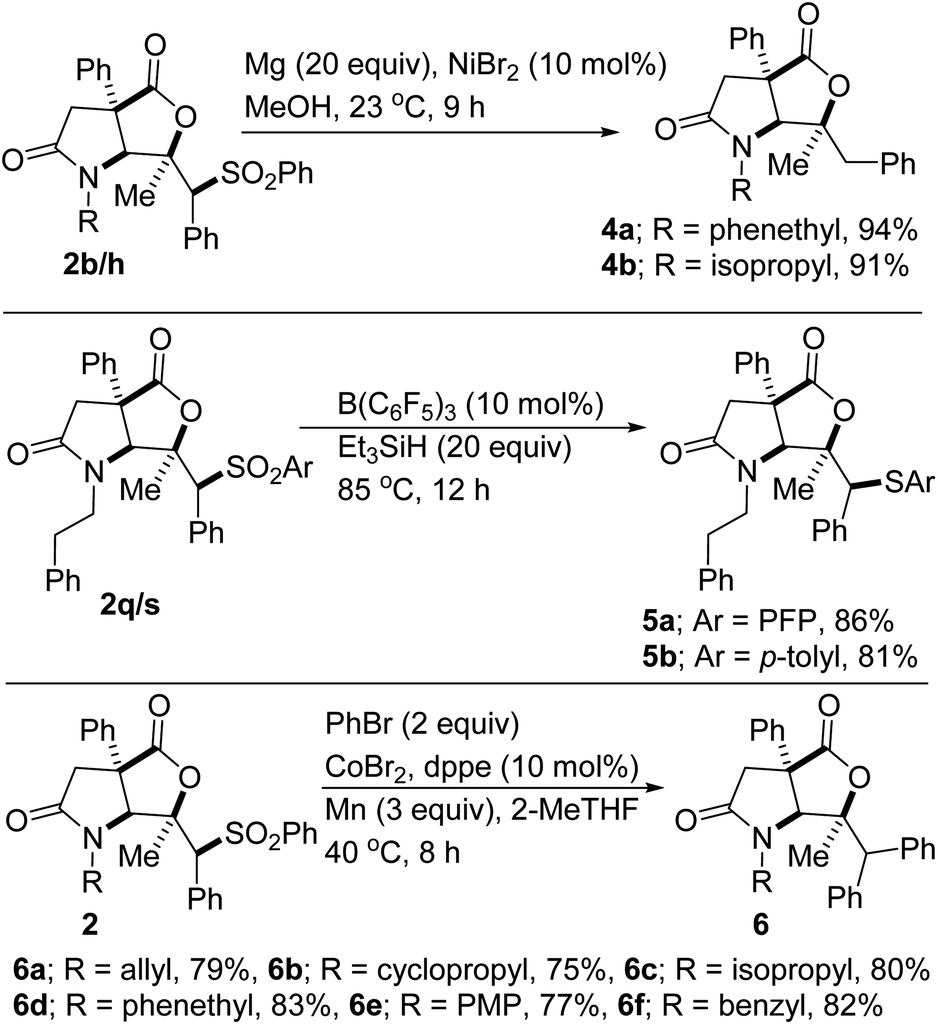
In the synthesis of potential drug candidates, scalability is often a significant factor as it serves to provide sufficient amounts for clinical tests. A potentially beneficial aspect of this methodology is therefore the scalable nature of the reactions given that products such as 2a/b/h/k/p/r have been prepared in gram scale, with little to no compromise in efficiency. This has set the stage for post-diversification studies. For example, Mg-mediated reductive hydro desulfonylation of sulfones 2b and 2h affords formal hydrolactonization products 4a and 4b, respectively (Scheme 3). When sulfones 2p and 2r are subjected to B(C6F5)3-catalyzed deoxygenation, thioether-bearing lactam–lactones 5a and 5b are obtained, respectively. Sulfones are often used as traceless linchpins for the construction of synthetically attractive molecular frameworks. Accordingly, several transition-metal-catalyzed and free-radical-based desulfonylative cross-couplings have emerged.20,21 Ni- and Co-catalyzed cross-electrophile couplings have been particularly useful in C(sp3)-C(sp2) bond-forming processes.22 In this electrophile–electrophile coupling mode, traditionally difficult functional groups (e.g., ester, ketone, nitrile, or alcohol groups) are impressively tolerated and the need for pregeneration of expensive or difficult-to-handle organometallic reagents is obviated. Accordingly, we decided to explore the amenability of the alkyl sulfones prepared herein to reductive cross-coupling with bromobenzene. If successfully implemented, this would represent the discovery and development of the first Co-catalyzed reductive cross-electrophile coupling reaction between alkyl sulfones and an aryl bromide.23 In the event, we were pleased to find that desulfonative phenylation of several sulfones with bromobenzene proceeds smoothly and affords formal aryllactonization products 6a–f. Efforts to extend this novel mode of reactivity to other aryl bromides are underway.
Conclusions
In summary, the site-selective, diastereoselective, and scalable synthesis of sulfone-tethered lactam–lactones has been accomplished, through the deployment of γ-lactam-tethered alkenoic acids in a sulfonyllactonization protocol. With respect to regioselectivity, trisubstituted alkenoic acids display a preference for 5-exo-trig cyclization whereas disubstituted alkenoic acids undergo exclusive 6-endo-trig cyclization. These sp3-rich sulfonylated bicycles bear medicinally relevant quaternary and contiguous stereocenters. We anticipate that this practical, cost-effective, and scalable strategy would expand the 3D-structural space for the discovery of new lactam–lactones with medicinal value. Post-modification of these versatile N,O-heterocycles has led to the unveiling of the first Co-catalyzed reductive cross-coupling of alkyl aryl sulfones with bromobenzene. Efforts to unambiguously determine the relative configuration of 2a or any of the products depicted in Scheme 1 through X-ray crystallographic analysis are ongoing. So far, the crystals obtained are not of sufficient quality. We have also initiated efforts to develop an enantioselective version of the sulfonyl lactonization reaction described herein. These findings will be disclosed in due course.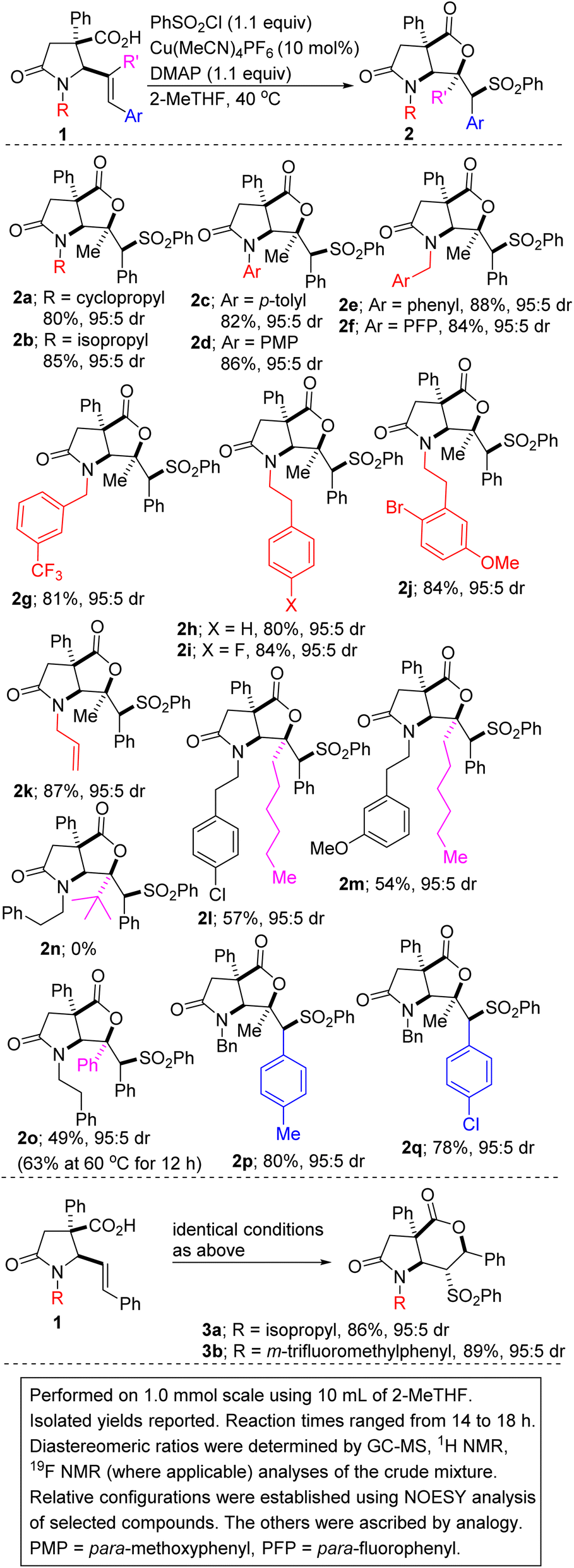 | ||
| Scheme 1 Scope of lactam-tethered alkenoic acid. | ||
Experimental
All experiments involving air and moisture-sensitive reagents were carried out under an inert atmosphere of nitrogen and using freshly distilled solvents. 2-MeTHF was distilled from sodium benzophenone ketyl. Column chromatography was performed on silica gel (230–400 mesh). Thin-layer chromatography (TLC) was performed using Silicycle SiliaplateTM glass backed plates (250 μm thickness, 60 Å porosity, F-254 indicator) and visualized using UV (254 nm) or CAM, p-anisaldehyde, or KMnO4 stain. All reported temperatures were internal to a reaction vessel. Unless otherwise indicated, 1H, 13C, and DEPT-135 spectra were acquired using CDCl3 as solvent, at room temperature. Chemical shifts are quoted in parts per million (ppm). HRMS-EI+ data were obtained using either electrospray ionization (ESI) or electron impact (EI) techniques. High-resolution ESI was obtained on an LTQ-FT (ion trap; analyzed using Excalibur). High resolution EI was obtained on an Autospec (magnetic sector; analyzed using MassLynx). Brine solutions are saturated solutions of aqueous sodium chloride.General procedure A: sulfonyllactonization
An oven-dried vial equipped with a Teflon-coated magnetic stir bar was charged with tetrakis(acetonitrile)copper(I) hexafluorophosphate (37 mg, 0.10 mmol, 10 mol%), the aryl sulfonyl chloride (1.1 mmol, 1.1 equiv.), DMAP (1.1 mmol, 1.1 equiv.), and alkenoic acid 1 (1.0 mmol, 1.0 equiv.). The reaction tube was sealed with a septum screw-cap and connected to a Schlenk line through a needle. The reaction tube was then briefly evacuated and backfilled with argon (for total of three times). Anhydrous 2-MeTHF (10 mL) was added to the tube via syringe and the argon pressure was removed. The reaction mixture was stirred at room temperature for 12 h (TLC and GC-MS monitoring). The reaction mixture was diluted with saturated aqueous sodium bicarbonate solution (20 mL) and ethyl acetate (20 mL). The aqueous layer was separated and extracted with ethyl acetate (20 mL × 3). The combined organic layers was dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was analyzed by 1H NMR spectroscopy and GC-MS to determine the diastereomeric ratio. Purification by flash column chromatography on silica gel afforded the pure sulfone-tethered lactam–lactones.General procedure B: desulfonylation
To a well-stirred solution of sulfone 2 (0.5 mmol) in dry MeOH (10 mL) was added Mg turnings (5.0 mmol, 10 equiv.) and NiBr2 (10 mol%) at 0 °C under Ar. The mixture was stirred at room temperature for 3 h prior to the addition of another portion of Mg turnings (5.0 mmol, 10 equiv.) and dry MeOH (10 mL). Stirring was continued for another 6 h (TLC and GC-MS monitoring). Upon completion, the reaction mixture was filtered through Celite and the residue was thoroughly washed with MeOH. The filtrate was concentrated under reduced pressure and dissolved in EtOAc. The solution was washed with a saturated solution of aqueous NH4Cl, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to give the crude desulfonylated product. Purification by flash column chromatography on silica gel afforded the pure lactam–lactone.General Procedure C: reduction of sulfones to thioethers
An oven-dried 5 mL screw-capped sealed tube equipped with a magnetic stir bar was charged with B(C6F5)3 (10 mol%), Et3SiH (20 equiv.), and the sulfone-tethered lactam–lactone (0.50 mmol), under an argon atmosphere. The tube was sealed properly and transferred to an oil bath thermostatted at 85 °C. After 12 h (TLC and GC-MS monitoring), the reaction was cooled to room temperature and passed through a small plug of silica gel using EtOAc. The crude material was dried (Na2SO4) and filtered, and the solvent was removed under reduced pressure. The mixture was then subjected to high vacuum at 70 °C until the unreacted hydrosilane was removed from the system. The residue was purified further by flash column chromatography (silica gel, hexanes/EtOAc 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 50:50) to afford the desired thioether-tethered lactam–lactones.
:10 to 50:50) to afford the desired thioether-tethered lactam–lactones.
General Procedure D: reductive desulfonylative cross-coupling with bromobenzene
To a solution of CoBr2 (22 mg, 0.10 mmol, 10 mol%), bis-1,2- diphenylphosphinopropane (41.2 mg, 0.10 mmol, 10 mol%), and manganese powder (165 mg, 3 mmol, 3 equiv.) in 2-MeTHF (5 mL) was added bromobenzene (2 mmol, 2 equiv.) at 40 °C. A solution of the sulfone (1 mmol, 1 equiv.) in 2-MeTHF (5 mL) was added slowly (2 mL h−1). After completion (as judged by TLC and GC-MS), the reaction mixture was treated with a mild acid such as 10% H3PO4 (aq) and extracted with EtOAc. The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure to afford the desired coupling product as an oil. Purification was carried out by flash column chromatography on silica, eluting with Hexanes/EtOAc.Synthesis of sulfone 2a
Prepared in 1.0 mmol scale using General Procedure A. Purification: Flash chromatography on silica eluting with hexane/EtOAc (50:50). Yellowish oil. Yield = 400.8 mg, 80%, 95:5 dr (syn:anti). 1H NMR (400 MHz, CDCl3) δ 7.54 (s, 1H), 7.28–7.19 (m, 3H), 7.23–7.12 (m, 7H), 7.00–6.97 (m, 3H), 6.25 (s, 1H), 4.73 (s, 1H), 4.03 (s, 1H), 3.00 (d, J = 14.8 Hz, 1H), 2.67 (d, J = 14.8 Hz, 1H), 2.35 (tt, J = 7.4, 4.1 Hz, 1H), 1.60 (s, 3H), 0.88 (dq, J = 9.9, 6.6 Hz, 1H), 0.63 (dtd, J = 10.6, 6.5, 3.9 Hz, 1H), 0.33 (dq, J = 9.9, 6.8 Hz, 1H), 0.00 (dq, J = 9.9, 6.8 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 174.3, 173.1, 139.2, 133.8, 129.7, 129.6, 129.3, 128.9, 128.8, 128.7, 128.3, 125.8, 88.2, 77.7, 71.9, 51.8, 45.1, 26.4, 21.8, 9.3, 5.1. FTIR (KBr): 2965.4, 1727.5, 1696.3, 1604.9, 1511.0, 1448.5, 1414.7, 1384.9, 1357.4, 1298.7, 1247.5, 1179.3, 1135.9, 1031.8, 905.8, 839.0. HRMS-EI+ (m/z): calc for C29H27NO5S [M]+ 501.1610, found 501.1618.
Note: All sulfones depicted in Schemes 1 and 2 were prepared as described above. Spectroscopic data can be found in the ESI.†
 | ||
| Scheme 2 Scope of sulfonyl chloride. | ||
Synthesis of lactam–lactone 4a
Prepared in 0.50 mmol scale using General Procedure B. Purification: flash chromatography on silica eluting with hexane/EtOAc (75:25). Yellowish oil. Yield = 200 mg, 94%. 1H NMR (400 MHz, CDCl3) δ 7.31–7.21 (m, 9H), 7.00–6.98 (m, 2H), 6.91–6.89 (m, 2H), 6.65 (dd, J = 7.7, 1.9 Hz, 2H), 4.23 (dt, J = 13.2, 6.0 Hz, 1H), 4.02 (s, 1H), 3.08 (d, J = 17.7 Hz, 1H), 2.99–2.74 (m, 5H), 2.66 (qd, J = 12.7, 7.0 Hz, 1H), 1.49 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 175.7, 172.3, 139.3, 137.9, 133.9, 130.9, 129.1, 128.8, 128.7, 127.8, 127.5, 126.7, 125.7, 87.4, 71.5, 53.1, 47.3, 44.5, 42.9, 33.1, 22.9. HRMS-EI+ (m/z): calc for C28H27NO3 [M]+ 425.1991, found 425.1995.
Note: Lactam–lactone 4b was prepared as described above. Spectroscopic data can be found in the ESI.†
Synthesis of thioether 5a
Prepared in 0.50 mmol scale using General Procedure C. Purification: flash chromatography on silica eluting with hexane/EtOAc (50:50). Yellowish oil. Yield = 237.2 mg, 86%. 1H NMR (400 MHz, CDCl3) δ 7.35–7.18 (m, 9H), 7.15–6.96 (m, 6H), 6.89 (t, J = 8.5 Hz, 2H), 6.69 (d, J = 7.8 Hz, 2H), 4.31 (s, 1H), 4.28 (dt, J = 13.8, 6.9 Hz, 1H), 4.05 (s, 1H), 3.14–3.01 (m, 2H), 2.93–2.78 (m, 2H), 2.69 (dt, J = 13.8, 6.7 Hz, 1H), 1.56 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.9, 172.3, 163.8, 161.4, 139.3, 137.6, 136.6, 134.8, 134.7, 129.9, 129.0, 128.8, 128.7, 128.3, 127.9, 126.7, 125.8, 116.4, 116.2, 89.4, 71.1, 64.6, 52.6, 44.0, 43.3, 33.0, 22.0. HRMS-EI+ (m/z): calc for C34H30FNO3S [M]+ 551.1930, found 551.1937.
Note: Thioether 5b was prepared as described above. Spectroscopic data can be found in the ESI.†
Synthesis of diarylmethane-tethered lactam–lactone 6a
Prepared in 0.50 mmol scale using General Procedure D. Purification: Flash chromatography on silica eluting with hexane/EtOAc (50:50). Greenish-yellow oil. Yield = 172.8 mg, 79%. 1H NMR (400 MHz, CDCl3) δ 7.34–7.06 (m, 15H), 5.66–5.52 (m, 1H), 5.13–5.04 (m, 2H), 4.69–4.56 (m, 2H), 4.14 (s, 1H), 3.40 (dd, J = 15.0, 8.4 Hz, 1H), 3.13 (d, J = 18.1 Hz, 1H), 3.00 (d, J = 18.1 Hz, 1H), 1.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.7, 172.1, 139.6, 137.5, 134.6, 131.9, 131.0, 129.7, 129.1, 129.0, 128.6, 128.1, 128.0, 127.8, 125.7, 120.2, 89.0, 69.9, 64.3, 52.2, 45.4, 43.5, 22.5. HRMS-EI+ (m/z): calc for C29H27NO3 [M]+ 437.1991, found 437.1995.
Note: All desulfonylative arylation products depicted in Scheme 3 were prepared as described above. Spectroscopic data can be found in the ESI.†
 | ||
| Scheme 3 Elaboration of the annulation products. | ||
Author contributions
T. K. B. – conceptualization, project administration, supervision, writing – original draft, internal funding acquisition. J. E. –investigation, data curation, validation; J. F. – investigation, methodology; K. Q. – investigation, methodology; S. S. – investigation, methodology; N. J. – investigation, methodology; M. D. – investigation, methodology.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Central Washington University for financial support through startup funds. J. F., K. Q., and S. S. thank Provost Denbeste for research fellowships. The school of graduate studies is thanked for a research fellowship to T. K. B. We also thank the Office of Undergraduate Studies (OUR) for research fellowships to J. E., J. F., and K. Q.Notes and references
- (a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (b) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed; (c) M. Feng, B. Tang, H. S. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed; (d) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed; (e) W.-M. Xu, F.-F. Han, M. He, D.-Y. Hu, J. He, S. Yang and B.-A. Song, J. Agric. Food Chem., 2012, 60, 1036 CrossRef CAS PubMed.
- (a) S. B. Madasu, N. A. Vekariya, M. N. V. D. Hari-Kiran, B. Gupta, A. Islam, P. S. Douglas and K. R. Babu, Beilstein J. Org. Chem., 2012, 8, 1400–1405 CrossRef CAS PubMed; (b) C. F. Sturino, G. O'Neill, N. Lachance, M. Boyd, C. Berthelette, M. Labelle, L. Li, B. Roy, J. Scheigetz, N. Tsou, Y. Aubin, K. P. Bateman, N. Chauret, S. H. Day, J.-F. Levesque, C. Seto, J. H. Silva, L. A. Trimble, M.-C. Carriere, D. Denis, G. Greig, S. Kargman, S. Lamontagne, M.-C. Mathieu, N. Sawyer, D. Slipetz, W. M. Abraham, T. Jones, M. McAuliffe, H. Piechuta, D. A. Nicoll-Griffith, Z. Wang, R. Zamboni, R. N. Young and K. M. Metters, J. Med. Chem., 2007, 50, 794–806 CrossRef CAS PubMed.
- (a) Y. I. Zhu and M. J. Stiller, J. Am. Acad. Dermatol., 2001, 45, 420–434 CrossRef CAS PubMed; (b) X. Codony, J. M. Vela and M. J. Ramírez, Curr. Opin. Pharmacol., 2011, 11, 94–100 CrossRef CAS PubMed; (c) G. La Regina, A. Coluccia, A. Brancale, F. Piscitelli, V. Gatti, G. Maga, A. Samuele, C. Pannecouque, D. Schols, J. Balzarini, E. Novellino and R. Silvestri, J. Med. Chem., 2011, 54, 1587–1598 CrossRef CAS PubMed; (d) P. Prasit, Z. Wang, C. Brideau, C. C. Chan, S. Charleson, J. Y. Gauthier, R. Gordon, J. Guay, M. Gresser, S. Kargman, B. Kennedy, Y. Leblanc, S. Leger, J. Mancini, G. P. O'Neill, M. Ouellet, M. D. Percival, H. Perrier, D. Riendeau, I. Rodger, P. Tagari, M. Therien, P. Vickers, E. Wong, L. J. Xu, R. N. Young and R. Zamboni, Bioorg. Med. Chem. Lett., 1999, 9, 1773 CrossRef CAS PubMed; (e) R. L. Lopez de Compadre, R. A. Pearlstein, A. J. Hopfinger and J. K. Seyde, J. Med. Chem., 1987, 30, 900 CrossRef CAS PubMed; (f) S. Leucht, G. Pitschel-Walz, R. R. Engel and W. Kissling, Am. J. Psychiatry, 2002, 159, 180 CrossRef PubMed; (g) W. Fischli and co-workers, Hypertension, 1991, 18, 22–31 CrossRef; (h) S. Doswald and co-workers, Bioorg. Med. Chem., 1994, 2, 403–410 CrossRef; (i) M. Teall and Co-Workers, Bioorg. Med. Chem. Lett., 2005, 15, 2685–2688 CrossRef PubMed.
- H. Sasabe, Y. Seino, M. Kimura and J. Kido, Chem. Mater., 2012, 24, 1404 CrossRef CAS.
- (a) Y.-S. Xiong, B. Zhang, Y. Yu, J. Weng and G. Lu, J. Org. Chem., 2019, 84, 13465–13472 CrossRef CAS PubMed; (b) W.-H. Rao, Q. Li, F.-Y. Chen, L.-L. Jiang, P. Xu, X.-W. Deng, M. Li, G.-D. Zou and X. Cao, J. Org. Chem., 2021, 86, 11998–12007 CrossRef CAS; (c) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 8069–8077 CrossRef CAS; (d) Y. Wang, L. Deng, J. Zhou, X. Wang, H. Mei, J. Han and Y. Pan, Adv. Synth. Catal., 2018, 360, 1060–1065 CrossRef CAS; (e) J. Zhang, K. Zhou and J. Wu, Org. Chem. Front., 2018, 5, 813–816 RSC.
- (a) J. Zhang, Y. An and J. Wu, Chem.– Eur. J., 2017, 23, 9477–9480 CrossRef CAS PubMed; (b) T. Liu, Y. Li, L. Lai, J. Cheng, J. Sun and J. Wu, Org. Lett., 2018, 20, 3605–3608 CrossRef CAS PubMed; (c) X. Wang, M. Yang, W. Xie, X. Fan and J. Wu, Chem. Commun., 2019, 55, 6010–6013 RSC; (d) W.-H. Rao, L.-L. Jiang, X.-M. Liu, M.-J. Chen, F.-Y. Chen, X. Jiang, J.-X. Zhao, G.-D. Zou, Y.-Q. Zhou and L. Tang, Org. Lett., 2019, 21, 2890–2893 CrossRef CAS PubMed; (e) J.-J. Wang and W. Yu, Org. Lett., 2019, 21, 9236–9240 CrossRef CAS PubMed; (f) Y. Xiong, Y. Sun and G. Zhang, Org. Lett., 2018, 20, 6250–6254 CrossRef CAS PubMed; (g) L.-J. Wang, J.-M. Chen, W. Dong, C.-Y. Hou, M. Pang, W.-B. Jin, F.-G. Dong, Z.-D. Xu and W. Li, J. Org. Chem., 2019, 84, 2330–2338 CrossRef CAS PubMed; (h) L. Song, L. Zhu, Z. Zhang, J.-H. Ye, S.-S. Yan, J.-L. Han, Z.-B. Yin, Y. Lan and D.-G. Yu, Org. Lett., 2018, 20, 3776–3779 CrossRef CAS PubMed; (i) M. Ito, A. Osaku, A. Shiibashi and T. Ikariya, Org. Lett., 2007, 9, 1821–1824 CrossRef CAS PubMed.
- (a) I. Pavlinov, E. M. Gerlach and L. N. Aldrich, Org. Biomol. Chem., 2019, 17, 1608–1623 RSC; (b) K.-J. Liu, Z. Wang, L.-H. Lu, J.-Y. Cheng, Y.-W. Lin, Z. Cao, X. Yu and W.-M. He, Green Chem., 2021, 23, 496–500 RSC; (c) X. Wang, Q. Wang, Y. Xue, K. Sun, L. Wu and B. Zhang, Chem. Commun., 2020, 56, 4436–64439 RSC; (d) Y. Zhao, X. Niu, H. Yang, J. Yang, Z. Wang and Q. Wang, Chem. Commun., 2022, 58, 8576–8579 RSC; (e) S. Jin, L. Wang, H. Han, X. Liu, Z. Bu and Q. Wang, Chem. Commun., 2021, 57, 395–362 RSC; (f) Q. H. Shi, Y.-H. Wang, X.-R. Wang, W.-H. Zhang, F.-L. Tian, L.-J. Peng, Y. Zhou and X.-L. Liu, Chem. Front., 2023, 10, 3307–3312 RSC.
- C. J. Gerry and S. L. Schreiber, Nat. Rev. Drug Discovery, 2018, 17, 333–352 CrossRef CAS PubMed.
- (a) T. Flagstad, G. Min, K. Bonnet, R. Morgentin, D. Roche, M. H. Clausen and T. E. Nielsen, Org. Biomol. Chem., 2016, 14, 4943–4946 RSC; (b) D. J. Foley, A. Nelson and S. P. Marsden, Angew. Chem., Int. Ed., 2016, 55, 13650–13657 CrossRef CAS; (c) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS.
- (a) H. Braunstein, S. Langevin, M. Khim, J. Adamson, K. Hovenkotter, L. Kotlarz, B. Mansker and T. K. Beng, Org. Biomol. Chem., 2016, 14, 8864–8872 RSC; (b) T. K. Beng and A. Moreno, New J. Chem., 2020, 44, 4257–4261 RSC; (c) T. K. Beng, M. Bauder, M. J. Rodriguez and A. Moreno, New J. Chem., 2018, 42, 16451–16455 RSC; (d) K. Hovenkotter, H. Braunstein, S. Langevin and T. K. Beng, Org. Biomol. Chem., 2017, 15, 1217–1221 RSC; (e) T. K. Beng and A. Moreno, RSC Adv., 2020, 10, 8805–8809 RSC; (f) T. K. Beng, M. Rodriguez and C. Borg, RSC Adv., 2022, 12, 17617–17620 RSC; (g) J. Garcia, J. Eichwald, J. Zesiger and T. K. Beng, RSC Adv., 2022, 12, 309–318 RSC; (h) T. K. Beng, C. Borg and M. Rodriguez, RSC Adv., 2022, 12, 28685–28691 RSC; (i) T. K. Beng, J. Garcia, J. Eichwald and C. Borg, RSC Adv., 2023, 13, 14355–14360 RSC.
- (a) D. S. Hamilton and D. A. Nicewicz, J. Am. Chem. Soc., 2012, 134, 18577–18580 CrossRef CAS PubMed; (b) H. Shigehisa, M. Hayashi, H. Ohkawa, T. Suzuki, H. Okayasu, M. Mukai, A. Yamazaki, R. Kawai, H. Kikuchi, Y. Satoh, A. Fukuyama and K. Hiroya, J. Am. Chem. Soc., 2016, 138, 10597–10604 CrossRef CAS PubMed; (c) L. Ferrand, Y. Tang, C. Aubert, L. Fensterback, V. Mouriès-Mansuy, M. Petit and M. Amatore, Org. Lett., 2017, 19, 2062–2065 CrossRef CAS PubMed.
- (a) H. Nakatsuji, Y. Sawamura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2014, 53, 6974–6977 CrossRef CAS; (b) H. Egami, J. Asada, K. Sato, D. Hashizume, Y. Kwato and Y. Hamashima, J. Am. Chem. Soc., 2015, 137, 10132–10135 CrossRef PubMed; (c) M. Wilking, C. G. Daniliuc and U. Hennecke, Chem.–Eur. J., 2016, 22, 18601–21807 CrossRef CAS PubMed; (d) S. E. Denmark, P. Ryabchuk, M. T. Burk and B. B. Gilbert, J. Org. Chem., 2016, 81, 10411–10423 CrossRef CAS; (e) J. D. Griffin, C. L. Cavanaugh and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 2097–2100 CrossRef CAS PubMed.
- (a) D. Karila, L. Leman and R. H. Dodd, Org. Lett., 2011, 13, 5830–6583 CrossRef CAS PubMed; (b) B. N. Hemric, K. Shen and Q. Wang, J. Am. Chem. Soc., 2016, 138, 5813–5816 CrossRef CAS; (c) J. Xie, Y.-W. Wang, L.-W. Qi and B. Zhang, Org. Lett., 2017, 19, 1148–1151 CrossRef CAS PubMed.
- (a) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462–12465 CrossRef CAS; (b) R. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 12655–12658 CrossRef CAS PubMed.
- A. R. Alcantara and P. D. de Maria, Curr. Green Chem., 2018, 5, 88 Search PubMed.
- T. K. Beng and R. E. Gawley, J. Am. Chem. Soc., 2010, 132, 12216 CrossRef CAS.
- (a) X. Qiao, D. L. Cheney, R. S. Alexander, A. M. Smallwood, S. R. King, K. He, A. R. Rendina, J. M. Luettgen, R. M. Knabb, R. R. Wexler and P. Y. S. Lam, Bioorg. Med. Chem. Lett., 2008, 18, 4118–4123 CrossRef; (b) M. A. Larsen, E. T. Hennessy, M. C. Deem, Y. Lam, J. Sauri and A. C. Sather, J. Am. Chem. Soc., 2020, 142, 726–732 CrossRef CAS.
- A. Calcaterra, L. Mangiardi, G. D. Monache, D. Quaglio, S. Balducci, S. Berardozzi, A. Iazzetti, R. Franzini, B. Botta and F. Ghirga, Molecules, 2020, 25, 414 CrossRef CAS PubMed.
- (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS; (b) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef; (c) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. L. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (d) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS; (e) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (f) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS.
- For selected examples, see (a) Y. Zhao, C. Ni, F. Jiang, B. Gao, X. Shen and J. Hu, ACS Catal., 2013, 3, 631–634 CrossRef CAS; (b) J. Gui, Q. Zhou, C.-M. Pan, Y. Yabe, A. C. Burns, M. R. Collins, M. A. Ornelas, Y. Ishihara and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 4853–4856 CrossRef CAS PubMed; (c) H. Watanabe, M. Takemoto, K. Adachi, Y. Okuda, A. Dakegata, T. Fukuyama, I. Ryu, K. Wakamatsu and A. Orita, Chem. Lett., 2020, 49, 409–412 CrossRef CAS.
- For selected examples, see (a) S. E. Denmark and A. J. Cresswell, J. Org. Chem., 2013, 78, 12593–12628 CrossRef CAS PubMed; (b) M. Nambo and C. M. Crudden, Angew. Chem., Int. Ed., 2014, 53, 742–746 CrossRef CAS PubMed; (c) M. Nambo, Z. T. Ariki, D. Canseco Gonzalez, D. D. Beattie and C. M. Crudden, Org. Lett., 2016, 18, 2339–2342 CrossRef CAS PubMed; (d) M. Nambo, E. C. Keske, J. P. G. Rygus, J. C.-H. Yim and C. M. Crudden, ACS Catal., 2017, 7, 1108–1112 CrossRef CAS; (e) Z. T. Ariki, Y. Maekawa, M. Nambo and C. M. Crudden, J. Am. Chem. Soc., 2018, 140, 78–81 CrossRef CAS PubMed; (f) R. R. Merchant, J. T. Edwards, T. Qin, M. M. Kruszyk, C. Bi, G. Che, D.-H. Bao, W. Qiao, L. Sun, M. R. Collins, O. O. Fadeyi, G. M. Gallego, J. J. Mousseau, P. Nuhant and P. S. Baran, Science, 2018, 360, 75–80 CrossRef CAS PubMed; (g) W. Miao, Y. Zhao, C. Ni, B. Gao, W. Zhang and J. Hu, J. Am. Chem. Soc., 2018, 140, 880–883 CrossRef CAS PubMed; (h) F. Takahashi, K. Nogi and H. Yorimitsu, Org. Lett., 2018, 20, 6601–6605 CrossRef CAS PubMed; (i) J. C.-H. Yim, M. Nambo, Y. Tahara and C. M. Crudden, Chem. Lett., 2019, 48, 975–977 CrossRef CAS; (j) M. Nambo, Y. Tahara, J. C.-H. Yim and C. M. Crudden, Chem.–Eur. J., 2019, 25, 1923–1926 CrossRef CAS PubMed; (k) T.-Y. Yu, Z.-J. Zheng, J.-H. Bai, H. Fang and H. Wei, Adv. Synth. Catal., 2019, 361, 2020–2024 CrossRef CAS; (l) J. M. E. Hughes and P. S. Fier, Org. Lett., 2019, 21, 5650–5654 CrossRef CAS; (m) Y. Maekawa, Z. T. Ariki, M. Nambo and C. M. Crudden, Org. Biomol. Chem., 2019, 17, 7300–7303 RSC; (n) M. Nambo, J. C.-H. Yim, L. B. O. Freitas, Y. Tahara, Z. T. Ariki, Y. Maekawa, D. Yokogawa and C. M. Crudden, Nat. Commun., 2019, 10, 4528 CrossRef PubMed; (o) P. Chatelain, A. Sau, C. N. Rowley and J. Moran, Angew. Chem., Int. Ed., 2019, 58, 14959–14963 CrossRef CAS PubMed; (p) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940–12944 CrossRef CAS PubMed; (q) Y. Xia and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9836–9840 CrossRef CAS; (r) M. Nambo, Y. Maekawa and C. M. Crudden, ACS Catal., 2022, 12, 3013–3032 CrossRef CAS; (s) M. Nambo, Y. Maekawa and C. M. Crudden, ACS Catal., 2022, 12, 3013–3032 CrossRef CAS.
- (a) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed; (b) T. Moragas, A. Correa and R. Martin, Chem.–Eur. J., 2014, 20, 8242–8258 CrossRef CAS PubMed; (c) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and von W. Jacobi, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed; (d) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS; (e) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC; (f) E. Richmond and J. Moran, Synthesis, 2018, 50, 499–513 CrossRef CAS; (g) L.-C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef CAS PubMed; (h) A. H. Cherney and S. E. Reisman, J. Am. Chem. Soc., 2014, 136, 14365–14368 CrossRef CAS; (i) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS PubMed; (j) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146–6159 CrossRef CAS PubMed; (k) D. P. Bassler, A. Alwali, L. Spence, O. Beale and T. K. Beng, J. Organomet. Chem., 2015, 780, 6 CrossRef CAS; (l) T. K. Beng, K. Sincavage, A. W. V. Silaire, A. Alwali, D. P. Bassler, L. E. Spence and O. Beale, Org. Biomol. Chem., 2015, 13, 5349–5353 RSC.
- For examples of reductive cross-coupling of alkyl aryl sulfones with aryl halides under nickel catalysis, see (a) J. M. E. Hughes and P. S. Fier, Org. Lett., 2019, 21, 5650–5654 CrossRef CAS; (b) W. Miao, C. Ni, P. Xiao, R. Jia, W. Zhang and J. Hu, Org. Lett., 2021, 23, 711–715 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data. See DOI: https://doi.org/10.1039/d3ra03800a |
| This journal is © The Royal Society of Chemistry 2023 |