DOI:
10.1039/D3RA03797E
(Paper)
RSC Adv., 2023,
13, 30855-30868
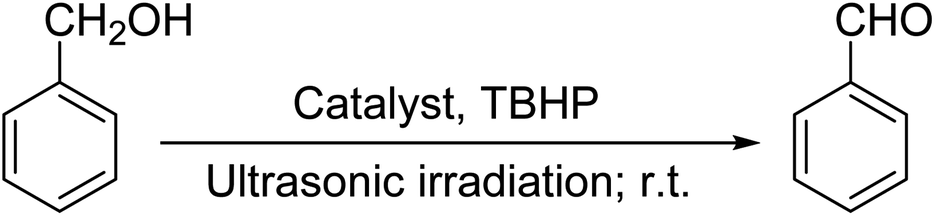
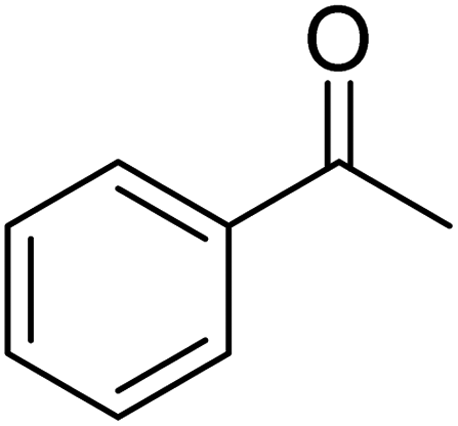
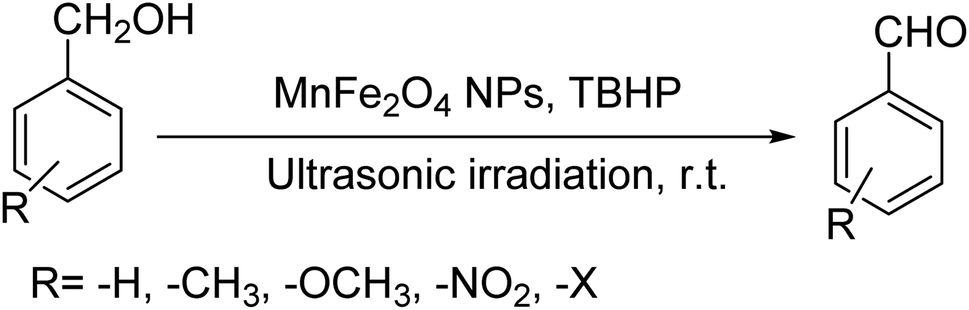
Ultrasonic irradiation-assisted MnFe2O4 nanoparticles catalyzed solvent-free selective oxidation of benzyl alcohol to benzaldehyde at room temperature†
Received
6th June 2023
, Accepted 13th October 2023
First published on 20th October 2023
Abstract
Magnetic nanoparticles (NPs) play a vital role in heterogeneous catalysis because of their easy separation, and effective recyclability. Herein, we report the synthesis of MnFe2O4 NPs for use as catalysts in the selective oxidation of benzyl alcohol to benzaldehyde under mild conditions. The MnFe2O4 NPs have been synthesized by precipitation method followed by hydrothermal ageing at 180 °C for 4.0 h. We have investigated the effect of chitosan and carboxymethyl cellulose on the size or morphology of the formed MnFe2O4 NPs. The X-ray diffraction study confirms the formation of pure and crystalline MnFe2O4 with varying average crystallite sizes ranging from 18 to 28 nm based on the type of additive used. The electron microscopy study reveals that the additive plays a significant role in controlling the morphology of the formed MnFe2O4 NPs. These MnFe2O4 NPs exhibit superparamagnetic behaviour at room temperature and can effectively catalyze the solvent-free selective oxidation of benzyl alcohol to benzaldehyde in the presence of tert-butyl hydroperoxide at room temperature under ultrasonic irradiation. The developed protocol can be extended to various substituted benzyl alcohols having both the electron withdrawing and electron donating groups to afford moderate to excellent yield of the products. The catalyst is magnetically retrievable, highly stable, and can be reused up to the sixth run without significant loss of catalytic activity.
1. Introduction
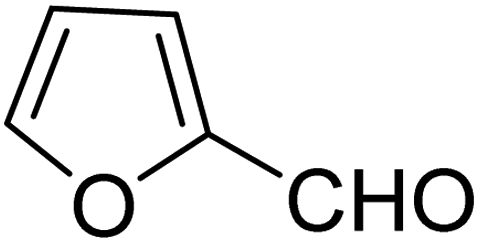
Selective oxidation of benzyl alcohol into benzaldehyde is an important research area as benzaldehyde is a noteworthy and useful synthetic intermediate in the synthesis of dyes, perfumery, agrochemicals, and pharmaceuticals.1 Traditionally, benzaldehyde is prepared by the hydrolysis of benzal halides,2 oxidation of benzyl chloride,3,4 liquid-phase oxidation of toluene,5,6 oxidation of styrene7,8 and the carbonylation of benzene.9 The presence of a chlorinated product, poor selectivity towards benzaldehyde, use of toxic or corrosive reagents and harsh reaction conditions are the major drawbacks of these processes.5,7,10 As a result, researchers adopted several pathways over the recent years for the synthesis of benzaldehyde selectively under milder conditions. Among different processes, the selective oxidation of benzyl alcohol catalyzed by both homogeneous and heterogeneous catalysts is the most popular technique. Although homogeneous catalyst possesses advantages such as readily accessible active sites under mild conditions, and good selectivity, their isolation from the reaction mixture for recycling, high cost and low stability are the major scientific challenges for their industrial applications.11–13 Whereas, despite low activity heterogeneous catalysts are preferred over homogeneous catalysts due to their environmental friendliness, operational simplicity, easy synthesis, easy separation and recyclability.14–18 Accordingly, several research groups reported the use of heterogeneous catalysts for the selective oxidation of benzyl alcohol using various metal-based oxidants.19–21 However most of these catalyst-oxidant systems failed to fulfil the demands for large-scale production because of the high cost, the toxicity of the oxidants and the production of toxic byproducts. To minimize the environmental hazards and cost, several research groups have developed heterogeneous catalysts based selective oxidation of benzyl alcohol using low-toxic oxidants such as O2, tert-butyl hydroperoxide (TBHP), H2O2, and so forth.22–26 However, most of the reported methods required high temperatures, prolonged reaction time and solvents which are again not beneficial for technological applications. Further, the recovery of the conventional catalyst for reuse is a daunting task in most cases because of the loss of the catalyst during purification by conventional filtration or centrifugation methods. It was observed that the substitution of conventional catalysts by magnetic ferrite nanoparticles (NPs) can overtake these problems due to their easy handling and recovery by a simple magnet for successive uses.13,14,16,24–27 Furthermore, the interesting physicochemical properties, low toxicity, low preparation cost, and simple synthetic approach, are some other added advantages of using such a catalyst.14,16 Accordingly, several research groups have reported the use of ferrite-based magnetic NPs for selective oxidation of benzyl alcohol using low-toxicity oxidants such as O2, TBHP, H2O2, etc.22–25,28 However, the use of a high amount of catalyst, low selectivity of benzaldehyde, high reaction time as well as temperature are the main drawbacks in some of the reported methods.23,24,29 So, a systematic study is required to further simplify the existing protocols and develop an active, selective, and recyclable catalyst that can work efficiently under solvent-free conditions for the selective oxidation of benzyl alcohols into corresponding benzaldehyde with high conversion.
Among the different ferrites, MnFe2O4 are widely used in different areas such as in catalysis, pollutant remediation from water, electrochemical, biomedical, sensors, and others due to their excellent soft magnetic properties, biocompatibility, and outstanding chemical and structural stability.27,30–37 In addition to the other catalytic applications, MnFe2O4 NPs have been utilized as a catalyst in the selective oxidation of benzyl alcohol to benzaldehyde by Jacintha et al. in 2015.28 They observed a maximum of 87.6% conversion with 100% selectivity at 80 °C for 10 h in presence of H2O2 as oxidant. A careful investigation suggests that there are no more reports on MnFe2O4 NPs catalyzed oxidation of benzyl alcohol to benzaldehyde. Thus, it is highly desirable to investigate if the process for the targeted reactions can be improved further using hydrothermally synthesized MnFe2O4 NPs in terms of reaction time and conversion under milder reaction conditions. Accordingly, the objective of this study is to synthesize MnFe2O4 NPs for use as the catalyst in the selective oxidation of benzyl alcohol into benzaldehyde under mild conditions. Herein, we report the synthesis of MnFe2O4 NPs via the precipitation method coupled with hydrothermal ageing in the presence of biodegradable polymers. We have also investigated the effect of additives on the morphology of MnFe2O4 NPs. The catalytic activity of the as-synthesized MnFe2O4 NPs has been investigated in the solvent-free selective oxidation of benzyl alcohol to benzaldehyde using TBHP as the oxidant at room temperature under ultrasonic irradiation. We have further extended the protocol to different substituted benzyl alcohols with both the electron-donating and electron-withdrawing groups.
2. Experimental section
2.1 Materials and methods
Anhydrous (anh.) ferric chloride (FeCl3, ≥98%), manganese(II) chloride tetrahydrate (MnCl2·4H2O, ≥98%), benzyl alcohol (≥99%), and NH4OH (25 wt%) were purchased from Merck, India. Chitosan (200–600 mPa s) and tert-butyl hydroperoxide (75% in water) were purchased from TCI Chemicals, India and Sigma-Aldrich, India respectively. The substituted benzylic, aliphatic, and heteroaromatic alcohols were purchased from Sigma-Aldrich, TCI Chemicals and Alfa-Aesar, India with 97–99% purity. All the reagents used throughout the experiments were of analytical grade (AR). Freshly prepared aqua-regia solution (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1, v/v) was used to clean all the glassware followed by washing with detergent and rinsing thoroughly with double distilled water and drying in an oven. All the solutions for materials synthesis were prepared in double-distilled water.
:HNO3 = 3:1, v/v) was used to clean all the glassware followed by washing with detergent and rinsing thoroughly with double distilled water and drying in an oven. All the solutions for materials synthesis were prepared in double-distilled water.
2.2 Synthesis of MnFe2O4 NPs
The synthesis of MnFe2O4 NPs was carried out by precipitation method followed by hydrothermal ageing. Typically, to an aqueous solution of chitosan (160 mg in 100 mL) taken in a glass vessel was added under continuous stirring an aqueous solution of FeCl3 (648.8 mg in 20 mL) and MnCl2·4H2O (395.8 mg in 20 mL) at room temperature which was varying from 25 to 30 °C. After 5 min of stirring an aqueous solution of NH4OH (4.5 mL of 25 wt% NH4OH diluted to 20 mL) was added dropwise to the reaction mixture and the stirring was continued for another 10 min. After that, the reaction mixture was transferred into a Teflon-lined autoclave of volume 200 mL and aged the reaction mixture at 180 °C for 4.0 h followed by cooling the reaction mixture to room temperature naturally. The product was isolated by centrifugation at 7000 rpm (REMI CPR 24 Plus) for 20 min. The isolated product was washed with double distilled water several times followed by centrifugation till the pH of the supernatant reached 7.0.
Finally, the solid product was dried in a vacuum oven at 60 °C overnight, and the obtained product was labelled as MnF-1 (Table 1). A similar set of reactions was carried out in the presence of carboxymethyl cellulose (CMC) keeping all other conditions identical and the products were labelled as MnF-2 (Table 1). As a controlled experiment, another sample (MnF-3) was also prepared without using any additives keeping all the reaction conditions identical to the previous two samples (Table 1). All the synthesized materials were analyzed by different characterization tools as discussed in detail in the ESI.†
Table 1 Reaction recipes for the synthesis of MnFe2O4 NPs
| Sample code |
Anh. FeCl3 (M) |
MnCl2·4H2O (M) |
NH4OH (M) |
Additive (wt%) |
| MnF-1 |
0.025 |
0.0125 |
0.375 |
Chitosan (0.1) |
| MnF-2 |
0.025 |
0.0125 |
0.375 |
CMC (0.1) |
| MnF-3 |
0.025 |
0.0125 |
0.375 |
No additive |
2.3 General procedure for the oxidation of benzyl alcohol
A mixture of 15.0 mg of MnFe2O4 NPs (MnF-1), benzyl alcohol (1.0 mmol), and TBHP (1.5 mmol), were taken in a 10 mL round bottom flask and then subjected to ultrasonic irradiation at room temperature which was varying from 25 to 30 °C. The progress of the reaction was monitored by thin-layer chromatography (TLC; Merck Germany made TLC plate with silica gel 60 coated on the aluminium sheet). After the completion of the reaction, the catalyst was recovered by using a simple bar magnet and the reaction mixture was dissolved in a mixture of 50 mL ethyl acetate and water (1:1 volume ratio) solution. The organic layer containing the crude product was then separated by extraction in a separating funnel, washed with brine (5 mL) solution, dried over anhydrous sodium sulphate, and then filtered. The crude product was obtained by removing the solvent under reduced pressure in a rotary evaporator (Buchi RII). The crude product was then purified by column chromatography using a solution of 5 vol% ethyl acetate in hexane and silica gel 100–200 mesh as the stationary phase to get the desired product. The purified products were identified by melting point and further characterized by 1H NMR and 13C {1H} NMR spectroscopy.
3. Results and discussion
3.1 X-ray diffraction studies
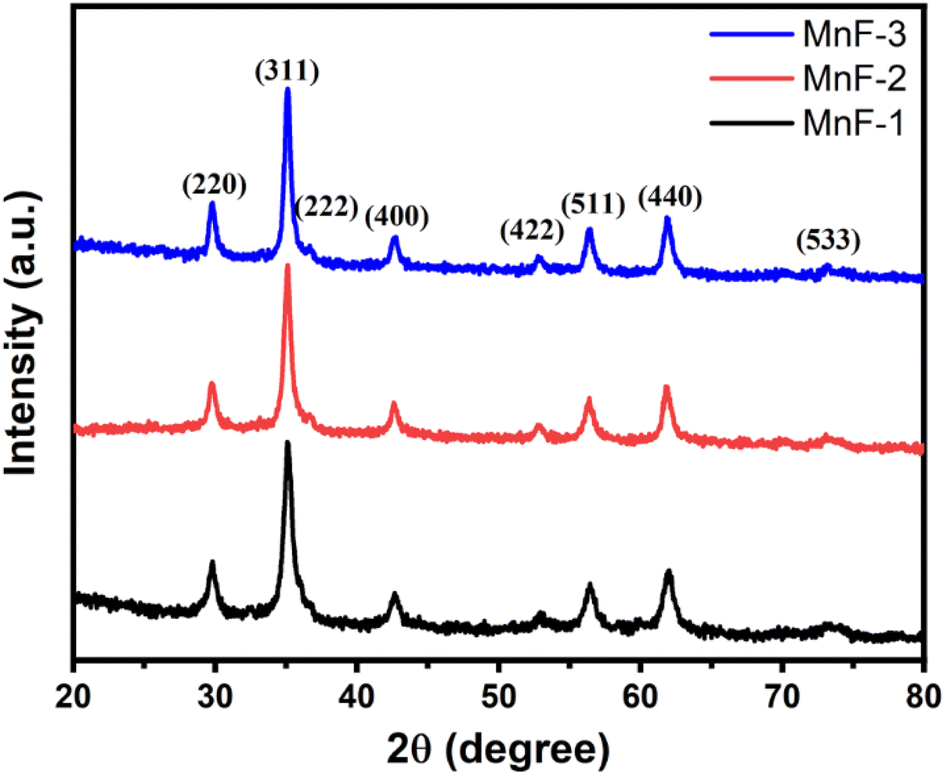
Fig. 1 shows the XRD pattern of different samples which exhibit diffraction peaks at 2θ = 29.7, 35.0, 36.7, 42.7, 52.8, 56.4, 61.9, and 73.1°. These peaks correspond to (220), (311), (222), (400), (422), (511), (440) and (533) planes of cubic spinel structures of MnFe2O4 with Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and are well matched with the standard data (JCPDS card no. 10-0319).27 The absence of any peak due to impurity confirmed the formation of high-purity single-phase crystalline MnFe2O4. The average crystallite size as calculated using Scherer's formula is 18.0, 24.0, and 28.0 nm for samples MnF-1, MnF-2, and MnF-3 respectively. The differences in the crystallite size might be attributed to the use of different additives in different samples. As chitosan and CMC have different functional groups, their presence during the synthesis greatly affects the nucleation rate and thereby controls crystallite size. While sample MnF-3 was prepared in the absence of an additive where uncontrolled nucleation results in particles of larger crystallite size. Considering the planes (220), (311), (400), (511) and (440), the mean lattice parameter, a of all the samples was calculated using the equation
m space group and are well matched with the standard data (JCPDS card no. 10-0319).27 The absence of any peak due to impurity confirmed the formation of high-purity single-phase crystalline MnFe2O4. The average crystallite size as calculated using Scherer's formula is 18.0, 24.0, and 28.0 nm for samples MnF-1, MnF-2, and MnF-3 respectively. The differences in the crystallite size might be attributed to the use of different additives in different samples. As chitosan and CMC have different functional groups, their presence during the synthesis greatly affects the nucleation rate and thereby controls crystallite size. While sample MnF-3 was prepared in the absence of an additive where uncontrolled nucleation results in particles of larger crystallite size. Considering the planes (220), (311), (400), (511) and (440), the mean lattice parameter, a of all the samples was calculated using the equation 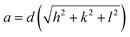 and the average value for different samples was estimated to be 8.47 Å, which is close to the theoretical value of 8.499 Å.
and the average value for different samples was estimated to be 8.47 Å, which is close to the theoretical value of 8.499 Å.
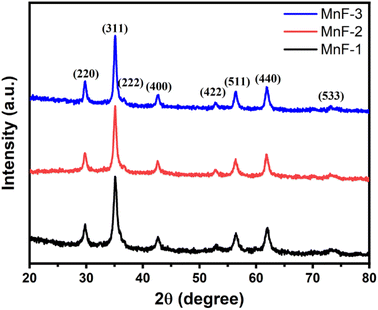 |
| | Fig. 1 XRD pattern of different samples of MnFe2O4 NPs. | |
3.2 Electron microscopy analysis
To study the surface topography of the synthesized MnFe2O4 NPs, the field emission scanning electron microscopy (FESEM) study was undertaken. Fig. 2 shows that spherical particles of sizes below 50 nm are formed in all the samples. However, the exact information about the particle size could not be established due to the overlapping of the particles in the micrographs. The energy dispersive X-ray (EDX) spectra as well as the elemental distribution were recorded from all the samples and are presented in Fig. S1.† The EDX spectra display the peaks due to elemental Mn, Fe and O confirming that the samples are composed of Mn, Fe and O. The elemental ratios of Mn:Fe:O is 1:1.83:3.97, 1:1.76:3.94 and 1:1.93:4.6 for samples MnF-1, MnF-2 and MnF-3 respectively. The elemental distribution results of respective individual elements (Fig. S1†) in the samples further support these findings. It is to be noted that the amount of O element in different samples is different which is due to the reason that the estimation of light elements, such as O in oxides cannot be accurately determined by direct measurement of O peak.38
 |
| | Fig. 2 SEM images of the synthesized MnFe2O4 NPs: samples (a) MnF-1, (b) MnF-2 and (c) MnF-3. | |
To get an idea about morphological details, the transmission electron microscopy (TEM) study was undertaken. The TEM image of sample MnF-1 (Fig. 3a) prepared with 0.1 wt% chitosan reveals the formation of nearly spherical particles. The sizes of these particles are varied from 9 to 16 nm. The high-resolution TEM image (Fig. 3b) of an individual particle confirmed that the particles are single crystalline with interplane space, d value of 0.5 nm corresponding to (111) plane of cubic MnFe2O4. The selected area electron diffraction (SAED) pattern presented in Fig. 3c indicates the polycrystalline nature of the particles which might be attributed to the overlapping of several particles in the TEM grid. Fig. 3d represents the TEM image of sample MnF-2 prepared in the presence of 0.1 wt% CMC which shows the formation of assembled particles that look like flowers and are composed of smaller-sized spherical particles. The sizes of these assembled structures are varied from 34 nm to 65 nm while the sizes of the spherical particles forming these assembled structures range from 10 to 30 nm. The high-resolution TEM image (Fig. 3e) of an individual particle again confirmed the single crystalline nature of the particles with interplane space, d value of 0.5 nm corresponding to (111) plane of cubic MnFe2O4. These particles are also polycrystalline as confirmed by the respective SAED pattern shown in Fig. 3f. Fig. 3g displays the TEM image of sample MnF-3 which shows the formation of particles of mixed morphology comprising mostly cube and spherical shapes. The sizes of these particles vary from 12 to 36 nm in which the spherical particles are relatively smaller than the cube-shaped particles. The individual particles are also single crystalline as the lattice planes are perfectly aligned in the high-resolution TEM image (Fig. 3h) with interplane space, d value of 0.5 nm. The SAED pattern recorded from a portion of the TEM image indicated the formation of well-crystalline MnFe2O4 also in the absence of any additive. It can be inferred from the TEM results that chitosan plays the role of stabilizer which controls the particles' growth thereby forming only spherical particles. While CMC though plays the role of stabilizer, it also helps in forming assembled structures quite similar to flower-like morphology. While in the absence of any additive uncontrolled growth of particles resulted in the formation of particles of mixed morphology. Besides, the additive also controls the crystallite sizes of the particles formed as discussed in the previous section 3.1.
 |
| | Fig. 3 TEM images of MnFe2O4 NPs: samples (a and b) MnF-1, (d and e) MnF-2, and (g and h) MnF-3 and (c, f, and i) represent the SAED patterns of the respective samples. | |
3.3 N2 gas adsorption–desorption study
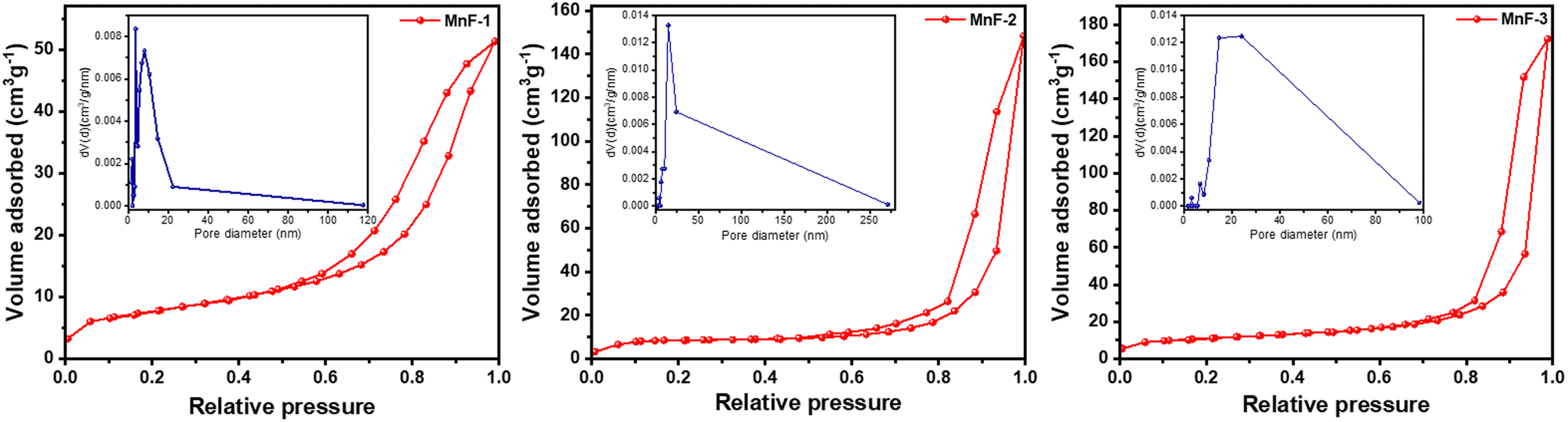
To understand the surface properties, N2 gas adsorption–desorption study was undertaken on all the samples. Fig. 4 depicts the nitrogen gas adsorption–desorption isotherms which indicate that all the samples exhibit a type IV isotherm with H3 hysteresis.39 These types of isotherms are usually exhibited by mesoporous materials.39 The pore size distribution curves of all the samples are displayed in the insets of Fig. 4. The average pore diameter and pore volume was calculated using the well-known Barrett–Joyner–Halenda (BJH) method and is presented in Table 2. The average pore diameters vary from 3.7 to 24.0 nm which further confirms the existence of mesopores in the particles in all the samples. The multi-point Brunauer–Emmett–Teller (BET) surface area analysis (Table 2) indicates that sample MnF-3 exhibited the highest surface area while sample MnF-1 possessed the lowest surface area of 27.2 m2 g−1. The different assembling patterns in the samples might be responsible for the variation of surface areas of the particles.
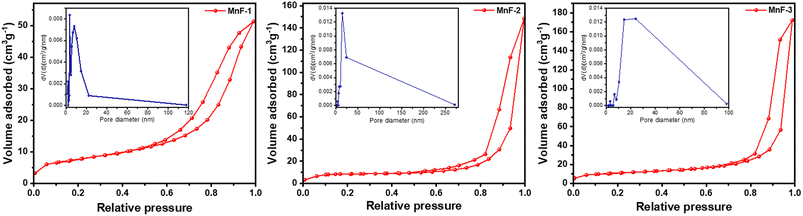 |
| | Fig. 4 N2 gas adsorption–desorption isotherms of different samples of MnFe2O4 NPs. The figure in the insets represents the BJH pore size distribution curve of the respective samples. | |
Table 2 Specific surface area and pore properties of the as-prepared MnFe2O4 NPs
| Sample |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Pore diameter (nm) |
| MnF-1 |
27.2 |
0.09 |
3.7 |
| MnF-2 |
32.5 |
0.24 |
14.9 |
| MnF-3 |
39.0 |
0.28 |
24.0 |
3.4 Magnetic property study
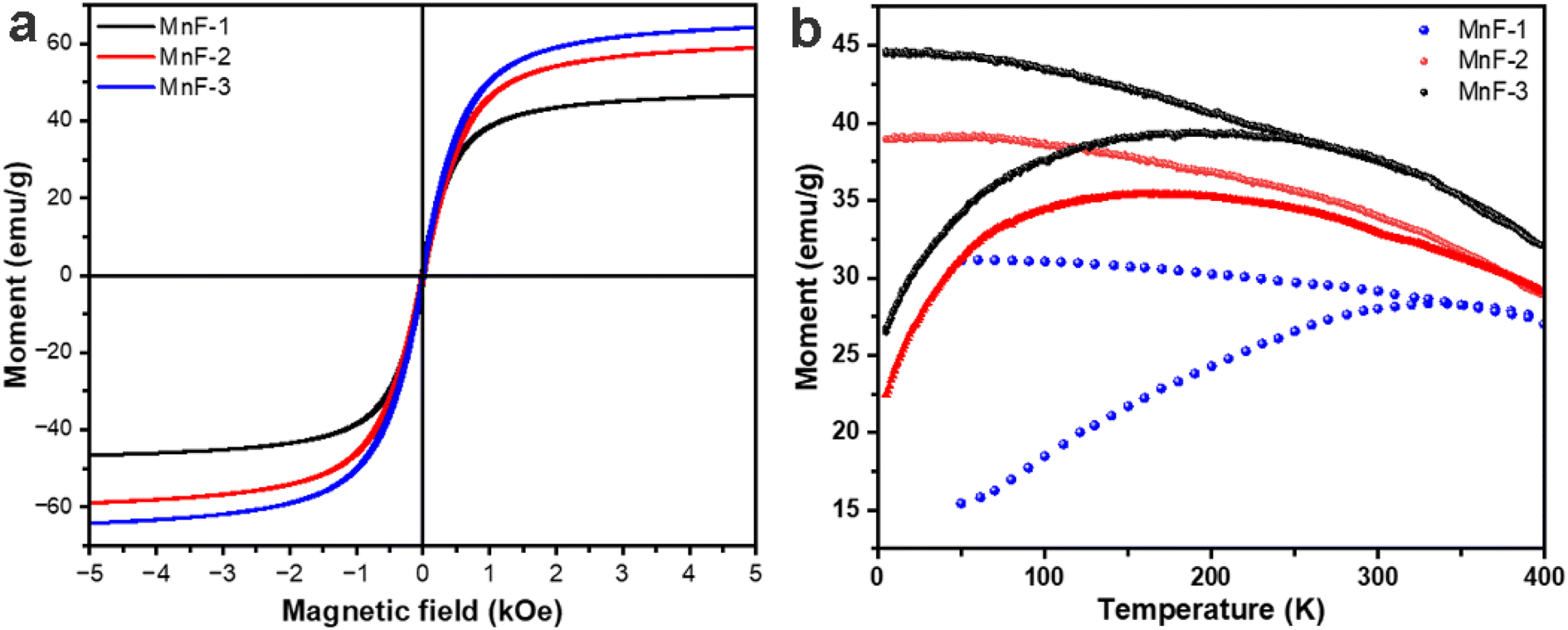
The magnetic properties of MnFe2O4 NPs were measured by vibrating sample magnetometry (VSM). Fig. 5a shows the variation of magnetization with the magnetic field (M–H curves) at 300 K. The absence of significant hysteresis loops in the M–H curves of all the samples confirmed the superparamagnetic behaviour of MnFe2O4 NPs at room temperature. The saturation magnetization (Ms) values were found to be 46.1, 58.9 and 64.1 emu g−1 for samples MnF-1, MnF-2, and MnF-3 respectively. These values are lower than the bulk values of 80 emu g−1 which is attributed to the decrease in the particle size.40 Further the trend of increase in the Ms values of different samples is in accordance to their average crystallite size discussed in section 3.1. Also, the presence of non-magnetic polymeric components on the surface of MnFe2O4 NPs in samples MnF-1 and MnF-2 might contribute to the lower Ms value compared to that of sample MnF-3 prepared in the absence of any additive.41,42
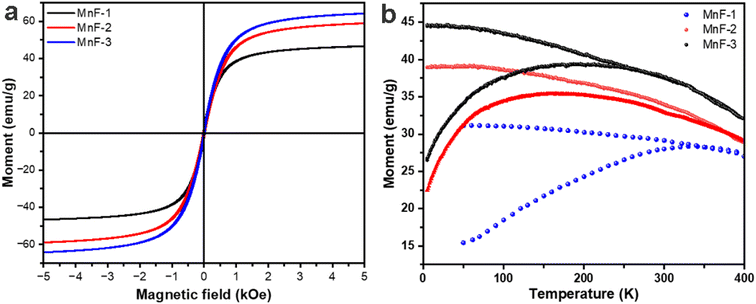 |
| | Fig. 5 (a) Magnetization versus applied magnetic field measured at 300 K and (b) zero-filed cooled (ZFC) and field cooled (FC) temperature dependence magnetization of different samples at an applied magnetic field of 500 Oe. | |
Fig. 5b displays the temperature-dependent magnetization curves (M versus T) of all the samples recorded at an applied field of 500 Oe. It was observed that with increasing the temperature, the ZFC magnetization increases up to a maximum point and then begins to fall as the temperature increases further. The blocking temperature, TB was found to be 331 K, 167 K, and 197 K for samples MnF-1, MnF-2, and MnF-3 respectively. Further, the ZFC-FC curves of samples MnF-1, MnF-2, and MnF-3 exhibit irreversibility temperatures of 341 K, 366 K, and 261 K respectively. This temperature dependence ZFC and FC magnetisation curve further confirms that the MnFe2O4 NPs exhibit superparamagnetic behaviour at higher temperatures.
3.5 X-ray photoelectron spectroscopy study
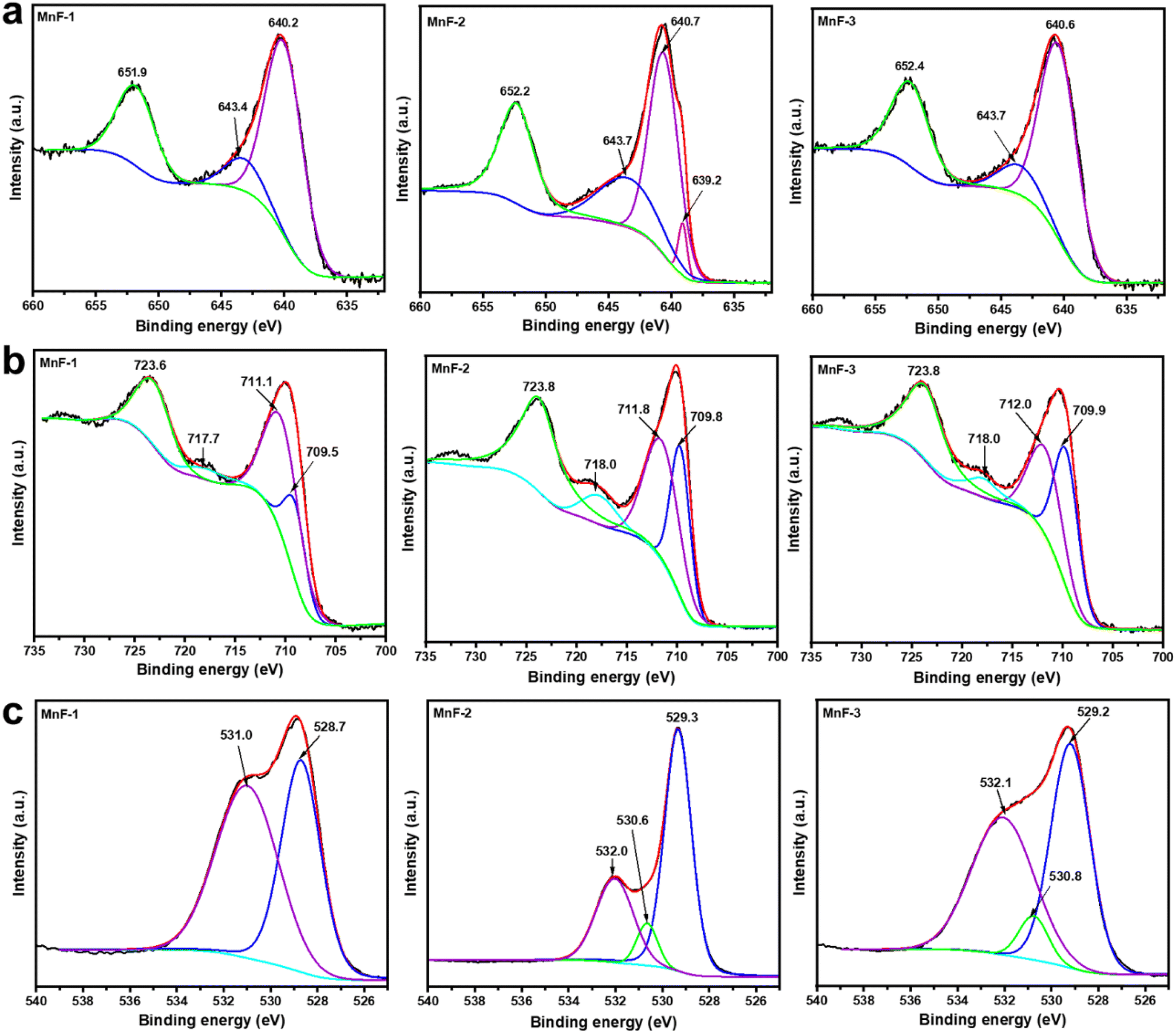
To ascertain the elemental composition and the oxidation state of the individual element in the samples, the X-ray photoelectron spectroscopy (XPS) study was conducted. The survey scan spectra (Fig. S2a†) reveal the presence of elemental peaks due to Mn 2p, Fe 2p, and O 1s in all the samples which indicates that the samples are composed of Mn, Fe and O. Besides, the presence of a peak due to N 1s in sample MnF-1 is attributed to the N present in the chitosan molecules used as additive during the synthesis. Additionally, the C 1s peak was also observed in all the samples which is attributed to the presence of polymer molecules in MnF-1 and MnF-2 and conductive carbon tape used to hold the samples to make them conductive. Further, the peak in sample MnF-3 might arise due to the adventitious carbon which is often made up of hydrocarbons and is present on the sample surface due to charging effects.43 However, the reason for appearing intense peak in sample MnF-3 is not known but it might be due to the different factors associated with the analysis. The high-resolution XPS spectra of the core level Mn 2p region (Fig. 6a) exhibit two prominent peaks at binding energies of 640.0–640.7 eV and 651.9–652.4 eV respectively which are assigned to the Mn 2p3/2 and Mn 2p1/2 level of Mn2+ in the samples. The fitted Mn 2p3/2 curves further exhibited peaks at binding energy 640.2–640.7 eV which is assigned to Mn2+ in the samples.44,45 While the broad shoulder peaks at around 643.4–643.7 eV correspond to the existence of Mn4+ in the samples.45,46 Additionally, a weak peak at around 639.2 eV could be noticed in the fitted Mn 2p3/2 peak in sample MnF-2 which is also attributable to the contribution from Mn2+.44 The high-resolution XPS spectra of the core level Fe 2p region (Fig. 6b) also display two prominent peaks at binding energies 723.6–723.8 eV and 710.0–710.4 eV due to the Fe 2p1/2 and Fe 2p3/2 level respectively. The Fe 2p3/2 peaks can be effectively fitted into two peaks at 709.5–709.9 eV and 711.1–712.0 eV, which are attributed to the presence of Fe3+ ions located in the octahedral and tetrahedral sites, respectively as reported earlier.46,47 Similarly, the Fe 2p1/2 peaks at 723.6–723.8 eV confirm the presence of Fe3+ ions in the octahedral sites of the spinel. Additionally, the presence of a satellite peak in the region 717.7–718.0 eV confirms the existence of Fe in the Fe3+ state in all samples. The fitted high-resolution XPS spectra of the O 1s region (Fig. 6c) display a peak in the binding energy region 528.7–529.3 eV which is assigned to the lattice oxygen (O2−).46 While the peaks at 530.6–531.0 eV and 532.0–532.1 eV correspond to the hydroxyl oxygen species or oxygen-related vacancy and a surface-adsorbed water molecule.46,47 The high-resolution XPS spectrum of the N 1s region (Fig. S2b†) displays a peak at binding energy 398.5 eV in sample MnF-1 which was prepared in the presence of chitosan. The peak can be assigned to the –NH2 group in the chitosan molecule.48 This indicates the adsorption of chitosan molecules on the surface of MnFe2O4 NPs in sample MnF-1 while these peaks are absent in the other two samples namely MnF-2 and MnF-3 prepared in the presence of CMC prepared and in the absence of any additive respectively.
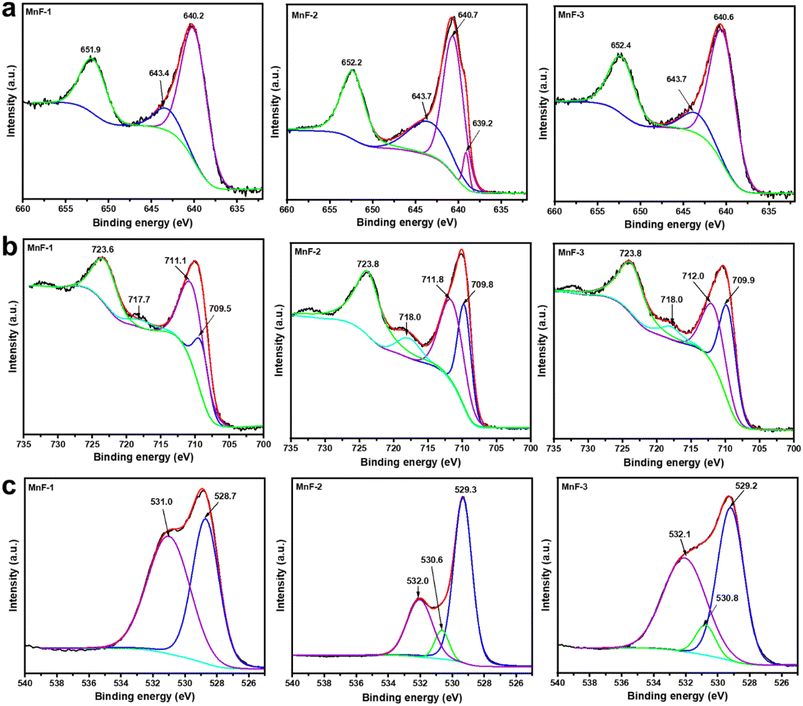 |
| | Fig. 6 High-resolution X-ray photoelectron spectra of (a) Mn 2p region, (b) Fe 2p region, and (c) O 1s region of different samples. | |
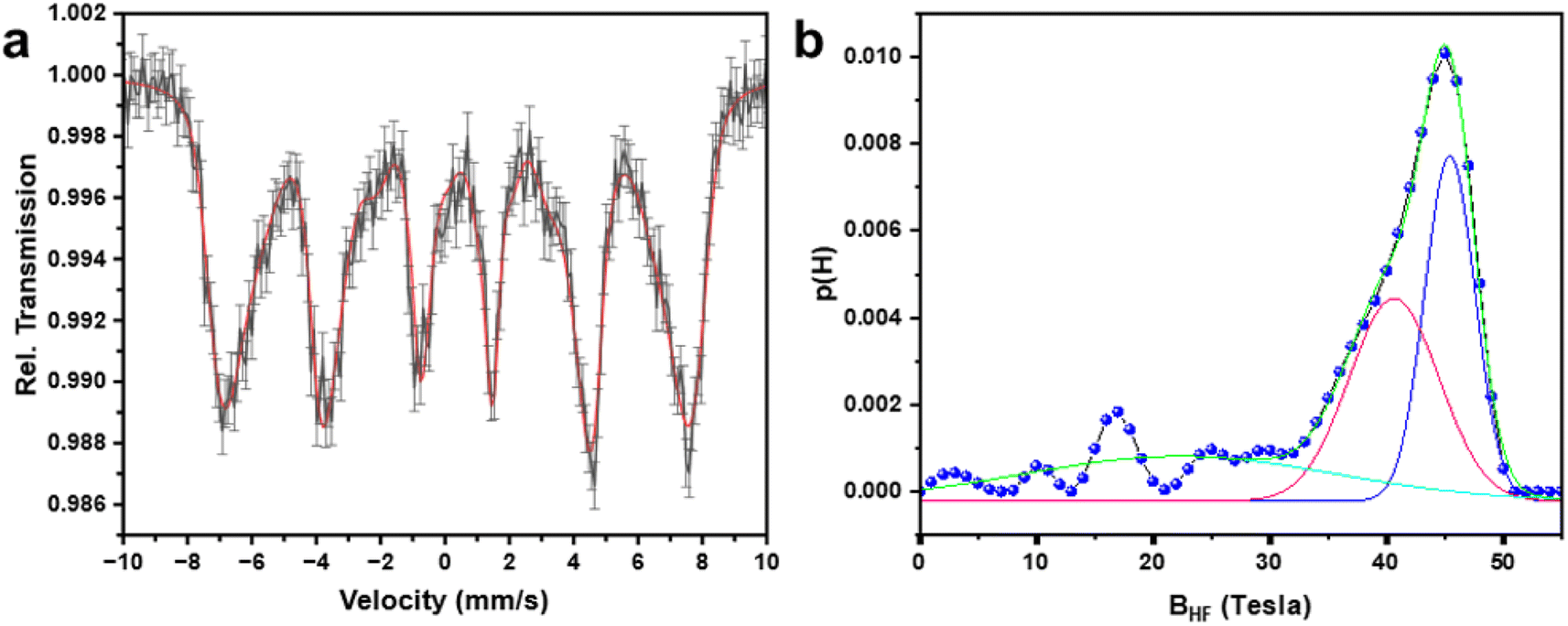
3.6 Mössbauer spectroscopy study
To understand the cationic distribution and magnetic properties of MnFe2O4 NPs, the room temperature 57Fe Mössbauer spectroscopy study was carried out on sample MnF-1. Fig. 7a depicts the Mössbauer spectrum which exhibits a magnetically split six-line hyperfine pattern confirming that the structure is magnetically ordered. As the peaks are broad, the data was analyzed considering the distribution of hyperfine fields.49 The obtained probability distribution of hyperfine fields is shown in Fig. 7b which shows two peaks at 16.8, and 44.9 Tesla indicating that two magnetic sextets correspond to A-, and B-sites of the spinel phase. The site with higher hyperfine field distribution is assigned to the octahedral (B) sites. The fitted curve at higher hyperfine field distribution produced two peaks with a relative spectral area of about 1:1 which gives a relative contribution of octahedral and tetrahedral Fe-ions in the sample. This is in agreement with the standard value of MnFe2O4 spinel.
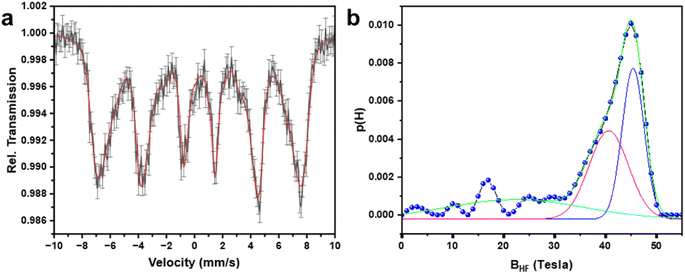 |
| | Fig. 7 (a) Room temperature 57Fe Mössbauer spectrum of sample MnFe2O4 NPs (MnF-1) and (b) the corresponding hyperfine field distribution. | |
3.7 Catalytic activity study
3.7.1 Optimization of the reaction condition. To evaluate the catalytic potential of the as-synthesized MnFe2O4 NPs, the oxidation of benzyl alcohol was chosen as a model reaction. In the initial study, benzyl alcohol (1.0 mmol) and TBHP (1.5 mmol) were allowed to react at room temperature under magnetic stirring in the absence of a solvent and catalyst for 3.0 h. This reaction produced only 16% of the desired product (Table 3; entry 1). However, the addition of 10.0 mg of MnFe2O4 NPs (MnF-1) as the catalyst to the reaction mixture yielded 52% product under identical reaction conditions (Table 3; entry 2). To assess the catalytic activity of the other two MnFe2O4 NPs namely sample MnF-2 and MnF-3, we repeated the same reaction under identical reaction conditions and observed 46% and 39% product yield respectively within the same time (Table 3; entries 3–4). The results indicate that sample MnF-1 exhibits better performance as catalysts over the other two but the overall catalytic activity is not satisfactory under the condition of investigation. It is reported that ultrasonic irradiation is an effective tool to carry out some organic transformations very efficiently.50 Further, our research group has also reported recently the use of ultrasonic irradiation to carry out aza-Michael reaction and C–B bond cleavage reactions very effectively in a milder way.51,52 To check if the activity of these catalysts under consideration can be enhanced under ultrasonic irradiation, we repeated the catalytic reaction under ultrasonic irradiation keeping other reaction parameters identical. Surprisingly, we noticed that the yield of the products increased significantly with all three catalysts under ultrasonic irradiation (Table 3; entries 5–7). However, sample MnF-1 offered the maximum product yield of 85% compared to the other two catalysts. The reason behind the enhanced conversion under ultrasonic irradiation might be the cavitation phenomenon caused by ultrasonic irradiation which may supply energy to the reaction medium by creating a repeating pattern of compressions and rarefaction.53,54 This results in an increase in the rate of the reaction and the yield. Further, it is obvious from Table 3 that the catalyst synthesized in the presence of additives offers higher catalytic activity which might be due to the different surface properties of the particles in different samples. For a systematic investigation, we performed several controlled reactions with bulk manganese chloride, ferric chloride, γ-Fe2O3, MnO2, and a mixture of γ-Fe2O3 and MnO2 (Table 3; entries 8–12). However, the product yield was not very satisfactory compared to the MnFe2O4 NPs. The better catalytic performance of MnFe2O4 NPs might be attributed to the synergistic effect of both the component and crystallinity of the ferrite compared to the individual monometallic oxide and the bulk metal salts. Accordingly, we chose sample MnF-1 as the catalyst to extend the optimization further for other reaction parameters such as solvent, oxidant, temperature and catalyst amount under ultrasonic irradiation to achieve the best optimum condition. Firstly, the impact of different solvents such as CH3CN, water, toluene, acetone, and a binary mixture of different solvents was examined (Table S1;† entries 2–7). It was observed that under the solvent-free condition, the reaction offered a higher yield of benzaldehyde with 100% selectivity (Table S1;† entry 1). The reason for the lower yield in a solvent might be attributed to the solvation effect of catalysts and reactants.55 In the absence of a solvent, the reactants approach the catalyst surface quickly thereby enhancing the reaction rate. On the other hand, in the presence of a solvent, the reactants are distributed in the solvent, which delays the interaction with the catalyst resulting in a lowering of the reaction rate. In the next assessment, we tested the reaction with different catalyst loading of 10.0 mg, 15.0 mg, and 20.0 mg (Table S1:† entries 1, 8, and 9), and we observed that increasing the amount of the catalyst from 10.0 mg to 15.0 mg increased the product yield from 85% to 90% but further increase in the catalyst loading to 20.0 mg did not improve the yield of the product significantly. Hence, the catalyst loading was fixed at 15.0 mg for further study. We also investigated the effect of the amount of oxidant (Table S1;† entries 8, 10 and 11) and observed that 1.5 mmol of TBHP is optimum for offering the maximum yield. Further, the impact of temperature on the catalytic performance of MnFe2O4 NPs (MnF-1) was also monitored by considering 15.0 mg of catalyst, 2.0 h of reaction time, and 1.5 mmol of TBHP under ultrasonic irradiation. We observed that the increase of the temperature from room temperature (25–30 °C) to 40 °C decreases the yield of benzaldehyde (Table S1;† entry 12), which might be due to the formation of benzoic acid as a by-product at the higher temperature as confirmed by the NMR spectroscopy (Fig. S18 and S19†). So, the room temperature was considered as the optimum temperature for the current protocol. We also recorded that when the reaction time was increased, no significant increase in product yield was observed (Table S1;† entry 13). In our final assessment, different oxidants such as O2, and hydrogen peroxide (H2O2) were tested (Table S1;† entries 14–15) but the yield was not satisfactory. The screening results confirmed that for 1.0 mmol of benzyl alcohol; 1.5 mmol of TBHP as oxidant, and 15.0 mg of MnFe2O4 NPs (MnF-1) as the catalyst in solvent-free condition at room temperature for 2.0 h is the optimum reaction conditions for the selective oxidation of benzyl alcohol to benzaldehyde.
Table 3 Comparison of the efficiency of different catalysts in the oxidation of benzyl alcohol to benzaldehydea
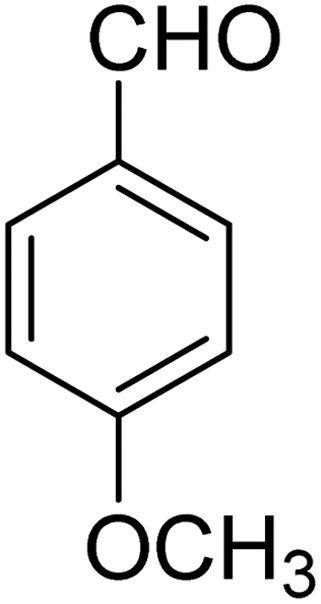
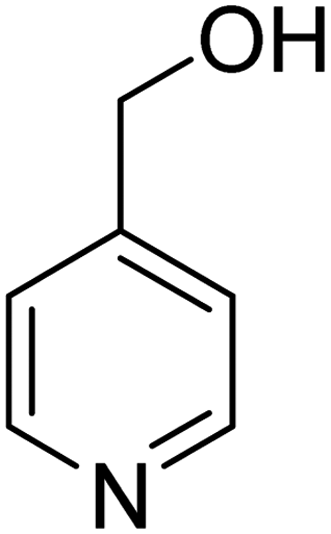
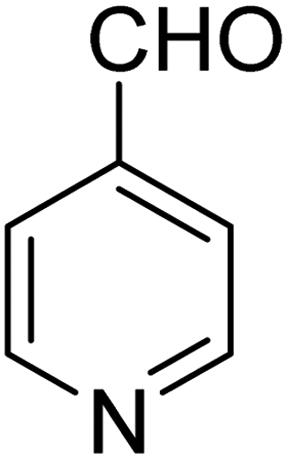
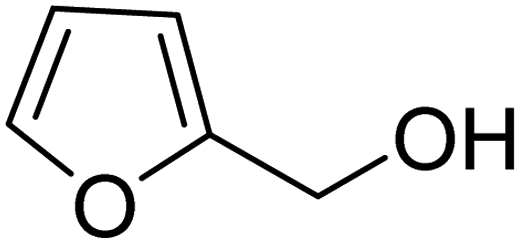
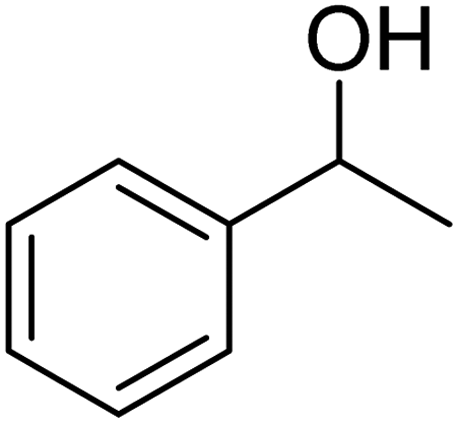
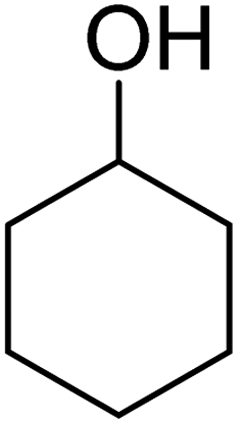
3.7.2 Substrate scope. With the optimized reaction conditions in hand, we expanded the oxidation reaction to different substituted benzyl alcohols, aliphatic alcohols, and heteroaromatic alcohols. As listed in Table 4, all the substituted benzyl alcohols are easily converted into corresponding aldehydes with 100% selectivity. No further oxidation to carboxylic acid was detected in the reaction system. We noticed that the substituted benzyl alcohol with electron-donating groups is more reactive (Table 4; entries 9–10) than benzyl alcohol-bearing electron-withdrawing groups (Table 4; entries 2–8). This might be due to the enhancement of electron density on the reactive site of the reactant by an electron-donating group making the oxidation easier.56 These results are also consistent with the reported observations by Chen et al.57 Further, the substituents at para-position of benzyl alcohol (Table 4; entries 3, 5, 8, 9, and 10) offer better yield than ortho (Table 4; entries 2, 4, and 6) and meta-substituted benzyl alcohols (Table 4; entry 7) which might be due to the minimum steric hindrance in the para position than the ortho and meta positions.56 On the other hand, heteroaromatic alcohols and aliphatic alcohols (Table 4; entries 11–15) offered a low yield of their corresponding oxidized product which might be due to the fact that the reaction was accomplished under mild conditions. It is reported that such heterocyclic alcohols have a tendency for strong coordination with the active metal centre thereby deactivating the activity of the catalyst.21 Similarly, aliphatic alcohols are very difficult to oxidize traditionally because of the absence of conjugation in the β-position of the hydroxyl group and hence their poor reactivity.56,58 Therefore, we conclude that the prepared MnFe2O4 NPs offer excellent catalytic activity towards different benzyl alcohols under mild reaction conditions at room temperature.
Table 4 Substrate scope for oxidation reaction under ultrasonic irradiationa
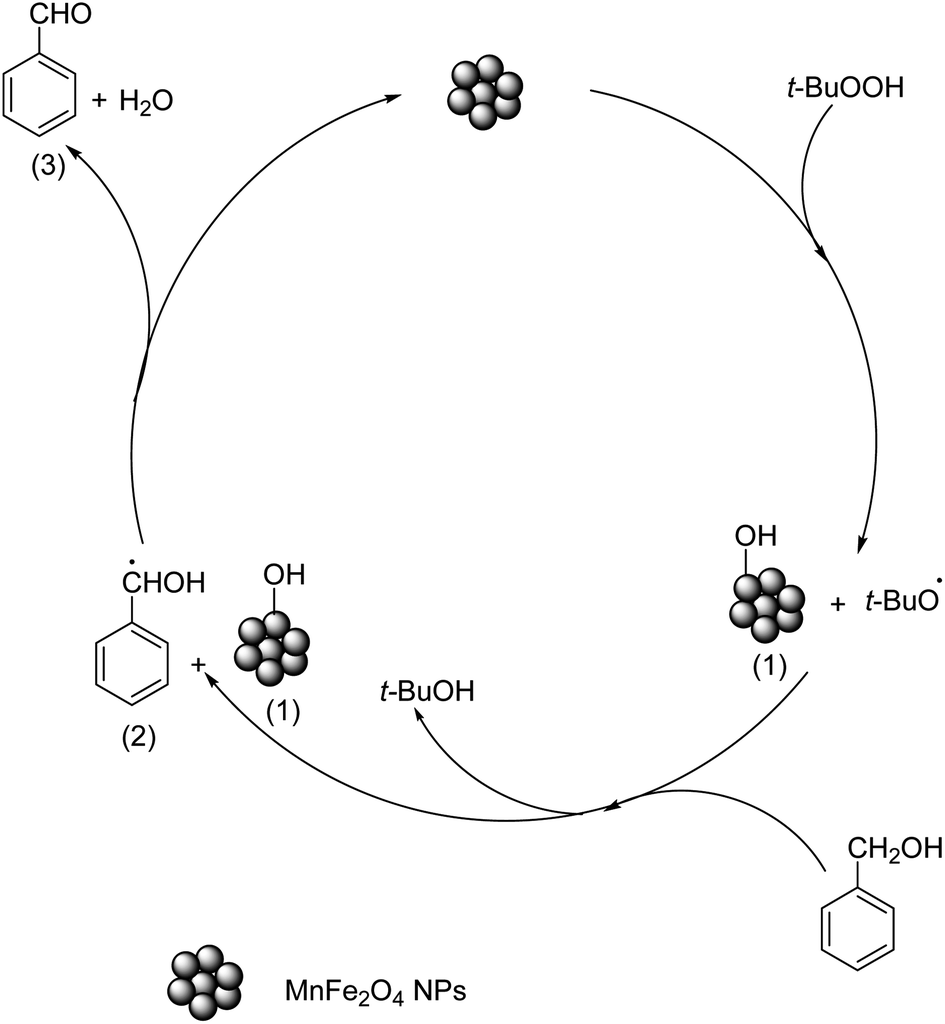
3.7.3 Reaction mechanism. Based on the literature reports,59,60 and our observations, a hypothesized mechanism for the formation of benzaldehyde from benzyl alcohol is proposed (Scheme 1). We believe that in the initial step, TBHP is homolytically cleaved on the surface of the catalyst at the O–O link via a free radical mechanism, resulting in the formation of active species t-BuO˙ and HO˙. The electrostatic interaction between HO˙ and the catalyst surface produces intermediate 1. The next step is the removal of hydrogen from benzyl alcohol to produce intermediate 2. Finally, the hydrogen transfer from intermediate 2 to intermediate 1 results in the formation of product 3 accompanied by the release of H2O as a byproduct, and the catalyst is regenerated. The involvement of the free radicals during the reaction was proved by doing a quick test using hydroquinone (Merck, India, ≥99%), an efficient free radical scavenger.23 When a small amount of hydroquinone (0.5 mmol) was added to the reaction medium no significant product formation was noticed in 3.0 h. This proves that the reaction proceeds via the formation of free radicals. Further to get an the idea of which component of the catalyst was involved in the catalyzed oxidation, the XPS study was carried out on the recovered sample. The fitted high-resolution XPS spectrum of the Mn 2p region (Fig. S3a†) shows the presence of peaks at binding energies 639.2 eV, 640.5 eV, 641.8 eV and 644.8 eV which corresponds to Mn(II), Mn(III) and Mn(IV) respectively.44,45 The additional peak at binding energy 641.6 eV due to Mn(III) compared to the fresh MnF-1 catalyst confirmed the conversion of Mn(II) to Mn(III) during the reaction. This might be due to the oxidation of Mn(II) by TBHP or Fe(III) mediated transition of Mn(II) to Mn(III) in the medium.61 While a slight shifting in peak position for Fe 2p was observed in the fitted XPS spectrum of the Fe 2p region (Fig. S3b†) which might be due to the change in the environment during the reaction. In the high-resolution XPS spectrum of the O 1s region (Fig. S3c†), an additional peak at binding energy 530.3 eV was observed compared to the parent sample which is attributable to the presence of hydroxyl oxygen species or oxygen-related vacancy.46 No characteristic changes were detected in the high-resolution XPS spectrum of the N 1s region (Fig. S3d†). From these observations, we assume that the change in the oxidation state from Mn2+ to Mn3+ during the catalysis reaction promotes the reaction compared to other bulk and individual mono metallic oxide NPs. Additionally, the literature reports suggest that the transition between Mn2+ to Mn3+ might also cause the conversion of Fe3+ to Fe2+ in MnFe2O4 in the presence of TBHP thereby generating highly active peroxy radicals and thus enhancing the catalytic activity of MnFe2O4 NPs compared to other tested catalysts .61
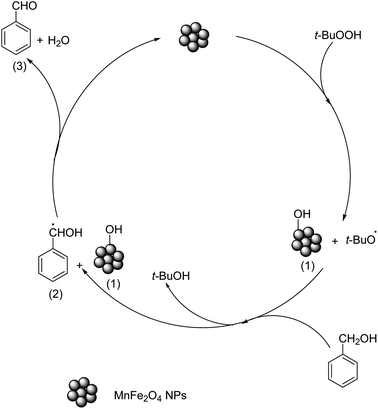 |
| | Scheme 1 Proposed mechanism for the MnFe2O4 NPs catalyzed oxidation of benzyl alcohol. | |
3.7.4 Comparison of catalytic activity with other reported catalysts. To examine the efficiency of the present protocol, the result of the current study is compared with other reported ferrite and non-ferrite-based catalyst systems (Table 5). The results indicate that our catalyst system has many advantages over other reported methods in terms of reaction time, yields, reaction temperature, and the use of toxic organic solvents.
Table 5 Comparison of catalytic activity of MnFe2O4 NPs with reported catalysts for the synthesis of benzaldehyde
| Entry |
Catalyst |
Oxidant |
Temperature (°C) |
Solvent |
Time (h) |
Yield (%) |
Selectivity (%) |
| 1 |
CuxFe3−xO4 (ref. 22) |
TBHP |
110 |
— |
5 |
>73 |
100 |
| 2 |
NiFe2O4 (ref. 23) |
TBHP |
60 |
CH3CN |
3 |
85 |
100 |
| 3 |
CoFe2O4 (ref. 24) |
H2O2 |
110 |
— |
5 |
>99 |
100 |
| 4 |
CuFe2O4 (ref. 26) |
O2/TEMPO |
100 |
Water |
24 |
95 |
— |
| 5 |
MnFe2O4 (ref. 28) |
H2O2 |
80 |
CH3CN |
10 |
87.6 |
100 |
| 6 |
CoCu62 |
TBHP |
r.t. |
CH3CN |
6 |
88 |
— |
| 7 |
Fe3O4/C/MnO2 (ref. 63) |
O2 |
100 |
Toluene |
4 |
78 |
100 |
| 8 |
NiFe2O4 nanorods (ref. 29) |
H2O2 |
110 |
Water |
6 |
86 |
77 |
| 9 |
PEG-NiFe2O4 NPs (ref. 64) |
H5IO6 |
r.t. |
Water |
1.5 |
98 |
99 |
| 10 |
La0.95Ce0.05MnO3 (ref. 65) |
O2 |
120 |
Toluene |
12 |
40 |
99 |
| 11 |
MnFe2O4 NPs (this work) |
TBHP |
r.t. |
— |
2 |
90 |
100 |
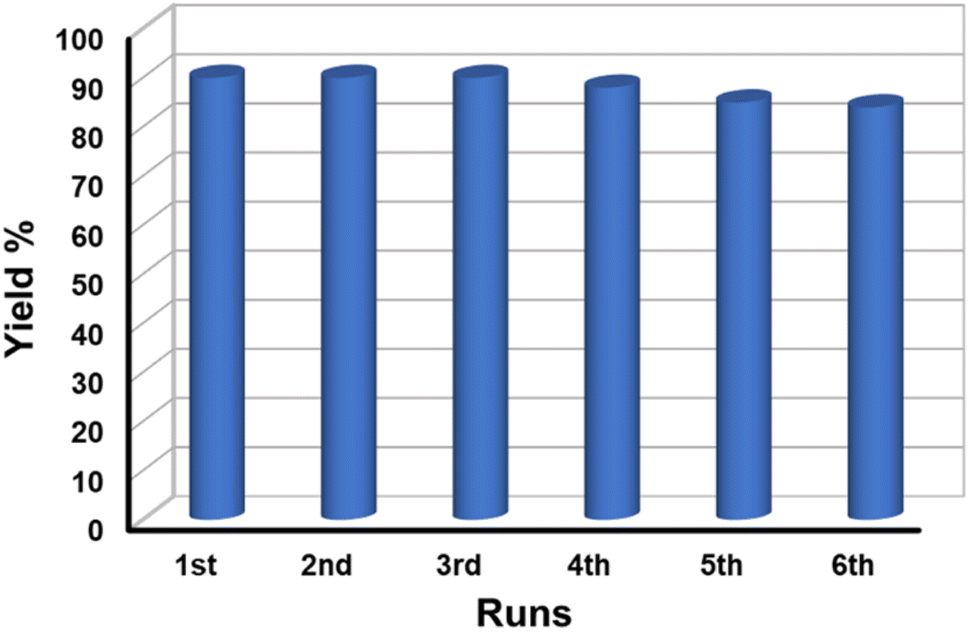
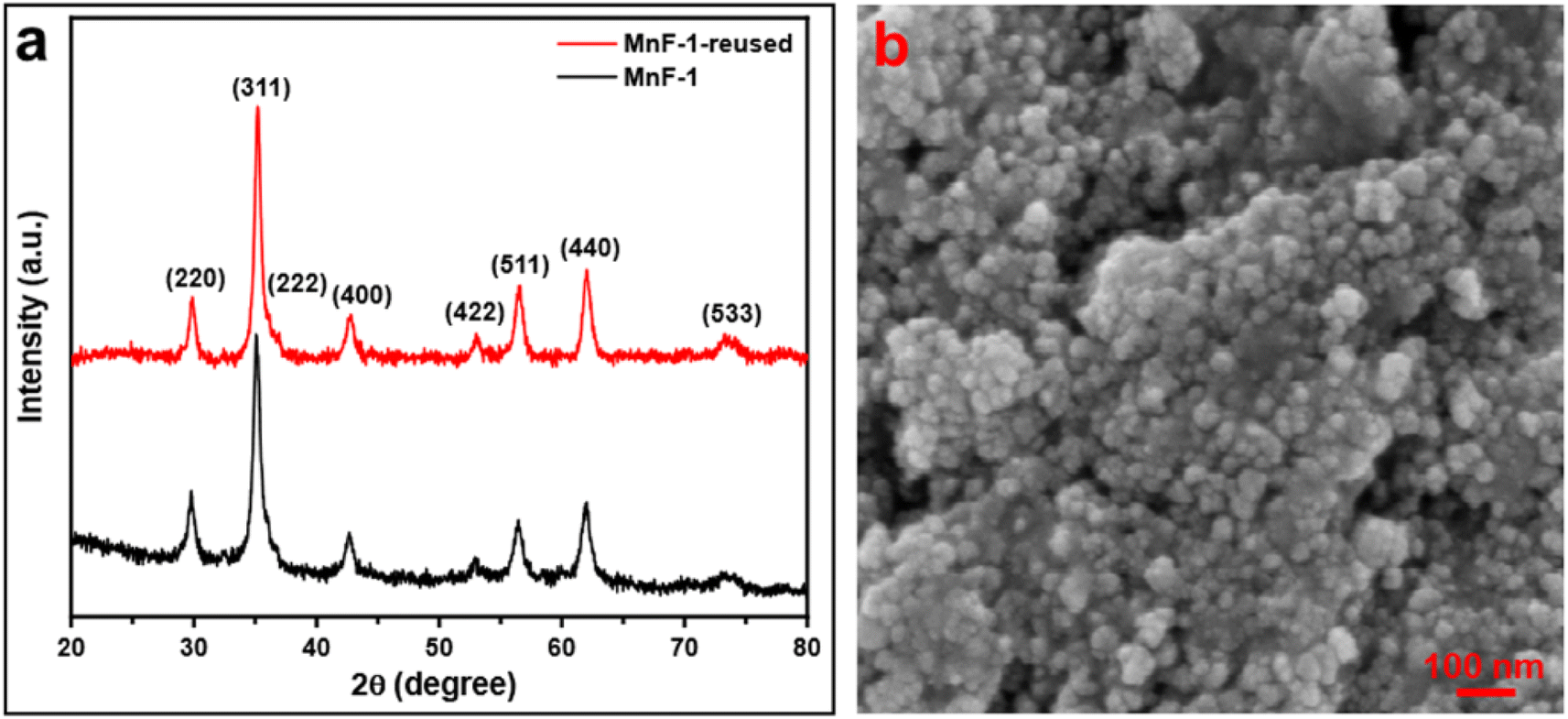
3.7.5 Reusability test of the catalyst. The reusability of a catalyst is vital for industrial applications. Therefore, to check the reusability of the catalyst, a series of tests were performed under similar reaction conditions. After the first cycle of the reaction, the catalyst was recovered conveniently from the reaction mixture by using an ordinary bar magnet. The isolated products were purified by washing with water and water–ethanol mixture to get rid of any organic components absorbed on the surface and the isolated products were dried in an oven at 60 °C under vacuum overnight. The recovered catalyst was used for another batch of reactions under identical reaction conditions. The process of isolation of the catalyst and their reusability was tested up to the 6th cycle. We observed that the catalyst could be reused up to the 6th cycle without significant loss of catalytic activity (Fig. 8). We noticed a gradual decrease in the catalytic activity after the 6th cycle which might be due to the adsorption of oxidized product on the surface of MnFe2O4 NPs resulting in the deactivation of the catalyst or physical loss of the catalyst during the purification process. Furthermore, we executed the elemental analysis of the residue collected after the reaction to explore if the reaction is heterogeneously catalyzed and the loss is due to any leaching of the metal during catalysis, (details are provided in ESI†). We recorded the presence of 0.02% Mn and 0.04% Fe in the residue which is an insignificant amount to contribute to the catalysis. This further signifies the stability, and effectiveness of the catalyst in the selective oxidation of benzyl alcohol to benzaldehyde. To check the stability of the catalyst, the recovered catalyst was examined by microscopy and XRD techniques. Fig. 9a represents the XRD pattern of sample MnF-1 before and after catalysis which shows that no change in the crystallinity of the catalyst. Moreover, the morphology of the MnF-1 catalyst remains intact after catalysis which is confirmed by the SEM study (Fig. 9b).
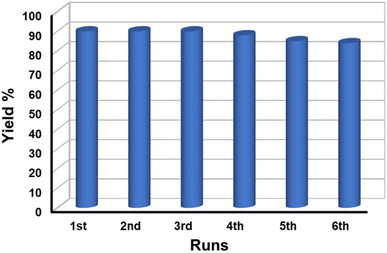 |
| | Fig. 8 Recyclability study of MnFe2O4 NPs for benzyl alcohol oxidation reaction. | |
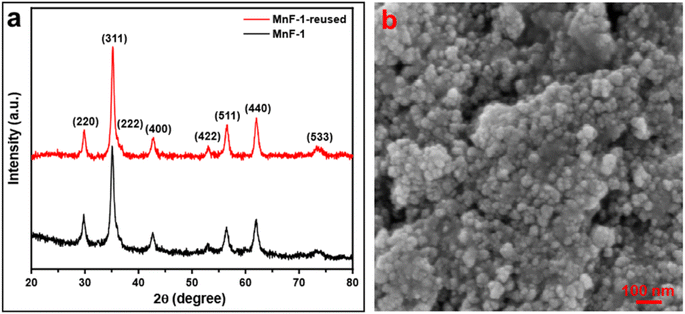 |
| | Fig. 9 (a) XRD pattern of sample MnF-1 before and after catalysis and (b) SEM image of sample MnF-1 after catalysis. | |
4. Conclusion
We have successfully synthesized MnFe2O4 NPs via the precipitation method followed by hydrothermal ageing both in the absence and presence of biodegradable polymers. The additives control the crystallite size as well as the morphology of the formed MnFe2O4 NPs. The as-prepared MnFe2O4 NPs are pure and crystalline exhibiting superparamagnetic behaviour. The MnFe2O4 NPs prepared in the presence of chitosan exhibit excellent catalytic activity in the solvent-free selective oxidation of benzyl alcohol to benzaldehyde at room temperature under ultrasonic irradiation with 100% selectivity. The catalysts are magnetically retrievable, stable and can be reused for up to 6th runs without a significant loss in the catalytic activity.
Data availability
All data generated or analysed during this study are included in this published article and its ESI files.†
Author contributions
All authors contributed to the study's conception and design. BK carried out the experiments, formal analysis, investigation, and writing of the original draft. SI carried out the initial experiments on catalyst synthesis. XB helped with the analysis of data and MHR supervised the research, provided conceptualization, and did the editing. All authors reviewed the manuscript.
Conflicts of interest
The authors have no relevant financial and non-financial interests to declare.
Acknowledgements
The authors acknowledge the Department of Science and Technology, Government of India for partial financial support under the DST-PURSE programme (SR/PURSE/2021/58). The authors also thank NEIST Jorhat, CNN-JMI Delhi, NECBH IIT Guwahati, SAIF-GU, Guwahati, MARC, Bengaluru and UGC-DAE CSR (Dr V. R. Reddy and Dr U. Deshpande of Indore Centre; Dr S. Hussain and Dr S. Chakravarty of Kalpakkam Node) for providing instrumental facilities used in this study. We are also thankful to Dr V. R. Reddy of UGC-DAE CSR, Indore Centre for his help with the analysis of Mössbauer spectroscopy data.
References
- J. A. B. Satrio and L. K. Doraiswamy, Chem. Eng. J., 2001, 82, 43–56 CrossRef CAS.
- D. Bankston, Synthesis, 2004, 2004, 283–289 CrossRef.
- S. W. Kshirsagar, N. R. Patil and S. D. Samant, Tetrahedron Lett., 2008, 49, 1160–1162 CrossRef CAS.
- G. D. Yadav and B. V. Haldavanekar, React. Funct. Polym., 1997, 32, 187–194 CrossRef CAS.
- H. V. Borgaonkar, S. R. Raverkar and S. B. Chandalia, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 455–458 CrossRef CAS.
- F. Wang, J. Xu, X. Li, J. Gao, L. Zhou and R. Ohnishi, Adv. Synth. Catal., 2005, 347, 1987–1992 CrossRef CAS.
- V. Hulea and E. Dumitriu, Appl. Catal., A, 2004, 277, 99–106 CrossRef CAS.
- J. Liu, Z. Wang, P. Jian and R. Jian, J. Colloid Interface Sci., 2018, 517, 144–154 CrossRef CAS PubMed.
- G. L. Zhuo and X. Z. Jiang, Catal. Lett., 2003, 87, 225–227 CrossRef CAS.
- V. R. Choudhary, P. A. Chaudhari and V. S. Narkhede, Catal. Commun., 2003, 4, 171–175 CrossRef CAS.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- M. N. Kopylovich, A. P. C. Ribeiro, E. C. B. A. Alegria, N. M. R. Martins, L. M. D. R. S. Martins and A. J. L. Pombeiro, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2015, ch. 3, vol. 63, pp. 91–174 Search PubMed.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- M. Amiri, K. Eskandari and M. Salavati-Niasari, Adv. Colloid Interface Sci., 2019, 271, 101982 CrossRef CAS PubMed.
- P. Losch, W. Huang, E. D. Goodman, C. J. Wrasman, A. Holm, A. R. Riscoe, J. A. Schwalbe and M. Cargnello, Nano Today, 2019, 24, 15–47 CrossRef CAS.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746–2762 RSC.
- H. Yan, M. Zhao, X. Feng, S. Zhao, X. Zhou, S. Li, M. Zha, F. Meng, X. Chen, Y. Liu, D. Chen, N. Yan and C. Yang, Angew. Chem., Int. Ed., 2022, 61, e202116059 CrossRef CAS PubMed.
- H. Goksu and F. Sen, Sci. Rep., 2020, 10, 5731 CrossRef CAS PubMed.
- Q. Han, X.-X. Guo, X.-T. Zhou and H.-B. Ji, Inorg. Chem. Commun., 2020, 112, 107544 CrossRef CAS.
- K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657–10666 CrossRef CAS PubMed.
- A. Arjunan, M. Manikandan, R. Vijayaraghavan and P. Sangeetha, ChemistrySelect, 2023, 8, e202202776 CrossRef CAS.
- S. Iraqui, S. S. Kashyap and M. H. Rashid, Nanoscale Adv., 2020, 2, 5790–5802 RSC.
- M. Nasrollahzadeh, M. Bagherzadeh and H. Karimi, J. Colloid Interface Sci., 2016, 465, 271–278 CrossRef CAS PubMed.
- B. Paul, D. D. Purkayastha and S. S. Dhar, Mater. Chem. Phys., 2016, 181, 99–105 CrossRef CAS.
- X. Zhu, D. Yang, W. Wei, M. Jiang, L. Li, X. Zhu, J. You and H. Wang, RSC Adv., 2014, 4, 64930–64935 RSC.
- J. Chen, W. Wen, L. Kong, S. Tian, F. Ding and Y. Xiong, Ind. Eng. Chem. Res., 2014, 53, 6297–6306 CrossRef CAS.
- A. M. Jacintha, A. Manikandan, K. Chinnaraj, S. A. Antony and P. Neeraja, J. Nanosci. Nanotechnol., 2015, 15, 9732–9740 CrossRef CAS PubMed.
- B. Iqbal, A. Laybourn, A. ul-Hamid and M. Zaheer, Ceram. Int., 2021, 47, 12433–12441 CrossRef CAS.
- N. Akhlaghi and G. Najafpour-Darzi, J. Ind. Eng. Chem., 2021, 103, 292–304 CrossRef CAS.
- M. M. Baig, M. A. Yousuf, P. O. Agboola, M. A. Khan, I. Shakir and M. F. Warsi, Ceram. Int., 2019, 45, 12682–12690 CrossRef CAS.
- D. Carta, M. F. Casula, P. Floris, A. Falqui, G. Mountjoy, A. Boni, C. Sangregorio and A. Corrias, Phys. Chem. Chem. Phys., 2010, 12, 5074–5083 RSC.
- J. Gao, F. Si, F. Wang, Y. Li, G. Wang, J. Zhao, Y. Ma, R. Yu, Y. Li, C. Jin and D. Li, Solid State Sci., 2023, 135, 107066 CrossRef CAS.
- R. Ghahremanzadeh, Z. Rashid, A. H. Zarnani and H. Naeimi, Appl. Catal., A, 2013, 467, 270–278 CrossRef CAS.
- S. R. Patade, D. D. Andhare, S. B. Somvanshi, S. A. Jadhav, M. V. Khedkar and K. M. Jadhav, Ceram. Int., 2020, 46, 25576–25583 CrossRef CAS.
- S. Tang, M. Zhao, D. Yuan, X. Li, X. Zhang, Z. Wang, T. Jiao and K. Wang, Sep. Purif. Technol., 2021, 255, 117690 CrossRef CAS.
- J. Wang, Y. Liu, W. Yin, Y. Cao, J. Hou, S. Wang and X. Wang, J. Mol. Liq., 2023, 371, 120845 CrossRef CAS.
- A. H. Cahyana, A. R. Liandi, Y. Yulizar, Y. Romdoni and T. P. Wendari, Ceram. Int., 2021, 47, 21373–21380 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- P. Iranmanesh, S. Saeednia, M. Mehran and S. R. Dafeh, J. Magn. Magn. Mater., 2017, 425, 31–36 CrossRef CAS.
- H. Fan, X. Ma, S. Zhou, J. Huang, Y. Liu and Y. Liu, Carbohydr. Polym., 2019, 213, 39–49 CrossRef CAS PubMed.
- A. Taavoni-Gilan, J. Chin. Chem. Soc., 2019, 66, 600–607 CrossRef CAS.
- A. Fujimoto, Y. Yamada, M. Koinuma and S. Sato, Anal. Chem., 2016, 88, 6110–6114 CrossRef CAS PubMed.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- H. W. Nesbitt and D. Banerjee, Am. Mineral., 1998, 83, 305–315 CrossRef CAS.
- A. Kumar, M. K. Gora, G. Lal, B. L. Choudhary, P. L. Meena, R. S. Dhaka, R. K. Singhal, S. Kumar and S. N. Dolia, Environ. Sci. Pollut. Res., 2023, 30, 18820–18842 CrossRef CAS PubMed.
- S. Iraqui, B. Kalita, R. Star, M. Gupta and M. H. Rashid, New J. Chem., 2023, 47, 10564–10575 RSC.
- G. K. Sarma, R. Sharma, R. Saikia, X. Borgohain, S. Iraqui, K. G. Bhattacharyya and M. H. Rashid, Environ. Sci. Pollut. Res., 2020, 27, 30067–30080 CrossRef CAS PubMed.
- M. Sumalatha, S. S. K. Reddy, M. S. Reddy, S. Sripada, M. M. Raja, C. G. Reddy, P. Y. Reddy and V. R. Reddy, J. Magn. Magn. Mater., 2021, 523, 167561 CrossRef CAS.
- Y. Hu, Y. Zou, H. Wu and D. Shi, Ultrason. Sonochem., 2012, 19, 264–269 CrossRef CAS PubMed.
- T. C. Saikia, X. Borgohain, S. Iraqui and M. H. Rashid, ACS Omega, 2022, 7, 42126–42137 CrossRef PubMed.
- T. C. Saikia, S. Iraqui and M. H. Rashid, Sustainable Chem. Pharm., 2022, 25, 100613 CrossRef CAS.
- M. I. D. Mardjan, M. F. Hariadi, I. M. Putri, N. A. Musyarrofah, M. Salimah, Priatmoko, B. Purwono and L. Commeiras, RSC Adv., 2022, 12, 19016–19021 RSC.
- S. Sadjadi and M. Eskandari, Ultrason. Sonochem., 2013, 20, 640–643 CrossRef CAS PubMed.
- D. B. Bankar, R. R. Hawaldar, S. S. Arbuj, M. H. Moulavi, S. T. Shinde, S. P. Takle, M. D. Shinde, D. P. Amalnerkar and K. G. Kanade, RSC Adv., 2019, 9, 32735–32743 RSC.
- M. E. Assal, M. R. Shaik, M. Kuniyil, M. Khan, A. Al-Warthan, M. R. H. Siddiqui, S. M. A. Khan, W. Tremel, M. N. Tahir and S. F. Adil, RSC Adv., 2017, 7, 55336–55349 RSC.
- Y. Chen, H. Lim, Q. Tang, Y. Gao, T. Sun, Q. Yan and Y. Yang, Appl. Catal., A, 2010, 380, 55–65 CrossRef CAS.
- E. Assady, B. Yadollahi, M. Riahi Farsani and M. Moghadam, Appl. Organomet. Chem., 2015, 29, 561–565 CrossRef CAS.
- T. M. Hafshejani, S. Saeednia, M. H. Ardakani and Z. P. Parizi, Transition Met. Chem., 2018, 43, 579–589 CrossRef.
- V. Mahdavi and M. Mardani, Res. Chem. Intermed., 2015, 41, 8907–8927 CrossRef CAS.
- A. B. Gawade, A. V. Nakhate and G. D. Yadav, Catal. Today, 2018, 309, 119–125 CrossRef CAS.
- B. J. Borah, A. Mahanta, M. Mondal, H. Gogoi, Y. Yamada and P. Bharali, ChemistrySelect, 2018, 3, 9826–9832 CrossRef CAS.
- E. Prathibha, R. Rangasamy, A. Sridhar and K. Lakshmi, ChemistrySelect, 2020, 5, 988–993 CrossRef CAS.
- B. Paul, D. D. Purkayastha and S. S. Dhar, Appl. Surf. Sci., 2016, 370, 469–475 CrossRef CAS.
- A. A. Ansari, N. Ahmad, M. Alam, S. F. Adil, S. M. Ramay, A. Albadri, A. Ahmad, A. M. Al-Enizi, B. F. Alrayes, M. E. Assal and A. A. Alwarthan, Sci. Rep., 2019, 9, 7747 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization techniques used, Table S1, Fig. S1 (EDX spectra), Fig. S2 (XPS survey scan spectra), Fig. S3 (XPS spectra of reused catalyst), and relevant 1H and 13C NMR spectra (Fig. S4–S19) of different products. See DOI: https://doi.org/10.1039/d3ra03797e |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Saddam Iraqui
,
Saddam Iraqui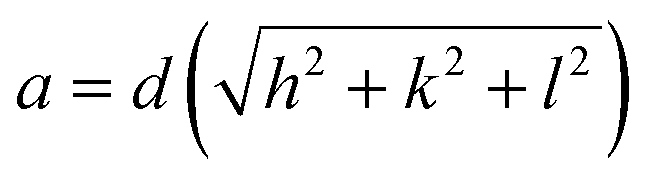 and the average value for different samples was estimated to be 8.47 Å, which is close to the theoretical value of 8.499 Å.
and the average value for different samples was estimated to be 8.47 Å, which is close to the theoretical value of 8.499 Å.